

Abstract
Reversing HIV-1 latency allows killing of infected cells in cure strategies, but no single latency reversing agent (LRA) or LRA combination have been shown to effectively reduce HIV-1 latent reservoir size in persons living with HIV-1 (PLWH). Here we describe an approach to systematically identify synergistic LRA combinations to reactivate latent HIV-1 using novel genome-wide CRISPR screens. Screens on cells treated with suboptimal concentrations of an LRA can identify host genes whose knockout enhances viral gene expression. Therefore, inhibitors of these genes should synergize with the LRA. We tested this approach using AZD5582, an activator of the non-canonical NF-kappa B (ncNF-κB) pathway, as an LRA and identified HDAC2, a histone deacetylation complex, and BRD2, part of the Bromodomain and Extra-Terminal motif (BET) protein family targeted by BET inhibitors. Using CD4+ T cells from PLWH, we confirmed synergy between AZD5582 and several HDAC inhibitors and between AZD5582 and the BET inhibitor JQ1. Remarkably, a reciprocal screen using suboptimal concentrations of an HDAC inhibitor as an LRA identified BRD2 and ncNF-κB regulators, especially BIRC2, as synergistic candidates for use in combination with HDAC inhibition. Moreover, we identified and validated several novel synergistic drug candidates in latency cell line cells and primary lymphocytes isolated from PLWH. Specifically, the knockout of genes CYLD or YPEL5 displayed synergy with the existing LRAs in inducing HIV mRNAs. Our studies provide novel insights into the roles of host factors in HIV-1 reactivation and validate a system for finding drug combinations for HIV-1 latency reversal.
One sentence summary:
We present the first comprehensive genome-wide screen for synergistic combinations of drugs targeting the latent HIV-1 reservoir to produce a cure.
Introduction
human immunodeficiency virus type 1 (HIV-1) persisting in a latent form in resting CD4+ T cells despite effective antiretroviral therapy (ART) is the major barrier to cure (1–4). A promising therapeutic approach known as “shock and kill” seeks to achieve cure by sequentially reversing latency in infected cells and then killing the productively infected cells (5–13). Latency reversing agents (LRAs) that act through a variety of proposed mechanisms have been identified using in vitro models of HIV-1 latency (9, 14–20). To date, however, no single LRA (or LRA combination) has been shown to reduce latent reservoir size in PLWH (9, 17, 21), and combinations of LRAs will likely be required to overcome multiple blocks to HIV-1 gene expression in resting CD4+ T cells (11).
Different genetic and epigenetic factors block the transcription of latent HIV-1 proviruses at several different stages, making it difficult to efficiently reactivate HIV-1 transcription with a single drug (reviewed in (4, 10, 11, 13, 22)). In resting CD4+ T cells, there are low level of active nuclear forms of essential host transcription initiation factors, including NF-kB and NFAT (23–27). Even with sufficient transcription initiation factors, transcription can be blocked at the trans-activation response (TAR) region by pausing of RNA polymerase II due to the lack of positive transcription elongation factor (P-TEFb), a complex of cyclin T1 and CDK9. The reactivation of latent HIV-1 may be limited by the availability of P-TEFb, which is present only at low level in resting CD4+ T cells for reasons that are not completely clear (28, 29). In addition to the availability of essential initiation and elongation factors, epigenetic modifications may create another barrier to successful reactivation of latent HIV-1 proviruses, including increased DNA methylation and the deposition of repressive histone marks, leading to reduced accessibility to the transcription machinery (22, 30–32). Epigenetic drugs can reverse HIV-1 latency by removing reversible restrictions to the chromatinized provirus (9, 10, 31, 33, 34).
In ex vivo studies with CD4+ T cells from treated individuals, the most effective LRAs are protein kinase C (PKC) agonists such as bryostatin, prostratin, and phorbol myristate acetate (PMA) (17). These compounds induce HIV-1 gene expression through activating the NF-κB pathway (17, 35). However, their toxicity is a major barrier to clinical use. Administration of histone deacetylase (HDAC) inhibitors (HDACi) disrupts latency in PLWH on ART, although these drugs have not yet been shown to reduce the HIV-1 reservoir (9, 21, 33, 34). Recent studies (19, 20) have demonstrated impressive latency reversal by agents that activate the noncanonical NF-κB (ncNF-κB) pathway, which is limited to fewer cellular processes and cell types (6, 14, 36). A repressor of the ncNF-κB pathway, the cellular inhibitor of apoptosis protein 1 (cIAP1), was identified as a therapeutic target for HIV-1 latency reversal by RNAi screening (18). Mimetics of the second mitochondrial-derived activator of caspases (SMAC) inhibit cIAP1 and thereby activate the ncNF-κB pathway (and latent HIV-1) (18). Compared to PKC agonists, SMAC mimetics have limited toxicity and cause more targeted cellular activation (18, 19) in cell lines and in animal models (19, 20). Furthermore, SMAC mimetics induce autophagy-dependent apoptosis of HIV-1-infected resting CD4+ T cells (37). Despite these promising developments, it remains unclear whether any single LRA (or LRA combination) can effectively drive the elimination of the latent reservoir (38).
Pharmacologic synergy among different LRAs could not only allow use of lower dosage of each LRA, but also induce greater HIV-1 expression due to effects on multiple pathways. Some attempts at combining existing LRAs have been described (39, 40), but the development of new and improved LRAs (or LRA combinations) requires a comprehensive understanding of the molecular control of HIV-1 latency. Functional genetic screening, especially CRISPR/Cas9-based screening, provides a global unbiased approach to understand molecular aspects of HIV-1 infection including HIV-1 entry and replication (41–45), interferon type 1-mediated inhibition of HIV-1 replication (46), and novel HIV-1 latency promoting genes (47–49). Although these advances support the application of genome-wide screens in discovering novel LRAs, few studies focused on systematically identifying LRA combinations that overcome the limitations of individual LRAs.
Here we describe the construction of a novel polyclonal model of HIV-1 latency and its use in genome-wide CRISPR screens to identify synergistic combinations of LRAs. We first identified factors that are required for latent HIV-1 expression in T cells under various stimuli. Next, we performed screens using suboptimal doses of a known LRA. Our approach was based on the concept that host genes whose knockout enhances latency reversal in these screens are potential targets for synergistic drugs. We validated this concept in reciprocal screens, and identified LRA combinations as well as novel host factors whose knockout synergistically activates viral gene expression in model systems and in primary cells from PLWH.
Results
Genome-wide CRISPR screens on a HIV-1 latency cell line model
Latently infected cells are present in vivo at very low frequencies (1/106 cells as detected by the quantitative viral outgrowth assay) (50), and therefore screening and mechanistic studies cannot be readily performed with cells from PLWH. Instead, cell line models satisfy the requirements of high transfection efficiency and large cell number needed for the genome-wide analysis of factors involved in latency maintenance or reversal. The existing Jurkat-derived latency model J-lat is widely used (51), but the clonal nature of these cells makes them less suitable to address the common determinants that govern proviral reactivation regardless of specific integration sites. To overcome this limitation, we establish a polyclonal in vitro model for HIV-1 latency (Fig. 1A). Briefly, we infected Jurkat cells with a VSV-G pseudotyped, replication-defective HIV-1 reporter virus carrying a sequence for a destabilized form of GFP in place of the nef gene (52). This virus contains Rev and a previously described, partially attenuated Tat mutants (52). With this virus, we were able to generate a latency model in which the majority of cells could be induced to express viral genes upon stimulation. Sorted GFP+ cells were then transduced with Cas9 and cultured under selection to yield a population of cells carrying an HIV-1 provirus and the Cas9 gene. After a series of carefully designed stimulation, sorting, and incubation steps (Fig. 1A), we obtained a population of latently infected (GFP−), Cas9-expressing cells to serve as a polyclonal latency model. HIV-1 gene expression can be re-induced in >90% of this population by TNF-α as evidenced by GFP expression (Fig. 1B).
Figure 1.
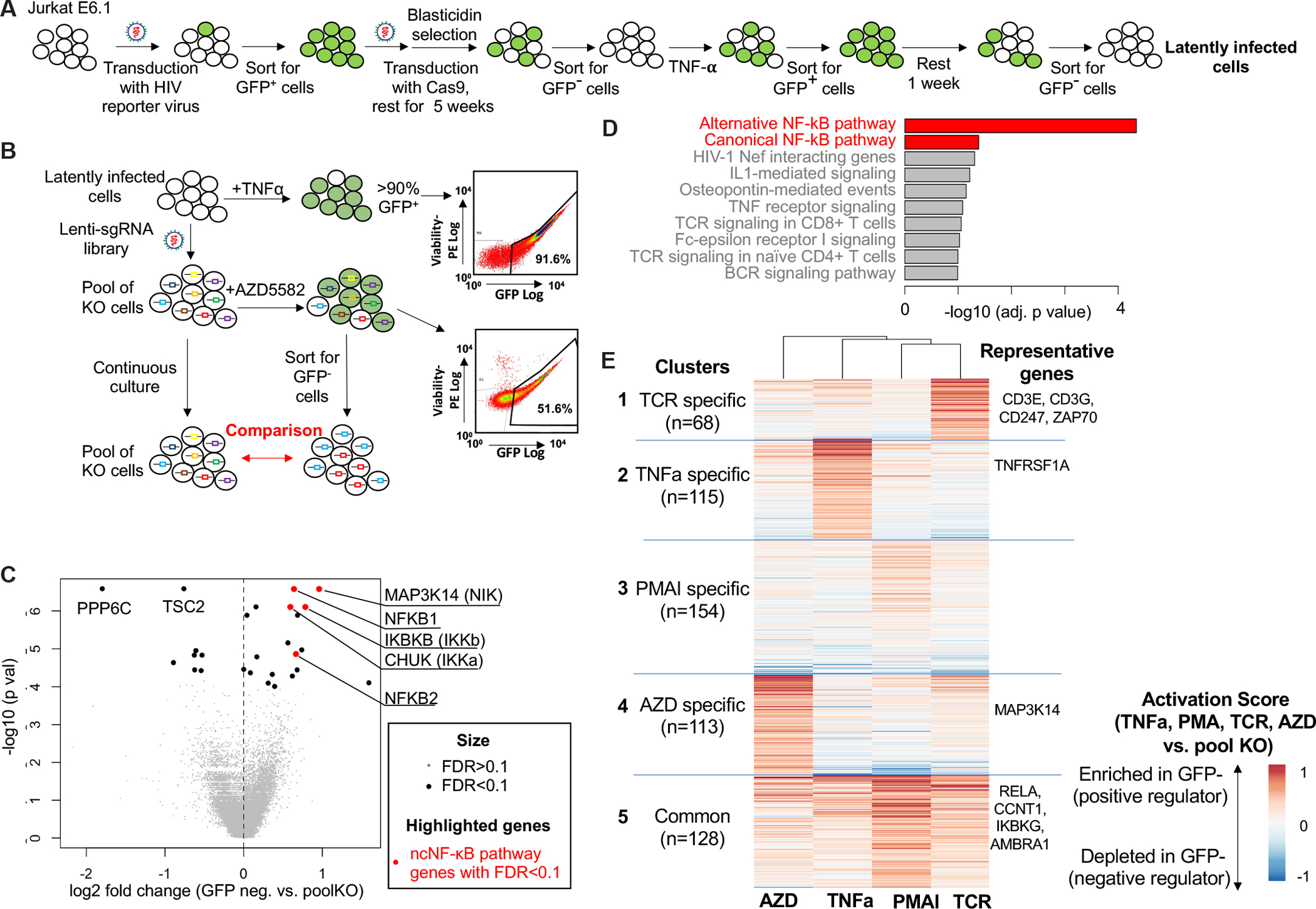
Overview of the HIV-1 latency model and the CRISPR screening for genes involved in latency reversal by SMAC mimetics. A, The construction of Jurkat HIV-1 latency model. The indicated series of transduction, incubation, stimulation, and sorting steps give rise to population of Cas9+ cells carrying a latent HIV-1 provirus. B, Screening strategy. Latently infected cells were transduced with a CRISPR sgRNA screening library, stimulated with the SMAC mimetic AZD5582 to induce GFP expression, and sorted for GFP-negative cells. Continuously cultured cells (pooled KO cells) were used as controls. C, Top hits in the GFP-negative population vs. pooled knockout population. MAGeCK software was used to calculate the log fold changes and associated p values for each gene. D, Pathways significantly enriched in the GFP− population. The adjusted p values of the top enriched pathways are shown. The enrichment analysis was performed using Enrichr (89). E, A unsupervised clustering of statistically significant genes (FDR<0.1) in any of the four stimulations (AZD,TNFa, PMAI, TCR). The activation score of each gene is defined as the normalized beta score in each stimulus vs. control. The beta score is estimated from the MAGeCK-VISPR software (85).
We then used this in vitro HIV-1 latency model to perform genome-wide CRISPR screens. Recent studies suggested that SMAC mimetics effectively reverse SIV latency via ncNF-κB signaling in vivo (18–20). However, there was a weaker effect ex vivo on CD4+ T cells from HIV-1-infected donors on ART (19). To investigate host factors that enable the induction of HIV-1 gene expression by the SMAC mimetic AZD5582, we conducted CRISPR screens in our latency model using AZD5582 as an LRA (Fig. 1B). The latently infected Jurkat-Cas9 cells were transduced with a genome-wide CRISPR single-guide RNA (sgRNA) library to generate a pool of knockout cells each expressing a different sgRNA (53). After treatment with AZD5582, which activates the ncNF-κB pathway, GFP− cells were isolated by cell sorting (Fig. 1). Pooled cells that were continuously cultured without AZD5582 stimulation were used as controls. The abundance of each sgRNA was then estimated using high-throughput sequencing, and the MAGeCK algorithm (54) was used for data analysis and statistical evaluation (see Methods).
To identify genes required for the reversal of HIV-1 latency by AZD5582, we first compared sgRNAs that were enriched in GFP− cells vs control cells (Fig. 1B). Genes identified in this manner are potential positive regulators of HIV-1 gene expression or latency reversal by AZD5582, as cells with a knockout of these genes fail to turn on GFP expression following stimulation. Using false discovery rate (FDR) of 10% as cutoff, we identified 19 genes with statistically significant enrichment in the GFP− cells (Fig. 1C, Data File S2). Genes that encode critical components in the ncNF-κB pathway, including MAP3K14 (encoding NIK), CHUK (encoding IKK-α), and NFKB2 (encoding p52), were among top hits, consistent with the expected major role of the ncNF-κB pathway in the response to AZD5582. In addition, several genes in canonical NF-κB pathway are also selected, including NFKB1 (encoding p105) and IKBKB (encoding IKK-β). A functional annotation of these genes identified major pathways involved in the induction of HIV-1 gene expression, including NF-κB pathways (canonical and alternative), HIV-1 Nef interacting genes, and TNF receptor signaling pathway. (Fig. 1D).
We also investigated negative regulators, or genes whose knockout led to increased GFP expression and hence the depletion of associated sgRNAs in GFP− cells. Two genes (TSC2 and PPP6C) were identified as negative regulators with statistical significance (FDR<0.01; Fig. 1C; Data File S2). TSC2 (TSC complex subunit 2) and PPP6C (Protein Phosphatase 6 Catalytic Subunit) are negative regulators of mTOR signaling and NF-κB signaling pathways (55), respectively. Both pathways are reported to be critical in latent HIV-1 induction in vitro (56), albeit the mTOR inhibitor rapamycin does not block reactivation of latent HIV-1 ex vivo. Collectively, these results demonstrated the feasibility of genome-wide CRISPR screens to identify critical factors for HIV-1 latency reversal.
Host factors required for HIV latency reversal across different stimulations
The genes identified in the screen described above could include those essential for latency reversal by any stimulus, as well as those that are unique to stimulation by SMAC mimetics. To investigate the common genes and pathways that are critical for HIV latency reversal across different stimulations, we performed similar genome-wide CRISPR screens using three other stimuli: TNF-α, PMA/I and TCR crosslinking. These stimuli are commonly used for T cell activation and induction of HIV transcription in various latency models (15, 17). An integration of these screens over four stimuli revealed 578 genes that are required in at least 1 stimulation condition (FDR<10%; Fig. 1E; Data File S3; see Methods). Unsupervised clustering of these 578 genes across different stimuli revealed five clusters of genes with similar functions (Fig. 1E). Clusters 1–4 contain genes that are specifically required for each stimulation (TNF-alpha, TCR, AZD5582, PMA/I), respectively. Cluster 5 contains genes required for all modes of stimulation. This analysis provided strong confirmation for the specificity of the screening protocol in that genes known to be necessary for signal transduction for given stimulus showed a high activation score in that condition. For example, CD3E/G, CD247 (CD3ζ) and ZAP70 are integral components of TCR signaling and showed a high activation score under the TCR condition (cluster 1). Similarly, TNFRSF1A, the receptor for TNF-α, had a high activation score only in TNF-α stimulated sample (cluster 2). Most importantly, a cluster of 128 genes (cluster 5) were required for all stimulations. This cluster included genes that are known to be involved in HIV transcription such as CCNT1 (Cyclin T1), a major subunit of positive transcription elongation factor b (p-TEFb) which is critical for HIV-1 transcription (57–60); RELA (p65), and IKBKG (Inhibitor Of Nuclear Factor Kappa B Kinase Regulatory Subunit Gamma) (61).
Functional analysis of these clusters revealed known pathways involved in each stimulation (fig. S1A–D): genes in cluster 1 (TCR specific cluster) are enriched in TCR signaling, TNFR1 signaling genes are enriched in cluster 2 (TNF-alpha specific cluster), and alternative NFκB pathway genes are enriched in cluster 4 (AZD5582 specific cluster). In cluster 5, which includes genes involved in latency reversal by all stimuli, there was enrichment in genes involved in canonical NFκB, T cell receptor and TNF signaling pathways. To further investigate the interactions of these regulators, we constructed a protein-protein interaction (PPI) network of these genes (and their interacting genes), using the STRING protein interactome database (62) (fig. S1E). This approach may also recover important genes that are missed in the screens (i.e., false negatives), as interacting genes are likely to function together. The PPI network identified several pathways and protein complexes that have strong interactions with each other, including the NFκB pathway (NFKB1, RELA, IKBKB, MAP3K7, etc.), the mediator complex (MED6/7/18/27/31) that is critical for the transcription initiation of HIV genes (63), and members of ribosomal subunit (RPL7/8/19) whose interaction with multiple HIV proteins (e.g., Nef, Gag) is critical for HIV-1 mRNA translation (64, 65).
A novel strategy to search for synergistic drug combinations using CRISPR screening
Given that HIV-1 latency may be influenced by multiple positive and negative regulatory factors, it is likely that no single LRA will fully induce latent HIV-1 proviruses. To identify candidate drug targets that potentially synergize with existing LRAs, we performed genome-wide CRISPR knockouts followed by induction of HIV-1 gene expression in latently infected cells using a suboptimal dose of a selected LRA, and then sorted for GFP+ cells directly (Fig. 2A). A gene whose knockout leads to enhanced GFP expression may emerge as a key negative regulator and the candidate drug target for synergy with the stimulating LRA if an inhibitor of the gene’s function exists.
Figure 2.

Searching synergistic drug combinations with SMAC mimetics or SAHA using CRISPR screening. A, Screening strategy using suboptimal AZD5582 dosage (5 nM). B, Top hits in GFP+ population vs. pooled knockout population. The MAGeCK software is used to calculate the log fold changes and associated p values of each gene. C-D, The significantly enriched pathways associated with depleted (C) or enriched (D) genes in the screens, respectively. Previously reported HIV-1 associated pathways are highlighted in red. The adjusted p values of the top enriched pathways are shown. The enrichment analysis is performed using Enrichr. E, Screening strategy using suboptimal SAHA dosage (0.1 μM). F, Top hits in GFP positive population vs. pooled knockout population. The MAGeCK software is used to calculate the log fold changes and associated p values of each gene.
We first tested this strategy by screening for genes whose knockout enhanced GPF expression in response to the SMAC mimetic AZD5582. We performed genome-wide CRISPR screens on cells stimulated with AZD5582 at a suboptimal concentration (5nM; Fig. 2A; fig. S2). Latently infected, Cas9 expressing cells cultured without sorting (“pooled KO”) were used as negative controls, similar to the GFP− screens (Fig. 1A). The screen identified 175 genes whose knockout led to the accumulation of GFP+ cells with statistical significance (FDR<0.01, Data File S4). Negative regulators of NF-κB pathways, including NFKBIA (encoding IκBα) and CYLD (66) were enriched in GFP+ cells (Fig. 2B). On the other hand, knocking out core members of NF-κB pathway (including CHUK and IKBKB) led to the depletion of the associated sgRNAs in GFP+ cells (Fig. 2B). These results are consistent with the role of the NF-κB pathway in response to AZD5582. Interestingly, knocking out proteasome subunits also greatly activated latent HIV-1 expression. Proteasome inhibitors are potential LRAs since proteasome inhibition reverses latency by promoting Tat-transactivation in cell line models (67). On the other hand, proteasome would promote HIV gene expression by degrading IκB (68). Proteosome inhibitors may also reverse latency through heat-shock stress related pathways in vitro (69). Functional analysis of genes in the screen revealed the enrichment of MYC signaling and the expected depletion of non-canonical NF-κB signaling genes in sorted GFP+ cells (Fig. 2C–D).
To further explore the possibility of drug synergy between HDAC inhibitors and suppression of host factors, we performed an independent genome-wide CRISPR screen using a suboptimal concentration (0.1 μM) of the HDAC inhibitor SAHA as an LRA (Fig. 2E). SAHA was previously shown to have effects on HIV-1 latency reversal in vitro and in vivo (9, 31). This screen identified 90 negative regulatory genes whose knockout led to statistically significant (FDR<0.1) accumulation of associated sgRNAs in GFP+ cells (Fig. 2F, Data File S5). There were an additional 22 positive regulators whose knockout led to statistically significant (FDR<0.1) depletion of associated sgRNAs in GFP+ cells. Among the negative regulators were inhibitory genes in the NF-κB signaling pathway (Fig. 2F), including NFKBIA (IκB), a member of the NF-κB inhibitor family. This was one of the strongest hits for accumulation in GFP+ cells. Other canonical and non-canonical NF-κB regulators, including CYLD, TRAF2/3, BIRC2 (encoding cIAP-1), were among the top hits. Functional analysis of genes in the screen revealed the expected depletion of HDAC signaling and enrichment of non-canonical NF-κB signaling genes in sorted GFP+ latent cells, respectively (fig. S3). Collectively, SAHA stimulated CRISPR screens established a potential synergy between the inhibition of HDAC enzyme, an epigenetic regulator, and the activation of NFKB, a transcription factor, in reversing HIV latency.
Identification of combinations of repurposed drugs for HIV latency reversal
To systematically prioritize potentially synergistic drug targets, we scored all genes with potential synergy with AZD5582 by considering two criteria. First, as an indication of minimal on-target toxicity, their knockout should have little effect on cell viability in control cell population; and second, their knockout should strongly induce GFP expression upon suboptimal AZD5582 stimulation. We used a rank product algorithm to aggregate the ranks of all genes from both criteria (Fig. 3A; table S1 and Data File S6; also see Methods), and identified 98 genes with statistical significance (FDR<0.1). Next, we categorize genes in four groups according to their mRNA expression and their “druggability” (70) (Fig. 3B). Druggability was based on targeting by known drugs in a drug-gene interaction database (70). Among all the significant hits, we focus on 13 (13%) Group 1 genes (“druggable” genes (70) with high expression in CD4+ T cells), as these genes directly inform potential LRA candidates that would work synergistically with AZD5582. The top genes in this group include HDAC2 (encoding histone deacetylase 2, ranked 7th), a target of several HDAC inhibitors including vorinostat (SAHA), romidepsin, and panobinostat. FKBP3 (encoding FKBP Prolyl Isomerase 3 or FKBP-25), a member of the immunophilin protein family, ranked 1st among all the druggable genes (Fig. 3B). Interestingly, FKBP3 modulates the signaling events of HDAC class 1 by directly interacting with HDAC1 and HDAC2 (71), and FKBP3 knockdown decreases HDAC2 expression (72). All these genes have little or no effect on on-target toxicity (measured in pooled KO cells; x axis in Fig. 3C) while inducing GFP expression upon perturbation (measured in GFP sorted cells; y axis in Fig. 3C). These results, based on an unbiased genome wide screen, imply that HDAC complexes, especially HDAC2, serve as a potentially synergistic drug targets for use in association with ncNF-κB activators like AZD5582. Compared with other HDAC protein perturbations that decrease cell viability (e.g., HDAC1, HDAC3), HDAC2 perturbation has minimal effect on cell viability (Fig. 3C; fig. S4).
Figure 3.

Identification of top hits in the CRISPR screens searching for synergistic drug combinations with existing LRAs. A, Procedures of ranking synergistic drug targets using CRISPR screening and gene expression data. B, The scores of genes in AZD5582 screen, generated by the rank product algorithm, that simultaneously measure the effect on inducing GFP expression and toxicity. A gene with smaller score is more likely to satisfy both conditions. The top scoring genes with FDR values <0.1 are divided into four tractability groups depending on their druggability and expression in CD4 cells. See table S1 and Data File S6 for more details. C, The beta scores of all genes generated from MAGeCK in comparing the behavior of gene knockout in control (pooled knockout, x axis) and AZD5582 treated cells (y axis). In control cells (x axis), genes with a beta score close to zero can be knocked out with minimum effect on viability, while in AZD5582-treated cells (y axis), a positive beta score indicates a stronger enrichment in GFP+ cells. Top genes in the group 1 are highlighted in red. D, The scores of all genes in SAHA screens, similar with the scores in B. See Table S2 and Data File S7 for more details. E, The enrichment (or depletion) scores of genes, comparing SAHA treated cells vs. pooled knockout cells, in ncNF-κΒ pathway. Colors indicate the differences of beta scores in two conditions (SAHA and pooled knockout). F, The beta scores of all genes in comparing the behavior of gene knockout in control (pooled knockout cells, x axis) and SAHA treated (y axis) cells, similar with the AZD5582 screens in C. G, the diagram that summarized the reciprocal findings in the CRISPR screens.
We performed similar analysis for SAHA-stimulated CRISPR screens (Fig. 3D; Data File S7). Among the 68 synergistic drug targets identified from the rank product algorithm with statistical significance (FDR<0.1), 14 (or 20.1%) belong to Group 1 (“druggable” genes that are expressed in CD4+ T cells; Table S2 and Data File S7). BIRC2, a regulator of non-canonical NF-κB pathway, is ranked 2nd among all druggable genes (Fig. 3D), consistent with the screening analysis (Fig. 2F). These data indicate negative regulators of ncNF-kb pathway, especially BIRC2, are potential synergistic drug targets with HDAC inhibitors like SAHA (Fig. 3E). These genes demonstrated a strong induction of GFP expression while maintaining minimal on-target toxicity (Fig. 3F). Other interesting druggable genes include TNFAIP3 (TNF Alpha Induced Protein 3, or A20), a negative regulator of NF-κB signaling pathway (73). Collectively, two independent CRISPR screens revealed reciprocal findings on the synergistic effects of ncNF-κΒ activation and HDAC inhibition (Fig. 3G). Interestingly, both screens identified BRD2 (bromodomain containing 2) as one the top candidates (Fig. 3B, 3D). BRD2 is one of the targets of bromodomain inhibitors that serve as potential LRA drugs (16), suggesting bromodomain inhibitors may synergize with SAHA and with AZD5582 in inducing the latent HIV-1 reservoir (74).
In vitro validations in cell lines confirmed findings from CRISPR screens
We next validated the predictions of gene knockout from two CRISPR screens that had potential synergy with AZD5582 or HDAC inhibitor in our Jurkat cell line model. We selected 13 genes, including genes whose knockout leads to an enrichment of GFP+ cells at both of these screens as positive controls (e.g., the negative regulators NELFCD, NFKBIA), novel targets that appear at least one of the screens, or genes that ranked top in our target analysis (e.g., HDAC2; Table S3). We first designed three guides that target each of these genes (sequences of sgRNAs listed in Data File S8), electroporated ribonucleoprotein (RNP) complexes formed from these guide RNAs and Cas9 protein into latently infected cells, and measured the percentage of GFP+ cells after 72 hours. Knockout efficiencies were evaluated using flow cytometry (fig. S5). We observed strong reductions in protein expression for these gene knockouts except for PHF5A, TRRAP and PRMT1. Therefore, these three genes were excluded from further examination. For the remaining ten genes with high knockout efficiencies, synergy was evaluated using Bliss independence model, which quantitatively estimates the synergy of drug and gene knockout assuming independent mechanisms of action (Fig. 4A and fig. S6; see Methods for more details). An interaction between a drug and a knockout was considered synergistic if the median synergy score (Δfaxy) of three replicates was greater than zero for at least one drug dose. All the hits were experimentally confirmed to be synergistic with at least one the drugs (Fig. 4A; a summary is provided in Table S3). For example, top hits in both screens, including NFKBIA, NELFCD, CYLD, and YPEL5, demonstrated synergistic effects with multiple concentrations of AZD5582 and SAHA (Fig. 4B–G). PPP6C, a target whose knockout synergizes with AZD5582 only, demonstrated a much stronger synergy in AZD5582 than in SAHA. Collectively, the high validation rate in the Jurkat cell line model confirmed the ability of the CRISPR screening strategy to identify synergistic combinations of drug targets.
Figure 4.
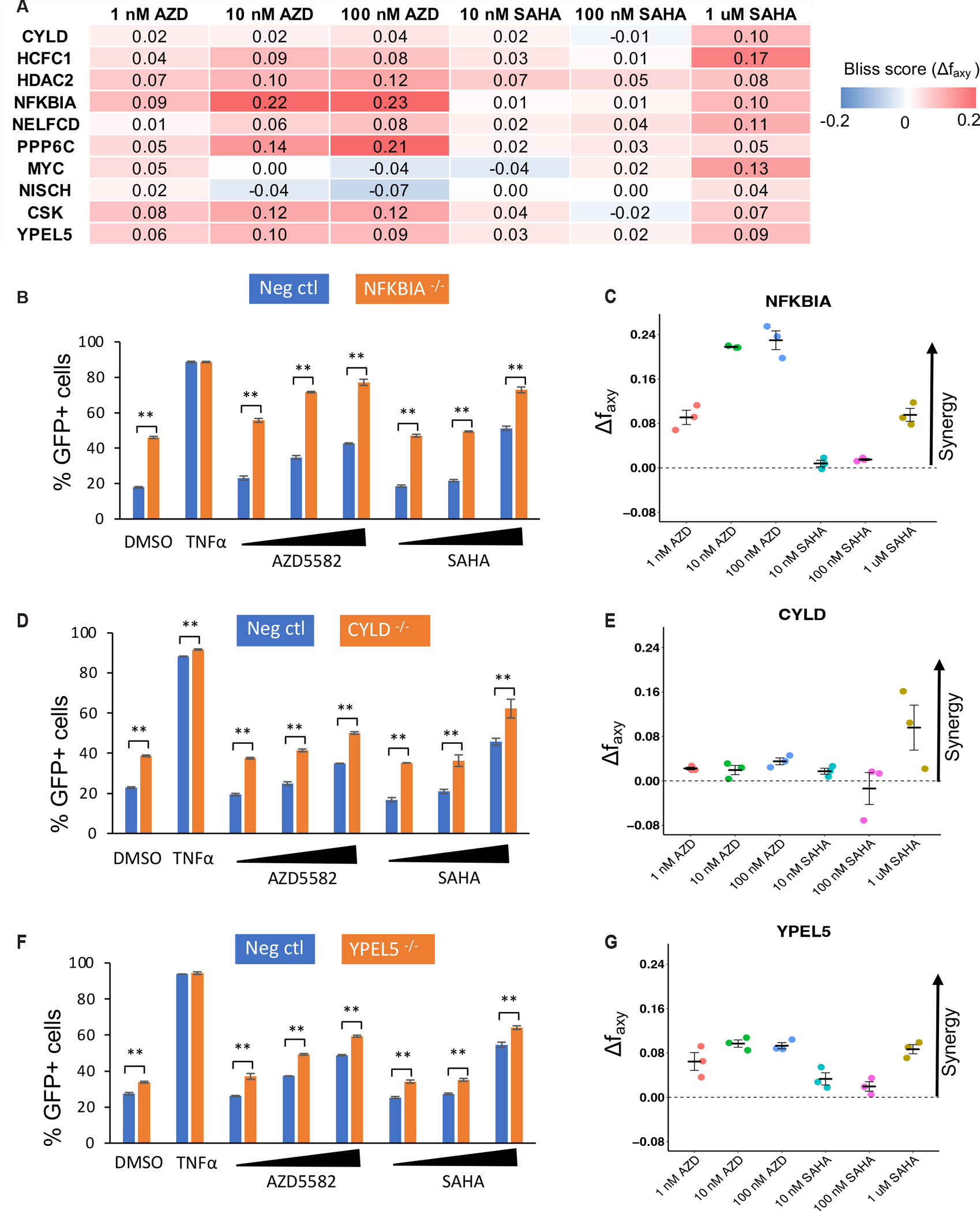
Validation of top hits in the CRISPR screens for synergistic interactions with drugs used in the screen. The fraction of GFP+ cells after knockout and drug treatment was assessed using the Jurkat latency model as described in the Methods. A, The average Bliss synergy score for all gene knockouts (rows) and drugs (columns). A score greater than zero (red color) indicates synergy between gene knockout and drug treatment. B, D and F, the percentages of GFP+ cells in the latency model cells following LRA treatments were compared in cells knocked out for the indicated gene and negative control cells. C, E, and G, the calculated synergy score for NFKBIA (C), CYLD (E) and YPEL5 (G). The concentrations of AZD5582 from low to high were 1 nM, 10 nM and 100 nM. The concentrations of SAHA from low to high were 10 nM, 100 nM, and 1 μM. P values were calculated using Student T test. n = 3. (*P < 0.05, **P < 0.01)
We next investigated whether combinations of AZD5582, SAHA and JQ1, predicted from our druggable gene analysis, would reverse HIV-1 latency. In the Jurkat cells used for CRISPR screens, the combination of suboptimal concentrations of SAHA and AZD5582 resulted in a higher percentage of GFP+ cells than stimulation with either LRA alone, consistent with results of the reciprocal screens (Fig. 5A). The combination of JQ1 with either SAHA or AZD5582 achieved a similar synergistic effect (Fig. 5B). The strongest effect came from the combination of AZD5582 + JQ1 (high dose) which induced GFP expression in 81.5% of the cells, an effect similar to that seen with PMA/I stimulation (85.7%). The synergistic effect of two drugs was further evaluated using Bliss independence model which quantitatively estimates the synergy of drugs with independent mechanisms of action (Fig. 5C). In this analysis, all drug combinations had synergistic effects, but AZD5582 + JQ1 (high or low doses) demonstrated the highest degree of synergy, independently confirming the results predicted by our CRISPR screens.
Figure 5.
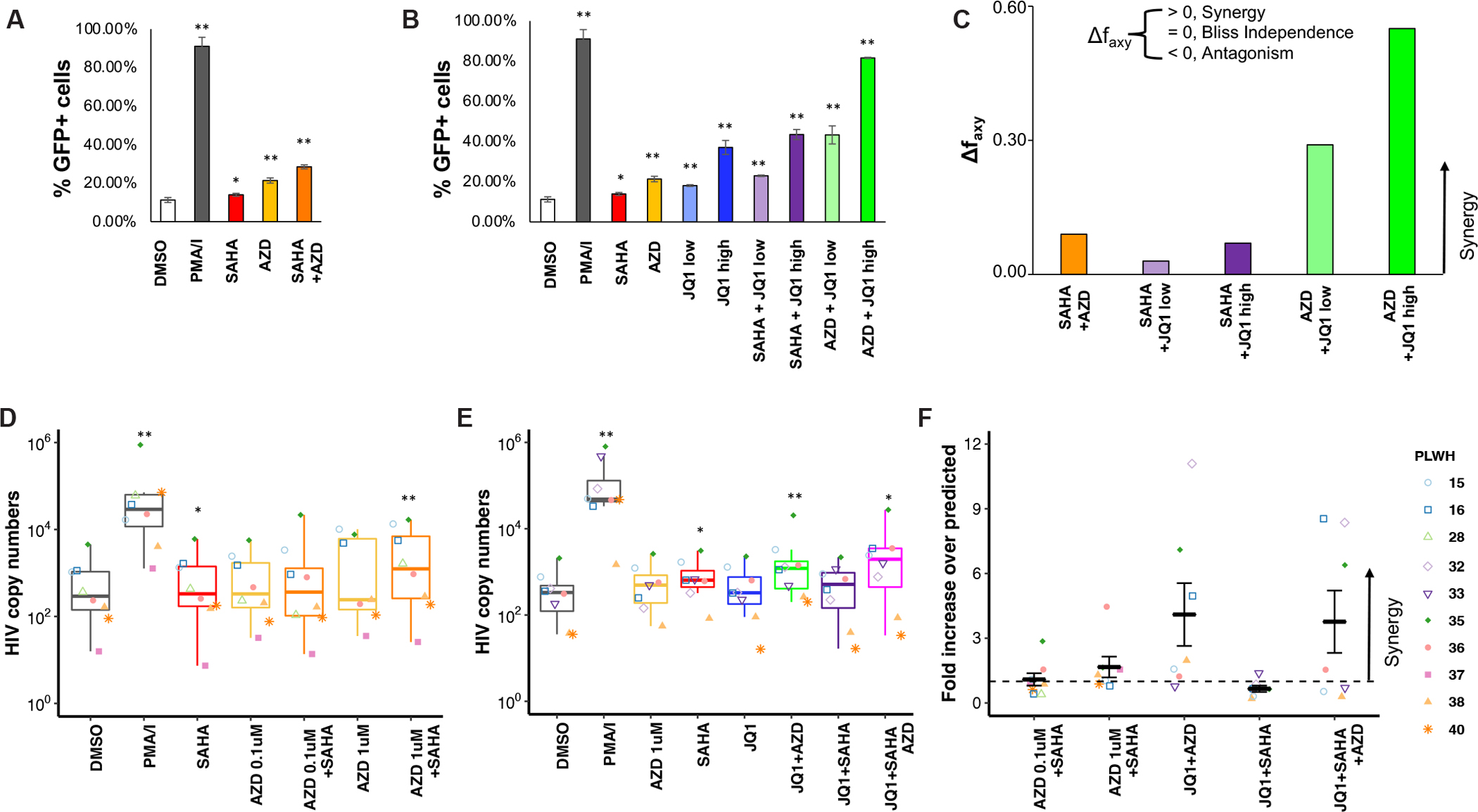
Predicted synergies between LRAs are validated in vitro and ex vivo. A-C, Combinations of AZD5582 (5 nM) and SAHA (0.1 μM). A, and of AZD5582 or SAHA, with two different concentrations of JQ1 (low, 0.1 μM; high, 1 μM). B, induced synergistic reactivation of latent HIV-1 as assessed by the fraction of GFP+ cells in the latency cell model. For A and B, P values were calculated using Student T test, and n = 3. C, Synergy estimation on drug combinations using Bliss independence model (90). Quantitative analysis of synergy (Δ faxy), calculated as described previously (91), for the combinations in a and b. D-E, Combination of AZD5582 (concentration indicated in the figure) and SAHA (2 μM). D, and AZD5582 (1 μM), SAHA (2 μM) and JQ1 (1 μM). E, that induced HIV mRNA production in cells from HIV-1 infected individuals on ART. Intracellular HIV-1 mRNA level in LRA-treated resting CD4+ T cells purified from infected donors presented as fold change relative to DMSO control. F, Synergy estimation on drug combinations using the fold increase over predicted method. For d-f, P values were calculated using Wilcoxon test, and n = 8. (*P < 0.05, **P < 0.01)
Combination of synergistic drug targets promoted HIV latency reversal ex vivo and in vivo
To determine whether the synergistic drug combinations discovered through CRISPR screening with cell lines would have similar effects ex vivo in cells from PLWH, we isolated resting CD4+ T cells from individuals on suppressive ART, incubated them with the relevant LRAs, and measured level of cell associated HIV-1 RNA using a previously described method (17) that detects all polyadenylated HIV-1 mRNA species. The LRAs and LRA combinations tested induced only minimal level of CD4+ T cell activation as assessed by the level of HLA-DR, CD69, and CD25 (fig. S7A). Although the combination of SAHA and AZD5582 induced a low level of apoptosis, neither concentration of AZD5582 causes a higher level of apoptosis than SAHA alone (fig. S7B). Stimulating resting CD4+ T cells with either SAHA or AZD5582 alone did not increase HIV-1 mRNA expression, consistent with our previous results (17, 69). However, the combination of SAHA and 1 μM AZD5582 significantly increased HIV-1 mRNA (Fig. 5D). AZD5582 also demonstrated a synergistic combination effect with other HDAC inhibitors, including panobinostat and romidepsin (fig. S8A–B). Similarly, AZD5582 combined with JQ1 increased HIV-1 mRNA level ex vivo in cells from PLWH by an average of 5.02 fold (Fig. 5E). The JQ1 and SAHA combination, which had the weakest synergy effect in our in vitro model (Fig. 5C), did not induce HIV-1 mRNA expression ex vivo (Fig. 5E).
We also evaluated the degree of synergy between different drug combinations. As HIV-1 copy numbers are reported in PLWH cells, we measured HIV copy numbers from drug combinations, and used the fold increase over predicted copy numbers (assuming no synergy exists between two drugs) as a measurement of synergy (Fig. 5F). A fold increase greater than 1 indicates a potential synergy. As is generally observed with LRAs and LRA combinations, the induction of HIV-1 RNA by AZD5582 and JQ1 was less than that caused by PMA/I (Fig. 5E). Nevertheless, the synergistic effects were observed in AZD5582 combined with either SAHA or JQ1, as well as three drug combination of SAHA, AZD5582, and JQ1, but not JQ1+SAHA (Fig. 5F).
We further performed single-cell RNA sequencing analysis to investigate the possible mechanism of synergy between AZD5582 and JQ1 (fig. S9A–B). Resting in CD4+ T cells isolated from one PLWH’s blood were stimulated with either AZD5582+JQ1 or anti-CD3/CD28-coated beads. AZD5582/JQ1 caused upregulation of many fewer genes in CD4+ T cells (35 genes with log2 fold change >0.5 and adjusted p value <0.1), in comparison with aCD3/CD28 stimulation (983 genes using the same cutoff; fig. S9C–D). These genes up-regulated by AZD5582/JQ1 treatment were enriched in ncNF-κB pathways (fig. S9E), but not cell-cycle related genes, which are up-regulated in aCD3/CD28 stimulation (fig. S9F).
Finally, we tested the combination effect of AZD5582 and JQ1 in vivo as this combination demonstrated the highest synergy in both cell line models and in cells from PLWH (Fig. 5C, F). Four SIVmac251-infected, macaques on suppressive cART were divided into two groups and treated with either AZD5582 or both AZD5582 and JQ1. Drugs were administered intravenously once per week for ten weeks, and were well-tolerated. Because the frequency and magnitude of “blips” of detectable plasma viral RNA induced by latency reversing agents are likely to depend on the size of the latent reservoir, we measured reservoir size prior to LRA treatment in study animals using the intact proviral DNA assay (IPDA) (75). The frequency of intact proviruses in peripheral blood ranged from 33.6 to 848 per million CD4+ T cells (fig. S10A). Both treatments induced transient low-level viremia in the animals, which had all previously had viral loads below the limit of detection for at least 32 weeks prior to the start of the study (fig. S10B). The animals treated with AZD5582 alone (KI6 and DEL5) had 5 and 8 incidences of transient low-level viremia, respectively, over the course of 23 viral load measurements each, with the transient peaks lasting for one to two timepoints (fig. S10B). The two animals treated with AZD5582 and JQ1, PZB and DEAB, had 5 and 4 incidences of transient low-level viremia, respectively, over the same number of measurements (fig. S10B).
The highest peaks of viremia were observed in AZD5582-treated animal DEL5, which also had the largest latent reservoir. Interestingly, the combination of AZD5582 and JQ1 induced four instances of low-level viremia in animal DEAB despite a 25-fold lower amount of the intact provirus compared to DEL5 (fig. S10). While this small study does not have the statistical power to determine if the combination of AZD5582 and JQ1 is superior to AZD5582 alone, the induction of transient low-level viremia in DEAB as well as the similar responses of KI6 and PZB suggest that the LRA combination may have synergistic effects in vivo. Further studies in larger cohorts of animals on long term ART will be required to determine whether the combination shows the same synergistic reaction of latent HIV observed here in vitro and ex vivo.
Validating novel drug targets in PLWH
Having demonstrated the efficacy of repurposed drugs with synergistic latency reversing activity (ncNF-KB activator, HDAC inhibitor, and bromodomain inhibitor), we next focused on screening hits that are not the targets of existing drugs. We selected seven genes that were validated in cell line model (NFKBIA, NELFCD, PPP6C, CYLD, NCFC1, CSK, YPEL5), none of which are the direct targets of existing drugs (FDA approved or in phase I/II clinical trial). We isolated resting CD4+ T cells from PLWH samples, and knocked out target genes without pre-activating cells. We then measured HIV copy numbers using the same approach we used to test the drugs (Fig. 6 and fig. S11). We estimated the fold increase over predicted copy numbers (assuming drug and gene knockout independently activate latent HIV), the same approach we used to estimate drug synergy (Fig. 5F), and evaluated whether the synergy was fully validated (fold increase greater than one in cells from at least two PLWH tested), partially validated (fold increase greater than one in only one participated), and not validated at all in any of the PLWH samples (no fold increase observed; table S3). The knockout efficiencies of each gene were measured at the protein level using flow cytometry (fig. S11).
Figure 6.

Novel synergistic LRA targets validated ex vivo using primary CD4+ T cells isolated from PLWH on ART, including NFKBIA (A-B), CYLD (C-D) and YPEL5 (E-F). The intracellular HIV copy numbers in primary CD4+ T cells following LRA treatments, AZD5582 (1 μM) or SAHA (2 μM) were compared between cells with knockout of the indicated gene and negative control cells (A, C, E), and the fold increase (gene KO + drug) over predicted copy numbers (B, D, F) assuming gene knockouts and drug treatments are independently activating latent HIV are shown.
Among the seven genes tested, three were fully validated to synergize with one or both drugs (NFKBIA, CYLD, YPEL5; Fig. 6), three were partially validated with evidence from one patient sample (NELFCD, HCFC1, CSK), and two were not validated in any of the patient samples (PPP6C) (fig. S12). Overall, 86% (6 out of 7) hits are fully or partially validated, including regulators of NF-kb pathways (NFKBIA and CYLD) as well as components of NELF complex that counteract with P-TEFb kinase complex (NELFCD). A remarkably high level of synergy with SAHA and AZD5582, reaching 10-fold in some PLWH, was observed for NFKBIA, which is an inhibitor of NF-κB (Fig. 6A–B). Strong synergy was also seen for YPEL5 (Yippee Like 5), a component of the CTLH E2 ubiquitin-protein ligase complex, is the only hit that was fully validated to synergize with both AZD5582 and SAHA (Fig. 6E–F).
It is not surprising that the validation rate in PLWH samples is lower than in cell line models (86% vs 100%), and synergies are not observed in all PLWH samples, reflecting variations between different PLWH. The latter may introduce false negatives; for example, NELFCD knockout showed strong synergy with both AZD5582 and SAHA in patient # 2274, but not in patient # 12 (fig. S12). Nevertheless, our results convincingly show that depletion of the novel factors identified in our CRISPR screens alone or together with existing LRAs effectively reverse HIV-1 latency and that development of novel drugs to these hits may lead to potent and effective LRA combinations for use in cure strategies.
Discussion
This study represents the first comprehensive search for synergistic LRA combinations using genome-wide CRISPR screens. Here we searched for drugs with potential synergy with an existing LRA by computationally prioritizing candidate drug targets in CRISPR screens. Screens on AZD5582-treated cells revealed HDAC2 as the target of potentially synergistic candidate LRAs, while screens on HDAC inhibitor-stimulated cells identified ncNF-κB regulators, as the reciprocal candidates (Fig. 3G). Validation on primary CD4+ cells from infected individuals using pharmacological intervention confirmed the synergy between an HDAC inhibitor and a ncNF-κB pathway activator. Importantly, neither an HDAC inhibitor (SAHA) nor AZD5582 alone led to increased HIV-1 gene expression in ex vivo analysis using cells from infected individuals, demonstrating the necessity of combining both HDAC inhibitors and AZD5582 in clinical settings. Interestingly, both of these reciprocal screens independently identified BRD2 as candidate, suggestion potential synergies with the bromodomain inhibitor JQ1. Furthermore, we identified and validated several novel host factors that regulate HIV-1 latency reversal in both the model system and ex vivo. Among them, CYLD and YPEL5 were first identified as the potential drug target for LRAs.
Numerous host factors have been claimed to influence HIV latency persistence and reversal. Our screening and integrative analysis identified 128 genes required in HIV-1 latency reversal mediated by multiple stimuli, including PMA/I, TNFα, αCD3/CD28, and AZD5582 (Fig. 1E). Other than well studies molecules cyclin T1 and regulators of NF-κB pathway (CHUK, MAP3K7, NFKB1, RELA), AMBRA1 is an essential factor of the autophagic core machinery(76). Enhanced autophagy mediated by AMBRA1 was observed in elite controller PBMC (77). In addition, viral reactivation was shown to be promoted by inhibition of autophagy, and was specific for HIV-infected cells (78). Here we for the first time suggested the essential role of AMBRA1 in HIV-1 latency reversal using CRISPR screens.
The drug targets identified in our synergy screen were targets of some previously described latency reversing agents LRAs. This result largely reflects our screening criteria and analysis pipeline (Fig. 3) which were designed to identify existing drugs that could be repurposed as LRAs. Importantly, a genome wide screen that is filtered to focus on the targets of existing drugs identified targets of known LRAs. This implies that of all existing drugs targeting diverse physiologic pathways, the only ones with potential synergy were previously discovered LRAs. In other words, there are not likely to be useful LRAs among existing drugs and that the focus of the field should be on novel targets for which there are not yet available drugs.
The synergies between AZD5582 and SAHA, and between AZD5582 and JQ1 were validated in both the latency cell line model and resting CD4+ T cells from PLWH (Fig. 5C and 5F). The combination of SAHA and JQ1 exhibited synergy in the cell line model but not in resting CD4+ T cells from PLWH (Fig. 5F). This result might reflect differences between the latency model and primary T cells such as higher level of cyclin T1 in model cells (69). In addition, the inhibition of top hits suggested by CRISPR screens needs to be potent enough to mimic the effects seen in the knockout screens. Although JQ1 did not synergize with SAHA in latency reversal in our study, a more potent BETi might serve the purpose (74).
It is also important to consider the heterogeneity in CD4+ T cells harboring replication-competent virus, as illustrated in recent findings (79–81). The effective reversal of latency without global T cell activation might require a precision medicine approach that varies from person to person, and with different phases of disease progression. In addition, recent studies combined pharmacological latency reversal with immune modulation that facilitates immune recognition (82); for example, transient CD8+ T cell depletion together with interleukin-15 superagonist N-803 led to a robust reactivation of SIV and HIV (20).Together with immune modulation, combination strategies in our study might reach full potential.
Given the fact that no single LRA (or LRA combination) reduces latent reservoir size in infected individuals (9, 17, 21), the development of novel LRAs for new targets is urgently needed. Our study offers a road map for mechanism-based LRA development, by categorizing top hits in their stages of pharmacological development, physiological relevance, and functions in HIV-1 latency reversal (Fig. 3C and 3F). In our study, less than 30% of the synergistic candidates identified in our AZD5582 and SAHA screens are targeted by existing drugs (table S1 and S2). The majority of the candidates, which demonstrated strong potential for synergy and minimal toxicity when knocked out, are not targeted by any drugs. These genes, once confirmed and validated with follow-up studies, may serve as the targets of new LRAs that can combine with existing drugs to reduce the HIV-1 reservoir.
Interestingly, we overserved that YPEL5 knockout alone in primary cells isolated from PLWH induced higher level of HIV copy number even without applying any LRA (Fig. 6E). The similar effect existed in the knockout of NFKBIA and CYLD in cells from one donor (Fig. 6A and C). The results suggest that inhibition of YPEL5 alone might enhance HIV-1 latency reversal, and that YEPL5 should be a target for development of LRAs. Moreover, these results indicated that our screening strategy may reveal host factors that regulate HIV latency on their own.
Although our study identified and validated potential combinatorial LRA drug targets from multiple genome-wide CRISPR screens, there are potential limitations to the approaches we used. First, the screening may have inherent false positives and false negatives, due to the limited number of guides in the library (4 per gene for Brunello library) and the potential off-target effect of these guides. Second, cell-line models were used for CRISPR screens. Since cell-line models and human primary T cells have intrinsically different transcriptomic and epigenetic profiles, evidenced in the previous studies (11, 13, 15, 69), extensive validations are required, especially using primary cell latency models and primary cells isolated from PWLH. Looking forward, a combination of screens in in vitro models with in vivo and clinical validations will be crucial to better understand the mechanism of latent HIV reactivation, and to find LRA drug combinations to eliminate latent HIV virus.
In summary, this study represents the first comprehensive identification of synergistic LRA combinations using genome-wide CRISPR screens. Looking forward, our approach that combines in vitro screens with in vivo and clinical validations will be crucial to better understand the mechanism of latent HIV-1 reactivation, and to find the combinations of existing and novel LRAs to eliminate latent HIV-1 virus.
Materials and Methods
Study design
This study aimed to identify synergistic LRA combinations to reactivate latent HIV-1 using a novel in vitro HIV-1 latency model and genome-wide CRISPR screens. We have performed several CRISPR screens using various LRA with different concentrations to address these objectives. Both cell line cells and primary CD4+ T cells isolated from PLWH were used for the validation of hits identified in the CRISPR screens by gene knockout via CRISPR/Cas9. The effect of each gene knockout was compared with the one of the non-targeting negative control using the same cells line cells or CD4 T+ cells isolated from the same individual.
We obtained deidentified leukapheresis samples from 2 PLWH on ART who were enrolled in the University of California San Francisco SCOPE cohort. The study was approved by Institutional Review Board (IRB) of the University of California San Francisco. Another 11 PLWH on ART were enrolled at the University of Pennsylvania. Selection criteria included viral suppression for >6 mo and generally undetectable plasma HIV-1 RNA levels (<50 copies per mL). Participants underwent leukapheresis under a protocol approved by the University of Pennsylvania IRB. All participants provided written informed consent. deidentified leukapheresis products were shipped to the Johns Hopkins School of Medicine for processing and analysis under a protocol approved by a Johns Hopkins University School of Medicine IRB. Experimenters were not blinded during the study.
Genome-wide CRISPR-Cas9 screens
The human CRISPR knockout pooled library (Brunello, two vector system) purchased from Addgene was used for the genome-wide CRISPR screens. The standard procedures in the screens were performed as described previously (83, 84). The CRISPR sgRNA library virus was packaged by transfecting HEK293T cells with library DNA, pMD2.G and a packaging vector pC-Help. After latent cells described in Figure 1A was sorted, 2.4 × 108 of the cells were infected at a low multiplicity of infection (0.3~0.5) to ensure that most cells receive only 1 viral construct with high probability. Two days after transduction, cells were selected in medium containing 1 μg ml−1 puromycin for two weeks to obtain the pooled knockout cells of latently HIV-infected cells. Then, 5 × 107 pooled knockout modified Jurkat cells were stimulated with either 0.1 μM SAHA or 5 nM AZD5582, as suboptimal doses, for 16 hours in experiments presented in Figure 2 and 3. To screen for regulators of HIV-1 latency reversal induced by LRA, the same number of cells were treated with one of the following LRA, 1 μM SAHA, 0.1 μM AZD5582, 10 ng/ml TNFα, or PMA/I (50ng/ml PMA in combination with 1 μM Ionomycin), for 16 hours. GFP+ cells were sorted by FACS. The sorted cells and and the control cells (transduced with the sgRNA library, cultured in parallel but without infection/sorting) were immediately lysed for genomic DNA extraction. The PCR to amplify the sgRNA was performed by using primers suggested by Brunello library inventors. The PCR products were purified by gel extraction, and the samples were deep-sequenced by Illumina Nextseq. Each library was sequenced at 30~40 million reads to achieve ~300X average coverage over the CRISPR library.
Computational analysis of the screens
We use MAGeCK algorithm (54) to identify genes whose knockout lead to the enrichment of GFP− cells compared with pooled controls (Fig. 1C and Fig. 2A). The MAGeCK algorithm builds a linear model to estimate the variance of gRNA read counts, and assigns a p-value using the Negative Binomial (NB) model. Next, the selection of genes is evaluated from the rankings of gRNAs (by their p-values) using the α-RRA (α-Robust Rank Aggregation) algorithm. A detailed description of the MAGeCK algorithm can be found in the original study (54).
We use the “β score”, generated from MAGeCK-VISPR algorithm (85), as a measurement of “activation score” in Figure 1e to compare genes across different conditions. “β score” is similar to the term of ‘log fold change’ in differential expression analysis, and β>0 (or <0) means the corresponding gene is positively (or negatively) selected, respectively. MAGeCK-VISPR uses a maximum likelihood (MLE) model to model all gRNA read counts of all samples, and iteratively estimate β scores using the Expectation-Maximization (EM) algorithm. A detailed description of the MAGeCK-MLE algorithm can be found in the original study (72). For the comparison of different stimulus (Fig. 1E), a further quantile normalization is applied to the β scores to ensure the distribution of scores across conditions is the same.
Identification of synergistic druggable genes
“Druggable genes”, or genes that have known targeting drugs, are extracted from the Drug-Gene Interaction Database (DGIdb) (70) (date of extraction: May 2020; category: “Druggable Genome”). To identify ideal synergistic druggable genes, we rank genes (1) whose knockout leads to an enrichment of GFP+ cells in SAHA or AZD5582 treatment; and (2) whose knockout has minimal selections in pooled control cells. Therefore, for each gene g, we define its synergistic score SLg using a rank-product algorithm (86), where
Where βLRA and βpool are the β scores (calculated from MAGeCK-VISPR) for screens with LRA stimulated cells or pooled knockout controls, respectively. rank(·) is the rank function (converted to uniform distributed values between [0,1]). A lower SLg score indicates this gene knockout leads to the activation of GFP+ cells (larger βLRA), and has minimal effect in pooled cells (smaller |βpool| and larger βLRA/|βpool|). Since genes with small βpool values usually have large p values from MAGeCK-VISPR (i.e., small βpool scores are likely not distinguishable from random noise), we did not rank |βpool| directly in the rank-product algorithm, but instead rank βLRA/|βpool| to increase the rank of genes with small |βpool| values. To avoid the extremely small values of |βpool| that leads to extreme values of βLRA/|βpool|, the value of |βpool| is adjusted to 0.1 if it is smaller than 0.1. The p values of the synergistic score are calculated using the methods described previously (86). The adjusted p values are calculated using Benjamini-Hochberg Procedure (FDR).
We further filter out genes with low expression in CD4+ T cells, using a public RNA-seq dataset on primary human CD4+ cells (data source: GSE142774). For each gene, we calculated the average log2 normalized counts from the CD4+ T cells of four individual donors (DMSO treated, 2 replicates per individual) as the expression of the gene. Highly expressed genes are defined as genes whose expression is greater than the median expression of all genes (Fig. 2F, 3C and table S1 and S2).
Statistical analysis
Student’s t-test was used to determine statistical significance where indicated except for the analysis in Figure 5D and 5E. In those two panels, we used Wilcoxon test for measure significant differences. We considered P < 0.05 to be statistically significant. Samples from all patients were handled in the same way for each experiment, thus there was no randomization or blinding. *P < 0.05, **P < 0.01. Error bars in the figures indicate mean ± s.e.m.
Data availability
Genome-wide screening data will be deposited at NCBI Short Read Archive (SRA) under the accession number (PRJNA689546).
Supplementary Material
Fig. S1. Pathways and networks of the clusters.
Fig. S2. Sorting strategy of GFP+ latent cells post AZD5582 stimulation.
Fig. S3. SAHA stimulated CRISPR screens.
Fig. S4. HDAC as a potential synergistic drug target in AZD5582 stimulated CRISPR screens.
Fig. S5. The knockout efficiencies in latency cell line cells.
Fig. S6. Validation of top hits in the CRISPR screens for the combinational strategy in the Jurkat latency cell line.
Fig. S7. The activation and viability of primary CD4+ T cells following the combinational LRAs treatment.
Fig. S8. Validating AZD and HDACi combination predictions.
Fig. S9. Single-cell RNA-seq analysis.
Fig. S10. Transient low-level viremia is induced in cART-suppressed Rhesus macaques following AZD5582 and JQ1 treatment.
Fig. S11. The knockout efficiencies in primary CD4+ T cells from one representative PLWH were shown at gene expression level.
Fig. S12. Validation of top hits in the CRISPR screens for the combinational strategy ex vivo using primary CD4+ T cells isolated from PLWH.
Table S1. The group of genes that synergize with AZD5582 with statistical significance (FDR<0.1) using the rank product algorithm.
Table S2. The group of genes that synergize with SAHA with statistical significance (FDR<0.1) using the rank product algorithm.
Table S3. Genes that are selected for validation, and their validation status in cell line model and in the T cells of PLWH.
Data File S1. Individual level data for figures and supplementary figures.
Data File S2. The MAGeCK analysis results of genome-wide CRISPR screens on AZD5582 stimulated GFP− cells vs. pooled control cells (Fig. 1c).
Data File S3. The normalized scores in 578 genes and their cluster assignments in Figure 1e.
Data File S4. The MAGeCK analysis results of genome-wide CRISPR screens on AZD5582 stimulated GFP+ cells vs. pooled control cells (Fig. 4b).
Data File S5. The MAGeCK analysis results of genome-wide CRISPR screens on SAHA stimulated GFP+ cells vs. pooled control cells (Supplementary Fig. 6a).
Data File S6. The rank product of all genes in AZD5582 stimulated GFP+ cells.
Data File S7. The rank product of all genes in SAHA stimulated GFP+ cells.
Data File S8. The sequences of sgRNAs used in single-gene knockout in cell line and primary cells.
Acknowledgements
This work was supported by the NIH Martin Delaney I4C (UM1 AI126603), Beat-HIV (UM1 AI126620) and DARE (UM1 AI126611) Collaboratories, by the Johns Hopkins Center for AIDS Research (P30AI094189), and by the Howard Hughes Medical Institute, and the Bill and Melinda Gates Foundation (OPP1115715). W.L. is supported by National Institute of Health (NIH)/National Human Genome Research Institute (R01HG010753), the PhRMA Foundation, and the startup funds of Children’s National Hospital. We thank Dr. Jonathan Karn for sharing the reagents. We thank all the members of Siliciano lab and Li lab for the help, advice and discussions.
Footnotes
Competing interests
W.L. is a paid consultant to Tavros Therapeutics Inc. J.D.S. is on the Gilead Sciences Scientific Advisory Board for HIV Cure. All other authors declare that they have no competing interests.
References
- 1.Chun TW, Stuyver L, Mizell SB, Ehler LA, Mican JA, Baseler M, Lloyd AL, Nowak MA, Fauci AS, Presence of an inducible HIV-1 latent reservoir during highly active antiretroviral therapy. Proc. Natl. Acad. Sci. U. S. A. 94, 13193–13197 (1997). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Finzi D, Hermankova M, Pierson T, Carruth LM, Buck C, Chaisson RE, Quinn TC, Chadwick K, Margolick J, Brookmeyer R, Gallant J, Markowitz M, Ho DD, Richman DD, Siliciano RF, Identification of a reservoir for HIV-1 in patients on highly active antiretroviral therapy. Science. 278, 1295–1300 (1997). [DOI] [PubMed] [Google Scholar]
- 3.Wong JK, Hezareh M, Günthard HF, Havlir DV, Ignacio CC, Spina CA, Richman DD, Recovery of replication-competent HIV despite prolonged suppression of plasma viremia. Science. 278, 1291–1295 (1997). [DOI] [PubMed] [Google Scholar]
- 4.Siliciano RF, Greene WC, HIV latency. Cold Spring Harb. Perspect. Med. 1, a007096 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Brooks DG, Hamer DH, Arlen PA, Gao L, Bristol G, Kitchen CMR, Berger EA, Zack JA, Molecular characterization, reactivation, and depletion of latent HIV. Immunity. 19, 413–423 (2003). [DOI] [PubMed] [Google Scholar]
- 6.Williams SA, Chen L-F, Kwon H, Fenard D, Bisgrove D, Verdin E, Greene WC, Prostratin antagonizes HIV latency by activating NF-kappaB. J. Biol. Chem. 279, 42008–42017 (2004). [DOI] [PubMed] [Google Scholar]
- 7.Contreras X, Schweneker M, Chen C-S, McCune JM, Deeks SG, Martin J, Peterlin BM, Suberoylanilide hydroxamic acid reactivates HIV from latently infected cells. J. Biol. Chem. 284, 6782–6789 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Deeks SG, HIV: Shock and kill. Nature. 487 (2012), pp. 439–440. [DOI] [PubMed] [Google Scholar]
- 9.Archin NM, Liberty AL, Kashuba AD, Choudhary SK, Kuruc JD, Crooks AM, Parker DC, Anderson EM, Kearney MF, Strain MC, Richman DD, Hudgens MG, Bosch RJ, Coffin JM, Eron JJ, Hazuda DJ, Margolis DM, Administration of vorinostat disrupts HIV-1 latency in patients on antiretroviral therapy. Nature. 487, 482–485 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Shirakawa K, Chavez L, Hakre S, Calvanese V, Verdin E, Reactivation of latent HIV by histone deacetylase inhibitors. Trends Microbiol. 21, 277–285 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Mbonye U, Karn J, Transcriptional control of HIV latency: cellular signaling pathways, epigenetics, happenstance and the hope for a cure. Virology. 454–455, 328–339 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Margolis DM, Garcia JV, Hazuda DJ, Haynes BF, Latency reversal and viral clearance to cure HIV-1. Science. 353, aaf6517 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Dutilleul A, Rodari A, Van Lint C, Depicting HIV-1 transcriptional mechanisms: A summary of what we know. Viruses. 12, 1385 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Bartholomeeusen K, Xiang Y, Fujinaga K, Peterlin BM, Bromodomain and extra-terminal (BET) bromodomain inhibition activate transcription via transient release of positive transcription elongation factor b (P-TEFb) from 7SK small nuclear ribonucleoprotein. J. Biol. Chem. 288, 12214 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Spina CA, Anderson J, Archin NM, Bosque A, Chan J, Famiglietti M, Greene WC, Kashuba A, Lewin SR, Margolis DM, Mau M, Ruelas D, Saleh S, Shirakawa K, Siliciano RF, Singhania A, Soto PC, Terry VH, Verdin E, Woelk C, Wooden S, Xing S, Planelles V, An in-depth comparison of latent HIV-1 reactivation in multiple cell model systems and resting CD4+ T cells from aviremic patients. PLoS Pathog. 9, e1003834 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Li Z, Guo J, Wu Y, Zhou Q, The BET bromodomain inhibitor JQ1 activates HIV latency through antagonizing Brd4 inhibition of Tat-transactivation. Nucleic Acids Res. 41, 277–287 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Bullen CK, Laird GM, Durand CM, Siliciano JD, Siliciano RF, New ex vivo approaches distinguish effective and ineffective single agents for reversing HIV-1 latency in vivo. Nat. Med. 20, 425–429 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Pache L, Dutra MS, Spivak AM, Marlett JM, Murry JP, Hwang Y, Maestre AM, Manganaro L, Vamos M, Teriete P, Martins LJ, König R, Simon V, Bosque A, Fernandez-Sesma A, Cosford NDP, Bushman FD, Young JAT, Planelles V, Chanda SK, BIRC2/cIAP1 is a negative regulator of HIV-1 transcription and can be targeted by smac mimetics to promote reversal of viral latency. Cell Host Microbe. 18, 345–353 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Nixon CC, Mavigner M, Sampey GC, Brooks AD, Spagnuolo RA, Irlbeck DM, Mattingly C, Ho PT, Schoof N, Cammon CG, Tharp GK, Kanke M, Wang Z, Cleary RA, Upadhyay AA, De C, Wills SR, Falcinelli SD, Galardi C, Walum H, Schramm NJ, Deutsch J, Lifson JD, Fennessey CM, Keele BF, Jean S, Maguire S, Liao B, Browne EP, Ferris RG, Brehm JH, Favre D, Vanderford TH, Bosinger SE, Jones CD, Routy J-P, Archin NM, Margolis DM, Wahl A, Dunham RM, Silvestri G, Chahroudi A, Garcia JV, Systemic HIV and SIV latency reversal via non-canonical NF-κB signalling in vivo. Nature. 578, 160–165 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.McBrien JB, Mavigner M, Franchitti L, Smith SA, White E, Tharp GK, Walum H, Busman-Sahay K, Aguilera-Sandoval CR, Thayer WO, Spagnuolo RA, Kovarova M, Wahl A, Cervasi B, Margolis DM, Vanderford TH, Carnathan DG, Paiardini M, Lifson JD, Lee JH, Safrit JT, Bosinger SE, Estes JD, Derdeyn CA, Garcia JV, Kulpa DA, Chahroudi A, Silvestri G, Robust and persistent reactivation of SIV and HIV by N-803 and depletion of CD8+ cells. Nature. 578, 154–159 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Elliott JH, Wightman F, Solomon A, Ghneim K, Ahlers J, Cameron MJ, Smith MZ, Spelman T, McMahon J, Velayudham P, Brown G, Roney J, Watson J, Prince MH, Hoy JF, Chomont N, Fromentin R, Procopio FA, Zeidan J, Palmer S, Odevall L, Johnstone RW, Martin BP, Sinclair E, Deeks SG, Hazuda DJ, Cameron PU, Sékaly R-P, Lewin SR, Activation of HIV transcription with short-course vorinostat in HIV-infected patients on suppressive antiretroviral therapy. PLoS Pathog. 10, e1004473 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Cary DC, Fujinaga K, Peterlin BM, Molecular mechanisms of HIV latency. J. Clin. Invest. 126, 448–454 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Osborn L, Kunkel S, Nabel GJ, Tumor necrosis factor alpha and interleukin 1 stimulate the human immunodeficiency virus enhancer by activation of the nuclear factor kappa B. Proc. Natl. Acad. Sci. U. S. A. 86, 2336–2340 (1989). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Duh EJ, Maury WJ, Folks TM, Fauci AS, Rabson AB, Tumor necrosis factor alpha activates human immunodeficiency virus type 1 through induction of nuclear factor binding to the NF-kappa B sites in the long terminal repeat. Proc. Natl. Acad. Sci. U. S. A. 86, 5974–5978 (1989). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Lusic M, Marcello A, Cereseto A, Giacca M, Regulation of HIV-1 gene expression by histone acetylation and factor recruitment at the LTR promoter. EMBO J. 22, 6550–6561 (2003). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Brooks DG, Arlen PA, Gao L, Kitchen CMR, Zack JA, Identification of T cell-signaling pathways that stimulate latent HIV in primary cells. Proc. Natl. Acad. Sci. U. S. A. 100, 12955–12960 (2003). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Bosque A, Planelles V, Induction of HIV-1 latency and reactivation in primary memory CD4+ T cells. Blood. 113, 58–65 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Marshall RM, Salerno D, Garriga J, Graña X, Cyclin T1 expression is regulated by multiple signaling pathways and mechanisms during activation of human peripheral blood lymphocytes. J. Immunol. 175, 6402–6411 (2005). [DOI] [PubMed] [Google Scholar]
- 29.Budhiraja S, Famiglietti M, Bosque A, Planelles V, Rice AP, Cyclin T1 and CDK9 T-loop phosphorylation are downregulated during establishment of HIV-1 latency in primary resting memory CD4+ T cells. J. Virol. 87, 1211–1220 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Verdin E, Paras P Jr, Van Lint C, Chromatin disruption in the promoter of human immunodeficiency virus type 1 during transcriptional activation. EMBO J. 12, 3249–3259 (1993). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Archin NM, Keedy KS, Espeseth A, Dang H, Hazuda DJ, Margolis DM, Expression of latent human immunodeficiency type 1 is induced by novel and selective histone deacetylase inhibitors. AIDS. 23, 1799–1806 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Battivelli E, Dahabieh MS, Abdel-Mohsen M, Svensson JP, Tojal Da Silva I, Cohn LB, Gramatica A, Deeks S, Greene WC, Pillai SK, Verdin E, Distinct chromatin functional states correlate with HIV latency reactivation in infected primary CD4+ T cells. Elife. 7 (2018), doi: 10.7554/elife.34655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Rasmussen TA, Tolstrup M, Brinkmann CR, Olesen R, Erikstrup C, Solomon A, Winckelmann A, Palmer S, Dinarello C, Buzon M, Lichterfeld M, Lewin SR, Østergaard L, Søgaard OS, Panobinostat, a histone deacetylase inhibitor, for latent-virus reactivation in HIV-infected patients on suppressive antiretroviral therapy: a phase 1/2, single group, clinical trial. Lancet HIV. 1, e13–21 (2014). [DOI] [PubMed] [Google Scholar]
- 34.Søgaard OS, Graversen ME, Leth S, Olesen R, Brinkmann CR, Nissen SK, Kjaer AS, Schleimann MH, Denton PW, Hey-Cunningham WJ, Koelsch KK, Pantaleo G, Krogsgaard K, Sommerfelt M, Fromentin R, Chomont N, Rasmussen TA, Østergaard L, Tolstrup M, The depsipeptide romidepsin reverses HIV-1 latency in vivo. PLoS Pathog. 11, e1005142 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Mbonye U, Leskov K, Shukla M, Valadkhan S, Karn J, Biogenesis of P-TEFb in CD4+ T cells to reverse HIV latency is mediated by protein kinase C (PKC)-independent signaling pathways. PLoS Pathog. 17, e1009581 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Sun S-C, The noncanonical NF-κB pathway. Immunol. Rev. 246, 125–140 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Campbell GR, Bruckman RS, Chu Y-L, Trout RN, Spector SA, SMAC mimetics induce autophagy-dependent apoptosis of HIV-1-infected resting memory CD4+ T cells. Cell Host Microbe. 24, 689–702.e7 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Dahabieh MS, Battivelli E, Verdin E, Understanding HIV latency: the road to an HIV cure. Annu. Rev. Med. 66, 407–421 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Laird GM, Bullen CK, Rosenbloom DIS, Martin AR, Hill AL, Durand CM, Siliciano JD, Siliciano RF, Ex vivo analysis identifies effective HIV-1 latency-reversing drug combinations. J. Clin. Invest. 125, 1901–1912 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Ait-Ammar A, Kula A, Darcis G, Verdikt R, De Wit S, Gautier V, Mallon PWG, Marcello A, Rohr O, Van Lint C, Current status of latency reversing agents facing the heterogeneity of HIV-1 cellular and tissue reservoirs. Front. Microbiol. 10, 3060 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Brass AL, Dykxhoorn DM, Benita Y, Yan N, Engelman A, Xavier RJ, Lieberman J, Elledge SJ, Identification of host proteins required for HIV infection through a functional genomic screen. Science. 319, 921–926 (2008). [DOI] [PubMed] [Google Scholar]
- 42.Zhou H, Xu M, Huang Q, Gates AT, Zhang XD, Castle JC, Stec E, Ferrer M, Strulovici B, Hazuda DJ, Espeseth AS, Genome-scale RNAi screen for host factors required for HIV replication. Cell Host Microbe. 4, 495–504 (2008). [DOI] [PubMed] [Google Scholar]
- 43.Yeung ML, Houzet L, Yedavalli VSRK, Jeang K-T, A genome-wide short hairpin RNA screening of jurkat T-cells for human proteins contributing to productive HIV-1 replication. J. Biol. Chem. 284, 19463–19473 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Park RJ, Wang T, Koundakjian D, Hultquist JF, Lamothe-Molina P, Monel B, Schumann K, Yu H, Krupzcak KM, Garcia-Beltran W, Piechocka-Trocha A, Krogan NJ, Marson A, Sabatini DM, Lander ES, Hacohen N, Walker BD, A genome-wide CRISPR screen identifies a restricted set of HIV host dependency factors. Nat. Genet. 49, 193–203 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Ndzinu JK, Takeuchi H, Saito H, Yoshida T, Yamaoka S, eIF4A2 is a host factor required for efficient HIV-1 replication. Microbes Infect. 20, 346–352 (2018). [DOI] [PubMed] [Google Scholar]
- 46.OhAinle M, Helms L, Vermeire J, Roesch F, Humes D, Basom R, Delrow JJ, Overbaugh J, Emerman M, A virus-packageable CRISPR screen identifies host factors mediating interferon inhibition of HIV. Elife. 7 (2018), doi: 10.7554/eLife.39823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Huang H, Kong W, Jean M, Fiches G, Zhou D, Hayashi T, Que J, Santoso N, Zhu J, A CRISPR/Cas9 screen identifies the histone demethylase MINA53 as a novel HIV-1 latency-promoting gene (LPG). Nucleic Acids Res. 47, 7333–7347 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Rathore A, Iketani S, Wang P, Jia M, Sahi V, Ho DD, CRISPR-based gene knockout screens reveal deubiquitinases involved in HIV-1 latency in two Jurkat cell models. Sci. Rep. 10, 5350 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Li Z, Hajian C, Greene WC, Identification of unrecognized host factors promoting HIV-1 latency. PLoS Pathog. 16, e1009055 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Eriksson S, Graf EH, Dahl V, Strain MC, Yukl SA, Lysenko ES, Bosch RJ, Lai J, Chioma S, Emad F, Abdel-Mohsen M, Hoh R, Hecht F, Hunt P, Somsouk M, Wong J, Johnston R, Siliciano RF, Richman DD, O’Doherty U, Palmer S, Deeks SG, Siliciano JD, Comparative analysis of measures of viral reservoirs in HIV-1 eradication studies. PLoS Pathog. 9, e1003174 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Jordan A, Bisgrove D, Verdin E, HIV reproducibly establishes a latent infection after acute infection of T cells in vitro. EMBO J. 22, 1868–1877 (2003). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Tyagi M, Pearson RJ, Karn J, Establishment of HIV latency in primary CD4+ cells is due to epigenetic transcriptional silencing and P-TEFb restriction. J. Virol. 84, 6425–6437 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Doench JG, Hartenian E, Graham DB, Tothova Z, Hegde M, Smith I, Sullender M, Ebert BL, Xavier RJ, Root DE, Rational design of highly active sgRNAs for CRISPR-Cas9-mediated gene inactivation. Nat. Biotechnol. 32, 1262–1267 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Li W, Xu H, Xiao T, Cong L, Love MI, Zhang F, Irizarry RA, Liu JS, Brown M, Liu XS, MAGeCK enables robust identification of essential genes from genome-scale CRISPR/Cas9 knockout screens. Genome Biol. 15, 554 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Kajino T, Ren H, Iemura S-I, Natsume T, Stefansson B, Brautigan DL, Matsumoto K, Ninomiya-Tsuji J, Protein phosphatase 6 down-regulates TAK1 kinase activation in the IL-1 signaling pathway. J. Biol. Chem. 281, 39891–39896 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Besnard E, Hakre S, Kampmann M, Lim HW, Hosmane NN, Martin A, Bassik MC, Verschueren E, Battivelli E, Chan J, Svensson JP, Gramatica A, Conrad RJ, Ott M, Greene WC, Krogan NJ, Siliciano RF, Weissman JS, Verdin E, The mTOR complex controls HIV latency. Cell Host Microbe. 20, 785–797 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Mancebo HS, Lee G, Flygare J, Tomassini J, Luu P, Zhu Y, Peng J, Blau C, Hazuda D, Price D, Flores O, P-TEFb kinase is required for HIV Tat transcriptional activation in vivo and in vitro. Genes Dev. 11, 2633–2644 (1997). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Yang X, Gold MO, Tang DN, Lewis DE, Aguilar-Cordova E, Rice AP, Herrmann CH, TAK, an HIV Tat-associated kinase, is a member of the cyclin-dependent family of protein kinases and is induced by activation of peripheral blood lymphocytes and differentiation of promonocytic cell lines. Proc. Natl. Acad. Sci. U. S. A. 94, 12331–12336 (1997). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Peng J, Zhu Y, Milton JT, Price DH, Identification of multiple cyclin subunits of human P-TEFb. Genes Dev. 12, 755–762 (1998). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Wei P, Garber ME, Fang S-M, Fischer WH, Jones KA, A novel CDK9-associated C-type cyclin interacts directly with HIV-1 tat and mediates its high-affinity, loop-specific binding to TAR RNA. Cell. 92, 451–462 (1998). [DOI] [PubMed] [Google Scholar]
- 61.Khoury G, Darcis G, Lee MY, Bouchat S, Van Driessche B, Purcell DFJ, Van Lint C, The molecular biology of HIV latency. Adv. Exp. Med. Biol. 1075, 187–212 (2018). [DOI] [PubMed] [Google Scholar]
- 62.Franceschini A, Szklarczyk D, Frankild S, Kuhn M, Simonovic M, Roth A, Lin J, Minguez P, Bork P, von Mering C, Jensen LJ, STRING v9.1: protein-protein interaction networks, with increased coverage and integration. Nucleic Acids Res. 41, D808–15 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Ruiz A, Pauls E, Badia R, Riveira-Muñoz E, Clotet B, Ballana E, Esté JA, Characterization of the influence of mediator complex in HIV-1 transcription. J. Biol. Chem. 289, 27665–27676 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Mekdad HE, Boutant E, Karnib H, Biedma ME, Sharma KK, Malytska I, Laumond G, Roy M, Réal E, Paillart J-C, Moog C, Darlix JL, Mély Y, de Rocquigny H, Characterization of the interaction between the HIV-1 Gag structural polyprotein and the cellular ribosomal protein L7 and its implication in viral nucleic acid remodeling. Retrovirology. 13 (2016), doi: 10.1186/s12977-016-0287-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Abbas W, Dichamp I, Herbein G, The HIV-1 Nef protein interacts with two components of the 40S small ribosomal subunit, the RPS10 protein and the 18S rRNA. Virol. J. 9, 103 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Kovalenko A, Chable-Bessia C, Cantarella G, Israël A, Wallach D, Courtois G, The tumour suppressor CYLD negatively regulates NF-kappaB signalling by deubiquitination. Nature. 424, 801–805 (2003). [DOI] [PubMed] [Google Scholar]
- 67.Li Z, Wu J, Chavez L, Hoh R, Deeks SG, Pillai SK, Zhou Q, Reiterative Enrichment and Authentication of CRISPRi Targets (REACT) identifies the proteasome as a key contributor to HIV-1 latency. PLOS Pathogens. 15, 1–21 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Chen ZJ, Ubiquitin signalling in the NF-kappaB pathway. Nat. Cell Biol. 7, 758–765 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Timmons A, Fray E, Kumar M, Wu F, Dai W, Bullen CK, Kim P, Hetzel C, Yang C, Beg S, Lai J, Pomerantz JL, Yukl SA, Siliciano JD, Siliciano RF, HSF1 inhibition attenuates HIV-1 latency reversal mediated by several candidate LRAs In Vitro and Ex Vivo. Proc. Natl. Acad. Sci. U. S. A. 117, 15763–15771 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Cotto KC, Wagner AH, Feng Y-Y, Kiwala S, Coffman AC, Spies G, Wollam A, Spies NC, Griffith OL, Griffith M, DGIdb 3.0: a redesign and expansion of the drug-gene interaction database. Nucleic Acids Res. 46, D1068–D1073 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Yang WM, Yao YL, Seto E, The FK506-binding protein 25 functionally associates with histone deacetylases and with transcription factor YY1. EMBO J. 20, 4814–4825 (2001). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Zhu W, Li Z, Xiong L, Yu X, Chen X, Lin Q, FKBP3 promotes proliferation of non-small cell lung cancer cells through regulating Sp1/HDAC2/p27. Theranostics. 7, 3078–3089 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Lee EG, Boone DL, Chai S, Libby SL, Chien M, Lodolce JP, Ma A, Failure to regulate TNF-induced NF-kappaB and cell death responses in A20-deficient mice. Science. 289, 2350–2354 (2000). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Huang H, Liu S, Jean M, Simpson S, Huang H, Merkley M, Hayashi T, Kong W, Rodríguez-Sánchez I, Zhang X, Yosief HO, Miao H, Que J, Kobie JJ, Bradner J, Santoso NG, Zhang W, Zhu J, A novel bromodomain inhibitor reverses HIV-1 latency through specific binding with BRD4 to promote tat and P-TEFb association. Front. Microbiol. 8, 1035 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Bender AM, Simonetti FR, Kumar MR, Fray EJ, Bruner KM, Timmons AE, Tai KY, Jenike KM, Antar AAR, Liu P-T, Ho Y-C, Raugi DN, Seydi M, Gottlieb GS, Okoye AA, Del Prete GQ, Picker LJ, Mankowski JL, Lifson JD, Siliciano JD, Laird GM, Barouch DH, Clements JE, Siliciano RF, The landscape of persistent viral genomes in ART-treated SIV, SHIV, and HIV-2 infections. Cell Host Microbe. 26, 73–85.e4 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Fimia GM, Stoykova A, Romagnoli A, Giunta L, Di Bartolomeo S, Nardacci R, Corazzari M, Fuoco C, Ucar A, Schwartz P, Gruss P, Piacentini M, Chowdhury K, Cecconi F, Ambra1 regulates autophagy and development of the nervous system. Nature. 447, 1121–1125 (2007). [DOI] [PubMed] [Google Scholar]
- 77.Nardacci R, Amendola A, Ciccosanti F, Corazzari M, Esposito V, Vlassi C, Taibi C, Fimia GM, Del Nonno F, Ippolito G, D’Offizi G, Piacentini M, Autophagy plays an important role in the containment of HIV-1 in nonprogressor-infected patients. Autophagy. 10, 1167–1178 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Li M, Liu W, Bauch T, Graviss EA, Arduino RC, Kimata JT, Chen M, Wang J, Clearance of HIV infection by selective elimination of host cells capable of producing HIV. Nat. Commun. 11, 4051 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Passaes CPB, Bruel T, Decalf J, David A, Angin M, Monceaux V, Muller-Trutwin M, Noel N, Bourdic K, Lambotte O, Albert ML, Duffy D, Schwartz O, Sáez-Cirión A, Ultrasensitive HIV-1 p24 assay detects single infected cells and differences in reservoir induction by latency reversal agents. J. Virol. 91 (2017), doi: 10.1128/JVI.02296-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Bradley T, Ferrari G, Haynes BF, Margolis DM, Browne EP, Single-cell analysis of quiescent HIV infection reveals host transcriptional profiles that regulate proviral latency. Cell Rep. 25, 107–117.e3 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Golumbeanu M, Cristinelli S, Rato S, Munoz M, Cavassini M, Beerenwinkel N, Ciuffi A, Single-cell RNA-seq reveals transcriptional heterogeneity in latent and reactivated HIV-infected cells. Cell Rep. 23, 942–950 (2018). [DOI] [PubMed] [Google Scholar]
- 82.Singh V, Dashti A, Mavigner M, Chahroudi A, Latency reversal 2.0: Giving the immune system a seat at the table. Curr. HIV/AIDS Rep. 18, 117–127 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Joung J, Konermann S, Gootenberg JS, Abudayyeh OO, Platt RJ, Brigham MD, Sanjana NE, Zhang F, Genome-scale CRISPR-Cas9 knockout and transcriptional activation screening. Nat. Protoc. 12, 828–863 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Xiao T, Li W, Wang X, Xu H, Yang J, Estrogen-regulated feedback loop limits the efficacy of estrogen receptor–targeted breast cancer therapy. Proceedings of the (2018) (available at https://www.pnas.org/content/115/31/7869.short). [DOI] [PMC free article] [PubMed]
- 85.Li W, Köster J, Xu H, Chen C-H, Xiao T, Liu JS, Brown M, Liu XS, Quality control, modeling, and visualization of CRISPR screens with MAGeCK-VISPR. Genome Biol. 16, 281 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Breitling R, Armengaud P, Amtmann A, Herzyk P, Rank products: a simple, yet powerful, new method to detect differentially regulated genes in replicated microarray experiments. FEBS Lett. 573, 83–92 (2004). [DOI] [PubMed] [Google Scholar]
- 87.Jilek BL, Zarr M, Sampah ME, Rabi SA, Bullen CK, Lai J, Shen L, Siliciano RF, A quantitative basis for antiretroviral therapy for HIV-1 infection. Nat. Med. 18, 446–451 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Borducchi EN, Liu J, Nkolola JP, Cadena AM, Yu W-H, Fischinger S, Broge T, Abbink P, Mercado NB, Chandrashekar A, Jetton D, Peter L, McMahan K, Moseley ET, Bekerman E, Hesselgesser J, Li W, Lewis MG, Alter G, Geleziunas R, Barouch DH, Antibody and TLR7 agonist delay viral rebound in SHIV-infected monkeys. Nature. 563, 360–364 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Kuleshov MV, Jones MR, Rouillard AD, Fernandez NF, Duan Q, Wang Z, Koplev S, Jenkins SL, Jagodnik KM, Lachmann A, McDermott MG, Monteiro CD, Gundersen GW, Ma’ayan A, Enrichr: a comprehensive gene set enrichment analysis web server 2016 update. Nucleic Acids Res. 44, W90–7 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Bliss CI, The toxicity of poisons applied jointly1. Ann. Appl. Biol. 26, 585–615 (1939). [Google Scholar]
- 91.Jiang G, Mendes EA, Kaiser P, Wong DP, Tang Y, Cai I, Fenton A, Melcher GP, Hildreth JEK, Thompson GR, Wong JK, Dandekar S, Synergistic reactivation of latent HIV expression by ingenol-3-angelate, PEP005, targeted NF-kB signaling in combination with JQ1 induced p-TEFb activation. PLoS Pathog. 11, e1005066 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Fig. S1. Pathways and networks of the clusters.
Fig. S2. Sorting strategy of GFP+ latent cells post AZD5582 stimulation.
Fig. S3. SAHA stimulated CRISPR screens.
Fig. S4. HDAC as a potential synergistic drug target in AZD5582 stimulated CRISPR screens.
Fig. S5. The knockout efficiencies in latency cell line cells.
Fig. S6. Validation of top hits in the CRISPR screens for the combinational strategy in the Jurkat latency cell line.
Fig. S7. The activation and viability of primary CD4+ T cells following the combinational LRAs treatment.
Fig. S8. Validating AZD and HDACi combination predictions.
Fig. S9. Single-cell RNA-seq analysis.
Fig. S10. Transient low-level viremia is induced in cART-suppressed Rhesus macaques following AZD5582 and JQ1 treatment.
Fig. S11. The knockout efficiencies in primary CD4+ T cells from one representative PLWH were shown at gene expression level.
Fig. S12. Validation of top hits in the CRISPR screens for the combinational strategy ex vivo using primary CD4+ T cells isolated from PLWH.
Table S1. The group of genes that synergize with AZD5582 with statistical significance (FDR<0.1) using the rank product algorithm.
Table S2. The group of genes that synergize with SAHA with statistical significance (FDR<0.1) using the rank product algorithm.
Table S3. Genes that are selected for validation, and their validation status in cell line model and in the T cells of PLWH.
Data File S1. Individual level data for figures and supplementary figures.
Data File S2. The MAGeCK analysis results of genome-wide CRISPR screens on AZD5582 stimulated GFP− cells vs. pooled control cells (Fig. 1c).
Data File S3. The normalized scores in 578 genes and their cluster assignments in Figure 1e.
Data File S4. The MAGeCK analysis results of genome-wide CRISPR screens on AZD5582 stimulated GFP+ cells vs. pooled control cells (Fig. 4b).
Data File S5. The MAGeCK analysis results of genome-wide CRISPR screens on SAHA stimulated GFP+ cells vs. pooled control cells (Supplementary Fig. 6a).
Data File S6. The rank product of all genes in AZD5582 stimulated GFP+ cells.
Data File S7. The rank product of all genes in SAHA stimulated GFP+ cells.
Data File S8. The sequences of sgRNAs used in single-gene knockout in cell line and primary cells.
Data Availability Statement
Genome-wide screening data will be deposited at NCBI Short Read Archive (SRA) under the accession number (PRJNA689546).