

Abstract
Background and Objective
Glepaglutide is a novel, long-acting, glucagon-like peptide-2 analogue in a stable aqueous formulation for subcutaneous dosing to treat patients with short bowel syndrome. This study was conducted primarily to characterise the pharmacokinetics of glepaglutide in healthy subjects.
Methods
In this open-label, partially randomised, parallel-group study, healthy subjects were evenly randomised to glepaglutide 5 or 10 mg dosed subcutaneously once weekly for 6 weeks or to a single intravenous infusion of glepaglutide 1 mg. Each group comprised 15 subjects. Blood samples were drawn to determine plasma concentrations of the parent drug and its two main metabolites. Concentrations of glepaglutide were calculated as the sum of these three analytes. Citrulline was included as a pharmacodynamic biomarker. Safety was assessed throughout the study.
Results
From a comparison of pharmacokinetic parameters following subcutaneous versus intravenous dosing, it is concluded that the pharmacokinetics of glepaglutide following subcutaneous dosing are primarily determined by slow release of the two main glepaglutide metabolites from a subcutaneous depot. For subcutaneous dosing once weekly, the two main metabolites accounted for >98% of the overall glepaglutide exposure at steady state, with the parent drug contributing to less than 1% of exposure. The estimated mean (95% confidence interval) effective half-life for glepaglutide 5 and 10 mg was 124 (73–185) h and 88 (31–146) h, respectively. There was an increase in the citrulline concentration for both glepaglutide subcutaneous dose levels. No safety issues were identified.
Conclusions
Slow release of active metabolites following subcutaneous dosing leads to a significantly protracted pharmacokinetic profile for glepaglutide. These results support that once- or twice-weekly subcutaneous dosing of glepaglutide could be an efficacious therapy for intestinal rehabilitation.
Clinicaltrials.gov Identifier
Supplementary Information
The online version contains supplementary material available at 10.1007/s40261-022-01210-1.
Key Points
| For the intended subcutaneous route of administration, the pharmacokinetics of glepaglutide is primarily determined by the slow release of the two main glepaglutide metabolites from a subcutaneous depot. The parent drug contributed to less than 1% of exposure at steady state. |
| The pharmacokinetic profile of glepaglutide is significantly protracted following subcutaneous dosing, with an estimated half-life of several days. The results support that once-weekly subcutaneous dosing of glepaglutide could be an efficacious therapy for intestinal rehabilitation in patients with short bowel syndrome. |
Introduction
Patients with short bowel syndrome (SBS) are characterised by having a reduced intestinal absorptive surface area, mainly due to extensive surgical bowel resection [1–4]. Glucagon-like peptide-2 (GLP-2) is a specific intestinal growth factor that plays a key role in enhancing intestinal mucosal morphology, function, and integrity under normal and pathophysiological conditions [5]. Pharmacological GLP-2 treatment aims accordingly to improve intestinal function, thereby reducing and potentially eliminating the need for parenteral administration of nutrients and fluids in patients with SBS who are dependent on parenteral support.
In response to nutrient intake, GLP-2 is secreted by L cells located mainly in the ileum and oral part of the colon. These intestinal segments are frequently resected in patients with SBS [6]. Degradation of native GLP-2 by the circulating enzyme dipeptidyl peptidase-IV occurs rapidly, with subsequent clearance via the kidney [7]. The short half-life of 5–7 min for circulating native GLP-2 [8] is a major drawback for its use in a therapeutic setting, which is improved for the currently marketed GLP-2 analogue teduglutide with a half-life in circulation of approximately 2 h [9].
Glepaglutide is a novel long-acting GLP-2 analogue, which is tested in phase III studies using a once-weekly or twice-weekly dosing regime to treat patients with SBS. Nine amino acid substitutions have been introduced in the glepaglutide polypeptide compared to native GLP-2, and a C-terminal structure inducing probe tail consisting of six lysines has been added. The result is a linear peptide with 39 natural l-amino acids that is stable in aqueous solution, enabling the glepaglutide medical product intended for the market to be dosed via a convenient ready-to-use autoinjector device.
Glepaglutide is catabolised via the same catabolic pathways as endogenous peptides into amino acids that are reutilised in the endogenous amino acid pool for the de novo biosynthesis of structural or functional body proteins and peptides (unpublished data). Unpublished data have indicated that release of the two main glepaglutide metabolites (M1 and M2, resulting from proteolytic cleavage at the C-terminal structure inducing probe tail; Table 1) from the injection site into the systemic circulation is the rate-limiting step that governs overall plasma exposure after subcutaneous (SC) dosing of glepaglutide. The human GLP-2 receptor potencies of the parent drug, M1 and M2 are in the same range (Table 1), and the pharmacokinetic and pharmacodynamic profile of glepaglutide can thus be conceptually understood as a composite of these three analytes. We report on a study to characterise the pharmacokinetic properties of glepaglutide in healthy subjects.
Table 1.
In vitro potency and amino acid sequence of glepaglutide analytes and hGLP-2

EC50 half maximal effective concentration, hGLP-2 human glucagon-like peptide-2, M1 main glepaglutide metabolite 1, M2 main glepaglutide metabolite 2
Red letters represent amino acid substitutions; green letters represent amino acid additions
aIn vitro potencies as determined by quantifying cAMP accumulation in a cell line stably overexpressing the hGLP-2 receptor
Methods
Study Design
This was an open-label, partially randomised, parallel-group study to characterise the pharmacokinetic profile of glepaglutide administered as SC or intravenous (IV) doses to healthy subjects. The study consisted of three parts. In part 1, subjects were evenly randomised to either SC glepaglutide 1 mg or 5 mg dosed daily for 7 days. As the dosing frequency tested in part 1 differs from the once-weekly or twice-weekly dosing regime intended for clinical use, the results are not described further herein. In part 2, subjects were evenly randomised to either SC glepaglutide 5 mg or 10 mg dosed once weekly for 6 weeks. In part 3, additional subjects were recruited into the study to receive a single IV infusion of glepaglutide 1 mg over 15 min. Part 3 of the study was included to enable a comparison of the pharmacokinetic properties of glepaglutide when dosed subcutaneously versus intravenously, thereby demonstrating the impact of a SC depot formation on glepaglutide pharmacokinetics.
Each dose group included 15 randomised subjects to ensure at least 12 completers per group. The study was conducted at Covance Clinical Research Unit Inc., Dallas, TX, USA in accordance with the Declaration of Helsinki (and its amendments) and applicable Good Clinical Practice guidelines, laws and regulations. The protocol was approved by an independent ethics committee, and all patients provided written informed consent.
Study Population
Healthy male and female subjects of any ethnic origin were eligible for the study. Subjects were required to be between 18 and 60 years of age, inclusive, to have a body mass index between 18 and 30 kg/m2, inclusive, and to be in good health as confirmed by no clinically significant findings from a medical history, physical examination, electrocardiogram, vital signs measurements, and clinical laboratory evaluations. The selection of healthy subjects was consistent with standard practices for clinical pharmacology studies.
Study Procedures
Subjects randomised to once-weekly dosing of either glepaglutide 5 mg or 10 mg were admitted to the research clinic the day before receiving the first of 6-weekly SC doses of glepaglutide. The subjects stayed at the clinic until day 4 after the initial dose, after which they returned on days 5, 6, 8, 15, 22 and 29 for dosing (days 8, 22 and 29) and study procedures. Subjects were again re-admitted to the clinic during days 36–38 for administration of the last dose and returned for outpatient visits on days 39, 41, 43, 50, 57 and 64 for study procedures and follow-up. The final follow-up visit occurred on day 71.
For each group, 2-mL blood samples were collected to determine plasma concentrations of the parent drug and its two main metabolites (M1 and M2). Consistent with the expected protraction of the pharmacokinetic profile for glepaglutide when dosed subcutaneously, pharmacokinetic blood sampling in subjects receiving 5 and 10 mg of glepaglutide once weekly occurred at the day 1 visit (pre-dose and 0.5, 1, 2, 4, 8, 12, 16, 20, 24, 36, 48, 60, 72, 96 and 120 h post-dose); pre-dose on days 8, 15, 22 and 29; and in connection with the last dosing visit on day 36 (pre-dose and 0.5, 1, 2, 4, 8, 12, 16, 20, 24, 36, 48, 72, 120, 168, 336, 504, 672 and 840 h post-dose). Blood sampling (2-mL sample) for citrulline, a biomarker for a small bowel enterocyte mass [10], was performed pre-dose on days 1, 3, 5 and 7 as well as on days 8, 10, 12, 14, 21, 28, 35 and 42.
Subjects enrolled to receive a single IV infusion of 1 mg of glepaglutide were admitted into the clinic on the day before dosing and stayed in-house until 2 days after dosing, when they were discharged. The subjects returned to the clinic for final study procedures on day 22 after dosing. The IV infusion of glepaglutide was to occur at a rate of 4 mg/hour for 15 min. Blood sampling for a pharmacokinetic analysis in these subjects was conducted pre-dose and at 5, 10, 15, 20, 25, 35, 45, 60, 75 and 90 min and 2, 3, 4, 8, 12, 16, 20, 24, 36 and 48 h after the start of the IV infusion, and again on day 22. Safety and tolerability were assessed throughout the study by means of adverse event (AE) recording, vital signs measurements, electrocardiograms, clinical laboratory evaluations including anti-glepaglutide antibodies, physical examinations and monitoring for injection-site reactions.
Bioanalysis
Venous blood samples for a pharmacokinetic analysis were drawn in tubes containing K2EDTA placed on ice until processing (within 15 min). Plasma was prepared at 4 °C (range 2–8 °C) in a refrigerated centrifuge at a 1200–1500 relative centrifugal force for 15 min. The plasma samples were stored at −70 °C until analysed. Parent drug and metabolites (M1 and M2) were quantified at the Charles River Laboratories Edinburgh Ltd (UK) using immunoaffinity extraction followed by liquid chromatography-tandem mass spectroscopy; for further details, see Electronic Supplementary Material. The assay was validated according to applicable guidelines for a bioanalysis of plasma samples. The assay had a lower limit of quantification of 25.0 pmol/L (parent drug and M1) and 50.0 pmol/L (M2), and an upper limit of detection of 2500 pmol/L (parent drug and M1) and 5000 pmol/L (M2). Plasma citrulline concentrations were measured by LabCorp, Burlington, NC, USA using liquid chromatography-tandem mass spectroscopy.
Outcome Measures
Pharmacokinetic parameters were calculated for the parent drug, the two main metabolites (M1 and M2) and glepaglutide using a non-compartmental approach. For glepaglutide, the concentration–time profile was constructed as the sum of concentrations of the parent drug, M1, and M2 for the individual measuring timepoints, based on which pharmacokinetic parameters were then calculated. The human GLP-2 receptor potencies of the three component analytes are in the same range (Table 1), which is a prerequisite for the sum of the three analytes to be representative for the overall pharmacological effect of glepaglutide. Additionally, one analyte (M2) accounts for the vast majority of the total exposure, whereas one analyte (parent) contributes very little (Table 3), which further reduces the impact of subcomponent potency differences on the conceptual glepaglutide analyte.
Table 3.
Pharmacokinetic parameters at steady state (day 36) for glepaglutide and individual analytes following once-weekly dosing
| Parameter | Glepaglutide | Individual analytes | ||
|---|---|---|---|---|
| Parent | M1 | M2 | ||
| SC glepaglutide 5 mg after 6 once-weekly doses | ||||
| AUClast (h × pmol/L) | 119,000 (56.6%) [15] | 516 (87.2%) [13] | 6590 (113%) [15] | 109,000 (57.8%) [15] |
| AUCτ (h × pmol/L) | 106,000 (53.2%) [15] | 833 (47.5%) [6] | 7650 (86.7%) [15] | 96,600 (54.4%) [15] |
| Cmax (pmol/L) | 2830 (66.9%) [15] | 223 (52.4%) [15] | 371 (98.7%) [15] | 2510 (71.4%) [15] |
| tmax (h); median (min-max) | 12.0 (8.00–24.1) [15] | 0.52 (0.45–1.03) [15] | 8.00 (0.93–12.0) [15] | 24.0 (8.02–24.1) [15] |
| t1/2, z (h), mean (SD) | 228 (474) [14] | 3.8 (2.5) [6] | 17.0 (6.53) [15] | 231 (475) [14] |
| CL/F (L/h) | 1390 (47.5%) [6] | |||
| MRAUCτ | 6.82 (151%) [6] | 95.4 (62.3%) [6] | ||
| RAAUC | 1.64 (51.0%) [15] | 0.649 (67.7%) [3] | 0.932 (53.9%) [13] | 1.75 (54.5%) [15] |
| t1/2, eff (h); estimated mean (95% CI) | 123.7 (73.3–184.9) [15] | |||
| SC glepaglutide 10 mg after 6 once-weekly doses | ||||
| AUClast (h × pmol/L) | 281000 (86.8%) [14] | 1040 (79.3%) [14] | 14300 (117%) [14] | 259000 (90.1%) [14] |
| AUCτ (h × pmol/L) | 232000 (94.7%) [14] | 1450 (74.6%) [10] | 15200 (108%) [14] | 211000 (99.7%) [14] |
| Cmax (pmol/L) | 5940 (97.1%) [14] | 432 (34.2%) [14] | 716 (102%) [14] | 5320 (106%) [14] |
| tmax (h), median (min–max) | 12.1 (4.00–36.0) [14] | 0.53 (0.47–2.05) [14] | 8.00 (4.00–12.0) [14] | 18.1 (4.00–36.0) [14] |
| t1/2, z (h); mean (SD) | 254 (198) [8] | 2.8 (2.1) [10] | 37.7 (44.2) [14] | 255 (251) [9] |
| CL/F (L/h) | 1600 (74.6%) [10] | |||
| MRAUCτ | 9.93 (134%) [10] | 153 (61.0%) [10] | ||
| RAAUC | 1.37 (53.3%) [14] | 0.695 (42.7%) [9] | 0.829 (68.5%) [14] | 1.43 (57.0%) [14] |
| t1/2, eff (h), estimated mean (95% CI) | 88.3 (30.6–146.3) [14] | |||
Geometric mean (CV%) is presented unless otherwise specified. Numbers in square brackets denote the number of subjects included in the analysis
AUClast area under the plasma concentration–time curve from time zero to the time of the last measurable concentration, AUCτ area under the plasma concentration–time curve over a dosing interval, CI confidence interval, CL/F apparent total plasma clearance, Cmax maximum observed plasma concentration, CV coefficient of variation, h hours, max maximum, min minimum, MRAUCτ metabolite: parent ratio based on AUCτ, RAAUC accumulation index based on the area under the curve over the dosing interval, SD standard deviation, t1/2, eff effective half-life, t1/2, z apparent plasma terminal elimination half-life, tmax time of the maximum observed plasma concentration
The following rules were applied for the determination of pharmacokinetic parameters as defined in Table 3. The area under the plasma concentration–time curve from time zero to the time of the last measurable concentration was not calculated for subjects with fewer than three consecutive quantifiable post-dose analyte concentrations available in the pharmacokinetic concentration–time profile. The apparent plasma terminal elimination half-life (t1/2,z) was not calculated if a subject’s terminal phase coefficient of determination was less than 0.7. A regression analysis was performed without weighting; therefore, a minimum value of 0.7 for R2 adjusted was necessary for acceptance of linear regression. Values below the lower limit of quantification were set to missing for endpoint derivations. Late positive concentration values were set to missing if following at least two consecutive values below the lower limit of quantification. Further, quantifiable pre-dose concentrations > 5% of the maximum observed plasma concentration were excluded from the pharmacokinetic analysis and mean concentration figures. Nominal times were used where actual times were missing. In addition to the terminal half-life, the effective half-life for accumulation [11] was calculated. This was to account for the rate of drug loss across the entire dosing interval, as plasma concentrations of glepaglutide decline in a multi-phasic manner. The mean effective half-life, t1/2,eff, was calculated for glepaglutide as: t1/2,eff = ln[2]×τ/ln[RAAUC/(RAAUC − 1)], where τ is the dosing interval and RAAUC is the geometric mean of the individual ratios AUCτ (sixth dose)/AUCτ (first dose). Plasma citrulline concentrations were assessed in subjects receiving SC glepaglutide.
Statistical Analysis
No formal sample size calculations were done. Pharmacokinetic parameters and other endpoints were summarised descriptively. All pharmacokinetic parameters except the mean effective half-life were calculated using Phoenix WinNonlin, Version 6.4 (Certara USA, Princeton, NJ, USA).
Results
Demographic and Baseline Characteristics
The three dose groups (SC glepaglutide 5 mg once weekly, SC glepaglutide 10 mg once weekly and a single IV glepaglutide 1-mg injection) each comprised 15 healthy subjects. Of these 45 subjects, 43 completed the study. The two non-completers concerned a subject receiving glepaglutide 10 mg once weekly who was withdrawn on day 29 because of a positive drug screen and a subject in the IV 1 mg dose group who did not attend the follow-up visit at day 22. All 45 subjects contributed to the pharmacokinetic and safety analyses. Demographic characteristics were comparable across groups (Table 2).
Table 2.
Demographic characteristics
| Variable | Glepaglutide dose group | ||
|---|---|---|---|
| SC 5 mg once weekly | SC 10 mg once weekly | IV 1 mg single dose | |
| Age (years), mean (SD) | 37 (12) | 42 (12) | 40 (11) |
| Sex, n (%) | |||
| Male | 7 (47) | 10 (67) | 10 (67) |
| Female | 8 (53) | 5 (33) | 5 (33) |
| Race, n (%) | |||
| Black or African American | 5 (33) | 4 (27) | 7 (47) |
| White | 10 (67) | 10 (67) | 8 (53) |
| Multiple | – | 1 (7) | – |
| Ethnicity, n (%) | |||
| Hispanic or Latino | 2 (13) | 4 (27) | 4 (27) |
| Not Hispanic or Latino | 13 (87) | 11 (73) | 11 (73) |
| Weight (kg), mean (SD) | 76.4 (11.6) | 78.8 (10.8) | 74.8 (8.2) |
| BMI (kg/m2), mean (SD) | 26.2 (2.4) | 26.6 (2.4) | 26.2 (1.8) |
BMI body mass index, IV intravenous, SC subcutaneous, SD standard deviation
Pharmacokinetics
Pharmacokinetic results presented below pertain to steady-state conditions, i.e. after the sixth weekly SC dose of glepaglutide, unless otherwise specified. Attainment of steady state after the sixth SC dose is supported by an evaluation of trough concentrations, which reached a steady plateau from the third weekly SC dose and onwards.
Mean plasma concentration profiles are shown for the parent drug and the two main metabolites (M1 and M2) for patients receiving once-weekly SC doses of glepaglutide 5 mg (Fig. 1a) and glepaglutide 10 mg (Fig. 1b). Pharmacokinetic parameters for parent, M1, M2 and for the conceptual glepaglutide analyte (the sum of parent, M1 and M2) are presented in Table 3. The maximal plasma concentration after SC administration of either 5 or 10 mg occurred after approximately 0.5, 8 and 18–24 h for the parent durg, M1 and M2, respectively. For both the SC 5-mg and 10-mg once-weekly dose groups, M2 was the predominant metabolite representing 91% of the total exposure. M1 accounted for approximately 7% of total exposure, whereas the parent drug contributed less than 1%. Consequently, time to maximum observed plasma concentration of glepaglutide was driven by the metabolites and occurred at 12 h.
Fig. 1.

Pharmacokinetic profile of parent drug and two main metabolites (M1 and M2) after six once-weekly subcutaneous doses of glepaglutide 5 mg (a) and glepaglutide 10 mg (b). Nominal times used
In contrast, following IV infusion, the plasma concentration of the parent drug was characterised by a tri-phasic decline, entering the elimination phase after 24 h with a terminal half-life of 11.7 h. Of the overall exposure, 92% was attributable to the parent drug and approximately 2% and 7% to M1 and M2, respectively (Fig. 2 and Table 4). Similar to observations following SC administration, the time to maximum observed plasma concentration of M1 (0.63 h) occurred before the time to maximum observed plasma concentration of M2 (1.50 h). For IV infusion, the steady-state distribution volume of the parent drug was 14.0 L and corresponded to approximately 20% of the bodyweight of the study participants. The clearance of the parent obtained from the IV group was 5.72 L/hour.
Fig. 2.
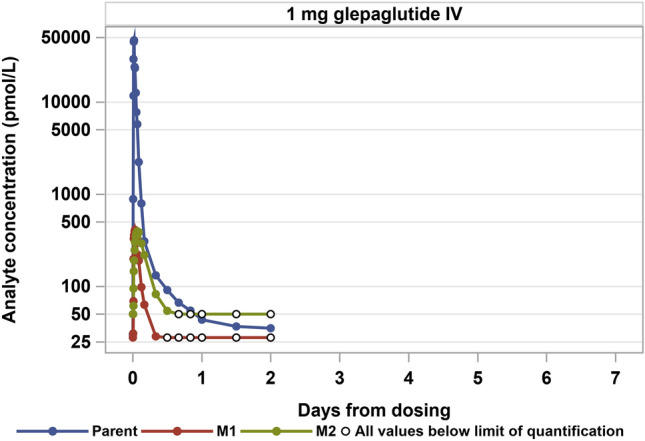
Pharmacokinetic profile of parent drug and two main metabolites (M1 and M2) after a single intravenous (IV) infusion of glepaglutide 1 mg. Nominal times used
Table 4.
Pharmacokinetic parameters for glepaglutide and metabolites following intravenous infusion of 1 mg
| Individual analytes | ||||
|---|---|---|---|---|
| Parameter | Glepaglutide | Parent | M1 | M2 |
| AUClast (h × pmol/L) | 40,800 (35.3%) [15] | 37,100 (38.8%) [15] | 782 (103%) [15] | 1580 (108%) [15] |
| AUCinf (h × pmol/L) | 44,200 (33.7%) [13] | 40,500 (39.8%) [12] | 1010 (70.4%) [14] | 2930 (59.3%) [9] |
| Cmax (pmol/L) | 54,900 (46.3%) [15] | 54,100 (46.9%) [15] | 446 (84.5%) [15] | 419 (79.9%) [15] |
| tmax (h), median (min-max) | 0.42 (0.28–0.58) [15] | 0.42 (0.28–0.58) [15] | 0.63 (0.35–1.02) [15] | 1.50 (1.25–3.00) [15] |
| t1/2,z (h), mean (SD) | 10.4 (8.93) [13] | 11.7 (8.10) [12] | 1.2 (0.24) [15] | 2.6 (0.90) [14] |
| CL (L/h) | 5.72 (39.8%) [12] | |||
| Vss (L) | 14.0 (120%) [12] | |||
| MRAUCinf | 0.0260 (97.3%) [11] | 0.0812 (94.2%) [8] | ||
Geometric mean (CV%) is presented unless otherwise specified. Numbers in square brackets denote the number of subjects included in the analysis
AUCinf area under the plasma concentration–time curve from time zero to infinity, AUClast area under the plasma concentration–time curve from time zero to the time of the last measurable concentration, CL clearance, Cmax maximum observed plasma concentration, CV coefficient of variation, h hours, max maximum, min minimum, MRAUCinf metabolite: parent ratio based on AUCinf, SD standard deviation, t1/2,z apparent plasma terminal elimination half-life, tmax time of the maximum observed plasma concentration, Vss volume of distribution at steady state
Based on an analysis of the steady-state plasma concentration curve following SC dosing, glepaglutide (constructed as the sum of the concentrations of the parent drug, M1 and M2) appeared to decline in a multi-phasic manner, with the terminal elimination half-life, t½,z, on average exceeding 200 h (Table 3). However, the prolonged terminal elimination phase does not contribute to the accumulation of the drug, as only a small fraction of the drug is eliminated during the terminal phase. The overall elimination kinetics for SC dosing is therefore more appropriately characterised by the effective half-life, t1/2eff, in line with regulatory guidance [11–13]. The estimated mean (95% CI) effective half-life for glepaglutide 10 mg was calculated to 88 (31–146) h based on the observed increase in area under the concentration–time curve by a factor of 1.37 at steady state compared with the first weekly dose of glepaglutide. The corresponding half-life for the 5-mg dose group was in a similar range (Table 3).
Pharmacodynamics
Mean plasma concentrations of the pharmacodynamic biomarker citrulline increased during the glepaglutide treatment period and remained elevated above baseline levels for the remainder of the study period (Fig. 3), which indicates some degree of persistency in the improvement of enterocyte mass beyond the glepaglutide treatment phase. Based on this limited set of observations, there was no clear dose response in the plasma citrulline concentration between the 5-mg and 10-mg once-weekly dosing groups.
Fig. 3.
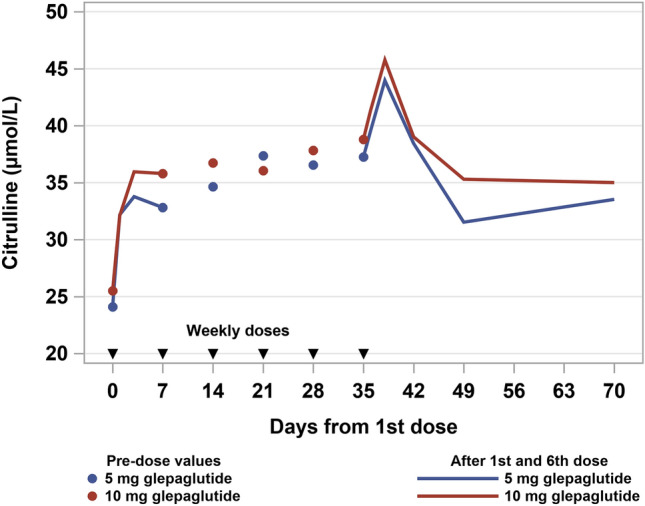
Mean plasma citrulline concentrations pre-dose and after the first and sixth dose in subjects receiving subcutaneous glepaglutide 5 mg and 10 mg once weekly
Safety and Tolerability
Overall, glepaglutide was well tolerated. No deaths or serious AEs were reported. The majority of AEs were mild in severity. Injection-site reactions were the most commonly reported AEs, with these events comprising 36 of 48 reported AEs in the 5-mg once-weekly dose group and 50 of 61 reported AEs in the 10-mg once-weekly dose group. Most injection-site reactions were reported by 0.5 h after dosing and were of transient duration. No AEs were reported in subjects receiving 1 mg of glepaglutide by an IV infusion.
Discussion
The pharmacokinetic profile of glepaglutide after an IV infusion was characterised by a fast distribution, fast elimination and a low volume of distribution. Metabolism of the parent drug was limited, and the metabolites only accounted for a small fraction of the total area under the concentration–time curve. In contrast, glepaglutide exhibited significantly protracted pharmacokinetics when injected subcutaneously, with an estimated mean effective half-life for 10 mg of glepaglutide of 88 h when administered once weekly. For both the SC 5-mg and 10 mg once-weekly dose groups, the two main metabolites accounted for the vast majority of the overall glepaglutide exposure. These results support that extensive metabolisation of the parent compound occurs in a SC depot following SC dosing and that the formed active metabolites (predominantly M1 and M2) are slowly released from the depot, thereby extending the effective exposure to glepaglutide. Another example of similar metabolisation is long-acting insulin glargine (Lantus®), for which the main active metabolite is slowly released from the subcutaneous depot and accounts for > 90% of plasma exposure [14]. The specific proteases involved are unknown, but the cleavage site on the C-terminal site of lysine is characteristic of trypsin-like proteases.
Time from dosing to maximum concentration was substantially longer for the two main metabolites than for the parent compound, which is consistent with gradual metabolite formation within, and subsequent systemic absorption from, the subcutaneous injection site. The mean absolute bioavailability of the parent drug following SC doses of 5 mg and 10 mg of glepaglutide was less than 1%, which reflects the retention and extensive metabolism within the subcutis. However, as significant amounts of M1 and particularly M2 enter into plasma following SC dosing, the functional bioavailability of glepaglutide is substantially higher. In fact, this study found that M2 accounted for > 90% of the overall glepaglutide exposure at steady state following SC dosing. With the in vitro potency of M2 on the human GLP-2 receptor being on par with that of the parent drug and M1, the vast majority of the pharmacodynamic effect of glepaglutide is driven by the M2 metabolite. Thus, the functional bioavailability of glepaglutide includes the active metabolites and is substantially higher than indicated by the absolute bioavailability.
Encouragingly, the pharmacodynamic biomarker citrulline [10] increased for both once-weekly dosing of 5 mg and 10 mg of glepaglutide in the current study, consistent with the mechanism of action of a GLP-2 receptor agonist in increasing a small bowel enterocyte mass. More definitive conclusions on the clinical long-term efficacy and safety of once-weekly dosing of glepaglutide are pending the completion of the phase III study programme.
Conclusions
The results of this clinical pharmacology study with the novel GLP-2 analogue glepaglutide show a significantly protracted pharmacokinetic profile at steady state following SC dosing. The estimated mean (95% CI) effective half-life for once-weekly dosing of 10 mg of glepaglutide was 88 (31–146) h, with results in a similar range for the 5-mg dose group. It is concluded that the pharmacokinetics of glepaglutide following SC dosing is primarily determined by the slow release of the two main glepaglutide metabolites from a SC depot. There was an increase in citrulline concentrations for both glepaglutide SC dose levels. Glepaglutide was well tolerated. These results support that once or twice-weekly SC dosing of glepaglutide could be an efficacious therapy for intestinal rehabilitation.
Supplementary Information
Below is the link to the electronic supplementary material.
Acknowledgements
The authors thank all study participants. Medical writing assistance was provided by Karsten Soendergaard of Zealand Pharma.
Declarations
Funding
The study was funded by Zealand Pharma.
Conflicts of interest/Competing interests
Mikkel Askjær Agersnap, Kim Mark Knudsen, Carsten Boye Knudsen and Mark Berner-Hansen are employed at Zealand Pharma. Kim Sonne was formerly employed at Zealand Pharma.
Ethics approval
Prior to the start of the study, the study protocol, protocol amendment, informed consent form and study recruitment materials were reviewed and approved by the applicable institutional review board (Schulman Associates Institutional Review Board, Cincinnati, OH, USA; approval number 201704938).
Consent to participate
All study participants gave written informed consent prior to undergoing any study-related procedures or assessments.
Consent for publication
Not applicable.
Availability of data and material
Data will be provided upon request to the corresponding author.
Code availability
The code will be provided upon request to the corresponding author.
Authors’ contributions
MAA, KS, CBK and MBH was scientifically responsible for the study, and KMK was responsible for the statistical considerations and analyses. CBK and KMK take full responsibility for the verification of the underlying data. All authors provided input to the manuscript and edited and approved the manuscript for submission. All authors had full access to the clinical study report and associated documents.
References
- 1.Buchman AL, Scolapio J, Fryer J. AGA technical review on short bowel syndrome and intestinal transplantation. Gastroenterology. 2003;124(4):1111–1134. doi: 10.1016/S0016-5085(03)70064-X. [DOI] [PubMed] [Google Scholar]
- 2.Jeppesen PB. Spectrum of short bowel syndrome in adults: intestinal insufficiency to intestinal failure. JPEN J Parenter Enteral Nutr. 2014;38(1 Suppl.):8S–13S. doi: 10.1177/0148607114520994. [DOI] [PubMed] [Google Scholar]
- 3.Pironi L, Arends J, Baxter J, Bozzetti F, Pelaez RB, Cuerda C, et al. ESPEN endorsed recommendations: definition and classification of intestinal failure in adults. Clin Nutr. 2015;34(2):171–180. doi: 10.1016/j.clnu.2014.08.017. [DOI] [PubMed] [Google Scholar]
- 4.Ladefoged K, Hessov I, Jarnum S. Nutrition in short-bowel syndrome. Scand J Gastroenterol Suppl. 1996;216:122–131. doi: 10.3109/00365529609094567. [DOI] [PubMed] [Google Scholar]
- 5.Drucker DJ, Erlich P, Asa SL, Brubaker PL. Induction of intestinal epithelial proliferation by glucagon-like peptide 2. Proc Natl Acad Sci U S A. 1996;93(15):7911–7916. doi: 10.1073/pnas.93.15.7911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Drucker DJ. Gut adaptation and the glucagon-like peptides. Gut. 2002;50(3):428–435. doi: 10.1136/gut.50.3.428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Tavares W, Drucker DJ, Brubaker PL. Enzymatic- and renal-dependent catabolism of the intestinotropic hormone glucagon-like peptide-2 in rats. Am J Physiol Endocrinol Metab. 2000;278(1):E134–E139. doi: 10.1152/ajpendo.2000.278.1.E134. [DOI] [PubMed] [Google Scholar]
- 8.Hartmann B, Johnsen AH, Orskov C, Adelhorst K, Thim L, Holst JJ. Structure, measurement, and secretion of human glucagon-like peptide-2. Peptides. 2000;21(1):73–80. doi: 10.1016/S0196-9781(99)00176-X. [DOI] [PubMed] [Google Scholar]
- 9.Kim ES, Keam SJ. Teduglutide: a review in short bowel syndrome. Drugs. 2017;77(3):345–352. doi: 10.1007/s40265-017-0703-7. [DOI] [PubMed] [Google Scholar]
- 10.Crenn P, Coudray-Lucas C, Thuillier F, Cynober L, Messing B. Postabsorptive plasma citrulline concentration is a marker of absorptive enterocyte mass and intestinal failure in humans. Gastroenterology. 2000;119(6):1496–1505. doi: 10.1053/gast.2000.20227. [DOI] [PubMed] [Google Scholar]
- 11.Gidal BE, Clark AM, Anders B, Gilliam F. The application of half-life in clinical decision making: Comparison of the pharmacokinetics of extended-release topiramate (USL255) and immediate-release topiramate. Epilepsy Res. 2017;129:26–32. doi: 10.1016/j.eplepsyres.2016.10.020. [DOI] [PubMed] [Google Scholar]
- 12.Boxenbaum H, Battle M. Effective half-life in clinical pharmacology. J Clin Pharmacol. 1995;35(8):763–766. doi: 10.1002/j.1552-4604.1995.tb04117.x. [DOI] [PubMed] [Google Scholar]
- 13.US Food and Drug Administration. Guidance for industry: clinical pharmacology section of labeling for human prescription drug and biological products: content and format. 2016. Available from: https://www.fda.gov/regulatory-information/search-fda-guidance-documents/clinical-pharmacology-labeling-human-prescription-drug-and-biological-products-content-and-format. Accessed 17 Oct 2022.
- 14.Lucidi P, Porcellati F, Rossetti P, Candeloro P, Andreoli AM, Cioli P, et al. Metabolism of insulin glargine after repeated daily subcutaneous injections in subjects with type 2 diabetes. Diabetes Care. 2012;35(12):2647–2649. doi: 10.2337/dc12-0271. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.