

Abstract
Spinal cord injury (SCI) is a severe disease of the nervous system that causes irreparable damage and loss of function, for which no effective treatments are available to date. Engineered extracellular vesicles (EVs) carrying therapeutic molecules hold promise as an alternative SCI therapy depending on the specific functionalized EVs and the appropriate engineering strategy. In this study, we demonstrated the design of a drug delivery system of peptide CAQK-modified, siRNA-loaded EVs (C-EVs-siRNA) for SCI-targeted therapy. The peptide CAQK was anchored through a chemical modification to the membranes of EVs isolated from induced neural stem cells (iNSCs). CCL2-siRNA was then loaded into the EVs through electroporation. The modified EVs still maintained the basic properties of EVs and showed favorable targeting and therapeutic effects in vitro and in vivo. C-EVs-siRNA specifically delivered siRNA to the SCI region and was taken up by target cells. C-EVs-siRNA used the inherent anti-inflammatory and neuroreparative functions of iNSCs-derived EVs in synergy with the loaded siRNA, thus enhancing the therapeutic effect against SCI. The combination of targeted modified EVs and siRNA effectively regulated the microenvironmental disturbance after SCI, promoted the transformation of microglia/macrophages from M1 to M2 and limited the negative effects of the inflammatory response and neuronal injury on functional recovery in mice after SCI. Thus, engineered EVs are a potentially feasible and efficacious treatment for SCI, and may also be used to develop targeted treatments for other diseases.
Keywords: Extracellular vesicles, Spinal cord injury, siRNA, Microglia/macrophages polarization, Induced neural stem cells
Graphical abstract
Highlights
-
•
Engineered EVs carrying therapeutic molecules hold promise as an alternative SCI therapy.
-
•
C-EVs-siRNA can specifically deliver siRNA to SCI areas and be taken up by target cells.
-
•
C-EVs-siRNA used the inherent neuroreparative functions of EVs and acted synergistically with the loaded siRNA.
-
•
C-EVs-siRNA modulated microenvironmental disturbance and promoted motor function recovery after SCI.
-
•
C-EVs-siRNA were a potentially viable and efficacious treatment for SCI.
1. Introduction
Spinal cord injury (SCI) is a severe disease that can lead to irreversible loss of neurological function [1]. Although many diseases are being overcome with the continual development of medical technology, no effective treatments are available for SCI, which has plagued human society for hundreds of years [2]. SCI can be classified as traumatic or non-traumatic, depending on its cause. Traumatic SCI is often caused by spinal fractures, such as those resulting from traffic accidents or other accidents, whereas non-traumatic SCI injury is caused by conditions including tumors, inflammation or infection [3]. SCI is divided into two processes: primary injury and secondary injury. Primary injury is mainly caused by the loss of neurons, whereas secondary injury is the main factor that aggravates tissue damage and impairs the recovery process, mainly through inflammation-related injury [4]. Therefore, anti-inflammatory and neuroprotective therapy remains the current focus of drug treatment for SCI [5]. Although therapeutic drugs have been investigated to counteract microglial inflammation and neuronal damage in secondary lesions, their therapeutic efficacy is often limited by the short residence time, poor accumulation and lack of controlled release of drugs in the lesioned tissue [6]. In addition, some drugs do not exert strong neuroprotective effects, mainly because they do not act effectively on target cells in the microenvironment of SCI [7]. Therefore, new safe drugs must be developed to target injured spinal cords.
Extracellular vesicles (EVs) have received substantial attention as an alternative nanocarrier for stem cell therapy in drug delivery and targeted therapy. Increasing studies show that EVs have promising applications in improving drug delivery and therapeutic efficacy, owing to their high biocompatibility, low cytotoxicity and immune inertness [8,9]. EVs are nanoscale membrane vesicles secreted by cells that carry or transport a variety of bioactive substances (e.g., proteins, lipids or RNAs) [10]. Neural stem cells/neural progenitor cells (NSCs/NPCs)-derived EVs have a variety of functions, including regulating the adjacent microenvironment, and acting as independent functional metabolic units and microglia morphogens [[11], [12], [13]]. Bian et al. have demonstrated that EVs from NPCs attenuated photoreceptor apoptosis by inhibiting microglial activation, thereby preserving photoreceptors during retinal degeneration [14]. Cao et al. have shown that EVs derived from NPCs promoted hair follicle growth by delivering miR-100 [15]. In addition, owing to their anti-inflammatory and neuroprotective properties, NSCs/NPCs-derived EVs have been used to treat a range of neurodegenerative diseases. For example, Tian et al. have demonstrated that NPCs-derived EVs elicited targeted anti-inflammatory effects after cerebral ischemia [16]. Ma et al. have shown that EVs released from NPCs regulated neurogenesis through miR-21a [17]. Our previous studies have also demonstrated that EVs secreted by NSCs inhibited apoptosis and inflammatory responses, thereby promoting motor function recovery after SCI [18,19]. Notably, a new NSCs subtype called iNSCs (derivatives of induced pluripotent stem cells (iPSCs)) combines the advantages of NSCs and iPSCs [20]. Because of the strong self-renewal ability of iPSCs, iNSCs can be continually obtained from iPSCs, thus avoiding the invasive collection procedures of traditional NSCs [20,21].
CC chemokine ligand 2 (CCL2) is a chemokine that, after binding to its receptor CC chemokine receptor 2 (CCR2), plays an important role in neuroinflammation, which is closely associated with the infiltration and activation of inflammatory cells [22]. Tian et al. have shown that blocking chemokine CCL2-CCR2 signaling inhibited microglial activation and neuronal cell death caused by pro-inflammatory mediators, and ameliorated neuroinflammatory damage after status epilepticus [23]. Another study has demonstrated that astrocyte derived CCL2 mediated M1 polarization of microglia, and the use of CCR2 inhibitors significantly decreased astrocyte produced CCL2-induced microglial activation and M1 polarization, as well as the expression of inflammatory factors [24]. In agreement with the above results, our previous study has also indicated that up-regulated CCL2 played a key role in microglial activation and neuronal apoptosis after SCI [25]. Specifically, activated astrocytes after SCI release CCL2, which acts on microglia and neurons through the paracrine pathway and promotes microglial activation and neuronal apoptosis by binding CCR2 receptors [25]. Therefore, downregulation of CCL2 expression may be a promising novel therapeutic strategy to modulate microglial activation and neuronal apoptosis after SCI.
Small interfering RNA (siRNA) can selectively suppress specific genes and has shown great potential in gene therapy [26,27]. However, clinical applications of siRNA are impeded by the lack of a safe delivery technology that targets only the desired tissues [28]. At present, the combination of nanocarrier delivery and RNA interference technology has become a new gene drug delivery system, which can effectively deliver siRNA into the human body. Because of their favorable stability and safety, EVs are an emerging biomaterial for delivery among the many nannocarriers widely used for various therapeutic drugs, including proteins, RNAs and chemical drugs; therefore, EVs have excellent prospects as delivery vehicles [9,29]. Engineered EVs can efficiently and safely deliver small RNAs to specific cellular environments [30]. Xu et al. have obtained EVs of mesenchymal stem cells (MSCs) with high CXCR4 expression through genetic engineering as targeted gene drug delivery vectors for cancer treatment [31]. Han et al. have also shown that targeted inhibition of SIRT6 by engineered EVs impairs prostate cancer tumorigenesis and metastasis [32]. As biological delivery vehicles, EVs have facilitated the development of RNA interference therapeutics in neurodegenerative diseases, considering that EVs can cross the blood-brain barrier and avoid the degradation of the carried siRNA by nucleases [33]. For example, Izco et al. have confirmed that rabies virus glycoprotein (RVG)-modified EVs prevented Parkinson's disease by delivering shRNA minicircles [34]. Another study has shown that intranasal administration of EVs derived from MSCs and loaded with PTEN-siRNA improved physiological function in rats after SCI [35]. Therefore, EVs have great promise for targeted drug delivery and SCI treatment, owing to their excellent target specificity and ability to penetrate biological barriers.
The clinical application of EVs is limited, because they mainly accumulate in, and are cleared by, the liver and spleen, thus resulting in a short biological half-life in vivo after systemic administration [36]. In general, EV surfaces are engineered through genetic manipulation or chemical modification to address their poor in vivo distribution [37]. To overcome these obstacles, we constructed novel EVs bearing a peptide CAQK modification and encapsulating siRNA. CAQK selectively binds chondroitin sulfate proteoglycans (CSPGs), which are upregulated in lesions after SCI [38]. Notably, this modification not only delivers EVs to the site of SCI but also targets reactive astrocytes at the site of SCI, because CSPGs are mainly expressed by activated astrocytes at the site of the lesion after SCI.
In conclusion, we demonstrate the design of a drug delivery system of peptide CAQK-modified, siRNA-loaded EVs (C-EVs-siRNA) for SCI-targeted therapy. The spinal cord-targeting peptide CAQK was anchored to the EV membrane through copper-free click chemistry, and CCL2-siRNA was then loaded into the EVs by electroporation. The physicochemical characteristics and biocompatibility of C-EVs-siRNA were extensively studied to ensure their applicability in SCI. Furthermore, in vivo and in vitro tests validated the targeting of CAQK-modified EVs. To explore the profound effects of C-EVs-siRNA in vivo and in vitro, we conducted multiple comprehensive assessments, including histological, imaging and functional investigation. Importantly, phenotypes including axonal, myelin and microglial polarization were tested to elucidate the detailed mechanisms through which C-EVs-siRNA promotes motor function recovery. Overall, C-EVs-siRNA targeted the injured spinal cord in SCI mice and elicited significant functional recovery. This discovery had important clinical therapeutic implications for SCI as well as other neurological diseases associated with inflammation.
2. Materials and methods
2.1. Extraction and identification of EVs
The collected iNSC culture supernatant was centrifuged at 300×g for 10 min, 2,000×g for 10 min and 10,000×g for 30 min at 4 °C to remove cells and debris, and then filtered through a 0.22-μm sterile filter (Millipore). The filtrate was centrifuged at 140,000×g for 70 min at 4 °C in an Optima L-100 XP ultracentrifuge (Beckman Coulter), and the pellet was resuspended in PBS and then ultracentrifuged again at 140,000×g for 70 min. The protein concentration was determined with the BCA method. The morphology of the obtained EVs was observed through transmission electron microscopy (TEM). The diameter size, distribution and zeta potential of EVs were assessed with nanoparticle tracking analysis (NTA). Western blot was used to determine EV-specific surface markers such as CD9, CD63, CD81, Alix and TSG101.
2.2. Modification of EVs with peptide CAQK
Following previous research methods [39], we used a heterobifunctional cross-linking agent to introduce reactive dibenzyl ethylene oxide (DBCO) into amine-containing molecules on EVs. Specifically, 3 mM dibenzocyclooctyl-N-hydroxysuccinimide ester (DBCO-sulfo-NHS) (Sigma-Aldrich, St. Louis, MO, USA) was added to 0.5 mg/mL EVs in PBS and allowed to react in a rotary mixer at room temperature for 4 h. Unbound DBCO-sulfo-NHS was removed through four washing steps on 100 kDa ultrafiltration tubes (Millipore). Then DBCO-conjugated EVs (DBCO-EVs) were attached to azide-containing molecules through copper-free click chemistry. Specifically, 0.3 mM CAQK-N3 (SynthbioCo,. Ltd) or Cy5.5-azide was added to DBCO-EVs in PBS. The reaction was performed in a rotating mixer at 4 °C for 12 h and pH 7.4. The EVs were then floated on a 30% sucrose/D2O cushion and centrifuged at 140,000 g for 70 min to remove unbound ligand. After being washed with PBS, the modified EVs were resuspended and stored at −80 °C until use. To assess the successful binding of EVs to azide ligands, we stained Cy5.5-bound EVs with DiO and examined them under a laser confocal microscope (Stellaris STED; LEICA, Wetzlar, Germany).
2.3. Loading siRNA into EVs through electroporation
Electroporation of EVs loaded with siRNA was performed on the basis of previous studies [40]. Briefly, EVs were mixed with siRNA at a ratio of 1:1 (wt/wt) and diluted to 0.5 mg/mL in electroporation buffer. The mixture was added to a 0.4 mm frozen tube and electroporated in a Gene Pulser Xcell instrument (Bio-Rad, Hercules, CA, USA) at 400 mV and a capacitance of 125 μF (pulse time 10–15 ms). To remove free siRNA/miRNA, we washed the mixture twice with cold PBS, ultracentrifuged it at 140,000 g for 70 min at 4 °C and resuspended the pellet in PBS.
2.4. Serum stability assays
To determine the serum stability of the siRNA delivery system, we incubated Free siRNA, EVs-siRNA or C-EVs-siRNA with 50% fetal bovine serum (FBS). Equal amounts of samples for each group of different incubation times were electrophoresed on agarose gels and then visualized with an UV illuminator.
2.5. In vitro cytotoxicity assays
Astrocytes were cultured at 1.5 × 104/mL in 96-well plates for 24 h. After treatment with EVs, C-EVs, EVs-siRNA and C-EVs-siRNA (100 μg/mL), 100 μL of fresh medium containing Cell Counting Kit-8 (CCK-8, Dojindo, Kumamoto, Japan) solution was added. After incubation at 37 °C for 2 h, the absorbance was measured at 450 nm with an absorbance microplate reader (ELx800; Bio-Tek, Winooski, VT USA).
After astrocytes were treated with C-EVs or C-EVs-siRNA (100 μg/mL), the cells were harvested by centrifugation at 300×g for 5 min at 4 °C. Cells were then resuspended in fluorescein isothiocyanate-labeled Annexin V (5 μL; BD Biosciences) and PI (5 μL; BD Biosciences) for 10 min in the dark. Apoptosis rates were detected with a flow cytometer (FACSCalibur; BD Biosciences).
2.6. In vivo imaging
In vivo imaging was performed with Cy5.5-labeled EVs. At 6 h after tail vein administration, mice were imaged with an in vivo imaging system (IVIS) (PerkinElmer, Waltham, MA, USA) to assess in vivo fluorescence signal distribution. Then the mice were sacrificed by cervical dislocation, and the heart, liver, spleen, lungs, kidneys and spinal cord were removed for fluorescence imaging. Cy5.5-related fluorescence signals were analyzed in Living Image software (PerkinElmer).
2.7. Immunohistochemistry
Hematoxylin and eosin (H&E) staining or Luxol fast blue (LFB) staining were used to observe spinal cord tissue repair or myelin damage. The methods and criteria were as in previous studies [18,41]. Images were taken under an optical microscope (Zeiss Axioscope; Zeiss, Dublin, CA, USA).
2.8. Nissl staining
After preparation, spinal cord slices were stained with 0.05% cresyl violet at 40 °C for 10 min. Next, sections were differentiated, dehydrated and removed from 95% alcohol, 100% alcohol and xylene. The researchers, blinded to the experimental groups, acquired images under an optical microscope.
2.9. In vivo toxicity evaluation
After SCI, groups of mice were treated with PBS, EVs, EVs-siRNA or C-EVs-siRNA, and the blood from each mouse was isolated for serum biochemical assays. Major organs, including the heart, liver, spleen, lungs and kidneys, were used for H&E staining.
2.10. DHE staining
Freshly prepared frozen spinal cord slices were incubated with 2 μmol/L of the fluorescent dye dihydroethidium (DHE; Thermo Fisher Scientific, Waltham, MA, USA) for 30 min in a humidified chamber at 37 °C and protected from the light. A Leica microscope (DMi8 Thunder) was used to capture images and quantify fluorescence intensity. After the DHE staining was completed, we randomly selected three slices from each group and three different fields of view per slice.
2.11. TUNEL staining
For TUNEL staining, slices were treated with 0.3% Triton X-100 for 15 min. After being washed with PBS, the slices were incubated with a TUNEL staining kit (Beyotime, China) according to the manufacturer's instructions. Images were captured with a Leica microscope (DMi8 Thunder), and TUNEL positive cells were quantified in ImageJ software (NIH, Bethesda, MD, USA). Three sections were selected from each group, and three fields of view were randomly selected from each section, and tunel-positive cells were counted.
2.12. Immunofluorescence staining
Cells or slices were permeabilized with 0.3% Triton X-100, blocked with 5% bovine serum albumin and incubated with primary antibodies overnight at 4 °C. Primary antibodies to the following were used: GFAP (Servicebio, Wuhan, China; 1:200), NF200 (Abcam, UK; 1:200), NeuN (Abcam, 1:400), F4/80 (Abcam, 1:200), iNOS (Abcam, 1:500), Arg1 (Cell Signaling Technology, Danvers, MA, USA; 1:50) and CCL2 (Serotec, Oxford, UK; 1:250). Conjugated secondary antibodies (Alexa Fluor 488 and Alexa Fluor 594; 1:200; Jackson ImmunoResearch, West Grove, PA, USA) were then added, and counterstaining was performed with DAPI (1:100; Beyotime). Images were captured with a Leica microscope (DMi8 Thunder).
2.13. Western blot
The total protein of cells or spinal cord tissue was extracted, and the protein concentration was determined with the BCA method (Thermo Fisher Scientific). The proteins were separated by SDS-PAGE and transferred onto PVDF membranes (Millipore, Burlington, MA, USA). Membranes were blocked with 5% bovine serum albumin for 1 h at room temperature, incubated with primary antibodies overnight at 4 °C and then incubated with secondary antibodies for 1 h. Bands were visualized with enhanced chemiluminescence reagent (Thermo Fisher Scientific). Band density in three independent experiments was quantified in ImageJ software, and protein levels were normalized to those of GAPDH. Primary antibodies to the following were used: CD9 (Santa Cruz Biotechnology, Santa Cruz, CA, USA; 1:500), CD63 (Abcam, 1:2,000), CD81 (Santa Cruz Biotechnology, 1:500), Alix (Santa Cruz Biotechnology, 1:1,000), TSG101 (Abcam, 1:2,000), Calnexin (Cell Signaling Technology, 1:1,000), MAP2 (Proteintech, 1:10,000), NF200 (Abcam, 1:5,000), GFAP (Servicebio, 1:1,000), Cleaved caspase-3 (Cell Signaling Technology, 1:1,000), Bax (Affinity, 1:1,000), Bcl-2 (Abcam, 1:1,000), Caspase-3 (Cell Signaling Technology, 1:1,000), iNOS (Abcam, 1:1,000), Arg1 (Cell Signaling Technology, 1:1,000), CCL2 (Proteintech, 1:2,000), MBP (Proteintech, 1:500) and GAPDH (Proteintech, 1:20000).
2.14. Statistical analysis
Data are presented as mean ± standard deviation for at least three independent biological replicates. GraphPad software 8.0 was used for statistical analysis. Colocalization analysis of confocal microscopy data and quantifications of immunoblotting results were conducted using ImageJ software. Student's t-test was used for comparisons between two groups. For multiple group comparisons, one-way or two-way analysis of variance with Tukey's post hoc test was used. A value of P < 0.05 were considered statistically significant.
3. Results
3.1. Preparation and characterization of C-EVs
The iPSCs were differentiated into iNSCs according to established protocols [42]. EVs were isolated from iNSCs-conditioned medium by ultracentrifugation (Fig. S1A). The purified unmodified iNSCs-EVs were identified by TEM, NTA and western blot. TEM images showed that the EVs exhibited a typical cup-like morphology (Fig. 1A). NTA revealed that the EVs had an average diameter of 120 nm (Fig. 1B). Western blot analysis revealed that CD9, CD63, CD81, Alix and TSG101 (known markers of EVs) were enriched in EVs, whereas Calnexin, which is expressed in the endoplasmic reticulum, was not detected in EVs (Fig. 1F). These data indicated that EVs were successfully isolated from iNSCs conditioned medium.
Fig. 1.

Preparation and characterization of C-EVs and in vitro targeting studies. (A) Morphology of EVs and C-EVs under TEM. (B) NTA analysis showing the particle size distribution of EVs and C-EVs. (C) Stability analysis of C-EVs in PBS and 10% FBS. (D) Number of particles per milliliter of EVs and C-EVs. (E) Zeta potential of EVs and C-EVs. (F) Western blot analysis of the EV biomarkers CD9, CD63, CD81, Alix and TSG101, and negative control Calnexin. (G, H) The carboxy terminus of the peptide was labeled with the Cy5.5 fluorophore, and the modified EVs were labeled with DiO dye. The co-localization of Cy5.5 fluorescence and DiO fluorescence was observed under a laser confocal microscope. (I) Co-culture of TGF-β-treated astrocytes with Cy5.5-labeled EVs and Cy5.5-labeled C-EVs, followed by observation of the uptake efficiency of EVs and C-EVs by astrocytes. (J) Uptake efficiency of C-EVs by astrocytes treated with TGF-β for the indicated times. *P < 0.05; **P < 0.01 (n = 3 per group).
For spinal cord-targeted modification of EVs, EVs were combined with the peptide CAQK (Fig. S1B). The morphology and size distribution of CAQK-modified EVs (C-EVs) were assessed with TEM and NTA. Specifically, C-EVs exhibited a typical cup-like morphology (Fig. 1A). NTA indicated that the constructed C-EVs displayed a slightly larger mean diameter than EVs and were well dispersed in PBS (pH 7.4) and 10% FBS, with long-term stability (Fig. 1B and C). EVs and C-EVs also did not significantly differ in the number of nanoparticles per mL (Fig. 1D). In addition, the surface zeta potential of EVs averaged −25.4 mV, and the surface zeta potential of C-EVs increased to an average of −21.4 mV (Fig. 1E). Western blot analysis indicated that the peptide CAQK used for targeted modification had no significant effects on EV marker proteins such as CD9, CD63, CD81, Alix and TSG101 (Fig. 1F). To verify the successful modification of EVs, we also conjugated Cy5.5-azide with DBCO groups, and used DiO dyes to label the modified EVs. The Cy5.5 fluorescence observed under confocal fluorescence microscopy strongly colocalized with the DiO fluorescence, thus indicating that the peptide CAQK was successfully anchored on the surfaces of the EVs (Fig. 1G and H). Therefore, C-EVs may have potential for in vitro and in vivo studies.
3.2. In vitro targeting studies of C-EVs
Next, we assessed whether the peptide CAQK used for targeted modification might affect the viability of cultured astrocytes in vitro. Flow cytometry results indicated that EVs modified with the peptide CAQK did not affect astrocyte viability (Figs. S1C and D). CCK-8 results further demonstrated that C-EVs had no significant effects on the viability of cultured astrocytes in vitro (Fig. S1E). TGF-β treatment induces astrocytes to produce CSPGs [43]. To verify the targeting of EVs modified by the peptide CAQK, we co-cultured C-EVs with TGF-β-treated (10 ng/ml) astrocytes to observe the uptake efficiency of C-EVs by astrocytes. Confocal microscopy based on the fluorescence intensity of C-EVs was then used to analyze the level of internalization. C-EVs co-localized more with TGF-β-treated astrocytes than did EVs, and the signal increased with treatment time (Fig. 1I). Fig. 1I shows that the uptake efficiency of C-EVs was significantly higher than that of EVs after astrocytes were treated with TGF-β for 72 h. In addition, Fig. 1J shows that the uptake efficiency of C-EVs by astrocytes increased significantly with TGF-β treatment time. These results indicated that C-EVs are targeted for the internalization of activated astrocytes, thereby further verifying the presence of CAQK peptide on EVs. Consequently, this biochemical method efficiently binds CAQK peptides to the surfaces of EVs, thereby yielding C-EVs with high affinity/specificity for cells expressing CSPGs.
3.3. The ability of C-EVs to target the injured spinal cord
Biodistribution is a fundamental method for probing the accumulation properties of nanoformulations in vivo. The targeted distribution of nanocarriers in the SCI area can enhance the therapeutic effects of drugs. To test whether C-EVs specifically bind the lesion site after SCI, we performed a multi-targeted binding experiment. EVs or C-EVs labeled with the fluorescent molecule Cy5.5 were intravenously injected into a SCI mouse model 72 h after injury. An IVIS was used to assess the tissue distribution of EVs or C-EVs and detect the fluorescence intensity of Cy5.5 in major organs. According to IVIS images, 6 h after tail vein injection of C-EVs, relatively high fluorescence was observed at the spinal cord lesions, whereas lower fluorescence was observed at the lesion site in the EVs group (Fig. 2A and B). In addition, the spinal cord was dissected and analyzed through IVIS. The fluorescence intensity of the spinal cord lesions was significantly higher after administration of C-EVs than unmodified EVs (Fig. 2C and D). These results indicated that the CAQK modification significantly enhanced the targeting of EVs to the SCI region. Next, various organs of mice were dissected and quantitatively analyzed through IVIS to study C-EVs biodistribution. For better image contrast, we imaged each organ separately. EVs mainly accumulated in the liver, followed by the lungs (Fig. 2E and F). Interestingly, C-EVs injection decreased the signal in the liver, spleen and kidneys compared with the EVs-treated group (Fig. 2E and F). The greater spinal cord targeting of C-EVs than EVs was consistent with previous findings in CAQK-modified polymeric micelles [41,44].
Fig. 2.

The ability of C-EVs to target the injured spinal cord. (A) Fluorescence images of Cy5.5-labeled EVs or Cy5.5-labeled C-EVs in mice. (B) Quantification of the fluorescence intensity of lesions in mice. (C) Representative IVIS images of the spinal cord in mice treated with PBS, Cy5.5-labeled EVs or Cy5.5-labeled C-EVs. (D) Quantification of fluorescence intensity in SCI areas in vitro. (E) Representative IVIS images of anatomical organs in mice treated with PBS, Cy5.5-labeled EVs or Cy5.5-labeled C-EVs. (F) Quantification of fluorescence intensity in the indicated organs. (G) Distribution of DIO-labeled EVs and DIO-labeled C-EVs at the center of spinal cord lesions. (H, I) Colocalization analysis of Cy5.5-labeled EVs and Cy5.5-labeled C-EVs with astrocytes at the center of spinal cord lesions. *P < 0.05; **P < 0.01 (n = 3 per group).
For further histological studies, we performed immunofluorescence staining analysis of spinal cord lesions. Prominent DIO-labeled fluorescent spots were observed in the EVs group, and the signal level was higher in the C-EVs group (Fig. 2G), in agreement with the IVIS findings. In addition, to verify the mechanism underlying the accumulation in diseased tissue, we analyzed the affinity of C-EVs for astrocytes. The co-localization of Cy5.5 and GFAP (an astrocyte marker) was lower in the EVs group, whereas the co-localization of Cy5.5 and GFAP in the C-EVs group was significantly higher (Fig. 2H and I), thus indicating extensive interaction between C-EVs and astrocytes. Together, these results demonstrated that modification with the peptide CAQK enhanced the ability of EVs to target spinal cord lesions.
3.4. Characterization of siRNA-loaded C-EVs
Among potential treatments for SCI, gene therapy, particularly siRNA therapy, are promising. However, despite extensive research and development, siRNAs’ lack of tissue specificity, induction of immune responses and cytotoxicity limit their clinical efficacy. Our previous study suggested that down-regulating CCL2 expression in reactive astrocytes may be a promising new strategy for the treatment of microglial activation and neuronal apoptosis following SCI [25]. Therefore, CCL2-siRNA was selected to evaluate the therapeutic effects of C-EVs-mediated siRNA delivery with SCI. CCL2-siRNA was loaded into EVs by electroporation (Fig. S1F). First, the ratio of siRNA to EVs was determined to establish optimal conditions for maximal association. To determine the loading efficiency and release kinetics of the loaded RNA, cel-miRNA-67 was chosen as the siRNA counterpart to minimize experimental background [45,46]. The miRNA loading efficiency of C-EVs was determined to be 17% by Q-PCR. The siRNA-loaded EVs or C-EVs were then subjected to TEM, NTA and western blot analysis. C-EVs-siRNA was similar to EVs-siRNA in morphology, zeta potential and the number of nanoparticles per milliliter, except that it had a slightly larger average diameter (Fig. 3A–D). Importantly, C-EVs-siRNA, as compared with C-EVs, also had no significant effects on morphology, -size and zeta potential (Fig. 3A and B VS Fig. 1A and B). Western blot results additionally confirmed that the final engineered EVs retained the protein markers of EVs (Fig. 3E). Overall, the general characteristics of EVs remained unchanged after siRNA loading.
Fig. 3.

Characterization of siRNA-loaded C-EVs. (A) Morphology of EVs-siRNA and C-EVs-siRNA under TEM. (B) NTA analysis of particle size distribution of EVs-siRNA and C-EVs-siRNA. (C) Number of particles per milliliter of EVs-siRNA and C-EVs-siRNA. (D) Zeta potential of EVs-siRNA and C-EVs-siRNA. (E) Western blot analysis of the EV biomarkers CD9, CD63, CD81, Alix and TSG101, and negative control Calnexin. (F) Analysis of the serum stability of free siRNA, EVs-siRNA and C-EVs-siRNA by gel electrophoresis. (G) Kinetics of siRNA release from C-EVs-siRNA. (H) CCK-8 evaluation of the effects of EVs-siRNA and C-EVs-siRNA on the viability of cultured astrocytes in vitro. (I) Flow cytometry detection of the effects of EVs-siRNA and C-EVs-siRNA on apoptosis of cultured astrocytes in vitro (n = 3 per group).
Protecting siRNA from degradation is an important reason for choosing EVs as a delivery system. To examine the tolerance to serum-mediated siRNA degradation, we incubated C-EVs-siRNA, EVs-siRNA and free siRNA with FBS for a series of time intervals. As shown in Fig. 3F, the band representing naked siRNA completely disappeared after 6 h of incubation, whereas siRNAs encapsulated in EVs and C-EVs were still detected at 12 h. These results suggested that the membranes of EVs maintain siRNA integrity, thereby protecting siRNA from nucleases in FBS after modification and electroporation.
3.5. Toxicity studies of C-EVs-siRNA in vitro and in vivo
The release kinetics and effects on cell viability of siRNA-loaded C-EVs were next investigated. After 2 h of incubation, the efflux of siRNA was approximately 9% in PBS and approximately 10% in 50% FBS. After 24 h of incubation, the release was approximately 93% in PBS and approximately 95% in FBS (Fig. 3G). CCK-8 results indicated that C-EVs-siRNA had no significant effect on cell viability (Fig. 3H). Furthermore, flow cytometry was used to assess the effects of C-EVs-siRNA on apoptosis at different time periods. After the addition of C-EVs-siRNA, the degree of apoptosis did not change significantly among time periods (Fig. 3I). The above results demonstrated that the engineered EVs had similar biosafety to native EVs. We then performed in vivo experiments to further confirm this finding. EVs, EVs-siRNA, C-EVs-siRNA or the same volume of PBS was administered to mice by tail vein injection, and the mice were sacrificed after continuous administration. The results of blood biochemical tests indicated no significant changes in liver function (ALT, AST, ATP and TP), renal function (BUN and CREA) and cardiac function indexes (LDH and CK) in each group of mice (Figs. S2A–H). The results of H&E staining showed no clear lesions in the organs and tissues of each group (Fig. S2I). Therefore, C-EVs-siRNA treatment was well tolerated without causing substantial systemic toxicity.
Next, we assessed the knockdown efficiency of C-EVs-siRNA on CCL2 by inducing astrocyte activation with TNF-α in vitro. Briefly, astrocytes were first stimulated with TNF-α (10 ng/mL) for 15 min, then replaced with normal medium and incubated with EVs, EVs-con-siRNA, EVs-siRNA or C-EVs-siRNA for 24 h. Western blot results indicated that CCL2 expression in astrocytes increased after TNF-α stimulation, whereas EVs-siRNA treatment inhibited the TNF-α-induced increase in CCL2 expression (Figs. S3A and B). Furthermore, C-EVs-siRNA further reduced CCL2 expression compared with EVs-siRNA treatment (Figs. S3A and B), suggesting that TNF-α may stimulate astrocytes to produce CSPGs, leading to increased uptake of CAQK-modified EVs by astrocytes. ELISA analysis further confirmed the western blot results (Fig. S3C). In conclusion, the above treatment with siRNA-loaded EVs inhibited TNF-a-induced CCL2 expression and release. Similarly, western blot results of spinal cord tissue indicated that EVs-siRNA effectively inhibited the expression of CCL2, whereas C-EVs-siRNA inhibited the expression to a greater extent (Figs. S3D and E). The co-localization of immunofluorescence staining of the astrocyte marker GFAP and CCL2 in vivo also revealed that activated astrocytes released CCL2 after SCI, and EVs-siRNA inhibited the expression of CCL2 (Figs. S3F–H). Interestingly, in agreement with the in vitro findings, C-EVs-siRNA further decreased CCL2 expression compared with EVs-siRNA (Figs. S3F–H). These in vitro and in vivo results demonstrated not only the effectiveness of siRNA-loaded EVs in knocking down CCL2 expression but also the targeting of C-EVs-siRNA.
3.6. C-EVs-siRNA promotes motor function recovery after SCI in mice
The neuroprotective effects of C-EVs-siRNA were assessed by evaluation of tissue damage and motor function recovery in a mouse model of SCI. PBS, EVs, EVs-siRNA or C-EVs-siRNA was administered immediately after SCI. At 28 days after SCI, morphological changes in the injured spinal cord were observed by GFAP staining. After EVs, EVs-siRNA or C-EVs-siRNA treatment, the lesion area was significantly smaller than that in the PBS group (Figs. S4A and B). Moreover, the lesion area in the C-EVs-siRNA group was significantly smaller than those in the EVs and EVs-siRNA groups (Figs. S4A and B). To provide additional evidence of motor function recovery, we used MRI to evaluate pathological changes in each group. MRI is an effective noninvasive method for the diagnosis and assessment of SCI, and was used to assess tissue repair 28 days after SCI. As shown in Figs. S4C and D, MRI images indicated low signal intensity at the injury site, which appeared as a fluid-filled cyst [47]. EVs-siRNA and C-EVs-siRNA treatment ameliorated this pathology. Specifically, EVs-siRNA and C-EVs-siRNA-treated spinal cords were more intact, with smaller cysts and better treatment effects than those receiving PBS treatment, in agreement with the results in Figs. S4A and B. In addition, C-EVs-siRNA treatment more significantly promoted the recovery of injured spinal cord and normal morphology.
Functional tests (e.g., BMS score, footprint test, rotarod test and MEP) are used to measure whether these imageable pathological effects translate into motor function recovery (Fig. 4A). Following SCI, animals exhibit flaccid paralysis followed by a modest time-dependent recovery. However, we observed that the degree of recovery varied among groups. As shown in Fig. 4B and C, the BMS scores of mice receiving EVs, EVs-siRNA and C-EVs-siRNA on day 7 post-injury were higher than those in the PBS group. From 14 to 28 days after injury, the BMS score in the C-EVs-siRNA group was significantly higher than those in the EVs and EVs-siRNA groups. Footprint tests demonstrated that mice treated with EVs-siRNA, compared with EVs, showed significantly faster gait recovery and improved motor coordination, and this effect was enhanced in the C-EVs-siRNA group (Fig. 4D and E), as demonstrated in rotarod assays (Fig. 4F). Together, these results demonstrated that C-EVs-siRNA treatment has an additive therapeutic effect on motor function in SCI mice beyond those of EVs and EVs-siRNA treatment. To further study motor function behavioral recovery, we performed electrophysiological analysis. As shown in Fig. 4G and H, on the 28th day after injury, the MEP amplitude in the C-EVs-siRNA group was higher than those in the EVs and EVs-siRNA groups, thus indicating that the electrophysiological function of the hindlimbs showed better recovery after C-EVs-siRNA administration. Together, these results suggested that EVs, EVs-siRNA and C-EVs-siRNA treatment all promoted functional behavioral recovery after SCI in mice, and these beneficial effects were more pronounced after C-EVs-siRNA treatment.
Fig. 4.

C-EVs-siRNA promotes motor function recovery after SCI in mice. (A) Schematic diagram of experimental design. (B, C) BMS scores of mice treated with PBS, EVs, EVs-siRNA or C-EVs-siRNA during the recovery period 28 days after injury. (D, E) Typical diagrams of footprint tests in mice treated with PBS, EVs, EVs-siRNA or C-EVs-siRNA on day 28 after injury. (F) Rotarod test on day 28 after injury in the indicated groups. (G) Electrophysiological assessment with MEP analysis on day 28 post-injury (arrows indicate onset of evoked potentials). (H) Quantification of peak-to-peak MEP amplitude and latency in mice in the indicated treatment groups. *P < 0.05; **P < 0.01; ***P < 0.001 (n = 6 per group).
3.7. C-EVs-siRNA promotes axonal regeneration and remyelination necessary for locomotion
We then tested how C-EVs-siRNA produced this promising effect. From the perspective of motor function recovery, axonal regeneration and remyelination are essential. Therefore, the density or state of axons in the lesioned core of the injured spinal cord was next assessed to elucidate the anatomical basis of the observed functional recovery. Neurofilament-200 (NF200) was used to assess axonal regeneration. In the PBS group, few NF200+ axons were observed in the lesions, whereas many NF200+-labeled fibers crossed the glial scar in the EVs and EVs-siRNA-treated groups, which showed axonal extensions (Fig. 5A and B). Interestingly, the C-EVs-siRNA group had more extended neurofilaments than the other two treatment groups, thus indicating that the combination of targeted modified EVs and siRNA was more effective (Fig. 5A and B). To confirm these results, we used the neuronal marker NeuN to stain live neurons in a specific region (Z1-Z4) in the injured spinal cord in both groups of mice, which was located at a specified distance from the lesion boundary, as previously described [48]. The number of surviving NeuN+ neurons in the Z1-Z3 region of the C-EVs-siRNA group was significantly higher than that in the EVs and EVs-siRNA groups (Fig. 5C and D). In addition, the Western blot results of NF-200 and MAP-2 also confirmed the trend between groups in immunofluorescence staining (Fig. 5E–H). Collectively, these results suggested that administration of C-EVs-siRNA was more effective in promoting functional behavioral recovery and post-injury axonal regeneration than EVs or EVs-siRNA alone.
Fig. 5.

C-EVs-siRNA promotes axonal regeneration necessary for locomotion. (A) Representative immunofluorescence images of NF200+ in injured spinal cords on day 28 after injury. (B) Quantitative analysis of the percentage of NF200+ areas at the indicated distances from the center of SCI lesions to the total area of distant uninjured axons. (C) Representative immunofluorescence images of NeuN+ neurons in the Z1-Z4 region adjacent to the lesion core on day 28 post-injury. (D) Quantification of NeuN+ neurons in the Z1-Z4 region adjacent to the lesion core on day 28 post-injury. (E-H) Western blot detection and quantitative analysis of NF200, MAP2 and GFAP proteins in spinal cord tissue on the 28th day after injury. *P < 0.05; **P < 0.01; ***P < 0.001 (n = 6 per group).
Axonal demyelination and damage to myelin structures persist after SCI. Therefore, we assessed the level of axonal myelination and the myelin ultrastructure at the injury site through LFB staining and TEM. First, LFB staining indicated that the residual amount of myelin in the EVs, EVs-siRNA and C-EVs-siRNA groups was significantly higher than that in the PBS group, and the C-EVs-siRNA treatment effect was better than those in the EVs and EVs-siRNA groups (Figs. S5A and B). TEM was used to assess the ultrastructure of myelin sheaths in mice receiving different treatments. The G-ratio (the ratio of axon inner diameter to outer diameter) and myelin diameter were also analyzed. TEM indicated that the myelin sheaths in the sham group were arranged in a tight and orderly manner, with uniform thickness and a clear layered structure. The myelin ultrastructure in the mice was most severely damaged after SCI. However, improvements in myelin after treatment with EVs alone were very limited. The number, diameter and thickness of myelin sheaths significantly increased after EVs-siRNA and C-EVs-siRNA treatments (Figs. S5C–E). In conclusion, both LFB staining and TEM results indicated a positive effect of C-EVs-siRNA treatment on demyelination. MBP is an important component of myelination during nerve regeneration after SCI [49]. Western blot analysis of MBP indicated that although both EVs and EVs-siRNA treatments increased MBP expression beyond that in the PBS group, C-EVs-siRNA treatment increased MBP expression to a greater extent (Figs. S5F and G). Together, our findings suggested that both EVs and siRNA promoted axonal growth, and that targeting modified EVs combined with siRNA administration had the greatest effect on axonal regeneration and myelin protection.
3.8. C-EVs-siRNA decreases apoptosis and promotes neuronal repair by affecting the inflammatory response and microglial polarization
Next, we aimed to explore how C-EVs-siRNA induced axonal regeneration. Neuronal survival is a prerequisite for axon regeneration. To investigate the regulatory effect of C-EVs-siRNA on neuronal apoptosis after SCI, we performed TUNEL staining on neurons 7 days after SCI. As shown in Fig. 6A and B, the number of TUNEL-positive cells in the PBS group significantly increased, whereas EVs, EVs-siRNA and C-EVs-siRNA treatment significantly decreased neuronal apoptosis. In contrast, C-EVs-siRNA elicited the best neuronal protection, showing fewest TUNEL-positive cells. Nissl staining was then used to assess the number of spinal motor neurons and the degree of damage. The number of neurons in the EVs, EVs-siRNA and C-EVs-siRNA groups was significantly higher than that in the PBS group 7 days after SCI. Interestingly, in agreement with the TUNEL staining results, the C-EVs-siRNA group showed the most neurons and the least damage (Fig. 6C and D). In addition, western blot of spinal cord tissue indicated that the expression of the pro-apoptotic markers Cleaved caspase-3 and Bax in the EVs, EVs-siRNA and C-EVs-siRNA groups was diminished, and the expression of the anti-apoptotic protein Bcl-2 was elevated (Fig. 6E and F). The C-EVs-siRNA group showed the best inhibitory effect on apoptosis (Fig. 6E and F). Together, the above results suggested that EVs and EVs-siRNA decreased neuronal apoptosis and promoted neuronal recovery, whereas targeted modification plus loaded siRNA enhanced the effects of EVs.
Fig. 6.

C-EVs-siRNA promotes neuronal repair by decreasing neuronal apoptosis in the SCI area. (A) On the 7th day after injury, TUNEL staining was used to detect cell apoptosis. (B) Quantitative analysis of TUNEL positive cells. (C) Nissl staining to assess the number and degree of injury of spinal motor neurons. (D) Quantitative analysis of the percentage of Nissl bodies at the indicated distances from the center of SCI lesions. (E) Western blot detection of pro-apoptotic and anti-apoptotic-related protein expression in spinal cord tissue on the 7th day after injury. (F) Quantitative analysis of pro-apoptotic and anti-apoptotic proteins. *P < 0.05; **P < 0.01; ***P < 0.001 (n = 6 per group).
To explore the mechanism through which C-EVs-siRNA decreases neuronal apoptosis, we detected inflammatory cells, such as microglia/macrophages, which invade injury sites and regulate “secondary injury” after SCI. First, we assessed oxidative stress levels in the SCI area through DHE staining. EVs or EVs-siRNA treatment inhibited reactive oxygen species (ROS) levels in the injured area of SCI, whereas C-EVs-siRNA treatment promoted this therapeutic effect (Fig. 7A and B). Next, we measured the concentrations of the proinflammatory cytokines TNF-α, IL-1β and IL-6, and the anti-inflammatory cytokines TGF-β, IL-4 and IL-10 in the spinal cord with ELISA. The administration of EVs, EVs-siRNA or C-EVs-siRNA, compared with PBS, significantly decreased the concentrations of pro-inflammatory cytokines and increased the concentrations of anti-inflammatory cytokines (Fig. 7C and D). However, treatment with C-EVs-siRNA, compared with EVs or EVs-siRNA alone, greatly promoted anti-inflammatory cytokine secretion and inhibited pro-inflammatory cytokine secretion (Fig. 7C and D). Microglia/macrophages can have two different phenotypes: pro-inflammatory microglia/macrophages (M1), which cause secondary damage, and anti-inflammatory microglia/macrophages (M2), which initiate tissue repair (Fig. 7E). We therefore wondered whether administration of EVs or siRNA might polarize microglia/macrophages from the M1 to the M2 phenotype after SCI. Gene expression of M1 (iNOS, TNF-α and IL-1β) and M2 (Arg1, CD206 and YM1/2) markers was analyzed by qRT-PCR. As shown in Fig. 7F and G, compared with that in the PBS group, the expression of M2 genes was significantly higher in the EVs, EVs-siRNA and C-EVs-siRNA groups, whereas the expression of M1 genes was lower. Meanwhile, compared with the EVs group, the EVs-siRNA group had higher M2 gene expression and lower M1 gene expression, and the effect of C-EVs-siRNA in promoting the conversion of M1 to M2 was clearer. Western blot analysis also confirmed the qRT-PCR results (Fig. 7H and I). We further assessed the characteristic polarization of microglia/macrophages after SCI in different groups with representative M1-associated iNOS and M2-associated Arg1 markers together with F4/80, which can be used to detect microglia/macrophages in the injured spinal cord. F4/80+ microglia/macrophages significantly increased at the injury site after SCI, and EVs and EVs-siRNA treatments significantly decreased the inflammatory response at the injury site after SCI. However, treatment with C-EVs-siRNA, compared with EVs or EVs-siRNA, greatly decreased the F4/80+ microglia/macrophages (Fig. 7J-M). Furthermore, the relative level of Arg1 protein increased with C-EVs-siRNA treatment, beyond that with EVs and EVs-siRNA alone, whereas the level of iNOS protein decreased (Fig. 7J-M). Thus, these results suggested that EVs-siRNA had a significant effect on the ratio of anti-inflammatory to pro-inflammatory phenotypes after SCI and could shift the microglia/macrophage polarization from the M1 to the M2 phenotype. This effect was more pronounced after peptide CAQK-targeted modification.
Fig. 7.

C-EVs-siRNA affects the inflammatory response and microglial polarization. (A) On the 7th day after injury, DHE staining was used to detect ROS levels in the injured spinal cords in mice. (B) Qualitative analysis of the fluorescence intensity of DHE staining. (C, D) On the 7th day after injury, ELISA was used to detect the concentrations of pro-inflammatory and anti-inflammatory cytokines in the PBS, EVs, EVs-siRNA and C-EVs-siRNA groups. (E) Schematic illustration of C-EVs-siRNA enhancing anti-inflammatory cytokine release from M2 microglia/macrophages and inhibiting pro-inflammatory cytokine release from M1 microglia/macrophages. (F, G) mRNA expression levels of M1 and M2-related genes, detected by qRT-PCR. (H, I) Detection of protein levels of M1 and M2-related genes by western blot analysis. (J, K) Representative immunostaining images of F4/80 (red) and iNOS (green) in the SCI area on day 7 after injury, and analysis of iNOS+ microglia/macrophages in the traumatic injury area. (L, M) Representative immunostaining images of F4/80 (red) and Arg1 (green) in the SCI area on day 7 post-injury and analysis of Arg1+ microglia/macrophages in the traumatic injury area. *P < 0.05; **P < 0.01; ***P < 0.001 (n = 6 per group).
3.9. C-EVs-siRNA reverses LPS-induced ROS generation and microglial polarization
To determine the effects of C-EVs-siRNA on the inflammatory response and microglial polarization, we added LPS to the culture system for 24 h before the addition of PBS, EVs, EVs-siRNA or C-EVs-siRNA (100 μg/mL, 24 h) to induce an inflammatory microenvironment mimicking SCI in vivo. First, we examined LPS-induced changes in ROS levels, another characteristic of M1 microglia/macrophages. EV, EVs-siRNA or C-EVs-siRNA treatment effectively inhibited the increase in cellular ROS levels after LPS stimulation, and EVs-siRNA had a stronger inhibitory effect than EVs (Figs. S6A and B). However, C-EVs-siRNA did not show a stronger antioxidant effect than EVs-siRNA (Figs. S6A and B). ELISA analysis indicated that both EVs and EVs-siRNA suppressed the concentrations of TNF-α, IL-1β and IL-6, and promoted the secretion of TGF-β, IL-4 and IL-10 in vitro (Figs. S6C and D). Furthermore, EVs-siRNA exerted more beneficial effects than EVs (Figs. S6C and D). qRT-PCR indicated that both EVs and EVs-siRNA decreased the expression of M1 markers and increased the expression of M2 markers. Moreover, EVs-siRNA was more powerful in converting M1 microglia to M2 microglia than EVs alone (Figs. S6E and F), similarly to the in vivo results. Flow cytometry revealed that the percentage of F4/80+ iNOS+ cells was lower, and the percentage of F4/80+ CD206+ cells was higher, in the EVs-siRNA group than the EVs group (Figs. S6G–J). These results were confirmed by western blot analysis (Fig. S6K and L). In addition, qRT-PCR, flow cytometry and western blot results all indicated that C-EVs-siRNA did not show a stronger effect than EVs-siRNA in promoting M1 to M2 conversion (Fig. S6E-L), indicating that the targeting effect of CAQK modification was mainly exerted in reactive astrocytes in vivo.
These findings confirmed the results observed in vivo, demonstrating that C-EVs-siRNA effectively decreased M1 microglia and drives M2 microglia polarization, thereby suppressing inflammation.
4. Discussion
The effects of SCI can be severe, resulting in motor dysfunction and poor prognoses [50]. At present, the main research focus is on decreasing neuronal apoptosis in the injured area and inhibiting the inflammatory response involving microglia invading the lesion site, thereby creating a favorable microenvironment for neurogenesis and axonal regeneration [51]. In this study, we constructed siRNA-loaded engineered EVs, which were modified with the spinal cord targeting peptide CAQK for SCI treatment (Fig. 8). Our results indicated that the physicochemical characteristics of C-EVs-siRNA were not significantly different from those of EVs, and it had good biocompatibility. In vitro and in vivo toxicity assessment experiments indicated that C-EVs-siRNA treatment was well tolerated, without causing significant systemic toxicity. Importantly, C-EVs-siRNA tail vein injection not only reached the SCI area but also targeted activated astrocytes. Tissue damage and functional evaluation experiments in the SCI mouse model indicated that the spinal cord was more intact after C-EVs-siRNA treatment, the lesion area was diminished, and the pathological changes were ameliorated. In addition, the motor function recovery of mice with SCI was clearer after C-EVs-siRNA treatment, as indicated by a higher BMS score, better gait recovery and motor coordination ability, increased rod rotation time and speed, decreased latency and increased amplitude of the electromyogram. Furthermore, C-EVs-siRNA promoted neuronal repair, mainly by affecting the inflammatory response and microglial polarization, and decreasing apoptosis, which in turn promoted axonal regeneration and remyelination necessary for movement.
Fig. 8.

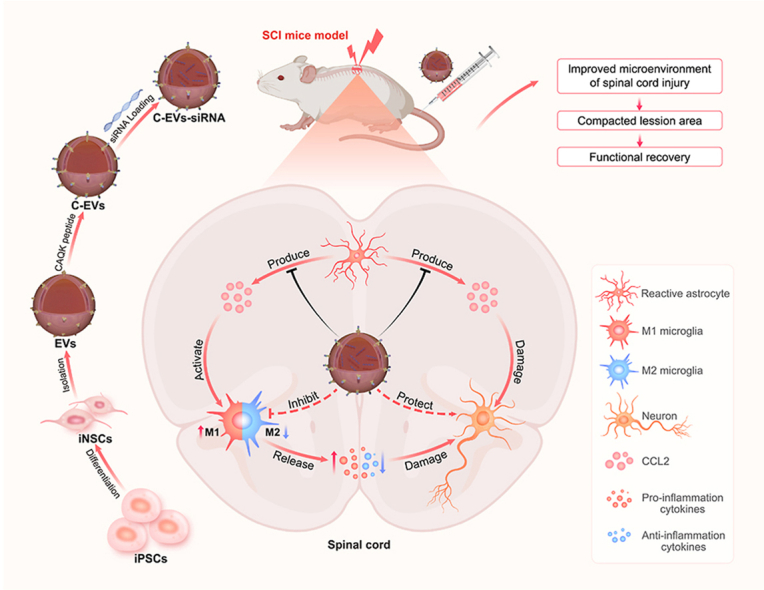
Preparation of C-EVs-siRNA and the therapeutic mechanism. Human iPSCs were induced to differentiate into iNSCs, and then the peptide CAQK was combined with EVs isolated from iNSC using a chemical modification method, and CCL2-siRNA was then loaded into the EVs by electroporation. C-EVs-siRNA can specifically deliver siRNA to SCI areas and be taken up by target cells. C-EVs-siRNA used the inherent anti-inflammatory and neuroreparative functions of iNSC-derived EVs and acted synergistically with the loaded siRNA, thus enhancing the therapeutic effects on SCI. The combination of targeted modified EVs and siRNA effectively modulated microenvironmental disturbance after SCI, altered the polarization state of microglia, and limited the effects of inflammatory response and neuronal injury on functional recovery in mice with SCI.
In addition to having relatively low immunogenicity and biodegradability, EVs have low toxicity and provide strong cargo protection, thus making them ideal for treating SCI [52,53]. Our previous studies in multiple animal models have demonstrated that EVs promote motor function recovery after SCI through different mechanisms of action [18,48,54,55]. However, unmodified EVs pose a challenge for intravenous infusion into SCI because of their poor targeting capabilities. Many previous studies have emphasized engineering of EVs to enhance therapeutic capabilities. Kim et al. have demonstrated that cardiac targeting peptide-modified EVs are more than 2-fold more efficient than unmodified EVs at delivering siRNA to the heart [56]. You et al. have reported refined surface editing of MSCs-EVs through metabolic glycoengineering of adipose-derived stem cells to target activated macrophages in rheumatoid arthritis [57]. Tian et al. have shown that after intravenous injection of RGD-modified NPCs-derived EVs, RGD-EVs targeted the diseased area of the ischemic brain and strongly inhibit the inflammatory response [16].
Mann et al. have demonstrated that the peptide CAQK selectively binds proteoglycan complexes in damaged mouse and human brains. When conjugated to CAQK, systemically administered molecules accumulate at injury sites [38]. Subsequently, Wang et al. constructed CAQK-modified liposomes, exploiting the mechanism through which CAQK selectively binds CSPGs for targeted drug delivery in the injured spinal cord [44]. Additionally, Wang et al. have developed a spinal cord-targeted, esterase-responsive nanocarrier based on CAQK for the treatment of spinal cord injuries [41]. However, no published studies have examined the use of CAQK-modified EVs in SCI. Therefore, we constructed C-EVs for targeted therapy for SCI. Briefly, we first conjugated the peptide CAQK to the surfaces of EVs through chemical modification and verified the successful modification of EVs. TEM, NTA and western blot results indicated that CAQK modification did not affect the general physical characteristics of EVs. In addition, our in vitro experiments demonstrated that C-EVs were targeted to activated astrocytes, thus further validating that CAQK successfully modifies EVs. Immunofluorescence results indicated that the targeting ability of C-EVs was mediated through the recognition of CSPGs released by activated astrocytes. This targeting approach involves efficient binding of the peptide CAQK to the surfaces of EVs, thus yielding C-EVs with high affinity/specificity for cells expressing CSPGs. In vivo IVIS also confirmed that C-EVs substantially accumulated in the lesion area of SCI after intravenous injection into mice receiving SCI.
As mediators of a cascade of events following SCI, microglia are important in activating and regulating neuroinflammation [58]. Microglia/macrophages have two phenotypes. The M1 phenotype secretes proinflammatory cytokines including TNF-α, IL-6 and IL-1β. The M2 phenotype secretes anti-inflammatory cytokines, including TGF-β, IL-10 and IL-4 [54,59]. After SCI, microglial activation is dominated by the M1 phenotype; beyond releasing proinflammatory mediators, microglia respond to proinflammatory signals released by other cells. CCL2 may play a critical role in the immune response in this context [60]. Zhang et al. have found that intrathecal administration of CCL2 activates microglia in wild-type mice but not CCR2-deficient mice [61]. Our previous studies have also demonstrated that CCL2 released by activated astrocytes promotes microglial activation and neuronal apoptosis after SCI, whereas activated microglia release proinflammatory cytokines including IL-1β, which act on neurons and further aggravate their apoptosis [25]. According to these findings, in this study, we constructed CCL2-siRNA to silence the expression of the CCL2 gene in activated astrocytes after SCI, thereby blocking the CCL2-mediated microglial inflammatory response and neuronal apoptosis.
Because EVs are nanoscale and membrane permeable, research has yielded insights into the possibility of using EVs as nucleic acid or drug delivery vehicles. Guo et al. have shown that EVs loaded with PTEN-siRNA attenuated PTEN expression after intranasal administration. Furthermore, treatment with siRNA-loaded EVs has been found to partially improve the structure and electrophysiological function, significantly causing functional recovery in rats with complete SCI [35]. Here, we loaded CCL2-siRNA into EVs by electroporation. TEM, NTA and western blot analysis indicated that the general characteristics of EVs remained unchanged after siRNA loading. Specifically, siRNA-loaded C-EVs had similar morphology and size to those of unloaded C-EVs. Western blot analysis also confirmed that the final engineered EVs still retained the protein markers of native EVs. In siRNA degradation and safety assays, the membranes of EVs maintained siRNA integrity, thereby protecting siRNA from nucleases in FBS after modification and electroporation. The siRNA-loaded C-EVs had comparable biological safety to natural EVs. Furthermore, in vitro and in vivo results confirmed the effectiveness of siRNA-loaded EVs in knockdown of CCL2 gene expression. Most importantly, the successful construction of C-EVs loaded with siRNA indicated synergistic and comprehensive therapeutic effects of SCI, including inhibiting neuronal apoptosis, influencing microglial polarization and promoting neuronal axon regeneration. Additionally, studies have indicated that siRNA loading on EVs appears to be dependent on the siRNA cargo, whereas EVs merely act as carriers. In contrast, C-EVs-siRNA exploits the inherent anti-inflammatory and neuroreparative functions of iNSCs-derived EVs and acts synergistically with the loaded siRNA to enhance efficacy. However, we acknowledge that complexity and technical challenges may limit the current clinical translation of C-EVs-siRNA.
Encouragingly, we found that C-EVs-siRNA treatment significantly improved functional recovery in SCI mice. The histological morphology of the spinal cord indicated a significant improvement in the condition of the spinal cord; moreover, the lesion area decreased, and the pathological changes were diminished. The anatomical basis for functional recovery is better understood with MRI. Our results revealed that C-EVs-siRNA treatment significantly contributed to the recovery of the damaged spinal cord. In addition, the tissue integrity after C-EVs-siRNA treatment was better than that after treatment with EVs or EVs-siRNA. As described above, this positive effect might have been due to the combined effect of EVs and siRNA in targeting multiple aspects of SCI. Interestingly, both EVs and siRNA promoted axonal growth, and the combination of targeted modified EVs and siRNA administration had the greatest effect on axonal regeneration and myelin protection. After SCI, M1 microglia/macrophages induce a significant inflammatory response by releasing proinflammatory cytokines and ROS, leading to neuronal death and axonal retraction [62,63]. Our data suggested that EVs may contribute to tissue repair by promoting the transformation of M1 microglia to M2 microglia. In addition, the improved inflammatory response increased cell survival, prevented axonal contraction, and increased neuronal growth potential and plasticity. The functional assessment results indicated that the BMS scores of the EVs and EVs-siRNA groups were higher than those in the SCI group at 28 days after injury; however, the EVs-siRNA group had better effects. The BMS score of the C-EVs-siRNA group was significantly higher than those of the EVs and C-EVs groups. These results suggested that EVs alone improved motor function in mice with SCI, whereas a targeting modification or loading of siRNA to silence the CCL2 gene significantly increased the effects of EVs on motor recovery. Other functional tests (footprint test, rotarod test and MEP) further confirmed the results of BMS.
According to our data, intravenous injection of C-EVs-siRNA was an acceptable route of administration and could effectively inhibit microglial inflammatory response and neuronal apoptosis in the area of SCI. Furthermore, we used C-EVs as an efficient siRNA delivery vehicle for SCI treatment. Other therapeutic drugs could also be loaded into C-EVs for further targeted delivery to the diseased area of SCI. Many efficient methods are capable of loading therapeutic small molecules, RNAs (siRNA and miRNA) and even proteins into EVs, including electroporation, sonication, passive incubation with permeabilizers and incubation with hydrophobically modified RNAs [30,39,40,64]. Electroporation is currently an effective method of loading specific cargo into EVs and it has little impact on the endogenous cargo of EVs. In this study, we loaded siRNA into EVs by electroporation, and the modification of the peptide CAQK did not affect the loading efficiency of electroporation. One possible problem is the accumulation of EVs in the liver and spleen. Studies have shown that intravenously administered EVs are cleared in large numbers by macrophages residing in the liver and spleen [10,65,66]. Thus, an effective method for decreasing the clearance of EVs in the liver and spleen is needed. Our results showed that EVs accumulated abundantly in the liver after injection via the tail vein, whereas the CAQK-modified EVs decreased this accumulation. Although modification with the peptide CAQK decreased the accumulation and clearance of EVs in the liver, spleen and kidney, many EVs remained in the liver and lung. Therefore, the liver and lung toxicity of C-EVs-siRNA should also be fully evaluated before clinical application. Here, we confirmed the safety of C-EVs-siRNA treatment. Our study revealed no tissue damage or loss of function in the liver and lung after intravenous administration of C-EVs-siRNA. In vivo blood biochemistry and H&E staining results of other major organs also indicated that C-EVs-siRNA treatment was well tolerated and had no clear systemic toxicity.
In principle, this method for targeted delivery of siRNA by modifying EVs is a safe, effective and risk-free gene therapy that overcomes the limitations of clinical gene therapy [67]. However, several limitations exist to obtaining sufficient EVs through NSCs. An important consideration is that harvesting NSCs from adult tissues is an invasive procedure. Furthermore, because NSCs reside in the subventricular zone lining the lateral ventricles [68], obtaining them from patients is challenging. The availability of NSCs-EVs for clinical treatment would be limited by these factors. Consequently, developing EV targeted for other tissues and improving their yield and efficiency would allow them to be applied in a wider range of applications, thereby increasing the effectiveness of this gene delivery technology. In recent years, advances in stem cell technology have enabled patient-specific iPSCs, which can differentiate into expandable progenitor and mature cells, to be generated from adult human cells [69]. Moreover, the use of EVs derived from iPSCs for autologous therapy does not raise ethical issues or immune rejection [70,71]. Cui et al. have developed an EV delivery system based on EVs secreted from MSCs differentiated from human iPSCs. Engineered EVs and loaded siRNA play a synergistic role in the treatment of osteoporosis [46]. In addition, somatic cells can be obtained from patients and transformed into iPSCs with induced pluripotent stem cell reprogramming techniques. iPSCs can then be differentiated into specific cells that could be used as EVs for production, thus avoiding the risk of ethical controversy or immune rejection [46]. Upadhya et al. have also isolated biologically active EVs from hiPSC-induced iNSCs containing multiple miRNAs and proteins associated with brain injury or post-disease repair, and observed anti-inflammatory and neurogenic properties [42]. In our study, we induced the differentiation of human iPSCs into iNSCs and then used a chemical modification method to successfully bind the spinal cord-targeting peptide CAQK to EVs isolated from iNSC. The modified EVs still maintained the basic properties of native EVs and showed good targeting and therapeutic effects in vitro and in vivo.
5. Conclusions
In conclusion, in this study, we designed iNSCs-based engineered EVs to promote functional recovery after SCI. C-EVs-siRNA can specifically deliver siRNA to SCI areas and be taken up by target cells. C-EVs-siRNA used the inherent anti-inflammatory and neuroreparative functions of iNSCs-derived EVs and acted synergistically with the loaded siRNA, thus enhancing the therapeutic effects on SCI. The combination of targeted modified EVs and siRNA effectively modulated microenvironmental disturbance after SCI, altered the polarization state of microglia, and limited the effects of inflammatory response and neuronal injury on functional recovery in mice with SCI. In conclusion, engineered EVs were a potentially viable and efficacious treatment for SCI, and may also be used to develop targeted treatments for other diseases.
Ethics approval and consent to participate
We confirm that any aspect of the work covered in this manuscript that has involved experimental animals has been conducted with the ethical approval of all relevant bodies and that such approvals are acknowledged within the manuscript.
CRediT authorship contribution statement
Yuluo Rong: Conceptualization, Methodology, Formal analysis, Validation, Investigation, Writing – original draft. Zhuanghui Wang: Methodology, Formal analysis, Validation. Pengyu Tang: Formal analysis, Investigation. Jiaxing Wang: Methodology, Validation, Formal analysis. Chengyue Ji: Methodology, Validation. Jie Chang: Resources, Visualization. Yufeng Zhu: Visualization, Investigation. Wu Ye: Methodology, Investigation. Jianling Bai: Methodology, Formal analysis. Wei Liu: Formal analysis, Investigation. Guoyong Yin: Visualization, Data curation. Lipeng Yu: Methodology, Formal analysis, Validation. Xuhui Zhou: Funding acquisition, Methodology. Weihua Cai: Conceptualization, Methodology, Funding acquisition, Writing – review & editing, Supervision, Data curation, Project administration.
Declaration of competing interest
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
Acknowledgments
This work was sponsored by the National Natural Science Foundation of China (grant No. 81974335, 82172426). We would like to thank the Core Facility of the First Affiliated Hospital of Nanjing Medical University for its help in the experiment.
Footnotes
Peer review under responsibility of KeAi Communications Co., Ltd.
Supplementary data to this article can be found online at https://doi.org/10.1016/j.bioactmat.2022.11.011.
Contributor Information
Yuluo Rong, Email: rongylspine@163.com.
Lipeng Yu, Email: lipeng_yu@aliyun.com.
Xuhui Zhou, Email: zhouxuhui@smmu.edu.cn.
Weihua Cai, Email: caiwhspine@sina.com.
Appendix A. Supplementary data
The following is the Supplementary data to this article:
References
- 1.McDonald J.W., Sadowsky C. Spinal-cord injury. Lancet (London, England) 2002;359(9304):417–425. doi: 10.1016/S0140-6736(02)07603-1. [DOI] [PubMed] [Google Scholar]
- 2.Eckert M.J., Martin M.J. Trauma: spinal cord injury. Surg. Clin. 2017;97(5):1031–1045. doi: 10.1016/j.suc.2017.06.008. [DOI] [PubMed] [Google Scholar]
- 3.Eli I., Lerner D.P., Ghogawala Z. Acute traumatic spinal cord injury. Neurol. Clin. 2021;39(2):471–488. doi: 10.1016/j.ncl.2021.02.004. [DOI] [PubMed] [Google Scholar]
- 4.Chhabra H.S., Sarda K. Clinical translation of stem cell based interventions for spinal cord injury - are we there yet? Adv. Drug Deliv. Rev. 2017;120:41–49. doi: 10.1016/j.addr.2017.09.021. [DOI] [PubMed] [Google Scholar]
- 5.Beattie M.S. Inflammation and apoptosis: linked therapeutic targets in spinal cord injury. Trends Mol. Med. 2004;10(12):580–583. doi: 10.1016/j.molmed.2004.10.006. [DOI] [PubMed] [Google Scholar]
- 6.Kwon B.K., Okon E., Hillyer J., Mann C., Baptiste D., Weaver L.C., Fehlings M.G., Tetzlaff W. A systematic review of non-invasive pharmacologic neuroprotective treatments for acute spinal cord injury. J. Neurotrauma. 2011;28(8):1545–1588. doi: 10.1089/neu.2009.1149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Song Y.H., Agrawal N.K., Griffin J.M., Schmidt C.E. Recent advances in nanotherapeutic strategies for spinal cord injury repair. Adv. Drug Deliv. Rev. 2019;148:38–59. doi: 10.1016/j.addr.2018.12.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Liao W., Du Y., Zhang C., Pan F., Yao Y., Zhang T., Peng Q. Exosomes: the next generation of endogenous nanomaterials for advanced drug delivery and therapy. Acta Biomater. 2019;86:1–14. doi: 10.1016/j.actbio.2018.12.045. [DOI] [PubMed] [Google Scholar]
- 9.Liang Y., Duan L., Lu J., Xia J. Engineering exosomes for targeted drug delivery. Theranostics. 2021;11(7):3183–3195. doi: 10.7150/thno.52570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Kalluri R., LeBleu V.S. The biology, function, and biomedical applications of exosomes. Science (New York, N.Y.) 2020;(6478):367. doi: 10.1126/science.aau6977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Cossetti C., Iraci N., Mercer T.R., Leonardi T., Alpi E., Drago D., Alfaro-Cervello C., Saini H.K., Davis M.P., Schaeffer J., Vega B., Stefanini M., Zhao C., Muller W., Garcia-Verdugo J.M., Mathivanan S., Bachi A., Enright A.J., Mattick J.S., Pluchino S. Extracellular vesicles from neural stem cells transfer IFN-γ via Ifngr1 to activate Stat1 signaling in target cells. Molecular cell. 2014;56(2):193–204. doi: 10.1016/j.molcel.2014.08.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Webb R.L., Kaiser E.E., Scoville S.L., Thompson T.A., Fatima S., Pandya C., Sriram K., Swetenburg R.L., Vaibhav K., Arbab A.S., Baban B., Dhandapani K.M., Hess D.C., Hoda M.N., Stice S.L. Human neural stem cell extracellular vesicles improve tissue and functional recovery in the murine thromboembolic stroke model. Translational stroke research. 2018;9(5):530–539. doi: 10.1007/s12975-017-0599-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Vogel A., Upadhya R., Shetty A.K. Neural stem cell derived extracellular vesicles: attributes and prospects for treating neurodegenerative disorders. EBioMedicine. 2018;38:273–282. doi: 10.1016/j.ebiom.2018.11.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Bian B., Zhao C., He X., Gong Y., Ren C., Ge L., Zeng Y., Li Q., Chen M., Weng C., He J., Fang Y., Xu H., Yin Z.Q. Exosomes derived from neural progenitor cells preserve photoreceptors during retinal degeneration by inactivating microglia. J. Extracell. Vesicles. 2020;9(1) doi: 10.1080/20013078.2020.1748931. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Cao L., Tian T., Huang Y., Tao S., Zhu X., Yang M., Gu J., Feng G., Ma Y., Xia R., Xu W., Wang L. Neural progenitor cell-derived nanovesicles promote hair follicle growth via miR-100. J. Nanobiotechnol. 2021;19(1):20. doi: 10.1186/s12951-020-00757-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Tian T., Cao L., He C., Ye Q., Liang R., You W., Zhang H., Wu J., Ye J., Tannous B.A., Gao J. Targeted delivery of neural progenitor cell-derived extracellular vesicles for anti-inflammation after cerebral ischemia. Theranostics. 2021;11(13):6507–6521. doi: 10.7150/thno.56367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Ma Y., Li C., Huang Y., Wang Y., Xia X., Zheng J.C. Exosomes released from neural progenitor cells and induced neural progenitor cells regulate neurogenesis through miR-21a. Cell Commun. Signal. : CCS. 2019;17(1):96. doi: 10.1186/s12964-019-0418-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Rong Y., Liu W., Wang J., Fan J., Luo Y., Li L., Kong F., Chen J., Tang P., Cai W. Neural stem cell-derived small extracellular vesicles attenuate apoptosis and neuroinflammation after traumatic spinal cord injury by activating autophagy. Cell Death Dis. 2019;10(5):340. doi: 10.1038/s41419-019-1571-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Rong Y., Liu W., Lv C., Wang J., Luo Y., Jiang D., Li L., Zhou Z., Zhou W., Li Q., Yin G., Yu L., Fan J., Cai W. Neural stem cell small extracellular vesicle-based delivery of 14-3-3t reduces apoptosis and neuroinflammation following traumatic spinal cord injury by enhancing autophagy by targeting Beclin-1. Aging. 2019;11(18):7723–7745. doi: 10.18632/aging.102283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Xie C., Liu Y.Q., Guan Y.T., Zhang G.X. Induced stem cells as a novel multiple sclerosis therapy. Curr. Stem Cell Res. Ther. 2016;11(4):313–320. doi: 10.2174/1574888x10666150302110013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Nagoshi N., Okano H. iPSC-derived neural precursor cells: potential for cell transplantation therapy in spinal cord injury. Cell. Mol. Life Sci. : CMLS. 2018;75(6):989–1000. doi: 10.1007/s00018-017-2676-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Bose S., Cho J. Role of chemokine CCL2 and its receptor CCR2 in neurodegenerative diseases. Arch Pharm. Res. (Seoul) 2013;36(9):1039–1050. doi: 10.1007/s12272-013-0161-z. [DOI] [PubMed] [Google Scholar]
- 23.Tian D.S., Peng J., Murugan M., Feng L.J., Liu J.L., Eyo U.B., Zhou L.J., Mogilevsky R., Wang W., Wu L.J. Chemokine CCL2-CCR2 signaling induces neuronal cell death via STAT3 activation and IL-1β production after status epilepticus. J. Neurosci. : the official journal of the Society for Neuroscience. 2017;37(33):7878–7892. doi: 10.1523/JNEUROSCI.0315-17.2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Xu J., Dong H., Qian Q., Zhang X., Wang Y., Jin W., Qian Y. Astrocyte-derived CCL2 participates in surgery-induced cognitive dysfunction and neuroinflammation via evoking microglia activation. Behav. Brain Res. 2017;332:145–153. doi: 10.1016/j.bbr.2017.05.066. [DOI] [PubMed] [Google Scholar]
- 25.Rong Y., Ji C., Wang Z., Ge X., Wang J., Ye W., Tang P., Jiang D., Fan J., Yin G., Liu W., Cai W. Small extracellular vesicles encapsulating CCL2 from activated astrocytes induce microglial activation and neuronal apoptosis after traumatic spinal cord injury. J. Neuroinflammation. 2021;18(1):196. doi: 10.1186/s12974-021-02268-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Hu B., Zhong L., Weng Y., Peng L., Huang Y., Zhao Y., Liang X.J. Therapeutic siRNA: state of the art. Signal Transduct. Targeted Ther. 2020;5(1):101. doi: 10.1038/s41392-020-0207-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Kaur I.P., Chopra K., Rishi P., Puri S., Sharma G. Small RNAs: the qualified candidates for gene manipulation in diverse clinical pathologies. Crit. Rev. Ther. Drug Carrier Syst. 2014;31(4):305–329. doi: 10.1615/critrevtherdrugcarriersyst.2014007943. [DOI] [PubMed] [Google Scholar]
- 28.Du Rietz H., Hedlund H., Wilhelmson S., Nordenfelt P., Wittrup A. Imaging small molecule-induced endosomal escape of siRNA. Nat. Commun. 2020;11(1):1809. doi: 10.1038/s41467-020-15300-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Armstrong J.P.K., Stevens M.M. Strategic design of extracellular vesicle drug delivery systems. Adv. Drug Deliv. Rev. 2018;130:12–16. doi: 10.1016/j.addr.2018.06.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Zhou Y., Zhou G., Tian C., Jiang W., Jin L., Zhang C., Chen X. Exosome-mediated small RNA delivery for gene therapy. Wiley interdisciplinary reviews. RNA. 2016;7(6):758–771. doi: 10.1002/wrna.1363. [DOI] [PubMed] [Google Scholar]
- 31.Xu S., Liu B., Fan J., Xue C., Lu Y., Li C., Cui D. Engineered mesenchymal stem cell-derived exosomes with high CXCR4 levels for targeted siRNA gene therapy against cancer. Nanoscale. 2022;14(11):4098–4113. doi: 10.1039/d1nr08170e. [DOI] [PubMed] [Google Scholar]
- 32.Han Q., Xie Q.R., Li F., Cheng Y., Wu T., Zhang Y., Lu X., Wong A.S.T., Sha J., Xia W. Targeted inhibition of SIRT6 via engineered exosomes impairs tumorigenesis and metastasis in prostate cancer. Theranostics. 2021;11(13):6526–6541. doi: 10.7150/thno.53886. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Duan L., Xu L., Xu X., Qin Z., Zhou X., Xiao Y., Liang Y., Xia J. Exosome-mediated delivery of gene vectors for gene therapy. Nanoscale. 2021;13(3):1387–1397. doi: 10.1039/d0nr07622h. [DOI] [PubMed] [Google Scholar]
- 34.Izco M., Blesa J., Schleef M., Schmeer M., Porcari R., Al-Shawi R., Ellmerich S., de Toro M., Gardiner C., Seow Y., Reinares-Sebastian A., Forcen R., Simons J.P., Bellotti V., Cooper J.M., Alvarez-Erviti L. Systemic exosomal delivery of shRNA minicircles prevents parkinsonian pathology. Mol. Ther. : the journal of the American Society of Gene Therapy. 2019;27(12):2111–2122. doi: 10.1016/j.ymthe.2019.08.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Guo S., Perets N., Betzer O., Ben-Shaul S., Sheinin A., Michaelevski I., Popovtzer R., Offen D., Levenberg S. Intranasal delivery of mesenchymal stem cell derived exosomes loaded with phosphatase and tensin homolog siRNA repairs complete spinal cord injury. ACS Nano. 2019;13(9):10015–10028. doi: 10.1021/acsnano.9b01892. [DOI] [PubMed] [Google Scholar]
- 36.Wiklander O.P., Nordin J.Z., O'Loughlin A., Gustafsson Y., Corso G., Mäger I., Vader P., Lee Y., Sork H., Seow Y., Heldring N., Alvarez-Erviti L., Smith C.I., Le Blanc K., Macchiarini P., Jungebluth P., Wood M.J., Andaloussi S.E. Extracellular vesicle in vivo biodistribution is determined by cell source, route of administration and targeting. J. Extracell. Vesicles. 2015;4 doi: 10.3402/jev.v4.26316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Lai C.P., Mardini O., Ericsson M., Prabhakar S., Maguire C., Chen J.W., Tannous B.A., Breakefield X.O. Dynamic biodistribution of extracellular vesicles in vivo using a multimodal imaging reporter. ACS Nano. 2014;8(1):483–494. doi: 10.1021/nn404945r. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Mann A.P., Scodeller P., Hussain S., Joo J., Kwon E., Braun G.B., Mölder T., She Z.G., Kotamraju V.R., Ranscht B., Krajewski S., Teesalu T., Bhatia S., Sailor M.J., Ruoslahti E. A peptide for targeted, systemic delivery of imaging and therapeutic compounds into acute brain injuries. Nat. Commun. 2016;7 doi: 10.1038/ncomms11980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Tian T., Zhang H.X., He C.P., Fan S., Zhu Y.L., Qi C., Huang N.P., Xiao Z.D., Lu Z.H., Tannous B.A., Gao J. Surface functionalized exosomes as targeted drug delivery vehicles for cerebral ischemia therapy. Biomaterials. 2018;150:137–149. doi: 10.1016/j.biomaterials.2017.10.012. [DOI] [PubMed] [Google Scholar]
- 40.Alvarez-Erviti L., Seow Y., Yin H., Betts C., Lakhal S., Wood M.J. Delivery of siRNA to the mouse brain by systemic injection of targeted exosomes. Nat. Biotechnol. 2011;29(4):341–345. doi: 10.1038/nbt.1807. [DOI] [PubMed] [Google Scholar]
- 41.Wang J., Li D., Liang C., Wang C., Zhou X., Ying L., Tao Y., Xu H., Shu J., Huang X., Gong Z., Xia K., Li F., Chen Q., Tang J., Shen Y. Scar tissue-targeting polymer micelle for spinal cord injury treatment. Small. 2020;16(8) doi: 10.1002/smll.201906415. [DOI] [PubMed] [Google Scholar]
- 42.Upadhya R., Madhu L.N., Attaluri S., Gitaí D.L.G., Pinson M.R., Kodali M., Shetty G., Zanirati G., Kumar S., Shuai B., Weintraub S.T., Shetty A.K. Extracellular vesicles from human iPSC-derived neural stem cells: miRNA and protein signatures, and anti-inflammatory and neurogenic properties. J. Extracell. Vesicles. 2020;9(1) doi: 10.1080/20013078.2020.1809064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Asher R.A., Morgenstern D.A., Fidler P.S., Adcock K.H., Oohira A., Braistead J.E., Levine J.M., Margolis R.U., Rogers J.H., Fawcett J.W. Neurocan is upregulated in injured brain and in cytokine-treated astrocytes. J. Neurosci. : the official journal of the Society for Neuroscience. 2000;20(7):2427–2438. doi: 10.1523/JNEUROSCI.20-07-02427.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Wang Q., Zhang H., Xu H., Zhao Y., Li Z., Li J., Wang H., Zhuge D., Guo X., Xu H., Jones S., Li X., Jia X., Xiao J. Novel multi-drug delivery hydrogel using scar-homing liposomes improves spinal cord injury repair. Theranostics. 2018;8(16):4429–4446. doi: 10.7150/thno.26717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Wang Y., Chen X., Tian B., Liu J., Yang L., Zeng L., Chen T., Hong A., Wang X. Nucleolin-targeted extracellular vesicles as a versatile platform for biologics delivery to breast cancer. Theranostics. 2017;7(5):1360–1372. doi: 10.7150/thno.16532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Cui Y., Guo Y., Kong L., Shi J., Liu P., Li R., Geng Y., Gao W., Zhang Z., Fu D. A bone-targeted engineered exosome platform delivering siRNA to treat osteoporosis. Bioact. Mater. 2022;10:207–221. doi: 10.1016/j.bioactmat.2021.09.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Wang Y., Wu M., Gu L., Li X., He J., Zhou L., Tong A., Shi J., Zhu H., Xu J., Guo G. Effective improvement of the neuroprotective activity after spinal cord injury by synergistic effect of glucocorticoid with biodegradable amphipathic nanomicelles. Drug Deliv. 2017;24(1):391–401. doi: 10.1080/10717544.2016.1256003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Liu W., Tang P., Wang J., Ye W., Ge X., Rong Y., Ji C., Wang Z., Bai J., Fan J., Yin G., Cai W. Extracellular vesicles derived from melatonin-preconditioned mesenchymal stem cells containing USP29 repair traumatic spinal cord injury by stabilizing NRF2. J. Pineal Res. 2021;71(4) doi: 10.1111/jpi.12769. [DOI] [PubMed] [Google Scholar]
- 49.Hauben E., Butovsky O., Nevo U., Yoles E., Moalem G., Agranov E., Mor F., Leibowitz-Amit R., Pevsner E., Akselrod S., Neeman M., Cohen I.R., Schwartz M. Passive or active immunization with myelin basic protein promotes recovery from spinal cord contusion. J. Neurosci. : the official journal of the Society for Neuroscience. 2000;20(17):6421–6430. doi: 10.1523/JNEUROSCI.20-17-06421.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Oyinbo C.A. Secondary injury mechanisms in traumatic spinal cord injury: a nugget of this multiply cascade. Acta Neurobiol. Exp. 2011;71(2):281–299. doi: 10.55782/ane-2011-1848. [DOI] [PubMed] [Google Scholar]
- 51.Anjum A., Yazid M.D., Fauzi Daud M., Idris J., Ng A.M.H., Selvi Naicker A., Ismail O.H.R., Athi Kumar R.K., Lokanathan Y. Spinal cord injury: pathophysiology, multimolecular interactions, and underlying recovery mechanisms. Int. J. Mol. Sci. 2020;21(20) doi: 10.3390/ijms21207533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Feng J., Zhang Y., Zhu Z., Gu C., Waqas A., Chen L. Emerging exosomes and exosomal MiRNAs in spinal cord injury. Front. Cell Dev. Biol. 2021;9 doi: 10.3389/fcell.2021.703989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Dutta D., Khan N., Wu J., Jay S.M. Extracellular vesicles as an emerging frontier in spinal cord injury pathobiology and therapy. Trends in neurosciences. 2021;44(6):492–506. doi: 10.1016/j.tins.2021.01.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Liu W., Rong Y., Wang J., Zhou Z., Ge X., Ji C., Jiang D., Gong F., Li L., Chen J., Zhao S., Kong F., Gu C., Fan J., Cai W. Exosome-shuttled miR-216a-5p from hypoxic preconditioned mesenchymal stem cells repair traumatic spinal cord injury by shifting microglial M1/M2 polarization. J. Neuroinflammation. 2020;17(1):47. doi: 10.1186/s12974-020-1726-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Ge X., Tang P., Rong Y., Jiang D., Lu X., Ji C., Wang J., Huang C., Duan A., Liu Y., Chen X., Chen X., Xu Z., Wang F., Wang Z., Li X., Zhao W., Fan J., Liu W., Yin G., Cai W. Exosomal miR-155 from M1-polarized macrophages promotes EndoMT and impairs mitochondrial function via activating NF-κB signaling pathway in vascular endothelial cells after traumatic spinal cord injury. Redox Biol. 2021;41 doi: 10.1016/j.redox.2021.101932. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Kim H., Mun D., Kang J.Y., Lee S.H., Yun N., Joung B. Improved cardiac-specific delivery of RAGE siRNA within small extracellular vesicles engineered to express intense cardiac targeting peptide attenuates myocarditis. Mol. Ther. Nucleic Acids. 2021;24:1024–1032. doi: 10.1016/j.omtn.2021.04.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.You D.G., Lim G.T., Kwon S., Um W., Oh B.H., Song S.H., Lee J., Jo D.G., Cho Y.W., Park J.H. Metabolically engineered stem cell-derived exosomes to regulate macrophage heterogeneity in rheumatoid arthritis. Sci. Adv. 2021;7(23) doi: 10.1126/sciadv.abe0083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Zhou X., He X., Ren Y. Function of microglia and macrophages in secondary damage after spinal cord injury. Neural Regeneration Res. 2014;9(20):1787–1795. doi: 10.4103/1673-5374.143423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Crain J.M., Nikodemova M., Watters J.J. Microglia express distinct M1 and M2 phenotypic markers in the postnatal and adult central nervous system in male and female mice. J. Neurosci. Res. 2013;91(9):1143–1151. doi: 10.1002/jnr.23242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Mayo L., Trauger S.A., Blain M., Nadeau M., Patel B., Alvarez J.I., Mascanfroni I.D., Yeste A., Kivisäkk P., Kallas K., Ellezam B., Bakshi R., Prat A., Antel J.P., Weiner H.L., Quintana F.J. Regulation of astrocyte activation by glycolipids drives chronic CNS inflammation. Nat. Med. 2014;20(10):1147–1156. doi: 10.1038/nm.3681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Zhang J., Shi X.Q., Echeverry S., Mogil J.S., De Koninck Y., Rivest S. Expression of CCR2 in both resident and bone marrow-derived microglia plays a critical role in neuropathic pain. J. Neurosci. : the official journal of the Society for Neuroscience. 2007;27(45):12396–12406. doi: 10.1523/JNEUROSCI.3016-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.David S., Kroner A. Repertoire of microglial and macrophage responses after spinal cord injury. Nat. Rev. Neurosci. 2011;12(7):388–399. doi: 10.1038/nrn3053. [DOI] [PubMed] [Google Scholar]
- 63.Gensel J.C., Zhang B. Macrophage activation and its role in repair and pathology after spinal cord injury. Brain Res. 2015;1619:1–11. doi: 10.1016/j.brainres.2014.12.045. [DOI] [PubMed] [Google Scholar]
- 64.Didiot M.C., Hall L.M., Coles A.H., Haraszti R.A., Godinho B.M., Chase K., Sapp E., Ly S., Alterman J.F., Hassler M.R., Echeverria D., Raj L., Morrissey D.V., DiFiglia M., Aronin N., Khvorova A. Exosome-mediated delivery of hydrophobically modified siRNA for huntingtin mRNA silencing. Mol. Ther. : the journal of the American Society of Gene Therapy. 2016;24(10):1836–1847. doi: 10.1038/mt.2016.126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.He C., Zheng S., Luo Y., Wang B. Exosome theranostics: biology and translational medicine. Theranostics. 2018;8(1):237–255. doi: 10.7150/thno.21945. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Vader P., Mol E.A., Pasterkamp G., Schiffelers R.M. Extracellular vesicles for drug delivery. Adv. Drug Deliv. Rev. 2016;106(Pt A):148–156. doi: 10.1016/j.addr.2016.02.006. [DOI] [PubMed] [Google Scholar]
- 67.Xiao Y., Tian J., Wu W.C., Gao Y.H., Guo Y.X., Song S.J., Gao R., Wang L.B., Wu X.Y., Zhang Y., Li X. Targeting central nervous system extracellular vesicles enhanced triiodothyronine remyelination effect on experimental autoimmune encephalomyelitis. Bioact. Mater. 2022;9:373–384. doi: 10.1016/j.bioactmat.2021.07.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Morshead C.M., Reynolds B.A., Craig C.G., McBurney M.W., Staines W.A., Morassutti D., Weiss S., van der Kooy D. Neural stem cells in the adult mammalian forebrain: a relatively quiescent subpopulation of subependymal cells. Neuron. 1994;13(5):1071–1082. doi: 10.1016/0896-6273(94)90046-9. [DOI] [PubMed] [Google Scholar]
- 69.Hirschi K.K., Li S., Roy K. Induced pluripotent stem cells for regenerative medicine. Annu. Rev. Biomed. Eng. 2014;16:277–294. doi: 10.1146/annurev-bioeng-071813-105108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Zhang J., Guan J., Niu X., Hu G., Guo S., Li Q., Xie Z., Zhang C., Wang Y. Exosomes released from human induced pluripotent stem cells-derived MSCs facilitate cutaneous wound healing by promoting collagen synthesis and angiogenesis. J. Transl. Med. 2015;13:49. doi: 10.1186/s12967-015-0417-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Hu G.W., Li Q., Niu X., Hu B., Liu J., Zhou S.M., Guo S.C., Lang H.L., Zhang C.Q., Wang Y., Deng Z.F. Exosomes secreted by human-induced pluripotent stem cell-derived mesenchymal stem cells attenuate limb ischemia by promoting angiogenesis in mice. Stem Cell Res. Ther. 2015;6(1):10. doi: 10.1186/scrt546. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.