

Abstract
Objective:
Despite the clinical importance of chronic and severe irritability, there is a paucity of controlled trials for its pharmacological treatment. Here, we examine the effects of adding citalopram (CTP) to methylphenidate (MPH) in the treatment of chronic severe irritability in youth using a double-blind randomized placebo-controlled design.
Method:
After a lead-in phase of open treatment with stimulant, 53 youth meeting criteria for severe mood dysregulation (SMD) were randomly assigned to receive CTP or placebo (PBO) for 8 weeks. A total of 49 participants, 48 of them (98%) meeting disruptive mood dysregulation disorder (DMDD) criteria, were included in the intent-to-treat analysis. The primary outcome measure was the proportion of response based on improvements of irritability at the week 8 of the trial.
Results:
At the end of the trial, a significantly higher proportion of response was seen in those participants randomly assigned to CTP+MPH compared to PBO+MPH (35% CTP+MPH versus 6% PBO+MPH; odds ratio = 11.70, 95% CI = 2.00–68.16, p = 0.006). However, there were no differences in functional impairment between groups at the end of the trial. No differences were found in any adverse effect between treatment groups, and no trial participant exhibited hypomanic or manic symptoms.
Conclusion:
Adjunctive CTP might be efficacious in the treatment of chronic severe irritability in youth resistant to stimulant treatment alone.
Clinical trial registration information:
A Controlled Trial of Serotonin Reuptake Inhibitors Added to Stimulant Medication in Youth With Severe Mood Dysregulation; https://clinicaltrials.gov; NCT00794040.
Keywords: irritability, disruptive mood dysregulation disorder, citalopram, methylphenidate, RCT
Chronic irritability is among the most common reasons for referral to child psychiatric services1,2 and is associated with substantial concurrent and future impairment.3–6 As a result, the American Psychiatric Association recently introduced the diagnosis of Disruptive Mood Dysregulation Disorder (DMDD) into the DSM-5 to capture youth with severe chronic irritability.7 Yet, there is a paucity of clinical trials for DMDD.2,8 In this article, we present the results of a randomized controlled trial (RCT) of citalopram (CTP) versus placebo (PBO) as addons to open-label stimulant optimization for the treatment of irritability in severe mood dysregulation (SMD), which became the basis for the DSM-5 DMDD.
Despite the increasing recognition of the importance of irritability, rigorous testing of possible treatments is still at a nascent state. In terms of psychological therapy, there is encouraging preliminary evidence of efficacy from a small number of trials,9,10 but further work is needed.11–13 Because pharmacological treatments have a clear role in most common psychiatric disorders in youth, including anxiety,14 depression,15,16 and attention-deficit/hyperactivity disorder (ADHD),17,18 and may work synergistically with psychological treatments,14 it is also imperative to identify pharmacological agents for the treatment of children experiencing chronic irritability.2
Although prior literature suggested a therapeutic effect of lithium on aggression,19,20 a small trial (N = 24)21 found lithium to be ineffective for the treatment of severe irritability in children and adolescents with SMD.22 There has been no RCT of antipsychotic medication specifically targeting irritability in children other than those with autism spectrum disorder (ASD). However, an open trial used risperidone in youth with SMD and showed a reduction in irritability.23
Further evidence comes from treatment trials with stimulant in youth with ADHD. A post hoc analysis of the Multimodal Treatment Study of Children with ADHD24 suggested that stimulant treatment of irritability in those with ADHD may be effective. Waxmonsky et al.,25 in a retrospective analysis of data from a crossover placebo-controlled trial of methylphenidate (MPH), reported a significant reduction of irritability/aggression in ADHD. Recently, evidence from several open-label trials with stimulant in youth with ADHD and comorbid SMD/DMDD has emerged. Waxmonsky et al.26 found that stimulant combined with parent management training (PMT) and cognitive-behavioral therapy might be efficacious to treat irritability in this population. The same group has also shown that stimulant alone might decrease behavioral and mood symptoms in youth with ADHD and comorbid DMDD.27 In addition, a recent small open-label trial (N = 22) has demonstrated reductions in irritability with medium to large effect sizes in these children following stimulant monotherapy.28 Nevertheless, a significant proportion (~50%) of those with aggression and ADHD remain refractory to stimulant medication even when combined with parent training/behavioral treatments.25,29
Focusing on the related construct of aggression, the Treatment of Severe Childhood Aggression (TOSCA) study group found that add-on risperidone and placebo were no different from each other when combined with parent training plus stimulant medication in children with ADHD30; similar results were obtained for the 12-week follow up of this study.31 In terms of other mood-stabilizing medication, Blader et al. reported that add-on valproate reduced aggression for a small (n = 14) cohort of children with ADHD who did not respond to optimized stimulant plus family education.32
Thus far, there has been no RCT of serotonin reuptake inhibitors (SRIs) specifically targeting chronic irritability. Indirect evidence derived from adult samples indicates that SRIs may be efficacious in the treatment of irritability in those who are depressed,33 as well as patients with intermittent explosive disorder34 and those with premenstrual dysphoria.35 A review on the effect of antidepressants on irritability in young people36 identified two uncontrolled studies of SRIs that reported on irritability as an outcome37,38; both indicated improvement of irritability with SRI treatment. In addition, a recent metanalysis has shown that irritability may be a specific and robust predictor of future anxiety and depressive disorders.3 Moreover, findings from genetically informative studies suggest shared pathophysiological mechanisms among irritability, anxiety and depression,39,40 thus indicating that agents effective for these disorders could also be useful for irritability. Taken together, these data make SRIs a promising candidate for the treatment of children with severe chronic irritability. Yet, this remains to be tested.
To address this gap in the literature, we conducted an RCT of CTP, an SRI, plus MPH against PBO plus MPH in youth who were originally recruited because they fulfilled criteria for SMD. Before randomization, children took part in an open-label stimulant optimization lead-in phase. This lead-in phase was motivated by the observation that a majority of youth with DMDD also suffer from ADHD41 and that stimulants appear to be efficacious for irritability in ADHD.24 Thus, our RCT was designed to provide a rigorous test of SRI effects over and above stimulant optimization in youth with severe irritability.
METHOD
Subjects
This study was conducted at the National Institute of Mental Health Division of Intramural Research Programs (NIMH DIRP) from November 2008 until January 2018, and was approved by the National Institute of Health’s Combined Neuro-Science Institutional Review Board (CNS-IRB). Before participation, the study was explained to parents and patients. All children gave written assent and parents gave written informed consent. Subjects (aged 7–17 years) were recruited through advertisements that were placed in local parenting magazines, on support groups’ websites, and distributed to psychiatrists nationwide. Also, information about the study was provided in talks to local practitioners and to advocacy/parent groups. The study was registered on ClinicalTrials.gov (Identifier: NCT00794040).
Screening
All participants met criteria for SMD, which were designed to capture those youth with nonepisodic irritability symptoms who were frequently diagnosed with bipolar disorder in clinical settings.22 Based on almost a decade of research on SMD, DMDD was introduced in DSM-5 after this study began. All but one randomized participant (n = 48, 98%) in this study met criteria for DMDD; one participant did not meet DMDD criteria because onset of severe irritability was after 10 years of age but was before age 12. Inclusion criteria for the trial were as follows: (1) irritability operationally defined as markedly increased reactivity to negative emotional stimuli manifest verbally or behaviorally at least three times weekly; (2) abnormal mood (anger or sadness), present at least half of the day most days; (3) three hyperarousal symptoms (insomnia, agitation, distractibility, racing thoughts or flight of ideas, pressured speech, intrusiveness); (4) symptoms cause severe impairment in at least one setting (home, school, or peers) and at least mild impairment in a second setting; (5) symptom onset before age 12 and currently present for at least 12 months without any symptom-free period greater than 2 months; (6) failing treatment as defined by current Children’s Global Assessment of Severity score <60, and outpatient treatment provider agrees that the child’s response to treatment is no more than minimal; and (7) on the basis of record review and interviews with child and parent, the research team agrees that the child’s response to his/her current treatment is no more than minimal. Having a score of >12 on the irritability subscale of the Aberrant Behavior Checklist (ABC)42 was initially an inclusion criterion, but we stopped collecting the ABC data for several reasons (see Supplement 1, available online). All the patients had histories of failed response to treatment, either pharmacological or psychological or both. None of patients were medication naive at the time of enrollment. For all patients, their providers in the community endorsed their patients’ participation in the study and acknowledged that the current treatment was only minimally successful and that other options should be explored.
Exclusion criteria for the trial were as follows: (1) presence of cardinal bipolar symptoms of elevated, expansive mood, grandiosity, inflated self-esteem, or episodically decreased need for sleep; (2) distinct episodes of hypo/manic symptoms greater than 1 day; (3) current major depressive disorder; (4) autism spectrum disorder; (5) psychosis; (6) posttraumatic stress disorder; (7) substance abuse within 3 months; (8) medical illnesses (eg, liver, seizure, renal, platelet disorder) that require medications, are chronic, unstable, could cause SMD symptoms, or are contraindications to treatment with serotonin reuptake inhibitor (SRI) or stimulant, or that require medications that are contraindicated with SRIs or MPH; (9) intelligence quotient (IQ) <70; (10) pregnant, lactating, or sexually active without barrier method of contraception; (11) failed adequate trial (defined as 4 weeks of consecutive treatment with no less than 20 mg citalopram or 10 mg escitalopram) or severe ill effects while on citalopram or escitalopram; (12) history of hypersensitivity or severe adverse reaction to methylphenidate or serious adverse reactions (psychosis, severely increased activation compared to baseline) to methylphenidate or amphetamines.
Comorbid ADHD, oppositional defiant disorder (ODD), or anxiety disorders were not exclusionary. Full Scale Intelligence Quotient (FSIQ) was measured using the Wechsler Abbreviated Scale of Intelligence (WASI) for all subjects.43
Following a telephone interview to screen for relevant inclusion or exclusion criteria, record review, and consultation with the child’s treating clinician, candidates for the study were invited to the NIMH DIRP (n = 311). On-site screening included the Kiddie Schedule for Affective Disorders–Present and Lifetime Version (K-SADS-PL)44 with an additional supplement for SMD, designed in collaboration with Joan Kaufman, PhD, to ascertain whether children met criteria for this syndrome. All diagnostic measures were administered to the parent and child individually by trained postgraduate-level clinicians with established interrater reliability (κ = 0.9, including distinguishing SMD subjects from those with distinct hypo/manic episodes).22,44 Diagnoses were based on best-estimate procedures45 generated in a consensus conference led by two psychiatrists with extensive experience evaluating children with mood disorders.
Enrollment
Figure S1, available online, provides the trial design overview and Figure 1 the Consolidated Standards of Reporting Trials (CONSORT) diagram. After on-site screening, subjects who met inclusion criteria were offered enrollment in the RCT. To maximize safety and observational data collection while they were tapered off medication and began open treatment with MPH, patients received inpatient care on a research child psychiatric unit at the NIH Clinical Center. Upon admission to the hospital, participants were assessed as described below (Baseline at admission) and then gradually tapered off psychotropic medications (Phase I). They were monitored closely for clinical deterioration and side effects; weekend passes with family were offered weekly or every other weekend depending on proximity to the NIH. Medication tapering (median = 3 weeks) was individually tailored and prioritized medications with longer half-lives (eg, atypical neuroleptics, antidepressants) before those with short half-lives (eg, psychostimulants). Care in the hospital for all patients included weekday on-site school, weekday rounds meeting with a psychiatrist or nurse practitioner, weekly meetings with parent(s), individual and group activities, and milieu treatment that are common in inpatient pediatric psychiatry units. Concomitant therapy and any other psychological treatment were not allowed during participation in the protocol. During medication taper, 12 participants were discharged for meeting exclusion criteria (see Supplement 1, available online) (Figure 1).
FIGURE 1. CONSORT Diagram of the Trial.
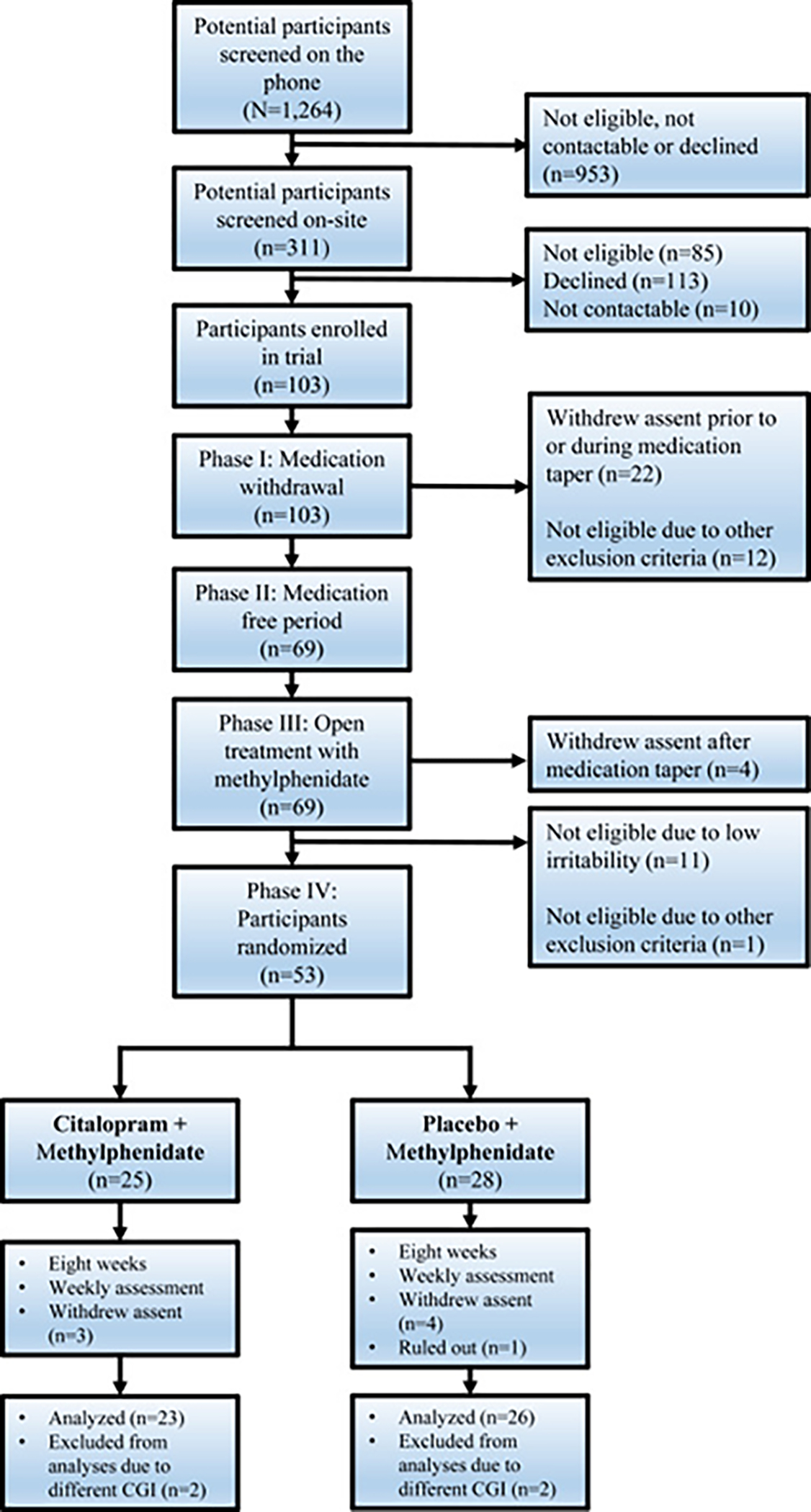
Note: Of the randomized participants (n = 53), 4 participants were excluded from analysis because the Clinical Global Impression (CGI) measure used was a different version and thus not comparable. Of the 49 participants included in the analyses, 41 completed the trial; 7 withdrew assent before week 8, and one was ruled out of the study by the experimenters because of high levels on liver function test. Please note color figures are available online.
Treatment of ADHD Symptoms
After completing up to two medication-free weeks (Phase II), participants completed up to 5 weeks of open treatment with MPH to achieve optimal control of ADHD symptoms (Phase III). For details on dosage, see Supplement 1, available online.
Blinded, Randomized Add-On Citalopram or Placebo
Before randomization, those who no longer met SMD criteria because of decreased irritability (n = 11) or met other exclusion criteria (n=1) were discharged (see Supplement 1, available online) (Figure 1). Those who continued to meet SMD criteria while on MPH (n = 53) had a second set of baseline measures collected (Baseline at randomization) and then were randomized to add-on treatment with either CTP or PBO for an 8-week double-blind RCT (Phase IV) (Figure S1, available online). Treatment allocation was randomized by the Pharmaceutical Development Service of the National Institutes of Health Clinical Center Pharmacy Department using a random numbers table, which was randomized in alternating blocks of six and four in a 1:1 ratio.
During the randomized phase, every prescriber and everyone who came into contact with the children was blinded to their treatment assignment. Commercial preparations of CTP were obtained from local distributors and investigational (blinded) capsules were compounded and supplied by the NIH Clinical Center Pharmaceutical Development Section.
The CTP/PBO dosing schedule began with 1 capsule (5 mg) daily; at 5-day intervals, the dose could increase by 1 capsule. After roughly 3 weeks of the 8-week trial (mean = 3.1 weeks, SD = 1.5, with no difference between groups, p = .899), when patients were receiving 4 capsules (equivalent to 20 mg/d) and clinicians decided it was safe to do so, they were discharged from inpatient care and returned home. Once home, they received study medication and were monitored with weekly clinical ratings, as described below, by blinded raters who were not prescribers. These ratings were done by telephone, alternating with weekly outpatient visits. Based on clinical judgment of minimal side effects and ongoing symptoms, weekly increments were permitted until a maximum dose of 8 capsules per day (equivalent to 40 mg) was achieved. The average dose achieved was 28.33 mg/day (range 5–40 mg/day) or 29.23 mg/day (range 20–40 mg/day) if one participant who withdrew 4 days after randomization is omitted. During the RCT, lorazepam was available as necessary for agitation; however, no SMD subject received lorazepam during the RCT.
Side Effects
Side effects were ascertained by checklists as described in the Supplement 1, available online. Information on suicidality and manic symptoms was collected by asking directly to participants and parents.
Assessment
Two graduate-level, highly experienced clinicians completed weekly the Clinical Global Impression’s Improvement (CGI-I) and Severity (CGI-S) scales46 independently of each other (as described in Supplement 1, available online), as well as the Children’s Global Assessment of Severity (CGAS),47 the Pediatric Anxiety Rating Scale (PARS),48 and the Children’s Depression Rating Scale (CDRS).49 Finally, ratings were reached by consensus in a case conference with a senior child and adolescent psychiatrist (K.T.); all participants were blinded to the treatment condition.
Our primary categorical clinical outcome was treatment response defined as a CGI-I score of 2 (much improved), or 1 (symptom free), consistent with the original protocol and common practice.16,50 Sensitivity analyses were also conducted using the CGI-S, as in other studies (see Supplement 1, available online, for detailed information).50
The ABC42 was initially designated as a primary outcome measure; however, it proved inappropriate for the population that we studied, as it showed minimal variability (see Supplement 1, available online, for detailed information). Thus, we stopped collecting data from it early on. Secondary outcomes were functional impairment (measured with CGAS), improvement in anxiety symptoms (measured with PARS), and improvement in depressive symptoms (measured with CDRS).
Statistical Analysis
A statistical analysis plan was written after the end of data collection but before the group-label unblinding of the analyst and statistician (P.V-R. and A.P.). All children who completed at least one postrandomization assessment were included in intent-to-treat analyses.
In addition, to examine the effect of stimulant optimization before randomization on irritability severity, we compared changes in CGI-S between baseline at admission and baseline at randomization with a paired t test, in all randomized subjects.
Primary and secondary outcomes after randomization were analyzed using multilevel models estimated by maximum likelihood enabling the inclusion of participants incompletely observed under the missing-at-random assumption. This is the recommended analytic approach for repeated measures data51 over other approaches such as last observation carried forward (LOCF). However, a sensitivity analyses was carried out using the LOCF method (see Supplement 2, available online).
For our primary categorical outcome (ie, treatment response as defined by CGI-I score <3), an efficient estimate was obtained by fitting a growth curve model for the repeated binary response fitted by maximum likelihood in the Stata program gsem, with binomial family and logit link function. The model included a random intercept. The fixed part of the model included treatment group, number of weeks as measure of time, and the group-by-week interaction (a test for a quadratic term was also carried out). The postestimation lincom was used to estimate the group difference at the week 8 and its 95% confidence interval (CI). To assist in interpretation, this conditional or subject-specific effect estimate (and its 95% CI) was translated to an approximate, but more easily understood, marginal mean estimate.52 The estimate from the group-by-week interaction in the model provided a measure of difference in response rate between groups across the 8 weeks of the trial. Estimated proportions of response were plotted with estimates provided by command margins. (See Supplement 1, available online, for analysis of continuous outcomes.)
Frequencies of the most common adverse effects are reported if present in more than one subject in either study group. These were compared using a two-sided Fisher exact test.
RESULTS
Participant Sample Derivation and Characteristics
As shown in the CONSORT diagram in Figure 1, of the 53 participants eligible for randomization, 25 were allocated to receive adjunctive citalopram (CTP) and 28 to receive adjunctive placebo (PBO). All subjects completed at least one post-randomization assessment. However, the first four participants recruited (two from each group) were excluded from the analysis because of technical problems with data collection. Of the remaining 49 participants included in the intent-to-treat analysis, 41 completed the trial. One participant was withdrawn by the experimenters at week 7 due to increased levels on the liver function test; specifically, alanine transaminase and aspartate transaminase were elevated to three times the upper limit of normal, but these normalized after cessation of methylphenidate, while the patient continued on open citalopram. Another 7 participants withdrew assent before week 8. There were no differences between groups in the number of weeks in inpatient care before participants were discharged to home (CTP+MPH: mean = 3.1, SD = 1.1; PBO+MPH: mean = 3.1, SD = 1.8 [t(45) = −0.13], p = .899). Table 1 contains the demographic and clinical characteristics of the 49 participants included in the intent-to-treat analysis. All of the participants in the trial had at least one comorbid diagnosis.
TABLE 1.
Sample Demographic and Clinical Characteristics
| MPH + CTP n = 23 SMD | MPH + PBO n = 26 SMD | |
|---|---|---|
| Sex, Female, n (%) | 10 (43) | 6 (23) |
| Age, y, Mean (SD), range | 11.4 (2.5), 7–15 | 11.7 (2.1), 8–14 |
| Ethnicity, n (%) | ||
| White | 19 (83) | 20 (77) |
| Black or African American | 0 (0) | 3 (12) |
| Asian | 1 (4) | 0 (0) |
| Multiple races | 3 (13) | 2 (8) |
| Unknown | 0 (0) | 1 (4) |
| IQ, Mean (SD) | 109.3 (14.8) | 113 (9.2) |
| Co-occurring Diagnoses, n (%) | ||
| Any disorder | 23 (100) | 26 (100) |
| ADHD | 20 (87) | 24 (92) |
| ODD | 17 (74) | 22 (85) |
| CD | 0 (0) | 1 (4) |
| Any anxiety disorder | 13 (57) | 15 (58) |
| Medicated at admission, n (%) | 21 (91) | 23 (84) |
| Number of Medications at Admission, n (%) | ||
| None | 2 (9) | 3 (12) |
| 1 | 4 (17) | 5 (19) |
| 2 | 5 (22) | 3 (12) |
| 3 or more | 12 (52) | 15 (57) |
| Type of Medication, n (%) | ||
| Atypical antipsychotic | 14 (60) | 17 (65) |
| Typical antipsychotic | 0 (0) | 1 (4) |
| Stimulant | 8 (35) | 9 (35) |
| Nonstimulant (Ato, Gua, Clo) | 9 (39) | 12 (46) |
| SRI | 7 (30) | 7 (27) |
| Other antidepressant | 3 (13) | 3 (12) |
| Anticonvulsant | 10 (43) | 7 (27) |
| Lithium | 4 (17) | 3 (12) |
| Prior Medication Treatments, Mean (SD), Range | 4.1 (3.5), 0–13 | 5.3 (3.1), 1–13 |
| Prior Hospitalizations, n (%) | ||
| None | 15 (65) | 15 (58) |
| 1 | 2 (9) | 5 (19) |
| 2 or more | 6 (26) | 6 (23) |
| Educational Programs, n (%) | ||
| Mainstream school (MS) | 11 (48) | 4 (15) |
| MS with accommodations | 1 (4) | 1 (4) |
| In-home school | 1 (4) | 1 (4) |
| Special education | 10 (43) | 20 (77) |
| Baseline Admission, Mean (SD) | ||
| CGI-Sa | 4.4 (0.6) | 4.6 (0.5) |
| CGAS | 44 (6.1) | 41.7 (2.2) |
| CDRS | 29.7 (5.9) | 32.0 (7.6) |
| PARS | 15.7 (4.5) | 14.8 (4.7) |
| Baseline Randomization, Mean (SD) | ||
| CGI-S | 4.0 (0.5) | 4.3 (0.6) |
| CGASb | 44.4 (3.3) | 42.9 (3.6) |
| CDRSc | 29.4 (5.7) | 34.6 (8.6) |
| PARSd | 13.3 (5.9) | 15.8 (5.2) |
Note: Any anxiety disorder includes separation anxiety disorder, generalized anxiety disorder, social anxiety disorder, and panic disorder. ADHD = attention-deficit/hyperactivity disorder; Ato = atomoxetine; CD = conduct disorder; CDRS = Children’s Depression Rating Scale; CGAS = Children’s Global Assessment Severity; CGI = Clinical Global Impression; CGI = Clinical Global Impression—Severity; Clo = clonidine; CTP = citalopram; Gua = guanfacine; MDD = major depressive disorder; MPH = methylphenidate; ODD = oppositional defiant disorder; PARS = Pediatric Anxiety Rating Scale; PBO = placebo; SMD = severe mood dysregulation; SRI = serotonin reuptake inhibitor.
Missing data:
n = 1 in MPH+CTP, n = 1 in MPH+PBO.
n = 1 in MPH+CTP.
n = 7 in MPH+CTP, n = 8 in MPH+PBO.
n = 1 in MPH+PBO.
Response to Open-Label Stimulant Optimization
Severity of irritability decreased from admission to randomization (effect size [ES] = 0.60, 95% CI = 0.30–0.89, p < .001) in the sample included in the intent-to-treat analysis (Figure 2, depicted as the black dotted line). However, of the randomized participants, only one was a responder based on CGI-I score. This is because, as per protocol, those who responded and no longer met irritability threshold criteria (n = 11) were withdrawn before randomization. As expected, stimulant medication had a strong effect on hyperarousal symptoms (ES = 1.10, p < .001) given the overlap with ADHD symptoms, but only a moderate effect on temper outbursts (ES = 0.54, p = .001) and no effect on mood between outbursts (ES = −0.08, p > .05). We present the detailed results and relevant graphs for separate SMD symptom domains in Supplement 2, available online.
FIGURE 2. Change in Irritability Severity Before and After Randomization in the Sample Included in the Intent-to-Treat Analysis.
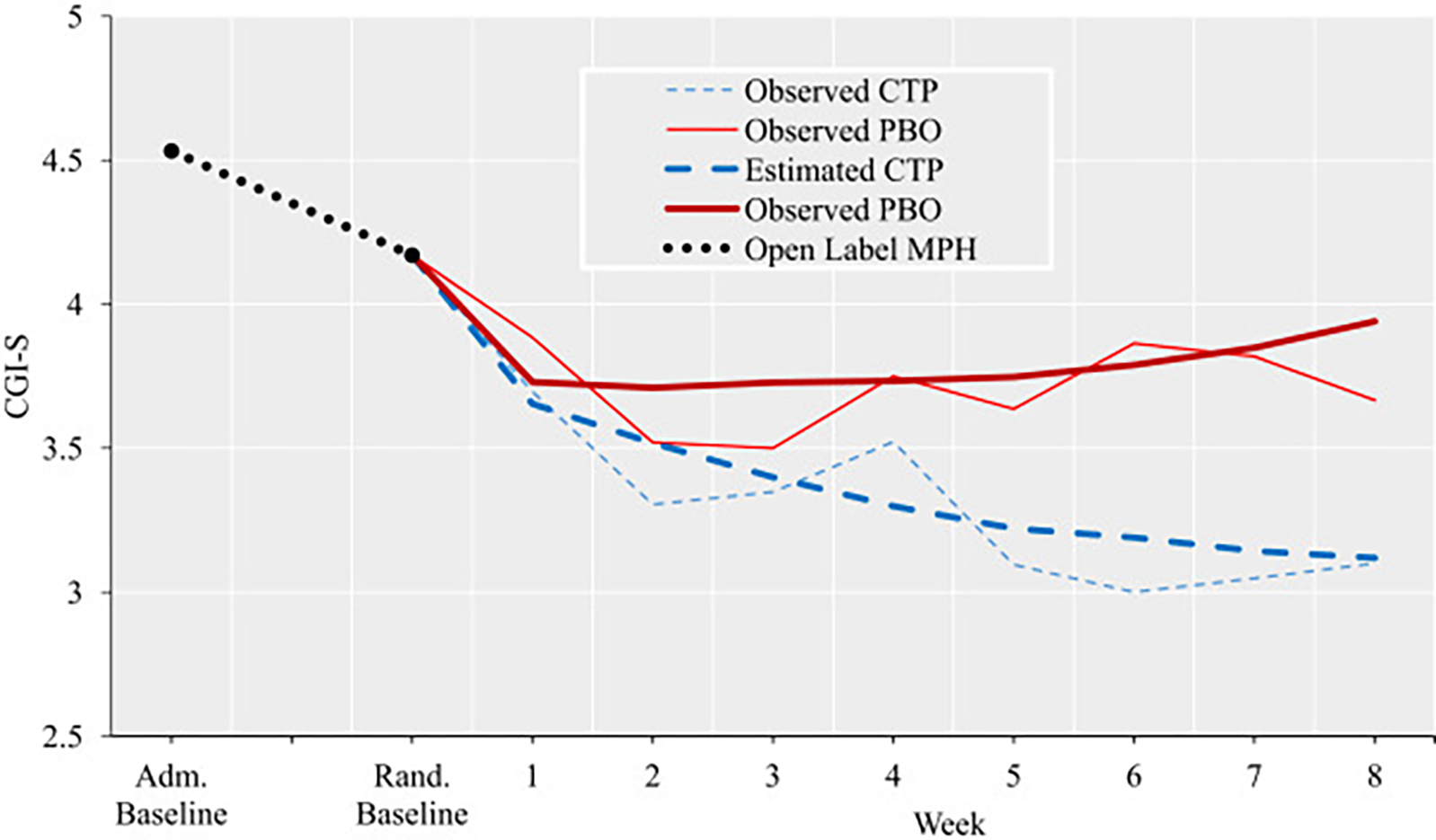
Note: The period between baseline at admission (Adm. Baseline) and baseline at randomization (Rand. Baseline) was variable, and included the washout period (Phase I), medication-free period (Phase II), and open-label lead phase with stimulant optimization (Phase III). Before randomization (depicted as the black dotted line), change in irritability severity is shown for the entire sample (n = 49). After randomization, both observed and estimated severity (provided by command margins in Stata) are displayed by week and treatment group (citalopram [CTP], n = 23; placebo [PBO], n = 26). MPH = methylphenidate. Please note color figures are available online.
Response to Citalopram Versus Placebo
For our primary outcome (ie, rates of response to treatment as measured with CGI-I), estimated proportions of response differed at week 8 of treatment between citalopram and placebo groups (35% CTP+MPH versus 6% PBO+MPH, difference of 29%, p = .005; OR = 11.70, 95% CI = 2.00–68.16, p = .006) (Table 2), with a number of patients needed to treat (NNT) of three. In addition, the group-by-week interaction for the estimated proportions of response across the 8 weeks of trial was also significant (b = 0.43, 95% CI = 0.120–74, p = .006) (Table 2). Figure 3 depicts the estimated proportion of response for all participants included in the analyses against the observed proportion of responses for individuals with available data.
TABLE 2.
Summary of Primary and Secondary Outcomes of the Randomized Controlled Trial With Adjunctive Citalopram Versus Placebo
|
Between-Group Difference
|
||||||
|---|---|---|---|---|---|---|
| Outcome | Baseline Randomization a | Week 8 of Trial b | Group by Week Interaction | At Week 8 | ||
| CTP | PBO | CTP | PBO | p | p | |
| % (SE) n | % (SE) n | % (SE) n | % (SE) n | |||
| CGI-I Responsec | ||||||
| Estimated | — | — | 35 (10) 8 | 6 (4) 2 | .006 | .006 |
| Observed | — | — | 25 (10) 5 | 10 (7) 2 | — | — |
| Mean (SE) n | Mean (SE) n | Mean (SE) | Mean (SE) | p | p | |
| CGI-S | 4.0 (0.4) 23 | 4.3 (0.4) 26 | 3.1 (0.3) | 3.9 (0.3) | .046 | .085 |
| CGAS | 44.4 (0.2) 22 | 42.9 (0.2) 26 | 52.6 (2.3) | 47.2 (2.1) | .109 | .124 |
| CDRS | 29.4 (0.5) 16 | 34.6 (0.7) 18 | 28.6 (1.8) | 30.1 (1.8) | .680 | .993 |
| PARS | 13.3 (1.2) 23 | 15.8 (1.0) 25 | 12.0 (1.2) | 13.4 (1.2) | .598 | .283 |
Note: CDRS = Children’s Depression Rating Scale; CGAS = Children’s Global Assessment Severity; CGI = Clinical Global Impression; CTP = citalopram; MPH = methylphenidate; PARS = Pediatric Anxiety Rating Scale; PBO = placebo; SE = standard error.
Baseline descriptive statistics are based in observed data.
Descriptive statistics at week 8 of the trial and p values of differences in the randomized controlled trial are based on model-based estimates of the intent-to-treat analysis.
For CGI-I response, both estimated (n = 49) and observed (n = 41) data are provided.
FIGURE 3. Proportion of Treatment Response by Week and Treatment Group.
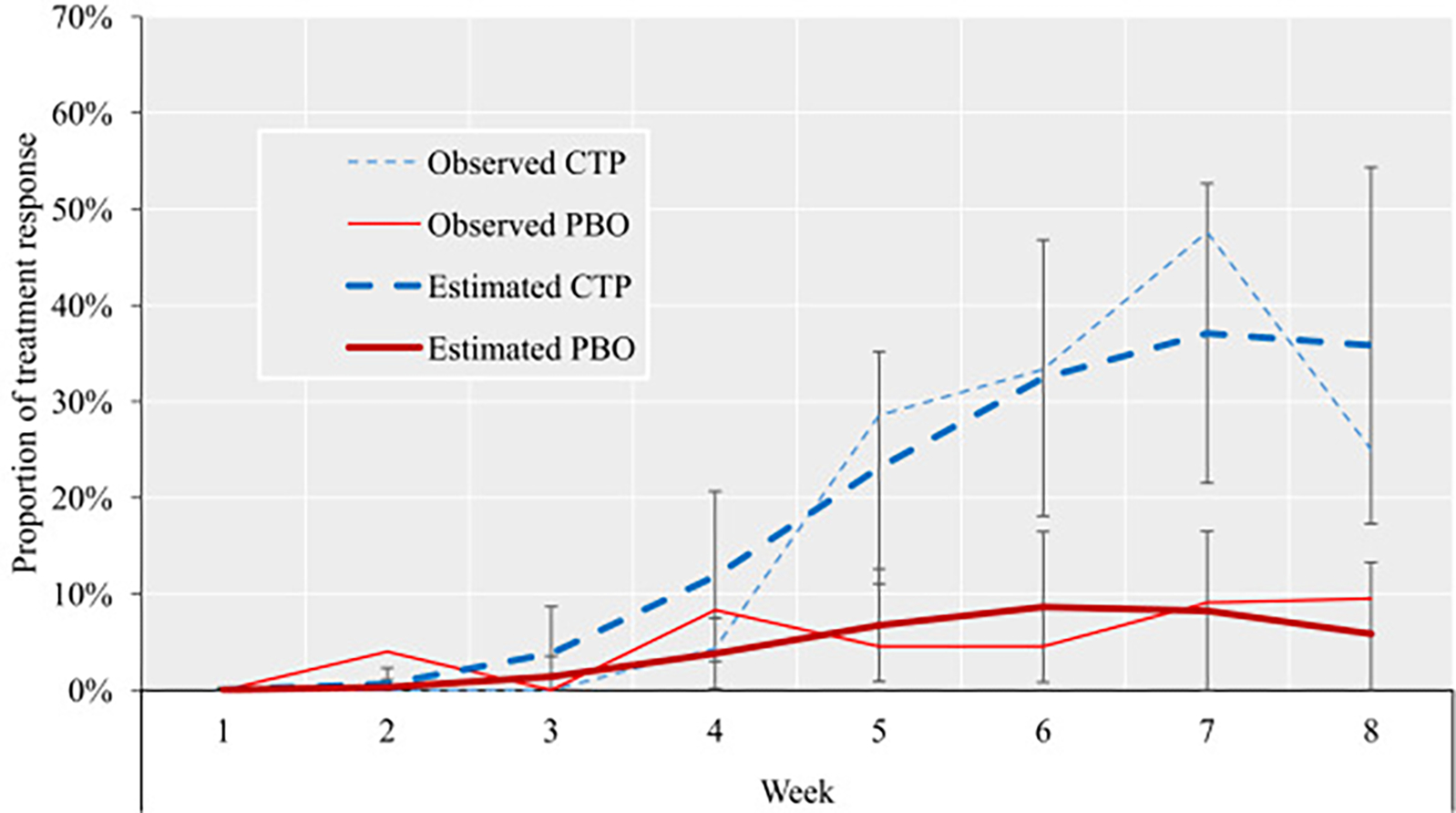
Note: Both observed and estimated proportions of response are displayed. Bars represent 95% CI. Estimated proportions were extracted with command margins in Stata and are based on n = 49. Observed proportions are based on n = 49 at week 1; n = 48 at week 2; n = 47 at weeks 3 and 4; n = 43 at weeks 5, 6, and 7; and n = 41 at week 8. CTP = citalopram; PBO = placebo. Please note color figures are available online.
The difference in irritability severity between groups, as measured with the CGI-S, at week 8 of the trial was a nonsignificant trend (b = −0.62, 95% CI = −1.32 to 0.09, p = .085, ES = −1.11, 95% CI = −2.38 to 0.15). However, a significant group-by-week interaction for the CGI-S emerged across the 8 weeks of the trial (b = −0.11, 95% CI = −0.21 to −0.002, p = .046) (Table 2 and Figure 2). This difference was driven primarily by changes in the severity of temper outbursts (Figure S2, available online) and, to a lesser extent, by irritable mood between outbursts (Figure S3, available online). No difference was observable for hyperarousal symptoms (Figure S4, available online).
Groups did not differ in functional impairment as measured with the CGAS at week 8 of the trial (b = 4.72, 95% CI = −1.30 to 10.74, p = .124, ES = 1.36, 95% CI = −0.37 to 3.09), and the group-by-week interaction was not significant (b = 0.75, 95% CI = −0.17 to 1.66, p = .109) (Table 2).
Groups did not differ in severity of depressive symptoms as measured with the CDRS at the week 8 of the trial (b = 0.02, 95% CI = −4.76 to 4.80, p = .993, ES = 0.00, 95% CI = −0.62 to 0.62), and the group-by-week interaction was not significant (b = −0.15, 95% CI −0.87 to 0.57, p = .680) (Table 2). However, the CDRS irritability item resulted in a nonsignificant trend at week 8 (b = 0.77, 95% CI = −0.02 to 1.56, p = .057, ES = 0.50, 95% CI = −0.02 to 1.02). Similarly, groups did not differ in severity of anxiety symptoms as measured with the PARS at week 8 of the trial (b = −1.02, 95% CI = −4.23 to 2.19, p = .534, ES = −0.19, 95% CI = −0.79 to 0.41), and the group-by-week interaction was not significant (b = 0.02, 95% CI = −0.40 to 0.45, p = .909).
Distribution of lowest level residuals was close to normal, so no transformations were required. Results and estimates of the sensitivity analyses, which used the lastobservation-carried-forward method, were very similar to the results described above (see Supplement 2, available online). Finally, the number of weeks in inpatient care did not have an impact in the outcomes.
Tolerability and Adverse Effects
Table S1, available online, shows rates of adverse effects in each of the treatment groups. No differences were found in any adverse effect or the total number of adverse effects between treatment groups (see Supplement 3, available online).
DISCUSSION
This is the first RCT of an SRI for the treatment of irritability in youth. Specifically, we report the findings of an 8-week, flexible-dose, double-blind, placebo-controlled trial of CTP as an add-on to open-label MPH in children with severe irritability. Our results provide some support for the efficacy of CTP+MPH in the treatment of irritability in children with severe irritability who are already receiving methylphenidate. However, it is important to note that there were no differences in functional impairment between groups at the end of the trial. Of note, although participants were initially recruited because they fulfilled SMD criteria, all but one participant also fulfilled DMDD criteria.
As expected, open-label treatment with MPH led to a significant reduction in SMD symptoms, specifically hyperarousal and temper outburst, with effect sizes consistent with what has previously been reported.29 The RCT’s primary outcomes are suggestive of citalopram’s efficacy. That is, at week 8, a significantly higher percentage of patients on citalopram improved, compared to those who received placebo; this was also true when taking all time points into account. At week 8, the reduction in the continuous score of irritability severity did not reach significance (p = 0.085), although the week-by-group interaction across the 8 weeks did (p = .046). The differentiation between citalopram and placebo appeared to emerge around week 5 (for observed data, between-group differences were as follows: week 5, p = .046; week 6, p = .021; and week 7, p = .007). This time course is consistent with the timing of action of SRIs.53 Importantly, the confidence intervals of the effect sizes vary from relatively small to large, indicating considerable uncertainty about the magnitude of the effect.
We note that the magnitude of the response to SRIs in this study was relatively low compared to that in anxiety and depression trials. In addition, the placebo response rate was also extremely low (ie, ~5%). Together, these two observations explain the very favorable number needed to treat (NNT) of three. However, although the response rate to active medication may appear low in comparison to effects observed in non–add-on trials, it is consistent with what is observed in add-on trials. For example, the remission rate in STAR*D for add-on treatment was 30%,54,55 and in CO-MED56 it was 38%. Similarly, in a meta-analytic study, add-on treatment for ADHD showed smaller effect sizes than monotherapies.57 Thus, it is unclear whether the relatively low response rate to CTP+MPH treatment (and placebo) in irritability in this trial, compared to those for depression or anxiety, is due to differences in the target phenotype or in trial design.
At the end of the trial, there were no differences in the CGAS scores between groups, with participants still showing moderate functional impairment after completion (or severe in at least one area, range 40–50). This suggests that improvements in irritability appear not to have translated into reductions in overall impairment with siblings, peers, and at school within the time scale of the trial. This interpretation would be in keeping with what has been described previously, in that it is common to see a dissociation between symptoms and impairment levels.58 However, it is important to note that the CGAS focuses on the lowest level of function during the period under consideration, and thus may be suboptimal as an outcome measure47 for disorders such as DMDD. As the functioning of a chronically irritable child during the previous week could include even short bursts of dysregulation, the CGAS might be expected to lag behind a measure of modal functioning.
We also observed no significant differences in CDRS scores. We note that depressive symptoms were low throughout the trial for most individuals, which is not surprising, given that individuals with ongoing MDD were ineligible. In addition, no significant differences were observed for anxiety symptoms as measured with PARS. The PARS scores were in the mid-range and barely changed in either group during the trial. It is well known that irritability frequently overlaps with depression and anxiety in patients and that they share etiological underpinnings.39,40 Our results suggest that improvements in irritability with SRI treatment may be seen in the absence of change in depression and anxiety.
The rate of side effects did not differ between CTP+MPH and PBO+MPH. However, the absolute level of side effects was high in both groups. This may be a consequence of directly asking about side effects and the detailed observations that trained nursing staff offered during the inpatient phase of the study. In addition, nurses and parents were instructed to rate what they observed without reference to changes from baseline irritability or time of day, and thus even low levels of severity were reported in the checklist. Only one patient was withdrawn by experimenters because of adverse effects: specifically, at week 7, because of elevated liver function test results, which normalized with methylphenidate discontinuation. We observed no hypomania or mania in any participant at any phase of the trial. This is consistent with previous findings from studies showing that chronic irritability is not a presage of bipolar disorder type I or type II6,59–61 and that chronically irritable youth are not at elevated risk for manic switches. However, the number of participants exposed to citalopram in this trial was too small to draw firm conclusions about the risk for medication-induced mania in this population.
Our study has several strengths. It used a rigorous double-blind, placebo-controlled approach in a well-phenotyped sample of youth to investigate the effects of a CTP+MPH in DMDD. It also used an initial open-label stimulant optimization phase, which resembles common clinical practice and reflects a rational approach to treatment of irritability in the presence of ADHD. The design of the study should also be placed in the context of where the field was when it began a decade ago. The design maximized human subjects’ protections by using hospitalization for safety and standardizing treatment and environmental care before randomization.
Our study also has limitations. First, the sample size was smaller than initially planned. In the 10-year duration of the study, we pursued nationwide recruitment using local resources, sending letters to every practicing child and adolescent psychiatrist in the American Academy of Child and Adolescent Psychiatry, and giving local talks and presentations across the country. Although the initial response to the study was positive, over time, the response declined steadily, and a corresponding increase in the use of SRIs, as well as MPH, for chronic irritability took place in clinical practice. Yet, our experience in recruitment was not unique.62,63 There are several potential explanations for this, including the increasing familiarization of community practitioners with prescribing for youth with chronic irritability, especially after the publication of studies showing that chronic irritability does not predict bipolar disorder. Nonetheless, we note that at the current sample size, the study had a power of approximately 75% to detect a difference at p < .05. Clearly, further study with a larger sample would be warranted. A second limitation of this study is that it did not use a broad array of dimensional measures of irritability or other psychopathology. The study preceded the psychometric development of the Affective Reactivity Index (ARI),64 or the Multidimensional Assessment of Preschool Disruptive Behavior (MAP-DB) scales, which would have been useful adjuncts. To date, however, there are still no validated self- or parent-report scales for assessing change, as opposed to trait measurement, of irritability. Future trials should use more comprehensive multi-source, multi-method approaches to the measurement of the clinical phenotype. A third limitation is that the sample in this study may not be generalized to all children with DMDD, as we initially recruited a population with SMD, which requires hyperarousal symptoms (eg, insomnia, agitation, distractibility) and might represent a more severe subgroup of the DMDD population. Furthermore, there are limitations of generalizing from subjects coming to research at NIMH to other populations because of the design of the study. Indeed, the length of the study, the compulsory inpatient interval, and the requirement that patients come off all psychotropic medication, were all obstacles to participation and limit the generalizability of our findings. Finally, the design of this trial required a period of hospitalization in an inpatient unit, which some studies have linked to initial improvement of irritability-related behaviors65,66 and might also explain the discordance among different informants.67 However, all participants were subjected to this potential phenomenon, and therefore it does not explain the distinct treatment effects seen after randomization.
In summary, we find that, among those unresponsive to stimulant alone, severe irritability symptoms improved in significantly more children who received add-on citalopram than those receiving add-on placebo. However, our results do not show evidence of effect on impairment in those patients.
Supplementary Material
Acknowledgments
This study represents independent research funded by Intramural Research Program (ZIAMH002786) of the National Institute of Mental Health (NIMH) with analysis supported by the UK National Institute for Health Research (NIHR) Biomedical Research Centre at South London and Maudsley NHS Foundation Trust and King’s College London. The views expressed are those of the author(s) and not necessarily those of the UK NHS, NIHR, or the Department of Health and Social Care of United Kingdom.
Drs. Vidal-Ribas and Pickles served as the statistical experts for this research.
The authors wish to acknowledge the skill, dedication, and effort of the 1 SW Nursing Staff from the NIH Clinical Center, Bethesda, MD, without whom this study would not have been possible. They gratefully acknowledge the children and families who participated in this trial and made this research possible.
Footnotes
Disclosure: Drs. Towbin, Vidal-Ribas, Brotman, Pickles, Overman, McNeil, Haring, Pine, Leibenluft, and Stringaris and Mss. Miller, Kaiser, Vitale, Engel, Davis, Lee, Wheeler, Yokum, Roule, Wambach, and Sharif-Askary have reported no biomedical financial interests or potential conflicts of interest.
Contributor Information
Kenneth Towbin, Mood Brain and Development Unit, Emotion and Development Branch, National Institute of Mental Health, National Institutes of Health, Bethesda, MD..
Pablo Vidal-Ribas, Mood Brain and Development Unit, Emotion and Development Branch, National Institute of Mental Health, National Institutes of Health, Bethesda, MD.; Institute of Psychiatry, Psychology & Neuroscience, King’s College London, UK.
Melissa A. Brotman, Neuroscience and Novel Therapeutics, Emotion and Development Branch, National Institute of Mental Health, National Institutes of Health, Bethesda, MD..
Andrew Pickles, Institute of Psychiatry, Psychology & Neuroscience, King’s College London, UK..
Katherine V. Miller, Mood Brain and Development Unit, Emotion and Development Branch, National Institute of Mental Health, National Institutes of Health, Bethesda, MD..
Ariela Kaiser, Mood Brain and Development Unit, Emotion and Development Branch, National Institute of Mental Health, National Institutes of Health, Bethesda, MD..
Aria D. Vitale, Mood Brain and Development Unit, Emotion and Development Branch, National Institute of Mental Health, National Institutes of Health, Bethesda, MD..
Chana Engel, Mood Brain and Development Unit, Emotion and Development Branch, National Institute of Mental Health, National Institutes of Health, Bethesda, MD..
Gerald P. Overman, National Institutes of Health, Bethesda, MD..
Mollie Davis, Section on Mood Dysregulation and Neuroscience, Emotion and Development Branch, National Institute of Mental Health, National Institutes of Health, Bethesda, MD..
Beth Lee, College of Nursing, University of Arizona, Tuscon..
Cheri McNeil, Section on Mood Dysregulation and Neuroscience, Emotion and Development Branch, National Institute of Mental Health, National Institutes of Health, Bethesda, MD..
Wanda Wheeler, Section on Mood Dysregulation and Neuroscience, Emotion and Development Branch, National Institute of Mental Health, National Institutes of Health, Bethesda, MD..
Catherine H. Yokum, Section on Mood Dysregulation and Neuroscience, Emotion and Development Branch, National Institute of Mental Health, National Institutes of Health, Bethesda, MD..
Catherine T. Haring, University of Michigan, Ann Arbor..
Alexandra Roule, Penn State University, State College, PA..
Caroline G. Wambach, Georgetown University School of Medicine, Washington, DC..
Banafsheh Sharif-Askary, Duke University School of Medicine, Durham, NC..
Daniel S. Pine, Section on Development and Affective Neuroscience, Emotion and Development Branch, National Institute of Mental Health, National Institutes of Health, Bethesda, MD..
Ellen Leibenluft, Section on Mood Dysregulation and Neuroscience, Emotion and Development Branch, National Institute of Mental Health, National Institutes of Health, Bethesda, MD..
Argyris Stringaris, Mood Brain and Development Unit, Emotion and Development Branch, National Institute of Mental Health, National Institutes of Health, Bethesda, MD..
REFERENCES
- 1.Mikita N, Stringaris A. Mood dysregulation. Eur Child Adolesc Psychiatry. 2013;22(Suppl 1):S11–S16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Stringaris A, Vidal-Ribas P, Brotman MA, Leibenluft E. Practitioner review: Definition, recognition, and treatment challenges of irritability in young people. J Child Psychol Psychiatry Allied Discip. 2017;59:721–739. [DOI] [PubMed] [Google Scholar]
- 3.Vidal-Ribas P, Brotman MA, Valdivieso I, Leibenluft E, Stringaris A. The status of irritability in psychiatry: a conceptual and quantitative review. J Am Acad Child Adolesc Psychiatry. 2016;55:556–570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Althoff RR, Verhulst FC, Rettew DC, Hudziak JJ, van der Ende J. Adult outcomes of childhood dysregulation: a 14-year follow-up study. J Am Acad Child Adolesc Psychiatry. 2010;49:1105–1116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Copeland WE, Angold A, Costello EJ, Egger H. Prevalence, comorbidity, and correlates of DSM-5 proposed disruptive mood dysregulation disorder. Am J Psychiatry. 2013;170:173–179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Copeland WE, Shanahan L, Egger H, Angold A, Costello EJ. Adult diagnostic and functional outcomes of DSM-5 disruptive mood dysregulation disorder. Am J Psychiatry. 2014;171:668–674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.American Psychiatric Association. Diagnostic and Statistical Manual of Mental Disorders (5th ed.). Washington, DC: American Psychiatric Association;2013. [Google Scholar]
- 8.Benarous X, Consoli A, Guile JM, Garny de La Riviere S, Cohen D, Olliac B. Evidence-based treatments for youths with severely dysregulated mood: a qualitative systematic review of trials for SMD and DMDD. Eur Child Adolesc Psychiatry. 2017;26:5–23. [DOI] [PubMed] [Google Scholar]
- 9.Perepletchikova F, Nathanson D, Axelrod SR, et al. Randomized clinical trial of dialectical behavior therapy for preadolescent children with disruptive mood dysregulation disorder: feasibility and outcomes. J Am Acad Child Adolesc Psychiatry. 2017;56:832–840. [DOI] [PubMed] [Google Scholar]
- 10.Scott S, O’Connor TG. An experimental test of differential susceptibility to parenting among emotionally-dysregulated children in a randomized controlled trial for oppositional behavior. J Child Psychol Psychiatry Allied Discip. 2012;53:1184–1193. [DOI] [PubMed] [Google Scholar]
- 11.Stoddard J, Sharif-Askary B, Harkins EA, et al. An open pilot study of training hostile interpretation bias to treat disruptive mood dysregulation disorder. J Child Adolesc Psychopharmacol. 2016;26:49–57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Kircanski K, Clayton ME, Leibenluft E, Brotman MA. Psychosocial treatment of irritability in youth. Curr Treat Options Psychiatry. 2018;5:129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Sukhodolsky DG, Smith SD, McCauley SA, Ibrahim K, Piasecka JB. Behavioral interventions for anger, irritability, and aggression in children and adolescents. J Child Adolesc Psychopharmacol. 2016;26:58–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Walkup JT, Albano AM, Piacentini J, et al. Cognitive behavioral therapy, sertraline, or a combination in childhood anxiety. N Engl J Med. 2008;359:2753–2766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Walkup JT. Antidepressant efficacy for depression in children and adolescents: industry- and NIMH-funded studies. Am J Psychiatry. 2017;174:430–437. [DOI] [PubMed] [Google Scholar]
- 16.March J, Silva S, Petrycki S, et al. Fluoxetine, cognitive-behavioral therapy, and their combination for adolescents with depression: Treatment for Adolescents With Depression Study (TADS) randomized controlled trial. JAMA. 2004;292:807–820. [DOI] [PubMed] [Google Scholar]
- 17.Group TMC. A 14-month randomized clinical trial of treatment strategies for attention-deficit/hyperactivity disorder. Multimodal Treatment Study of Children with ADHD. Arch Gen Psychiatry. 1999;56:1073–1086. [DOI] [PubMed] [Google Scholar]
- 18.Cortese S, Adamo N, Del Giovane C, et al. Comparative efficacy and tolerability of medications for attention-deficit hyperactivity disorder in children, adolescents, and adults: a systematic review and network meta-analysis. Lancet Psychiatry. 2018;5:727–738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Campbell M, Adams PB, Small AM, et al. Lithium in hospitalized aggressive children with conduct disorder: a double-blind and placebo-controlled study. J Am Acad Child Adolesc Psychiatry. 1995;34:445–453. [PubMed] [Google Scholar]
- 20.Malone RP, Delaney MA, Luebbert JF, Cater J, Campbell M. A double-blind placebo-controlled study of lithium in hospitalized aggressive children and adolescents with conduct disorder. Arch Gen Psychiatry. 2000;57:649–654. [DOI] [PubMed] [Google Scholar]
- 21.Dickstein DP, Towbin KE, Van Der Veen JW, et al. Randomized double-blind placebo-controlled trial of lithium in youths with severe mood dysregulation. J Child Adolesc Psychopharmacology. 2009;19:61–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Leibenluft E, Charney DS, Towbin KE, Bhangoo RK, Pine DS. Defining clinical phenotypes of juvenile mania. Am J Psychiatry. 2003;160:430–437. [DOI] [PubMed] [Google Scholar]
- 23.Krieger FV, Pheula GF, Coelho R, et al. An open-label trial of risperidone in children and adolescents with severe mood dysregulation. J Child Adolesc Psychopharmacol. 2011;21:237–243. [DOI] [PubMed] [Google Scholar]
- 24.Fernandez de la Cruz L, Simonoff E, McGough JJ, Halperin JM, Arnold LE, Stringaris A. Treatment of children with attention-deficit/hyperactivity disorder (ADHD) and irritability: results from the Multimodal Treatment Study of Children with ADHD (MTA). J Am Acad Child Adolesc Psychiatry. 2015;54:62–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Waxmonsky J, Pelham WE, Gnagy E, et al. The efficacy and tolerability of methylphenidate and behavior modification in children with attention-deficit/hyperactivity disorder and severe mood dysregulation. J Child Adolesc Psychopharmacology. 2008;18:573–588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Waxmonsky JG, Waschbusch DA, Belin P, et al. A randomized clinical trial of an integrative group therapy for children with severe mood dysregulation. J Am Acad Child Adolesc Psychiatry. 2016;55:196–207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Baweja R, Belin PJ, Humphrey HH, et al. The effectiveness and tolerability of central nervous system stimulants in school-age children with attention-deficit/hyperactivity disorder and disruptive mood dysregulation disorder across home and school. J Child Adolesc Psychopharmacol. 2016;26:154–163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Winters DE, Fukui S, Leibenluft E, Hulvershorn LA. Improvements in irritability with open-label methylphenidate treatment in youth with comorbid attention deficit/hyperactivity disorder and disruptive mood dysregulation disorder. J Child Adolesc Psychopharmacol. 2018;28:298–305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Blader JC, Pliszka SR, Jensen PS, Schooler NR, Kafantaris V. Stimulant-responsive and stimulant-refractory aggressive behavior among children with ADHD. Pediatrics. 2010;126:e796–e806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Aman MG, Bukstein OG, Gadow KD, et al. What does risperidone add to parent training and stimulant for severe aggression in child attention-deficit/hyperactivity disorder? J Am Acad Child Adolesc Psychiatry. 2014;53:47–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Findling RL, Townsend L, Brown NV, et al. The Treatment of Severe Childhood Aggression Study: 12 weeks of extended, blinded treatment in clinical responders. J Child Adolesc Psychopharmacol. 2017;27:52–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Blader JC, Schooler NR, Jensen PS, Pliszka SR, Kafantaris V. Adjunctive divalproex versus placebo for children with ADHD and aggression refractory to stimulant monotherapy. Am J Psychiatry. 2009;166:1392–1401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Fava M, Rosenbaum JF. Anger attacks in patients with depression. J Clin Psychiatry. 1999;60:21–24. [PubMed] [Google Scholar]
- 34.Coccaro EF, Lee RJ, Kavoussi RJ. A double-blind, randomized, placebo-controlled trial of fluoxetine in patients with intermittent explosive disorder. J Clin Psychiatry. 2009;70:653–662. [DOI] [PubMed] [Google Scholar]
- 35.Dimmock PW, Wyatt KM, Jones PW, O’Brien PS. Efficacy of selective serotonin-reuptake inhibitors in premenstrual syndrome: a systematic review. Lancet. 2000;356:1131–1136. [DOI] [PubMed] [Google Scholar]
- 36.Kim S, Boylan K. Effectiveness of antidepressant medications for symptoms of irritability and disruptive behaviors in children and adolescents. J Child Adolesc Psychopharmacol. 2016;26:694–704. [DOI] [PubMed] [Google Scholar]
- 37.Garland EJ, Weiss M. Case study: obsessive difficult temperament and its response to serotonergic medication. J Am Acad Child Adolesc Psychiatry. 1996;35:916–920. [DOI] [PubMed] [Google Scholar]
- 38.Armenteros JL, Lewis JE. Citalopram treatment for impulsive aggression in children and adolescents: an open pilot study. J Am Acad Child Adolesc Psychiatry. 2002;41:522–529. [DOI] [PubMed] [Google Scholar]
- 39.Stringaris A, Zavos H, Leibenluft E, Maughan B, Eley TC. Adolescent irritability: phenotypic associations and genetic links with depressed mood. Am J Psychiatry. 2012;169:47–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Savage J, Verhulst B, Copeland W, Althoff RR, Lichtenstein P, Roberson-Nay R. A genetically informed study of the longitudinal relation between irritability and anxious/depressed symptoms. J Am Acad Child Adolesc Psychiatry. 2015;54:377–384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Deveney CM, Hommer RE, Reeves E, et al. A prospective study of severe irritability in youths: 2- and 4-year follow-up. Depress Anxiety. 2015;32:364–372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Aman MG, Singh NN, Stewart AW, Field CJ. The aberrant behavior checklist: a behavior rating scale for the assessment of treatment effects. Am J Ment Defic. 1985;89:485–491. [PubMed] [Google Scholar]
- 43.Wechsler D Wechsler Abbreviated Scale of Intelligence. San Antonio, TX: Psychological Corporation; 1999. [Google Scholar]
- 44.Kaufman J, Birmaher B, Brent D, et al. Schedule for Affective Disorders and Schizophrenia for School-Age Childrene–Present and Lifetime Version (K-SADS-PL): initial reliability and validity data. J Am Acad Child Adolesc Psychiatry. 1997;36:980–988. [DOI] [PubMed] [Google Scholar]
- 45.Leckman JF, Sholomskas D, Thompson WD, Belanger A, Weissman MM. Best estimate of lifetime psychiatric diagnosis: a methodological study. Arch Gen Psychiatry. 1982;39:879–883. [DOI] [PubMed] [Google Scholar]
- 46.Spearing MK, Post RM, Leverich GS, Brandt D, Nolen W. Modification of the Clinical Global Impressions (CGI) Scale for use in bipolar illness (BP): the CGI-BP. Psychiatry Res. 1997;73:159–171. [DOI] [PubMed] [Google Scholar]
- 47.Shaffer D, Gould MS, Brasic J, et al. A Children’s Global Assessment Scale (CGAS). Arch Gen Psychiatry. 1983;40:1228–1231. [DOI] [PubMed] [Google Scholar]
- 48.Riddle MD, Ginsburg GS, Walkup JT, et al. The Pediatric Anxiety Rating Scale (PARS): development and psychometric properties. J Am Acad Child Adolesc Psychiatry. 2002;41:1061–1069. [DOI] [PubMed] [Google Scholar]
- 49.Poznanski E, Mokros H. Children’s Depression Rating Scale–Revised (CDRS-R). Los Angeles, CA: WPS; 1996. [Google Scholar]
- 50.Brent D, Emslie G, Clarke G, et al. Switching to another SSRI or to venlafaxine with or without cognitive behavioral therapy for adolescents with SSRI-resistant depression: the TORDIA randomized controlled trial. JAMA. 2008;299:901–913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Hamer RM, Simpson PM. Last observation carried forward versus mixed models in the analysis of psychiatric clinical trials. Am J Psychiatry. 2009;166:639–641. [DOI] [PubMed] [Google Scholar]
- 52.Szmaragd C, Clarke P, Steele F. Subject specific and population average models for binary longitudinal data: a tutorial. Longitudinal Life Course Stud. 2013;4:147–165. [Google Scholar]
- 53.Henssler J, Kurschus M, Franklin J, Bschor T, Baethge C. Trajectories of acute antidepressant efficacy: how long to wait for response? A systematic review and meta-analysis of long-term, placebo-controlled acute treatment trials. J Clin Psychiatry. 2018;79. pii:17r11470. [DOI] [PubMed] [Google Scholar]
- 54.Trivedi MH, Fava M, Wisniewski SR, et al. Medication augmentation after the failure of SSRIs for depression. N Engl J Med. 2006;354:1243–1252. [DOI] [PubMed] [Google Scholar]
- 55.Rush AJ. Limitations in efficacy of antidepressant monotherapy. J Clin Psychiatry. 2007;68(Suppl 10):8–10. [PubMed] [Google Scholar]
- 56.Rush AJ, Trivedi MH, Stewart JW, et al. Combining Medications to Enhance Depression Outcomes (CO-MED): acute and long-term outcomes of a single-blind randomized study. Am J Psychiatry. 2011;168:689–701. [DOI] [PubMed] [Google Scholar]
- 57.Hirota T, Schwartz S, Correll CU. Alpha-2 agonists for attention-deficit/hyperactivity disorder in youth: a systematic review and meta-analysis of monotherapy and add-on trials to stimulant therapy. J Am Acad Child Adolesc Psychiatry. 2014;53:153–173. [DOI] [PubMed] [Google Scholar]
- 58.Stringaris A, Goodman R. The value of measuring impact alongside symptoms in children and adolescents: a longitudinal assessment in a community sample. J Abnorm Child Psychol. 2013;41:1109–1120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Stringaris A, Baroni A, Haimm C, et al. Pediatric bipolar disorder versus severe mood dysregulation: risk for manic episodes on follow-up. J Am Acad Child Adolesc Psychiatry. 2010;49:397–405. [PMC free article] [PubMed] [Google Scholar]
- 60.Axelson D, Findling RL, Fristad MA, et al. Examining the proposed disruptive mood dysregulation disorder diagnosis in children in the Longitudinal Assessment of Manic Symptoms study. J Clin Psychiatry. 2012;73:1342–1350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Althoff RR, Crehan ET, He JP, Burstein M, Hudziak JJ, Merikangas KR. Disruptive mood dysregulation disorder at ages 13–18: results from the National Comorbidity Survey–Adolescent Supplement. J Child Adolesc Psychopharmacol. 2016;26:107–113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.McGough JJ. Chronic non-episodic irritability in childhood: current and future challenges. Am J Psychiatry. 2014;171:607–610. [DOI] [PubMed] [Google Scholar]
- 63.McGough J Characterization and Sequential Pharmacotherapy of Severe Mood Dysregulation (ClinicalTrials.gov Identifier: NCT01714310). 2012. Available at: https://clinicaltrials.gov/ct2/show/NCT01714310. Accessed July 29, 2018.
- 64.Stringaris A, Goodman R, Ferdinando S, et al. The Affective Reactivity Index: a concise irritability scale for clinical and research settings. J Child Psychol Psychiatry Allied Discip. 2012;53:1109–1117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Malone RP, Luebbert JF, Delaney MA, et al. Nonpharmacological response in hospitalized children with conduct disorder. J Am Acad Child Adolesc Psychiatry. 1997;36:242–247. [DOI] [PubMed] [Google Scholar]
- 66.Blader JC, Abikoff H, Foley C, Koplewicz HS. Children’s behavioral adaptation early in psychiatric hospitalization. J Child Psychol Psychiatry Allied Discip. 1994;35:709–721. [DOI] [PubMed] [Google Scholar]
- 67.Margulies DM, Weintraub S, Basile J, Grover PJ, Carlson GA. Will disruptive mood dysregulation disorder reduce false diagnosis of bipolar disorder in children? Bipolar Disord. 2012;14:488–496. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.