

Abstract
Background
The ability of malaria rapid diagnostic tests (RDTs) to effectively detect active infections is being compromised by the presence of malaria strains with genomic deletions at the hrp2 and hrp3 loci, encoding the antigens most commonly targeted in diagnostics for Plasmodium falciparum detection. The presence of such deletions can be determined in publically available P. falciparum whole genome sequencing (WGS) datasets. A computational approach was developed and validated, termed Gene Coverage Count and Classification (GC3), to analyse genome-wide sequence coverage data and provide informative outputs to assess presence and coverage profile of a target locus in WGS data. GC3 was applied to detect deletions at hrp2 and hrp3 (hrp2/3) and flanking genes in different geographic regions and across time points.
Methods
GC3 uses Python and R scripts to extract locus read coverage metrics from mapped WGS data according to user-defined parameters and generates relevant tables and figures. GC3 was tested using WGS data for laboratory reference strains with known hrp2/3 genotypes, and its results compared to those of a hrp2/3-specific qPCR assay. Samples with at least 25% of coding region positions with zero coverage were classified as having a deletion. Publicly available sequence data was analysed and compared with published deletion frequency estimates.
Results
GC3 results matched the expected coverage of known laboratory reference strains. Agreement between GC3 and a hrp2/3-specific qPCR assay reported for 19/19 (100%) hrp2 deletions and 18/19 (94.7%) hrp3 deletions. Among Cambodian (n = 127) and Brazilian (n = 20) WGS datasets, which had not been previously analysed for hrp2/3 deletions, GC3 identified hrp2 deletions in three and four samples, and hrp3 deletions in 10 and 15 samples, respectively. Plots of hrp2/3 coding regions, grouped by year of sample collection, showed a decrease in median standardized coverage among Malawian samples (n = 150) suggesting the importance of a careful, properly controlled follow up to determine if an increase in frequency of deletions has occurred between 2007–2008 and 2014–2015. Among Malian (n = 90) samples, median standardized coverage was lower in 2002 than 2010, indicating widespread deletions present at the gene locus in 2002.
Conclusions
The GC3 tool accurately classified hrp2/3 deletions and provided informative tables and figures to analyse targeted gene coverage. GC3 is an appropriate tool when performing preliminary and exploratory assessment of locus coverage data.
Supplementary Information
The online version contains supplementary material available at 10.1186/s12936-022-04376-3.
Keywords: Malaria, Rapid Diagnostic Test, hrp2, hrp3, Deletion, Gene coverage, Genomics, Bioinformatics
Background
From 2010 to 2020, national malaria control programmes (NMCPs) distributed 2.2 billion rapid diagnostic tests (RDTs) for malaria and 3.1 billion RDTs were sold by manufacturers, the majority of these going to malaria-endemic countries in sub-Saharan Africa [1]. RDTs are an integral part of nearly all NMCP’s clinical and field interventions since they provide quick and effective malaria diagnosis. These RDTs include a small cassette detecting Plasmodium-specific antigens in the blood of an infected individual and are user-friendly and affordable [2]. Predominantly, RDTs detect the Plasmodium falciparum-specific antigen histidine-rich protein 2 (HRP2), which is released into the bloodstream in large quantities when infected red blood cells lyse [3]. Plasmodium falciparum accounts for vast majority of the 241 million reported human malaria cases in 2020 and is the primary parasite causing malaria-related mortality and morbidity [1]. Due to considerable sequence similarity between the two proteins, (HRP3 is a truncated protein of HRP2 [4] and the two are encoded by similar loci), HRP3 can also bind to the monoclonal antibody on HRP2-based RDTs, but becomes more apparent in high-density infections [5]. As evidence of their effectiveness, 94% of WHO-qualified RDTs are either HRP2-based or based on a combination of HRP2 and a partner antigen, such as parasite lactate dehydrogenase or aldolase [6–8]. HRP2-based RDTs are an essential diagnostic tool for NMCPs to scale surveillance operations and adequately assess infection, leading to proper treatment administration, measure intervention progress and identify malaria reservoirs.
Recently, however, the effectiveness of HRP2-based RDTs is becoming compromised due to the emergence of deletions in the hrp2 and hrp3 (hrp2/3) loci that prevent the expression of a detectable protein [6, 8–16]. In particular, full deletions, as well as some partial deletions, in one or both of these genes eliminate HRP2 and/or HRP3 signal on RDTs, preventing accurate malaria diagnosis. Previous estimates of hrp2/3 deletion prevalence report higher frequencies in South and Central America, followed by Africa, then Asia and Oceania [17]. Low-transmission areas with high treatment rates, characteristics often found in elimination settings, are especially at risk for the spread of strains with hrp2/3 gene deletions, as models show that, under those conditions, strains with hrp2/3 deletions have a strong fitness advantage over those with intact genes [18]. Therefore, as NMCPs continue to control and move toward elimination, it is critical to monitor the presence and spread of hrp2/3 deletions. Without fully understanding the dynamics of hrp2/3 deletions, and spread of those deletions in particular, undiagnosed infections may lead to an increase in malaria prevalence and mortality, and hinder global progress towards control and elimination.
A computational tool that facilitates detection and classification of deletions in hrp2/3 (e.g. partial vs. complete deletions) in published whole genome sequencing (WGS) datasets will enable rapid and detailed analysis of deletions within datasets, and comparisons between datasets. The development of baseline values as well as the comparison of deletion prevalences across current samples sets as well as temporal comparison between these and previously published datasets may be particularly informative. Previous studies have performed analyses using WGS data [19–21]; however, implementation of the methods used in these studies requires a strong understanding of bioinformatics tools and packages. The development of a more user-friendly computational tool would expand the ability to assess the presence of locus deletions based on WGS coverage data to a wider audience investigating copy number variations, including deletions, in hrp2/3 or other target genes. This work aimed to fill this gap, by developing a computational tool, termed “Gene Coverage Count and Classification”, or GC3, to provide translatable results on the presence of hrp2/3 deletions and their classification, based on short-read WGS data, among global P. falciparum samples for which WGS data is available.
Methods
Samples
The WGS data used were generated either by direct sequencing of total DNA extracted from each isolate or by sequencing post selective whole genome amplification (sWGA) of extracted DNA, and were reported previously [22]. Some of the WGS datasets were generated as part of the MalariaGEN project [23] and downloaded from the Sequence Read Archive (SRA). A selection of field samples representing 19 different countries from Africa (n = 9), South America (n = 5), Asia (n = 4) and Oceania (n = 1), for a total of 1120 datasets (1114 global samples + 6 reference strains), were evaluated for general results (Additional file 1: Figure S1). The following laboratory reference strains with known hrp2 and hpr3 genotype were used for developing and testing GC3: NF54 (West Africa) – hrp2 and hrp3 present, 7G8 (Brazil) – hrp2 and hrp3 present, NF135.C10 (Cambodia) – hrp2 and hrp3 present, NF166 (Guinea) – hrp2 and hrp3 present, Dd2 (Laos) – hrp2 absent/hrp3 present and HB3 (Honduras) – hrp2 present/hrp3 absent.
Read coverage files were generated by aligning raw reads in fastq format to the Pf3D7 reference genome assembly (PlasmoDB release v24) using bowtie2 (v2.2.9 and above). Alignment files in BAM (Binary sequence Alignment/Map) format were processed according to GATK’s (Genome Analysis Toolkit) Best Practices documentation. Genome-wide coverage per site was recovered using bedtools’ genomecov function [22, 24]. The resulting BED (Browser Extensible Data) file (a tab-delimited text file) is used as the initial input to GC3. However, any delimited file with columns for molecule identifier (e.g. Pf3D7_08_v3), chromosomal position and coverage value is acceptable. When comparing sample datasets, coverage values per base pair (bp) were standardized by dividing ‘locus coverage’ by ‘subtelomeric mean coverage’ to account for differences in sequencing depth among samples.
Computational tool framework and algorithm
For GC3 to function properly, Python v3.0 and R v4.1.1 (or later versions) with the following libraries must be installed: readxl, writexl, dplyr, reshape2, and ggplot2. GC3 uses a Python-based script to extract read coverage information for genomic coordinates set by the user and processes these output files using an R script (Fig. 1). Following the framework, the user is required to provide input parameters at two junctions.
Fig. 1.

GC3 framework. GC3 extracts read coverage information and processes it into a metric database and descriptive tables/figures. Ovals denote initial/intermediate input(s). Orange rectangles denote scripts for data processing. User input parameters are needed at two junctions in the process and are listed (required and optional). (1) Python script extracts coverage data either using a “sliding window”, or coverage at every locus between user-defined start and end coordinates. Overall mean coverage between start and end coordinates can be extracted using a separate function. Output files from the python script (i.e. intermediate output) become the input into the R script, which generates metrics and relevant tables/figures. (2) User input into the R-script is required to define path (directory) to intermediate output as well as the file name, target gene coordinates, intron coordinates (if necessary), coordinates of regions of interest (e.g. flanking genes), and definition of subgroups (optional). Output from R script is Excel versions of intermediate outputs, metrics database, position descriptive database, and relevant figures
Extracting target coverage data – Python-based script
Within the Python script, the user is required to provide parameter inputs depending on the desired output. To use the “sliding window” option, the user must provide: (1) name of input file, (2) start coordinate, (3) end coordinate, (4) molecule identifier containing target locus (e.g. Pf3D7_08_v3), (5) interval size (i.e. window length, in base pairs), and (6) step size (i.e. shift between windows, in base pairs). In the initial window, defined by start coordinate and interval size, the average coverage is obtained by adding read coverage across all positions and dividing by interval size. The start position is then updated by adding step size to the previous start coordinate and the process is repeated until the end coordinate is reached. The output file will report an interval’s start and end coordinates separated by a colon and flanked by apostrophes and the interval’s average read coverage separated from the coordinates by a colon (e.g. '1,290,240:1,290,740': 294.228).
If individual coverage of all positions in the interval of interest is desired, then the interval size should be set to 1, and GC3 will extract values for each coordinate between start and end coordinates, inclusively (step size is automatically set to 1). Output file will report the position and respective coverage (e.g. '1,372,236': 387). The intermediate output is a text file with position(s) and corresponding read coverage values.
Additionally, the user can calculate mean coverage between start and end coordinates using a separate GC3 function. User parameters needed are (1) name of input file, (2) start coordinate, (3) end coordinate, and (4) molecule identifier. This function is needed if the user desires to know, for example, the mean coverage over a wider region or to standardize coverage between different sets of samples (i.e., sample subgroups).
Coverage data processing—R-based script
The user will need to input intermediate output files into the separate GC3’s R script to clean, and generate sample metrics and descriptive plots. At the start of the R script, the user will define (1) path to the intermediate files, (2) name of the intermediate file(s), (3) target locus’ coordinates in reference genome, (4) gene’s intron coordinates (if necessary), in reference genome, (5) position coordinates of interest (e.g. flanking gene positions), (6) list of subgroup sample identifiers and subgroup name (optional). If read coverage is to be standardized relative to coverage in a reference chromosome or chromosomal segment, then a file of mean read coverage per chromosome or segment (obtained as described above) per sample should also be defined. The GC3 R script will output several files, namely, (i) Excel version of intermediate text files, (ii) summary metrics: sample identifier, overall mean coverage—if mean coverage file included, mean target gene coverage, proportion of gene positions with coverage, proportion of smaller regions of interest (including coding regions, exons), deletion classification, and count of positions with 0X coverage, (iii) read coverage information for target gene (number of positions with zero coverage, and mean and median coverage per position over all samples), and (iv) descriptive plots: sliding window coverage over region of interest (i.e. subtelomeric region), all coordinates coverage over target gene positions (i.e. hrp2 and hrp3), and proportion of positions with zero coverage.
Python and R scripts can be found at the Silva group’s GitHub (https://github.com/igs-jcsilva-lab) as well as a README file with detailed instructions and input examples.
Detection of deletions in hrp2, hrp3 and flanking regions
The gene structure of hrp2 and hrp3 in the reference 3D7 strain was obtained from PlasmoDB (www.plasmodb.org). Both hrp2 and hrp3 consist of two coding exons. Exon1 is 69 bp in length for both genes, and exon2 is 848 bp long in hrp2 and 758 bp in hrp3 (Fig. 2). The analysis of WGS data focused on subtelomeric regions of chromosome 8 (P. falciparum 3D7 reference strain coordinates 1,290,240–1,443,449, for a total of 153,209 bp), containing the hrp2 coding DNA sequence (CDS) and intervening intron, and of chromosome 13 (P. falciparum 3D7 reference strain coordinates 2,731,041–2,892,340, for 161,299 bp), containing the hrp3 CDS and intron. Subtelomeric coordinates were chosen to include the closest “essential” gene [25] downstream of hrp2 or hrp3 and farthest upstream functional gene (i.e. PfEMP1-encoding var gene).
Fig. 2.

Schematic of target genomic regions, encoding hrp2/3. A Schematic of the hrp2-containing chromosomal region plus flanking genes (unlabeled light-blue boxes represent pseudogenes), labelled with their respective gene products, and coding regions (exon1 and exon2), their base pair length and direction of transcription. Red highlighted areas denote locations of hrp2/3-specific qPCR forward primer (1) and reverse primer (2), and the purple highlighted area denotes the location of the qPCR probe (P). Chromosome 08 PfEMP1 reference – Pf3D7_0833500 (multi-copy gene). B Same as A, but for hrp3. Chromosome 13 PfEMP1 reference – Pf3D7_1373500 (multi-copy gene)
Metrics were generated to classify samples by presence or absence of full or partial deletions in each locus of interest. If ≤ 25% of the CDS was missing (i.e. at most 25% of the reference CDS had zero coverage) the locus was considered present with a “small deletion of uncertain functional impact” (SDUFI). If > 25% (but not 100%) of the reference CDS positions had zero coverage the sample was classified as having a partial deletion (25% < %-positions-with-zero-coverage < 100%); it was classified as having a complete deletion if all CDS positions have zero coverage. This classification is partly informed by Sepúlveda and colleagues [19], who implemented an algorithm to perform deletion calling without having to analyse the coverage profile of the entire genome. They classified deletions based on a 75% threshold of positions with ≤ 2X coverage, but may be too stringent and decreased GC3 thresholds as explained above to account for “SDUFIs” or partial deletions less than the 75% threshold that might impact protein detection. Deletions of flanking genes were assigned to samples if > 25% of the flanking gene’s positions reported zero coverage. Intergenic regions were excluded to reduce the effect of variable read coverages in non-coding regions.
GC3 agreement with qPCR assay
A previously described hrp2/3-specific qPCR assay capable of detecting locus deletions in mono- and poly-clonal infections [26] was utilized to compare with the hrp2/3 deletion genotype inferred by GC3. In summary, primer sequences were adapted from conventional PCR [27] to bind to conserved regions of hrp2, hrp3 and an apicomplexan-specific single copy gene used as positive control, rnr2e2 (ribonucleotide reductase R2_e2, [28]). The computational approach used by GC3 for the detection of hrp2/3 deletions was compared to this hrp2/3-specific qPCR assay [26], using the following samples:
NF54—Positive control
7G8—Positive control
Dd2—hrp2 absent control
HB3—hrp3 absent control
17 global samples (see Additional file 1: Table S1 for details)
The presence and classification of hrp2/3 deletions is reported for the four laboratory reference strains mentioned above and for 17 global samples from Brazil (n = 3), Cambodia (n = 6), Mali (n = 3), Malawi (n = 4) and Thailand (n = 1). Global samples with accessible DNA material were randomly selected to represent the following GC3-inferred genotype subgroups: samples with no deletions, hrp2 deletion (complete), hrp3 deletion (complete), double hrp2/3 deletion, low overall sample mean read coverage (< 20X), possible discordant pairs (partial deletion, with non-zero coverage in qPCR primer binding sites), and PCR primer site deletions (samples with zero coverage in qPCR primer binding site – either in hrp2 or hrp3). Accession ID and subgroup stratification of global samples can be found on Additional file 1: Table S1.
Statistical analyses
When measuring correlation between mean coverage in hrp2/3 positions and subtelomeric or upstream/downstream gene, Spearman’s rank correlation method was used (Additional file 3). Spearman’s method accounts for non-parametric distribution and, therefore, mean coverages were not standardized [29]. R v4.1.1 program was used to conduct statistical analysis.
Results
Sample read coverages by sliding windows of 1000 bp intervals and 500 bp step size were generated over the subtelomeric regions of chromosome 8 and chromosome 13 (sum of coverage across all positions in interval/interval length). Additionally, coverage at every position (interval = 1) was generated at every position between coordinates 1,372,236 to 1,377,299 on chromosome 8 and 2,835,756 to 2,847,557 on chromosome 13. These positions corresponded to hrp2 and hrp3 coordinates plus 2000 bp on either end of their respective coding regions.
Demonstrating GC3 features using laboratory strains of known genotype
WGS data from reference laboratory strains were analysed to estimate hrp2/3 coverage per bp and the proportion of positions with coverage by at least one read (≥ 1X coverage) at hrp2 and hrp3 coordinates, and ultimately evaluate the validity of results from GC3. Expected coverage was estimated using each respective subtelomeric region as reference. Overall, for each lab strain reference, excellent concordance was found between coverage values in each locus and the respective subtelomeric chromosomal regions (Table 1). Mean subtelomeric coverage of the Dd2 strain (with hrp2 deletion genotype) was high (chromosome 8: 29X; chromosome 13: 48X), and, as expected, mean coverage at the hrp2 positions was 0X, while hrp3 mean coverage was 45X (with 100% of CDS coordinates with coverage > 0). The HB3 strain (hrp3 deletion genotype) was sequenced to ~ 145X coverage (chromosome 08 and chromosome 13 subtelomeric regions with 159X and 131X coverage, respectively). Mean coverage at the hrp3 positions was 0X (50% proportional coverage) and while hrp2 was similar to genome-wide coverage (142X, with 100% proportional coverage of gene positions). Residual coverage may have occurred at the hrp3 gene of HB3 despite its known deletion due to mapping of some reads originating from hrp2 and mapping to similar but non-orthologous locations. GC3 correctly identified HB3 as having a hrp3 gene deletion (Table 1). These results are similar to previously described coverage profiles of Dd2 and HB3 [19].
Table 1.
Subtelomeric coverage, gene coverage, and coding region proportional coverage among known reference strains
| Reference Strain | Mean subtelomeric read coverage (sum of coverage/bp) | Mean gene coverage (sum of coverage/bp) | Proportion of coding positions with ≥ 1X coverage | |||
|---|---|---|---|---|---|---|
| Chromosome 08 | Chromosome 13 | hrp2 | hrp3 | hrp2 | hrp3 | |
| NF54 | 156.5 | 173.7 | 125.2 | 134.8 | 100% | 100% |
| 7G8 | 201.9 | 236.3 | 247.0 | 269.9 | 100% | 100% |
| NF135.C10 | 35.3 | 42.7 | 36.9 | 41.7 | 100% | 97% |
| NF166 | 280.0 | 328.4 | 295.7 | 344.6 | 100% | 100% |
| Dd2 | 29.3 | 48.3 | 0.0 | 45.0 | 0% | 100% |
| HB3 | 159.3 | 130.5 | 142.2 | 0.35 | 100% | 50% |
Plotting of read coverage in subtelomeric region of select reference strains
GC3 can create plots of the sliding window findings in order to provide a visual perspective of the target region. To illustrate the coverage data provided in Table 1, the subtelomeric regions containing hrp2 and hrp3 of reference strains Dd2, HB3 and NF54 were plotted (Fig. 3). Results were normalized on a log scale to better visualize large fluctuations in coverage generated by whole genome shotgun sequencing. Sliding window plots confirm validation results of Dd2, HB3 and NF54, and clearly illustrate coverage for each respective strain. On chromosome 08, coverage of the Dd2 strain decreases to zero for several thousand base pairs that include the hrp2 locus, whereas NF54 and HB3 have high coverage in the same region. Noticeably, in the HB3 strain, a section of the subtelomeric region upstream of the hrp2 CDS has poor coverage (near position 1,400,000 on chromosome 8). This section of poor coverage would not impact hrp2 presence/absence, and could be due to the presence of one or more deletion(s), or to poor mapping. Poor read mapping can occur in the subtelomeric regions for several reasons, including the presence of multiple members of highly variable multigene families (var, stevor and rifin) that differ between strains or to the presence of low complexity regions. On chromosome 13, it is the HB3 strain that has several thousand base pairs with little or no coverage, including the hrp3 locus, whereas NF54 and Dd2 coverage remains high. GC3 visuals showed no deletions in NF54, a complete hrp2 deletion in Dd2 and a large section of little to no coverage at the hrp3 locus in HB3, respectively.
Fig. 3.
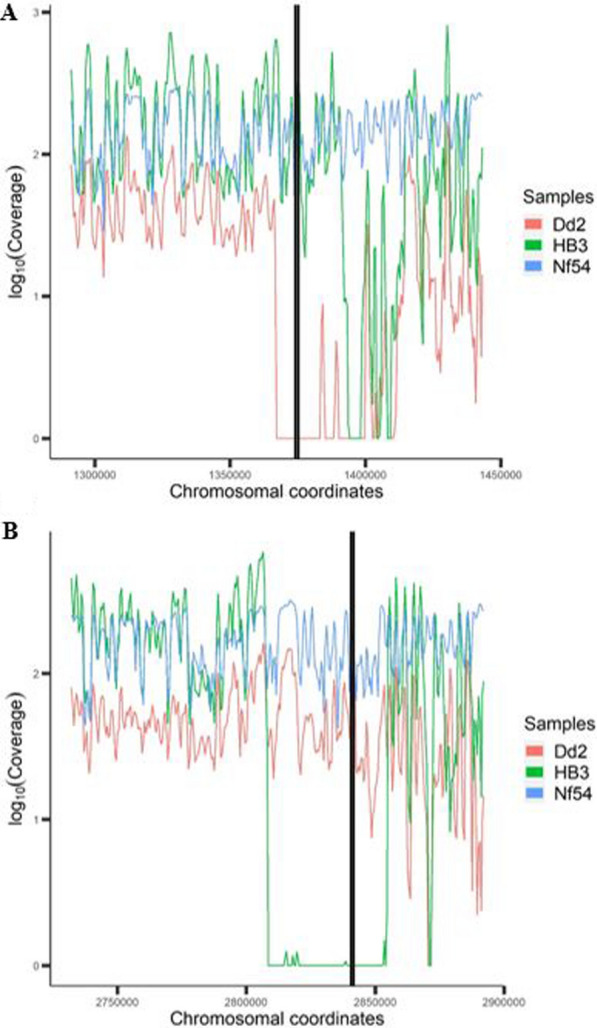
Subtelomeric read coverage distribution plots. Log10 of subtelomeric read coverage for reference strains NF54 (hrp2 and hrp3 present), Dd2 (hrp2 absent) and HB3 (hrp3 absent). Coverage was measured using a 1000 bp sliding window (i.e. interval) with a 500 bp “step” between windows. A. Chromosome 08 subtelomeric region coverage (black lines denotes hrp2 coding positions 1,374,236–1,375,299). B. Chromosome 13 subtelomeric region coverage (black lines denotes hrp3 coding positions 2,840,727–2,841,703)
GC3 agreement with a hrp2/3-specific qPCR assay on field samples
A subset of global samples (n = 17) and reference strains (n = 4) underwent qPCR specific for hrp2 and hrp3 to compare with GC3 results. Two among the selected global samples were excluded due to low parasitaemia resulting in very low or no detection of the positive control gene by qPCR (Cq threshold cutoff = 37.5). There was very good agreement between GC3 (computational) and qPCR assay results. Out of four references strains and remaining 15 global samples, GC3 matched qPCR results 19/19 (100%) for hrp2 and 18/19 (94.7%) for hrp3 (Table 2). Only one sample (IGS-CBD-099) had a discordant result between methods. In particular, for this sample, GC3 classified it as having a partial deletion at the hrp3 locus, and coverage assessment with base-pair granularity suggested partial lack of read coverage, including the exon 2 primer binding region, between 2,841,390—2,841,412 (Fig. 4). It is noteworthy that the average coverage in this region is very low (~ 1X), however coverage is high for the corresponding chromosomal subtelomeric and core regions (~ 124× and ~ 143×, respectively). On the other hand, the qPCR assay was positive for hrp3 (Cq = 25.4). Taken together, the results suggest the sample has a partial deletion at the hrp3 locus, which does not encompass the qPCR primer binding regions, but that is possibly close enough to the binding site of the primer in exon 2 to interfere with read mapping in that region.
Table 2.
Agreement between GC3 deletion assessment results on global samples and hrp2/3-specific qPCR assaya
| Sample name | Country | Phenotype subgroup | hrp2 b | hrp3 b | rnr2e2 |
|---|---|---|---|---|---|
| (control gene) | |||||
| GC3/PCR | GC3/PCR | PCR | |||
| 7G8 | Reference (Brazil) | Control | Present/Present | Present/Present | Present |
| NF54 | Reference | Control | Present/Present | Present/Present | Present |
| Dd2 | Reference (Laos) | Control—hrp2 deletion | Absent/Absent | Present/Present | Present |
| HB3 | Reference (Honduras) | Control—hrp3 deletion | Present/Present | Absent/Absent | Present |
| IGS-BRA-017sA | Brazil | No deletions | Present/Present | Present/Present | Present |
| IGS-THL-017 | Thailand | No deletions | Present/Present | Present/Present | Present |
| IGS-BRA-021 | Brazil | No deletions | Present/Present | Present/Present | Present |
| IGS-CBD-026 | Cambodia | No deletions | Present/Present | Present/Present | Present |
| IGS-CBD-031 | Cambodia | hrp2 deletion (complete) | Absent/Absent | Present/Present | Present |
| IGS-MLI-036 | Mali | hrp3 deletion (complete) | Present/Present | Absent/Absent | Present |
| IGS-BRA-001sA | Brazil | Double hrp2/3 deletion | Absent/Absent | Absent/Absent | Present |
| IGS-CBD-008 | Cambodia | Low coverage sample | Present/Present | Present/Present | Present |
| IGS-MWI-254sA | Malawi | Low coverage sample | Present/Present | Present/Present | Present |
| IGS-MWI-251sA | Malawi | Low coverage sample | Present/Present | Present/Present | Present |
| IGS-MLI-039 | Mali | hrp2 discordant pair | Present/Present | Present/Present | Present |
| IGS-MLI-031 | Mali | hrp3 discordant pair | Present/Present | Present/Present | Present |
| IGS-CBD-034 | Cambodia | hrp2 PCR primer deletion | Present/Present | Present/Present | Present |
| IGS-CBD-094 | Cambodia | hrp3 PCR primer deletion | Present/Present | Present/Present | Present |
| IGS-CBD-099 | Cambodia | hrp3 PCR primer deletion | Present/Present | Absent/Present | Present |
a Cells in purple and blue denote agreement between GC3 and qPCR results (i.e. "PCR"); red denotes disagreement between methods
Fig. 4.
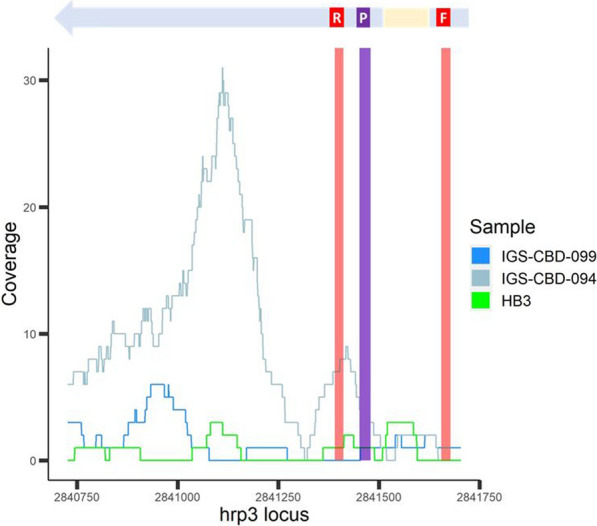
Coverage of hrp3 among select validated samples. Coverage plot of hrp3 locus of validated samples IGS-CBD-099, IGS-CBD-094 and strain HB3 (known hrp3 deletion genotype) with hrp3 schematic representation above the plot. Red areas denote the primer binding sites, and purple area denotes the probe binding site of the hrp2/3-specific qPCR assay. GC3 assigned IGS-CBD-099 as hrp3-absent; however the 5′ end of the gene was amplified by qPCR. As a comparator, IGS-CBD-094 is a similar sample (apparent deletion at qPCR primer binding site in exon 1) that GC3 assigned as hrp3-present. HB3 is a laboratory reference strain known to be missing the hrp3 locus. It is important to note some position with non-zero coverage in HB3, which suggests that non-orthologous reads from HB3 map the hrp3 locus of Pf3D7
Comparison of GC3 output using Kenyan and Peruvian sample sets previously genotyped for hrp2/3 deletions
Previously, a subset of Kenyan samples (n = 27) was genotyped for hrp2/3 deletions, with two and one deletions identified in hrp2 and hrp3, respectively [19] (Table 3). In addition, Sepúlveda and colleagues also identified no hrp2 deletions among twelve Peruvian samples and two samples with deletions in hrp3 [19]. In that study, the criterion used to call deletions was > 75% of gene positions with ≤ 2X coverage [19]. To determine how GC3 performed on a similar set of samples SRA samples were downloaded from the same time point from Kenya (n = 57, including 24 from [19]) and Peru (n = 11, including 7 from [19]). For hrp3, the same number of deletions were identified as reported previously [19]. However, the hrp2 were discordant. One complete deletion was identified among Kenyan samples, and one partial hrp2 deletion among the Peruvian samples, which differs from previous reporting. Among the Kenyan samples, a previous study reported two hrp2 deletions [19], one of which was also identified by GC3. Whereas the discordant Peruvian sample was only identified to have a deletion by GC3. The difference in assessment is likely due to differences in the criteria used between GC3 and that used by Sepúlveda and colleagues to call deletions.
Table 3.
Deletion identification and classification of previously genotyped samples
| Country | n | hrp2 | |||||
|---|---|---|---|---|---|---|---|
| No Deletion | GC3 Deletion Classification (Partial/Complete)a | ex1-/ex2 + b | ex1 + /ex2-b | ex1-/ex2-b | Previous Genotype (Deletion/Total)c | ||
| Kenya | 59 | 58 | 0/1 | 0 | 0 | 1 | 2/27 |
| Peru | 11 | 10 | 1/0 | 1 | 0 | 0 | 0/12 |
| Country | n | hrp3 | |||||
|---|---|---|---|---|---|---|---|
| No Deletion | GC3Deletion Classification (Partial/Complete)a | ex1-/ex2 + b | ex1 + /ex2-b | ex1-/ex2-b | Previous Genotype (Deletion/Total)c | ||
| Kenya | 59 | 58 | 1/0 | 0 | 0 | 1 | 1/27 |
| Peru | 11 | 9 | 2/0 | 0 | 1 | 1 | 2/12 |
ahrp2/3 deletions assigned to isolates with > 25% CDS positions with zero coverage
bExon absence (-) assigned to isolates if > 25% exon positions have zero coverage. ( +) signifies exon is present
cPrevious hrp2/3 deletion genotype results from Sepúlveda and colleagues. Deletions called for samples with > 75% of coding region with ≤ 2X coverage [19]
Analysis of novel samples for hrp2 and hrp3 deletions
All global samples used in the study (n = 1114) were examined for hrp2/3 deletions (Additional file 1: Table S2). Cambodian (n = 127) and Brazilian (n = 20) samples were further visualized in more detail at the subtelomeric regions of interest (Additional file 1: Figure S2) and examined for hrp2/3 exon presence/absence since they have not previously been described (Table 4). Although hrp2/3 have not been described for these samples, computational results are comparable to previous estimates in each respective region, where deletions have been previously observed [19, 30]. Among Cambodian samples collected in 2009–2011, there were one hrp2 deletion, eight hrp3 deletions, and two hrp2/3 double deletions (both hrp2 and hrp3), with frequencies in this sample set of 0.8%, 6.3% and 1.6%, respectively. All hrp2 deletions corresponded to absent exons (> 25% zero coverage positions on both exons), but were classified as two partial and one complete hrp2 deletion as two samples still had coverage in a low proportion on hrp2 positions. Deletions of hrp3 among Cambodian samples were classified as seven partial and three complete deletions. Among Brazilian samples collected in 2016, four had hrp2/3 double deletions, and eleven with hrp3 deletions, corresponding to frequencies of 20% and 55% respectively. Of the four hrp2 deletions, two were partial deletions, and two were complete hrp2 deletions. Of the 15 hrp3 deletions, one was a partial deletion on exon 2, and 14 were complete hrp3 deletions.
Table 4.
Deletion identification and classification of undescribed samples
| Country | n | hrp2 | ||||
|---|---|---|---|---|---|---|
| No Deletion | Deletion Classification (Partial/Complete)a | ex1-/ex2 + b | ex1 + /ex2-b | ex1-/ex2-b | ||
| Cambodia | 127 | 124 | 2/1 | 0 | 0 | 3 |
| Brazil | 20 | 16 | 2/2 | 0 | 1 | 3 |
| Country | n | hrp3 | ||||
|---|---|---|---|---|---|---|
| No Deletion | Deletion Classification (Partial/Complete)a | ex1-/ex2 + b | ex1 + /ex2-b | ex1-/ex2-b | ||
| Cambodia | 127 | 117 | 7/3 | 0 | 2 | 8 |
| Brazil | 20 | 5 | 1/14 | 0 | 1 | 14 |
ahrp2/3 deletions assigned to isolates with > 25% CDS positions with zero coverage
bExon absence (-) assigned to isolates if > 25% exon positions have zero coverage. ( +) signifies exon is present
Quantifying flanking region deletions among Cambodian samples
Deletions at the hrp2/3 positions may extend to flanking genes, possibly with additional impact on overall parasite fitness. Therefore, an option in the GC3 R-script was built in to assign deletions of flanking genes. To determine whether deletions extend into these flanking coding regions, a table was generated for Cambodian samples, where samples with > 25% gene positions with zero coverage in upstream and downstream flanking regions were classified as having a locus deletion (Table 5). Of note are the observations that the presence of a hrp2/3 deletion is not always associated with deletions in flanking genes and, conversely, deletions in flanking genes are not always associated with hrp2/3 deletions. A subset of Cambodian samples has been plotted to illustrate flanking gene coverage as it relates to hrp2 or hrp3 (Additional file 1: Figure S3). Results suggest that deletions can occur independently in hrp2 (or hrp3) and their respective flanking genes.
Table 5.
Frequency of deletions in hrp2/3 flanking genes, among Cambodian samples
| Flanking geneb (Upstream/Downstream)c |
hrp2a | hrp3a | ||
|---|---|---|---|---|
| Present | Absent | Present | Absent | |
| Present/Present | 124 | 2 | 113 | 3 |
| Absent/Present | 0 | 1 | 4 | 0 |
| Present/Absent | 0 | 0 | 0 | 1 |
| Absent/Absent | 0 | 0 | 0 | 6 |
| Total | 124 | 3 | 117 | 10 |
aDeletion assigned to samples if > 25% of coding region positions had zero coverage
bDeletion of flanking genes assigned to samples with > 25% gene positions with zero coverage
cFor hrp2, an upstream gene was a STEVOR family gene, and a downstream gene encoded heat shock protein 70. For hrp3, upstream and downstream genes were PHIST-encoding genes of unknown function
Temporal comparison of standardized coverage of hrp2/3 positions
Coverage plots of hrp2/3 coordinates were generated for Cambodian, Malawian, and Malian samples to demonstrate: (1) magnified plots of only hrp2/3 positions and (2) differences in relative depth of coverage between samples collected at different time points (Fig. 5). Cambodian samples were collected in Battambang, Pailin, Koh Kong, Kampot, Kampong Speu, Oddar Meanchey, Preah Sihanouk, and Preah Vihear districts from volunteers aged 18–65 years in 2010 and 2011, and then sequenced at IGS with 100 bp paired-end Illumina reads [31]. NF135.C10 was cultured in the laboratory and sequenced at IGS with 150 bp paired-end reads. Two datasets of Malawian samples were collected in 2007–08 and 2014–16. Samples from 2007–08 were collected during a malaria drug study in Ndirande, outside Blantyre, from children 6 months to 5 years of age and sequenced at IGS with 150 bp paired-end reads [32]. Samples collected in 2014–16 are from a cohort study of malaria incidence in Chikwawa, south of Blantyre, where samples from volunteers aged 2–8 years were sequenced at IGS with 150 bp paired-end reads [22]. Comparisons were also made between two Malian datasets from 2002 and 2010, both collected in Bandiagara, Mali. Samples from 2002 are from a case/control study of severe malaria among 3 months to 14 year old volunteers and were sequenced at IGS with 100 bp paired-end reads [33]. Malian samples collected in 2010 are from a cohort study of malaria incidence among volunteers aged 1–5 years and sequenced at IGS using 150 bp paired-end reads [34]. It should be noted that comparing these datasets are for illustrative purposes only, since the extent to which relatively small samples sizes and potential confounders (including sampling location and strategy, sample independence, sequencing approach, read length) impact observed deletion frequency is unknown. To account for differences in sequencing depths between samples, coverage values were standardized. For each sample, site or locus coverage were divided by the expected coverage, obtained from mean coverage in subtelomeric region in which each locus is located (see Methods for coordinates). Standardized coverage of ~ 1 shows locus coverage similar to subtelomeric mean coverage. A strong, positive correlation between subtelomeric and hrp2/3 gene coverage justifies the use of this standardization approach (Additional file 1: Figures S4 and S5). A decrease in standardized coverage over time would suggest an increase in frequency in hrp2/3 deletions. To avoid undue impact of outlier standardized values, median standardized coverage was plotted per group.
Fig. 5.

Median of standardized coverage between longitudinal subgroups. Median of standardized coverage [SUM (Coverage/Subtelomeric Mean Coverage)/ Total Samples] in hrp2 and hrp3 grouped by year of sample collection (Year). Countries include A. Cambodian hrp2 and hrp3 positions (n = 127), B. Malawi hrp2 and hrp3 positions (n = 150), C. Mali hrp2 and hrp3 positions (n = 90). Tan shading marks intron positions of target gene, whereas unshaded areas are exon positions (hrp2—> exon 1: 1,375,299–1,385,231; intron: 1,375,230–1,375,085; exon 2: 1,375,084–1,374,236; hrp3—> exon 1: 2,841,703–2,841,635; intron: 2,841,634–2,841,486; exon 2: 2,841,485–2,840,727). Analysis and figures were generated using R v4.1.1
Standardized read coverage for Cambodian samples was plotted alongside standardized coverage for the geographically representative strain NF135.C10 (Fig. 5A), and showed that the uneven standardized coverage of NF135.C10 is mirrored in the clinical samples. This suggests there are sequence-inherent properties that impact sequencing or mapping success. (see Additional file 2: Table S3 for descriptive coverage of each sample). In contrast, Malawian samples collected in 2014–2016 had lower median standardized coverage than the sample set collected in 2007–2008 (Fig. 5B), everything else being equal, this would suggest an increase in hrp2 and hrp3 deletions between the two time points. Interestingly, median standardized coverage is low (< 1) in both time points, showing that read coverage in the target genes is half of that in the respective subtelomeric regions. The majority of Malawian samples underwent sWGA (n = 139) prior to sequencing (Additional file 2: Table S4) which may explain the lower standardized coverage as compared to standardized coverage of directly sequenced samples (Additional file 1: Figure S6). Finally, Mali samples from 2002 showed lower standardized coverage than 2010 samples on both hrp2 and hrp3 positions (Fig. 5C). All Malian WGS data was obtained by direct sequencing of total DNA from venous blood, using a similar protocol [22, 33] with high sequence coverage in the core genomes (Additional file 2: Table S5), a strong suggestion that the quality of WGS data did not contribute to the observed difference. However, it remains unclear if the datasets are directly comparable as differences in, for example, sample strategy/bias and read length could distort observed frequencies [22, 33, 34]. Mali results highlight the potential impact of read length in some of the observed results. Lower coverage may be due to mapping ambiguity in smaller read datasets. Overall, figures offer a visual perspective between different time points as monitoring of hrp2/3-deletions become crucial in the possibility of their expansion; however, caution should be applied since the factors that influence WGS coverage need to be considered as part of any interpretation.
Count of positions with zero coverage
To provide a clearer illustration of the proportional frequency of gene coordinates with zero coverage and the location of those positions along the locus, a view of hrp2 and hrp3 gene positions by proportional counts of no coverage (0X coverage) vs. coverage (≥ 1X coverage) was generated for Cambodian samples (Fig. 6). Additional file 1: Figure S7 provides the same proportional counts of zero coverage per position for Malawian and Mali samples. Among Cambodian samples, there is a clear increase in 0X coverage positions at the intron regions (hrp2 intron: 1,375,232–1,375,083; hrp3 intron: 2,841,636–2,841,484) relative to exon coverage. This is to be expected, as the length (145–148 bp) and the nucleotide composition of these Pf introns (hrp2 AT%: 91%; hrp3 AT%: 91.2%) prevent unambiguous mapping of 101 bp-long reads centered in the middle of the intron. On hrp3 positions, there are also two spikes in zero coverage positions on either side of coordinate 2,841,250, likely due to differences in Cambodia samples compared to Pf3D7 reference, such as indels or rapidly evolving sequence motifs among genetically similar Cambodian strains, which prevent read mapping in a subset of samples. That read mapping pattern is also observed in the troughs in hrp3 coverage plot in Fig. 5A, for samples collected in 2010 and 2011 (but curiously not in Pf NF135).
Fig. 6.
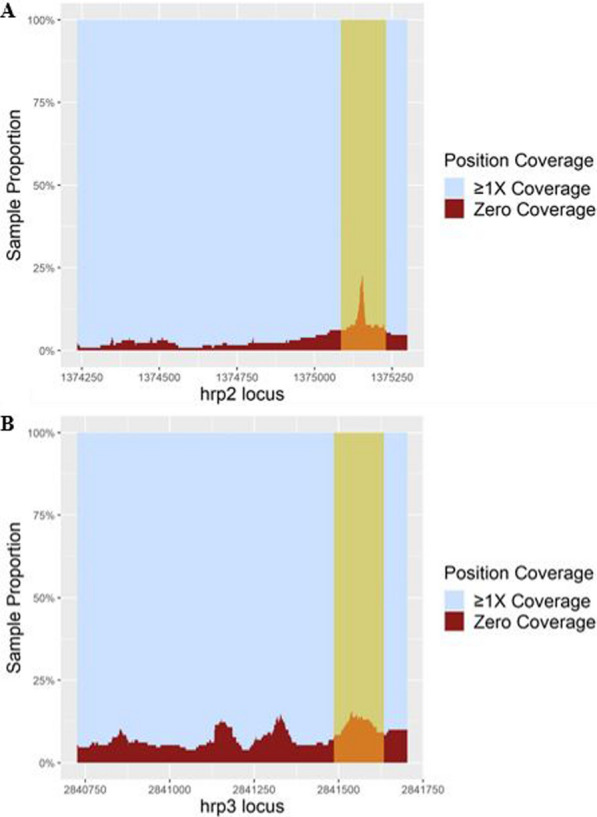
Proportional count of zero coverage positions at the hrp2/3 positions Proportional sample count of hrp2 and hrp3 gene positions with zero coverage vs. positions with ≥ 1X coverage among Cambodian samples (n = 127). A. shows count of zero coverage positions among hrp2 positions, and B. shows count of zero coverage positions among hrp3 positions. The tan section on each plot represents the intron region of each respective gene. Analysis and figures were generated using R v4.1.1
Discussion
Next-generation short-read WGS data has the capability to provide detailed genotype information, but often necessitates a good understanding and use of bioinformatics tools and packages. GC3 was developed to be a user-friendly computational tool to (1) extract coverage profiles of target genome regions, (2) provide interpretable results regarding location and frequency of deletions, (3) classify samples according to the type of gene deletions, and (4) validate large-scale, qPCR-based, studies conducted to inform NMCPs concerning frequency of copy number variants (including deletions) in genes of translational importance. In this study, it is demonstrated that GC3 can be used for these purposes by applying it to Plasmodium falciparum genome segments, specifically the regions containing hrp2 and hrp3 genes. Most NMCPs in malaria-endemic settings rely on HRP2-based RDTs for day-to-day diagnosis in both clinical and field settings. There is evidence of recent expansions of P. falciparum strains lacking HRP2, a cause for concern as stated by the WHO [1, 20, 35, 36]. Given the continuing decrease in sequencing costs and the widespread generation of WGS data, computational tools, such as GC3, that take advantage of such data to efficiently assess the presence of hrp2- or hrp3-deletion strains, provides a valuable monitoring resource to researchers and public health professionals concerned with malaria RDT effectiveness.
This work demonstrated the validity and utility of the GC3 tool to assess hrp2 and hrp3 deletion frequencies in P. falciparum sample sets, as well as to carefully characterize those deletions in individual samples. The sliding window capability of GC3 provided a wider view of large deletions in reference lab strains (i.e. Dd2 and HB3) while adjusting for fluctuations in subtelomeric read coverage and allowed for visually-friendly figures. The option of extracting and plotting every chromosome position within an interval allowed for a magnified view of target loci, and better illustrated details in coverage within the target gene. GC3’s genotype results were validated against previously reported genotypes of P. falciparum laboratory reference and representative strains, and similar, publicly available sample sets from Kenya and Peru, where hrp2/3 deletions have been observed [12, 19, 37]. Analysis of samples from Cambodia and Brazil demonstrate GC3’s capability to process novel WGS data from regions other than West Africa, the location of origin of PfNF54, the parental isolate from which the reference 3D7 was cloned [38, 39]. Results are consistent with previous estimates of hrp2/3 deletions among each respective country [20, 30, 40]. Of note is the high prevalence of all deletions, and especially hrp3 deletions among Brazilian samples. Deletions in hrp2/3, and reports of high deletion prevalence, were first observed in the American continent [8, 9, 17, 19, 40–42], especially in the hrp3 locus [8, 43]. Overall, GC3 can appropriately process and analyse publicly available WGS datasets from a variety of genomic studies.
Additional comparison against a hrp2/3-specific qPCR assay demonstrated very good reliability of GC3’s capability. Although there was one discordant result between tools, this may be a reflection of GC3’s sensitivity and the qPCR assay’s difficulty to detect partial deletions, as these are only detected by the qPCR-based assay if they overlap the primer-binding or amplicon sites. A potential challenge for GC3 are the samples with a very low amount of parasite DNA resulting in genomic libraries of substandard quality and overall low depth of coverage and uneven representation of the loci of interest in the genomic library and/or among the WGS data, leading to significant regions of the hrp2/3 loci with zero coverage (and then a ‘deletion’ assessment by GC3), despite the loci being present in the genome. However, this situation was not observed in this study. In the specific case of discordant results in a sample from Cambodia, the sample had very high coverage at the core and subtelomeric regions. In general, partial deletions present a challenge since some cases have shown a qPCR assay can amplify part of hrp2/3, but corresponds to false negative RDT diagnoses [44, 45]. Ultimately, there were very few such samples, so their impact on overall results is considered minimal. Although beyond the scope of this study, further examination of hrp2/3 partial deletions, their specific location within the locus and their respective RDT diagnosis may provide valuable information regarding the most appropriate criteria and thresholds to accurately identify gene deletions with a functional phenotype, i.e., those deletions that abrogate protein expression.
The application of GC3 to describe and visualize patterns of partial deletions makes it a valuable resource for research purposes. By providing exact genomic coordinates that lack read coverage, GC3 may inform on genomic sequence backgrounds more prone to such mutations and eventually deletions that prevent RDT detection. Defining exact locations of deletions allows the user to determine whether the observed deletions can be explained by a single deletion event followed by lineage expansion, or whether multiple events need to be invoked to explain the observations. This examination of the evolutionary history of gene deletions can provide insights into mechanisms and rate of origin of indels and allow for improved monitoring of target genes.
Among Cambodian samples, read coverage in flanking genes was further analysed and demonstrated that hrp2/3 deletions can be restricted to just the locus proper, or extend to flanking genes, but without a discernible pattern. These results are consistent to previous reports [19, 20, 43, 46, 47]. Overall, GC3 reported similar results in deletion frequency and classification trends within and among global malaria-endemic regions.
Utilizing the function of GC3 to extract all positions (i.e. interval = 1, step size = 1) and plotting hrp2/3 coding positions only, coverage is clearer and subsets of samples can be compared, if desired. The greater difference in median relative coverage was visualized between the two sample sets collected in Bandiagara, Mali. Samples collected in 2002 had lower standardized coverage than those collected in 2010, in both hrp2 and hrp3. Were these samples comparable, this result would be unexpected, since the frequency of deletions is expected to have increased over time, due to the selection imposed by parasite detection by RDT. Interestingly, hrp2 deletions had already been observed in Mali in the late 1990s prior to significant RDT use in the country, indicative of deletion recurrence or persistence in the population at the time despite the absence of pressure from RDT usage [48]. In fact, random polymorphisms occur naturally particularly in the subtelomeric region, including large deletions, without evolutionary pressure [49]. Further, the fitness cost associated with hrp2 loss is not significant, although a small cost appears to be associated with hrp3 deletions [3, 19]. However, too many potentially confounding factors exist between the two sample sets to appropriately interpret results as being indicative of a significant change in frequency of these gene deletions. Even slight differences, apparently innocuous, between sample sets, like read length (100 bp in Mali 2002 samples and 150 bp in Mali 2010 samples), could have an impact when comparing sample sets. Other factors include how malaria-positive cases were detected, study objective (case–control study vs. cohort study), and sample collection strategy (passive vs. active). A carefully controlled study using large sample sizes would be necessary to draw definitive conclusions. Ultimately, this observation showcases GC3’s capability to visualize coverage patterns between sample sets and how other factors can impact deletion rate at these loci.
Closer examination of the proportional coverage at each hrp2/3 position among Cambodian samples, revealed how mapping artifacts can result in no coverage, and potentially confounding results. On hrp2, zero coverage positions increase and then spike around the intron region which, as mentioned before, is likely due to the intron’s high AT content [50] that can cause challenges for read mapping. Interestingly, there were two spikes in zero coverage positions on the hrp3 locus. Examination of a previous whole genome sequence-based hierarchical cluster analysis of the same Cambodian samples [31] revealed that the majority of samples contributing to one or both peaks belong to the same Cambodian subpopulation and hence share a similar genetic background. Ultimately, the figure offers a useful preliminary view of the hrp2/3 genes and their characteristics.
Some limitations exist when interpreting GC3’s results, particularly when comparing sample sets. In this case, it is critical to ensure that sample sets are directly comparable (similar sampling location, collection design and protocol, sample processing and sequencing approach, etc.) or else that interpretation of results is robust to potential confounding factors. In such studies, GC3 is most useful when WGS data is all that is available, and biological material has been exhausted. Pertaining to GC3’s results, the quality of computational results is influenced by the depth of the P. falciparum sequencing data, as measured by the total number of reads mapped to the reference genome. In the case of P. falciparum, it is considered good coverage data if the percentage of genome with coverage asymptotes at ~ 12 million 100-bp reads mapped to the parasite genome, averaging ~ 52X coverage genome-wide. It is also recommended that GC3 is used to calculate mean coverage over the broader region/chromosome where the target gene is located to estimate expected coverage. Further, high-quality WGS data results obtained with well-described DNA extraction methods and sequencing methods, either by direct sequencing or sWGA [22, 24], and established quality control and filtering protocols should be used when comparing samples from different studies. When comparing standardized coverage between direct and sWGA sequence data, direct sequencing achieves more uniform coverage due to the inefficient amplification on the subtelomeric region by sWGA primers [24], but sWGA still provides good coverage at hrp2/3 gene positions (Additional file 1: Figure S6). Even with high-quality data, polyclonality adds another layer of complexity, especially in high transmission settings, where these are most common, since the presence of multiple strains can mask the lack of coverage at a target gene absent in some but not all strains [18]. This factor would need to be considered particularly if GC3 is the only method being used to assess for the presence of deletions. The deletion criteria can be easily adjusted by the user to be more stringent depending on their purposes, much like the deletion criteria used in the comparator study [19]. Despite the limitations, GC3 appropriately processed hrp2/3 coverage data and classified deletions. Its utility can be extended to analyse and visualize coverage data of any target gene on any pathogen.
Summary statement
Overall, validation of GC3 to extract and process WGS data was successful when comparing with expected results using reference strains, well-described samples and a hrp2/3-specific qPCR assay. Following the criteria for identifying deletions, the results agreed with previous estimations of hrp2/3 deletion frequency in each respective country. Apparent in the results is the level of detail that can be extracted from short-read WGS data and viewed using a comprehensive computational tool. Although challenges persist in ensuring high-quality WGS data and achieving similar coverage among low parasitaemia samples using sWGA, GC3’s results are expected to be fairly robust. Further investigation of the partial deletions threshold that results in a false negative RDT diagnosis is needed to validate the deletion criteria. Ultimately, groups investigating a target gene’s coverage can use GC3 to efficiently generate translatable results and figures to understand and interpret broad patterns using hundreds to thousands of previously generated genomic datasets.
Supplementary Information
Additional file 1: Figure S1. Distribution of global isolates including reference strains NF54 (West Africa), 7G8 (Brazil), NF135.C10 (Cambodia), NF166 (Guinea), DD2 (Laos) and HB3 (Honduras). Figure created using Mapchart.net. Figure S2. Subtelomeric read coverage distribution plots. Figure S3. Cambodian sample subset of hrp2/3 and flanking genes. Figure S4. Scatter plots of subtelomeric mean coverage vs. mean coverage of respective HRP-encoding locus. Figure S5. Scatter plots of mean downstream/upstream gene coverage vs. mean coverage of respective HRP-encoding locus. Figure S6. Median standardized coverage by direct sequencing vs sWGA. Figure S7. Proportion of hrp2/3 positions with 0X vs ≥1X coverage for Malawi and Mali samples. Table S1. List of samples that underwent hrp2/3-specific qPCR assay. Table S2. GC3 deletion assignments for hrp2/3 per country.
Additional file 2: Table S3. Summaries of Cambodian sample gene and chromosomal coverages grouped by year of collection. Table S4. Summaries of Malawian sample gene and chromosomal coverages grouped by year of collection. Table S5. Summaries of Malian sample gene and chromosomal coverages grouped by year of collection.
Additional file 3. Samples used for analyses in the manuscript "“Gene Coverage Count and Classification” (GC3), a coverage assessment tool, and its application to identify and classify histidine-rich protein 2 and 3 deletions in Plasmodium falciparum using short-read whole genome sequencing data".
Acknowledgements
We would like to thank the study teams that supported the collection of samples in the Brazil, Malawi, Mali, and Myanmar and Thailand studies. Specifically, we would like to acknowledge the generosity of the UMB PIs from each respective study, namely Dr. Miriam Laufer, Dr. Shannon Takala-Harrison and Dr. Mark Travassos, as well as the University of São Paulo PI, Dr. Marcelo Urbano Ferreira. Parasite isolate DNA from Cambodia and Thailand was derived from clinical samples collected by the Department of Bacterial and Parasitic Diseases at the Armed Forces Research Institute of Medical Sciences in collaboration with Dr. Shannon Takala-Harrison and had undergone whole genome sequencing as part of a previous study [51]. Malawian isolates were generated from samples by the Mfera Health Center in the Chikhwawa district from a previous investigation [52]. Malian samples were collected by the Bandiagara Malaria Project, led by Professor Mahamadou A. Thera. Dr. Drissa Coulibaly supervised the sample collection [53]. Brazilian samples were collected by the University of São Paulo Department of Public Health Practices [22]. Accession numbers for the samples are provided in Additional file 1: Table S1.
Abbreviations
- NMCP
National Malaria Control Programme
- RDTs
Rapid Diagnostic Tests
- hrp2/3
Loci encoding histidine-rich proteins 2 and 3
- WGS
Whole Genome Sequencing
- sWGA
Selective Whole Genome Amplification
- SRA
Sequence Read Archive
- BAM
Binary sequence Alignment/Map
- GATK
Genome Analysis Toolkit
- BED
Browser Extensible Data
- bp
Base pair
- CDS
Coding DNA sequence
Author contributions
TCS conceived the study, designed the bioinformatics toolkit, performed the analyses, interpreted the results and wrote the paper. AD supported the development of GC3, contributed to the interpretation of results, and edited the manuscript. JCS conceived and designed the study and bioinformatics toolkit, interpreted the results, contributed funding and resources, and wrote the paper. BS and SJ were responsible for all laboratory procedures and edited the paper. TS designed the hrp2/3-specific qPCR protocol and edited the paper. AO contributed to the interpretation of results and edited the paper. GG and CD contributed fundamental resources to the study and reviewed the paper. All authors read and approved the final version of the manuscript.
Funding
This work was funded in part by the National Institutes of Health (NIH) awards U19 AI110820 and R01 AI141900 to JCS. AO received funding from the National Heart, Lung and Blood Institute (1K01HL140285-01A1).
Availability of data and materials
The datasets from field studies are available from public repositories, with access information reported previously [22].
Declarations
Ethics approval and consent to participate
All WGS datasets used are publicly available through GenBank. P. falciparum samples for which DNA material was utilized to perform qPCR were collected during field studies in Brazil, Malawi, Mali, and Thailand. All samples were collected after recruitment/enrollment and with written informed consent from the subject or a parent/guardian, and assent obtained from children under 18 years old. Informed consent included authorization to generate parasite genomic data. The respective field studies were approved by the following Institutional review boards (IRBs): Institute of Biomedical Sciences, University of São Paulo (1368/17/CEPSH), for collection in Acre, Brazil; Malawi National Health Sciences Research Committee, and the IRB at the University of Maryland, Baltimore (FWA#00005976), for collection in Malawi; Comité D’Éthique de la FMPOS (Faculté de Médecine de Pharmacie et d’Odonto-stomatologie), Université des Sciences, des Techniques, et des Technologies de Bamako, Mali, and the IRB at the University of Maryland, Baltimore (HP-00041382), for collection on Mali; Ethics Review Committee of the Department of Medical Research, Human Subject Protection Branch (HSPB) of the Walter Reed Army Institute of Research, Silver Spring, MD (FWA# 00000015, IRB# 00000794), and Institute for the Development of Human Research Protection (IHRP), Ministry of Public Health, Thailand (FWA# 00017503, IRB#00006539), for collections in Thailand. This study conforms to the principles established in the Helsinki Declaration.
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
Footnotes
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Contributor Information
Thomas C. Stabler, Email: thomas.stabler@swisstph.ch
Joana C. Silva, Email: jcsilva@som.umaryland.edu
References
- 1.WHO. World malaria report 2021. Geneva: World Health Organization; 2021.
- 2.Mukkala AN, Kwan J, Lau R, Harris D, Kain D, Boggild AK. An update on malaria rapid diagnostic tests. Curr Infect Dis Rep. 2018;20:49. doi: 10.1007/s11908-018-0655-4. [DOI] [PubMed] [Google Scholar]
- 3.Poti KE, Sullivan DJ, Dondorp AM, Woodrow CJ. HRP2: transforming malaria diagnosis, but with caveats. Trends Parasitol. 2020;36:112–126. doi: 10.1016/j.pt.2019.12.004. [DOI] [PubMed] [Google Scholar]
- 4.Rock EP, Marsh K, Saul AJ, Wellems TE, Taylor DW, Maloy WL, et al. Comparative analysis of the Plasmodium falciparum histidine-rich proteins HRP-I, HRP-II and HRP-III in malaria parasites of diverse origin. Parasitology. 1987;95:209–227. doi: 10.1017/S0031182000057681. [DOI] [PubMed] [Google Scholar]
- 5.Kong A, Wilson SA, Ah Y, Nace D, Rogier E, Aidoo M. HRP2 and HRP3 cross-reactivity and implications for HRP2-based RDT use in regions with Plasmodium falciparum hrp2 gene deletions. Malar J. 2021;20:207. doi: 10.1186/s12936-021-03739-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Thomson R, Parr JB, Cheng Q, Chenet S, Perkins M, Cunningham J. Prevalence of Plasmodium falciparum lacking histidine-rich proteins 2 and 3: a systematic review. Bull World Health Organ. 2020;98:558–568F. doi: 10.2471/BLT.20.250621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Kozycki CT, Umulisa N, Rulisa S, Mwikarago EI, Musabyimana JP, Habimana JP, et al. False-negative malaria rapid diagnostic tests in Rwanda: impact of Plasmodium falciparum isolates lacking hrp2 and declining malaria transmission. Malar J. 2017;16:123. doi: 10.1186/s12936-017-1768-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Gendrot M, Fawaz R, Dormoi J, Madamet M, Pradines B. Genetic diversity and deletion of Plasmodium falciparum histidine-rich protein 2 and 3: a threat to diagnosis of P falciparum malaria. Clin Microbiol Infect. 2019;25:580–585. doi: 10.1016/j.cmi.2018.09.009. [DOI] [PubMed] [Google Scholar]
- 9.Agaba BB, Yeka A, Nsobya S, Arinaitwe E, Nankabirwa J, Opigo J, et al. Systematic review of the status of pfhrp2 and pfhrp3 gene deletion, approaches and methods used for its estimation and reporting in Plasmodium falciparum populations in Africa: review of published studies 2010–2019. Malar J. 2019;18:355. doi: 10.1186/s12936-019-2987-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Akinyi Okoth S, Abdallah JF, Ceron N, Adhin MR, Chandrabose J, Krishnalall K, et al. Variation in Plasmodium falciparum Histidine-Rich Protein 2 (Pfhrp2) and Plasmodium falciparum Histidine-Rich Protein 3 (Pfhrp3) Gene Deletions in Guyana and Suriname. PLoS ONE. 2015;10:e0126805. doi: 10.1371/journal.pone.0126805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Berzosa P, González V, Taravillo L, Mayor A, Romay-Barja M, García L, et al. First evidence of the deletion in the pfhrp2 and pfhrp3 genes in Plasmodium falciparum from Equatorial Guinea. Malar J. 2020;19:99. doi: 10.1186/s12936-020-03178-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Beshir KB, Sepúlveda N, Bharmal J, Robinson A, Mwanguzi J, Busula AO, et al. Plasmodium falciparum parasites with histidine-rich protein 2 (pfhrp2) and pfhrp3 gene deletions in two endemic regions of Kenya. Sci Rep. 2017;7:14718. doi: 10.1038/s41598-017-15031-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Funwei R, Nderu D, Nguetse CN, Thomas BN, Falade CO, Velavan TP, et al. Molecular surveillance of pfhrp2 and pfhrp3 genes deletion in Plasmodium falciparum isolates and the implications for rapid diagnostic tests in Nigeria. Acta Trop. 2019;196:121–125. doi: 10.1016/j.actatropica.2019.05.016. [DOI] [PubMed] [Google Scholar]
- 14.Kumar N, Pande V, Bhatt RM, Shah NK, Mishra N, Srivastava B, et al. Genetic deletion of HRP2 and HRP3 in Indian Plasmodium falciparum population and false negative malaria rapid diagnostic test. Acta Trop. 2013;125:119–121. doi: 10.1016/j.actatropica.2012.09.015. [DOI] [PubMed] [Google Scholar]
- 15.Thomson R, Beshir KB, Cunningham J, Baiden F, Bharmal J, Bruxvoort KJ, et al. pfhrp2 and pfhrp3 gene deletions that affect malaria rapid diagnostic tests for Plasmodium falciparum: analysis of archived blood samples from 3 African countries. J Infect Dis. 2019;220:1444–1452. doi: 10.1093/infdis/jiz335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Verma AK, Bharti PK, Das A. HRP-2 deletion: a hole in the ship of malaria elimination. Lancet Infect Dis. 2018;18:826–827. doi: 10.1016/S1473-3099(18)30420-1. [DOI] [PubMed] [Google Scholar]
- 17.Molina-de la Fuente I, Pastor A, Herrador Z, Benito A, Berzosa P. Impact of Plasmodium falciparum pfhrp2 and pfhrp3 gene deletions on malaria control worldwide: a systematic review and meta-analysis. Malar J. 2021;20:276. doi: 10.1186/s12936-021-03812-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Watson OJ, Slater HC, Verity R, Parr JB, Mwandagalirwa MK, Tshefu A, et al. Modelling the drivers of the spread of Plasmodium falciparum hrp2 gene deletions in sub-Saharan Africa. Life. 2017;6:e25008. doi: 10.7554/eLife.25008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Sepúlveda N, Phelan J, Diez-Benavente E, Campino S, Clark TG, Hopkins H, et al. Global analysis of Plasmodium falciparum histidine-rich protein-2 (pfhrp2) and pfhrp3 gene deletions using whole-genome sequencing data and meta-analysis. Infect Genet Evol. 2018;62:211–219. doi: 10.1016/j.meegid.2018.04.039. [DOI] [PubMed] [Google Scholar]
- 20.Gibbons J, Qin J, Malla P, Wang Z, Brashear A, Wang C, et al. Lineage-specific expansion of Plasmodium falciparum parasites with pfhrp2 deletion in the Greater Mekong Subregion. J Infect Dis. 2020;222:1561–1569. doi: 10.1093/infdis/jiaa250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Feleke SM, Reichert EN, Mohammed H, Brhane BG, Mekete K, Mamo H, et al. Emergence and evolution of Plasmodium falciparum histidine-rich protein 2 and 3 deletion mutant parasites in Ethiopia. medRxiv. 2021.
- 22.Moser KA, Drábek EF, Dwivedi A, Stucke EM, Crabtree J, Dara A, et al. Strains used in whole organism Plasmodium falciparum vaccine trials differ in genome structure, sequence, and immunogenic potential. Genome Med. 2020;12:6. doi: 10.1186/s13073-019-0708-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Malaria GEN Plasmodium falciparum Community Project. Genomic epidemiology of artemisinin resistant malaria. eLife. 2016;5:e08714. [DOI] [PMC free article] [PubMed]
- 24.Oyola SO, Ariani CV, Hamilton WL, Kekre M, Amenga-Etego LN, Ghansah A, et al. Whole genome sequencing of Plasmodium falciparum from dried blood spots using selective whole genome amplification. Malar J. 2016;15:597. doi: 10.1186/s12936-016-1641-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Zhang M, Wang C, Otto TD, Oberstaller J, Liao X, Adapa SR, et al. Uncovering the essential genes of the human malaria parasite Plasmodium falciparum by saturation mutagenesis. Science. 2018;360:e7847. doi: 10.1126/science.aap7847. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Schindler T, Deal AC, Fink M, Guirou E, Moser KA, Mwakasungula SM, et al. A multiplex qPCR approach for detection of pfhrp2 and pfhrp3 gene deletions in multiple strain infections of Plasmodium falciparum. Sci Rep. 2019;9:13107. doi: 10.1038/s41598-019-49389-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Abdallah JF, Okoth SA, Fontecha GA, Torres REM, Banegas EI, Matute ML, et al. Prevalence of pfhrp2 and pfhrp3 gene deletions in Puerto Lempira. Honduras Malar J. 2015;14:19. doi: 10.1186/s12936-014-0537-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Munro JB, Jacob CG, Silva JC. A novel clade of unique eukaryotic ribonucleotide reductase R2 subunits is exclusive to apicomplexan parasites. J Mol Evol. 2013;77:92–106. doi: 10.1007/s00239-013-9583-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Xiao C, Ye J, Esteves RM, Rong C. Using Spearman's correlation coefficients for exploratory data analysis on big dataset. Concurrency Computation. 2016;28:3866–3878. doi: 10.1002/cpe.3745. [DOI] [Google Scholar]
- 30.Rachid Viana GM, Akinyi Okoth S, Silva-Flannery L, Lima Barbosa DR, Macedo de Oliveira A, Goldman IF, et al. Histidine-rich protein 2 (pfhrp2) and pfhrp3 gene deletions in Plasmodium falciparum isolates from select sites in Brazil and Bolivia. PLoS ONE. 2017;12:e0171150. doi: 10.1371/journal.pone.0171150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Agrawal S, Moser KA, Morton L, Cummings MP, Parihar A, Dwivedi A, et al. Association of a novel mutation in the Plasmodium falciparum chloroquine resistance transporter with decreased piperaquine sensitivity. J Infect Dis. 2017;216:468–476. doi: 10.1093/infdis/jix334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Laufer MK, Thesing PC, Dzinjalamala FK, Nyirenda OM, Masonga R, Laurens MB, et al. A longitudinal trial comparing chloroquine as monotherapy or in combination with artesunate, azithromycin or atovaquone-proguanil to treat malaria. PLoS ONE. 2012;7:e42284. doi: 10.1371/journal.pone.0042284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Lyke KE, Burges RB, Cissoko Y, Sangare L, Kone A, Dao M, et al. HLA-A2 supertype-restricted cell-mediated immunity by peripheral blood mononuclear cells derived from malian children with severe or uncomplicated Plasmodium falciparum malaria and healthy controls. Infect Immun. 2005;73:5799–5808. doi: 10.1128/IAI.73.9.5799-5808.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Travassos MA, Coulibaly D, Laurens MB, Dembélé A, Tolo Y, Koné AK, et al. Hemoglobin C trait provides protection from clinical falciparum malaria in Malian children. J Infect Dis. 2015;212:1778–1786. doi: 10.1093/infdis/jiv308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Golassa L, Messele A, Amambua-Ngwa A, Swedberg G. High prevalence and extended deletions in Plasmodium falciparum hrp2/3 genomic loci in Ethiopia. PLoS ONE. 2020;15:e0241807. doi: 10.1371/journal.pone.0241807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Berhane A, Anderson K, Mihreteab S, Gresty K, Rogier E, Mohamed S, et al. Major threat to malaria control programs by Plasmodium falciparum lacking histidine-rich protein 2. Eritrea Emerg Incet Dis. 2018;24:462–470. doi: 10.3201/eid2403.171723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Gamboa D, Ho M-F, Bendezu J, Torres K, Chiodini PL, Barnwell JW, et al. A large proportion of P falciparum isolates in the Amazon Region of Peru lack pfhrp2 and pfhrp3: implications for malaria rapid diagnostic tests. PLoS ONE. 2010;5:e8091. doi: 10.1371/journal.pone.0008091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Walliker D, Quakyi IA, Wellems TE, McCutchan TF, Szarfman A, London WT, et al. Genetic analysis of the human malaria parasite Plasmodium falciparum. Science. 1987;236:1661–1666. doi: 10.1126/science.3299700. [DOI] [PubMed] [Google Scholar]
- 39.Preston MD, Campino S, Assefa SA, Echeverry DF, Ocholla H, Amambua-Ngwa A, et al. A barcode of organellar genome polymorphisms identifies the geographic origin of Plasmodium falciparum strains. Nat Commun. 2014;5:4052. doi: 10.1038/ncomms5052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Góes L, Chamma-Siqueira N, Peres JM, Nascimento JM, Valle S, Arcanjo AR, et al. Evaluation of histidine-rich proteins 2 and 3 gene deletions in Plasmodium falciparum in endemic areas of the Brazilian Amazon. Int J Environ Res Public Health. 2020;18:123. doi: 10.3390/ijerph18010123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Bhasin VK, Trager W. Gametocyte-forming and non-gametocyte-forming clones of Plasmodium falciparum. Am J Trop Med Hyg. 1984;33:534–537. doi: 10.4269/ajtmh.1984.33.534. [DOI] [PubMed] [Google Scholar]
- 42.Vera-Arias CA, Holzschuh A, Oduma CO, Badu K, Abdul-Hakim M, Yukich J, et al. Plasmodium falciparum hrp2 and hrp3 gene deletion status in Africa and South America by highly sensitive and specific digital PCR. medRxiv. 2021.
- 43.Wurtz N, Fall B, Bui K, Pascual A, Fall M, Camara C, et al. Pfhrp2 and pfhrp3 polymorphisms in Plasmodium falciparum isolates from Dakar, Senegal: impact on rapid malaria diagnostic tests. Malar J. 2013;12:34. doi: 10.1186/1475-2875-12-34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Houzé S, Hubert V, Pessec GL, Bras JL, Clain J. Combined deletions of pfhrp2 and pfhrp3 genes result in Plasmodium falciparum malaria false-negative rapid diagnostic test. J Clin Microbiol. 2011;49:2694–2696. doi: 10.1128/JCM.00281-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Nima MK, Hougard T, Hossain ME, Kibria MG, Mohon AN, Johora FT, et al. A case of Plasmodium falciparum hrp2 and hrp3 gene mutation in Bangladesh. Am J Trop Med Hyg. 2017;97:1155–1158. doi: 10.4269/ajtmh.16-0884. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Li P, Xing H, Zhao Z, Yang Z, Cao Y, Li W, Yan G, et al. Genetic diversity of Plasmodium falciparum histidine-rich protein 2 in the China-Myanmar border area. Acta Trop. 2015;152:26–31. doi: 10.1016/j.actatropica.2015.08.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Parr JB, Verity R, Doctor SM, Janko M, Carey-Ewend K, Turman BJ, et al. Pfhrp2-deleted Plasmodium falciparum parasites in the Democratic Republic of the Congo: a national cross-sectional survey. J Infect Dis. 2016;216:36–44. doi: 10.1093/infdis/jiw538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Koita OA, Doumbo OK, Ouattara A, Tall LK, Konaré A, Diakité M, et al. False-negative rapid diagnostic tests for malaria and deletion of the histidine-rich repeat region of the hrp2 gene. Am J Trop Med Hyg. 2012;86:194–198. doi: 10.4269/ajtmh.2012.10-0665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.de Bruin D, Lanzer M, Ravetch JV. The polymorphic subtelomeric regions of Plasmodium falciparum chromosomes contain arrays of repetitive sequence elements. Proc Natl Acad Sci USA.1994;91:619–623. [DOI] [PMC free article] [PubMed]
- 50.Hamilton WL, Claessens A, Otto TD, Kekre M, Fairhurst RM, Rayner JC, et al. Extreme mutation bias and high AT content in Plasmodium falciparum. Nucleic Acids Res. 2016;45:1889–1901. doi: 10.1093/nar/gkw1259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Takala-Harrison S, Jacob CG, Arze C, Cummings MP, Silva JC, Dondorp AM, et al. Independent emergence of artemisinin resistance mutations among Plasmodium falciparum in Southeast Asia. J Infect Dis. 2014;211:670–679. doi: 10.1093/infdis/jiu491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Buchwald AG, Sixpence A, Chimenya M, Damson M, Sorkin JD, Wilson ML, et al. Clinical implications of asymptomatic Plasmodium falciparum infections in Malawi. Clin Infect Dis. 2018;68:106–112. doi: 10.1093/cid/ciy427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Coulibaly D, Travassos MA, Kone AK, Tolo Y, Laurens MB, Traore K, et al. Stable malaria incidence despite scaling up control strategies in a malaria vaccine-testing site in Mali. Malar J. 2014;13:374. doi: 10.1186/1475-2875-13-374. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Additional file 1: Figure S1. Distribution of global isolates including reference strains NF54 (West Africa), 7G8 (Brazil), NF135.C10 (Cambodia), NF166 (Guinea), DD2 (Laos) and HB3 (Honduras). Figure created using Mapchart.net. Figure S2. Subtelomeric read coverage distribution plots. Figure S3. Cambodian sample subset of hrp2/3 and flanking genes. Figure S4. Scatter plots of subtelomeric mean coverage vs. mean coverage of respective HRP-encoding locus. Figure S5. Scatter plots of mean downstream/upstream gene coverage vs. mean coverage of respective HRP-encoding locus. Figure S6. Median standardized coverage by direct sequencing vs sWGA. Figure S7. Proportion of hrp2/3 positions with 0X vs ≥1X coverage for Malawi and Mali samples. Table S1. List of samples that underwent hrp2/3-specific qPCR assay. Table S2. GC3 deletion assignments for hrp2/3 per country.
Additional file 2: Table S3. Summaries of Cambodian sample gene and chromosomal coverages grouped by year of collection. Table S4. Summaries of Malawian sample gene and chromosomal coverages grouped by year of collection. Table S5. Summaries of Malian sample gene and chromosomal coverages grouped by year of collection.
Additional file 3. Samples used for analyses in the manuscript "“Gene Coverage Count and Classification” (GC3), a coverage assessment tool, and its application to identify and classify histidine-rich protein 2 and 3 deletions in Plasmodium falciparum using short-read whole genome sequencing data".
Data Availability Statement
The datasets from field studies are available from public repositories, with access information reported previously [22].