

Abstract
Haemophilus influenzae is a pathogen that causes invasive bacterial infections in humans. The highest prevalence lies in both young children and adults. Generally, there are no vaccines available that target all the strains of Haemophilus influenzae. Hence, the purpose of this research is to employ bioinformatics and immunoinformatics approaches to design a Multi-Epitope Vaccine candidate employing the pathogenic cell division protein FtsN that specifically combat all the Haemophilus influenzae strains. The current research focuses on developing subunit vaccine in contrast to vaccines generated from the entire pathogen. This will be accomplished by combining multiple bioinformatics and immunoinformatics approaches. As a result, prospective T cells (helper T lymphocyte and cytotoxic T lymphocytes) and B cells epitopes were investigated. The human leukocyte antigen allele having strong associations with the antigenic and overlapping epitopes were chosen, with 70% of the total coverage of the world population. To construct a linked vaccine design, multiple linkers were used. To increase the immunogenic profile, an adjuvant was linked using EAAAK linker. The final vaccine construct with 149 amino acids was obtained after adjuvants and linkers were added. The developed Multi-Epitope Vaccine has a high antigenicity as well as viable physiochemical features. The 3D conformation was modeled and undergoes refinement and validation using bioinformatics methods. Furthermore, protein–protein molecular docking analysis was performed to predict the effective binding poses of Multi-Epitope Vaccine with the Toll-like receptor 4 protein. Besides, vaccine underwent the codon translational optimization and computational cloning to verify the reliability and proper Multi-Epitope Vaccine expression. In addition, it is necessary to conduct experiments and research in the laboratory to demonstrate that the vaccine that has been developed is immunogenic and protective.
Keywords: Haemophilus influenzae, Epitope, Cell division protein, Bioinformatics, Linkers
Introduction
Haemophilus influenzae (Hi) is a Gram-negative coccobacillus which belongs to the Pasteurellaceae family (Soeters et al. 2018). It is tiny (0.3–1 µm), anaerobic, pleomorphic pathogenic bacteria. The infection caused by Hi is termed as “Haemophilus influenzae disease” (Suga et al. 2018). It can cause pneumonia, otitis media, epiglottitis, and meningitis in the upper respiratory tract (Slack et al. 2020). It usually gets into the lungs via aspiration or hematogenous dissemination. It is the source of an extensive range of bacterial illnesses (Butler and Myers 2018). They are divided into two types: encapsulated and non-encapsulated (Wilson et al. 2020). Non-typeable Hi (NTHi) strains do not express a polysaccharide capsule, but Hi serotypes (i.e., Hia, Hib, Hic, Hid, Hie, and Hif) do, with Type B being the most prevalent cause of pneumonia, infecting predominantly children and immunocompromised people (Wilson et al. 2020). However, NTHi is linked to both invasive and non-invasive illnesses. The Hi genome (Rd KW20) was sequenced first among the members of Pasteurellaceae family (Pinto et al. 2019). The genome size is 1,830,137 base pair with 38% GC content (López-López et al. 2021).
Hi adheres to host cells and exhibits pathogenicity via various modes of transmission (Chen et al. 2018). One of the crucial steps in infection and pathogenesis is the initial adhesion of Hi to the host cells (López-López et al. 2021). Hi adheres to host cells and exhibits pathogenicity such as the capsule, the adhesion proteins, the IgA1 protease, and, last but not least, the lipooligosaccharide (Huska et al. 2022; Vitovski et al. 2002; Kilian et al. 1983). The bacterial capsule confers antiphagocytic properties, and the absence of an anti-capsular antibody promotes bacterial propagation (Potts et al. 2019). As a result, they evade and change the host immune responses, as well as aid in the development of a biofilm-like colony, through comparable interactions (Mirzaei et al. 2020).
The Hi takes as little as a few days to elicit clinical signs but the exact incubation period is still unknown (Saadati et al. 2021). Infected persons spread the disease by coughing or sneezing, which causes minute respiratory droplets containing the bacteria (Soeters et al. 2018). If other people breathe in those droplets, they may become ill. The sickness caused by Hi affects predominantly children under the age of 5 and adults aged 65 and up (Butler and Myers 2018). American Indians, Alaska Natives, or people with specific medical issues are at risk (Soeters et al. 2018; Butler and Myers 2018). The infected organ, and the history and physical examination are the clinical manifestations of Hi. The most prominent symptoms of pneumonia are high-grade fever, chills as well as cough, fatigue, breathlessness, chest pain, and body aches (Wen et al. 2020).
Antibiotic-resistant bacteria has emerged due to the frequent use of antibiotics (Serwecińska 2020). Major challenges to developing a vaccine against Hi are: (i) lack of polysaccharide capsule in non-typeable Hi, necessitating the development of alternative vaccine antigens; (ii) non-typeable strains have a lot of genetic and antigenic diversity, whereas Type B encapsulated bacteria are usually a clonal population, and (iii) the pathogenesis of non-typeable. The contiguous spread of Hi infections from mucosal surfaces implies that a successful vaccination will demand the creation of an alternate vaccine generated by hematogenous dispersion (Vitovski et al. 2002). The use of vaccinology may be a viable strategy for preventing and controlling Hi infections (Kim et al. 2004). With the evolution of bioinformatics, computational tools have simplified computational evaluations for the vaccine candidates, greatly reducing the time required for preclinical and clinical operations (Ko et al. 2012). Advancements in bioinformatics and immunoinformatics have resulted in the rapid epitope evaluation and validation of proteins that could be used as vaccine targets (Colovos and Yeates 1993). To create a viable subunit vaccine, several antigenic markers and adjuvants must be chosen and added to create a vaccine construct. This vaccine construct can then be utilized to evaluate the simulated immunogenic response of the host using immunoinformatics techniques (“Designing a chimeric subunit vaccine for influenza virus, based on HA2, M2e and CTxB 2022).
Except vaccines for Hib, there are no vaccines available that target general Hi strains; therefore, in this research, bioinformatics and immunoinformatics tools are employed to computationally design a subunit Multi-Epitope Vaccine (MEV) against Hi (Fig. 1). This research puts emphasis on the development of subunit vaccinations as opposed to the vaccines generated from entire pathogens. Therefore, a highly conserved and antigenic cell division protein FtsN of Hi was targeted. Therefore, FtsN protein was selected and utilized for major histocompatibility class (MHC)-I and MHC class-II binding T cell and B cell epitopes prediction. These specific epitopes are responsible for inducing interferon-gamma (IFN-γ). The highly antigenic epitopes are therefore utilized for MEV construction using structure prediction to computationally assess the vaccine construct. The immunological and physicochemical attributes of the vaccine construct such as its stability, flexibility, and solubility, as well as its binding efficacy to the Toll-like receptor 4 (TLR4), were evaluated. The MEV was also simulated to evaluate the generation of immune response by the host. Lastly, codon translation enhancement and vaccine cloning (computational simulation) are performed to assure the successful construction and expression of the MEV construct.
Fig. 1.

Research methodology employed to develop a subunit MEV vaccine against Hi
Methodology
Sequence retrieval and structural analysis
The FASTA sequence for cell division protein FtsN was first obtained from the National Center of Biotechnology Information (NCBI) as a FASTA file (NCBI Resource Coordinators 2013). ProtParam was then used to predict the physical and chemical properties of FtsN (Gasteiger et al. 2005). VaxiJen v.2.0 tool determined the antigenicity of proteins (Doytchinova and Flower 2007). It uses an alignment-free method to predict the antigenicity from physicochemical data. With the help of SOPMA, prediction of the secondary structure was carried out (Geourjon and Deléage 1995). Researchers have not yet figured out the 3D conformation of the FtsN protein, which is crucial in the final stages of cell division. Therefore, the Robetta server estimates the 3D conformation of the FtsN protein (Kim et al. 2004). It employs two different approaches: comparative modeling and de novo 3D conformation prediction. We used GalaxyWEB to fine-tune the predicted 3D protein models (Ko et al. 2012). The SAVES server assessed the predicted models for a quality check (Colovos and Yeates 1993). Moreover, the Ramachandran plot was used to validate our predicted models on the RAMPAGE server (Lovell et al. 2003).
MHC class-I T cell epitopes identification and analysis
The IEDB-AR v.2.22 consensus technique server was employed to identify the 9-mer MHC class-I epitopes (Zhang et al. 2008; Calis et al. 2013). The FtsN protein sequence was input for prediction of T cell epitopes, and the respective alleles were preferred. To narrow down the field of epitopes for further study, those with consensus scores of fewer than two are deemed good binders. The 15-mer MHC class-II T cell epitopes were predicted using NetMHCIIpan 4.0 server (Reynisson et al. 2020). The epitopes were divided into strong, weak, or non-binding at the default threshold based on the percentage value. We chose 2%, 10%, and greater than 10% as the percentiles for determining whether or not a substance is strongly bound. Cytokines activate immune system including other cytotoxic T cells. Vaccine development involves the critical component of cytokines that induce helper T lymphocyte (HTL) and cytotoxic T lymphocytes (CTL). Using the IFN epitope server, IFN- γ epitopes were characterized. It uses the support vector machine (SVM) approach, hybrid motif method, and IFN- γ vs non-IFN-γ model (Dhanda et al. 2013a). In contrast, IL-4Pred and IL-10Pred predicted IL-4 and IL-10 enhancing qualities (Dhanda et al. 2013b; Nagpal et al. 2017). Both servers predicted interleukin-inducing peptides in-silico. According to the IEDB-AR MHC-I immunogenicity tool and the VaxiJen server, antigenicity and immunogenicity were evaluated (Doytchinova and Flower 2007). AlgPred 2.0 was used for allergenic profiling (Saha and Raghava 2006a). ToxinPred was employed to predict toxicity in the study (Gupta et al. 2013). In this way, each epitope was screened and tested.
Prediction and evaluation of B cell epitopes
The promising candidates for vaccine development are B cell epitopes because they induce adaptive immune response (Zhang et al. 2014). Conformational and linear are two subtypes of B cell epitopes. The ABCPred server was used for the prediction of linear B cell type 16-mer epitopes and those 0.5 prediction score were shortlisted (Saha and Raghava 2006b). For the prediction of discontinuous B cell epitopes, the Discotope server was utilized (Kringelum et al. 2012). VaxiJen 2.0, ToxinPred 2.0, and AlgPred 2.0 were used to analyze the antigenic, toxic, and allergic characteristics of the newly discovered B cell epitopes (Doytchinova and Flower 2007; Saha and Raghava 2006a; Gupta et al. 2013).
Shortlisting and conservancy check of identified epitopes
A conservancy check was run through the IEDB-AR v.2.22 for shortlisting of conserved T cell and B cell epitopes (Bui et al. 2006). We focused on epitopes that showed a 100% conservation. Cytokine-prompting capabilities were recognized as a crucial criterion in sorting out effective epitopes. IFN is known for its intrinsically safe reactions and its ability to prevent viral duplication. In addition to this, they can prepare both CTL and HTL, both of which can induce a range of immunological responses. The IFN stimulating potential of expected epitopes was analyzed using IFN epitope server which employs SVM algorithm (Dhanda et al. 2013a).
HLA population coverage analysis
Levels of dispersion and expression vary among HLA alleles based on ethnicities and regions around the world, which influences the development of a successful vaccination (Bui et al. 2006; Immunoinformatics Approach for Epitope-Based Peptide Vaccine Design and Active Site Prediction against Polyprotein of Emerging Oropouche Virus 2022; Nain et al. 2020; Hoover et al. 2003). IEDB-AR v.2.22 Population Coverage tool was employed for the population coverage analysis on the shortlisted epitopes (Bui et al. 2006). Population coverage was evaluated for each epitope for variety of geographic locations based on HLA alleles’ classification. The areas with great significance were identified with the Hi pathogen under the current research.
Designing of MEV
The MEV contains the highly antigenic epitopes that are non-allergenic, 100 percent conserved and overlapping, have extensive population coverage, and have a strong association with a common human allele. As a result, only those epitopes that met the aforementioned requirements were included in the MEV. Following inter-interaction compatibility validation and to enhance the immunological response, the first CTL epitope was linked to the adjuvant using EAAAK linker. This linkage decreases association with other biomolecules through efficient dissociation and enhances stability (Hoover et al. 2003; Mahram and Herbordt 2010; Bjellqvist et al. 1993). With an adjuvant, the immunogenicity of the vaccination may rise. AAY and GPGPG linkers were used for other epitopes to maintain their distinct immunogenicity. The primary goal in the development of MEV is to prevent the induction of junctional epitopes, and both AAY and GPGPG are able to achieve this purpose yet, they enhance immunization and epitope performance (Lovell, et al. 2003; McGuffin et al. 2000). The beta-defensin adjuvant having length of 45 amino acids was used in this investigation that serves as both an antibacterial agent and an immunomodulatory (Hoover et al. 2003; Wiederstein and Sippl 2007).
Primary and secondary structural analyses
The human proteome was examined with the BLASTp using its default settings to make certain that the generated MEV sequence was not similar to any other sequence (threshold: 10; word size: 6; matrix BLOSUM62) (Kuriata et al. 2018). A protein is considered non-homologous if it shares less than 37% of its amino acid sequence. ProtParam evaluated the physicochemical features of the vaccine construct (Gasteiger et al. 2005). Using the Vaxijen v.2.0 server, antigenicity predictions were made possible for vaccine. PSIPRED was employed to determine the 2D structure analysis of the MEV (McGuffin et al. 2000). It examined the extensive chain, coils, alpha-helices, and beta-strands (McGuffin et al. 2000; Kurcinski et al. 2019).
MEV construct 3D conformation prediction, refinement, and evaluation
For 3D conformation prediction of the MEV, Robetta with de novo modeling approach was employed to model because the vaccine is a collection of different epitopes. Secondly, there was not a suitable template with appropriate identity percentage to be used (Kim et al. 2004; Prediction of residues in discontinuous B-cell epitopes using protein 3D structures-Haste Andersen 2006). The projected 3D shape of our vaccine was subjected to a GalaxyWEB server to improve the structure quality (Ko et al. 2012). The Ramachandran plot evaluation of the refined 3D conformation of MEV was performed with the assistance of the RAMPAGE server (Lovell, et al. 2003). The Z-score for MEV was computed via the ProSA-web server (Wiederstein and Sippl 2007). With the help of the SAVES server, we were able to choose and test the best-predicted model (Colovos and Yeates 1993). Additionally, to assess the MEV structural flexibility, CABS-Flex v.2.0 server was used (Kuriata, et al. 2018). The flexibility of the constructed vaccine is critical to its function, and it provides complete picture stability of a protein by simulating its residues (Kurcinski et al. 2019).
Screening for B cell epitopes
The Discotope server is utilized to forecast discontinuous structural B cell epitopes with a threshold of -3.7 which is used for the construction of final MEV (Kringelum et al. 2012). For the prediction of linear B cell epitopes, ABCPred server was employed (Saha and Raghava 2006b).
Protein–protein docking between TLR4 receptor and MEV
If a vaccine reaches the target immune cells in the right way, the host will create an effective immune response. As a result, the MEV’s affinity for human immunological receptors was investigated using molecular docking. The antiviral immune responses are mostly attributed to TLR4 receptor. The TLR4 receptor protein (PDB ID: 4G8A) was docked against the vaccine construct using the ClusPro server, a popular technique for protein–protein docking (The ClusPro web server for protein–protein docking 2022; Mugunthan and Harish 2021). The docked adducts were visualized with PyMOLv.1 (Pymol: an open-source molecular graphics tool—ScienceOpen 2022). PDBsum was used to achieve the schematic diagrams demonstrating the interactions among the docked protein complex (Laskowski 2009).
Immune system simulation for vaccine efficacy
To observe the generated immunological responses by the designed MEV, computational immune simulation server C-ImmSim 10.1 was used (Pandey 2016). The server simulates the lymph node, thymus, and bone barrow. The number of simulation steps (100), injections (1 each), and random seed number (12,345) were set, whereas HLA-binding alleles such as, B0702, B0702, A0101, A0101, DRB1 0101, and DRB1 0101 and volume (10) were chosen.
In-silico E. coli cloning and codon optimization
Each species uses a different set of codons for the translation of the mRNAs into proteins. Therefore, specie to specie usage of codon varies. If codons are not optimal for the host species, gene expression may be lowered. The Java Codon Adaption Tool (JCAT) server was employed to optimize codons for E. Coli strain K12 (Pandey 2016; Grote et al. 2005). The cleavage sites of restriction enzymes and rho-independent transcription termination were chosen to avoid bacterial ribosome binding sites. The codon adaption index score and GC percentage content of the optimized DNA sequence were evaluated (Livingston et al. 2002).
Results
Sequence retrieval and structural analysis
The sequence for the cell division protein FtsN was obtained from NCBI with accession number WP_168769343.1 in FASTA format having 256 amino acids. VaxiJen version 2.0 was then used for antigenicity testing. The results indicated that protein was considerably allergenic with a score of 0.8390. The physicochemical parameters, such as half-life, stability profiling, theoretical pI of 9.92, molecular weight, and aliphatic index, were evaluated with the help of ProtParam (Table 1). The secondary structure analysis using SOPMA revealed more helixes in the protein secondary structure as compared to strands (Table 2, Fig. 2).
Table 1.
FtsN cell division antigenic protein physiochemical properties
| Molecular weight | Isoelectric point | Instability index | Stability profiling | Aliphatic index | Grand average of hydropathy | Half-life |
|---|---|---|---|---|---|---|
| 29,139.38 | 9.92 | 39.84 | Stable | 79.65 | − 0.761 |
30 h (mammalian reticulocytes, in vitro) > 20 h (yeast, in vivo) > 10 h (Escherichia coli, in vivo) |
Table 2.
SOPMA secondary structure prediction results of FtsN cell division protein
| Amino acids | Alpha-helix | Beta-turn | Coils |
|---|---|---|---|
| 256 | 52.73% | 1.56% | 35.94% |
Fig. 2.

SOPMA secondary structure prediction of FtsN protein. Yellow color bars indicate strands, pink bars are representing helices, and grey color lines are coils
The Robetta server and GalaxyWEB application predicted and refined the 3D model, respectively (Fig. 3A). Ramachandran plot assessed the estimated structure (Fig. 3B). The FtsN structure contained the 99.587% allowed amino acids. In SAVES, the optimized model score was 100. As a result, it was clear that the finalized model was of the highest possible quality.
Fig. 3.
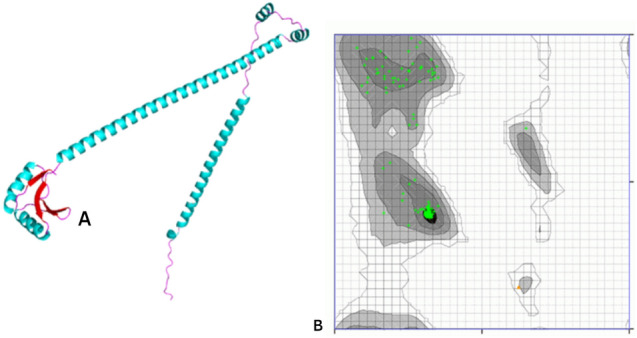
Prediction of FtsN 3D structure and its evaluation: A FtsN refined 3D structure, the cyan color represents the helices in the protein structure and red color represents the beta sheets whereas the pink color represents loops; B identification of favorable amino acid residues in FtsN protein
Identification and analysis of B cell and T cell epitopes
IEDB-AR v.2.22 predicted a total of 1,792 epitopes, whereas 72 HTL epitopes were identified using NetMHCIIpan 4.0 server. ABCPred server predicted a total of 25 linear B cell epitopes, while the Discotope server identified 224 discontinuous B cell epitopes. The IFN triggering capability of 16 predicted epitopes was predicted using the IFN epitope server. It employed SVM hybrid algorithms excluding motif to predict. Several interesting epitopes based on the criteria; 100 percent conserved among proteins have a significant association, do not overlap with human proteins, and be highly antigenic/immunogenic were selected. These CTL epitopes were analyzed for their cytokine-inducing capabilities (Interleukin-10, Interleukin-4, and IFN- γ), and a total of 55 CTL epitopes, 5 HTL epitopes, 5 linear B cell epitopes, and 12 discontinuous B cell epitopes were selected. After the conservancy analysis, eventually, 7 CTL epitopes and 2 discontinuous B cell epitopes were shortlisted as shown in Table 3. Whereas no HTL epitopes met the criteria for non-allergenicity, antigenicity, or toxicity, and none could elicit an IFN immunological response.
Table 3.
Selected CTL epitopes of from FtsN protein
| CTL epitopes | Conservancy | Toxicity | Allergenicity | Antigenicity | IL4pred | IL10pred | IFN epitope server | Alleles |
|---|---|---|---|---|---|---|---|---|
| IKALETRTV | 100.00% (21/21) | Non-toxin | Non-allergen | Antigenic | Inducer | Positive | Positive | HLA-B*08:01 |
| LETRTVPVD | 100.00% (21/21) | Non-toxin | Non-allergen | Antigenic | Inducer | Negative | Negative | HLA-B*40:01 |
| MAHRDFAAR | 100.00% (21/21) | Non-toxin | Non-allergen | Antigenic | Inducer | Positive | Positive | HLA-A*02:01 |
| RPEEVWSYI | 100.00% (21/21) | Non-toxin | Non-allergen | Antigenic | Inducer | Negative | Negative | HLA-A*11:01 |
| SVEQNMRLT | 100.00% (21/21) | Non-toxin | Non-allergen | Antigenic | Inducer | Negative | Negative | HLA-A*11:01 |
| SYIKALETR | 100.00% (21/21) | Non-toxin | Non-allergen | Antigenic | Inducer | Negative | Negative | HLA-A*24:02 |
| YIKALETRT | 100.00% (21/21) | Non-toxin | Non-allergen | Antigenic | Inducer | Negative | Negative | HLA-B*08:01 |
HLA population coverage analysis
A total of seven selected CTL epitopes with their corresponding HLA alleles were estimated for the population coverage collectively. HLA alleles vary according to ethnicity and geographical places. As a result, it has an impact on the development of an epitope-based vaccination. For selected epitopes, the analysis found cumulative coverage of 72% of the world population (Fig. 4). For the population of the Philippines, the highest coverage was reported. However, the least population coverage was found within the population of Scotland. HLA alleles are highly variable among ethnic groups and retain T cell epitope responses. T cell epitopes can bind to HLA alleles for better population coverage. For the estimation of the global distribution of HLA alleles, we used CTL and HTL epitopes. According to the results, in several geographical regions around the locations of the world, epitopes with the best coverage of their alleles were picked. According to our findings, population coverage was higher in regions in which Hi cases had been reported in the past. Furthermore, our findings revealed that specific epitopes had the potential to serve as candidates for MEV.
Fig. 4.

HLA alleles population coverage analysis of FtsN epitopes according to regions
MEV design and evaluation
All nine identified epitopes were subsequently utilized to create MEV construct. Beta-defensin was linked to the EAAAK linker to the MEV N-terminal. Beta-defensin is a 45-amino acid long adjuvant. EAAK linker boost the immune responses. AAY and GPGPG linkers were used to link other epitopes to preserve their different immunogenic activity. The final design of the vaccine contained 149 amino acids (Fig. 5).
Fig. 5.
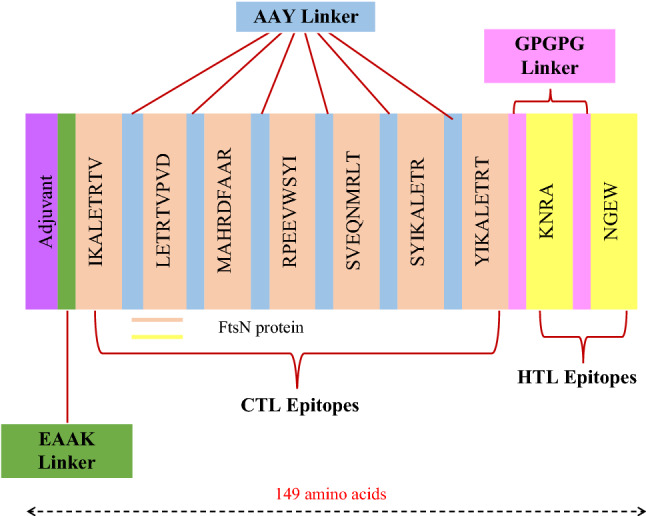
Sequence overview of the MEV construction. It has 149 amino acids, including an adjuvant (shown in purple) connected to the N-terminus of MEV via the EAAAK linker (shown in green). The AAY linker (shown in blue) was utilized to connect the CTL epitopes, whereas the GPGPG linker (in pink) connects the HTL epitopes
Physicochemical and immunogenic characterization
Immunogenic and physiochemical properties were evaluated after the vaccine design was created. The findings of evaluating the sequence homology of the created MEV were compared to the human proteome and found to have no resemblance with any part of the human proteome. The immunological analyses were then performed. It was found that the vaccine was non-allergic, non-toxic, and had a high antigenicity (0.7831 at the 0.4 percent threshold). ProtParam was used to analyze the physicochemical properties of the vaccine. The theoretical PI was 9.74 kDa while the molecular weight of the vaccination was 16,588.10 kDa. The designed vaccine has a mean half-life of 30 h in vitro, > 20 h in vivo (Yeast), and > 10 h in vivo (E. coli in-vivo). The GRAVY has been calculated to be -0.516; a negative sign indicates that the vaccine is hydrophilic. Hence, analysis revealed that FtsN protein is a potential vaccine candidate.
Structural analyses
SOPMA server was used to analyze the secondary structure of the vaccine construct (Fig. 6). The analysis revealed the overall secondary structure of the vaccine, which comprises an alpha-helix with 84 residues (56.38%), beta-strands with 8 residues (5.37%), and coils with 45 amino acids (30.20%).
Fig. 6.

SOPMA secondary structure prediction analysis of the vaccine construct. Yellow colors are indicating strands, helices are shown in pink color, and grey color lines are representing coils
Tertiary structure prediction and refinement
The ROBETTA server was utilized for tertiary structure prediction of the vaccine construct. The deep learning method called RoseTTAFold was employed for the 3D modeling of MEV (Fig. 7A). The GalaxyWEB server optimized the predicted structure. The Ramachandran plot analysis for refined structure revealed that 97.674% are in highly preferred observations, 2.326% are in preferred observations, and 0% are in the outlier region (Fig. 7B). The Z-score obtained is − 4.87 (Fig. 7C). The optimized model received a perfect score during the ERRAT quality check using SAVES. Thus, the refined model possesses good quality.
Fig. 7.
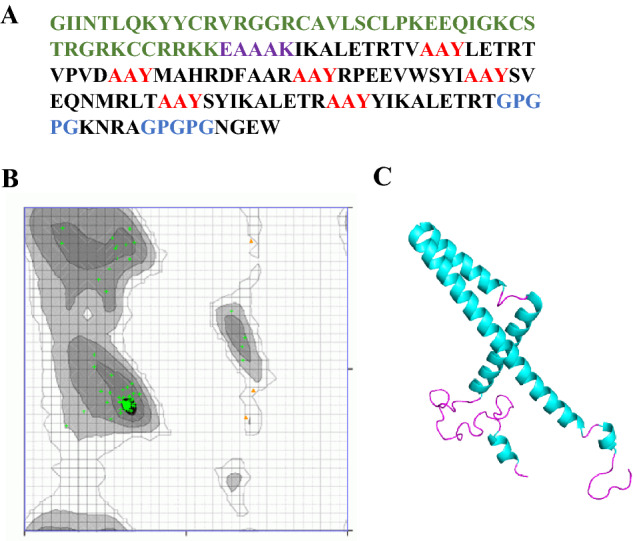
The 3D structure of the MEV structure prediction and validation: A epitopes are shown in black color in the MEV construct. The adjuvant sequence is shown in green color, EAAAK linker sequence is shown in purple color, the AAY linkers are shown in red color, and the GPGPG linkers are shown in blue colors; B identification of favorable amino acid residues using Ramachandran plot analysis; C MEV refined conformation (alpha-helix are in cyan color, and loops are shown in pink color)
Additionally, the CABS-flex 2.0 server ran 50 simulation cycles, at temperature of 1.4 °C to test the structural flexibility of MEV. Regions adjacent to the N-terminal showed the least variance among the ten final 3D conformations (Fig. 8A). Optimum residue interaction map for the last ten models was collected (Fig. 8B). Lastly, the root mean square variation (RMSF) plot revealed the variations of all amino acids (Fig. 8C). The existence of changes in the MEV structure is indicative of its high adaptability and supports its viability as a vaccine candidate.
Fig. 8.

CABS-flex 2.0 results: A cartoon representation of the top ten models; B residue contact map; C RMSF graph showing simulation-induced MEV residue variations
B cell epitopes screening
Besides making antibodies, B lymphocytes secrete cytokines, which are important for humoral immunity, and MEV should have B cell epitopes specific to its domain. A total of 26 discontinuous and 15 linear epitopes were predicted from the final MEV construct using ABCPred 2.0 and Discotope (Table 4). Discontinuous B cell epitopes of the MEV were visualized using PyMOL v1.3. The following illustration is shown in Fig. 9.
Table 4.
Final list of the linear B cell epitopes in the final MEV construct
| No | Peptide sequence | Start position | Score |
|---|---|---|---|
| 1 | LETRTGPGPGKNRAGP | 127 | 0.96 |
| 2 | DAAYMAHRDFAARAAY | 71 | 0.91 |
| 3 | PGPGKNRAGPGPGNGE | 133 | 0.89 |
| 4 | EEQIGKCSTRGRKCCR | 27 | 0.86 |
| 5 | YSYIKALETRAAYYIK | 110 | 0.83 |
| 6 | YCRVRGGRCAVLSCLP | 10 | 0.83 |
| 7 | PEEVWSYIAAYSVEQN | 88 | 0.82 |
| 8 | SVEQNMRLTAAYSYIK | 99 | 0.81 |
| 9 | AVLSCLPKEEQIGKCS | 19 | 0.81 |
| 10 | TRTVPVDAAYMAHRDF | 65 | 0.8 |
| 11 | CSTRGRKCCRRKKEAA | 33 | 0.78 |
| 12 | KIKALETRTVAAYLET | 50 | 0.75 |
| 12 | DFAARAAYRPEEVWSY | 79 | 0.72 |
| 13 | TRTVAAYLETRTVPVD | 56 | 0.67 |
| 14 | CRRKKEAAAKIKALET | 41 | 0.59 |
Fig. 9.
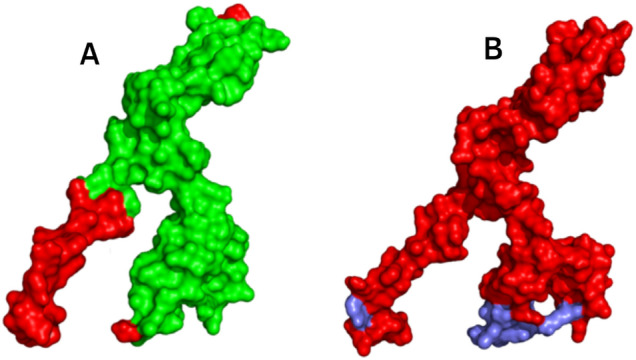
Surface representation of the MEV. A Discontinuous B cell epitopes in MEV design are highlighted in red color; B linear B cell epitopes are colored red in MEV
Protein–protein docking between TLR4 receptor and MEV
An immune response is activated as a result of suitable interaction between antigen molecules and immune receptor molecules. Consequently, the server ClusPro 2.0 is utilized for MEV protein–protein docking against the human immunological receptor TLR4. TLR4 possess the ability of eliciting an immune response in response to bacterial identification. The docking analysis revealed a significant interaction between MEV and TLR4. TLR4–MEV has a binding score of -1035.2 kcal/mol (Fig. 10).
Fig. 10.
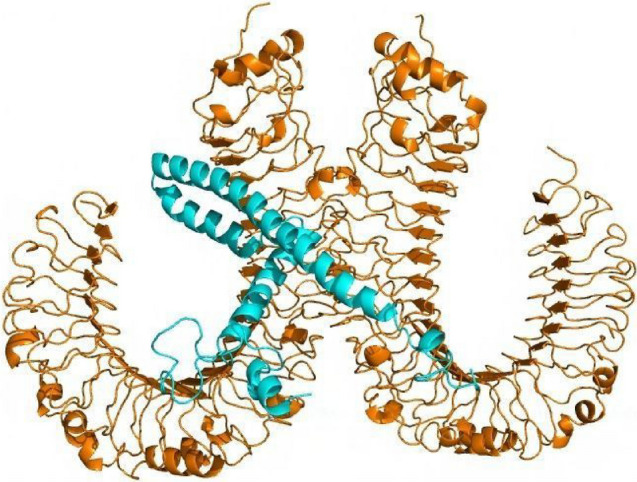
Molecular docking between the MEV and TLR4: cartoon representation of the docked TLR4–MEV complex. TLR4 is shown in orange–brown color and MEV is shown in cyan color
Interaction analysis between vaccine and TLR4
To activate immunological responsiveness, immune receptor molecules must form a suitable connection with the antigen molecule. Thus, the ClusPro v.2.0 server was utilized to execute protein–protein molecular docking of the MEV with human immune receptors TLR4. TLR4 is capable of inducing an immune response following the viral invasion. The docking analysis revealed that the MEV had robust interactions with both TLR4 chains A and B. TLR4–MEV binding score was − 1035.2 kcal cal/mol (Fig. 11A). MEV was shown to have 11 hydrogen bond interactions with TLR4 chain A and 9 hydrogen bond contacts with TLR4 chain B (Fig. 11B). Green sticks illustrate MEV residues hydrogen bonding to TLR4, whereas TLR4 residues interacting with MEV are displayed in hot-pink color stick representation.
Fig. 11.

A Interacting residues of MEV that interact with TLR4 are shown as a hot-pink color, whereas TLR4 residues that interact with MEV are shown as a green color stick representation. The hydrogen bonds are given as yellow dashed lines; B blue lines represent hydrogen bonds, red lines represent salt bridges, and orange lines represent other interactions. Different colored residues indicate amino acid properties (positive: blue color, negative: red color, aliphatic: grey color, aromatic: pink color, proline and glycine: orange color, and cystine residue: yellow color)
Immune system simulation of vaccine construction for vaccine efficacy
The immunogenic responses induced by the vaccine construct were predicted using C-ImmSim 10.1 server. The genuine immune system within the host consists of both secondary and primary immunological responses that contribute significantly to the pathogen invasion. The C-ImmSim server is utilized to perform the simulation of the host immune response to the antigen. It was observed that antigen reduces over time when passing through the secondary and primary phases. In secondary and primary responses, IgG + IgG and IgM concentrations were higher than IgM, IgG1 + IgG2, and IgG1 Fig. 12B. On the other hand, a strong cytokine reaction to the vaccine was observed which indicates that MEV may generate a successful immune response and clearance of the antigen in the host as it did in the simulation Fig. 12A.
Fig. 12.

Simulated immunological responses to MEV: A cytokine, interleukin production; B immunoglobulin synthesis and B cell isotypes after exposure to varied conditions
In-silico E. coli cloning and codon optimization
The JCAT server implemented the optimization of MEV codons. Codon optimization was executed to efficiently express and produce the MEV in the host E. coli system. According to JCAT results, MEV codons adapt due to codon usage of the E. coli K12 strain. One of the best findings was a CAI score of 1.0, with a GC content of 50.799% projected for the modified DNA sequence.
Discussion
Hi causes pneumonia, bacterial meningitis, and other serious infections, particularly among infants. These infections range from mild to serious, like bloodstream infections and ear infections (Haemophilus influenzae Infections in the H. influenzae Type b Conjugate Vaccine Era 2022). According to the World Health Organization, Hi causes 3 million cases of serious disease and 400,000 deaths worldwide each year (Haemophilus influenzae Infections in the H. influenzae Type b Conjugate Vaccine Era 2022). The majority of incidents occur in unimmunized children and in economically underdeveloped countries. This global health issue could be alleviated with a very effective vaccine. However, there is currently no vaccination available for Hi. One of the most basic and safest methods of preventing infectious diseases worldwide is vaccination (Vitovski et al. 2002; Potts et al. 2019; Haemophilus influenzae Infections in the H. influenzae Type b Conjugate Vaccine Era 2022). The current research focuses on subunit vaccinations in contrast to vaccines generated from the entire pathogen. Different immunogenic parts of pathogens help to build subunit vaccines; therefore, they can induce a strong immune response. The successful development of potent MEVs remains a considerable challenge due to difficulties in selecting epitopes, and adequate antigens (Potts et al. 2019; Fleischmann et al. 1995). As a result, forecasting relevant epitopes in an antigen is a critical step in constructing a MEV (Cripps et al. 2002).
Furthermore, due to the increase in antibiotic-resistant pathogenic bacteria and the limitations of conventional vaccines, it is important to focus on new strategies (Gilsdorf 1987). As a new approach, Multi-Epitope Vaccines (MEV) have been made using immunoinformatics methods, such as predicting epitopes with reliable servers, attaching epitopes with the appropriate linkers and adjuvants, and evaluating immunological, physicochemical, and structural properties with bioinformatics servers and tools (Qamar et al. 2021). These are the basic steps in making MEVs. MEVs stand out from other types of vaccines because they are more cost effective, safe, less time consuming to design, use natural adjuvants, and have good preclinical evaluations. So, MEVs are promising vaccines against invasive Hi infections (Designing a chimeric subunit vaccine for influenza virus, based on HA2, M2e and CTxB: a bioinformatics study 2022; Jalalvand and Riesbeck 2018, p.).
In this study, we aimed at utilizing the potential immunogenic epitopes obtained from the FTsN protein of Hi. Several computational tools were employed to create MEV that provokes both primary and secondary immune responses. IEDB-AR v.2.22, NetMHCIIpan 4.0, and ABCPred servers were used to predict the CTL, HTL, and LBL epitopes, respectively, to find potential vaccine candidates based on the parameters of toxicity, conservancy, antigenicity, allergenicity. B lymphocytes make antibodies, HTL causes both primary and secondary immune responses, and CTL stops the spread of viruses and bacteria by making certain cytokines and killing virally infected cells. Filters such as the epitopes that are immunogenic, antigenic, and non-toxic, have overlapping CTL and HTL epitopes, and most critically, have epitope conservancy. BLAST searches revealed that all of the epitopes were non-self, as they displayed no resemblance to the human proteome. A final MEV includes both the B cell and T cell epitopes for encouraging a complete system of immune reaction. This may help researchers to use the anticipated epitopes in the current study to computationally build therapeutic antibodies against Hi. Rather than depending on comprehensive functional screening from large libraries, they will be able to rationally create functional therapeutic antibodies.
The epitopic sequences linked with adjuvant as well as appropriate linkers make up the vaccine design. The adjuvant (beta-defensin) acts as an antibacterial and immunomodulator. EAAAK linkers which form helix were used to connect the adjuvant to the first CTL epitope of the FtsN protein. The EAAAK linker boosts bifunctional catalytic activity while decreasing protein toxicity. HTL epitopes were linked with GPGPG while remaining CTL epitopes were linked with AAY linkers to enhance immunogenicity. The use of linkers brings the pH of the construct closer to that of the physiological range. The final MEV design contains 149 amino acids and is non-allergenic, antigenic, and non-toxic (16,588.10 KDa). The final design was extremely basic (PI 9.74), suggesting it may enable prolonged normal pH contact. Furthermore, the expected instability index (25.92) represents protein stability even after expression, which increases its use. In addition, the calculated aliphatic index (A1) indicates its stability. Hydrophilicity was revealed through a negative GRAVY value (− 0.516), indicating water molecules form strong interactions with MEV. The 3D MEV conformation was also developed utilizing the Robetta server. Using the SAVES server, structure validation tools prove the accuracy of the predicted model with a 100-quality score. A higher score of − 155.51 predicted using AggreScan3D suggested a high solubility level of MEV. Whereas, the Ramachandran plot analysis revealed that 97.674% are in highly preferred observations. Protein–protein docking was performed between TLR4 immune receptor and MEV to explore the efficacy of this vaccine. If a vaccination interacts effectively with the target immune cells, the host creates an efficient immunological response. As a result, the interaction between the MEV and the immunological receptor TLR4 was investigated with a molecular docking approach. Thus, molecular docking analysis revealed that vaccine and receptor have a strong and stable binding with many hydrogen bond interactions.
In the current research, the immunoinformatic strategy for vaccine designing was utilized to generate MEV capable of eliciting immune responses against Hi. We believe that this vaccination will elicit both primary and secondary immune responses. The interactions and binding patterns between the TL4 and the MEV were highly stable. We may conclude that our vaccine design is capable of eliciting an immunological response in the real life. However, the real immune stimulation potential is needed to be validated to combat Hi, and further wet laboratory investigations are crucial to check their efficacy and safety.
Conclusion
Infection caused by Hi has remained mysterious in terms of treatment and underlying immunological concepts. Hi is now a global disease and issue. There are no proper treatments available for Hi that can treat the infection effectively; however, there are treatments available for management purposes. As such, there is no proper cure available for Hi. Multiple antibacterial medications have been investigated against Hi, but no feasible results have been found. These limitations led to the concept of developing a subunit MEV humoral vaccine in this study. The development is centered around bioinformatics and immunoinformatics analysis that can identify key vaccine candidates to generate a cellular immune response. MEV model was proposed using a combination of computational biology and immunology data to help design a possible vaccine against Hi infection. The analysis elucidates that the proposed vaccine model would be effective against the infection and in Hi treatment and research. This research employs a comprehensive computational approach for developing a subunit vaccine model. Therefore, additional experimental and laboratory research is required to test and analyze the specificity, efficacy, and safety of a vaccine.
Data availability
All data for this work are included in manuscript body.
Footnotes
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
References
- Bjellqvist B, et al. The focusing positions of polypeptides in immobilized pH gradients can be predicted from their amino acid sequences. Electrophoresis. 1993;14(1):1023–1031. doi: 10.1002/elps.11501401163. [DOI] [PubMed] [Google Scholar]
- Bui H-H, Sidney J, Dinh K, Southwood S, Newman MJ, Sette A. Predicting population coverage of T cell epitope-based diagnostics and vaccines. BMC Bioinform. 2006;7:153. doi: 10.1186/1471-2105-7-153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Butler DF, Myers AL. Changing epidemiology of Haemophilus influenzae in children. Infect Dis Clin. 2018;32(1):119–128. doi: 10.1016/j.idc.2017.10.005. [DOI] [PubMed] [Google Scholar]
- Calis JJA, et al. Properties of MHC class I presented peptides that enhance immunogenicity. PLoS Comput Biol. 2013;9(10):10. doi: 10.1371/journal.pcbi.1003266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen D, et al. Microbial virulence, molecular epidemiology and pathogenic factors of fluoroquinolone-resistant Haemophilus influenzae infections in Guangzhou, China. Ann Clin Microbiol Antimicrob. 2018;17(1):41. doi: 10.1186/s12941-018-0290-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Colovos C, Yeates TO. Verification of protein structures: patterns of nonbonded atomic interactions. Protein Sci Publ Protein Soc. 1993;2(9):9. doi: 10.1002/pro.5560020916. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cripps AW, Foxwell R, Kyd J. Challenges for the development of vaccines against Haemophilus influenzae and Neisseria meningitidis. Curr Opin Immunol. 2002;14(5):553–557. doi: 10.1016/S0952-7915(02)00373-4. [DOI] [PubMed] [Google Scholar]
- Designing a chimeric subunit vaccine for influenza virus, based on HA2, M2e and CTxB: a bioinformatics study|BMC Molecular and Cell Biology|Full Text. https://bmcmolcellbiol.biomedcentral.com/articles/10.1186/s12860-020-00334-6 Accessed 20 Sep 2022 [DOI] [PMC free article] [PubMed]
- Dhanda SK, Vir P, Raghava GPS. Designing of interferon-gamma inducing MHC class-II binders. Biol Direct. 2013;8:30. doi: 10.1186/1745-6150-8-30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dhanda SK, Gupta S, Vir P, Raghava GPS. Prediction of IL4 inducing peptides. Clin Dev Immunol. 2013;2013:263952. doi: 10.1155/2013/263952. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Doytchinova IA, Flower DR. VaxiJen: a server for prediction of protective antigens, tumour antigens and subunit vaccines. BMC Bioinform. 2007;8(1):1. doi: 10.1186/1471-2105-8-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fleischmann RD, et al. Whole-genome random sequencing and assembly of Haemophilus influenzae Rd. Science. 1995;269(5223):496–512. doi: 10.1126/science.7542800. [DOI] [PubMed] [Google Scholar]
- Gasteiger E, et al. Protein identification and analysis tools on the ExPASy server. In: Walker JM, et al., editors. The proteomics protocols handbook. Totowa: Humana Press; 2005. pp. 571–607. [Google Scholar]
- Geourjon C, Deléage G. SOPMA: significant improvements in protein secondary structure prediction by consensus prediction from multiple alignments. Comput Appl Biosci CABIOS. 1995;11(6):6. doi: 10.1093/bioinformatics/11.6.681. [DOI] [PubMed] [Google Scholar]
- Gilsdorf JR. Haemophilus influenzae non-type b infections in children. Am J Dis Child. 1987;141(10):1063–1065. doi: 10.1001/archpedi.141.10.1063. [DOI] [PubMed] [Google Scholar]
- Grote A, et al. JCat: a novel tool to adapt codon usage of a target gene to its potential expression host. Nucleic Acids Res. 2005 doi: 10.1093/nar/gki376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gupta S, et al. In silico approach for predicting toxicity of peptides and proteins. PLoS ONE. 2013;8(9):9. doi: 10.1371/journal.pone.0073957. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haemophilus influenzae infections in the H. influenzae type b conjugate vaccine era | J Clin Microbiol. https://journals.asm.org/doi/10.1128/JCM.05476-11 accessed 20 Sep 2022 [DOI] [PMC free article] [PubMed]
- Hoover DM, Wu Z, Tucker K, Lu W, Lubkowski J. Antimicrobial characterization of human beta-defensin 3 derivatives. Antimicrob Agents Chemother. 2003;47(9):9. doi: 10.1128/AAC.47.9.2804-2809.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huska B, Kubinec C, Sadarangani M, Ulanova M. Seroprevalence of IgG and IgM antibodies to Haemophilus influenzae type a in Canadian children. Vaccine. 2022;40(8):1128–1134. doi: 10.1016/j.vaccine.2022.01.029. [DOI] [PubMed] [Google Scholar]
- Immunoinformatics Approach for Epitope-Based Peptide Vaccine Design and Active Site Prediction against Polyprotein of Emerging Oropouche Virus. https://www.hindawi.com/journals/jir/2018/6718083/ accessed 20 Sep 2022 [DOI] [PMC free article] [PubMed]
- Jalalvand F, Riesbeck K. Update on non-typeable Haemophilus influenzae-mediated disease and vaccine development. Expert Rev Vaccines. 2018;17(6):503–512. doi: 10.1080/14760584.2018.1484286. [DOI] [PubMed] [Google Scholar]
- Kilian M, Thomsen B, Petersen TE, Bleeg H. Molecular biology of Haemophilus influenzae IgA1 proteases. Mol Immunol. 1983;20(9):1051–1058. doi: 10.1016/0161-5890(83)90046-9. [DOI] [PubMed] [Google Scholar]
- Kim DE, Chivian D, Baker D. Protein structure prediction and analysis using the Robetta server. Nucleic Acids Res. 2004 doi: 10.1093/nar/gkh468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ko J, Park H, Heo L, Seok C. GalaxyWEB server for protein structure prediction and refinement. Nucleic Acids Res. 2012 doi: 10.1093/nar/gks493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kringelum JV, Lundegaard C, Lund O, Nielsen M. Reliable B cell epitope predictions: impacts of method development and improved benchmarking. PLoS Comput Biol. 2012;8(12):12. doi: 10.1371/journal.pcbi.1002829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kurcinski M, Oleniecki T, Ciemny MP, Kuriata A, Kolinski A, Kmiecik S. CABS-flex standalone: a simulation environment for fast modeling of protein flexibility. Bioinformatics. 2019;35(4):694–695. doi: 10.1093/bioinformatics/bty685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuriata A, et al. CABS-flex 2.0: a web server for fast simulations of flexibility of protein structures. Nucleic Acids Res. 2018;46(W1):W1. doi: 10.1093/nar/gky356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laskowski RA. PDBsum new things. Nucleic Acids Res. 2009 doi: 10.1093/nar/gkn860. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Livingston B, et al. A rational strategy to design multiepitope immunogens based on multiple Th lymphocyte epitopes. J Immunol. 2002;168(11):5499–5506. doi: 10.4049/jimmunol.168.11.5499. [DOI] [PubMed] [Google Scholar]
- López-López N, Gil-Campillo C, Díez-Martínez R, Garmendia J. Learning from –omics strategies applied to uncover Haemophilus influenzae host-pathogen interactions: current status and perspectives. Comput Struct Biotechnol J. 2021;19:3042–3050. doi: 10.1016/j.csbj.2021.05.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lovell SC, et al. Structure validation by Calpha geometry: phi, psi and Cbeta deviation. Proteins. 2003;50(3):3. doi: 10.1002/prot.10286. [DOI] [PubMed] [Google Scholar]
- Mahram A, Herbordt MC (2010) Fast and accurate NCBI BLASTP: acceleration with multiphase FPGA-based prefiltering. In: Proceedings of the 24th ACM International Conference on Supercomputing, New York, NY, USA, 2010, pp. 73–82. 10.1145/1810085.1810099
- McGuffin LJ, Bryson K, Jones DT. The PSIPRED protein structure prediction server. Bioinforma Oxf Engl. 2000;16(4):4. doi: 10.1093/bioinformatics/16.4.404. [DOI] [PubMed] [Google Scholar]
- Mirzaei R, et al. The importance of intracellular bacterial biofilm in infectious diseases. Microb Pathog. 2020;147:104393. doi: 10.1016/j.micpath.2020.104393. [DOI] [PubMed] [Google Scholar]
- Mugunthan SP, Harish MC. Multi-epitope-Based vaccine designed by targeting cytoadherence proteins of Mycoplasma gallisepticum. ACS Omega. 2021;6(21):13742–13755. doi: 10.1021/acsomega.1c01032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nagpal G, et al. Computer-aided designing of immunosuppressive peptides based on IL-10 inducing potential. Sci Rep. 2017;7:42851. doi: 10.1038/srep42851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nain Z, Karim MM, Sen MK, Adhikari UK. Structural basis and designing of peptide vaccine using PE-PGRS family protein of Mycobacterium ulcerans—an integrated vaccinomics approach. Mol Immunol. 2020;120:146–163. doi: 10.1016/j.molimm.2020.02.009. [DOI] [PubMed] [Google Scholar]
- NCBI Resource Coordinators Database resources of the National Center for Biotechnology Information. Nucleic Acids Res. 2013;41(D1):D8–D20. doi: 10.1093/nar/gks1189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pandey R, et al. Exploring dual inhibitory role of febrifugine analogues against Plasmodium utilizing structure based virtual screening and molecular dynamic simulation. J Biomol Struct Dyn. 2016 doi: 10.1080/07391102.2016.1161560. [DOI] [PubMed] [Google Scholar]
- Pinto M, et al. Insights into the population structure and pan-genome of Haemophilus influenzae. Infect Genet Evol. 2019;67:126–135. doi: 10.1016/j.meegid.2018.10.025. [DOI] [PubMed] [Google Scholar]
- Potts CC, et al. Genomic characterization of Haemophilus influenzae: a focus on the capsule locus. BMC Genomics. 2019;20(1):733. doi: 10.1186/s12864-019-6145-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prediction of residues in discontinuous B cell epitopes using protein 3D structures—Haste Andersen—2006—Protein Science—Wiley Online Library.” https://onlinelibrary.wiley.com/doi/full/10.1110/ps.062405906 accessed 20 Sep 2022 [DOI] [PMC free article] [PubMed]
- Pymol: an open-source molecular graphics tool—ScienceOpen. https://www.scienceopen.com/document?vid=4362f9a2-0b29-433f-aa65-51db01f4962f accessed 20 Sep 2022
- Qamar M, et al. Designing multi-epitope vaccine against Staphylococcus aureus by employing subtractive proteomics, reverse vaccinology and immuno-informatics approaches. Comput Biol Med. 2021 doi: 10.1016/j.compbiomed.2021.104389. [DOI] [PubMed] [Google Scholar]
- Reynisson B, Barra C, Kaabinejadian S, Hildebrand WH, Peters B, Nielsen M. Improved prediction of MHC II antigen presentation through integration and motif deconvolution of mass spectrometry MHC eluted ligand data. J Proteome Res. 2020;19(6):6. doi: 10.1021/acs.jproteome.9b00874. [DOI] [PubMed] [Google Scholar]
- Saadati A, Kholafazad-Kordasht H, Ehsani M, Hasanzadeh M, Seidi F, Shadjou N. An innovative flexible and portable DNA based biodevice towards sensitive identification of Haemophilus influenzae bacterial genome: a new platform for the rapid and low cost recognition of pathogenic bacteria using point of care (POC) analysis. Microchem J. 2021;169:106610. doi: 10.1016/j.microc.2021.106610. [DOI] [Google Scholar]
- Saha S, Raghava GPS. AlgPred: prediction of allergenic proteins and mapping of IgE epitopes. Nucleic Acids Res. 2006 doi: 10.1093/nar/gkl343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saha S, Raghava GPS. Prediction of continuous B cell epitopes in an antigen using recurrent neural network. Proteins. 2006;65(1):1. doi: 10.1002/prot.21078. [DOI] [PubMed] [Google Scholar]
- Serwecińska L. Antimicrobials and antibiotic-resistant bacteria: a risk to the environment and to public health. Water. 2020;12(12):12. doi: 10.3390/w12123313. [DOI] [Google Scholar]
- Slack M, et al. Haemophilus influenzae type b disease in the era of conjugate vaccines: critical factors for successful eradication. Expert Rev Vaccines. 2020;19(10):903–917. doi: 10.1080/14760584.2020.1825948. [DOI] [PubMed] [Google Scholar]
- Soeters HM, et al. Current epidemiology and trends in invasive Haemophilus influenzae disease—United States, 2009–2015. Clin Infect Dis. 2018;67(6):881–889. doi: 10.1093/cid/ciy187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suga S, et al. A nationwide population-based surveillance of invasive Haemophilus influenzae diseases in children after the introduction of the Haemophilus influenzae type b vaccine in Japan. Vaccine. 2018;36(38):5678–5684. doi: 10.1016/j.vaccine.2018.08.029. [DOI] [PubMed] [Google Scholar]
- The ClusPro web server for protein–protein docking|Nature Protocols. https://www.nature.com/articles/nprot.2016.169 accessed 20 Sep 2022 [DOI] [PMC free article] [PubMed]
- Vitovski S, Dunkin KT, Howard AJ, Sayers JR. Nontypeable Haemophilus influenzae in carriage and disease a difference in IgA1 protease activity levels. JAMA. 2002;287(13):1699–1705. doi: 10.1001/jama.287.13.1699. [DOI] [PubMed] [Google Scholar]
- Wen S, Feng D, Chen D, Yang L, Xu Z. Molecular epidemiology and evolution of Haemophilus influenzae. Infect Genet Evol. 2020;80:104205. doi: 10.1016/j.meegid.2020.104205. [DOI] [PubMed] [Google Scholar]
- Wiederstein M, Sippl MJ. ProSA-web: interactive web service for the recognition of errors in three-dimensional structures of proteins. Nucleic Acids Res. 2007 doi: 10.1093/nar/gkm290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilson C, Mitchelmore P, Brown A. Culture-independent multilocus sequence typing (MLST) screening for Haemophilus influenzae cross-infection in non-cystic fibrosis bronchiectasis (NCFB) Access Microbiol. 2020;2(2):179. doi: 10.1099/acmi.fis2019.po0173. [DOI] [PubMed] [Google Scholar]
- Zhang Q, et al. Immune epitope database analysis resource (IEDB-AR) Nucleic Acids Res. 2008 doi: 10.1093/nar/gkn254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang J, Zhao X, Sun P, Gao B, Ma Z. Conformational B cell epitopes prediction from sequences using cost-sensitive ensemble classifiers and spatial clustering. BioMed Res Int. 2014;2014:689219. doi: 10.1155/2014/689219. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
All data for this work are included in manuscript body.