

Abstract
Objective
The aim of this study was to characterize the safety and efficacy of filgotinib, lanraplenib and tirabrutinib in patients with active SS.
Methods
This multicentre, double-blind study randomized patients with active primary or secondary SS [EULAR SS disease activity index (ESSDAI) ≥5) to receive filgotinib 200 mg (Janus kinase-1 inhibitor), lanraplenib 30 mg (spleen tyrosine kinase inhibitor), tirabrutinib 40 mg (Bruton’s tyrosine kinase inhibitor), or placebo. The composite primary end point was the week-12 proportion of patients fulfilling protocol-specified improvement criteria (based on CRP and SS-related symptoms). The EULAR SS patient-reported index (ESSPRI) and the ESSDAI change from baseline (CFB) were secondary end points. Exploratory end points included disease-related biomarkers. Treatment-emergent adverse events (AEs) represented safety outcomes.
Results
The mean of the baseline ESSDAI was 10.1, and of ESSPRI was 6.2 in the 150 patients who were treated; 125 completed the 24-week placebo-controlled treatment period. At week 12, 43.3% of the filgotinib group achieved the primary end point (P = 0.17 vs placebo) vs 42.3% (P = 0.16), 34.7% (P = 0.33), and 26.7% of lanraplenib, tirabrutinib, and placebo groups, respectively. Neither secondary end point was met. Biomarker reductions included immunoglobulins classically associated with SS disease activity. Filgotinib ESSDAI CFB appeared more pronounced in subgroups with baseline ESSDAI ≥14 or without DMARDs/CSs. Most AEs were Grade 1 or 2.
Conclusion
Three drugs with disparate mechanisms were tested, but no significant differences vs placebo in primary or secondary end points were observed. These results may be considered hypothesis-generating, given the drug tolerability, subgroup analysis, and biomarker findings.
Trial registration
ClinicalTrials.gov, https://clinicaltrials.gov, NCT03100942.
Keywords: Sjögren's syndrome, randomized trial, safety, efficacy, lanraplenib, filgotinib, tirabrutinib
Rheumatology key messages.
Filgotinib, lanraplenib and tirabrutinib were well tolerated in patients with Sjögren’s syndrome.
Neither the primary end point nor either secondary end point was met.
Biomarker assays suggested signs of biologic activity for tirabrutinib and filgotinib.
Introduction
SS is a systemic autoimmune disease primarily affecting exocrine glands, leading to symptoms of dry eyes and mouth [1]. Systemic manifestations are common, and health-related quality of life can be severely impacted [2, 3]. Sjögren’s may manifest alone or alongside another autoimmune disease [1]. SS is considered to be among the most common autoimmune disorders; however, due in part to varying diagnostic criteria, prevalence estimates across nations vary considerably (from 0.03% to as high as 2.7%) [1, 4].
To date, no disease-modifying treatment has been approved for the treatment of SS [5]. The results of small, open-label or controlled studies evaluating DMARDs or biologics used in related autoimmune diseases have been mixed [6–11]. Larger and controlled clinical trials have been infrequent and thus far have not identified an effective immunomodulatory treatment for the systemic or glandular manifestations [11–13]. Thus, treatment of SS is typically determined by symptoms; modalities may include saliva substitution for severe oral dryness, artificial tears for first-line ocular dryness, and analgesics for musculoskeletal pain. An unmet medical need for novel therapies persists [14].
In SS patients, proinflammatory cytokines, including Type I and Type II interferons, are overexpressed in glandular tissue and in the peripheral blood, and the Janus kinase (JAK)/signal transducer and activator of transcription proteins (STAT) pathway plays a pivotal role in their signal transduction [15, 16]. Two recent studies demonstrated an increased expression of both Bruton’s tyrosine kinase (BTK) mRNA and protein in B cells of patients with SS compared with B cells from healthy controls; systemic B cell hyperreactivity is a hallmark of SS. Additionally, spleen tyrosine kinase (SYK) and BTK play a key role in B cell receptor signalling [17, 18]. Therefore, JAK/STAT, SYK and BTK appear to be relevant therapeutic targets to evaluate in potential treatments for SS [19]. Filgotinib is a once-daily, oral JAK-1 preferential inhibitor approved in Japan and Europe for the treatment of moderately to severely active RA [20, 21]. Lanraplenib is a potent and selective oral inhibitor of SYK that is in development for the treatment of inflammatory and autoimmune diseases [22]. Tirabrutinib is a potent and selective inhibitor of BTK under development for the treatment of B cell malignancies and inflammatory diseases and is approved for the treatment of recurrent or refractory primary CNS lymphoma in Japan [23]. Here, we present the results of a Phase 2 study evaluating the safety and efficacy of filgotinib, lanraplenib and tirabrutinib in adult patients with active primary and secondary SS; the study also examined mechanistic effects via biomarkers [15, 24–26].
Methods
Study design
This multicentre, global, randomized, double-blind, placebo-controlled, Phase 2 trial was conducted at 35 study centres in the USA, 8 in Spain, 5 in the UK, and 3 in Poland. Patients were screened between 1 May 2017 and 2 October 2019.
All patients provided written informed consent, and all study procedures observed international scientific and ethical standards, including the International Council for Harmonization guideline for Good Clinical Practice and the original principles embodied in the Declaration of Helsinki. This study is registered in the ClinicalTrials.gov database, identification code NCT03100942. Ethics committees are listed in Supplementary Table S1, available at Rheumatology online.
Study participants
Eligible patients were aged 18–75 years and had active SS, either primary or associated with a concomitant systemic autoimmune disease, with EULAR SS disease activity index (ESSDAI) score ≥5 (representing moderately to severely active disease) [27] and seropositivity for SSA and/or SSB based on the American–European Consensus Group classification. Patients who had previously received B cell–depleting therapies were required to have documented return of CD19 cells. Baseline dosages of concomitant chronic medications, including CSs, DMARDs or immunomodulators, were stable and were continued at the stable dosage for at least the first 12 weeks of the study, although dose adjustments were permitted for toxicities. Following primary end point collection at week 12, dose adjustments of concomitant medications were permitted at the investigator’s discretion. Key exclusion criteria are detailed in the supplementary data available at Rheumatology online.
Interventions
Patients were randomized 1:1:1:1 in double-blind fashion to receive filgotinib 200 mg, lanraplenib 30 mg, tirabrutinib 40 mg, or placebo, each administered once daily. Randomization was stratified by concurrent use of immunomodulatory drugs at baseline (CSs or conventional synthetic DMARDs), and the sum of haematologic and biological component scores of the ESSDAI obtained at screening (combined score <2 or ≥2). Filgotinib, lanraplenib and tirabrutinib groups were dosed for up to 48 weeks (W), and patients returned for a follow-up 4 W after their last visit. The placebo group was dosed for 24 W, then re-randomized in a blinded fashion 1:1:1 to the other treatment groups for 24 W without further stratification (Fig. 1A). This article presents study results of the 24-W placebo-controlled period.
Fig. 1.

Study design and disposition
(A) Study schema. After screening, eligible patients were randomized 1:1:1:1 in a blinded fashion to receive FIL 200 mg QD (n = 38), LANRA 30 mg QD (n = 37), TIRA 40 mg QD (n = 39) or PBO (n = 36) for 24 weeks. Initial randomization was stratified by concurrent use of systemic CSs or csDMARDs and by ESSDAI haematologic + biological domain score <2 or ≥2. The primary end point was assessed at W12 and secondary end points at W12 and W24. The FIL, LANRA and TIRA groups continued treatment until W48. At W24, the PBO group was re-randomized 1:1:1 into other treatment groups until W48. All patients had 4-week follow-up at W52. (B) Study disposition by treatment phase. One hundred and fifty-two patients were randomized; 150 patients received at least one dose of a study drug and were included in the safety analysis set. Thirty-four (89.5%) and 30 (78.9%) of the patients in the FIL group completed treatment at W12 and W24, respectively. Thirty (81.1%) and 27 (73.0%) of the patients in the LANRA group completed treatment at W12 and W24, respectively. Thirty-six (92.3%) of the patients in the TIRA group completed treatment at W12 and W24. Thirty-four (94.4%) and 32 (88.9%) of the patients in the PBO group completed treatment at W12 and W24, respectively. BIO: biological component score of ESSDAI; csDMARD: conventional synthetic DMARD; ESSDAI: EULAR SS disease activity index; FIL: filgotinib; HEM: haematologic component score of ESSDAI; LANRA: lanraplenib; PBO: placebo; QD: once daily; TIRA: tirabrutinib; W: week.
End points and assessments
Primary end point
The primary efficacy end point (proportion of patients fulfilling protocol-specified response criteria at W12 vs baseline) was a composite of three components. The high-sensitivity CRP (hsCRP) response component was defined as ≥20% improvement in patients with high baseline hsCRP [≥1.5 × upper limit of normal (ULN)], while response in patients without elevated baseline hsCRP required hsCRP to remain at <1.5 × ULN. Additionally, primary end point response in patients with or without high baseline hsCRP required ≥20% improvement in at least three of five patient-reported, SS-related symptom assessments by visual analogue scale (VAS; patients’ assessments of global disease, pain, oral dryness, ocular dryness, and fatigue) as well as no worsening (i.e. no increase of >30 mm from baseline) of any of these VAS-measured symptoms.
Secondary and exploratory end points
Secondary end points included change from baseline in ESSDAI and in EULAR SS patient-reported index (ESSPRI) scores at W12 and W24. Exploratory efficacy end points included Schirmer’s test (mm/5 min) up to W24, salivary flow (g/min) up to W24 (unstimulated and stimulated), treatment response on specific ESSDAI domains, and ESSDAI score change from baseline in subgroups of patients.
Exploratory biomarker-related end points
Percentage change from baseline was calculated for selected peripheral biomarkers for each patient at W4, W12, and W24. IgA, IgG, IgM, RF and CRP were measured in Covance central laboratory panels; B cell and plasma cell subsets were measured in the Covance flow cytometry laboratory. Sample measurements below the lower limit of quantification (LLOQ) were excluded, and all samples for a given assay and patient were excluded when baseline measurements were below LLOQ. Methods of interferon signature assessments are described in the Supplementary Materials available at Rheumatology online.
Safety assessments
Safety assessments included recording of adverse events (AEs) and findings from clinical laboratory analyses, vital sign measurements, ECGs or physical examinations. Treatment-emergent adverse events (TEAEs) include events that began on the start date of the study drug to ≤30 days after the last day of the study drug. TEAEs were summarized by severity (Common Terminology Criteria for Adverse Events, version 4.03) and causality. TEAEs of interest were identified using either the Standardized Medical Dictionary for Regulatory Activities Queries or Medical Search Terms. These included all infections, serious infections, infections of special interest (Herpes zoster, active Mycobacteriumtuberculosis, opportunistic infections, and hepatitis B or C infections), venous thromboembolic events (VTEs, e.g. pulmonary embolism and deep vein thrombosis), malignancies, gastrointestinal perforations, elevations of liver transaminases, serious major adverse cardiovascular events (MACEs), bleeding or haemorrhage, cytopenias, non-infectious diarrhoea, and renal toxicity. VTEs and MACE events were not adjudicated but collected as reported.
The safety analysis set included all patients who received at least one dose of a study drug, according to the treatment received. Safety was reported up to W24 of the study.
Statistical methods and data analysis
Baseline demographics and patient characteristics were summarized with descriptive statistics. The primary efficacy end point in each active treatment group was compared with placebo using a superiority test at the two-sided 0.05 level. The analyses were performed using the Cochran–Mantel–Haenszel test adjusted for randomization stratification factors. This analysis was based on the treatment policy estimand, with missing values imputed using a multiple-imputation method. Secondary continuous end points were analysed using a mixed-effects model for repeated measures (MMRM), with treatment, randomization stratification factors, visit, and treatment-by-visit interaction as fixed effects, patients as a random effect, and the respective baseline value (ESSDAI or ESSPRI score) as a covariate. Each of the four end points (ESSDAI and ESSPRI scores at W12 and W24) were tested separately at the two-sided 0.05 level. Secondary end point analyses were not adjusted for multiplicity, and missing data were not imputed. Nominal P values were provided for exploratory continuous end points using an MMRM similar to that used for the secondary end points. The primary analysis population for efficacy assessments was the full analysis set, which consisted of all randomized and treated patients, according to the randomized treatment. Results of subgroup analyses (ESSDAI changes in patients with baseline ESSDAI ≤14 or without concomitant DMARDs/CSs) were summarized descriptively. Statistical analysis of biomarker data is discussed in the supplementary data available at Rheumatology online.
Results
Baseline patient population and characteristics
In total, 152 patients with SS were randomized, and 150 received ≥1 dose of a study drug (38, 37, 39 and 36 in the filgotinib, lanraplenib, tirabrutinib and placebo groups, respectively). All treatment groups had >80% of patients complete the study through W12, and >70% complete through W24 (Fig. 1B).
Table 1 shows baseline demographics and patient characteristics. A majority of patients were female (97.3%) and white (84.7%); the mean age was 54.4 years. Thirty-four percent of patients had a concomitant autoimmune disease at baseline, including 25.3% with SLE and/or RA. The mean ESSDAI score across the study population was 10.1, and 68.7% of patients were using conventional synthetic DMARDs or CSs at baseline. Only one patient in the lanraplenib group and two in the placebo group had baseline hsCRP ≥1.5 × ULN.
Table 1.
Baseline demographics and patient characteristics
| N (%) unless noted | FIL 200 mg (n = 38) | LANRA 30 mg (n = 37) | TIRA 40 mg (n = 39) | PBO (n = 36) | Total (N = 150) |
|---|---|---|---|---|---|
| Age, mean (s.d.), years | 52.2 (10.5) | 56.2 (9.7) | 55.8 (10.1) | 53.2 (10.3) | 54.4 (10.2) |
| ≥50 years | 25 (65.8) | 27 (73.0) | 29 (74.4) | 26 (72.2) | 107 (71.3) |
| <50 years | 13 (34.2) | 10 (27.0) | 10 (25.6) | 10 (27.8) | 43 (28.7) |
| Sex | |||||
| Female | 38 (100) | 36 (97.3) | 37 (94.9) | 35 (97.2) | 146 (97.3) |
| Race | |||||
| White | 32 (84.2) | 31 (83.8) | 34 (87.2) | 30 (83.3) | 127 (84.7) |
| Black | 5 (13.2) | 5 (13.5) | 4 (10.3) | 5 (13.9) | 19 (12.7) |
| Asian | 1 (2.6) | 0 | 1 (2.6) | 0 | 2 (1.3) |
| American Indian or Alaska native | 0 | 0 | 0 | 1 (2.8) | 1 (0.7) |
| Other | 0 | 1 (2.7) | 0 | 0 | 1 (0.7) |
| Ethnicity | |||||
| Not Hispanic or Latino | 34 (89.5) | 31 (83.8) | 38 (97.4) | 30 (83.3) | 133 (88.7) |
| Hispanic or Latino | 4 (10.5) | 6 (16.2) | 1 (2.6) | 6 (16.7) | 17 (11.3) |
| Duration of SS, mean (s.d.) | 5.3 (6.9) | 9.4 (9.4) | 8.1 (6.2) | 8.2 (8.0) | 7.7 (7.8) |
| Concomitant autoimmune diseasea | 10 (26.3) | 14 (37.8) | 14 (35.9) | 13 (36.1) | 51 (34.0) |
| SLE or RA | 8 (21.1) | 12 (32.4) | 8 (20.5) | 10 (27.8) | 38 (25.3) |
| SLE | 4 (10.5) | 8 (21.6) | 4 (10.3) | 6 (16.7) | 22 (14.7) |
| RA | 5 (13.2) | 7 (18.9) | 5 (12.8) | 6 (16.7) | 23 (15.3) |
| ESSDAI, mean (s.d.) | 10.2 (6.23) | 10.5 (4.89) | 10.4 (5.36) | 9.3 (3.96) | 10.1 (5.16) |
| Median (range) | 10.0 (0, 39) | 10.0 (0, 22) | 9.0 (0, 22) | 9.0 (0, 18) | 9.0 (0, 39) |
| Baseline haematological + biological Component Score <2 |
23 (60.5) | 20 (54.1) | 26 (66.7) | 22 (61.1) | 91 (60.7) |
| Baseline haematological + biological Component Score ≥2 |
15 (39.5) | 17 (45.9) | 13 (33.3) | 14 (38.9) | 59 (39.3) |
| ESSPRI, mean (s.d.) | 6.3 (2.3) | 6.6 (1.9) | 5.9 (2.4) | 5.9 (2.2) | 6.2 (2.2) |
| Median (range) | 7.0 (1.7, 9.7) | 7.0 (3.3, 9.7) | 6.7 (1.7, 9.7) | 5.8 (2.3, 9.3) | 6.7 (1.7, 9.7) |
| hsCRP, mg/dL, mean (s.d.) | 0.30 (0.28) | 0.41 (0.53) | 0.36 (0.36) | 0.34 (0.52) | 0.35 (0.43) |
| hsCRP ≥1.5 × ULN | 0 | 1 (2.7) | 0 | 2 (5.6) | 3 (2.0) |
| Both SSA and SSB positiveb | 21 (55.3) | 11 (29.7) | 22 (56.4) | 20 (55.6) | 74 (49.3) |
| SSA positive | 17 (44.7) | 25 (67.6) | 17 (43.6) | 16 (44.4) | 75 (50.0) |
| Use of concurrent immunomodulatory drugsc at baseline | 24 (63.2) | 26 (70.3) | 27 (69.2) | 26 (72.2) | 103 (68.7) |
| Use of concurrent csDMARD at baseline | 23 (60.5) | 25 (67.6) | 25 (64.1) | 24 (66.7) | 97 (64.7) |
| Use of concurrent systemic CSs at baseline | 7 (18.4) | 13 (35.1) | 4 (10.3) | 11 (30.6) | 35 (23.3) |
Data presented as n (%) unless otherwise noted. Baseline characteristics are reported for all randomized patients who received at least one dose of a study drug.
aIncluding any of the following: autoimmune thyroiditis, autoimmune thyroid disorder, coeliac disease, immune thrombocytopenic purpura, psoriasis, RA, rheumatoid nodule, scleroderma, scLE, SLE, SLE rash, SSc, type I diabetes mellitus, and vitiligo.
One patient in the lanraplenib subgroup was SSB positive only.
csDMARD or systemic CSs. csDMARD: conventional synthetic DMARD; ESSDAI: EULAR SS disease activity index; ESSPRI: EULAR SS patient-reported index; FIL: filgotinib; hsCRP: high-sensitivity CRP; LANRA: lanraplenib; PBO: placebo; TIRA: tirabrutinib; ULN: upper limit of normal.
Primary efficacy end point
The primary end point evaluated the proportion of responders at W12 using the treatment policy estimand. Response rates in the filgotinib, lanraplenib and tirabrutinib groups were 16.6% (P = 0.17), 15.6% (P = 0.16) and 8.1% (P = 0.33) higher, respectively, than in the placebo group (Fig. 2). Nominal P values were >0.05 for all comparisons vs placebo at all time points through W24 (Supplementary Fig. S1, available at Rheumatology online).
Fig. 2.
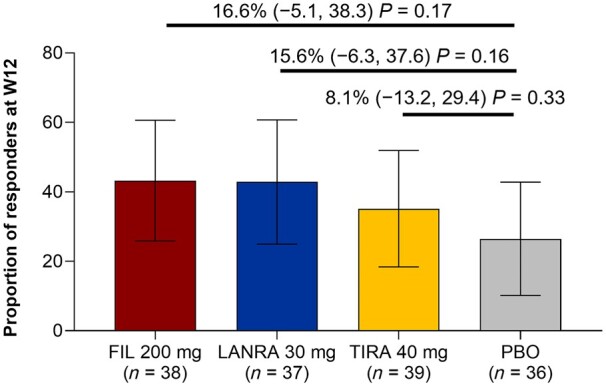
Primary end point, proportion of responders at W12
The primary end point is summarized using the full analysis set, which includes patients who were randomized and received at least one dose of the study drug. Error bars show 95% CIs. Numbers on graph show difference in response rate vs PBO, 95% CI of differences vs placebo, and the P-value of the active treatment group compared with PBO. FIL: filgotinib; LANRA: lanraplenib; PBO: placebo; TIRA: tirabrutinib; W: week.
Secondary efficacy and exploratory end points
ESSDAI total scores at W12 and W24 decreased in all study arms: the respective least-square (LS) mean (s.d.) changes from baseline were –4.7 (0.72), –2.5 (0.76), –3.2 (0.73) and –3.9 (0.76) for filgotinib, lanraplenib, tirabrutinib and placebo at W12, respectively, and –5.4 (0.75), –4.3 (0.81), –4.0 (0.75) and –4.2 (0.78) at W24, respectively (Fig. 3A). ESSDAI total scores decreased from baseline in all treatment groups at all other time points. Changes in ESSDAI domains by treatment group are shown in Supplementary Table S2 (available at Rheumatology online); patients showed mild to moderate activity in most domains other than the articular domain, and the proportions of patients with high activity in the articular domain decreased in all treatment groups from baseline to W24. ESSPRI total scores decreased from baseline at all time points and in all treatment groups. The LS mean (s.d.) changes from baseline in ESSPRI scores for filgotinib, lanraplenib, tirabrutinib and placebo were –1.4 (0.33), –1.0 (0.34), –1.4 (0.33) and –1.0 (0.34) at W12, respectively, and –0.8 (0.31), –1.1 (0.34), –1.2 (0.31) and –0.9 (0.33) at W24, respectively (Supplementary Fig. S2, available at Rheumatology online). At W12, 64.9%, 47.1%, 56.8% and 58.8% of patients in the filgotinib, lanraplenib, tirabrutinib and placebo groups, respectively, achieved the minimal clinically important improvement in ESSDAI score [27] of ≥3 points from baseline, and the corresponding proportions at W24 were 62.9%, 66.7%, 59.5% and 71.9% (Supplementary Fig. S3, available at Rheumatology online). At W12, 51.4%, 51.4%, 43.2% and 41.2% of patients in the filgotinib, lanraplenib, tirabrutinib and placebo groups, respectively, had an ESSPRI score improvement of ≥1 point from baseline; the corresponding W24 proportions were 42.9%, 50.0%, 35.1% and 34.4%.
Fig. 3.

ESSDAI score–adjusted mean change from baseline
(A) Overall study population. Error bars show 95% CI of LS mean. LS means, 95% CIs and P values were obtained from a mixed-effects model for repeated measures with the terms for baseline value, treatment, stratification factors, visit, and treatment-by-visit interaction. **P < 0.01 vs placebo. The baseline value was the last available value collected on or prior to first dose of the study drug (day 1). (B) ESSDAI score–adjusted mean change from baseline in patients with baseline ESSDAI ≥14. Markers indicate median; error bars indicate interquartile range. Numbers below the graph show numbers of patients. The baseline value was the last available value collected on or prior to the first dose of the study drug (day 1). (C) The ESSDAI score–adjusted mean change from baseline in patients not taking DMARDs/CSs at baseline. Markers indicate median; error bars indicate interquartile range. Numbers below the graph show numbers of patients. The baseline value was the last available value collected on or prior to the first dose of the study drug (day 1). ESSDAI: EULAR SS disease activity index; FIL: filgotinib; LANRA: lanraplenib; LS: least square; PBO: placebo; Q1: first quartile; Q3: third quartile; TIRA: tirabrutinib.
At W12, the LS mean (s.e.) differences from placebo in change from baseline in tear production, as assessed by Schirmer’s test (averaged between both eyes, measured as millimetres of moisture strip-wetting after 5 min), were 3.01 (2.037) in the filgotinib group (P = 0.14), 4.57 (2.050) in the lanraplenib group (P = 0.03) and 3.72 (2.021) in the tirabrutinib group (P = 0.07) (Supplementary Fig. S4, available at Rheumatology online). At W24, the LS mean (s.e.) differences from placebo in change from baseline in tear production were 2.92 (1.740; P = 0.10), 3.68 (1.764; P = 0.04) and 3.14 (1.712; P = 0.07) for filgotinib, lanraplenib and tirabrutinib, respectively. Unstimulated and stimulated salivary flow rates are summarized in the supplementary data, available at Rheumatology online.
Subgroup analyses
ESSDAI was analysed in two subpopulations: patients with severe disease activity (baseline ESSDAI score ≥14) and patients who were not receiving concomitant DMARDs/CSs. Patient numbers in these subgroups were small (<15 per treatment group). In these subgroups, median decreases in ESSDAI from baseline were numerically greater at W24 in patients receiving filgotinib compared with those receiving placebo (Fig. 3B and 3C).
Biomarker analyses
By W24, greater decreases in RF, IgM, IgG and IgA, along with increases in memory B cells, were seen in the filgotinib group compared with placebo (Fig. 4A). In contrast, in the lanraplenib group, biomarker levels remained similar to baseline at W4, W12 and W24. In the tirabrutinib group, decreases were seen in precursor plasma cells, mature plasma cells and regulatory B cells as early as W4, with P values <0.01 for precursor plasma cells and regulatory B cells at W24. Although significantly decreased CRP was observed in patients who received filgotinib, and a trend towards increased CRP was observed in those who received tirabrutinib, the vast majority of patients across study groups remained below the threshold of 1 mg/dL for CRP throughout the course of treatment (Supplementary Fig. S5, available at Rheumatology online).
Fig. 4.

Biomarker changes
(A) Median percentage change in biomarker activity after treatment. The heat map represents changes from baseline at Weeks 4, 12 and 24 (bottom labels) in each treatment group (top labels). At baseline, 50%, 71.4%, 52.6% and 71.4% of patients in the filgotinib, lanraplenib, tirabrutinib and placebo groups had RF values below the LLOQ; 2.6% of patients in the tirabrutinib group had IgM levels below LLOQ; no patients had IgG levels below LLOQ; and 8.3% of patients in the placebo group had CRP levels below LLOQ. Treated groups were compared with placebo by the Wilcoxon rank-sum test with Hommel’s method for multiplicity adjustment. ***P < 0.001, **P < 0.01, *P < 0.05. (B) Change in Type I IFN signature from baseline by treatment group. The y-axis shows the adjusted mean and 95% CI of change in the pathway activity score following treatment. The outlined circle is a statistically significant difference compared with baseline and placebo. LLOQ: lower limit of quantification.
Baseline Type I interferon activity was elevated in the whole blood of SS patients compared with that of healthy volunteers (Supplementary Fig. S6, available at Rheumatology online). Interferon activity was significantly reduced from baseline in the filgotinib group at W4 and W12 (Fig. 4B). In contrast, interferon signature activity was significantly increased from baseline in the lanraplenib group at W4 and W12. Both placebo and tirabrutinib had interferon signature activity similar to that of baseline through W12. Cytosolic DNA sensing and chemokine signalling pathways were reduced with filgotinib (Supplementary Fig. S7, available at Rheumatology online).
Safety
Treatment with filgotinib, lanraplenib and tirabrutinib was generally well tolerated. TEAEs occurring up to W24 are summarized in Table 2. The most common treatment-related TEAEs observed in this study were increased alanine aminotransferase and aspartate aminotransferase levels, which were observed in three patients in the lanraplenib group and one patient in the tirabrutinib group. During the 24-week placebo-controlled period, most TEAEs were Grade 1 or Grade 2. TEAEs of Grade ≥3 occurred in 7.9%, 10.8%, 2.6% and 5.6% of filgotinib, lanraplenib, tirabrutinib and placebo patients, respectively. Grade 3 or higher TEAEs related to a study drug were reported in two patients in the filgotinib group (ophthalmic Herpes zoster, renal failure) and one patient in the placebo group (sinusitis).
Table 2.
Summary of TEAEs (up to week 24)
| Data presented as n (%) | FIL 200 mg (n = 38) | LANRA 30 mg (n = 37) | TIRA 40 mg (n = 39) | PBO (n = 36) |
|---|---|---|---|---|
| Any TEAE | 32 (84.2) | 29 (78.4) | 29 (74.4) | 27 (75.0) |
| TEAE related to study drug | 10 (26.3) | 10 (27.0) | 5 (12.8) | 6 (16.7) |
| TEAE ≥ Grade 3 | 3 (7.9) | 4 (10.8) | 1 (2.6) | 2 (5.6) |
| TEAE related to study drug ≥ Grade 3 | 2 (5.3) | 0 | 0 | 1 (2.8) |
| TE serious AE | 3 (7.9) | 3 (8.1) | 1 (2.6) | 2 (5.6) |
| TE serious AE related to study drug | 1 (2.6) | 0 | 0 | 0 |
| TEAE leading to premature discontinuation of study drug | 4 (10.5) | 7 (18.9) | 0 | 0 |
| TEAE leading to premature discontinuation of study | 1 (2.6) | 5 (13.5) | 0 | 0 |
| Death | 0 | 0 | 0 | 0 |
TEAEs are AEs that began on/after the treatment start date and ≤30 days after last dose of study drug or led to premature treatment discontinuation. AE: adverse event; FIL: filgotinib; LANRA: lanraplenib; PBO: placebo; TE: treatment-emergent; TEAE: treatment-emergent adverse event; TIRA: tirabrutinib.
Treatment-emergent serious AEs were reported for nine patients: three (7.9%) patients in the filgotinib group, three (8.1%) in the lanraplenib group, one (2.6%) in the tirabrutinib group and two (5.6%) in the placebo group. Of these patients, only one in the filgotinib group had a serious TEAE reported as related to a study drug (renal failure; see below). Four (10.5%) patients in the filgotinib group and seven patients (18.9%) in the lanraplenib group had a TEAE leading to premature discontinuation of a study drug. No patients in the tirabrutinib or placebo groups had a TEAE that led to premature discontinuation of a study drug. No deaths occurred in this study.
TEAEs of interest up to W24 (regardless of causality and based on preferred terms as reported by the investigators) are reported in Supplementary Table S3, available at Rheumatology online. The most common TEAEs of interest were in the category of infections and infestations: these occurred in 17 patients in the filgotinib group (44.7%), 12 in the lanraplenib group (32.4%), 17 in the tirabrutinib group (43.6%) and 18 receiving placebo (50.0%). Of these patients, only one in the filgotinib group experienced a serious infection. One (2.6%) patient receiving filgotinib, five (13.5%) receiving lanraplenib and one (2.6%) receiving tirabrutinib experienced liver transaminase elevation reported as an AE; four of the lanraplenib patients discontinued the study or study drug, while the other patients continued. A serious MACE occurred in two patients over 24 weeks: one (2.6%) in the filgotinib group (a 54-year-old woman with a medical history of hypertension and chronic antihypertensive medications had renal failure and was treated with discontinuation of angiotensin-converting enzyme and diuretic) and one (2.7%) in the lanraplenib group (a 64-year-old woman hospitalized for unstable angina that resolved, considered related to a pre-existing condition). One other serious AE of interest over 24 weeks was an instance of diverticulitis in the filgotinib group requiring hospitalization (the event was assessed as unrelated to the study drug and was considered related to a pre-existing condition). No malignancy, active tuberculosis, gastrointestinal perforation, hepatitis B or C infection, opportunistic infection, renal toxicity, or VTE was reported.
Discussion
In this large, double-blind, placebo-controlled study, none of the study drugs investigated showed a statistically or clinically meaningful difference in treatment effect vs placebo in the primary or secondary end points. These results underscore the need for development of clear efficacy end points in this difficult-to-assess disease.
There are several notable characteristics of this study. Unlike those in previous randomized trials of therapies for SS, this study population was mixed in that it included patients with and without concomitant autoimmune diseases [11, 28]. All participants were required to have seropositivity for SS-related antigens at study entry, ensuring that an adaptive immune-mediated response was active in these patients. The primary end point measure was a composite outcome, which included patient-reported outcomes and change in hsCRP, a parameter that remained under LLOQ in most patients. Although numbers of patients in each group remained limited, the study was relatively large by the standards of prior SS studies and demonstrated that enrolling large, randomized trials for SS is feasible [29, 30].
Though study results showed few significant differences in clinical efficacy across drugs, our results may be considered hypothesis-generating, given the satisfactory tolerance of the evaluated drugs, stabilization of tear production, effect of filgotinib in patients with highly active systemic disease and substantial biomarker data, including those showing a significant effect of filgotinib and tirabrutinib. In patients with high ESSDAI scores at baseline, the suggestion of greater effect for filgotinib as well as the relatively lower placebo response may help direct future research. In exploratory analyses concerning the two most characteristic symptoms of SS, salivary flow rates remained similar to baseline throughout the 24-week placebo-controlled period, while stabilization or a trend towards stabilization of tear production using Schirmer’s test was seen in each study drug group up to week 24.
In this study, reductions in immunoglobulins classically associated with SS disease activity were also seen in patients who received filgotinib or tirabrutinib, despite an absence of clinical response. There was little biologic activity effect seen for lanraplenib in the present study. Filgotinib-treated patients also demonstrated a reduction in Type I interferon signature activity, and patients who received tirabrutinib showed significant decreases in circulating regulatory B and precursor plasma cells. Both observations are consistent with the expected mechanism of action of the respective study drug. Similarly, other recent randomized controlled trials of biologics showed a clear impact on disease-relevant biomarkers but failed to show clinical efficacy in SS. Notably, in a Phase 3 trial of abatacept, the mean changes in IgG, IgA, IgM-RF, kappa light chain, and C4 were significantly different in the abatacept group compared with the placebo group, although efficacy end points were not met [12].
Dosing over 24 weeks with filgotinib 200 mg, lanraplenib 30 mg and tirabrutinib 40 mg was generally well tolerated. Most AEs were Grade 1 or 2. The safety and tolerability of each study drug was consistent with its known safety profile, and no new safety signals emerged. Serious AEs were uncommon. Venous thrombosis is a known class effect with JAK inhibitors, and patients with SS or other autoimmune diseases may be at increased risk [31–33]; however, no deep vein thromboses or pulmonary emboli were observed. Liver function test elevations have been reported with the use of another SYK inhibitor, fostamatinib, in other studies [34], and some elevations were observed with lanraplenib treatment in this study; however, other safety concerns reported for fostamatinib—such as hypertension, diarrhoea and rashes—were not observed with the use of the highly selective SYK inhibitor lanraplenib. Bleeding has been described as a side effect of BTK inhibitors approved for use in B cell malignancies [35]; however, in this trial, no serious bleeding events were observed.
To date, only a few controlled clinical trials of novel investigational agents have been conducted in SS, with most not showing clinical benefit. This highlights the challenges of achieving clinically relevant treatment effect in SS, including determining the ideal clinical trial design and outcome measures. The ESSDAI was developed for use in clinical practice and trials, and recently, it has become the primary outcome measure used in most randomized controlled trials [36]. Several randomized trials to date have shown large decreases in ESSDAI in all treatment groups including placebo, similar to what we observed in this study [37]. High placebo responses in ESSDAI might be explained by natural variation in disease activity, therapeutic benefit the patient might be receiving via participation in a trial [38], increased adherence to background conventional synthetic DMARDs, or the methodologic challenges of the ESSDAI and its scoring in randomized controlled trials [36]. These methodologic challenges have led the scientific community to propose new outcome criteria, such as the Composite of Relevant End points for SS (CRESS), that could allow decrease of the placebo effect in future trials [39].
There are several limitations to this study that should be considered. The primary end point was a composite measure not validated or previously used for the evaluation of SS, chosen because the inclusion of patients with both primary and secondary SS would have made ESSDAI insufficient. The placebo response rates were high across numerous clinical outcome measures, except in the subgroup analyses, potentially limiting the differentiation of active treatments from placebo. It is difficult to assess to what degree the placebo response rate was due to the high proportion of patients who were receiving a concomitant conventional synthetic DMARD. Of the study patients, 70.7% were enrolled in the USA; thus, findings may not be generalizable to other parts of the world (results by country was not an included subgroup analysis). Geographic incidence of SS varies widely, with no clear understanding of which races and ethnicities may be more prone to developing SS or may be more likely to have more severe symptoms [40]. Given the complexities of SS, which shares pathogenetic features with other complex seropositive syndromes such as RA and SLE, it is possible that treatment with several concurrent therapies is required before a clinically meaningful benefit can be demonstrated.
Conclusion
This randomized trial evaluated the use of three oral immunomodulatory agents in patients with active SS, but none led to a higher response rate or sustained response in primary or secondary end points as compared with placebo. All three drugs were well tolerated over the study period, even in the context of concurrent DMARD and/or CS therapy. Intriguing signs of biologic activity were seen for tirabrutinib and filgotinib (including in B cell subsets, IgM, RF and Type I interferon signature), which reinforces the rationale of targeting BTK and JAK/STAT in SS. Exploratory analyses suggested potential symptoms or populations that might benefit from treatment. Tear production was increased in the lanraplenib group based on the Schirmer’s assay. In the subgroup with a high baseline ESSDAI score and the subgroup with no DMARD/CS use, the decrease in ESSDAI score from baseline appeared positive for the filgotinib group as compared with placebo. These observations, plus further interrogation of the biomarker sets and post-week 24 data and possible post hoc analysis using a newly emerging end point such as the CRESS, may provide additional avenues for future research. Future investigations may benefit from consideration of the tolerance of the evaluated drugs and the possible methodological challenges encountered in this study.
Supplementary Material
Acknowledgements
Medical writing support was provided by Gregory Suess, PhD, and Rob Coover, MPH, of AlphaScientia, LLC.
Funding: Funding for this study and medical writing support was provided by Gilead Sciences, Inc.
Disclosure statement: E.P. has no conflicts of interest to declare. M.B. reports receiving grant/research support from Medimmune and Janssen; serving as a consultant for Medimmune, GSK, and Janssen; and serving on a speaker’s bureau for Medimmune and UCB. A.K. is a shareholder of Amgen, Gilead Sciences, GlaxoSmithKline, Pfizer, and Sanofi; serving as a consultant for AbbVie, Boehringer Ingelheim, Flexion, Genzyme, Gilead Sciences, Janssen, Novartis, Pfizer, Regeneron, Sanofi, and Sun Pharma Advanced Research; serving as a paid instructor for Celgene, Genzyme, Horizon, Merck, Novartis, Pfizer, Regeneron, and Sanofi; and serving on a speaker’s bureau for AbbVie, Celgene, Flexion, Genzyme, Horizon, Merck, Novartis, Pfizer, Regeneron, and Sanofi. F.M. is a former employee of Gilead Sciences, Inc. and may hold shares. O.G., A.P., W.J., B.D., A.Ma., and A.Mo. are shareholders and employees of Gilead Sciences, Inc. N.M. is a current employee of Janssen Pharmaceuticals, is a former employee of Gilead Sciences, Inc., and may hold shares. J.E.G. reports receiving grant/research support from Bristol Myers Squibb and Pfizer; serving as a consultant for Bristol Myers Squibb, Sanofi Genzyme, and UCB; and serving on a speaker’s bureau for AbbVie, Eli Lilly and Co., Roche, Sanofi Genzyme, and UCB.
Contributor Information
Elizabeth Price, Department of Rheumatology, Great Western Hospital, Swindon.
Michele Bombardieri, Centre for Experimental Medicine and Rheumatology, William Harvey Research Institute, Queen Mary University of London, London.
Alan Kivitz, Altoona Center for Clinical Research, Duncansville, PA.
Franziska Matzkies, Clinical Research.
Oksana Gurtovaya, Biostatistics.
Alena Pechonkina, Clinical Research.
Wendy Jiang, Bioinformatics, Gilead Sciences, Inc., Foster City, CA.
Bryan Downie, Bioinformatics, Gilead Sciences, Inc., Foster City, CA.
Anubhav Mathur, Clinical Research.
Afsaneh Mozaffarian, Clinical Research.
Neelufar Mozaffarian, Immunology Research & Development, Janssen Pharmaceuticals, Cambridge, MA, USA.
J Eric Gottenberg, Hôpitaux Universitaires de Strasbourg et Université de Strasbourg, and Centre de Référence pour les Maladies Auto-Immunes Systémiques Rares, CNRS, IBMC, UPR3572, Strasbourg, France.
Data availability statement
Anonymized individual patient data will be shared upon request for research purposes dependent upon the nature of the request, the merit of the proposed research, the availability of the data, and its intended use. The full data sharing policy for Gilead Sciences, Inc., can be found at https://www.gilead.com/science-and-medicine/research/clinical-trials-transparency-and-data-sharing-policy.
Supplementary data
Supplementary data are available at Rheumatology online.
References
- 1. Patel R, Shahane A.. The epidemiology of Sjögren’s syndrome. Clin Epidemiol 2014;6:247–55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Miyamoto ST, Valim V, Fisher BA.. Health-related quality of life and costs in Sjögren’s syndrome. Rheumatology (Oxford) 2021;60:2588–601. [DOI] [PubMed] [Google Scholar]
- 3. Retamozo S, Acar-Denizli N, Rasmussen A. et al. Systemic manifestations of primary Sjögren’s syndrome out of the ESSDAI classification: prevalence and clinical relevance in a large international, multi-ethnic cohort of patients. Clin Exp Rheumatol 2019;37(Suppl 118):97–106. [PubMed] [Google Scholar]
- 4. Vivino FB, Bunya VY, Massaro-Giordano G. et al. Sjogren’s syndrome: an update on disease pathogenesis, clinical manifestations and treatment. Clin Immunol 2019;203:81–121. [DOI] [PubMed] [Google Scholar]
- 5. Felten R, Scher F, Sibilia J, Gottenberg JE, Arnaud L.. The pipeline of targeted therapies under clinical development for primary Sjögren’s syndrome: a systematic review of trials. Autoimmun Rev 2019;18:576–82. [DOI] [PubMed] [Google Scholar]
- 6. Skopouli FN, Jagiello P, Tsifetaki N, Moutsopoulos HM.. Methotrexate in primary Sjögren’s syndrome. Clin Exp Rheumatol 1996;14:555–8. [PubMed] [Google Scholar]
- 7. Devauchelle-Pensec V, Mariette X, Jousse-Joulin S. et al. Treatment of primary Sjögren syndrome with rituximab: a randomized trial. Ann Intern Med 2014;160:233–42. [DOI] [PubMed] [Google Scholar]
- 8. Bowman SJ, Everett CC, O’Dwyer JL. et al. Randomized controlled trial of rituximab and cost-effectiveness analysis in treating fatigue and oral dryness in primary Sjögren’s syndrome. Arthritis Rheumatol 2017;69:1440–50. [DOI] [PubMed] [Google Scholar]
- 9. Meijer JM, Meiners PM, Vissink A. et al. Effectiveness of rituximab treatment in primary Sjögren’s syndrome: a randomized, double-blind, placebo-controlled trial. Arthritis Rheum 2010;62:960–8. [DOI] [PubMed] [Google Scholar]
- 10. Demarchi J, Papasidero S, Medina MA. et al. Primary Sjögren’s syndrome: extraglandular manifestations and hydroxychloroquine therapy. Clin Rheumatol 2017;36:2455–60. [DOI] [PubMed] [Google Scholar]
- 11. Fisher BS, Ng W-F, Bombardieri M. et al. Assessment of the anti-CD40 antibody iscalimab in patients with primary Sjögren’s syndrome: a multicentre, randomised, double-blind, placebo-controlled, proof-of-concept study. Lancet Rheumatol 2020;2:e142–52. [DOI] [PubMed] [Google Scholar]
- 12. Baer AN, Gottenberg JE, St Clair EW. et al. Efficacy and safety of abatacept in active primary Sjögren’s syndrome: results of a phase III, randomised, placebo-controlled trial. Ann Rheum Dis 2021;80:339–48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. van Nimwegen JF, Mossel M, van Zuiden GS. et al. Abatacept treatment for patients with early active primary Sjögren’s syndrome: a single-centre, randomised, double-blind, placebo-controlled, phase 3 trial (ASAP-III study). Lancet Rheumatology 2020;2:e153–63. [DOI] [PubMed] [Google Scholar]
- 14. Ramos-Casals M, Brito-Zeron P, Bombardieri S. et al. ; EULAR-Sjögren Syndrome Task Force Group. EULAR recommendations for the management of Sjögren’s syndrome with topical and systemic therapies. Ann Rheum Dis 2020;79:3–18. [DOI] [PubMed] [Google Scholar]
- 15. Banerjee S, Biehl A, Gadina M, Hasni S, Schwartz DM.. JAK-STAT signaling as a target for inflammatory and autoimmune diseases: current and future prospects. Drugs 2017;77:521–46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Roescher N, Tak PP, Illei GG.. Cytokines in Sjögren’s syndrome. Oral Dis 2009;15:519–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Corneth OBJ, Verstappen GMP, Paulissen SMJ. et al. Enhanced Bruton’s tyrosine kinase activity in peripheral blood B lymphocytes from patients with autoimmune disease. Arthritis Rheumatol 2017;69:1313–24. [DOI] [PubMed] [Google Scholar]
- 18. Imgenberg-Kreuz J, Sandling JK, Bjork A. et al. Transcription profiling of peripheral B cells in antibody-positive primary Sjögren’s syndrome reveals upregulated expression of CX3CR1 and a type I and type II interferon signature. Scand J Immunol 2018;87:e12662. [DOI] [PubMed] [Google Scholar]
- 19. Fox RI, Fox CM, Gottenberg JE, Dorner T.. Treatment of Sjögren’s syndrome: current therapy and future directions. Rheumatology (Oxford) 2021;60:2066–74. [DOI] [PubMed] [Google Scholar]
- 20. Jyseleca® (Filgotinib maleate tablets). Japanese Package Insert. Gilead Sciences K.K. Tokyo, Japan. 2020. Available at: https://www.jyseleca.jp/-/media/Files/Filgotinib/product/basic/jys_if.pdf (5 April 2022, date last accessed).
- 21. Jyseleca® (filgotinib) 100 and 200 mg film-coated tablets. Summary of Product Characteristics. Gilead Sciences Ltd. London, UK. 2020. https://www.ema.europa.eu/documents/product-information/jyseleca-epar-product-information_en.pdf (5 April 2022, date last accessed). [Google Scholar]
- 22. Blomgren P, Chandrasekhar J, Di Paolo JA. et al. Discovery of Lanraplenib (GS-9876): a once-daily spleen tyrosine kinase inhibitor for autoimmune diseases. ACS Med Chem Letters 2020;11:506–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Dhillon S. Tirabrutinib: first approval. Drugs 2020;80:835–40. [DOI] [PubMed] [Google Scholar]
- 24. Bahjat FR, Pine PR, Reitsma A. et al. An orally bioavailable spleen tyrosine kinase inhibitor delays disease progression and prolongs survival in murine lupus. Arthritis Rheum 2008;58:1433–44. [DOI] [PubMed] [Google Scholar]
- 25. Hutcheson J, Vanarsa K, Bashmakov A. et al. Modulating proximal cell signaling by targeting Btk ameliorates humoral autoimmunity and end-organ disease in murine lupus. Arthritis Res Ther 2012;14:R243–R. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Rankin AL, Seth N, Keegan S. et al. Selective inhibition of BTK prevents murine lupus and antibody-mediated glomerulonephritis. J Immunol 2013;191:4540–50. [DOI] [PubMed] [Google Scholar]
- 27. Seror R, Bootsma H, Saraux A. et al. Defining disease activity states and clinically meaningful improvement in primary Sjögren’s syndrome with EULAR primary Sjögren’s syndrome disease activity (ESSDAI) and patient-reported indexes (ESSPRI). Ann Rheum Dis 2016;75:382–9. [DOI] [PubMed] [Google Scholar]
- 28. van der Heijden EB, Hillen MR, Lopes APP. et al. Leflunomide–hydroxychloroquine combination therapy in patients with primary Sjögren’s syndrome (RepurpSS-I): a placebo-controlled, double-blinded, randomised clinical trial. Lancet Rheumatol 2020;2:e260–9. [DOI] [PubMed] [Google Scholar]
- 29. Seror R, Bowman SJ, Brito-Zeron P. et al. EULAR Sjögren’s syndrome disease activity index (ESSDAI): a user guide. RMD Open 2015;1:e000022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Seror R, Ravaud P, Mariette X. et al. EULAR Sjögren’s Syndrome Patient Reported Index (ESSPRI): development of a consensus patient index for primary Sjögren’s syndrome. Ann Rheum Dis 2011;70:968–72. [DOI] [PubMed] [Google Scholar]
- 31. Avina-Zubieta JA, Jansz M, Sayre EC, Choi HK.. The risk of deep venous thrombosis and pulmonary embolism in primary Sjögren syndrome: a general population-based study. J Rheumatol 2017;44:1184–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Nash P, Kerschbaumer A, Dorner T. et al. Points to consider for the treatment of immune-mediated inflammatory diseases with Janus kinase inhibitors: a consensus statement. Ann Rheum Dis 2021;80:71–87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Bazzan M, Vaccarino A, Marletto F.. Systemic lupus erythematosus and thrombosis. Thromb J 2015;13:16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Rigel Pharmaceuticals Inc. TAVALISSE® (fostamatinib disodium hexahydrate) tablets [prescribing information]. 2020. https://www.accessdata.fda.gov/drugsatfda_docs/label/2018/209299lbl.pdf (5 April 2022, date last accessed).
- 35. Pharmacyclics LLC. IMBRUVICA® (ibrutinib) [prescribing information]. 2020. https://www.imbruvica.com/files/prescribing-information.pdf (5 April 2022, date last accessed).
- 36. de Wolff L, Arends S, van Nimwegen JF, Bootsma H.. Ten years of the ESSDAI: is it fit for purpose? Clin Exp Rheumatol 2020;38(Suppl 126):283–90. [PubMed] [Google Scholar]
- 37. Felten R, Gottenberg JE.. Advances in treatments for Sjögren’s syndrome: the glass is half full. Lancet Rheumatology 2020;2:e516–8. [DOI] [PubMed] [Google Scholar]
- 38. Benedetti F, Carlino E, Pollo A.. How placebos change the patient’s brain. Neuropsychopharmacology 2011;36:339–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Arends S. D, van Nimwegen JG, Verstappen GMPJ. et al. Composite of Relevant Endpoints for Sjögren’s Syndrome (CRESS): development and validation of a novel outcome measure. Lancet Rheumatol 2021;3:E553–62. [DOI] [PubMed] [Google Scholar]
- 40. Narváez J, Sánchez-Fernández SÁ, Seoane-Mato D, Díaz-González F, Bustabad S.. Prevalence of Sjögren’s syndrome in the general adult population in Spain: estimating the proportion of undiagnosed cases. Sci Rep 2020;10:10627. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Anonymized individual patient data will be shared upon request for research purposes dependent upon the nature of the request, the merit of the proposed research, the availability of the data, and its intended use. The full data sharing policy for Gilead Sciences, Inc., can be found at https://www.gilead.com/science-and-medicine/research/clinical-trials-transparency-and-data-sharing-policy.