

Abstract
Cerebrospinal fluid (CSF) soluble triggering receptor expressed on myeloid cells-2 (sTREM2) is an emerging biomarker of neuroinflammation in Alzheimer’s disease (AD). Yet, sTREM2 expression has not been systematically evaluated in relation to concomitant drivers of neuroinflammation. While associations between sTREM2 and tau in CSF are established, we sought to determine additional biological correlates of CSF sTREM2 during the prodromal stages of AD by evaluating CSF Aβ species (Aβx-40), a fluid biomarker of blood-brain barrier integrity (CSF/plasma albumin ratio), and CSF biomarkers of neurodegeneration measured in 155 participants from the Vanderbilt Memory and Aging Project. A novel association between high CSF levels of both sTREM2 and Aβx-40 was observed and replicated in an independent dataset. Aβx-40 levels, as well as the CSF/plasma albumin ratio, explained additional and unique variance in sTREM2 levels above and beyond that of CSF biomarkers of neurodegeneration. The component of sTREM2 levels correlated with Aβx-40 levels best predicted future cognitive performance. We highlight potential contributions of Aβ homeostasis and blood-brain barrier integrity to elevated CSF sTREM2, underscoring novel biomarker associations relevant to disease progression and clinical outcome measures.
Keywords: Alzheimer’s disease, sTREM2, CSF biomarkers, microglia, cerebrovascular injury, blood-brain barrier
1. Introduction
Alzheimer’s disease (AD) is a devastating neurodegenerative disease and the most common cause of dementia, affecting more than one in nine seniors in the U.S. ('Alzheimer's disease facts and figures' 2022). Nosologically-defined AD pathology consists of amyloid-β (Aβ) plaques and neurofibrillary tangles that are thought to drive neuroinflammation, blood-brain barrier (BBB) dysfunction, and neurodegeneration resulting in cognitive decline and clinical disease (Attems and Jellinger 2014; Sweeney, Sagare, and Zlokovic 2018). The prodrome of AD can be 20+ years, whereby neuropathology begins to deposit and brain changes occur years prior to the onset of clinical symptoms. For that reason, there has been an incredible focus on the development of biomarkers in AD, including both fluid and imaging biomarkers of AD neuropathology, neurodegeneration (Jack and Holtzman 2013; Jack et al. 2016), and more recently neuroinflammation and BBB dysfunction (Heslegrave et al. 2016; Hao et al. 2021; Bostrom et al. 2021). While cerebrospinal fluid (CSF) biomarkers of amyloid and tau are well-established, there is a pressing need to better characterize emerging biomarkers to understand their temporal dynamics and biological correlates.
One promising biomarker with a strong genetic and molecular basis is soluble triggering receptor expressed on myeloid cells-2 (sTREM2) measured in CSF. TREM2, encoding its transmembrane parent, was originally implicated as an AD risk gene through two large genome-wide association studies (GWAS), including the identification of the R47H missense mutation that confers an increased risk similar in magnitude to a single copy of the APOE-ε4 allele (Guerreiro et al. 2013; Jonsson et al. 2013). TREM2 is expressed preferentially on microglia in the brain and plays a critical role in the neuroinflammatory response to AD. More specifically, functional studies of TREM2 have revealed roles in the regulation of parenchymal Aβ plaque deposition (Jay et al. 2017; Kober et al. 2020; Song et al. 2018; Ulland et al. 2017; Wang et al. 2015; Wang et al. 2016; Yuan et al. 2016), progression of tau pathology (Bemiller et al. 2017; Gratuze et al. 2020; Jiang et al. 2018; Lee et al. 2021; Leyns et al. 2017), and BBB dysfunction (Wu et al. 2017; Wang, Yang, et al. 2020; Taylor et al. 2020). Beyond these roles in AD, TREM2 is also involved more generally in microglial activation (Hamerman et al. 2006; Turnbull et al. 2006), ischemia/hypoxia (Wu et al. 2017), oxidative stress responses (Linnartz-Gerlach et al. 2019; Liu, Chu, and Wang 2019), and transcriptional regulation of brain endothelial cells (Carbajosa et al. 2018), providing numerous potential avenues that could contribute to risk and progression in AD.
Cleavage of TREM2 ectodomain produces a soluble fragment (sTREM2), considered a biomarker of microglial activation, whereby increased protein levels have been reported to coincide with the transition of mild cognitive impairment (MCI) to AD dementia (Liu et al. 2018; Suarez-Calvet, Kleinberger, et al. 2016). Additionally, sTREM2 is signaling competent, thought to modulate inflammatory and phagocytic responses from microglia, and has also been shown in this manner to promote Aβ clearance in 5XFAD mice (Zhong et al. 2019). Increases in sTREM2 levels during AD may mark a transition in the neuroinflammatory state that correlates with neurodegeneration and clinical progression. Thus, it is not surprising that CSF sTREM2 is strongly correlated with CSF biomarkers of tau pathology that track closely with the neurodegenerative processes in AD (Suarez-Calvet, Kleinberger, et al. 2016; Henjum et al. 2016; Heslegrave et al. 2016; Suarez-Calvet et al. 2019). In contrast, CSF sTREM2 associations with Aβ and BBB dysfunction have been inconsistent (Heslegrave et al. 2016; Suarez-Calvet, Kleinberger, et al. 2016; Henjum et al. 2016; Suarez-Calvet et al. 2019; Bekris et al. 2018) and remain poorly understood. There is a need to fully characterize the biological correlates of CSF sTREM2, particularly a need to deconvolve the variance in sTREM2 levels that are explained by biomarkers of Aβ, tau, neurodegeneration, and BBB dysfunction, which are all thought to contribute to the neuroinflammatory milieu in AD.
It has been hypothesized that sTREM2 may be a complementary biomarker that could be useful in the context of aging and disease. Moreover, therapeutics targeting TREM2 are in active development, yet there is limited knowledge of the types of related biological processes and functions of sTREM2 itself. The goal of this manuscript is to clarify the biological correlates and thus types of biological processes that coincide with elevated sTREM2 in CSF. A comprehensive characterization of biomarkers in the CSF compartment related to sTREM2 elevation and therefore microglial activation will build understanding of potential early neuroimmune dynamics relevant to AD. First, we fully characterize the associations between CSF sTREM2 levels and well-established biomarkers of AD pathology, neurodegeneration, and BBB dysfunction. Second, we evaluate the unique contribution of each of these biomarkers to sTREM2 levels in competitive models, evaluating whether biomarkers of BBB and Aβ explain variance in sTREM2 levels above and beyond the well-established associations with CSF tau. Third, we relate residual variance in sTREM2 levels to measurements of longitudinal cognition evaluating potential clinical relevance of sTREM2 associations in CSF. Together, these analyses provide a more comprehensive picture of the biological underpinnings of elevated CSF sTREM2 in aging and disease and provide critical information for the application and interpretation of CSF sTREM2 levels in future biomarker studies.
2. Materials and Methods
2.1. Study cohort
Participants were drawn from the Vanderbilt Memory and Aging Project (VMAP) launched in 2012 in Nashville, TN. VMAP is a longitudinal study of vascular health and brain aging (Jefferson et al. 2016). A total of 335 participants, 60-92 years of age, were enrolled. This included 168 with mild cognitive impairment (MCI) and 167 age-, sex-, and race-matched cognitively normal controls (NC). MCI diagnosis was determined by the National Institute on Aging/Alzheimer’s Association Workgroup core clinical criteria (Albert et al. 2011). Briefly, this includes a CDR score 0≥0.5, concern of changes in cognition (reported by the participant, informant, or clinician), absence of dementia, relatively spared daily functioning, and neuropsychological functioning indicating objective impairment outside age-adjusted mean performance in one or more cognitive domains. Inclusion criteria required participants to speak English, have adequate auditory and visual acuity, and have a reliable study partner. Exclusion criteria included MRI contraindications, history of neurological disease or major psychiatric illness, heart failure, head injury with loss of consciousness >5 min, or a systemic or terminal illness. A subset of participants underwent fasting lumbar puncture for CSF collection at baseline. Written informed consent was obtained from all participants prior to data collection, and all protocols were approved by the Vanderbilt University Medical Center Institutional Review Board.
2.2. Neuropsychological composites
Participants underwent detailed neuropsychological assessment of various domains of cognitive performance at baseline and every 18 months. An episodic memory composite was calculated as a z-score from the following independent tests: California Verbal Learning Test Second Edition (CVLT-II) Total Immediate Recall, CVLT-II Delayed Recall, CVLT-II Recognition, Biber Figure Learning Test (BFLT) Total Immediate Recall, BFLT Delayed Recall, and BLFT Recognition. An executive functioning composite was calculated as a z-score from the following: Delis–Kaplan Executive Function System (D-KEFS) Number-Letter Switching Test, D-KEFS Color-Word Inhibition Test, and Letter Fluency Test (FAS). Assessments were reviewed to avoid floor and ceiling effects and composites were calculated from a latent variable model where each item was treated as a raw continuous variable loaded on a general factor, also as on a test-specific factor to reduce potential confounds (Jefferson et al. 2016; Kresge et al. 2018). Participants with longitudinal cognition data had up to five measurement timepoints (mean±sd=2.6±1.3 visits) and a mean follow-up period of (mean±sd=4.6±1.7 years).
2.3. Blood draw and albumin measurement
Participants underwent morning fasting venous blood draw and samples were immediately stored at −80°C. Whole blood was centrifuged at 2000g and 4°C for 15 min and plasma was extracted and stored in ten 0.5mL aliquots. Albumin levels (plasma and CSF) were measured by immunonephelometry on a Beckman Immage Immunochemistry system (Beckman Instruments, Beckman Coulter, Brea, CA, USA). The albumin ratio was calculated as CSF albumin (mg/L) / plasma albumin (g/L).
2.4. APOE genotyping
White blood cell extraction was performed on frozen whole blood. The TaqMan single nucleotide polymorphism genotyping assay from Applied Biosystems (Foster City, CA) was applied to determine APOE genotypes by identifying the two single-nucleotide polymorphisms that characterize alleles ε2, ε3, and ε4. Polymerase chain reaction (PCR) was performed as previously described (Jefferson et al. 2016). Genotyping efficiency was >99%.
2.5. Lumbar puncture and biochemical analyses
A maximum of 25mLs of CSF was drawn from a baseline, optional, fasting lumbar puncture procedure and collected with polypropylene syringes using a Sprotte 25-gauge spinal needle from an intervertebral lumbar space. CSF supernatant was immediately extracted and aliquoted in 0.5mL polypropylene tubes and stored at −80°C. Analysis of CSF total tau, p-tau181, Aβx-40, Aβx-42, Aβ1-42, and neurofilament light (NfL) was performed in batch using commercially available enzyme-linked immunosorbent assays (carboxy-terminal specific antibodies aided in quantification of Aβx-40 and Aβx-42 species with varying amino-terminal lengths). CSF sTREM2 concentration was measured using an in-house Meso Scale Discovery (MSD) assay (Rockville, MD), as previously described in detail (Jensen et al. 2019). Samples were processed in one round of experiments using one batch of reagents by board-certified laboratory technicians blinded to clinical information. Coefficients of variation for duplicate samples were <10% (mean 2.4%). Supplemental Table 1 contains assay kit information.
2.6. Replication of amyloid-β results using ADNI data
The Alzheimer’s Disease Neuroimaging Initiative (ADNI) is a longitudinal multisite study launched in 2004 focused on the development of biomarkers for AD early detection. Participant demographics are provided in Supplemental Table 2. Baseline CSF biomarker measurement of Aβ1-42, Aβ1-40, and Aβ1-38 were acquired utilizing 2D-UPLC tandem mass spectrometry. Each data point represents the average of duplicate 0.1mL aliquots from a single CSF sample. Methodology was previously validated for analysis of Aβ1-42 (Korecka et al. 2014) and then adapted for the additional peptides by including their internal standards and re-validation of the protocol (Korecka et al. 2020). A detailed summary of the analytical method including sample preparation, parameters, and standards is publicly available for download on the ADNI database (adni.loni.usc.edu). Tau positivity was determined by the previously defined cut-off value of 23pg/mL (Shaw et al. 2009).
An MSD platform-based assay was used for CSF sTREM2 measurement which has been previously established and validated by Christian Haass’ group and reported (Kleinberger et al. 2014; Suarez-Calvet, Araque Caballero, et al. 2016; Suarez-Calvet, Kleinberger, et al. 2016; Suarez-Calvet et al. 2019).
2.7. Statistical analyses
Statistical analyses were performed in R v.4.1.2 using R Studio IDE (https://www.rstudio.com/). Linear regression models were leveraged using CSF protein levels of AD biomarkers to predict CSF sTREM2 measures at baseline. Covariates included age, sex, education, and clinical diagnosis (MCI vs. NC). Following independent models for each biomarker, we performed competitive models leveraging a hierarchical linear regression approach to evaluate the unique contribution and variance explained by each significant predictor from the independent analyses. Model selection, aided by Akaike information criterion (AIC) and Bayesian information criterion (BIC) calculations, was performed using R packages AICcmodave and flemix, respectively. Residuals were then calculated from the cross-sectional models assessing variance in baseline CSF sTREM2 measurements and used to predict future cognitive performance using either a memory or executive functioning composite score as the outcome variable within a longitudinal linear mixed-effects regression.
Given the established association between CSF tau and CSF sTREM2, we performed post-hoc interaction analyses for the biomarkers that remained statistically significant in competitive hierarchical linear regression models to better understand whether the novel biomarker associations were modified according to tau status. All covariates remained the same as in our primary models above.
Sensitivity analyses included interaction models with sex, APOE-ε4 carrier status, and MCI diagnosis (Supplemental Table 3). Additional analyses included the date of CSF collection as a covariate to account for potential protein storage/degradation effects; however, accounting for this added variable did not have a significant impact on the main effects results (Supplemental Table 4) or competitive models (Supplemental Table 5). Further sensitivity analyses explored potential variation in results due to statistical and visual outliers as well as adjusting MCI diagnosis criteria to align with ADNI (Supplemental Tables 6-9). Scatter plots showing sTREM2 by additional biomarkers after outlier removal are provided in Supplementary Figures 1A-D.
All models were corrected for multiple comparisons using the Benjamini & Hochberg (1995) false discovery rate.
3. Results
3.1. Participant characteristics
The VMAP discovery cohort is divided fairly equally among individuals with MCI (46%) and NC (54%), comprised mostly of males (67%), also non-Hispanic White participants (94%), and is highly educated (mean: 16 years). Baseline age and APOE-ε4 carrier status was similar across diagnostic groups, but years of education differed with lower levels in MCI (mean: 15 years) compared to NC (mean: 17 years) shown in Table 1.
Table 1.
VMAP Cohort Demographics
| Characteristic | Clinical Diagnosis | Total (N=155) |
P-value | |
|---|---|---|---|---|
| Normal Cognition (N=83) |
Mild Cognitive Impairment (N=72) |
|||
| Male, no. (%) | 58 (70) | 46 (63) | 104 (67) | 0.535 |
| Age (baseline) | 72±6.50 | 72±6.18 | 72±6.33 | 0.458 |
| Education | 17±2.41 | 15±2.94 | 16±2.80 | 0.001 |
| APOE-ε4 carriers, no. (%) | 24 (29) | 27 (38) | 51 (33) | 0.334 |
| sTREM2 CSF pg/mL | 3530±1867.29 | 3817±1759.49 | 3667±1812.50 | 0.327 |
| p-tau181 CSF pg/mL (% p-tau positive) † | 56±21.92(17) | 67±28.59(26) | 61±25.70(21) | 0.212 |
| Aβ1-42 CSF pg/mL (% Aβ positive) †† | 760±229.54 (20) | 662±254.02 (40) | 714±245.40 (30) | 0.012 |
Values are presented as mean±standard deviation, unless otherwise indicated. A student’s t-test or a Pearson’s chi-squared test was used to compare continuous or categorical variables, respectively, between cognitive diagnoses. Bold represents statistical significance set to a priori threshold P<0.05. 6 participants are Black/African American; 2 American Indian/Alaska Native; 2 Asian.
p-tau positive ≥ 80pg/mL
Aβ positive ≤ 530pg/mL.
3.2. Biomarker associations with CSF sTREM2
Main effects of AD biomarkers on sTREM2 levels (sTREM2 ~ biomarker + base covariates) were examined. First, sTREM2 associations were characterized with respect to biomarkers of Aβ peptide abundance. CSF sTREM2 did not relate to Aβ1-42 (p=2.59e-01; Table 2; Figure 1A), consistent with previous work showing weak (Suarez-Calvet, Kleinberger, et al. 2016) or no association (Heslegrave et al. 2016; Suarez-Calvet et al. 2019). In contrast, higher levels of sTREM2 robustly related to higher CSF Aβx-40 (p=1.53e-09; Table 2; Figure 1B), and to a lesser degree related to higher levels of N-truncated Aβx-42 species (p=7.72e-03; Table 2; Figure 1C). Second, associations with biomarkers of tau pathology and axonal injury (NfL) were assessed. As expected, higher CSF sTREM2 was associated with higher levels of both total and phosphorylated tau (p=6.13e-07 and 1.81e-08, respectively; Table 2; Figures 1D-E). High levels of sTREM2 also associated with high NfL (p=3.18e-04; Table 2; Figure 1F). Next, associations of sTREM2 levels with a CSF biomarker of BBB integrity were investigated. Higher levels of sTREM2 protein in CSF associated with an increased CSF/plasma albumin ratio, indicating decreased BBB integrity (p=1.35e-07; Figure 2A). This association remained regardless of APOE-ε4 carrier status, an independent predictor of BBB permeability (Figure 2B) and interaction models between the CSF/plasma albumin ratio and APOE-ε4 carrier status on sTREM2 levels (sTREM2 ~ CSF/plasma albumin ratio*APOE-ε4 + base covariates) were insignificant (p=0.29; Supplemental Table 3). A correlation matrix of sTREM2 and additional biomarkers is provided in Supplemental Figure 2.
Table 2.
Main Effects of Baseline CSF Biomarkers on sTREM2 Measurement
| Predictor | β | SE | DF | P |
|---|---|---|---|---|
| Aβx-40 | 0.490 | 0.076 | 149 | 1.532e-09* |
| p-tau181 | 30.513 | 5.126 | 149 | 1.818e-08* |
| CSF/plasma albumin ratio | 327.552 | 59.077 | 145 | 1.355e-07* |
| t-tau | 3.146 | 0.604 | 149 | 6.137e-07* |
| NfL | 0.969 | 0.263 | 144 | 3.185e-04* |
| Aβx-42 | 1.448 | 0.536 | 149 | 7.728e-03* |
| Aβ1-42 | 0.678 | 0.599 | 149 | 2.599e-01 |
Bold represents statistical significance set to a priori threshold P<0.05. An asterisk indicates survival for multiple comparisons by FDR correction across each primary model (Benjamini & Hochberg 1995). Significance value (P), degrees of freedom (DF), standard error (SE) and estimate of coefficient (β) represented for each model.
Figure 1:
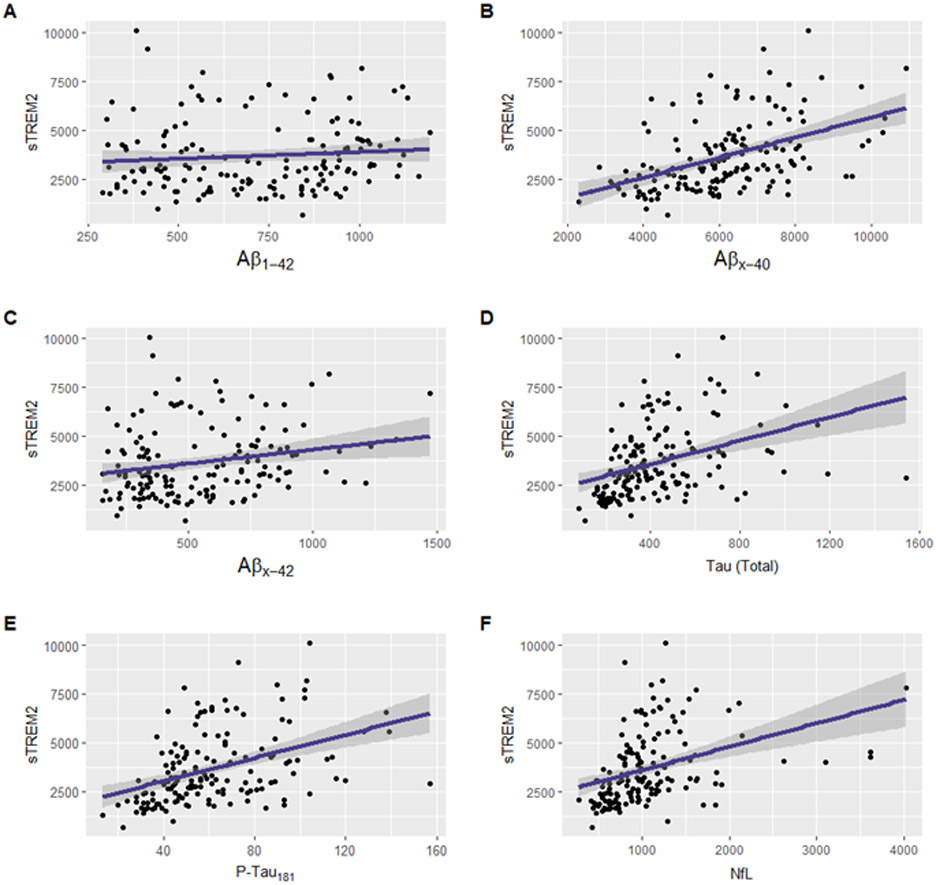
Unadjusted scatter plots showing the main effects of AD CSF Biomarkers (x axis) on CSF sTREM2 (y axis). Higher sTREM2 levels relate to increases in AD biomarkers of shorter amyloid-β peptides and neurodegeneration: (A) sTREM2 levels do not significantly relate to levels of full-length Aβ1-42 , p=2.59e-1. Higher sTREM2 levels significantly relate to increases in (B) Aβx-40, p=1.53e-9; (C) Aβx-42, p=7.72e-3; (D) total tau, p=6.13e-7; (E) phosphorylated tau, p=1.81e-8, and (F) NfL, p=3.18e-4. Protein measurements given in pg/mL.
Figure 2.
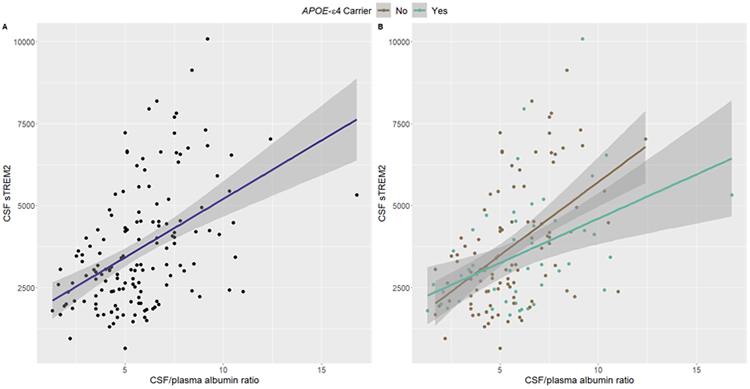
Unadjusted plots showing higher CSF sTREM2 levels relate to an increased CSF/plasma albumin ratio independent of APOE-ε4 carrier status: (A) Main effect of the CSF/plasma albumin ratio on sTREM2 (p=1.35e-07). (B) Interaction of CSF/plasma albumin* APOE-ε4 carrier status on sTREM2 (p.int.= 0.28). sTREM2 protein measurements given in pg/mL.
Due to the novelty of the Aβ species results, we replicated associations of sTREM2 with both Aβ1-40 and Aβ1-38 in ADNI (β=0.37, p=6.37e-38 and β=1.50, p=4.09e-34, respectively; Figures 3A-B), indicating broad elevations of Aβ peptide species concurrent with rises in sTREM2.
Figure 3.
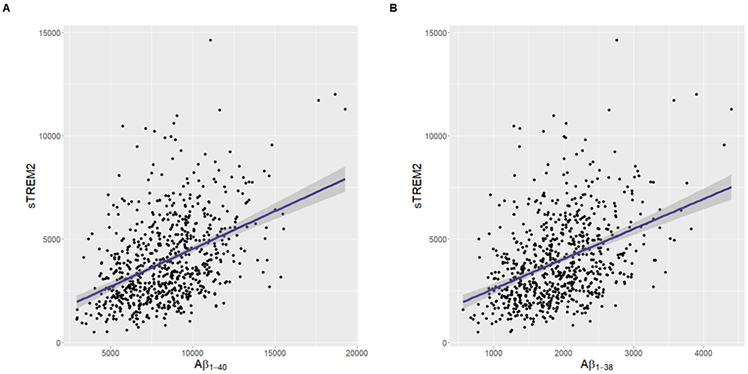
Unadjusted plots demonstrating main effects of shorter Aβ species on sTREM2 using ADNI data. Higher CSF sTREM2 levels relate to increases in CSF (A) Aβ1-40 and (B) Aβ1-38 (β=0.37, p=6.37e-38 and β=1.50, p=4.09e-34, respectively). Protein measurements given in pg/mL.
3.3. Competitive models highlight sTREM2 relationships beyond tau
To further deconvolve components of CSF sTREM2 signal in VMAP, competitive hierarchical linear regression models were utilized (Table 3). The base model (CSF sTREM2 ~ age + sex + education + cognitive diagnosis) explained 11.8% of variance in sTREM2 protein measurement. Independently, p-tau181 explained 17.7%, Aβx-40 explained 21.2% and the CSF/plasma albumin ratio explained 21.2% of variance in sTREM2 levels. For the purpose of model selection, p-tau was inputted first given the association is well-established in the literature. This allowed use of the hierarchical model to evaluate variance explained above and beyond p-tau and covariates. Model 1 includes p-tau181 as a predictor explaining an additional 16.9% of variance in sTREM2 levels above and beyond the base model. The addition of Aβx-40 in Model 2 explains an additional 4.6% of variance above and beyond Model 1. And the inclusion of the BBB marker in Model 3 explains 14.8% of variance above and beyond Model 2 (R2=0.4813). When including biomarkers (p-tau, Aβ, and the CSF/plasma albumin ratio) in Model 3 all three remained statistically significant. Together, p-tau181, Aβx-40, and the CSF/plasma albumin ratio explain 36% of the variance in CSF sTREM2 levels.
Table 3.
Competitive Hierarchical Linear Regression Results
| Model | Formula | DF | AIC | BIC | R2 | Adjusted R2 | ΔR2 |
|---|---|---|---|---|---|---|---|
| base | CSF sTREM2 ~ age + sex + education + cognitive diagnosis | 150 | 2778 | 2798 | 0.118 | 0.089 | N/A |
| 1 | CSF sTREM2 ~ base covariates + p-tau181 | 149 | 2747 | 2770 | 0.288 | 0.259 | 0.169 |
| 2 | CSF sTREM2 ~ base covariates + p-tau181 + Aβx-40 | 148 | 2738 | 2765 | 0.333 | 0.302 | 0.046 |
| 3 | CSF sTREM2 ~ base covariates + p-tau181 + Aβx-40 + CSF/plasma Albumin ratio | 143 | 2636 | 2664 | 0.481 | 0.452 | 0.148 |
ΔR2 = change in R2 from previous nested model. Akaike information criterion (AIC) and Bayesian information criterion (BIC) calculations derived as follows: AIC = 2K – 2ln(L); where K = number of model parameters, and ln(L) = model log-likelihood. BIC = (RSS+log(n)dσ 2) / n; where RSS = residual sum of squares, n = total observations, d = number of predictors, and σ = estimate of variance of the error associated with each response measurement.
3.4. Deconvolving tau, Aβ, and BBB associations with post-hoc interaction models
Given the known association between CSF tau and CSF sTREM2, we sought to better understand if the novel sTREM2 associations with Aβ and BBB differed by tau status. We did not observe statistically significant interactions between Aβx-40 and p-tau181 on sTREM2 (p=0.64) or between the CSF/plasma albumin ratio and p-tau181 (p=0.25), demonstrating associations were present regardless of tau status (Figures S3A-B). Similarly, no significant interaction between Aβ1-40 and p-tau181 positivity on sTREM2 in the larger ADNI dataset was observed (β=0.07, se=0.06, p=0.22; Figure S4).
3.5. Sensitivity analyses
Interactions between tau markers with cognitive diagnosis, as well as APOE-ε4 carrier status on sTREM2, survived correction for multiple comparisons suggesting a stronger association between sTREM2 and biomarkers of tau pathology amongst APOE-ε4 non-carriers compared to carriers, and among individuals with NC compared to those with MCI (Figures 4A-D). Additionally, nominal interactions between tau and sex (Supplemental Table 3) were observed. In contrast, neither interactions between Aβx-40 nor the CSF/plasma albumin ratio with sex, APOE-ε4 carrier status, or diagnosis (Figures 5A-D and Supplemental Table 3) were observed. In replication analyses using ADNI data we observed similar APOE-ε4 (p<0.02) and diagnosis interactions (p<2.0e-05; Supplemental Figure 5) with tau on sTREM2 levels and did not observe such interactions with Aβ species (p>0.16).
Figure 4.
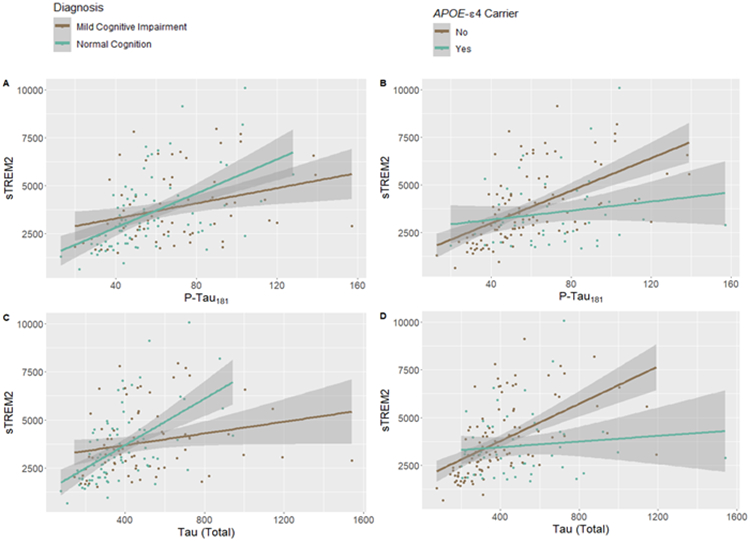
Unadjusted plots showing tau biomarkers interact with cognitive diagnosis as well as APOE-ε4 carrier status on sTREM2 levels in CSF: (A) phosphorylated tau*diagnosis, p.int.=0.033, (B) phosphorylated tau*APOE-ε4, p.int.=0.006, (C) total tau*diagnosis, p.int.=0.003, and (D) total tau* APOE-ε4, p.int.=0.002. Protein measurements given in pg/mL.
Figure 5.
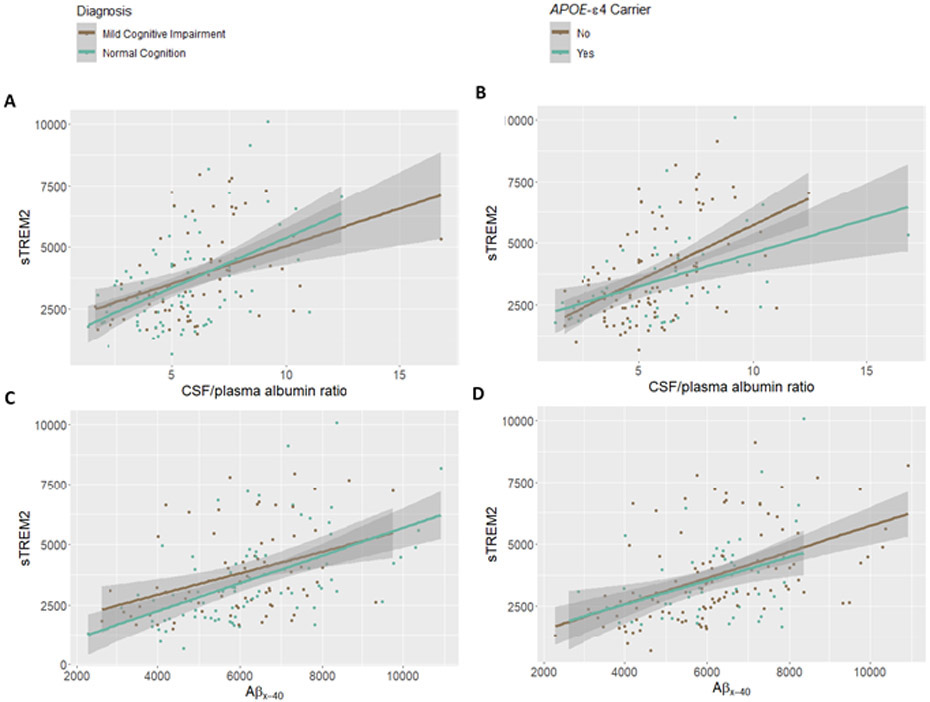
Unadjusted plot demonstrating Aβ, and BBB biomarkers do not significantly interact with cognitive diagnosis nor APOE-ε4 carrier status on sTREM2 levels in CSF: (A) CSF/plasma albumin ratio * diagnosis, p.int.=0.62, (B) CSF/plasma albumin ratio * APOE-ε4, p.int.=0.28, (C) Aβx-40 * diagnosis, p.int. =0.36, and (D) Aβx-40 * APOE-ε4, p.int.=0.78. Protein measurements given in pg/mL.
Lastly, sensitivity analyses assessed the impact of MCI diagnosis criteria between VMAP and ADNI as well as statistical and visual outliers. Specifically, VMAP MCI diagnosis criteria was updated to resemble more closely that of ADNI. This included the removal of 13 individuals with a Clinical Dementia Rating (CDR) of 0 or a Montreal Cognitive Assessment (MoCA) score of 17 or less (a MoCA score of 18 corresponds to an MMSE score of 24) (Trzepacz et al. 2015). These analyses yielded similar results and are provided in Supplemental Tables 6-9.
3.6. Associations with cognition and clinical progression
Given that CSF p-tau181, CSF Aβx-40, and the CSF/plasma album ratio all explain independent variance in sTREM2, next, it was explored which component of the sTREM2 variance is also associated with cognitive performance. This was evaluated by regressing out variance in sTREM2 associated with each biomarker sequentially and assessing the association between sTREM2 residual variance (when accounting for a given biomarker) with cognition. If regressing out a biomarker alters the association with cognition, it was concluded that the variance in sTREM2 associated with that particular biomarker is also relevant to cognitive performance. Baseline CSF sTREM2 levels predicted longitudinal memory in VMAP (β=1.44e-05, se=6.80e-06, p=0.03), similar to previous reports (Ewers et al. 2019; Edwin et al. 2020) but not longitudinal executive functioning (β= −3.22e-06, se=7.32e-06, p=0.66). Due to the well-established association between tau levels and cognitive decline, we wanted to determine whether this association was due simply to covariance with tau. When regressing out the variance in sTREM2 associated with p-tau181, the residual variance in sTREM2 remained associated with longitudinal memory decline (β=1.68e-05, se=7.28e-06, p=0.02). Similarly, when regressing out the variance in sTREM2 associated with both the CSF/plasma albumin ratio and p-tau181 the residual variance remained significantly associated with longitudinal memory decline (β=2.21e-05, se=8.15e-06, p=6.83e-03). In contrast, when regressing out the variance in sTREM2 associated with CSF Aβx-40, the residual variance was not associated with longitudinal cognitive performance (β=1.37e-05, se=7.41e-06, p=0.06). These results suggest that variance in sTREM2 that is related to Aβ species may indeed be relevant to cognitive trajectory.
4. Discussion
The present results provide an in-depth characterization of CSF sTREM2 expression in non-demented older adults. We provide strong evidence that fluid biomarkers of tau pathology, Aβ abundance, and BBB dysfunction independently relate to sTREM2 levels, and that together these biomarkers explain substantial variance in CSF sTREM2. Specifically, we recapitulate previously reported associations between CSF sTREM2 and both biomarkers of BBB integrity and tau pathology demonstrating for the first time that all three biomarkers (Aβx-40, the CSF/plasma albumin ratio, and p-tau) explain unique variance in sTREM2 using competitive models. Importantly, our results provide novel evidence that sTREM2 relates to elevated CSF Aβ species and that the variance in baseline CSF sTREM2 levels associated with Aβx-40 predicts future cognitive performance. Together, our results highlight the need to better understand sTREM2 in relation to the complex intersection of AD neuropathology, microglial activation, and BBB dysfunction to characterize its utility as both a dynamic biomarker of disease and therapeutic target.
4.1. Unique contribution of tau, BBB dysfunction, and Aβ abundance to sTREM2 levels
Together, findings suggest that a heterogeneous set of biological correlates in the aging brain likely contributes to sTREM2 changes in CSF, including independent associations with tau, BBB dysfunction, and the most abundant Aβ species (Table 2; Figures 1-2). Given the previously described associations between CSF biomarkers of tau pathology and CSF sTREM2, it is not surprising that CSF p-tau181 explained significant variance in sTREM2 levels. Previous work has demonstrated not only that sTREM2 relates to CSF p-tau, but also that the ratio of sTREM2 to p-tau is predictive of future cognitive decline (Ewers et al. 2019). Although this was not recapitulated in our smaller cohort, we do see a significant interaction between total tau and cognitive diagnosis on sTREM2 levels (sTREM2 ~ t-tau*diagnosis + base covariates) while p-tau performs similarly but does not reach statistical significance (Figure 4), demonstrating a stronger association between tau biomarkers and sTREM2 in cognitively normal individuals compared to those with MCI. Likewise, using ADNI data, the association between p-tau181 and sTREM2 differs by diagnosis where tight coupling is attenuated in both MCI and AD compared to cognitively normal individuals (Figure S5). Therefore, the sTREM2/p-tau ratio likely predicts longitudinal cognitive outcomes because of this decoupling of sTREM2 and tau as the disease progresses. In contrast to p-tau, Aβx-40 explains a slightly larger percentage of variance in sTREM2, but this association does not differ by diagnosis. Interestingly, even when removing the variance in sTREM2 that is due to p-tau, the variance in sTREM2 that is associated with Aβx-40 remains associated with future cognitive decline, suggesting that the complex interplay between Aβ abundance, tau pathology, and sTREM2 is needed to properly interpret the clinical relevance of this emerging biomarker. Finally, the CSF/plasma albumin ratio explained an additional 14.8% of variance in CSF sTREM2 beyond covariates, CSF p-tau181, and Aβx-40, highlighting the potential importance of the neurovascular unit (NVU) to changing CSF sTREM2 levels (Table 3). While previous work has discussed the potential drivers of tau associations with sTREM2 (Suarez-Calvet et al. 2019; Ulrich et al. 2017; Gratuze et al. 2020), less is known about the independent associations with BBB and Aβ abundance, so we expand our discussion of those two relationships below.
4.2. CSF sTREM2 relates to broad peptide species of CSF amyloid-β
CSF sTREM2 associations with Aβ have been inconsistent and focused on Aβ1-42 (Heslegrave et al. 2016; Suarez-Calvet, Kleinberger, et al. 2016; Henjum et al. 2016; Suarez-Calvet et al. 2019). However, we observed a robust, novel association between CSF sTREM2 and shorter species including truncated CSF Aβ (Figure 1B-C; Table 2) as well as Aβ1-40 and Aβ1-38 using ADNI data (Figure 3) that may explain some of the discrepant reports in the literature. One possibility is that the positive correlation between Aβx-40 and sTREM2 concentration reflects a direct beneficial role of sTREM2 in facilitating amyloid clearance described in the animal model literature. Specifically, injection of recombinant sTREM2 in Trem2-knockout and wild-type mice, as well as in culture, induced inflammatory responses in microglia, significantly increasing IL-1β, IL-10, IL-6, and TNF cytokine production and enhancing microglial survival (Zhong et al. 2017). Additionally, sTREM2 stereotaxic injection in the hippocampus has been shown to ameliorate Aβ plaque load in 5XFAD mice, suggesting a beneficial role for sTREM2 in AD (Zhong et al. 2019). However, we did not observe associations with Aβ1-42 or the Aβ42/40 ratio (Figure S2), both of which are thought to be the most sensitive markers of AD neuropathology, suggesting the associations with other Aβ species may not reflect brain amyloidosis. This is particularly interesting given previous evidence that sTREM2 levels decline in the early preclinical stages of AD in amyloid positive individuals before elevating later in the disease cascade (Suarez-Calvet et al. 2019), a pattern recapitulated in the present cohort. It may be that the tau association masks an independent Aβ1-42 association as pathology begins to emerge, but the data presented cannot speak to such a possibility.
Beyond a direct role in Aβ processing or clearance, it is also possible that the association between sTREM2 and Aβ reflects a compensatory alteration in Aβ abundance in response to glial activation and/or altered neuronal activity. Synaptic activity has indeed been linked to the regulation of soluble Aβ abundance in interstitial fluid (ISF) in vivo and in vitro (Bero et al. 2011; Cirrito et al. 2005). And this activity-dependent modulation of Aβ production has been proposed as a compensation to neuronal hyperactivity (Kamenetz et al. 2003; Wei et al. 2010). This compensation hypothesis aligns with our data suggesting the relationship of Aβx-40 to sTREM2 may indeed be important to cognitive functioning. It is possible that prior to the onset of neurodegeneration, sTREM2 is elevated concurrently with an inflammatory response that promotes glymphatic trafficking of free and abundant Aβ peptide. As Aβ40 is the most abundant species, this provides for a more sensitive window in which it is possible to detect alterations of abundance in the CSF compartment by regulators of ISF/CSF flow. Functional studies will be needed to evaluate these mechanistic hypotheses and understand the interplay more thoroughly between Aβ species and TREM2 proteins. Finally, it is also possible that sTREM2 levels rise concurrently with a parallel mechanism of Aβ abundance that is causally unrelated or driven by a third unmeasured variable. Regardless of the mechanism, our results provide strong evidence that sTREM2 levels are correlated with the abundance of Aβ species in a manner that does not appear to be specific to plaque deposition and that does not change with clinical disease.
4.3. CSF sTREM2 relates to BBB integrity
We also identified an association of the TREM2 axis with BBB permeability, recapitulating one other report in the literature (Bekris et al. 2018) whereby higher levels of CSF sTREM2 were associated with greater BBB permeability as indicated by the CSF/plasma albumin ratio (Figure 2A; Table 2). This association was independent of APOE-ε4 carrier status (Figure 2B), suggesting an alternative pathway of cerebrovascular injury coincides with elevations in CSF sTREM2. Notably, our analyses revealed that BBB integrity, as measured by the CSF/plasma albumin ratio, explained a substantial proportion of variance in CSF sTREM2 levels (Table 3). BBB breakdown allows blood-derived accumulation of toxic proteins (i.e., fibrin and thrombin), microbial agents, as well as peripheral immune cells within the brain parenchyma (Sweeney, Sagare, and Zlokovic 2018). In turn, this remodeling drives microglial alterations associated with elevated sTREM2. Moreover, it is possible that the high levels of CSF/plasma albumin and sTREM2 reflect a compensatory change in barrier permeability in response to deposition of amyloid as early neuroinflammation may also serve to stave off plaque formation. Similarly, the pro-inflammatory role of sTREM2 in activating microglia may drive cerebrovascular dysregulation as microglia are known to regulate critical NVU mechanisms such as the recruitment of peripheral immune cells and the integrity of tight junction proteins (da Fonseca et al. 2014). This balance of neuronal-glial-vascular communication is a critical component of AD pathophysiology and subsequent heterogeneity. A better understanding of the role of sTREM2 and BBB function is critical, particularly as modulation of TREM2 is being actively pursued as a treatment target for AD pathogenesis (Wang, Mustafa, et al. 2020). Future studies including longitudinal measurement of sTREM2, and markers of cerebrovascular injury are needed to determine whether early elevations of sTREM2 predict changes in other NVU processes over the course of disease.
4.4. Strengths and limitations
Our deeply characterized cohort provides rich timepoints early in the disease process providing a unique opportunity to understand changes in sTREM2 within the preclinical period. Moreover, we were able to replicate previous associations and provide independent replication for the novel associations reported herein. Despite these strengths, our focus on baseline biomarker measurements precludes the temporal resolution and experimental control needed to determine cause and effect. Finally, it should be noted that our sample is enriched for highly educated, non-Hispanic White individuals, limiting our ability to generalize to other populations.
4.5. Conclusions
Taken together, we demonstrate that CSF sTREM2 relates to biomarkers of concomitant pathological processes in AD including Aβ peptide abundance, tau pathology, and BBB dysfunction. We highlight multiple novel and independent pathways that are relevant to sTREM2 levels in aging and AD, enhancing its characterization as a biomarker and therapeutic target. Results suggest sTREM2 is relevant to cognitive progression with a heterogeneous etiology that must be further explored if it is going to have future clinical utility.
Supplementary Material
Unadjusted scatter plots showing CSF AD biomarkers (x axis) by CSF sTREM2 (y axis) after visual and statistical outlier removal. Statistical outliers were determined by values 4 standard deviations outside of the mean measurement.
Correlation matrix between sTREM2 and additional CSF biomarker measurements in VMAP. Non-significant Pearson’s correlation coefficients are denoted by an “X”.
Aβ and BBB biomarkers do not significantly interact with tau status on sTREM2 levels in CSF: (A) Aβx-40 * tau status, p.int.=0.64, and (B) CSF/plasma albumin ratio * tau status, p.int.=0.25. Protein measurements given in pg/mL.
Replication of Aβ40 * tau status interaction on sTREM2. Aβ1-40 does not significantly interact with tau status on sTREM2 levels in CSF using ADNI data: Aβ1-40 * tau status, p.int. =0.22. Protein measurements given in pg/mL.
Using ADNI data, sTREM2 decouples from tau as disease progresses. Protein measurements given in pg/mL.
Highlights.
We describe a novel association between sTREM2 and Aβx-40 levels in cerebrospinal fluid important to cognitive memory trajectory in an aged cohort.
Aβx-40 levels and a fluid biomarker of blood-brain barrier integrity explain sTREM2 signal above and beyond established associations with tau.
Aβx-40, the CSF/plasma albumin ratio, and p-tau independently relate to sTREM2 and jointly explain 36% of variance in sTREM2 levels.
Acknowledgments
This research was supported in part by RO1-AG059716, K01-AG049164, R21-AG059941, K24-AG046373, Alzheimer’s Association IIRG-08-88733, P20-AG068082 (Vanderbilt Alzheimer’s Disease Research Center), T32 AG058524, R01 AG034962, and the Vanderbilt Memory & Alzheimer’s Center. HZ is a Wallenberg Scholar supported by grants from the Swedish Research Council (#2018-02532), the European Research Council (#681712), Swedish State Support for Clinical Research (#ALFGBG-720931), the Alzheimer Drug Discovery Foundation (ADDF), USA (#201809-2016862), the AD Strategic Fund and the Alzheimer's Association (#ADSF-21-831376-C, #ADSF-21-831381-C and #ADSF-21-831377-C), the Olav Thon Foundation, the Erling-Persson Family Foundation, Stiftelsen för Gamla Tjänarinnor, Hjärnfonden, Sweden (#FO2019-0228), the European Union’s Horizon 2020 research and innovation programme under the Marie Skłodowska-Curie grant agreement No 860197 (MIRIADE), and the UK Dementia Research Institute at UCL. KB is supported by the Swedish Research Council (#2017-00915), the Alzheimer Drug Discovery Foundation (ADDF), USA (#RDAPB-201809-2016615), the Swedish Alzheimer Foundation (#AF-742881), Hjärnfonden, Sweden (#FO2017-0243), the Swedish state under the agreement between the Swedish government and the County Councils, the ALF-agreement (#ALFGBG-715986), the European Union Joint Program for Neurodegenerative Disorders (JPND2019-466-236), and the National Institute of Health (NIH), USA, (grant #1R01AG068398-01).
Data collection and sharing for this project was funded by the Alzheimer's Disease Neuroimaging Initiative (ADNI) (National Institutes of Health Grant U01 AG024904) and DOD ADNI (Department of Defense award number W81XWH-12-2-0012). ADNI is funded by the National Institute on Aging, the National Institute of Biomedical Imaging and Bioengineering, and through generous contributions from the following: AbbVie, Alzheimer’s Association; Alzheimer’s Drug Discovery Foundation; Araclon Biotech; BioClinica, Inc.; Biogen; Bristol-Myers Squibb Company; CereSpir, Inc.; Cogstate; Eisai Inc.; Elan Pharmaceuticals, Inc.; Eli Lilly and Company; EuroImmun; F. Hoffmann-La Roche Ltd and its affiliated company Genentech, Inc.; Fujirebio; GE Healthcare; IXICO Ltd.; Janssen Alzheimer Immunotherapy Research & Development, LLC.; Johnson & Johnson Pharmaceutical Research & Development LLC.; Lumosity; Lundbeck; Merck & Co., Inc.; Meso Scale Diagnostics, LLC.; NeuroRx Research; Neurotrack Technologies; Novartis Pharmaceuticals Corporation; Pfizer Inc.; Piramal Imaging; Servier; Takeda Pharmaceutical Company; and Transition Therapeutics. The Canadian Institutes of Health Research is providing funds to support ADNI clinical sites in Canada. Private sector contributions are facilitated by the Foundation for the National Institutes of Health (www.fnih.org). The grantee organization is the Northern California Institute for Research and Education, and the study is coordinated by the Alzheimer’s Therapeutic Research Institute at the University of Southern California. ADNI data are disseminated by the Laboratory for Neuro Imaging at the University of Southern California.
Abbreviations:
- CSF
Cerebrospinal fluid
- TREM2
Triggering Receptor Expressed on Myeloid Cells-2
- sTREM2
Soluble Triggering Receptor Expressed on Myeloid Cells-2
- AD
Alzheimer’s disease
- VMAP
Vanderbilt Memory and Aging Project
- ADNI
Alzheimer’s Disease Neuroimaging Initiative
- APOE-ε4
Apolipoprotein E epsilon 4
- BBB
Blood-brain barrier
- Aβ
Beta-amyloid
- NVU
Neurovascular unit
- MCI
Mild cognitive impairment
- NC
Normal cognition
- NfL
Neurofilament light chain protein
Footnotes
Conflicts of interest
HZ has served at scientific advisory boards and/or as a consultant for Alector, Eisai, Denali, Roche Diagnostics, Wave, Samumed, Siemens Healthineers, Pinteon Therapeutics, Nervgen, AZTherapies, CogRx and Red Abbey Labs, has given lectures in symposia sponsored by Cellectricon, Fujirebio, Alzecure and Biogen, and is a co-founder of Brain Biomarker Solutions in Gothenburg AB (BBS), which is a part of the GU Ventures Incubator Program (outside submitted work). KB has served as a consultant, at advisory boards, or at data monitoring committees for Abcam, Axon, Biogen, JOMDD/Shimadzu. Julius Clinical, Lilly, MagQu, Novartis, Prothena, Roche Diagnostics, and Siemens Healthineers, and is a co-founder of Brain Biomarker Solutions in Gothenburg AB (BBS), which is a part of the GU Ventures Incubator Program (all outside the work presented in this paper). TH is a member of the scientific advisory board for Vivid Genomics (also outside the work presented herein).
Data used in preparation of this article were obtained from the Alzheimer’s Disease Neuroimaging Initiative (ADNI) database (adni.loni.usc.edu). As such, the investigators within the ADNI contributed to the design and implementation of ADNI and/or provided data but did not participate in analysis or writing of this report. A complete listing of ADNI investigators can be found at: http://adni.loni.usc.edu/wp-content/uploads/how_to_apply/ADNI_Acknowledgement_List.pdf
Ethics approval and consent to participate
All protocols were conducted with approval by the Vanderbilt University Medical Center Institutional Review Board after written informed consent from all subjects was obtained.
Consent for publication
All authors have given consent to the publication of this manuscript.
Availability of data and material
VMAP data can be requested within our data sharing portal and will be made freely available to qualified investigators (https://www.vmacdata.org/vmap/data-requests).
Citations
- Albert MS, DeKosky ST, Dickson D, Dubois B, Feldman HH, Fox NC, Gamst A, Holtzman DM, Jagust WJ, Petersen RC, Snyder PJ, Carrillo MC, Thies B, and Phelps CH. 2011. 'The diagnosis of mild cognitive impairment due to Alzheimer's disease: recommendations from the National Institute on Aging-Alzheimer's Association workgroups on diagnostic guidelines for Alzheimer's disease', Alzheimers Dement, 7: 270–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 'Alzheimer's disease facts and figures'. 2022. Alzheimers Dement, 18: 700–89. [DOI] [PubMed] [Google Scholar]
- Attems J, and Jellinger KA. 2014. 'The overlap between vascular disease and Alzheimer's disease--lessons from pathology', BMC Med, 12: 206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bekris LM, Khrestian M, Dyne E, Shao Y, Pillai JA, Rao SM, Bemiller SM, Lamb B, Fernandez HH, and Leverenz JB. 2018. 'Soluble TREM2 and biomarkers of central and peripheral inflammation in neurodegenerative disease', J Neuroimmunol, 319: 19–27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bemiller SM, McCray TJ, Allan K, Formica SV, Xu G, Wilson G, Kokiko-Cochran ON, Crish SD, Lasagna-Reeves CA, Ransohoff RM, Landreth GE, and Lamb BT. 2017. 'TREM2 deficiency exacerbates tau pathology through dysregulated kinase signaling in a mouse model of tauopathy', Mol Neurodegener, 12: 74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bero AW, Yan P, Roh JH, Cirrito JR, Stewart FR, Raichle ME, Lee JM, and Holtzman DM. 2011. 'Neuronal activity regulates the regional vulnerability to amyloid-beta deposition', Nat Neurosci, 14: 750–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bostrom G, Freyhult E, Virhammar J, Alcolea D, Tumani H, Otto M, Brundin RM, Kilander L, Lowenmark M, Giedraitis V, Lleo A, von Arnim CAF, Kultima K, and Ingelsson M. 2021. 'Different Inflammatory Signatures in Alzheimer's Disease and Frontotemporal Dementia Cerebrospinal Fluid', J Alzheimers Dis, 81: 629–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carbajosa G, Malki K, Lawless N, Wang H, Ryder JW, Wozniak E, Wood K, Mein CA, Dobson RJB, Collier DA, O'Neill MJ, Hodges AK, and Newhouse SJ. 2018. 'Loss of Trem2 in microglia leads to widespread disruption of cell coexpression networks in mouse brain', Neurobiol Aging, 69: 151–66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cirrito JR, Yamada KA, Finn MB, Sloviter RS, Bales KR, May PC, Schoepp DD, Paul SM, Mennerick S, and Holtzman DM. 2005. 'Synaptic activity regulates interstitial fluid amyloid-beta levels in vivo', Neuron, 48: 913–22. [DOI] [PubMed] [Google Scholar]
- da Fonseca AC, Matias D, Garcia C, Amaral R, Geraldo LH, Freitas C, and Lima FR. 2014. 'The impact of microglial activation on blood-brain barrier in brain diseases', Front Cell Neurosci, 8: 362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Edwin TH, Henjum K, Nilsson LNG, Watne LO, Persson K, Eldholm RS, Saltvedt I, Halaas NB, Selbaek G, Engedal K, Strand BH, and Knapskog AB. 2020. 'A high cerebrospinal fluid soluble TREM2 level is associated with slow clinical progression of Alzheimer's disease', Alzheimers Dement (Amst), 12: e12128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ewers M, Franzmeier N, Suarez-Calvet M, Morenas-Rodriguez E, Caballero MAA, Kleinberger G, Piccio L, Cruchaga C, Deming Y, Dichgans M, Trojanowski JQ, Shaw LM, Weiner MW, Haass C, and Initiative Alzheimer's Disease Neuroimaging. 2019. 'Increased soluble TREM2 in cerebrospinal fluid is associated with reduced cognitive and clinical decline in Alzheimer's disease', Sci Transl Med, 11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gratuze M, Leyns CE, Sauerbeck AD, St-Pierre MK, Xiong M, Kim N, Serrano JR, Tremblay ME, Kummer TT, Colonna M, Ulrich JD, and Holtzman DM. 2020. 'Impact of TREM2R47H variant on tau pathology-induced gliosis and neurodegeneration', J Clin Invest, 130: 4954–68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guerreiro R, Wojtas A, Bras J, Carrasquillo M, Rogaeva E, Majounie E, Cruchaga C, Sassi C, Kauwe JS, Younkin S, Hazrati L, Collinge J, Pocock J, Lashley T, Williams J, Lambert JC, Amouyel P, Goate A, Rademakers R, Morgan K, Powell J, St George-Hyslop P, Singleton A, Hardy J, and Group Alzheimer Genetic Analysis. 2013. 'TREM2 variants in Alzheimer's disease', N Engl J Med, 368: 117–27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hamerman JA, Jarjoura JR, Humphrey MB, Nakamura MC, Seaman WE, and Lanier LL. 2006. 'Cutting edge: inhibition of TLR and FcR responses in macrophages by triggering receptor expressed on myeloid cells (TREM)-2 and DAP12', J Immunol, 177: 2051–5. [DOI] [PubMed] [Google Scholar]
- Hao J, Qiao Y, Li T, Yang J, Song Y, Jia L, and Jia J. 2021. 'Investigating Changes in the Serum Inflammatory Factors in Alzheimer's Disease and Their Correlation with Cognitive Function', J Alzheimers Dis, 84: 835–42. [DOI] [PubMed] [Google Scholar]
- Henjum K, Almdahl IS, Arskog V, Minthon L, Hansson O, Fladby T, and Nilsson LN. 2016. 'Cerebrospinal fluid soluble TREM2 in aging and Alzheimer's disease', Alzheimers Res Ther, 8: 17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heslegrave A, Heywood W, Paterson R, Magdalinou N, Svensson J, Johansson P, Ohrfelt A, Blennow K, Hardy J, Schott J, Mills K, and Zetterberg H. 2016. 'Increased cerebrospinal fluid soluble TREM2 concentration in Alzheimer's disease', Mol Neurodegener, 11: 3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jack CR Jr., Bennett DA, Blennow K, Carrillo MC, Feldman HH, Frisoni GB, Hampel H, Jagust WJ, Johnson KA, Knopman DS, Petersen RC, Scheltens P, Sperling RA, and Dubois B. 2016. 'A/T/N: An unbiased descriptive classification scheme for Alzheimer disease biomarkers', Neurology, 87: 539–47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jack CR Jr., and Holtzman DM. 2013. 'Biomarker modeling of Alzheimer's disease', Neuron, 80: 1347–58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jay TR, Hirsch AM, Broihier ML, Miller CM, Neilson LE, Ransohoff RM, Lamb BT, and Landreth GE. 2017. 'Disease Progression-Dependent Effects of TREM2 Deficiency in a Mouse Model of Alzheimer's Disease', J Neurosci, 37: 637–47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jefferson AL, Gifford KA, Acosta LM, Bell SP, Donahue MJ, Davis LT, Gottlieb J, Gupta DK, Hohman TJ, Lane EM, Libon DJ, Mendes LA, Niswender K, Pechman KR, Rane S, Ruberg FL, Su YR, Zetterberg H, and Liu D. 2016. 'The Vanderbilt Memory & Aging Project: Study Design and Baseline Cohort Overview', J Alzheimers Dis, 52: 539–59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jensen CS, Bahl JM, Ostergaard LB, Hogh P, Wermuth L, Heslegrave A, Zetterberg H, Heegaard NHH, Hasselbalch SG, and Simonsen AH. 2019. 'Exercise as a potential modulator of inflammation in patients with Alzheimer's disease measured in cerebrospinal fluid and plasma', Exp Gerontol, 121: 91–98. [DOI] [PubMed] [Google Scholar]
- Jiang T, Zhang YD, Gao Q, Ou Z, Gong PY, Shi JQ, Wu L, and Zhou JS. 2018. 'TREM2 Ameliorates Neuronal Tau Pathology Through Suppression of Microglial Inflammatory Response', Inflammation, 41: 811–23. [DOI] [PubMed] [Google Scholar]
- Jonsson T, Stefansson H, Steinberg S, Jonsdottir I, Jonsson PV, Snaedal J, Bjornsson S, Huttenlocher J, Levey AI, Lah JJ, Rujescu D, Hampel H, Giegling I, Andreassen OA, Engedal K, Ulstein I, Djurovic S, Ibrahim-Verbaas C, Hofman A, Ikram MA, van Duijn CM, Thorsteinsdottir U, Kong A, and Stefansson K. 2013. 'Variant of TREM2 associated with the risk of Alzheimer's disease', N Engl J Med, 368: 107–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kamenetz F, Tomita T, Hsieh H, Seabrook G, Borchelt D, Iwatsubo T, Sisodia S, and Malinow R. 2003. 'APP processing and synaptic function', Neuron, 37: 925–37. [DOI] [PubMed] [Google Scholar]
- Kleinberger G, Yamanishi Y, Suarez-Calvet M, Czirr E, Lohmann E, Cuyvers E, Struyfs H, Pettkus N, Wenninger-Weinzierl A, Mazaheri F, Tahirovic S, Lleo A, Alcolea D, Fortea J, Willem M, Lammich S, Molinuevo JL, Sanchez-Valle R, Antonell A, Ramirez A, Heneka MT, Sleegers K, van der Zee J, Martin JJ, Engelborghs S, Demirtas-Tatlidede A, Zetterberg H, Van Broeckhoven C, Gurvit H, Wyss-Coray T, Hardy J, Colonna M, and Haass C. 2014. 'TREM2 mutations implicated in neurodegeneration impair cell surface transport and phagocytosis', Sci Transl Med, 6: 243ra86. [DOI] [PubMed] [Google Scholar]
- Kober DL, Stuchell-Brereton MD, Kluender CE, Dean HB, Strickland MR, Steinberg DF, Nelson SS, Baban B, Holtzman DM, Frieden C, Alexander-Brett J, Roberson ED, Song Y, and Brett TJ. 2020. 'Functional insights from biophysical study of TREM2 interactions with apoE and Abeta1–42', Alzheimers Dement. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Korecka M, Figurski MJ, Landau SM, Brylska M, Alexander J, Blennow K, Zetterberg H, Jagust WJ, Trojanowski JQ, Shaw LM, and Initiative Alzheimer's Disease Neuroimaging. 2020. 'Analytical and Clinical Performance of Amyloid-Beta Peptides Measurements in CSF of ADNIGO/2 Participants by an LC-MS/MS Reference Method', Clin Chem, 66: 587–97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Korecka M, Waligorska T, Figurski M, Toledo JB, Arnold SE, Grossman M, Trojanowski JQ, and Shaw LM. 2014. 'Qualification of a surrogate matrix-based absolute quantification method for amyloid-beta(4)(2) in human cerebrospinal fluid using 2D UPLC-tandem mass spectrometry', J Alzheimers Dis, 41: 441–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kresge HA, Khan OA, Wagener MA, Liu D, Terry JG, Nair S, Cambronero FE, Gifford KA, Osborn KE, Hohman TJ, Pechman KR, Bell SP, Wang TJ, Carr JJ, and Jefferson AL. 2018. 'Subclinical Compromise in Cardiac Strain Relates to Lower Cognitive Performances in Older Adults', J Am Heart Assoc, 7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee SH, Meilandt WJ, Xie L, Gandham VD, Ngu H, Barck KH, Rezzonico MG, Imperio J, Lalehzadeh G, Huntley MA, Stark KL, Foreman O, Carano RAD, Friedman BA, Sheng M, Easton A, Bohlen CJ, and Hansen DV. 2021. 'Trem2 restrains the enhancement of tau accumulation and neurodegeneration by beta-amyloid pathology', Neuron, 109: 1283–301 e6. [DOI] [PubMed] [Google Scholar]
- Leyns CEG, Ulrich JD, Finn MB, Stewart FR, Koscal LJ, Remolina Serrano J, Robinson GO, Anderson E, Colonna M, and Holtzman DM. 2017. 'TREM2 deficiency attenuates neuroinflammation and protects against neurodegeneration in a mouse model of tauopathy', Proc Natl Acad Sci U S A, 114: 11524–29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Linnartz-Gerlach B, Bodea LG, Klaus C, Ginolhac A, Halder R, Sinkkonen L, Walter J, Colonna M, and Neumann H. 2019. 'TREM2 triggers microglial density and age-related neuronal loss', Glia, 67: 539–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu AH, Chu M, and Wang YP. 2019. 'Up-Regulation of Trem2 Inhibits Hippocampal Neuronal Apoptosis and Alleviates Oxidative Stress in Epilepsy via the PI3K/Akt Pathway in Mice', Neurosci Bull, 35: 471–85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu D, Cao B, Zhao Y, Huang H, McIntyre RS, Rosenblat JD, and Zhou H. 2018. 'Soluble TREM2 changes during the clinical course of Alzheimer's disease: A meta-analysis', Neurosci Lett, 686: 10–16. [DOI] [PubMed] [Google Scholar]
- Shaw LM, Vanderstichele H, Knapik-Czajka M, Clark CM, Aisen PS, Petersen RC, Blennow K, Soares H, Simon A, Lewczuk P, Dean R, Siemers E, Potter W, Lee VM, Trojanowski JQ, and Initiative Alzheimer's Disease Neuroimaging. 2009. 'Cerebrospinal fluid biomarker signature in Alzheimer's disease neuroimaging initiative subjects', Ann Neurol, 65: 403–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Song WM, Joshita S, Zhou Y, Ulland TK, Gilfillan S, and Colonna M. 2018. 'Humanized TREM2 mice reveal microglia-intrinsic and -extrinsic effects of R47H polymorphism', J Exp Med, 215: 745–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suarez-Calvet M, Araque Caballero MA, Kleinberger G, Bateman RJ, Fagan AM, Morris JC, Levin J, Danek A, Ewers M, Haass C, and Network Dominantly Inherited Alzheimer. 2016. 'Early changes in CSF sTREM2 in dominantly inherited Alzheimer's disease occur after amyloid deposition and neuronal injury', Sci Transl Med, 8: 369ra178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suarez-Calvet M, Kleinberger G, Araque Caballero MA, Brendel M, Rominger A, Alcolea D, Fortea J, Lleo A, Blesa R, Gispert JD, Sanchez-Valle R, Antonell A, Rami L, Molinuevo JL, Brosseron F, Traschutz A, Heneka MT, Struyfs H, Engelborghs S, Sleegers K, Van Broeckhoven C, Zetterberg H, Nellgard B, Blennow K, Crispin A, Ewers M, and Haass C. 2016. 'sTREM2 cerebrospinal fluid levels are a potential biomarker for microglia activity in early-stage Alzheimer's disease and associate with neuronal injury markers', EMBO Mol Med, 8: 466–76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suarez-Calvet M, Morenas-Rodriguez E, Kleinberger G, Schlepckow K, Araque Caballero MA, Franzmeier N, Capell A, Fellerer K, Nuscher B, Eren E, Levin J, Deming Y, Piccio L, Karch CM, Cruchaga C, Shaw LM, Trojanowski JQ, Weiner M, Ewers M, Haass C, and Initiative Alzheimer's Disease Neuroimaging. 2019. 'Early increase of CSF sTREM2 in Alzheimer's disease is associated with tau related-neurodegeneration but not with amyloid-beta pathology', Mol Neurodegener, 14: 1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sweeney MD, Sagare AP, and Zlokovic BV. 2018. 'Blood-brain barrier breakdown in Alzheimer disease and other neurodegenerative disorders', Nat Rev Neurol, 14: 133–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taylor X, Cisternas P, You Y, You Y, Xiang S, Marambio Y, Zhang J, Vidal R, and Lasagna-Reeves CA. 2020. 'A1 reactive astrocytes and a loss of TREM2 are associated with an early stage of pathology in a mouse model of cerebral amyloid angiopathy', J Neuroinflammation, 17: 223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trzepacz PT, Hochstetler H, Wang S, Walker B, Saykin AJ, and Initiative Alzheimer's Disease Neuroimaging. 2015. 'Relationship between the Montreal Cognitive Assessment and Mini-mental State Examination for assessment of mild cognitive impairment in older adults', BMC Geriatr, 15: 107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Turnbull IR, Gilfillan S, Cella M, Aoshi T, Miller M, Piccio L, Hernandez M, and Colonna M. 2006. 'Cutting edge: TREM-2 attenuates macrophage activation', J Immunol, 177: 3520–4. [DOI] [PubMed] [Google Scholar]
- Ulland TK, Song WM, Huang SC, Ulrich JD, Sergushichev A, Beatty WL, Loboda AA, Zhou Y, Cairns NJ, Kambal A, Loginicheva E, Gilfillan S, Cella M, Virgin HW, Unanue ER, Wang Y, Artyomov MN, Holtzman DM, and Colonna M. 2017. 'TREM2 Maintains Microglial Metabolic Fitness in Alzheimer's Disease', Cell, 170: 649–63 e13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ulrich JD, Ulland TK, Colonna M, and Holtzman DM. 2017. 'Elucidating the Role of TREM2 in Alzheimer's Disease', Neuron, 94: 237–48. [DOI] [PubMed] [Google Scholar]
- Wang Q, Yang W, Zhang J, Zhao Y, and Xu Y. 2020. 'TREM2 Overexpression Attenuates Cognitive Deficits in Experimental Models of Vascular Dementia', Neural Plast, 2020: 8834275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang S, Mustafa M, Yuede CM, Salazar SV, Kong P, Long H, Ward M, Siddiqui O, Paul R, Gilfillan S, Ibrahim A, Rhinn H, Tassi I, Rosenthal A, Schwabe T, and Colonna M. 2020. 'Anti-human TREM2 induces microglia proliferation and reduces pathology in an Alzheimer's disease model', J Exp Med, 217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Y, Cella M, Mallinson K, Ulrich JD, Young KL, Robinette ML, Gilfillan S, Krishnan GM, Sudhakar S, Zinselmeyer BH, Holtzman DM, Cirrito JR, and Colonna M. 2015. 'TREM2 lipid sensing sustains the microglial response in an Alzheimer's disease model', Cell, 160: 1061–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Y, Ulland TK, Ulrich JD, Song W, Tzaferis JA, Hole JT, Yuan P, Mahan TE, Shi Y, Gilfillan S, Cella M, Grutzendler J, DeMattos RB, Cirrito JR, Holtzman DM, and Colonna M. 2016. 'TREM2-mediated early microglial response limits diffusion and toxicity of amyloid plaques', J Exp Med, 213: 667–75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wei W, Nguyen LN, Kessels HW, Hagiwara H, Sisodia S, and Malinow R. 2010. 'Amyloid beta from axons and dendrites reduces local spine number and plasticity', Nat Neurosci, 13: 190–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu R, Li X, Xu P, Huang L, Cheng J, Huang X, Jiang J, Wu LJ, and Tang Y. 2017. 'TREM2 protects against cerebral ischemia/reperfusion injury', Mol Brain, 10: 20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yuan P, Condello C, Keene CD, Wang Y, Bird TD, Paul SM, Luo W, Colonna M, Baddeley D, and Grutzendler J. 2016. 'TREM2 Haplodeficiency in Mice and Humans Impairs the Microglia Barrier Function Leading to Decreased Amyloid Compaction and Severe Axonal Dystrophy', Neuron, 90: 724–39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhong L, Chen XF, Wang T, Wang Z, Liao C, Wang Z, Huang R, Wang D, Li X, Wu L, Jia L, Zheng H, Painter M, Atagi Y, Liu CC, Zhang YW, Fryer JD, Xu H, and Bu G. 2017. 'Soluble TREM2 induces inflammatory responses and enhances microglial survival', J Exp Med, 214: 597–607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhong L, Xu Y, Zhuo R, Wang T, Wang K, Huang R, Wang D, Gao Y, Zhu Y, Sheng X, Chen K, Wang N, Zhu L, Can D, Marten Y, Shinohara M, Liu CC, Du D, Sun H, Wen L, Xu H, Bu G, and Chen XF. 2019. 'Soluble TREM2 ameliorates pathological phenotypes by modulating microglial functions in an Alzheimer's disease model', Nat Commun, 10: 1365. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Unadjusted scatter plots showing CSF AD biomarkers (x axis) by CSF sTREM2 (y axis) after visual and statistical outlier removal. Statistical outliers were determined by values 4 standard deviations outside of the mean measurement.
Correlation matrix between sTREM2 and additional CSF biomarker measurements in VMAP. Non-significant Pearson’s correlation coefficients are denoted by an “X”.
Aβ and BBB biomarkers do not significantly interact with tau status on sTREM2 levels in CSF: (A) Aβx-40 * tau status, p.int.=0.64, and (B) CSF/plasma albumin ratio * tau status, p.int.=0.25. Protein measurements given in pg/mL.
Replication of Aβ40 * tau status interaction on sTREM2. Aβ1-40 does not significantly interact with tau status on sTREM2 levels in CSF using ADNI data: Aβ1-40 * tau status, p.int. =0.22. Protein measurements given in pg/mL.
Using ADNI data, sTREM2 decouples from tau as disease progresses. Protein measurements given in pg/mL.