

Achieving a thorough understanding of reactions in liquids requires measurements at the molecular level and with sufficient time-resolution to map the dynamics (1). These studies could resolve the structure-function correlation of these reactions (2). On page xxx of this issue, Lin et al. (3) used liquid-phase ultrafast electron diffraction (UED) to capture the transient structure of the short-lived OH(H3O+) radical-cation pairs produced in photoionization of water at atomic spatiotemporal resolution. The pairs dissociate into an OH radical and an H3O+ cation in ~250 fs. In addition, a concomitant ultrafast heating of the liquid water was recorded by tracing the structural change of the heated water. These observations reveal the transient reaction intermediates and the dynamic flow of energy from ionization centers to water, as can occur during radiolysis, a reaction that is ubiquitous in nature.
The elementary steps associated with the ionization of liquid water provide a framework for understanding radiation-matter interactions in chemistry and biology (4), which are essential to photosynthesis and other biological reactions. Ionization of liquid water forms hydrated electrons and water cations (H2O+). Subsequent ultrafast proton transfer from H2O+ to H2O produces a hydroxyl radical (OH) and a hydronium cation (H3O+). This process takes place in < 100 fs, with concurrent hydration, relaxation, and thermalization of the ionized electrons. At longer times, the hydrated and thermalized electrons recombine with the ion cores and OH radicals through either geminate or non-geminate recombination that proceeds on time scales between tens of picoseconds to nanoseconds (3). Previous time-resolved measurements and theoretical studies have reported the elementary steps involved and the lifetimes of transient species. However, direct measurements of these transient structures could not be accomplished given the limitations of time-resolved structural probes.
In general, direct measurement of transient molecular structures can identify the reaction intermediates and trace how reactants go through intermediates to final products (2). However, chemical bond formation and breakage must be followed on the femtosecond time scale and at sub-Ångström spatial resolution, which became possible only after the advent of femtosecond lasers in the early 1980s. Much like the function of a high-speed shutter and the fast flashlight in high-speed photography, femtosecond laser pulses can take snapshots of the motions of atoms, and transient optical spectroscopies based on ultrafast pulses have provided important insights into reaction dynamics. However, when applying these optical techniques to large and complex molecular systems, formidable obstacles are encountered in linking the transient spectroscopy to the dynamics of nuclear motions.
Capturing the subtle changes of bond distances in molecules undergoing an ultrafast chemical reaction requires diffraction methods with large momentum transfer and femtosecond temporal resolution, such as ultrafast electron (5–7) and x-ray diffraction techniques (8, 9). In contrast to optical spectroscopies, time-resolved diffraction can record all of the chemical species in a reaction and identify each individual piece by its diffraction fingerprint (2), and provide a global view of reaction dynamics. This capability is crucial to mapping solution-phase reaction dynamics, which involves not only reactants, intermediates, and products, but also solvent molecules that can alter the reaction dynamics in many aspects (1).
The development of liquid-phase megaelectron volt (MeV) UED (10) provides the much needed sub-Ångström spatial and femtosecond temporal resolution together with high detection sensitivity to trace solution-phase reaction dynamics. The much stronger electron scattering with atoms, about five orders of magnitude larger than that of x-ray (5), allows simultaneous measurement of the O··H and O··O intermolecular bonds. In contrast, x-ray diffraction is specifically sensitive only to the heavier-atom O··O bond. Probing the intermolecular O··H network is critical for understanding the reaction mechanisms involving the hydrogen-bond interaction in ionized water. Besides high detection sensitivity, relativistic effects start to set in at MeV beam energies and greatly ease the UED performance degradation instigated by the space-charge effect (7) and are crucial in maintaining time resolution.
Another important innovation in this study is the development of a sub-100 nanometer ultrathin liquid jet in the high-vacuum environment (11) and integrating it into the MeV UED system (10). Generation of ultrathin free-flowing liquid sheets with a microfluidic nozzle mitigates the strong water absorption and scattering, making it possible for interrogating aqueous systems with spectroscopic and structural probes. It also allows control over sample content and on-the-fly sample exchange, thus facilitating a high-throughput data acquisition.
The realization of real-time imaging of the short-lived (OHH3O+) pair at the atomic resolution in the photolysis of water revolutionizes the field of ultrafast electron diffraction. It opens doors to conducting measurements on more complex solution-based reactions. Similar studies of photosynthesis and other biological photoreactions can be performed to gain new insight for affordable renewable energy sources and potential drug therapeutics. One such complex system is the recently well-studied repair of UV-induced DNA damage by enzyme photolyase, where ten elementary steps in the catalytic reaction involving seven electron-transfer processes have been identified by transient spectroscopic measurements (see the figure). Under blue light, photolyase uses a redox photocycle between the flavin cofactor in the enzyme and the cyclobutane pyrimidine dimer in DNA to sequentially split two single bonds to restore normal bases on an ultrafast time scale (12). The many intermediates formed in this complicated repair process and their dynamic evolution could in theory be traced by ultrafast diffraction measurement, which would provide the molecular basis for development of anticancer drugs.
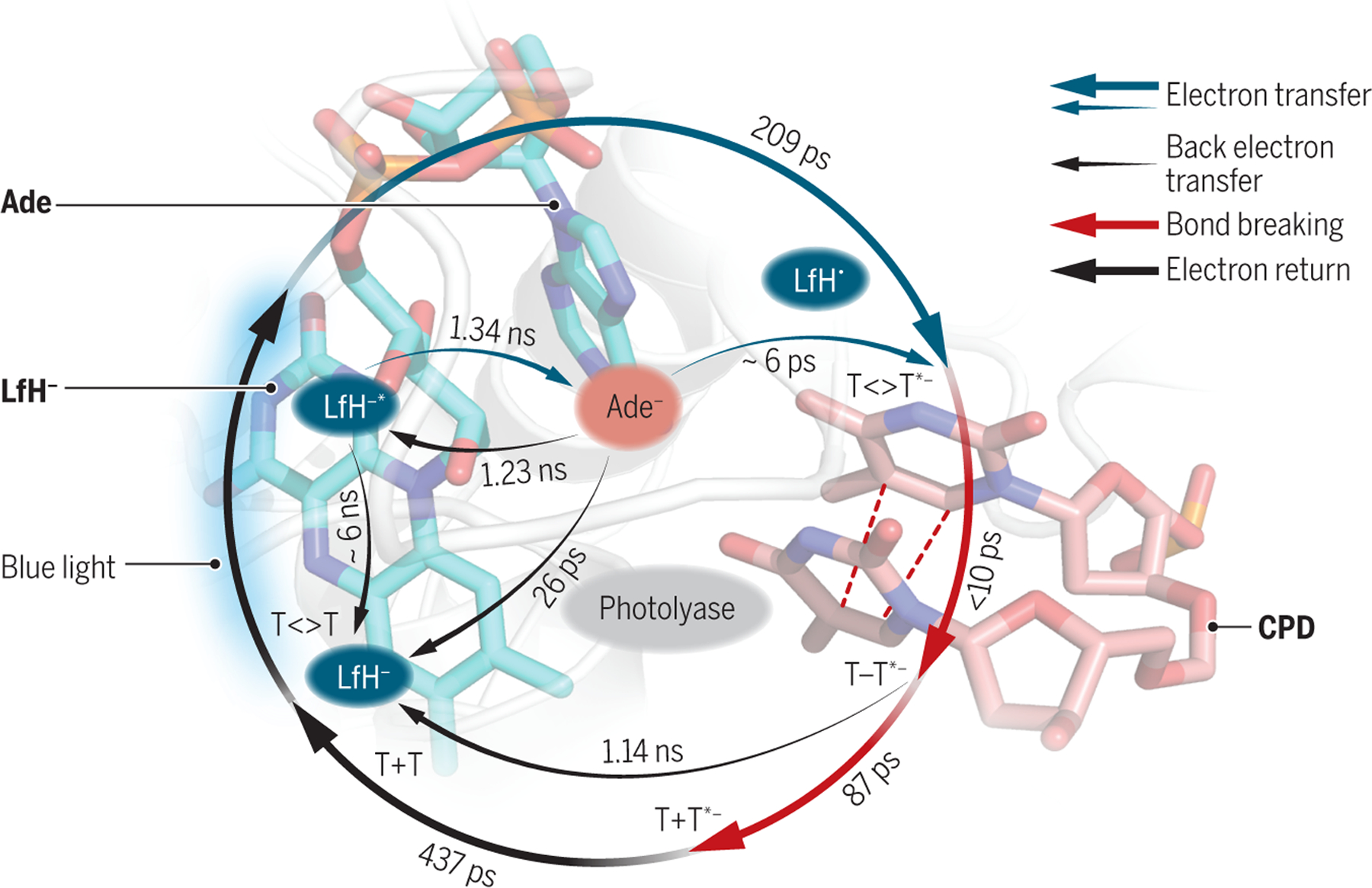
Ultrafast structure dynamics beyond water
Ultrafast solution-phase electron diffraction studies, as used by Lin et al. could be applied to other photoactivated reactions. For example, the structures of intermediates in the enzyme repair of the thymine dimer (T<>T) in DNA are largely unknown, although the time scales are known from ultrafast transient adsorption (12).
Photolyase repair
Blue light powers the redox cycle in photolyase between the flavin cofactor (LfH-) and the adenine (Ade) in the enzyme and the cyclobutane pyrimidine dimer (CPD) to sequentially split two single bonds (red dashed lines). The states that form from flavin are shown in blue ovals.
ACKNOWLEDGMENTS
We thank the National Natural Science Foundation of China for the support of the collaboration work on ultrafast science during the summer stays of J.C. and D.Z. at the Center. D.Z. also like to thank the support from National Institute of Health (GM144047).
REFERENCES AND NOTES
- 1.Orr-Ewing AJ, Chem. Soc. Rev 46, 7597 (2017). [DOI] [PubMed] [Google Scholar]
- 2.Zewail AH, Thomas JM, 4D Electron Microscopy: imaging in space and time (World Scientific Publishing, 2009). [Google Scholar]
- 3.Lin M-F et al. , Science 374, 92 (2020). [Google Scholar]
- 4.Loh ZH et al. , Science 367, 179 (2020). [DOI] [PubMed] [Google Scholar]
- 5.Cao J et al. , Appl. Phys. Lett 83, 1044 (2003). [Google Scholar]
- 6.Siwick BJ, Dwyer JR, Jordan RE, Miller RJD, Science 302, 1382 (2003). [DOI] [PubMed] [Google Scholar]
- 7.Wang XJ, Ihee H, Femto-seconds electron beam diffraction using photocathode RF gun. Chew J, Lucas P, Webber S, Eds., Proceedings of the 2003 Particle Accelerator Conference, Vols 1–5 (2003), pp. 420–422. [Google Scholar]
- 8.Zalden P et al. , Science 364, 1062 (2019). [DOI] [PubMed] [Google Scholar]
- 9.Yun JH et al. , Proc. Natl. Acad. Sci. U.S.A 118, e2020486118 (2021).33753488 [Google Scholar]
- 10.Nunes JPF et al. , Struct. Dyn 7, 024301 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Koralek JD et al. , Nat. Commun 9, 1353 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Zhang M, Wang LJ, Shu S, Sancar A, Zhong DP, Science 354, 209 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]