

ABSTRACT
RAS is the most frequently mutated oncogene in human cancer with nearly ~20% of cancer patients possessing mutations in one of three RAS genes (K, N or HRAS). However, KRAS is mutated in nearly 90% of pancreatic ductal carcinomas (PDAC). Although pharmacological inhibition of RAS has been challenging, KRAS(G12C)-specific inhibitors have recently entered the clinic. While KRAS(G12C) is frequently expressed in lung cancers, it is rare in PDAC. Thus, more broadly efficacious RAS inhibitors are needed for treating KRAS mutant-driven cancers such as PDAC. A RAS-specific tool biologic, NS1 Monobody, inhibits HRAS- and KRAS-mediated signalling and oncogenic transformation both in vitro and in vivo by targeting the α4–α5 allosteric site of RAS and blocking RAS self-association. Here, we evaluated the efficacy of targeting the α4-α5 interface of KRAS as an approach to inhibit PDAC development using an immunocompetent orthotopic mouse model. Chemically regulated NS1 expression inhibited ERK and AKT activation in KRAS(G12D) mutant KPC PDAC cells and reduced the formation and progression of pancreatic tumours. NS1-expressing tumours were characterized by increased infiltration of CD4 + T helper cells. These results suggest that targeting the #x3B1;4-#x3B1;5 allosteric site of KRAS may represent a viable therapeutic approach for inhibiting KRAS-mutant pancreatic tumours.
KEYWORDS: Pancreatic ductal adenocarcinoma, tumourigenesis, Monobody, T-cell
Introduction
RAS proteins are small GTPases that function as molecular switches that alternate between an active, GTP–bound form (RAS·GTP) and an inactive, GDP–bound form (RAS·GDP) [1,2]. Normally, RAS·GDP is converted to the active state by guanine nucleotide exchange factors (GEFs) that promote the release of GDP and subsequent uptake of GTP. RAS·GTP is converted to the inactive GDP-bound form through its intrinsic GTPase activity, which is enhanced by GTPase accelerating proteins (GAPs) [3–6]. Mutations in RAS are frequently observed in cancer, particularly at codons 12, 13 and 61, and promote the shift of RAS to the active, GTP-bound state [7–9]. Such mutations increase the engagement of RAS with downstream effectors to enhance cancer cell survival, proliferation and tumourigenesis. Furthermore, many tumours that lack RAS mutations nevertheless rely on RAS activity due to mutations in upstream RAS activators such as growth factor receptors [10].
Although many groups have isolated promising compounds that bind and inhibit RAS in preclinical settings, none have yet progressed to successful clinical trials and FDA approval [11]. However, the recent success with mutation-specific KRAS(G12C) inhibitors has revived hope for the possibility of direct pharmacological inhibition of KRAS. These G12C-specific compounds bind in a previously unrecognized pocket near the SW2 region and utilize chemical warheads that react with the thiol group of Cys12 of mutant KRAS [12–14]. Two such inhibitors, AMG510 (Sotorasib) and MRTX849 (Adagrasib), have entered clinical trials and show promising clinical efficacy thus far [15,16]. However, despite these promising results, these compounds are limited in their pharmacological efficacy to treating tumours harbouring KRAS(G12C) which is present in only 2.2% of all KRAS mutant cancers and particularly rare in PDACs [8]. Thus, there remains a significant unmet need to devise novel approaches to pharmacologically inhibit additional mutant KRAS proteins.
Previously, we identified a potent RAS inhibitory biologic called NS1 using Monobody technology, an engineered binding protein platform that has produced high-affinity and high-specificity binders to diverse targets [17–19]. NS1 selectively binds with low nanomolar affinity to KRAS and HRAS but not to NRAS. Further, NS1 specifically inhibits the biochemical and biological activity HRAS and KRAS but not other oncogenic GTPases or downstream oncogenic kinases. Biochemical and biophysical studies revealed that NS1 targets the ⍺4-⍺5 interface of RAS to inhibit dimerization and nanoclustering of RAS resulting in inhibition of RAS-induced RAF dimerization and activation [20–22]. Although NS1 potently inhibits oncogenic signalling and transformation in experimental models both in vitro and in vivo, an important remaining question is whether targeting the ⍺4-⍺5 interface represents a viable approach to block PDAC in vivo. Here, we demonstrate that NS1 expression reduced PDAC tumour development in an orthotopic, immunocompetent model of PDAC. These findings suggest that targeting the ⍺4-⍺5 allosteric site on RAS by drug-like NS1 mimetics may represent a viable approach to inhibit KRAS-mutant PDACs.
Results
NS1 binds and inhibits murine KRAS both in vitro and in cellulo
Although human and mouse KRAS are nearly identical, residue 132 differs between the two orthologs (Figure 1(a)). D132 (human) is located at the periphery of the NS1-RAS interface and its side chain directly contacts NS1 (Figure 1(a)). Given that arginine(R) 135 in HRAS/KRAS determines the specificity of NS1 and presence of lysine(K) at similar position in NRAS abolishes NS1 binding [20], we tested whether glutamic acid (E) at position 132 of the murine KRAS altered NS1 binding. Quantitative in vitro binding analysis using yeast surface display demonstrated that NS1 bound equivalently to both human (apparent KD = 37 ± 2 nM) and murine (apparent KD = 30 ± 5 nM) KRAS (Figure 1(b)). In addition, CFP-FLAG-tagged NS1 (hereafter referred to as CFP-NS1) interacted similarly with KRAS(G12V) harbouring either D132 (human) or E132 (murine) (Figure 1(c)). Furthermore, CFP-NS1 inhibited ERK-MAPK activation by both versions of KRAS(G12V) (Figure 1(d)). Consistent with these results, CFP-NS1 inhibited the biological transformation of NIH/3T3s by both human and murine KRAS(G12V) (Figure 1(e)). These results demonstrate that NS1 is equally potent at inhibiting signalling and biological transformation mediated by oncogenic human or murine KRAS.
Figure 1.
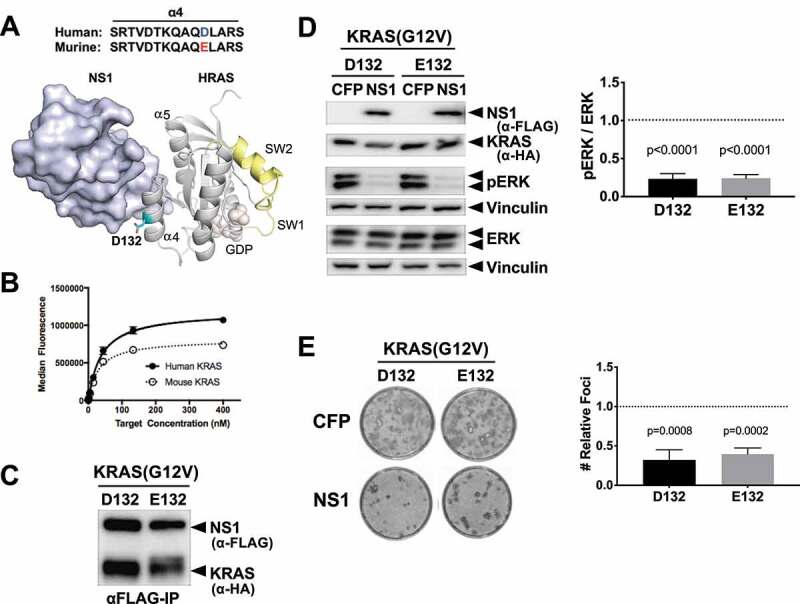
NS1 binds and potently inhibits murine RAS. A) The location of D132 in the crystal structure of the HRAS-NS1 complex (PDB 5E95). The ⍺4 and ⍺5 helices, switch 1 and 2, and GDP are also denoted. Alignment of the ⍺4 sequence of the human and murine KRAS is shown above the HRAS-NS1 structure. B) Yeast display binding of NS1 to human and murine KRAS isoforms bound to GDP. Affinity values (apparent KD) were as follows: human HRAS, 37 ± 2 nM; mouse KRAS, 30 ± 5 nM. Error bars represent s.d. of n = 3 independent binding experiments. a.u., arbitrary units. C) Coimmunoprecipitation of HA-tagged KRAS(G12V) orthologs with CFP-FLAG tagged NS1. Human (D132) and murine (E132) KRAS(G12V) were co-expressed with CFP-FLAG-NS1 (NS1) in HEK293 cells. D) Effects of NS1 on ERK-MAPK activation by human (D132) and murine (E132) KRAS(G12V) orthologs. HEK293 cells were co-transfected with KRAS(G12V) ortholog and either CFP or CFP-FLAG-NS1 (NS1). ERK activation by KRAS(G12V) was determined by Western blot of whole cell lysates with antibodies specific for phosphorylated ERK (pERK). Vinculin was used as a normalization control for protein loading. Quantification of relative ERK activation from 3 independent experiments ± s.d. is shown in the graph. p values correspond to the difference between pERK levels in CFP (dotted line) vs CFP-NS1 for each KRAS protein and were calculated by unpaired, two-tailed t test. E) NIH/3T3 cells were co-transfected with indicated KRAS(G12V) ortholog and either CFP- alone or CFP-FLAG-NS1. Representative plates of foci from each condition are shown. Quantification of relative foci number for each oncogene is shown in the graphs. Results represent the ratio of foci number in the presence of CFP-NS1 to that with CFP alone (dotted line) and are shown as mean ± s.d. of three independent experiments performed in triplicate. p values are shown above each column and were calculated by unpaired, two-tailed t test between CFP and CFP-NS1 for each KRAS protein.
NS1 inhibits KRAS mutant murine PDAC cells
Next, we examined whether NS1 inhibited PDAC cells derived from the KPC mouse model (KrasG12D/+;TP53R172H/+) [23]. We established a derivative of KPC cells (KPCNS1) that expressed CFP-NS1 following administration of doxycycline (DOX) (Figure 2). Consistent with our in vitro results (Figure 1(b,Figure 1c)), immunoprecipitation of CFP-NS1 revealed similar binding to endogenous KRAS from KPCNS1 cells vs human CFPAC1NS1 cells (Figure 2(a)). Further, DOX-induced expression of CFP-NS1 in KPCNS1 cells resulted in a robust decrease in both active ERK (Figure 2(b,c)) and active AKT (Figure 2(b,d) and Extended Figure 1(a)). Similar results for MAPK signalling were previously demonstrated in human PDAC cells [22].
Figure 2.
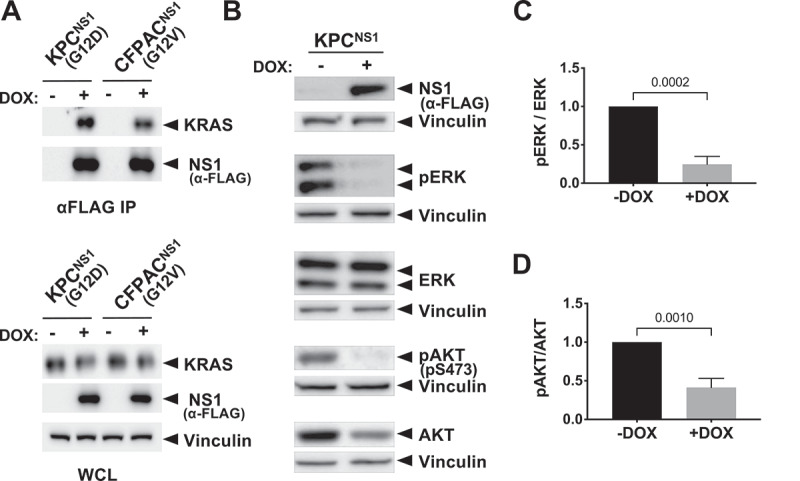
NS1 inhibits KRAS signalling in mouse and human PDAC cells. Mutant KRAS-expressing murine (KPC) and human (CFPAC) PDAC cell lines were infected with lentivirus encoding a doxycycline (DOX)-inducible NS1 expression construct to generate stable cell lines. A) Following DOX induction, NS1 was immunoprecipitated from both KPCNS1 and CFPACNS1 cells followed by Western blot with a KRAS antibody (OP24, Millipore Sigma). Panels below illustrate expression of proteins in whole cell lysates (WCL). B) Effects of DOX-induced NS1 expression on ERK and AKT activation in mouse KPCNS1 PDAC cells. ERK and AKT activation was measured by Western blot of cell lysates with phosphospecific antibodies. Vinculin was used as a normalization control for protein loading. C, D) Quantification of pERK (c) or pAKT (d) from KPCNS1 cell lysates in (b) was done using LI-COR Biosciences Image Studio Lite software (v.5.2.5) and presented as relative pERK or pAKT activation compared to no DOX lysates. Error bars represent s.d. from n = 3 independent experiments. p values were calculated by unpaired, two-tailed t test and are indicated on the graphs.
Given the ability of NS1 to disrupt KRAS recruitment and dimerization of RAF [20], we next evaluated the effects of NS1 on murine KRAS recruitment and heterodimerization of RAF. CRAF was immunoprecipitated from lysates of KPCNS1 and CFPACNS1 cells treated ±DOX. Consistent with our prior results in HEK293 cells [20], NS1 impaired both KRAS:CRAF association and BRAF:CRAF heterodimerization (Extended Figure 1(b,c,d)).
Given the potent inhibition of signalling in KPCNS1 following DOX treatment, we next evaluated the ability of CFP-NS1 to inhibit the growth of KPCNS1 cells. Indeed, the proliferation of KPCNS1 cells in 2D culture was significantly reduced upon DOX-induced CFP-NS1 expression (Figure 3(a)). Furthermore, DOX treatment of KPCNS1 cells resulted in a comparable inhibition of anchorage-independent growth in soft agar consistent with the effects that NS1 induces on signalling and cell proliferation in 2D (Figure 3(b,c)). As expected, treatment of parental KPC cells with DOX did not affect MAPK activation or proliferation in either 2D or 3D conditions (Extended Figure 2). These results demonstrate the efficacy of targeting the α4-α5 region of oncogenic KRAS(G12D) to inhibit the growth of PDAC cells in both 2D and 3D culture conditions.
Figure 3.
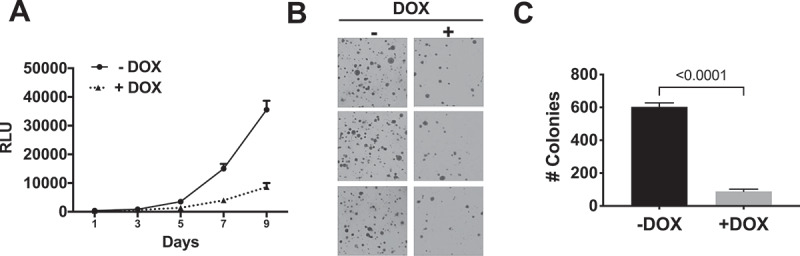
NS1 inhibits growth of KPC cells in vitro. A) Effects of DOX-induced NS1 expression on proliferation of KPCNS1 cells. Results at each time point represent the average of triplicate wells. B) Effect of DOX-induced NS1 expression on anchorage-independent growth of KPCNS1 cells. KPCNS1cells were plated in soft agar and grown ±DOX. C) Colony numbers from three independent wells for each condition were quantified using NIH ImageJ software. Graphs represent the average number of colonies per well ± s.d. p value was calculated by unpaired, two-tailed t test between -DOX and +DOX conditions and is indicated on the graphs.
Further, to rule out any ‘off-target effects’, we evaluated the effects of NS1 in various non-cancerous transformed model cell systems (HEK293NS1 and NIH/3T3NS1). Unlike RAS mutant KPC cells, expression of CFP-NS1 did not affect the proliferation of non-RAS mutant HEK293NS1 or NIH/3T3NS1 cells (Extended Figure 3(a,b)). To determine if NS1 expression in these lines was optimal to inhibit RAS signalling, we transfected HEK293NS1 with oncogenic HRAS(Q61L) (Extended Figure 3(c)). DOX-induced expression of CFP-NS1 inhibited ERK-MAPK activation mediated by HRAS(Q61L) (Extended Figure 3(c)).
In vivo antitumour activity of NS1 in a syngeneic mouse model
We previously demonstrated that NS1 inhibited growth of RAS mutant human tumour cells in an athymic xenograft mouse model suggesting that targeting the α4-α5 region was sufficient to inhibit tumourigenesis [22]. However, given that this model represents an immunocompromised environment, we next addressed whether these results could be translated to a model that more closely mimics human pancreatic tumour development. Thus, KPCNS1 cells (which also expressed enhanced firefly luciferase) were injected directly into the pancreas of immunocompetent C57Bl/6 syngeneic mice. Untreated KPCNS1 cells established tumours with similar kinetics as the parental KPC cells (data not shown), with tumours detected as early as 3 days post injection (PI) by bioluminescence imaging (Figure 4). Treatment of KPCNS1-injected mice with DOX beginning on day 1 (D1) post-injection (PI), resulted in a significant diminution of tumour progression compared to untreated control mice (Figure 4(a,b)).
Figure 4.
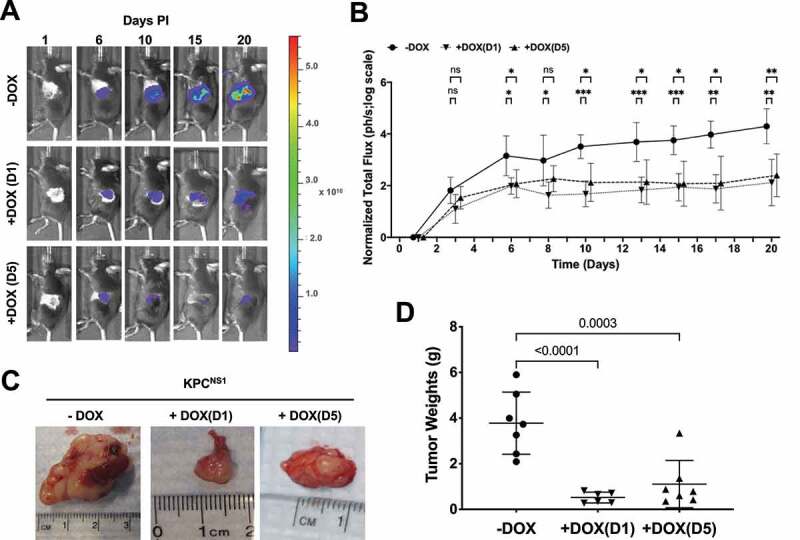
Targeting ⍺4-⍺5 interface of KRAS inhibits PDAC tumour growth. A) C57BL/6 J mice injected with KPCNS1 cells were segregated into three groups, no DOX treatment [(-DOX); n = 7], DOX treated at day 1 post injection (PI) [+DOX(D1); n = 6] or DOX treated at day 5 PI [+DOX(D5); n = 7]. A representative mouse is shown from each cohort. Colour scale (radiance; units are photons per sec per cm2 per steradian): Min = 2.00e8, Max = 5.60e10 B) Tumour growth kinetics in various cohorts of KPCNS1 mice based on the quantification of luciferase activity by IVIS imaging. p values were calculated using mixed-effects analysis with Dunnett’s multiple comparisons test comparing -DOX group to +DOX(D1) and +DOX(D5), respectively, for each time point. ***p < 0.001, **p < 0.01, *p < 0.05, ns = not significant. C) Representative images of pancreatic tumours. D) Quantification of tumour weights. The individual weights of tumours and mice body weights (Extended Figure 4a) from the cohorts were plotted and p values were calculated using one-way ANOVA with Dunnett’s multiple comparisons test and comparing each condition to -DOX control.
To further address whether NS1 might inhibit established tumours, we treated a separate cohort of KPCNS1-injected mice with DOX at day 5 PI (D5). As with early DOX treatment and NS1 induction, later expression of NS1 substantially inhibited growth of established pancreatic tumours compared to the untreated cohort (Figure 4(a,b)). Furthermore, NS1 expression resulted in a significant decrease in tumour burden as illustrated by the difference in tumour weights in both DOX-treated cohorts compared to untreated controls (Figure 4(c,d)). The expression of NS1 following DOX administration did not affect the body weights of +DOX(D5) cohort; however, a slight but significant (p = 0.0410) decrease in body weight was observed in +DOX(D1) group (Extended Figure 4(a)).
NS1 inhibits RAS signalling and induces apoptosis in pancreatic tumours
To determine the effect of NS1 on KRAS signalling in tumours, we examined whether expression of CFP-NS1 inhibited KRAS(G12D) activation of ERK-MAPK in vivo. Western blot analysis of tumour lysates indicated that CFP-NS1 expression was only observed in DOX treated cohorts (Figure 5a). NS1 inhibited KRAS activation of ERK-MAPK in the DOX treated cohorts (Figure 5(a) and Extended Figure 4(b)). In addition, we observed that CFP-NS1 induction resulted in activation of caspase-3 (Figure 5(b) and Extended Figure 4(c)). Comparison of luciferase levels between tumour samples indicated that samples were comprised of similar proportions of KPC cells. Thus, NS1 inhibited the kinetics of tumour growth by decreasing ERK-MAPK activation and increasing apoptosis, i.e., increased caspase-3 activation (Figure 5 and Extended Figure 4(b,c)).
Figure 5.
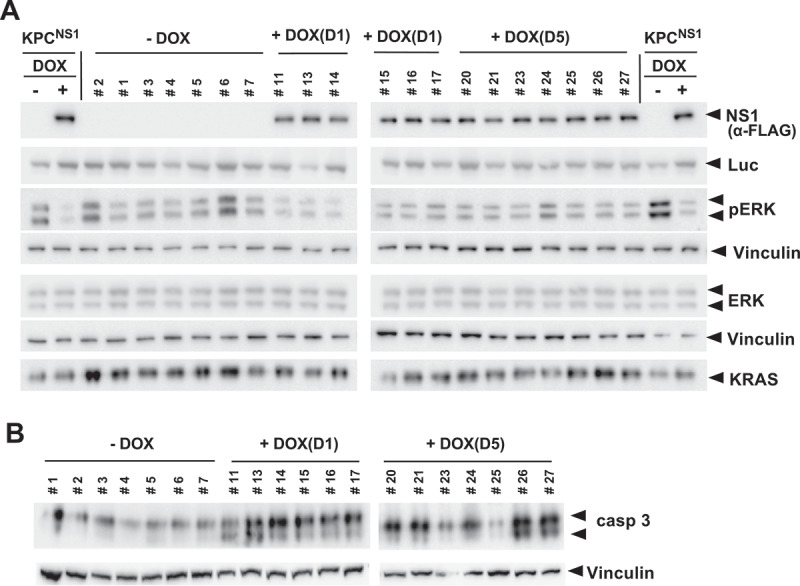
NS1 inhibits ERK phosphorylation and induces apoptosis in vivo. A) Lysates of KPCNS1 PDAC tumours were analysed by western blot for activation of ERK. The homogeneity of KPC cells in each tumour is illustrated by the anti-luciferase (Luc) blot. NS1 expression was seen only in DOX-treated samples. B) Caspase-3 activation was measured by Western blot using cleaved Caspase-3 (Asp175) antibody (casp3). Vinculin was used as a loading control for ERK, pERK and caspase-3 blots. Quantification of blots is shown in Extended Figure 4b and c.
KRAS inhibition by NS1 renders pancreatic tumours vulnerable to immune attack
PDAC tumours are characterized by an immunosuppressive microenvironment that is hostile to effector immune cells [24–26]. Recent studies suggest that KRAS inhibition in lung cancer models reconditioned the immunosuppressive tumour microenvironment to immune favouring anti-tumour environment mainly by improving T-cell mediated adaptive immunity [15,27]. To understand the effects of selective inhibition of KRAS by NS1 on the tumour immune response in PDACs, tumours from the various cohorts were immunophenotyped to evaluate the infiltration of different pools of T cells and antigen presenting cells (APCs). Immunohistochemical analysis revealed a significant increase in total CD4+ T helper cells with no substantial change in total cytotoxic CD8+ T cells (Figure 6). Using an established macrophage marker (F4/80), we did not observe a significant increase in tumour-infiltrating macrophages from mice treated at D1 or D5 compared to untreated control mice. Furthermore, the decrease in tumour favouring M2 macrophages (CD206+) in DOX-treated cohorts was not significant (Extended Figure 5.
Figure 6.
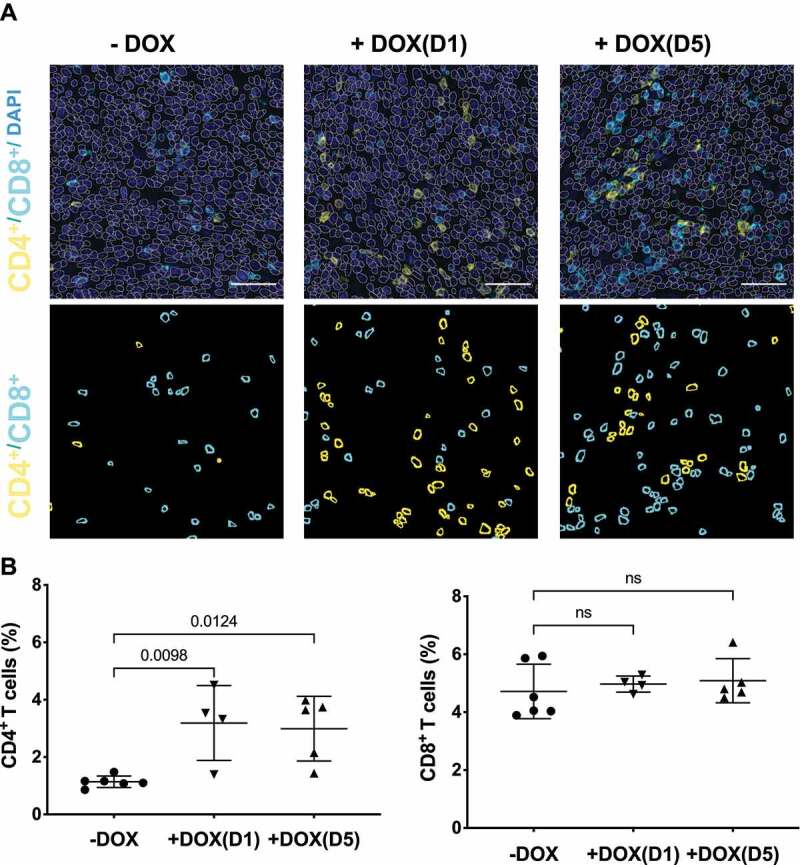
NS1 inhibition of KRAS stimulates an adaptive immune response. Tumours from KPCNS1 PDACs were immunophenotyped by immunohistochemistry for CD4+ or CD8+ (A) Images acquired from tumours in an untreated mouse, a mouse treated +DOX(D1), and a mouse treated +DOX(D5). First row: DAPI staining in blue, CD4 in yellow and CD8 in cyan. The segmented nuclei are overlaid as grey circles. The scale bar corresponds to 50 µm. Second row: segmentation masks showing identified CD4+ and CD8+ cells. B) Proportion of CD4+ and CD8+ cells for untreated, +DOX(D1), and +DOX(D5) mice. p values were calculated using one-way ANOVA with Dunnett’s multiple comparisons test comparing each condition to -DOX control.
Discussion
Since its discovery as an oncogene in the early 1980s, RAS has been implicated in the direct onset and progression of many human cancers, most notably pancreatic cancer, lung adenocarcinoma and colorectal cancer [8,9]. Direct pharmacological inhibition of RAS, however, has been challenging due in part to an apparent lack of amenable binding pockets for candidate compounds [28,29]. The recent success with covalent inhibitors that selectively target KRAS(G12C), such as AMG-510 and MRTX849, has demonstrated that direct RAS inhibition is indeed feasible [15,16]. However, the remaining KRAS mutant proteins lack the requisite chemical properties for the development of covalent inhibitors as seen with the (G12C) mutant [30]. Thus, alternative strategies are needed to directly inhibit more prominent KRAS mutation such as (G12D) and (G12V) that account for the majority of oncogenic KRAS mutations in PDACs [9,31].
NS1 Monobody binds the ⍺4-⍺5 interface to selectively inhibit HRAS and KRAS but not NRAS [20–22]. Although murine and human KRAS differ by a single amino acid in the ⍺4 helix, this difference does not affect the binding or inhibitory activity of NS1 towards murine KRAS. Indeed, when expressed in murine PDAC cells, NS1 inhibited signalling, proliferation (both in anchorage-dependent and independent conditions), and pancreatic tumour growth. The inhibition of KRAS(G12D) mutant PDAC tumours by NS1 resulted in an anti-tumour adaptive immune response that differed from the response seen with KRAS(G12C) inhibitors. Direct inhibition of KRAS(G12C) with AMG510 or MRTX849 reduced the immune suppressive environment in a KRAS(G12C)-mutant colorectal tumour model and enhanced anti-tumour immunoreactivity by significantly improving both helper CD4+ and cytotoxic CD8+ pools of T cells [15,16,27,32]. We observed an increase in CD4+ T cells in tumours following NS1 induction and a slight, but not statistically significant, difference in the number of tumourinfiltrating CD8+ T cells and macrophages. CD8+ cytotoxic T lymphocytes are a major force of adaptive immunity while CD4+ cells have classically been viewed as supporting cells in T cell mediated adaptive immune response. However, recent findings from both preclinical and clinical settings have demonstrated CD4+ T cells play a critical role in developing and sustaining effective anti-tumour immunity without engaging CD8+ T cells [33–39]. Interestingly, the colorectal cancer mouse model generated by DOX inducible KRAS(G12D) expression displayed low infiltration with CD4+ T cells and extinguishing KRAS signalling by DOX withdrawal led to increased CD4+ infiltration [40]. Thus, the difference in immune response with NS1 inhibition of KRAS vs inhibition with covalent (G12C) inhibitors may stem from the different tumour models or potentially different immunological responses to distinct KRAS mutant proteins. Indeed, recent studies illustrate that different KRAS mutations lead to distinct biochemical, cellular, and pathological responses in different tissues [41,42].
Despite potent decrease in MAPK signalling and induction of apoptosis, NS1 did not completely inhibit the tumour formation. The incomplete tumour reduction is likely due to at least two possibilities. First, the KPCNS1 cells represent a polyclonal population and not all cells will express sufficient levels of NS1 to inhibit KRAS. Further a subpopulation of cells may have lost CFP-NS1 expression or become resistant to the inhibitory effects of NS1. Indeed, we have observed development of resistance in prior xenograft experiments.
Although the role of dimerization in RAS function remains controversial, recent findings from several groups support a role for the α4-α5 interface (and dimerization) in RAS function. Mutations in the α4-α5 region disrupted KRAS dimerization/clustering and reduced KRAS-mediated oncogenicity [43]. Two KRAS-specific DARPins, K13 and K19, inhibited KRAS by binding the α3-α4 interface to disrupt dimerization and inhibit KRAS function [44]. Furthermore, recent work on DIRAS3, a RAS-family GTPase that antagonizes RAS function, suggested that DIRAS3 functioned in part as an endogenous RAS dimerization inhibitor [45]. Like NS1, DIRAS3 specifically bound HRAS and KRAS, but not NRAS, through interaction with the α5 helix, thereby decreasing KRAS dimerization/nanoclustering and function. Thus, these studies highlight the importance of targeting the α4-α5 interface of RAS as a potential avenue to inhibit KRAS addicted tumours such as PDACs.
Consistent with our previous findings, the inhibition of KRAS oncogenesis by NS1 Monobody in both human (CFPAC-1) and murine (KPC) PDAC cells is mediated in part by decreased KRAS-RAF association [20,21]. The decrease in KRAS-RAF engagement further decreased the CRAF-BRAF heterodimerization and consequently impaired the ERK-MAPK signalling. However, as we have reported previously, the inhibitory effects of NS1 on RAS function may be mediated by multiple modes and NS1 shows isoform-specific effects with regard to RAF binding and plasma membrane localization [21]. For oncogenic KRAS, NS1 inhibits self-association, effector engagement, and plasma membrane localization, whereas the inhibition of oncogenic HRAS is mainly driven by decreased RAS self-association [20,21]. However, NS1 reduces wild-type HRAS-GTP levels possibly either by interfering with GTP loading or enhancing GTP hydrolysis [21]. Further, the isoform-specific effect of NS1 on RAS:RAF association is dictated by the HVRs and not by G-domain [21]. Thus, targeting of the α4-α5 interface with NS1 may result in multiple levels of RAS inhibition. Whether similar levels of inhibition can be attained with small molecule NS1 mimetics remains to be seen.
Due to its large size and poor cell penetrance, NS1 in its current form is not a viable therapeutic. Nevertheless, our studies demonstrate NS1’s ‘drug-like’ activity as a genetically encodable reagent that blocks KRAS-driven signalling and transformation both in vitro and in vivo. Given the more than 130 different reported RAS missense mutations in human cancer [9], development of small molecules that target the KRAS α4-α5 interface may allow for inhibition of a wider range of mutant RAS proteins beyond KRAS(G12C), which is currently targeted by a variety of covalent inhibitors. Although such inhibitors would not be selective for a particular RAS mutation and would inhibit both wild type and mutant RAS potentially resulting in potential unwanted toxicities, we did not observe any inhibitory effect on the proliferation of fibroblasts (HEK293 or NIH/3T3) grown in culture. These results suggest that such α4-α5 interface inhibitors may not result in significant inhibitory effects in non-RAS mutant cells. However, this lack of toxicity may be due to the selectivity of NS1 for H/KRAS, allowing residual NRAS to drive proliferation in NS1-expressing cells. Further, KRAS mutation status can be heterogeneous within the same tumour and between primary and metastatic tumours [46–48]. This observation highlights the importance of developing an inhibitor that blocks RAS irrespective of mutation and lends support to the development of small molecule mimetics of NS1. Thus, targeting the α4-α5 interface of RAS by small molecule NS1 mimetics could prove useful in treating tumours addicted to mutant KRAS as well as tumours driven by upstream oncoproteins (e.g., mutant receptor tyrosine kinases) that rely on RAS signalling. Furthermore, the augmented CD4+ T-cell response observed upon inhibition of KRAS with NS1 further supports the combination of anti-RAS compounds with immunotherapy in treating immunologically cold tumours such as PDACs. Indeed, recent studies in lung cancer models demonstrated that KRAS (G12C) inhibitors synergized with immune checkpoint inhibitors and the combination was more potent in tumour regression than monotherapy with agents [15,27].
Materials and Methods
Generation of mouse ortholog from human KRAS
The point mutation for conversion of Aspartic acid (D) to Glutamic acid (E) at position 132 in human KRAS was generated by PCR. Briefly, KRAS was amplified by two separate PCRs using 5ʹ KRAS Gibson primer [(5ʹ GGAGGACCTTCTAGCGGATCCATGACTG AATATAAACTTGTGGTAGTTGGAGCT 3ʹ) and a 3ʹ mutagenic primer (5ʹ TCCATAACTTCTTGCTAATTCCTGAGCCTGTT TTGTGTC 3ʹ) in one reaction and 5ʹ mutagenic primer (5ʹ GACACAAAACAGGCTCAGGAATTAGCAAGAAGTTATGGA 3ʹ) and 3ʹ KRAS Gibson primer (5ʹ TCACCCTGAAGTTCTCAGGATCCTTACATAATTACACACTTTGTCTTTGACTTCTTTTTCTTCTTTTTACC 3ʹ) in the other. The two resulting amplicons (248 and 466 base pairs) were mixed with BamHI digested vector (pCGN) and ligated using Gibson Assembly Mix (New England Biolabs) and then used to transform E. coli. The resulting transformants were screened for inserts and sequenced to confirm mutants.
Affinity measurement of NS1 using yeast display
An expression vector for KRAS (residues 1–174) containing the D132E mutation was produced using Kunkel mutagenesis [49]. The protein was expressed and purified as described previously [20]. Quantitative measurements of NS1 affinity in the yeast display format [19,20] were performed as follows. Yeast EBY100 cells expressing the Aga2-NS1-V5 tag fusion protein on the cell surface were incubated with the biotinylated RAS protein of various concentrations as well as anti V5 antibody (Thermo Fisher, catalogue number MA5-15,253) in the BSS/Mg/DTT buffer (50 mM TrisCl, 150 mM NaCl, 20 mM MgCl2, 0.1 mM DTT, 1 mg/ml BSA (Gemini Bio, catalogue number 700–100P), pH 7.5), in a total volume of 20 µl in wells of 96-well plate (Greiner, catalogue number 650,261) at room temperature. After 30 min of incubation, the solutions were transferred to the wells of a 96-well filter plate (MultiScreenHTS, Millipore, catalogue number MSHVN4550) and vacuum was applied to collect cells and remove the solution. After washing cells with 150 µl of the BSST/Mg/DTT buffer BSS plus 0.1% Tween 20 (Thermo Fisher, catalogue number BP337-500) twice, cells were incubated with 20 µl each of the BSST/Mg2+/DTT buffer containing streptavidin-DyLight650 (Thermo Fisher, catalogue number 84,547) and anti-mouse IgG-FITC (Millipore Sigma, catalogue number F0257), each diluted 100 times, for 30 min at 4°C. Cells were washed with 150 µl of BSST/Mg2+/DTT twice, suspended in 100 µl BSS/Mg2+/DTT and fluorescence intensities were measured using an iQue screener (Sartorius). The median fluorescence intensity of DyLight650 of the top 75–95% population was determined, and the apparent dissociation constants were determined by nonlinear least squares fitting of the non-cooperative 1:1 binding function using Prism (GraphPad).
Cell culture and transfection
HEK-293 and HEK-293 T cells were cultured in DMEM with 10% foetal bovine serum. NIH/3T3 cells were cultured in DMEM with 10% calf serum. Murine KRAS(G12D) mutant pancreatic cancer cells (KPC) and human KRAS(G12V) mutant PDAC cells (CFPAC-1) were grown in DMEM with 10% FBS. All culture media were purchased commercially (Corning). Transient transfection in HEK-293, HEK-293 T and NIH/3T3 cells were done using polyethyleneimine (PEI). For HEK-293 and HEK-293 T, typically, 3 µl of 1 mg/ml PEI stock was used for each µg of DNA in Opti-MEM reduced serum media (Life Technologies). Opti-MEM-PEI mixture was incubated for 10 minutes at room temperature. Next, DNA was added to Opti-MEM: PEI cocktail and incubated for at least 20 minutes at room temperature. The Opti-MEM: PEI-DNA mixture was added to cells in serum-free media and incubated for 3 hours after which the media was replaced with fresh complete media. Transfections in NIH/3T3 cells were done with the same procedures; however, 10 µl of 1 mg/ml PEI stock was used for each µg of DNA and media was replenished after 5 hours.
Stable cell generation of inducible KPCNS1 cells
HEK-293 T cells were used to generate viral particles for DOX-inducible NS1 expression. The packaging HEK-293 T cells were transfected by calcium phosphate using pCW57.1-CFP-NS1(transfer plasmid) along with a plasmid encoding packaging plasmid (pCMVdR8.74) and the viral envelope (pMD2.G) in 4:3:1 ratio to generate viral particles. The following day packaging cells were placed in fresh media and on day 2 post-transfection, conditioned media from the HEK-293 T cells were collected, filtered, and used to infect KPC cells followed by selection in tetracycline free media containing puromycin (2 ug/ml). Following selection, colonies were pooled to generate a polyclonal cell line that was used for all subsequent analyses.
Immunoblotting and antibodies
Cell lysates were made by washing cells once in cold PBS followed by lysis PLC buffer (50 mM HEPES, pH 7.5, 150 mM NaCl, 10% glycerol, 1% Triton X-100, 1 mM EGTA, 1.5 mM magnesium chloride, 100 mM sodium fluoride supplemented with 1 mM vanadate, 10 μg/ml leupeptin and 10 μg/ml aprotinin). To generate tumour lysates, tumours were harvested, transferred to microfuge tubes, and snap-frozen by immersing in liquid nitrogen. Forty-50 mg of tissue was then homogenized in ~1 ml of cold PLC buffer on ice. Homogenate was passed through 70 µm cell strainer to clear the lysates then centrifuged at 13,000 rpm for 20 minutes at 4°C. Supernatant was collected and transferred to fresh tubes. The lysates were directly used for protein estimation and analysis or stored at −80°C for later use. The following antibodies were used: monoclonal HA (clone 16B12, Biolegend #90,154), polyclonal rabbit HA (Poly9023, Biolegend #923,502), monoclonal FLAG (Clone M2, Sigma #F1804), polyclonal rabbit FLAG (Sigma #F7425), phospho-ERK (Thr202/Tyr204, CST #9101), total ERK (CST #9102), phospho-AKT (Ser473, CST #9271 and T308, CST#40556), total AKT (CST #9272S), vinculin (SC #73,614), KRAS antibody (Millipore Sigma # OP24), cleaved Caspase-3 (Asp-175, CST #9661), CRAF (BD Biosciences # 610,151), BRAF (Santa Cruz #sc-9002), Luciferase (Santa Cruz #sc-74,548).
KRAS binding and signalling assays
Briefly, HEK-293 cells were transfected with the indicated constructs. Following immunoprecipitation of CFP-NS1 using FLAG antibody, samples were analysed by Western blot for co-precipitation of the HA-tagged RAS. For interaction of NS1 with endogenous mutant human or murine KRAS, human pancreatic cancer cells CFPAC-1 harbouring KRAS(G12V) mutation and murine pancreatic cancer cells KPC [KRAS(G12D)] were treated with doxycycline (DOX) for 48 hours and following immunoprecipitation of CFP-NS1 using FLAG antibody, samples were analysed by Western blot using the indicated antibodies for co-precipitation of the endogenous KRAS.
For transient cell signalling assays, HEK293 cells were transfected with indicated HA-tagged RAS mutant constructs or FLAG-tagged CFP-NS1 or CFP alone and analysed for effects on MAPK signalling as previously described [20]. For analysis of signalling in KPC cells, cells were treated with 4 µg/ml of DOX for 48 hours, serum starved overnight, then lysed in buffer and analysed as described previously using phosphospecific ERK (pERK) or AKT [pAKT(S473) or (T308)] antibodies [22]. Western blots were quantified with the software Image Studio Lite (v.5.2.5, LI-COR Biosciences) using the Analysis function. Ratio of pERK (normalized to vinculin) divided by total ERK (normalized to vinculin) was determined for each condition. The resulting values were divided by value for CFP alone for each protein.
NIH/3T3 transformation assays
Freshly revived NIH/3T3 cells were passaged no more than twice and then seeded in 60 mm dishes to a density of 2.5 × 105 cells in complete media. Cells were transfected with the indicated constructs and media changes were performed every 2 days. Foci were stained with 0.1% crystal violet and counted after 15 days. All assays were performed in triplicate and repeated three times.
Proliferation assays
Proliferation assays were performed using CellTiter Glow (Promega) as previously described [20,22]. Briefly, KPC cells were plated on 24-well plates in complete medium (DMEM with 10% FBS plus 1 μg/ml puromycin) then treated ± DOX (4 μg/ml) in triplicate for each condition. Luminescence was quantified using Clariostar (BMG, Labtech) 96-well microtiter plate luminometer following the manufacturer’s instructions.
Soft agar colony formation assays
Soft agar colony formation assays were performed essentially as described elsewhere [50]. Cells were fed 1–2x per week with media ± 4 µg/ml doxycycline (DOX) to induce expression of NS1. Two weeks after platting cells were stained using MTT (100 μl of 2 mg/ml per well). Colony numbers and average colony size were quantified using ImageJ.
Orthotopic pancreatic tumour assay
The procedures described herein are in compliance with protocols approved by the Institutional Animal Care and Use Committee (IACUC) at the Medical University of South Carolina. C57BL/6 J mice (female, age 5 weeks) were purchased from The Jackson Laboratory and acclimatized for 1 week. A 100 µl suspension of 5 × 104 KPC-luc cells in a 1:1 solution (v/v) of serum-free DMEM/Matrigel basement membrane matrix was injected directly into the pancreas of anaesthetized C57BL/6 J mice (2.5% isoflurane) [51]. The mice were injected (IP) with D-luciferin (150 µg/ml) and imaged 24 hours post-implantation of the KPC-luc cells to establish a baseline using a PerkinElmer Xenogen IVIS 200 bioluminescent imaging system in the Small Animal Imaging facility at the Medical University of South Carolina (P30 CA138313). Mice were then randomly assigned to control or treatment cohorts and subsequently imaged every 2 days for the course of the study. tumours were harvested after 3 weeks and collected separately either for biochemical analysis by western blotting or for immunohistochemical studies (IHC).
Multiplex immunohistochemistry
Tumours harvested from mice were fixed in neutral-buffered formalin for 24 hours, washed in 70% ethanol and then processed and embedded in paraffin using standard techniques. Subsequently, 4–5 µm sections of FFPE tissue on super frost plus slides were deparaffinized and stained using the Ventana Discovery Ultra-automated immunostainer (Roche Diagnostics Corp., Indianapolis, IN) and Akoya OPAL™ reagents (Akoya Biosciences, Marlborough, MA). Heat-induced epitope retrieval (HIER) was performed in EDTA buffer pH 9 (Cat. #S2367 Agilent/Dako Santa Clara, CA) for 32 minutes at 95°C and endogenous peroxidase was blocked with a solution of hydrogen peroxide after incubation of the first primary antibody. Antibodies used included CD4 (AbCam, clone EPR19514, 1:500), CD8 (Cell Signalling Technologies, clone D4W2Z, 1:200), FOXP3 (Cell Signalling Technologies, clone D6O8R, 1:200), F4/80 (Cell Signalling Technologies, clone D2S9R, 1:100), CD206 (NovusBio, NBP1-90,020 Rabbit polyclonal, 1:500) and cytokeratin 19 (DSHB, clone TROMA-III, 1:200). After incubation with primary and secondary antibodies, the Akoya Opal tyramide signal amplification reagents were used for fluorescence detection. The following fluorophores were used: OPAL 480, OPAL 520, OPAL 570, OPAL 620 and OPAL 780. DAPI was used for nuclear counterstaining. Between each sequential antibody staining step, slides were incubated in citrate buffer pH 6 (Cell Conditioning Solution (CC2) Cat. #980-223, Roche Diagnostics) at 90°C for 8 minutes to remove the previous primary and secondary antibody complexes. Stained slides are mounted with ProLong™ Gold Antifade Reagent (Cat. # P36934, ThermoFisher) and imaged using the Akoya Vectra® Polaris™ Automated Imaging system (Akoya Biosciences, Marlborough, MA). Whole slide scans were done at 20X magnification and subsequently eight regions of interest (ROIs) where chosen at random across each tumour for further analysis. Spectral unmixing and removal of autofluorescence were performed using the inForm® Software v2.4.10 (Akoya Biosciences, Marlborough, MA), and the resulting images were exported in TIFF format for further analysis.
Multiplex IHC image analysis
Image preparation – number of mice – number of images per mouse
The nuclei were segmented by using the deep learning-based segmentation Mask R-CNN [52] with a Resnet-101 architecture for the backbone network [53]. Twenty images of size 1868 × 1400 pixels showing nuclei stained with DAPI were manually annotated to define the training dataset. Data augmentation to increase the size of the training dataset by a factor of 100 was processed. A transfer learning with fine-tuning from a Mask R-CNN network trained on the coco dataset [54] was achieved. In the first epoch, only the region proposal network, the classifier and mask heads were trained. All the network was then trained for the next 3 epochs.
The ImageJ [55] plugin Annotater [56] was used to identify the nuclei positive for CD4, CD8, F4/80 and CD206. For each marker, about 50 positive and 50 negative nuclei were manually annotated over 5 different images to train a logistic regression classifier by using the average and standard deviation of the intensity in the nuclear membrane area, by using the nucleus size and the nucleus circularity. The trained classifiers were finally applied to identify the positive nuclei for each marker.
Statistical analyses
Statistical analyses were performed using GraphPad Prism v9 software for Mac. p values for the effects of NS1 on RAS signalling were calculated as described for each experiment. Significance is denoted as *p < 0.05, **p < 0.01, ***p < 0.001.
Supplementary Material
Acknowledgments
We wish to thank members of the O’Bryan and Ostrowski labs for comments on this work. This work was supported in part by a Merit Review Award (1I01BX002095) from the United States (US) Department of Veterans Affairs Biomedical Laboratory Research and Development Service to J.P.O., an NIH award to J.P.O. and S.K. (CA212608), a Chan Zuckerberg Initiative DAF grant to T.P. (2019-198009), and support from the Hollings Cancer Center. The in vivo imaging was supported in part by the Small Animal Imaging Unit of the Cell & Molecular Imaging Shared Resource, Hollings Cancer Center, Medical University of South Carolina (P30 CA138313). The contents do not represent the views of the US Department of Veterans Affairs or the United States Government.
Funding Statement
This work was supported by the National Institutes of Health [CA212608]; National Institutes of Health [P30 CA138313]; US Department of Veterans Affairs [1I01BX002095]; Chan Zuckerberg Initiative DAF grant [2019-198009].
Disclosure statement
S.K. and A.K. are inventors on a patent that covers designs of monobody libraries (US Patent 9,512,199 B2).
Supplemental Material
Supplemental data for this article can be accessed here.
Author Contributions
I.K., M.C.O. and J.P.O. designed the study; E.D., A.K. and S.K. performed the Monobody affinity experiments; I.K. and M.Z. performed the biochemical and cell biological studies; I.K., C.M-B., J.L., D.M.C. and A.-M.B. performed the animal studies; T.P. and C.T. performed the IHC analyses; I.K. and J.P.O. wrote the manuscript and all authors commented and approved the manuscript.
References
- [1].Karnoub AE, Weinberg RA.. Ras oncogenes: split personalities. Nat Rev Mol Cell Biol. 2008;9(7):517–531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2].Spencer-Smith R, O’Bryan JP.. Direct inhibition of RAS: quest for the Holy Grail? Semin Cancer Biol. 2019;54:138–148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Cox AD, Der CJ. Ras history: the saga continues. Small GTPases. 2010;1(1):2–27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Simanshu DK, Nissley DV, McCormick F. RAS Proteins and Their Regulators in Human Disease. Cell 2017;170(1):17–33 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Vigil D, Cherfils J, Rossman KL, et al. Ras superfamily GEFs and GAPs: validated and tractable targets for cancer therapy? Nat Rev Cancer. 2010;10(12):842–857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Cherfils J, Zeghouf M. Regulation of small GTPases by GEFs, GAPs, and GDIs. Physiol Rev. 2013;93(1):269–309. [DOI] [PubMed] [Google Scholar]
- [7].Cox AD, Fesik SW, Kimmelman AC, et al. Drugging the undruggable RAS: mission possible? Nat Rev Drug Discov. 2014;13(11):828–851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Zuberi M, Khan I, O’Bryan JP. Inhibition of RAS: proven and potential vulnerabilities. Biochem Soc Trans. 2020;48(5):1831–1841. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Hobbs GA, Der CJ, Rossman KL. RAS isoforms and mutations in cancer at a glance. J Cell Sci. 2016;129(7):1287–1292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Targeting DJ. RAS signalling pathways in cancer therapy. Nat Rev Cancer. 2003;3(1):11–22. [DOI] [PubMed] [Google Scholar]
- [11].O’Bryan JP. Pharmacological targeting of RAS: recent success with direct inhibitors. Pharmacol Res. 2018;139:503–511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Ostrem JM, Shokat KM. Direct small-molecule inhibitors of KRAS: from structural insights to mechanism-based design. Nat Rev Drug Discov. 2016;15(11):771–785. [DOI] [PubMed] [Google Scholar]
- [13].Lito P, Solomon M, Li LS, et al. Allele-specific inhibitors inactivate mutant KRAS G12C by a trapping mechanism. Science. 2016;351(6273):604–608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Janes MR, Zhang J, Li L-S, et al. Targeting KRAS Mutant Cancers with a Covalent G12C-Specific Inhibitor. Cell. 2018;172(3):578–589. e17. [DOI] [PubMed] [Google Scholar]
- [15].Canon J, Rex K, Saiki AY, et al. The clinical KRAS(G12C) inhibitor AMG 510 drives anti-tumour immunity. Nature. 2019;575(7781):217–223. [DOI] [PubMed] [Google Scholar]
- [16].Hallin J, Engstrom LD, Hargis L, et al. The KRAS(G12C) Inhibitor MRTX849 Provides Insight toward Therapeutic Susceptibility of KRAS-Mutant Cancers in Mouse Models and Patients. Cancer Discov. 2020;10(1):54–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Sha F, Salzman G, Gupta A, et al. Monobodies and other synthetic binding proteins for expanding protein science. Protein Sci. 2017;26(5):910–924. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Koide A, Koide S. Monobodies: antibody mimics based on the scaffold of the fibronectin type III domain. Methods Mol Biol. 2007;352:95–109. [pii]. [DOI] [PubMed] [Google Scholar]
- [19].Koide A, Wojcik J, Gilbreth RN, et al. Teaching an old scaffold new tricks: monobodies constructed using alternative surfaces of the FN3 scaffold. [pii] 10.1016/j.jmb.2011.12.019 J Mol Biol. 2012;4151:393–405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Spencer-Smith R, Koide A, Zhou Y, et al. Inhibition of RAS function through targeting an allosteric regulatory site. Nat Chem Biol. 2017;13(1):62–68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Spencer-Smith R, Li L, Prasad S, et al. Targeting the alpha4-alpha5 interface of RAS results in multiple levels of inhibition. 0 Small GTPases. 2017;105:378–387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Khan I, Spencer-Smith R, O’Bryan JP. Targeting the α4-α5 dimerization interface of K-RAS inhibits tumor formation in vivo. Oncogene. 2018. 10.1038/s41388-018-0636-y [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Hingorani SR, Wang L, Multani AS, et al. Trp53R172H and KrasG12D cooperate to promote chromosomal instability and widely metastatic pancreatic ductal adenocarcinoma in mice. Cancer Cell. 2005;7(5):469–483. [DOI] [PubMed] [Google Scholar]
- [24].McCormick F. Sticking it to KRAS: covalent Inhibitors Enter the Clinic. Cancer Cell. 2020;37(1):3–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Bonaventura P, Shekarian T, Alcazer V, et al. Cold Tumors: a Therapeutic Challenge for Immunotherapy. Front Immunol. 2019;10:168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Coelho MA, De Carné Trécesson S, Rana S, et al. Oncogenic RAS Signaling Promotes Tumor Immunoresistance by Stabilizing PD-L1 mRNA. Immunity. 2017;47(6):1083–99.e6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Briere DM. The KRASG12C inhibitor MRTX849 reconditions the tumor immune microenvironment and leads to durable complete responses in combination with anti-PD-1 therapy in a syngeneic mouse model. Mol Cancer Ther. 2019. Dec 2019. Published. 10.1158/1535-7163.TARG-19-LB-C09 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Khan I, Rhett JM, O’Bryan JP. Therapeutic targeting of RAS: new hope for drugging the “undruggable”. Biochim Biophys Acta Mol Cell Res. 2020;1867(2):118570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Moore AR, Rosenberg SC, McCormick F, et al. RAS-targeted therapies: is the undruggable drugged? Nat Rev Drug Discov. 2020;19(8):533–552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Rhett JM, Khan I, O’Bryan JP. Biology, pathology, and therapeutic targeting of RAS. Adv Cancer Res. 2020;148:69–146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Prior IA, Lewis PD, Mattos C. A comprehensive survey of Ras mutations in cancer. Cancer Res. 2012;72(10):2457–2467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Fell JB, Fischer JP, Baer BR, et al. Identification of the Clinical Development Candidate MRTX849, a Covalent KRAS(G12C) Inhibitor for the Treatment of Cancer. J Med Chem. 2020;63(13):6679–6693. [DOI] [PubMed] [Google Scholar]
- [33].Linnemann C, Van Buuren MM, Bies L, et al. High-throughput epitope discovery reveals frequent recognition of neo-antigens by CD4+ T cells in human melanoma. Nat Med. 2015;21(1):81–85. [DOI] [PubMed] [Google Scholar]
- [34].Kreiter S, Vormehr M, Van De Roemer N, et al. Mutant MHC class II epitopes drive therapeutic immune responses to cancer. Nature. 2015;520(7549):692–696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [35].Ott PA, Hu Z, Keskin DB, et al. An immunogenic personal neoantigen vaccine for patients with melanoma. Nature. 2017;547(7662):217–221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].Sahin U, Derhovanessian E, Miller M, et al. Personalized RNA mutanome vaccines mobilize poly-specific therapeutic immunity against cancer. Nature. 2017;547(7662):222–226. [DOI] [PubMed] [Google Scholar]
- [37].Zhang L, Yu X, Zheng L, et al. Lineage tracking reveals dynamic relationships of T cells in colorectal cancer. Nature. 2018;564(7735):268–272. [DOI] [PubMed] [Google Scholar]
- [38].Galaine J, Turco C, Vauchy C, et al. CD4 T cells target colorectal cancer antigens upregulated by oxaliplatin. Int J Cancer. 2019;145(11):3112–3125. [DOI] [PubMed] [Google Scholar]
- [39].Tay RE, Richardson EK, Toh HC. Revisiting the role of CD4(+) T cells in cancer immunotherapy-new insights into old paradigms. Cancer Gene Ther. 2020;28(1–2):5–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [40].Liao W, Overman MJ, Boutin AT, et al. KRAS-IRF2 Axis Drives Immune Suppression and Immune Therapy Resistance in Colorectal Cancer. Cancer Cell. 2019;35(4):559–572. e7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [41].Poulin EJ, Bera AK, Lu J, et al. Tissue-Specific Oncogenic Activity of KRAS A146T. Cancer Discov. 2019;9(6):738–755. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [42].Hobbs GA, Baker NM, Miermont AM, et al. Atypical KRAS(G12R) Mutant Is Impaired in PI3K Signaling and Macropinocytosis in Pancreatic Cancer. Cancer Discov. 2020;10(1):104–123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [43].Ambrogio C, Köhler J, Zhou Z-W, et al. KRAS Dimerization Impacts MEK Inhibitor Sensitivity and Oncogenic Activity of Mutant KRAS. Cell. 2018;172(4):857–868. e15. [DOI] [PubMed] [Google Scholar]
- [44].Bery N, Legg S, Debreczeni J, et al. KRAS-specific inhibition using a DARPin binding to a site in the allosteric lobe. Nat Commun. 2019;10(1):2607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [45].Sutton MN, Lu Z, Li YC, et al. DIRAS3 (ARHI) Blocks RAS/MAPK Signaling by Binding Directly to RAS and Disrupting RAS Clusters. Cell Rep. 2019;29(11):3448–59.e6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [46].Kordiak J, Szemraj J, Grabska-Kobylecka I, et al. Intratumor heterogeneity and tissue distribution of KRAS mutation in non-small cell lung cancer: implications for detection of mutated KRAS oncogene in exhaled breath condensate. J Cancer Res Clin Oncol. 2019;145(1):241–251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [47].Lamy A, Blanchard F, Le Pessot F, et al. Metastatic colorectal cancer KRAS genotyping in routine practice: results and pitfalls. Mod Pathol. 2011;24(8):1090–1100. [DOI] [PubMed] [Google Scholar]
- [48].Richman SD, Chambers P, Seymour MT, et al. Intra-tumoral heterogeneity of KRAS and BRAF mutation status in patients with advanced colorectal cancer (aCRC) and cost-effectiveness of multiple sample testing. Anal Cell Pathol (Amst). 2011;34(1–2):61–66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [49].Kunkel TA, Roberts JD, Zakour RA. Rapid and efficient site-specific mutagenesis without phenotypic selection. Methods Enzymol. 1987;154:367–382. [DOI] [PubMed] [Google Scholar]
- [50].Clark G, Cox AD, Graham SM, et al. Biological assays for Ras transformation. Methods Enzymol. 1995;255:395–412. [DOI] [PubMed] [Google Scholar]
- [51].Pitarresi JR, Liu X, Avendano A, et al. Disruption of stromal hedgehog signaling initiates RNF5-mediated proteasomal degradation of PTEN and accelerates pancreatic tumor growth. Life Sci Alliance. 2018;1(5):e201800190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [52].He K, Gkioxari G, Dollár P, et al. Mask r-cnn. arXiv:1703.06870v3 [Google Scholar]
- [53].He K, Zhang X, Ren S, et al. Deep residual learning for image recognition. CVPR Conference 2016. p. 770–778. New York City: IEEE. [Google Scholar]
- [54].Lin T-Y, Maire M, Belongie S. et al. Microsoft coco: common objects in context. arXiv:1405.0312v3 [Google Scholar]
- [55].Schindelin J, Rueden CT, Hiner MC, et al. The ImageJ ecosystem: an open platform for biomedical image analysis. Mol Reprod Dev. 2015;82(7–8):518–529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [56].Ollion J, Cochennec J, Loll F, et al. TANGO: a generic tool for high-throughput 3D image analysis for studying nuclear organization. Bioinformatics. 2013;29(14):1840–1841. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.