

ABSTRACT
The hexanucleotide repeat (GGGGCC) expansion in C9orf72 is accounted for a large proportion of the genetic amyotrophic lateral sclerosis (ALS) and frontotemporal dementia (FTD). The hypotheses of how the massive G4C2 repeats in C9orf72 destroy the neurons and lead to ALS/FTD are raised and improving. As a multirole player, C9orf72 exerts critical roles in many cellular processes, including autophagy, membrane trafficking, immune response, and so on. Notably, the partners of C9orf72, through which C9orf72 participates in the cell activities, have been identified. Notably, the structures of the C9orf72-SMCR8-WDR41 complex shed light on its activity as GTPase activating proteins (GAP). In this manuscript, we reviewed the latest research progress in the C9orf72-mediated ALS/FTD, the physiological functions of C9orf72, and the putative function models of C9orf72/C9orf72-containing complex.
KEYWORDS: DENN domain, autophagy, membrane trafficking, neurodegenerative, GAP, GEF, C9orf72, SMCR8, WDR41, lysosome, mTOR
Introduction
The massive GGGGCC (G4C2) repeat in the first intron of C9orf72 has been demonstrated to be one of the genetic hallmarks of amyotrophic lateral sclerosis (ALS) and frontotemporal dementia (FTD) [1–3], two lethal and untreatable neurodegenerative diseases. As a potential diagnostic and therapeutic target for genetic ALS and FTD, C9orf72 has been extensively researched, and significant progress has been achieved in the past decade.
First, a notable headway in discerning the pathogenic mechanism of C9orf72-mediated ALS and FTD has been made. Two main hypotheses, the toxic gain-of-function and loss-of-function of C9orf72, have been suggested to elucidate the pathogenic mechanism of how the G4C2 repeats in C9orf72 damage the neurons [4–19]. A recent study suggested that C9orf72 loss-of-function exacerbates the toxic gain-of-function phenotype [3].
In addition, the signalling pathways and physiological processes in which C9orf72 participates have been gradually deciphered. Dozens of reports have demonstrated that C9orf72 participates in the regulation of macroautophagy (hereafter referred to as autophagy) through different mechanisms, e.g., membrane trafficking and transcription [20–24]. Interestingly, the lysosome is a critical organelle that mediates C9orf72 function and is highly correlated with the C9orf72-ALS/FTD [25–27]. Intriguingly, substantial evidence indicates that the massive G4C2 repeat expansion of C9orf72 and thus the C9orf72 protein impacts immune activity [27–29]. Nevertheless, the specific function of C9orf72 is not clear.
During the exploration of the function of C9orf72, several protein partners of C9orf72 have been identified, including but not limited to SMCR8, WDR41, the ULK1 complex, and RAB39B [21,22,25,26]. Notably, C9orf72, SMCR8, and WDR41 can form a stable complex that exerts a regulatory function on lysosomes and autophagy pathways [21,24–27]. Intriguingly, both C9orf72 and SMCR8 belong to Differentially Expressed in Normal and Neoplastic cells (DENN) domain family. The most well-defined function of DENN domain family proteins is as guanine nucleotide exchange factors (GEF) for small GTPases [30–33]. Bioinformatic analysis indicates that C9orf72, SMCR8, FNIP1/2 (folliculin-interacting protein), and FLCN (folliculin) are subbranches of the DENN family [33–35]. However, the FNIP1/2-FLCN complex functions as a GTPase-activating protein (GAP) of RRAGC/D, which raises a new question: Does C9orf72 functions only as a GEF? A structural study of both the C9orf72-SMCR8 complex and FNIP2-FLCN complex demonstrated that these two complexes share a similar overall structure [36–40]. Remarkably, the C9orf72-SMCR8 complex has been proved to be a GAP for RAB8A, RAB11A, and ARF136 [38–40].
In this review, we summarize the relationship between C9orf72 and genetic ALS, present hypotheses of the relationship between abnormal C9orf72 and pathology, discuss the function of C9orf72, and explain the progress in exploring the structural biology of C9orf72 and related protein complexes.
C9orf72 and ALS disease
ALS and ALS-linked genes
Amyotrophic lateral sclerosis (ALS) is a devastating and fatal neurodegenerative disease characterized by progressive degeneration of both upper and lower motor neurons in the spinal cord and brain, leading to hyperreflexia, spasticity, muscular atrophy, paralysis, and ultimately death [41,42]. ALS typically starts focally, either in an upper limb or a lower limb or the bulbar region, and then spreads to other regions with time [41]. The disease usually progresses rapidly, with most patients dying of respiratory failure within 3–5 years of symptom onset [41]. Although there are currently no effective cures, three treatments for ALS have been approved, and many clinical trials are ongoing, including trials of antisense oligonucleotides (ASOs) targeting C9orf72 sense HRE-containing RNAs (ASO BIIB078, ClinicalTrials.gov identifier: NCT03626012) and other ALS-linked genes [43]. The three approved pharmacological treatments for ALS are Riluzole, Dextromethorphan, and Edaravone [44]. Riluzole is an antagonist of glutamate release, and it extends patient survival for approximately 3–19 months [45–47]. Dextromethorphan was approved to ameliorate ALS patients’ emotional lability and enhance speaking and swallowing function in patients [48,49]. Edaravone, a free radical scavenger, was recently shown to slow the decline in functional scores after six months of treatment in a phase III clinical study. Thus, it was approved for the treatment of early-stage ALS cases [50]. Unfortunately, these currently approved molecules only showed moderate improvement in ALS patients. Therefore, significantly improved treatments are greatly needed.
ALS is traditionally classified into familial and sporadic subtypes based on ALS presence in family members, whereas sporadic ALS comprises the most ALS patients, ~ 90% [41]. More than 20 genes have been implicated in familial ALS, sporadic ALS, or both, with the most common genes including SOD1, FUS, TARDBP/TDP-43, C9orf72, PFN1, and UBQLN2 [41,51–54].
In 2006, genetic studies, including the conventional linkage and genome-wide association studies, first linked ALS and frontotemporal dementia (FTD) to the chromosome 9 open reading frame 72 (C9orf72) gene [55,56], showing that a mutation in the noncoding region of C9orf72 is the most common genetic cause of ALS and FTD in a specific population. The C9orf72 gene, consisting of 11 exons, has three main alternatively spliced transcript variants and produces two protein isoforms (Figure 1). It was recently found that the expression of the long C9orf72 isoform, but not the short isoform, can partially rescue the dendritic arborization phenotype in hippocampal neurons from C9orf72-knockout mice, suggesting that the two C9orf72 isoforms might function differently [57]. To date, little is known about the functions of the short C9orf72 isoform; hence, in this review, C9orf72 refers to the long isoform.
Figure 1.
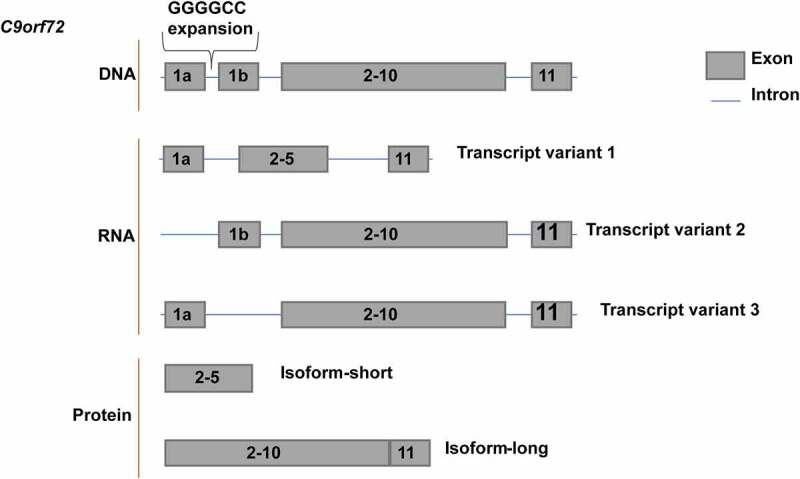
Gene products of C9orf72.
The three RNA transcripts of the C9orf72 gene eventually lead to two protein isoforms.
Roles of C9orf72 in ALS
G4C2 hexanucleotide repeat expansion (HRE) in the first intron of the C9orf72 gene may play a critical role in the process of ALS disease. Mechanisms of C9orf72 HRE-induced neurodegenerative changes have been proposed (Figure 2), which mainly consist of loss-of-function and gain-of-toxicity mechanisms, but their relative contribution to pathogenicity is still debated.
Figure 2.
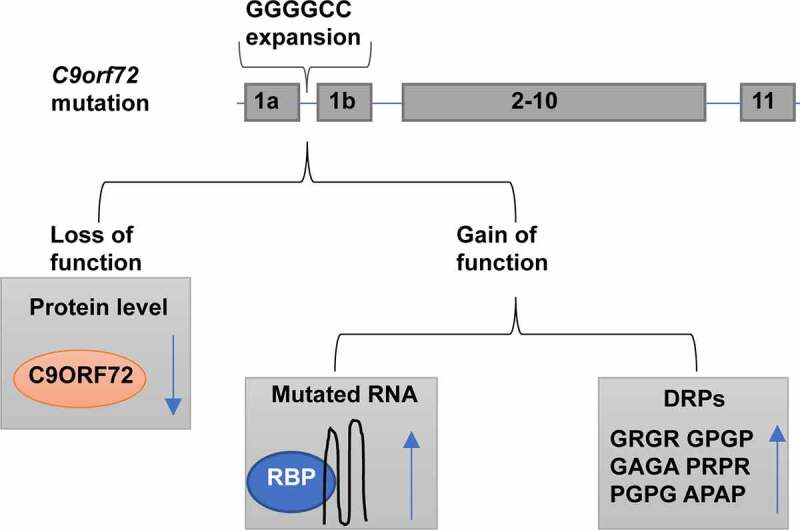
The hypothesized assaults caused by G4C2 repeat expansion in C9orf72.
The consequences of C9orf72 mutation include loss of C9orf72 function and gain-of-function with either RNAs that capture RNA binding proteins (RBPs) or dipeptides that disturb cellular homoeostasis.
Most evidence suggests that C9orf72 HRE-induced neurodegeneration depends mainly on a gain-of-toxicity mechanism [58–60], including RNA toxicity and dipeptide repeat protein (DPR) toxicity [29,61]. Because of G4C2 repeat expansion in the C9orf72 gene, higher-order DNA structure G-quadruplexes are formed, which can generate truncated RNA transcripts [62]. These aberrant RNA transcripts form G-quadruplexes and RNA hairpin structures. Additionally, HRE-containing RNA can hybridize with HRE-containing DNA to form R-loops, which are incredibly stable. During cellular processes, including DNA replication, transcription, translation, and telomere function, R-loops are transiently formed [63]. However, the persistent R-loops lead to adverse effects, including genome instability and DNA damage [63]. The G4C2 repeat sense and antisense RNAs transcribed from HREs can form nuclear RNA foci capable of sequestering vital RNA-binding proteins (RBPs) and thus disturb the normal functions of theses proteins [64,65]. On the other hand, repeat-associated non-ATG (RAN) translation generates five different dipeptide repeat proteins (DPRs): glycine-alanine (GA), glycine-arginine (GR), proline-alanine (PA), proline-arginine (PR), and glycine-proline (GP). Furthermore, these DPRs exhibit distinct biochemical properties, with arginine-rich DPRs apparently the most toxic, primarily impeding primarily nucleocytoplasmic transport and RNA processing [5,11,66].
Further studies observed overt motor neuron degeneration when the C9orf72 level was reduced in C.elegans and zebrafish. In addition, C9orf72-KO mice exhibited no neuron degeneration or motor deficit, suggesting that C9orf72 haploinsufficiency is insufficient to cause neurodegeneration in mammals [67–71].
The determination of whether gain-of-function or loss-of-function mechanisms induce the predominate G4C2-expanded C9orf72 effect was realized in a recent study [4]. In human-induced motor neurons (iMNs), the repeated expansion reduced C9orf72 expression, which caused neurodegeneration through the accumulation of glutamate receptors resulting from C9orf72 haploinsufficiency and blocked the degradation of neurotoxic DPR proteins resulting from the G4C2 expansion [4]. The neurodegeneration caused by cooperating gain-of-function and loss-of-function mechanisms was attenuated by the expression of constitutively active GTPase RAB54, indicating that dysfunctional vesicle trafficking is involved in C9orf72 ALS. This finding is in line with that the function of C9orf72 as a GAP to regulate GTPase activity, which is discussed in the subsequent sections. Indeed, recent studies have shown that a reduction in C9orf72 protein levels with a long HRE can enhance the early accumulation of DPRs, suggesting that the loss of C9orf72 has a synergistic effect with the repeat-dependent gain-of-toxicity [3].
Physiological functions of C9orf72 in model organisms
TDP-43 mutations and mouse models
The studies on TARDBP (gene encoding TDP-43) provided informative references for C9orf72 research. Mutations in TARDBP (gene encoding TDP-43) account for approximately 4% of familial ALS cases and a smaller percentage of sporadic ALS cases [72–75]. Tremendous research has been performed to elucidate the molecular genetic underpinnings of TDP-43 related ALS in mouse models [76–79], and the results may serve references for studying C9orf72. TDP-43 is a nucleic acid-binding protein that shuttles between the nucleus and the cytoplasm [80]. In ALS patients’ motor neurons, TDP-43 is mislocalized to the cytoplasm and self-aggregates after posttranslational modification [81]. Additionally, various mutations in TDP-43 have been discovered to be associated with ALS [41]. More than 50 mutations in TDP-43, including G298S, A315T, M337V, G348C, and A382T, have been demonstrated to cause or be linked with ALS [75,82,83].
To study the mechanism of TDP-43 toxicity, various model organisms have been generated, including yeast [84,85], Drosophila [85], Caenorhabditis elegans [86,87], zebrafish [88,89], and many mouse models [79,90,91]. Although it is impossible to clarify the unique features of TDP-43-induced ALS based on a phenotype-genotype correlation found in each mouse model of TDP-43 mutations, some certain clinical characteristics of human TDP-43 mutations can be reproduced in mouse models [79,90,91]. Since genetic knockout of TARDBP in mice causes embryonic lethality [92–94], transgenic mice overexpressing human TDP-43 (WT or mutated) are mostly used [79,90,91]. Correlations between mutations and phenotypes in mouse models can be determined based on protein expression levels and phenotypes [79,90,91]. Low hTDP-43 expression causes age-related motor dysfunction and cytosolic inclusions. Nevertheless, mice with low hTDP-43 expression were not paralysed and died in the normal age range, and those with high hTDP-43 expression showed early-onset and fast disease progression but no clear motor neuron loss or hTDP-43 inclusion bodies [90,95,96]. In mice, hTDP-43-WT overexpression only partially recapitulated ALS features with rapid onset, low survival, motor neuron loss, and muscle atrophy [90,97–99]. However, the double knock-in mice expressing hTDP-43-WT and hTDP-43-Q331K developed rapid progressive limb paralysis, died early, and exhibited p62-, ubiquitin-, and TDP-43-positive inclusion bodies [100]. hTDP-43A315T and hTDP-43G348C transgenic mice developed age-related progressive motor deficits and cognitive impairments with TDP-43 nuclear and cytosolic inclusion bodies in neurons [95,99,101,102]. hTDP-43 Q331K knock-in mice developed behavioural abnormalities but presented with no ALS motor phenotypes [103]. hTDP-43 M337V knock-in mice developed mutant dose-dependent ALS motor phenotypes and neuromuscular junction abnormalities [91,104]. In addition to mice harbouring the aforementioned coding mutations, mouse models expressing a cytoplasmic form of TDP-43 without a nuclear targeting sequence (TDP-43-ΔNLS) recapitulated multiple features of TDP-43 ALS, including progressive neurodegeneration, gliosis, and abnormalities in motor, cognitive and social domains [105–107].
Physiological functions of C9orf72 in model organisms
The in vivo physiological functions of C9orf72 have also been well studied in model organisms. Various animal models have been generated to elucidate the pathological mechanisms of C9orf72-ALS for decades, including yeast [66,108,109], worms [68,110–112], fruit flies [11,12,113–115], zebrafish [67,116–119], mice [7,9,120–124], and induced pluripotent stem neuron cells (iPSNs) derived from the ALS patients [113,125–130].
The nematode Caenorhabditis elegans has also been used as an ALS animal model to study the mechanisms and phenotypes of C9orf72 loss-of-function [110]. Deletion of the C. elegans C9orf72 orthologue, F18A1.6/alfa-1, caused severe paralysis, impaired cytoplasm-nuclear transportation, neurodegenerative motive defects, and early death [68]. The alfa-1 deletion also caused dysregulation in endosomal-lysosomal homoeostasis, which was partially rescued by the expression of human C9orf72 [111]. Interestingly, C. elegans alfa-1 cooperatively functions with smcr8 in regulating the function and re-formation of lysosomes [111]. In addition, deletion of alfa-1 in C. elegans caused enhanced nuclear translocation of transcription factor hlh-30 (an ortholog of human TFEB), leading to activated lipolysis and premature death under starvation conditions [112]. Thus, this study revealed novel functions of C9orf72/alfa-1 in nutrient sensing and metabolic pathways, which may indicate potential mechanisms by which C9orf72-loss disturbs cellular homoeostasis in neurons.
The C9orf72 ortholog in zebrafish is very similar to human C9orf72[67], and three different studies have been performed by either knocking down or deleting C9orf72 in zebrafish [116]. Antisense morpholino oligonucleotide (MOs) knockdown of zebrafish C9orf72 caused motor neuron axonopathy and shortened axons [67]. Disruption of only the functional DENN domain of C9orf72 in zebrafish without manipulation of the full-length gene, caused an altered neuronal network, deficient axon formation, and motor activities [117]. Interestingly, dramatically reduced GTPase activity was observed in this zebrafish model [117]. This finding is in line with the molecular function of C9orf72 as a GAP to regulate GTPase activity, which is discussed in the subsequent sections. Recently, a microRNA-based C9orf72-knockdown model was developed with zebrafish, and it featured phenotypes of motor neuron defects, muscle atrophy, and mislocalized TDP-43[118-119]. The observations of theses zebrafish models suggest that C9orf72 loss-of-function can contribute to ALS pathogenesis, although it is not an exclusive mechanism.
With a short generation time and a relatively less complex brain, Drosophila models offer great advantages for the study of neurodegenerative diseases [131]. Numerous studies using fly models of C9orf72-ALS have been established mostly by the overexpression of repeat sequences or DPRs, which is necessary since Drosophila lack a C9orf72 homolog [113]. However, C9orf72-knockout mice exhibited splenomegaly and lymphadenopathy but no the neurodegenerative phenotypes, as described above, although mouse C9orf72 is homologous to human C9orf72, and more phenotypes related to immunity and inflammation were observed [69,70,132]. Mice with deletion of one or two copies of the C9orf72 genes showed age-dependent lethality [132]. A deficiency of C9orf72 caused low mTOR activity and thereby upregulated levels of transcription factor EB (TFEB), which is repressed by mTOR and can promote the transcription of lysosomal and autophagy genes [132]. The deletion of C9orf72 or its partner SMCR8 caused obvious autoimmunity and inflammatory reactions [69,133,134]. Besides, deletion of the Toll-like receptors (TLRs) TLR3, TLR7, and TLR9 attenuated induced splenomegaly and lymphadenopathy, and activated circulating T cells in SMCR8 deleted mice [69]. Double-knockout (dKO of C9orf72 and SMCR8) mice showed more severe immune abnormalities than single-gene-knockout mice [70]. Further analysis found that the macrophages from the dKO mice exhibit blocked lysosomal degradation and exocytosis [70]. Interestingly, the dKO mice showed increased mTORC1 activity, and mTORC1 inhibition reversed macrophage dysfunction [70].
Synaptic dysfunction caused by C9orf72 deletion or mutations
Loss of normal communication between the presynaptic motor neurons and postsynaptic muscle is one of the hallmark features of ALS [135,136]. Synaptic dysfunction has been implicated in C9orf72-ALS [127,137]. The localization of C9orf72 in neurons provides clues for the link between its mutation and synaptic dysfunction [71]. It has been shown that C9orf72 is enriched at synapses [138,139] and localized to both the pre-and postsynaptic compartments [71]. Experiments with C9orf72-knockout mice suggested that C9orf72 is vital for maintaining postsynaptic receptor levels [71] and is required for synaptic plasticity and adult neurogenesis [120]. The overexpression of poly-GA peptides encoded by C9orf72 with G4C2 expansion in primary mouse cortical neurons led to decreased dendritic arborization, which supports the concept of synaptic dysfunction in C9orf72-ALS [121]. The overexpression of a 48 G4C2 repeat construct in rat spinal cord neurons reduced the number of primary dendritic branches [59]. In mice expressing 30 or more G4C2 repeats in C9orf72, a significant decrease in synaptic bouton number and synaptic quantal content, leading to attenuated evoked potentials, was observed [12,126]. A transgenic mouse line expressing poly-PR of C9orf72 with G4C2 expansion specifically in neurons, develops partial neuropathology of C9orf72-ALS and shows differential expression of genes related to synaptic transmission [17]. Poly-GA in the neurites of mouse models exhibited altered Ca [2]+ influx and synaptic vesicle release, causing a reduction in synaptic vesicle-associated protein 2 (SV2), which was also seen in cortical and motor neurons derived from patient induced cell lines [137].
Immune dysfunction caused by C9orf72 deletion or mutation
Multiple studies have verified that C9orf72 is related to immune system function; for example, immune system dysregulation was observed in homozygous C9orf72-knockout mice, including hyperplasia in the spleen and cervical lymph node, severe autoimmunity, T-cell activation, and increased expression of inflammatory cytokines and chemokines [133,134,140–142]. These C9orf72-null mice exhibited increased immune infiltration in lymphoid organs and elevated cytokines in plasma [133,134]. At the cellular level, bone marrow-derived macrophages (BMDMs) and microglia from these mice exhibited a proinflammatory state, and bone marrow transplantation (BMT) experiments were performed, indicating that the efficacy of C9orf72 in immune function [134,140]. Besides, a similar phenotype was reported in a recent retrospective study. Analysis of the cerebrospinal fluid (CSF) and plasma of 248 patients with motor neuron disease (MND) showed that 164 ALS patients carry mutations in SOD1 (n = 24), C9orf72 (n = 19), and in other ALS related genes (n = 119), which revealed that the differences in survival between patients with different ALS subtypes are correlated with cytokine levels, and more specifically, C9orf72-ALS patients had higher CSF interferon-alpha levels than the patients with one of the other aforementioned ALS subtypes, providing some progressive insights into the pathological mechanism of ALS [134].
A recent study showed that selectively ablating C9orf72 in mouse dendritic cells (DCs) caused marked early-activation of the type I interferon response [142]. In addition, C9orf72 deficiency in myeloid cells disrupted autophagy-mediated degradation of STING, which led to persistent activation of type I interferon, following enhanced autoimmunity, adaptive immune activation, and antitumor immunity [142]. Although these results collectively related to immune dysfunction, the inconsistency in the severity of the persistent autoimmunity outcomes reported by different groups was notable, with some reporting higher premature mortality [60,134,141] and others reporting no effect on survival [140], indicating that environmental contribution might occur in the field. Indeed, researchers supported a hypothesis that the environment, primarily the gut microbiota, plays a significant role in modulating autoimmunity, neuroinflammation, motor deficits, and survival of C9orf72-null mice [140]. Numerous studies have committed to discovering the connection among autophagy, immunity, and neurodegeneration through C9orf72 function and advancing our understanding of the pathological mechanism of C9orf72-induced ALS.
Mechanism of C9orf72 function in autophagy
Regulation of autophagy by C9orf72 and the related complex
The autophagic function of C9orf72 was implied in C9orf72, or SMCR8 deleted mice, which show dysregulation of mTOR or TFEB, which are closely related to autophagy [69,70,132]. The interaction between C9orf72 and SMCR8 was initially proposed due to their structural similarity to FNIP and FLCN, respectively, which are two DENN-containing proteins that are able to form a stable complex [34,35]. In 2016, following affinity purification and mass spectrometric (MS) analysis of anti-C9orf72 immunoprecipitants from different cell lines, three studies separately confirmed that the C9orf72 protein can form a tripartite complex with SMCR8 and WDR41 at both the endogenous and exogenous levels [21,22,25,26,132]. Additionally, the interaction between C9orf72 and SMCR8 was proven to be mediated by the DENN domain in C9orf72 [21]. C9orf72 interacted tightly with SMCR8, and the C9orf72-SMCR8 complex was found to be strong and even resistant to high amounts of salt and detergent [25]. Furthermore, the C9orf72 protein level declined when the expression of SMCR8 was suppressed through small interfering RNA (siRNA) [25,27], while neither cellular C9orf72 nor SMCR8 knockout led to notable downregulation of WDR41 protein levels [132]. Similarly, a recent study demonstrated that loss of SMCR8 and WDR41 significantly diminished C9orf72 protein levels, and SMCR8 levels were also decreased after C9orf72 and WDR41 loss [27,143]. Taken together, these results suggest that both SMCR8 and C9orf72 play a key role in maintaining the stabilization of each other, while WDR41 might stabilize the C9orf72-SMCR8 heterodimer.
A systematic proteomic analysis of the human autophagy system initially revealed SMCR8 as an interacting protein of FIP200 [144], a component of the ULK1 complex. FIP200, ATG13, ULK1, and ATG101 form the ULK1 kinase complex [145,146], the most upstream of the autophagy machinery regulating autophagy in response to nutrient deprivation. C9orf72 exhibited colocalization with both autophagosomes and lysosomes in N2a cells, indicating that C9orf72 might directly affect the autophagy machinery [20]. Accordingly, the evidence suggested a potential connection between C9orf72 and autophagy initiation. The interaction between the C9orf72-SMCR8-WDR41 complex and all the ULK1 complex subunits was then determined [21,23,24,26]. Besides, the interaction between C9orf72 and the ULK1 complex was enhanced under amino acid starvation conditions [21]. Additionally, the interaction between SMCR8 and FIP200 was much weaker than that of the C9orf72-SMCR8-WDR41 complex [21]. In summary, these results suggest that the C9orf72-SMCR8-WDR41 complex likely regulates autophagy through its interaction with the ULK1 complex (Figure 3).
Figure 3.
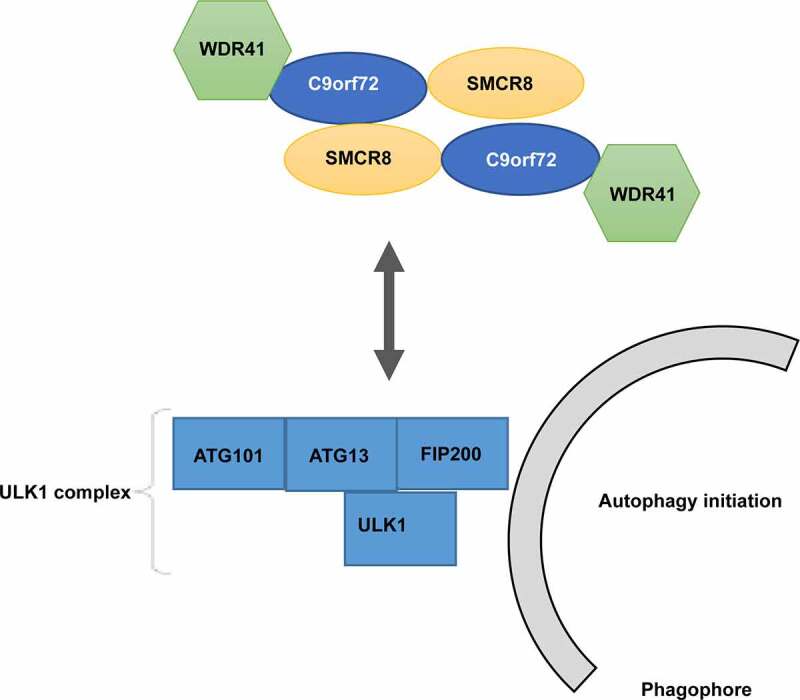
Regulation of the ULK1 complex by C9orf72-SMCR8-WDR41.
C9orf72-SMCR8-WDR41 binds to the ULK1 complex in an unclarified interaction mode, which leads to increased activity of the ULK1 complex and followed autophagy initiation.
Indeed, experiments showed that the decrease in either C9orf72 or SMCR8 caused impaired autophagy induction [21,24,147], which was the same circumstance in both the C9orf72 and SMCR8 knockdown cells [21,24,147]. Additionally, both ULK1 protein levels and phosphorylation at serine 318 of the ULK1 substrate ATG13 were markedly increased upon SMCR8 knockout. In contrast, an opposite effect was observed in the same study resulting from the depletion of C9orf72 or WDR41[21-24]. A recent study found inhibited autophagy and reduced ULK1 protein levels in neurons from C9orf72 depleted mice [147], supporting the notion that C9orf72 and SMCR8 might act against each other to regulate the autophagy initiation complex. Nevertheless, it was later discovered that the increased abundance of ULK1 protein was on due to elevated ULK1 mRNA levels caused by SMCR8 loss. This study also revealed that SMCR8 can regulate the expression of several autophagy genes, including ULK1 and WIPI2 [24]. Since the native functions of these proteins are multifaceted, the exact mechanism by which C9orf72-SMCR8-WDR41 regulates the ULK1 complex remains unclear (Figure 3).
The regulation of the autophagy inhibitor mTORC1 by C9orf72 and the related complexes
Intriguingly, C9orf72 and SMCR8 were reported to regulate mTORC1 activity (Figure 4), indicating that the ULK1 complex might be involved in a mTORC1-dependent regulatory mechanism of C9orf72 and SMCR8 [70,132,143]. Recent studies have shown that the levels of phosphorylated ribosomal protein S6 kinase 1 (S6K1) and mTORC1 are both increased in C9orf72- or SMCR8-deficient macrophages compared to WT controls [70,143]. In addition, several studies observed that C9orf72, SMCR8, and WDR41 colocalize at lysosomes, and this colocalization is enhanced upon starvation, such as amino acid deprivation [25,26,148,149]. It was further confirmed that WDR41 is indispensable for recruiting C9orf72 and SMCR8 to lysosomes during starvation [148]. More specifically, lysosomes were enlarged and compactly clustered in the perinuclear region in C9orf72 or SMCR8 deficient cells [148]. Furthermore, SMCR8-defective cells are generally accompanied by cellular hypertrophy [25,70,140,150]. Moreover, lysosomal defects in macrophages in C9orf72-deficient mice were observed, as indicated by the increased levels of lysosomal proteins such as progranulin (PGRN), prosaposin (PSAP), LAMP1, and cathepsin D (CathD) [26]. Similarly, SMCR8 deficiency caused reduced levels of LAMP1 [70,143]. Accordingly, we can conclude that C9orf72 and SMCR8 regulate mTORC1 activity and lysosomal biogenesis, despite in different ways, and raising the possibility that C9orf72-SMCR8-WDR41 may regulate the ULK1 kinase complex by mTORC1-dependent and mTORC1-independent mechanisms.
Figure 4.

Regulation of the mTOCRC1 complex by C9orf72-SMCR8-WDR41.
The WDR41 in the complex confers the lysosome localization, where it interacts with and regulates the kinase activity of the mTORC1 complex.
The structure of C9orf72 and the related complexes
The structure of C9orf72
The DENN domain-containing family is an ancient and highly conserved protein branch, and its members have the N-terminal Longin domain and the C-terminal DENN domain.
Structure-based model building suggested that C9orf72, together with SMCR8, FLCN, and FNIP1/2, compose a subfamily of DENN family proteins (Figure 5a) [33,34,151]. In the C9orf72-SMCR8-WDR41 complex structure, C9orf72 folds into a classic DENN family protein with an N-terminal Longin domain (uDENN domain) and a C-terminal DENN domain that are highly conserved to those in DENN1B-S (Fig. 5b, Fig. 6a) [31,38,39]. The Longin domain is composed of five central antiparallel β-strands (β1- β5) sandwiched between helix α1 and helices α3 and α4. Similarly, the core of the DENN domain consists of a β-sheet (β1ʹ- β5ʹ), which is layered by α6-α9 and appended by a helical bundle (α10-α15) (Figure 5b). The order of β strands in the Longin domain is β3, β4, β5, β1, and β2, whereas in the DENN domain is β2, β3, β1, β4, and β5 (Figure 5b). Of note, the linker between the Longin domain and the DENN domain of C9orf72 is highly flexible. Thus, C9orf72 may adopt an overall structure similar to that of DENN1B-S (Figure 6a).
Figure 5.
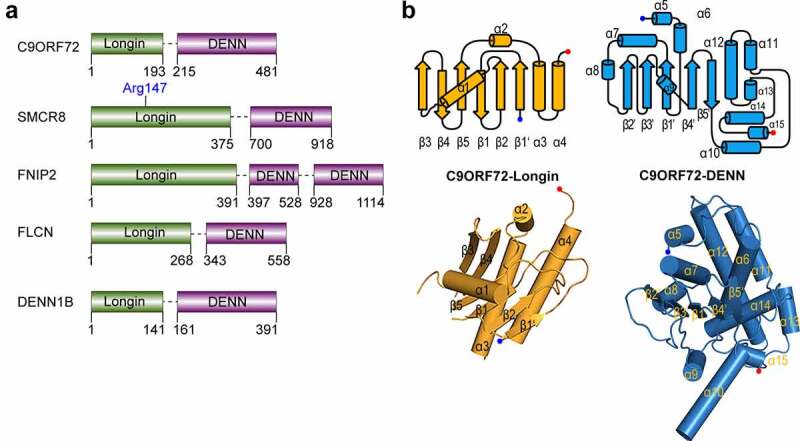
C9orf72 is a member of the DENN domain family.
a. The domain schematic of C9orf72, SMCR8, FNIP2, and FLCN.
b. The structural feature of the Longin domain and DENN domain. Upper: The topology of the Longin and DENN domain. Lower: The Longin domain (orange) and DENN domain (blue) of C9orf72 (PDB: 6LT0). All the structure models in this paper are generated using Pymol [182].
Figure 6.
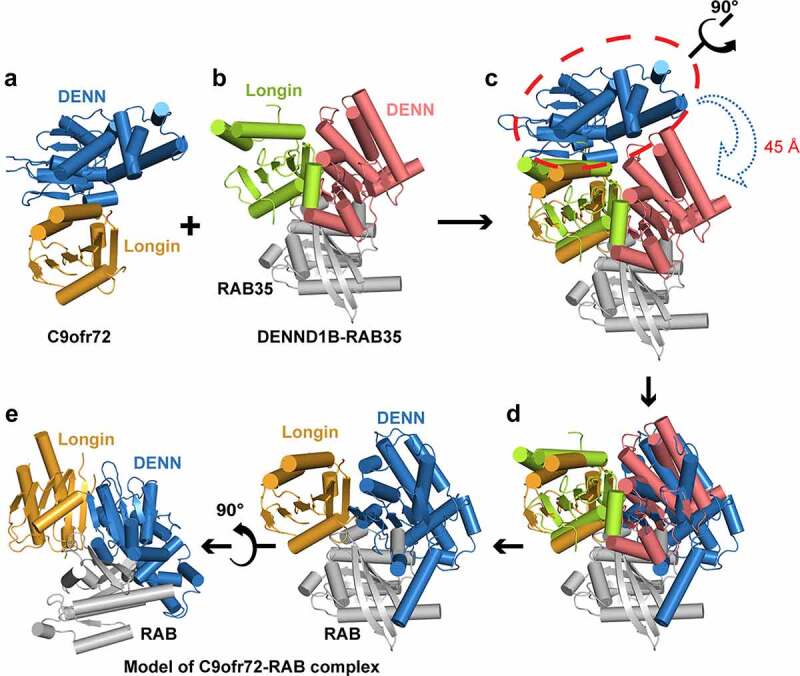
A putative model of the C9orf72-RAB complex.
a. The structure model of C9orf72 (PDB: 6LT0), Longin domain: orange, DENN domain: blue.
b. DENND1B-RAB35 (PDB: 3TW8), Longin domain: green, DENN domain: red, RAB35: grey.
c. Superimposition of the structure models of C9orf72 and DENND1B-RAB35. The colours are same as (A) and (B). The two structures will adopt a similar overall structure after that the DENN domain of C9orf72 rotates ~90° and moves ~45 Å towards the DENN of DENND1B.
d. The structure model of C9orf72 adopts similar folds to DENND1B.
e. The putative structure model of C9orf72-RAB is generated on the basis of the structure of DENND1B-RAB35.
The structure of the C9orf72-SMCR8 complex
C9orf72 and SMCR8 can form a stable complex that participates in the regulation of autophagy [21–27]. Biophysical analysis and cryo-EM analysis revealed that the C9orf72-SMCR8 complex is a heterodimeric dimer(Figure 7a, C) [38]; moreover, the monomeric structure of the C9orf72-SMCR8 complex was reported as well [39]. The discrepancy between the two studies might reflect three reasons: First, the C9orf72-SMCR8 dimer was overexpressed in insect cells [38], whereas the monomer was purified for HEK293 cells [39]. The posttranslational modifications in the two cell lines might be different, which might lead to two different statuses of the proteins. Second, the complex purified from the two systems might both be dimeric with most parts of the dimers broken during the process of cryo-EM sample preparation in work by Su et al [39]. The latter scenario is supported by observing monomeric particles of C9orf72-SMCR8 in the cryo-EM images in the paper by Tang et al [38]. Finally, and most possibly, C9orf72 and SMCR8 may bind to each other to form a heterodimer, and then the heterodimers dimerize to form a tetramer of C9orf72 and SMCR8 with a 2:2 stoichiometry. Thus, the C9orf72-SMCR8 complex might equilibrate between monomeric and dimeric states according to cellular activity. The two studies captured only the monomeric and dimeric status. Therefore, more work is needed to fully confirm the current data and gain understanding of the physiological significance of the dimerization of the C9orf72-SMCR8 protomer.
Figure 7.

The interface between DENN domains of the C9orf72 and SMCR8.
a. Left: the structure of the FLCN-FNIP2 complex (PDB: 6ULG). Longin domain: orange (FNIP2) and pink (FLCN), DENN domain: blue (FNIP2), and cyan (FLCN). The dash line oval indicates the interface between the FNIP2 DENN domain and the FLCN DENN domain. Right: the structure of C9orf72-SMCR8 protomer (PDB: 6LT0), Longin domain: orange (C9orf72), and pink (SMCR8), DENN domain: blue (C9orf72) and cyan (SMCR8). The dash line oval indicates the interface between the C9orf72 DENN domain and the SMCR8 DENN domain, which contributes to the formation of the C9orf72-SMCR8 protomer.
b. In the helix bundle on the C9orf72-DENN and the SMCR8-DENN interface, three different views are showed from left to right. This helix bundle shows a pseudo-centre symmetry.
c. The structure of the C9orf72-SMCR8 dimer complex. The colours are same as panel (A). The dash line ovals indicate the interface between the C9orf72 DENN domain and the SMCR8 DENN domain, while the dash line rectangles indicate the dimer interface between the C9orf72-SMCR8 protomers.
d. The schematic diagram of the structure of the C9orf72-SMCR8 dimer complex in panel (C).
Characterizations of DENN domains in C9orf72-SMCR8 complex
DENN family proteins are reported to be monomers in solution, e.g., DENN1A (PDB: 6EKK), and DENN1B [31]. However, in the cryo-EM structure model of the FLCN-FNIP2 complex, the DENN domains of FLCN and FNIP2 are dimerized with their C-terminal helices forming a helix bundle. This binding feature is confirmed in the structure of the C9orf72-SMCR8 complex (Figure 7a) [38,39], with structural homology to the FLCN-FNIP2 complex. The helix bundle of the C9orf72-SMCR8 interface consists of helices from α11 to α14 in C9orf72 and helices from α10 to α13 in SMCR8. (Figure 7a) [37,152]. Intriguingly, the eight helices show a pseudo-centre symmetry: α11 of C9orf72 to the α10 of SMCR8, the α12 of C9orf72 to the α11 of SMCR8, the α13 of C9orf72 to the α12 of SMCR8, the α14 of C9orf72 to α13 of SMCR8 (Figure 7a,B).
Unexpectedly, the dimerization of the two protomers in the C9orf72-SMCR8 complex is mediated by the most distal C-terminus motif of C9orf72 and the DENN domain of SMCR8 (Figure 7c, D) [38]. Although the electron density of this interface is obscure, the region of C9orf72 that binds to the SMCR8 DENN domain was identified by mutagenesis analysis and analytical ultracentrifugation [38]. This binding manner between DENN domains defines a novel organization mode of DENN domains [38,40]. Whether the interface mediated the dimerization of protomers is widespread among DENN family proteins has yet to be explored, and a bioinformatics analysis will be an effective way to make this determination.
C9orf72 is a regulator of the RAS superfamily
The RAS superfamily of small GTPases consists of more than150 members in humans and can be divided into six major subgroups based on the function and amino acid sequence: RAS, RHO, RAB, RAN, ARF, and RRAG [153,154]. The RAS members, sharing a conserved globular G-domain, function as binary molecular switches that are activated by binding GTP and inactivated when GTP is hydrolysed to GDP [153,155–157]. The GTP-GDP cycle of the RAS superfamily is controlled by GEFs, which facilitate the exchange of GDP to GTP, and by GAPs, which stimulate GTP hydrolysis [153,155–157]. RABs are the largest subfamily among the RAS superfamily and play a critical role in regulating membrane trafficking [155–158]. ARFs are ubiquitous in eukaryotic cells and vital regulators of vesicle biogenesis during intracellular trafficking [153]. RRAGs, the sixth branch of the RAS superfamily, are sensors of amino acids in mTOR signal pathway and function in heterodimers, e.g., RRAGA/B-RRAGC/D [37,152,154,159,160].
DENN-containing proteins have been shown to be GEF or GAP of RABs [20–23,26,34,35,38,40,151,161], ARFs [39,162], and RRAGs [37,152,154,159,160]. For example, DENN1B acts as a GEF for RAB35 by inducing conformational changes in the Switch I and Switch II motifs of RAB35 to facilitate the GDP dissociation [30,31,163]. The MON1-CCZ1 complex functions as a GEF for RAB7 by locking the conformation of the RAB7 Switch I motif, making it is incapable of binding with GDP [33,164].
As a DENN family protein member, C9orf72 is suggested to be a GEF for dozens of RAB GTPases [20–23,151]. Consistent with this hypothesis, C9orf72 was shown to interact and colocalize with a bunch of RAB proteins, including but not limited to RAB1A, RAB5A, RAB7A, RAB8A, RAB11A, and RAB39B [20–23,151]. These RABs are essential for cell migration, membrane receptor recycling, synaptic vesicle secretion, and cell polarization [158,165]. Hence, C9orf72 may plays a pivotal role in the RAB cascade and regulate these cellular processes via RABs. Intriguingly, C9orf72 was indicated as an effector protein of RAB1A because C9orf72 binds to activated RAB1A preferentially [23], and confirmation of this hypothesis would expand the spectrum of known functions of C9orf72 and the DENN family.
Further studies demonstrated that C9orf72 in complex with SMCR8 and WDR41 possesses GEF activity towards RAB8A and RAB39B, as shown in in vitro GDP/GTP exchange assays [21,22]. Moreover, the C9orf72-SMRC8 complex has been proven to be GAP for small GTPases [38–40], however, these studies do not rule out the possibility that C9orf72 alone might function as a GEF or other kind of regulator of small GTPases. As the study into C9orf72 advances, an increasing number of small GTPases will likely be identified as the partners of C9orf72.
The C9orf72-SMCR8 complex is a GAP for members of the RAS superfamily
Similar to C9orf72 alone, the C9orf72-SMCR8 complex functions as a GEF for several RABs in many studies [21–27,132]. Unexpectedly, the C9orf72-SMCR8 complex was also found to be able to accelerate the consumption rate of GTP by RAB8, RAB11, and ARF1, however, this complex cannot increase the nucleotide exchange rate of these small GTPases [38–40], The FNIP2 complex functions as a GAP for RRAGC/D on lysosomes [37,152,159,166], which indicates that the C9orf72-SMCR8 complex is not a GEF for these small GTPases. Similarly, the FLCN was wrongly suggested to be a GEF for RAB35 on the basis of an in vitro assay [167]. Of note, the structure of the C9orf72-SMCR8 protomer resembles that of the FLCN-FNIP2 complex. Strikingly, the structural superimposition suggests that C9orf72 is the structural homolog of FNIP2, while SMCR8 corresponds to FLCN [38–40]. Moreover, both bioinformatic analysis and structural comparisons show that the arginine finger of FLCN is conserved in SMCR8[38-40]. Mutagenesis analyses combined with biophysical analyses demonstrated that the C9orf72-SMCR8 complex acts as a GAP for RAB8A [38], RAB11A [38], and ARF1[39].
Unfortunately, the mechanisms by which the C9orf72-SMCR8 complex binds to small GTPases and the role of the SMCR8 arginine finger in facilitating the hydrolysis of GTP are unknown.
Proposed structural model of the C9orf72-RAB complex and C9orf72-SMCR8-RAB complex
The existing binding region in DENN family proteins
A comparative analysis indicated that three regions in the Longin domain, the A region, B region, and C region, are critical for both intra- and intermolecular binding [168]. The A region, formed by β3, α1 and part of β4/5, primarily mediates the dimerization of Longin domains and binding with the effectors (Figure 8a) [168]. The B region, consisting of α1 and the β1-β2 hairpin, is mainly involved in the recognition of small GTPases (Figure 8a) [168]. The function of the C region is the most variable and usually involves the α2, α3, and sometimes α4; however, this region also participates in binding with the partners (Figure 8a) [168]. The three regions partially overlap, and sometimes the binding of DENN family proteins with their partners requires cooperation between the A and C region; e.g., MON1-CCZ1 must form a complex to function as the GEF for RAB7 [164]; GATOR1 requires NPRL2 and NPRL3, both containing the Longin domain, to regulate the RRAGA/B GTPases as GAP [160]; and the FLCN-FNIP2 complex functions as a GAP of RRAGC/D GTPases via its Longin pair [37,152]. Both C9orf72 and the Longin domain of SMCR8 are necessary for the GAP activity of RAB8A/11A38[39]. In general, the DENN family proteins interact with their partners in multiple manners, bestowing regulatory ability of C9orf72 and related complex to modulate the RAS superfamily through different mechanisms.
Figure 8.
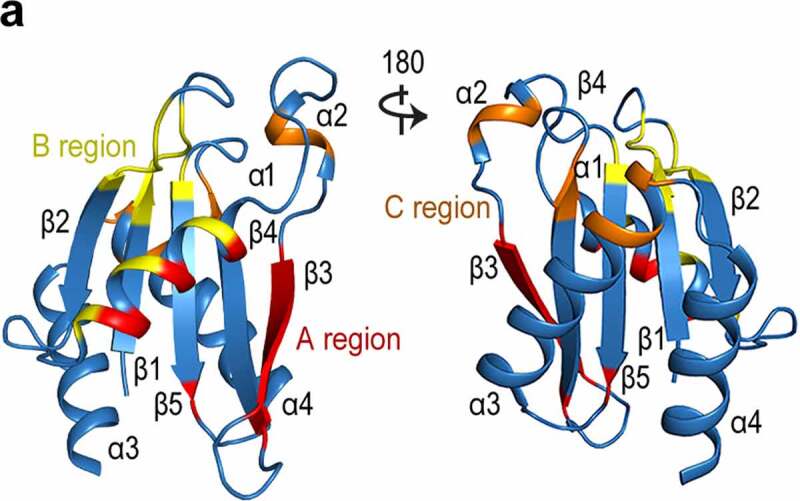
The existing binding regions on DENN family proteins.
a. The A region, Bregion, and C region, which are responsible for both intra- and inter-molecular binding, are coloured in red, yellow, and orange, respectively. Two views are showed here.
Structural model of the C9orf72-RAB complex
Radioactive assays showed that C9orf72 alone is able to bind to several RABs [21,23]. Hence, one of the remaining questions is how C9orf72 alone recognizes specific RABs.
Although this question cannot be addressed clearly before the structure of C9orf72-RAB is solved, based on the structure of DENN1B-RAB35, a speculative structural model of C9orf72-RAB was generated (Figure 6c-E). In this model, C9orf72 adopts a conformation that is similar to that of DENN1B-S, and the binding of C9orf72 and RAB is dependent on both the Longin domain and the DENN domain of C9orf72. However, the binding mode between C9orf72 and RABs is unclear, whether C9orf72 alone is a GEF for RABs is unknown, and the determination of both issues requires more functional, biochemical, and structural studies.
Structural model of the C9orf72-SMCR8-RAB complex
The structural model of the C9orf72-SMCR8-RAB complex has not yet been reported; however, we can obtain some clues regarding the model of the C9orf72-SMCR8-RAB complex by studying the existing structural model of GAP-RAB complexes or GEF-RAB complexes, e.g., the structure of the FLCN-FNIP2-RRAGA-RRAGC complex [37,152], the structure of the NPRL2-NPRL3 subcomplex [160], and the structure of the MON1-CCZ1-RAB7 complex [164]. Notably, in the structure of the MON1-CCZ1-RAB7 complex, MON1 and CCZ1 form a Longin pair, which shows a highly similar overall fold to that of the C9orf72-SMCR8 complex (Figure 9a, B). Attractively, the complex of C9orf72 and the Longin domain of SMCR8 shows almost full GAP activity for RAB8A and RAB11A [38]. Although the MON1-CCZ1 complex is a GEF for RAB7 and the C9orf72-SMCR8 complex is a GAP for RAB8/11, it is possible that the Longin pairs of the two complexes recognize RABs in a similar manner. Therefore, a putative structural model of the C9orf72-SMCR8-RAB complex is proposed here based on the structures of the MON1-CCZ1-RAB7 complex and the C9orf72-SMCR8 complex (Figure 9c-E). Since both RABs and ARFs belong to the RAS superfamily and share a highly conserved G domain, the C9orf72-SMCR8 complex probably binds to ARF1 in a manner similar to that shown in the structural model of the C9orf72-SMCR8-RAB complex. Nevertheless, verification of the true mechanism by which the C9orf72-SMCR8 complex recognizes its target RABs or ARFs requires data support.
Figure 9.
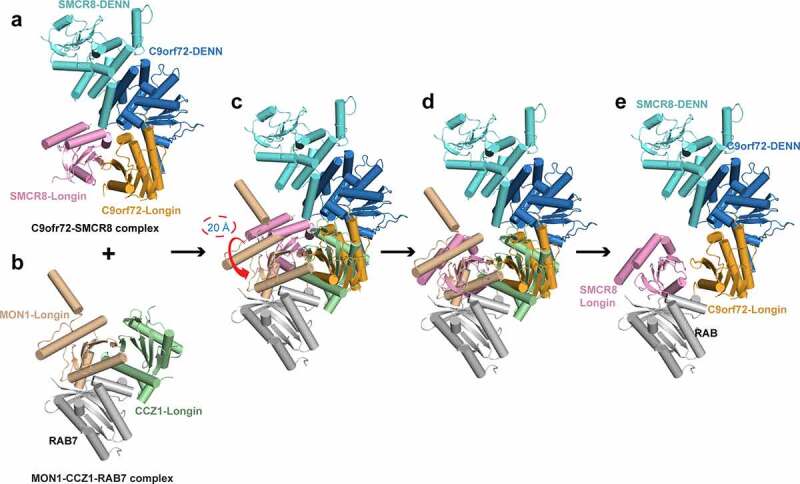
A putative model of the C9orf72-SMCR8-RAB complex.
a. The structure of the C9orf72-SMCR8 complex (PDB: 6LT0), Longin domain: orange (C9orf72), and pink (SMCR8), DENN domain: blue (C9orf72) and cyan (SMCR8).
b. The structure of the MON1-CCZ1-RAB7 complex (PDB: 5LDD). MON1-Longin: wheat, CCZ1-Longin: pale green, RAB7: grey.
c. And d. Superimposition of C9orf72-SMCR8 complex and MON1-CCZ1-RAB7 complex. The two Longin pairs will adopt a similar overall structure after the SMCR8-Longin moves ~20 Å towards the RAB7.
e. The putative structure model of the C9orf72-SMCR8-RAB complex.
Cellular localization of the C9orf72-SMCR8 complex is mediated by WDR41
WDR41, a WD40 repeat-containing protein, is one subunit of the C9orf72-SMCR8-WDR41 complex with its C-terminal helix embedded into the hydrophobic groove of the DENN domain of SMCR8 without directly contacting with C9orf72[38-40].
WDR41 expression is widespread in cells, including the cytoplasm, ER, Golgi, and lysosome [22,25,26]. Recent studies have shown that WDR41 localizes to the lysosomes upon interacting with SLC66A1/PQLC2 and therefore anchors the C9orf72-SMCR8 complex to the lysosomes, which modulates mTOC1 signalling [148,149]. The results from the Hurley lab indicate that the docking of the C9orf72-SMCR8-WDR41 complex to lysosomes sequestrates the GAP activity for ARF1[39]; moreover, the C9orf72-SMCR8 complex also functions as a GAP for RABs [38,40]. The latest results from Ferguson’s lab’s suggested that WDR41 binds to the inwards facing SLC66A1/PQLC2 through the 7 CD loop [169]. SLC66A1/PQLC2 might function as both a receptor of WDR41 and a transporter of cationic amino acids, which links the lysosome homoeostasis, the cationic amino acid level, and the function of the C9orf72-SMCR8-WDR41 complex [169].
Upon depletion of cationic amino acids, the C9orf72-SMCR8-WDR41 complex is recruited to a lysosome by SLC66A1/PQLC2 [148, 149, 169]. At this time point, the following scenarios might be possible: First, the C9orf72-SMCR8-WDR41 complex cannot inactivate the targeted small GTPases, including ARF1. Inactivation of ARF1 leads to the redistribution of the Golgi apparatus to the endoplasmic reticulum (ER) [170,171]. The ER-Golgi intermediate is critical to the biogenesis of autophagosomes [172]. Together, the recruitment of the C9orf72-SMCR8-WDR41 complex to lysosomes might be important for maintaining the activation state of ARFs, which is critical to the maintenance of ER-Golgi intermediates and the initiation of autophagy (Figure 10A). Second, there might be some unidentified effector proteins of the C9orf72-SMCR8-WDR41 complex on the lysosome. Interestingly, many studies indicate that C9orf72 participates in nutrient-sensing in lysosome that is independent of mTOR signalling [25,132,149,173]. Accordingly, the recruitment of the C9orf72-SMCR8-WDR41 complex to lysosomes might be part of a lysosome-associated pathway that participates in the cellular response to nutrient variation [148,169]. Of course, the recruitment of C9orf72 and the related complex to the lysosome also facilitates the interplay with mTOR [173,174]. Noteworthily, these two hypotheses may reflect functions that parallel each other and might represent only small portions of the C9orf72-related network.
Figure 10.

The proposed function models of the C9orf72-SMCR8-WDR41 complex.
a. The C9orf72-SMCR8-WDR41 complex acts as the GAP for ARF1 or other small GTPases in the cytoplasm, whereas the C9orf72-SMCR8-WDR41 complex cannot function as the GAP of ARF1 when it is sequestered by SLC66A1/PQLC2, which can sense the level of cationic amino acids and binds to WDR41 at the inwards open conformation. On the other hand, recruitment of the CSW complex to the lysosome is essential to the cross-talk between the CSW complex and its partners on the lysosome. The structure model of SLC66A1/PQLC2 is generated using human SLC26A9 (PDB:7CH1) as a template [183]. Grey: ARF1, orange, and teal: SLC66A1/PQLC2. Blue: C9orf72, pink: SMCR8, green: WDR41, grey: RABs. The arrows with a solid line indicate reported signal pathways, whereas the arrows with a dashed line indicate proposed connections.
b. Model of the CSW complex localizing on ER or Golgi apparatus: the CSW complex localizes on ER/Golgi via binding to receptors or lipidation. The labels and colours are the same as the panel(A). The brown lines with a circle head denote the lipidation.
C. The C9orf72-SMCR8-WDR41 complex tether two endosomes/vesicles and regulates the fusion through its GAP activity. The C9orf72-SMCR8-WDR41 complex is coloured as a panel(A).
The mechanism of WDR41 localization to the ER and Golgi remains unclear. Many receptors and transporters that are similar to SLC66A1/PQLC2 localize to the ER/Golgi as well and play a pivotal role in the sensing and regulation of nutrients and metabolites, e.g., SLC16A13[175] SLC30A7[175] and the SLC35 subfamily [176,177]. Based on the binding mode of WDR41 and SLC66A1/PQLC2, WDR41 might dock on the ER/Golgi by interacting with a specific receptor or transporter under certain conditions (Figure 10B). It would not be surprising if receptors for WDR41 were found on the ER/Golgi in future.
The localization of the C9orf72-SMCR8-WDR41 complex to the ER/Golgi might also be facilitated by lipidation modifications, such as prenylation [157,178], palmitoylation [179,180], and myristoylation [181]. Intriguingly, the localization of RABs that function on the ER/Golgi is regulated by prenylation [178]. The lipidation of the C9orf72-SMCR8-WDR41 complex might synergize with the prenylation of RABs and facilitate the colocalization of the C9orf72-SMCR8-WDR41 complex and RABs to the ER/Golgi, where the C9orf72-SMCR8-WDR41 complex takes part in the regulation of membrane trafficking as a GAP (Figure 10B).
Moreover, C9orf72 is able to regulate the fusion of endosomes via RABs [70,151,161]. The C9orf72-SMCR8-WDR41 heterotrimer complex can form dimers; therefore, the C9orf72-SMCR8-WDR41 complex can tether two endosomes close to each other using WDR41 as a membrane anchor and regulate the membrane fusion via its GAP activity for RABs or ARF1(Figure 10C). [38,40].
Perspectives
The G4C2 repeat expansion of C9orf72 accounts for almost 50% of genetic ALS cases and ~ 30% cases of genetic FTD cases. This historic finding highlights the future of C9orf72 as a therapeutic target for genetic ALS and FTD. The pathogenic mechanism has been revealed, and the major hypotheses converge. The final answer to this riddle awaits the discovery of more details of C9orf72 related cellular processes.
As a versatile gene, C9orf72 is critical to homoeostasis since its produce participates in the regulation of many signalling pathways, e.g., autophagy, membrane trafficking, lysosome function, and immune activity. Dozens of converging points that C9orf72 regulates the aforementioned pathways have been identified; however, we believe that they represent only a small part of the C9orf72-related network.
Despite many unanswered questions, the physiological function of the C9orf72-SMCR8-WDR41 complex is emerging, which deepens our understanding of the function of the C9orf72-SMCR8-WDR41 complex and sheds light on the pathogenic mechanism of C9orf72 abnormalities in the C9-ALS/FTD. To uncoil the remaining entangled details related to C9orf72, persistent multidisciplinary efforts and collaboration are required.
Acknowledgments
This work was supported by the National Key R&D Program of China grant 2017YFA0506300 (L. K.), 2018YFC1004601 (S.Q.), and NSFC grants 32071214 (Q. S.), 31770820 (L. K.).
Funding Statement
This work was supported by the National Key R&D Program of China [2017YFA0506300]; NSFC [31770820]; NSFC [32071214]; National Key R&D Program of China [2018YFC1004601].
Disclosure statement
The authors declare that they have no conflicts of interest with the contents of this article.
References
- [1].DeJesus-Hernandez M, Mackenzie I, Boeve B, et al. Expanded GGGGCC hexanucleotide repeat in noncoding region of C9ORF72 causes chromosome 9p-linked FTD and ALS. Neuron. 2011;72(2):245–256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2].Renton AE, Majounie E, Waite A, et al. A hexanucleotide repeat expansion in C9ORF72 is the cause of chromosome 9p21-linked ALS-FTD. Neuron. 2011;72:257–268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Zhu Q, Jiang J, Gendron TF, et al. Reduced C9ORF72 function exacerbates gain of toxicity from ALS/FTD-causing repeat expansion in C9orf72. Nat Neurosci. 2020;23:615–624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Shi Y, Lin S, Staats KA, et al. Haploinsufficiency leads to neurodegeneration in C9ORF72 ALS/FTD human induced motor neurons. Nat Med. 2018;24:313–325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Kwon I, Xiang S, Kato M, et al. Poly-dipeptides encoded by the C9orf72 repeats bind nucleoli, impede RNA biogenesis, and kill cells. Science. 2014;345:1139–1145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Haeusler AR, Donnelly CJ, Periz G, et al. C9orf72 nucleotide repeat structures initiate molecular cascades of disease. Nature. 2014;507:195–200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Hayes LR, Rothstein JD.. C9ORF72-ALS/FTD: transgenic mice make a come-BAC. Neuron. 2016;90:427–431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Conlon EG, Lu L, Sharma A, et al. The C9ORF72 GGGGCC expansion forms RNA G-quadruplex inclusions and sequesters hnRNP H to disrupt splicing in ALS brains. Elife. 2016;5: DOI: 10.7554/eLife.17820 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Balendra R, Isaacs AM.. C9orf72-mediated ALS and FTD: multiple pathways to disease. Nat Rev Neurol. 2018;14:544–558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Mori K, Weng S-M, Arzberger T, et al. The C9orf72 GGGGCC repeat is translated into aggregating dipeptide-repeat proteins in FTLD/ALS. Science. 2013;339(6125):1335–1338. [DOI] [PubMed] [Google Scholar]
- [11].Mizielinska S, Gronke S, Niccoli T, et al. C9orf72 repeat expansions cause neurodegeneration in Drosophila through arginine-rich proteins. Science. 2014;345:1192–1194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Freibaum BD, Lu Y, Lopez-Gonzalez R, et al. GGGGCC repeat expansion in C9orf72 compromises nucleocytoplasmic transport. Nature. 2015;525:129–133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Chew J, Gendron TF, Prudencio M, et al. Neurodegeneration. C9ORF72 repeat expansions in mice cause TDP-43 pathology, neuronal loss, and behavioral deficits. Science. 2015;348:1151–1154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Zhang YJ, Gendron TF, Grima JC, et al. C9ORF72 poly(GA) aggregates sequester and impair HR23 and nucleocytoplasmic transport proteins. Nat Neurosci. 2016;19:668–677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Lee KH, Zhang P, Kim HJ, et al. C9orf72 dipeptide repeats impair the assembly, dynamics, and function of membrane-less organelles. Cell. 2016;167:774–788 e717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Guo Q, Lehmer C, Martinez-Sanchez A, et al. In situ structure of neuronal C9orf72 poly-GA aggregates reveals proteasome recruitment. Cell. 2018;172:696–705 e612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Hao Z, Liu L, Tao Z, et al. Motor dysfunction and neurodegeneration in a C9orf72 mouse line expressing poly-PR. Nat Commun. 2019;10:2906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Wang R, Xu X, Hao Z, et al. Poly-PR in C9ORF72-related amyotrophic lateral sclerosis/frontotemporal dementia causes neurotoxicity by clathrin-dependent endocytosis. Neurosci Bull. 2019;35:889–900. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Zhang YJ, Guo L, Gonzales PK, et al. Heterochromatin anomalies and double-stranded RNA accumulation underlie C9orf72 poly(PR) toxicity. Science. 2019;363: DOI: 10.1126/science.aav2606 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Farg MA, Sundaramoorthy V, Sultana JM, et al. C9ORF72, implicated in amytrophic lateral sclerosis and frontotemporal dementia, regulates endosomal trafficking. Hum Mol Genet. 2014;23:3579–3595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Yang M, Liang C, Swaminathan K, et al. A C9ORF72/SMCR8-containing complex regulates ULK1 and plays a dual role in autophagy. Sci Adv. 2016;2:e1601167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Sellier C, Campanari ML, Corbier CJ, et al. Loss of C9ORF72 impairs autophagy and synergizes with polyQ Ataxin-2 to induce motor neuron dysfunction and cell death. Embo J. 2016;35:1276–1297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Webster CP, Smith EF, Bauer CS, et al. The C9orf72 protein interacts with Rab1a and the ULK1 complex to regulate initiation of autophagy. Embo J. 2016;35:1656–1676. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Jung J, Nayak A, Schaeffer V, et al. Multiplex image-based autophagy RNAi screening identifies SMCR8 as ULK1 kinase activity and gene expression regulator. Elife. 2017;6: DOI: 10.7554/eLife.23063 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Amick J, Roczniak-Ferguson A, Ferguson SM. C9orf72 binds SMCR8, localizes to lysosomes, and regulates mTORC1 signaling. Mol Biol Cell. 2016;27:3040–3051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Sullivan PM, Zhou X, Robins AM, et al. The ALS/FTLD associated protein C9orf72 associates with SMCR8 and WDR41 to regulate the autophagy-lysosome pathway. Acta Neuropathol Commun. 2016;4:51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Zhang Y, Burberry A, Wang JY, et al. The C9orf72-interacting protein Smcr8 is a negative regulator of autoimmunity and lysosomal exocytosis. Genes Dev. 2018;32:929–943. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Trageser KJ, Smith C, Herman FJ, et al. Mechanisms of immune activation by c9orf72-expansions in amyotrophic lateral sclerosis and frontotemporal dementia. Front Neurosci. 2019;13:1298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Lai JD, Ichida JK. C9ORF72 protein function and immune dysregulation in amyotrophic lateral sclerosis. Neurosci Lett. 2019;713:134523. [DOI] [PubMed] [Google Scholar]
- [30].Yoshimura S, Gerondopoulos A, Linford A, et al. Family-wide characterization of the DENN domain Rab GDP-GTP exchange factors. J Cell Biol. 2010;191:367–381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Wu X, Bradley MJ, Cai Y, et al. Insights regarding guanine nucleotide exchange from the structure of a DENN-domain protein complexed with its Rab GTPase substrate. Proc Natl Acad Sci U S A. 2011;108:18672–18677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Marat AL, Dokainish H, McPherson PS. DENN domain proteins: regulators of Rab GTPases. J Biol Chem. 2011;286:13791–13800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Levine TP, Danials RD, Wong LH, et al. Discovery of new Longin and Roadblock domains that form platforms for small GTPases in Ragulator and TRAPP-II. Small GTPases. 2013;4:62–69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Zhang D, Iyer LM, He F, et al. Discovery of novel DENN proteins: implications for the evolution of eukaryotic intracellular membrane structures and human disease. Front Genet. 2012;3:283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [35].Levine TP, Daniels RD, Gatta AT, et al. The product of C9orf72, a gene strongly implicated in neurodegeneration, is structurally related to DENN Rab-GEFs. Bioinformatics. 2013;29:499–503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].Levivier E, Goud B, Souchet M, et al. uDENN, DENN, and dDENN: indissociable domains in Rab and MAP kinases signaling pathways. Biochem Biophys Res Commun. 2001;287:688–695. [DOI] [PubMed] [Google Scholar]
- [37].Shen K, Rogala KB, Chou HT, et al. Cryo-EM structure of the human FLCN-FNIP2-Rag-ragulator complex. Cell. 2019;179:1319–1329 e1318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [38].Tang D, Sheng J, Xu L, et al. Cryo-EM structure of C9ORF72-SMCR8-WDR41 reveals the role as a GAP for Rab8a and Rab11a. Proc Natl Acad Sci U S A. 2020;117:9876–9883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [39].Su M-Y, Fromm SA, Zoncu R, et al. Structure of the C9orf72 ARF GAP complex that is haploinsufficient in ALS and FTD. Nature. 2020;585:251–255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [40].Tang D, Sheng J, Xu L, et al. The C9orf72-SMCR8-WDR41 complex is a GAP for small GTPases. Autophagy. 2020. DOI: 10.1080/15548627.2020.1779473 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [41].Brown RH, Al-Chalabi A. Amyotrophic lateral sclerosis. N Engl J Med. 2017;377:162–172. [DOI] [PubMed] [Google Scholar]
- [42].Pasinelli P, Brown RH. Molecular biology of amyotrophic lateral sclerosis: insights from genetics. Nat Rev Neurosci. 2006;7:710–723. [DOI] [PubMed] [Google Scholar]
- [43].Kim G, Gautier O, Tassoni-Tsuchida E, et al. Implications for future therapies. Neuron. 2020;108:822–842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [44].Norris SP, Likanje MN, Andrews JA. Amyotrophic lateral sclerosis: update on clinical management. Curr Opin Neurol. 2020;33:641–648. [DOI] [PubMed] [Google Scholar]
- [45].Bensimon G, Lacomblez L, Meininger V. A controlled trial of riluzole in amyotrophic lateral sclerosis. ALS/Riluzole Study Group. N Engl J Med. 1994;330:585–591. [DOI] [PubMed] [Google Scholar]
- [46].Lacomblez L, Bensimon G, Leigh PN, et al. A confirmatory dose-ranging study of riluzole in ALS. ALS/Riluzole Study Group-II. Neurology. 1996;47:S242–250. [DOI] [PubMed] [Google Scholar]
- [47].Andrews JA, Jackson CE, Heiman-Patterson TD, et al. Real-world evidence of riluzole effectiveness in treating amyotrophic lateral sclerosis. Amyotroph Lateral Scler Frontotemporal Degener. 2020;21:509–518. [DOI] [PubMed] [Google Scholar]
- [48].Smith R, Pioro E, Myers K, et al. Enhanced bulbar function in amyotrophic lateral sclerosis: the nuedexta treatment trial. Neurotherapeutics. 2017;14:762–772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [49].Brooks BR, Thisted RA, Appel SH, et al. Treatment of pseudobulbar affect in ALS with dextromethorphan/quinidine. A Randomized Trial. 2004;63:1364–1370. [DOI] [PubMed] [Google Scholar]
- [50].Writing G, Edaravone ALSSG. Safety and efficacy of edaravone in well defined patients with amyotrophic lateral sclerosis: a randomised, double-blind, placebo-controlled trial. Lancet Neurol. 2017;16:505–512. [DOI] [PubMed] [Google Scholar]
- [51].Hulisz D. Amyotrophic lateral sclerosis: disease state overview. Am J Manag Care. 2018;24:S320-S326. [PubMed]
- [52].Renton AE, Chio A, Traynor BJ. State of play in amyotrophic lateral sclerosis genetics. Nat Neurosci. 2014;17:17–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [53].van Rheenen W, Shatunov A, Dekker AM, et al. Genome-wide association analyses identify new risk variants and the genetic architecture of amyotrophic lateral sclerosis. Nat Genet. 2016;48:1043–1048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [54].Hulisz D. Amyotrophic lateral sclerosis: disease state overview. Am J Manag Care. 2018;24:S320–s326. [PubMed] [Google Scholar]
- [55].Vance C, Al-Chalabi A, Ruddy D, et al. Familial amyotrophic lateral sclerosis with frontotemporal dementia is linked to a locus on chromosome 9p13.2-21.3. Brain. 2006;129:868–876. [DOI] [PubMed] [Google Scholar]
- [56].Morita M, Al-Chalabi A, Andersen PM, et al. A locus on chromosome 9p confers susceptibility to ALS and frontotemporal dementia. Neurology. 2006;66:839–844. [DOI] [PubMed] [Google Scholar]
- [57].Jung J, Behrends C. Multifaceted role of SMCR8 as autophagy regulator. Small GTPases. 2020;11:53–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [58].Haeusler AR, Donnelly CJ, Rothstein JD. The expanding biology of the C9orf72 nucleotide repeat expansion in neurodegenerative disease. Nat Rev Neurosci. 2016;17:383–395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [59].Burguete AS, Almeida S, Gao FB, et al. GGGGCC microsatellite RNA is neuritically localized, induces branching defects, and perturbs transport granule function. Elife. 2015;4:e08881. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [60].Jiang J, Zhu Q, Gendron TF, et al. Gain of Toxicity from ALS/FTD-linked repeat expansions in C9ORF72 Is alleviated by antisense oligonucleotides targeting GGGGCC-containing RNAs. Neuron. 2016;90:535–550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [61].Gendron TF, Petrucelli L. Disease Mechanisms of C9ORF72 Repeat Expansions. Cold Spring Harb Perspect Med. 2018;8: DOI: 10.1101/cshperspect.a024224 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [62].Chan YA, Aristizabal MJ, Lu PY, et al. Genome-wide profiling of yeast DNA:RNA hybrid prone sites with DRIP-chip. PLoS Genet. 2014;10:e1004288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [63].Aguilera A, Garcia-Muse T. R loops: from transcription byproducts to threats to genome stability. Mol Cell. 2012;46:115–124. [DOI] [PubMed] [Google Scholar]
- [64].Zu T, Liu Y, Banez-Coronel M, et al. RAN proteins and RNA foci from antisense transcripts in C9ORF72 ALS and frontotemporal dementia. Proc Natl Acad Sci U S A. 2013;110:E4968–4977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [65].Babic Leko M, Zupunski V, Kirincich J, et al. Molecular mechanisms of neurodegeneration related to C9orf72 hexanucleotide repeat expansion. Behav Neurol. 2019;2019:2909168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [66].Jovicic A, Mertens J, Boeynaems S, et al. Modifiers of C9orf72 dipeptide repeat toxicity connect nucleocytoplasmic transport defects to FTD/ALS. Nat Neurosci. 2015;18:1226–1229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [67].Ciura S, Lattante S, Ber IL, et al. Loss of function of C9orf72 causes motor deficits in a zebrafish model of amyotrophic lateral sclerosis. Ann Neurol. 2013;74:180–187. [DOI] [PubMed] [Google Scholar]
- [68].Therrien M, Rouleau GA, Dion PA, et al. Deletion of C9ORF72 results in motor neuron degeneration and stress sensitivity in C. elegans. PLoS One. 2013;8:e83450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [69].McAlpine W, Sun L, Wang KW, et al. Excessive endosomal TLR signaling causes inflammatory disease in mice with defective SMCR8-WDR41-C9ORF72 complex function. Proc Natl Acad Sci U S A. 2018;115:E11523–E11531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [70].Shao Q, Yang M, Liang C, et al. C9orf72 and smcr8 mutant mice reveal MTORC1 activation due to impaired lysosomal degradation and exocytosis. Autophagy. 2019;1–16. DOI: 10.1080/15548627.2019.1703353 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [71].Xiao S, McKeever PM, Lau A, et al. Synaptic localization of C9orf72 regulates post-synaptic glutamate receptor 1 levels. Acta Neuropathol Commun. 2019;7:161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [72].Sreedharan J, Blair IP, Tripathi VB, et al. TDP-43 mutations in familial and sporadic amyotrophic lateral sclerosis. Science. 2008;319:1668–1672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [73].Van Deerlin VM, Leverenz JB, Bekris LM, et al. TARDBP mutations in amyotrophic lateral sclerosis with TDP-43 neuropathology: a genetic and histopathological analysis. Lancet Neurol. 2008;7:409–416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [74].Chio A, Calvo A, Mazzini L, et al. Extensive genetics of ALS: a population-based study in Italy. Neurology. 2012;79:1983–1989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [75].Corcia P, Valdmanis P, Millecamps S, et al. Phenotype and genotype analysis in amyotrophic lateral sclerosis with TARDBP gene mutations. Neurology. 2012;78:1519–1526. [DOI] [PubMed] [Google Scholar]
- [76].Sleigh JN, Tosolini AP, Gordon D, et al. Mice carrying ALS mutant TDP-43, but not mutant FUS, display in vivo defects in axonal transport of signaling endosomes. Cell Rep. 2020;30:3655–3662 e3652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [77].Wong P, Ho WY, Yen YC, et al. The vulnerability of motor and frontal cortex-dependent behaviors in mice expressing ALS-linked mutation in TDP-43. Neurobiol Aging. 2020;92:43–60. [DOI] [PubMed] [Google Scholar]
- [78].Sieverding K, Ulmer J, Bruno C, et al. Hemizygous deletion of Tbk1 worsens neuromuscular junction pathology in TDP-43(G298S) transgenic mice. Exp Neurol. 2021;335:113496. [DOI] [PubMed] [Google Scholar]
- [79].Tsao W, Jeong YH, Lin S, et al. Rodent models of TDP-43: recent advances. Brain Res. 2012;1462:26–39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [80].Ayala YM, Zago P, Ambrogio AD, et al. Structural determinants of the cellular localization and shuttling of TDP-43. J Cell Sci. 2008;121:3778–3785. [DOI] [PubMed] [Google Scholar]
- [81].Neumann M, Sampathu DM, Kwong LK, et al. Ubiquitinated TDP-43 in frontotemporal lobar degeneration and amyotrophic lateral sclerosis. Science. 2006;314:130–133. [DOI] [PubMed] [Google Scholar]
- [82].Ling SC. Synaptic Paths to Neurodegeneration: the Emerging Role of TDP-43 and FUS in Synaptic Functions. Neural Plast. 2018;2018:8413496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [83].Lattante S, Rouleau GA, Kabashi E. TARDBP and FUS mutations associated with amyotrophic lateral sclerosis: summary and update. Hum Mutat. 2013;34:812–826. [DOI] [PubMed] [Google Scholar]
- [84].Armakola M, Higgins MJ, Figley MD, et al. Inhibition of RNA lariat debranching enzyme suppresses TDP-43 toxicity in ALS disease models. Nat Genet. 2012;44:1302–1309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [85].Elden AC, Kim HJ, Hart MP, et al. Ataxin-2 intermediate-length polyglutamine expansions are associated with increased risk for ALS. Nature. 2010;466:1069–1075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [86].Ash PE, Zhang YJ, Roberts CM, et al. Neurotoxic effects of TDP-43 overexpression in C. elegans. Hum Mol Genet. 2010;19:3206–3218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [87].Liachko NF, Guthrie CR, Kraemer BC. Phosphorylation promotes neurotoxicity in a Caenorhabditis elegans model of TDP-43 proteinopathy. J Neurosci. 2010;30:16208–16219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [88].Kabashi E, Lin L, Tradewell ML, et al. Gain and loss of function of ALS-related mutations of TARDBP (TDP-43) cause motor deficits in vivo. Hum Mol Genet. 2010;19:671–683. [DOI] [PubMed] [Google Scholar]
- [89].Laird AS, Hoecke AV, Muynck LD, et al. Progranulin is neurotrophic in vivo and protects against a mutant TDP-43 induced axonopathy. PLoS One. 2010;5:e13368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [90].Picher-Martel V, Valdmanis PN, Gould PV, et al. From animal models to human disease: a genetic approach for personalized medicine in ALS. Acta Neuropathol Commun. 2016;4:70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [91].Gordon D, Dafinca R, Scaber J, et al. Single-copy expression of an amyotrophic lateral sclerosis-linked TDP-43 mutation (M337V) in BAC transgenic mice leads to altered stress granule dynamics and progressive motor dysfunction. Neurobiol Dis. 2019;121:148–162. [DOI] [PubMed] [Google Scholar]
- [92].Kraemer BC, Schuck T, Wheeler JM, et al. Loss of murine TDP-43 disrupts motor function and plays an essential role in embryogenesis. Acta Neuropathol. 2010;119:409–419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [93].Sephton CF, Good SK, Atkin S, et al. TDP-43 is a developmentally regulated protein essential for early embryonic development. J Biol Chem. 2010;285:6826–6834. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [94].Wu LS, Cheng WC, Hou SC, et al. TDP-43, a neuro-pathosignature factor, is essential for early mouse embryogenesis. Genesis. 2010;48:56–62. [DOI] [PubMed] [Google Scholar]
- [95].Swarup V, Phaneuf D, Bareil C, et al. Pathological hallmarks of amyotrophic lateral sclerosis/frontotemporal lobar degeneration in transgenic mice produced with TDP-43 genomic fragments. Brain. 2011;134:2610–2626. [DOI] [PubMed] [Google Scholar]
- [96].Arnold ES, Ling SC, Huelga SC, et al. ALS-linked TDP-43 mutations produce aberrant RNA splicing and adult-onset motor neuron disease without aggregation or loss of nuclear TDP-43. Proc Natl Acad Sci U S A. 2013;110:E736–745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [97].Xu YF, Gendron TF, Zhang YJ, et al. Wild-type human TDP-43 expression causes TDP-43 phosphorylation, mitochondrial aggregation, motor deficits, and early mortality in transgenic mice. J Neurosci. 2010;30:10851–10859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [98].Liu YC, Chiang PM, Tsai KJ. Disease animal models of TDP-43 proteinopathy and their pre-clinical applications. Int J Mol Sci. 2013;14:20079–20111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [99].Wegorzewska I, Bell S, Cairns NJ, et al. TDP-43 mutant transgenic mice develop features of ALS and frontotemporal lobar degeneration. Proc Natl Acad Sci U S A. 2009;106:18809–18814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [100].Mitchell JC, Constable R, So E, et al. Wild type human TDP-43 potentiates ALS-linked mutant TDP-43 driven progressive motor and cortical neuron degeneration with pathological features of ALS. Acta Neuropathol Commun. 2015;3(1):36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [101].Esmaeili MA, Panahi M, Yadav S, et al. Premature death of TDP-43 (A315T) transgenic mice due to gastrointestinal complications prior to development of full neurological symptoms of amyotrophic lateral sclerosis. Int J Exp Pathol. 2013;94(1):56–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [102].Hatzipetros T, Bogdanik LP, Tassinari VR, et al. C57BL/6J congenic Prp-TDP43A315T mice develop progressive neurodegeneration in the myenteric plexus of the colon without exhibiting key features of ALS. Brain Res. 2014;1584:59–72. [DOI] [PubMed] [Google Scholar]
- [103].White MA, Lin Z, Kim E, et al. Sarm1 deletion suppresses TDP-43-linked motor neuron degeneration and cortical spine loss. Acta Neuropathol Commun. 2019;7:166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [104].Ebstein SY, Yagudayeva I, Shneider NA. Mutant TDP-43 causes early-stage dose-dependent motor neuron degeneration in a TARDBP knockin mouse model of ALS. Cell Rep. 2019;26:364–373 e364. [DOI] [PubMed] [Google Scholar]
- [105].Walker AK, Spiller KJ, Ge G, et al. Functional recovery in new mouse models of ALS/FTLD after clearance of pathological cytoplasmic TDP-43. Acta Neuropathol. 2015;130:643–660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [106].Igaz LM, Kwong L, Lee EB, et al. Dysregulation of the ALS-associated gene TDP-43 leads to neuronal death and degeneration in mice. J Clin Invest. 2011;121:726–738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [107].Alfieri JA, Pino NS, Igaz LM. Reversible behavioral phenotypes in a conditional mouse model of TDP-43 proteinopathies. J Neurosci. 2014;34:15244–15259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [108].Chai N, Gitler AD. Yeast screen for modifiers of C9orf72 poly(glycine-arginine) dipeptide repeat toxicity. FEMS Yeast Res. 2018;18: DOI: 10.1093/femsyr/foy024 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [109].Furuta N, Tsukagoshi S, Hirayanagi K, et al. Suppression of the yeast elongation factor Spt4 ortholog reduces expanded SCA36 GGCCUG repeat aggregation and cytotoxicity. Brain Res. 2019;1711:29–40. [DOI] [PubMed] [Google Scholar]
- [110].Gois AM, Mendonca DMF, Freire MAM, et al. In vitro and in vivo models of amyotrophic lateral sclerosis: an updated overview. Brain Res Bull. 2020;159:32–43. [DOI] [PubMed] [Google Scholar]
- [111].Corrionero A, Horvitz HR, C9orf72 A. ALS/FTD ortholog acts in endolysosomal degradation and lysosomal homeostasis. Curr Biol. 2018;28:1522–1535 e1525. [DOI] [PubMed] [Google Scholar]
- [112].Ji YJ, Ugolino J, Zhang T, et al. C9orf72/ALFA-1 controls TFEB/HLH-30-dependent metabolism through dynamic regulation of Rag GTPases. PLoS Genet. 2020;16:e1008738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [113].Yuva-Aydemir Y, Almeida S, Gao FB. Insights into C9ORF72-Related ALS/FTD from Drosophila and iPSC Models. Trends Neurosci. 2018;41:457–469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [114].Olesnicky EC, Wright EG. Drosophila as a model for assessing the function of RNA-binding proteins during neurogenesis and neurological disease. J Dev Biol. 2018;6: DOI: 10.3390/jdb6030021 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [115].Tran H, Almeida S, Moore J, et al. Differential toxicity of nuclear RNA Foci versus dipeptide repeat proteins in a drosophila model of C9ORF72 FTD/ALS. Neuron. 2015;87:1207–1214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [116].Fortier G, Butti Z, Patten SA. Modelling C9orf72-related amyotrophic lateral sclerosis in zebrafish. Biomedicines. 2020;8: DOI: 10.3390/biomedicines8100440 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [117].Yeh TH, Liu HF, Li YW, et al. C9orf72 is essential for neurodevelopment and motility mediated by Cyclin G1. Exp Neurol. 2018;304:114–124. [DOI] [PubMed] [Google Scholar]
- [118].Butti Z, Patten K. GJ. Reduced C9orf72 function leads to defective synaptic vesicle release and neuromuscular dysfunction in zebrafish. Research Square. 2020. DOI: 10.21203/rs.3.rs-49118/v1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [119].Giacomotto J, Rinkwitz S, Becker TS. Effective heritable gene knockdown in zebrafish using synthetic microRNAs. Nat Commun. 2015;6:7378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [120].Ho WY, Navakkode S, Liu F, et al. Deregulated expression of a longevity gene, Klotho, in the C9orf72 deletion mice with impaired synaptic plasticity and adult hippocampal neurogenesis. Acta Neuropathol Commun. 2020;8:155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [121].May S, Hornburg D, Schludi MH, et al. C9orf72 FTLD/ALS-associated Gly-Ala dipeptide repeat proteins cause neuronal toxicity and Unc119 sequestration. Acta Neuropathol. 2014;128:485–503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [122].Morrice JR, Gregory-Evans CY, Shaw CA. Animal models of amyotrophic lateral sclerosis: A comparison of model validity. Neural Regen Res. 2018;13:2050–2054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [123].Moens TG, Partridge L, Isaacs AM. Genetic models of C9orf72: what is toxic? Curr Opin Genet Dev. 2017;44:92–101. [DOI] [PubMed] [Google Scholar]
- [124].Batra R, Lee CW. Mouse models of C9orf72 hexanucleotide repeat expansion in amyotrophic lateral sclerosis/frontotemporal dementia. Front Cell Neurosci. 2017;11:196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [125].Sareen D, O'Rourke JG, Meera P, et al. Targeting RNA foci in iPSC-derived motor neurons from ALS patients with a C9ORF72 repeat expansion. Sci Transl Med. 2013;5:208ra149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [126].Zhang K, Donnelly CJ, Haeusler AR, et al. The C9orf72 repeat expansion disrupts nucleocytoplasmic transport. Nature. 2015;525:56–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [127].Devlin AC, Burr K, Borooah S, et al. Human iPSC-derived motoneurons harbouring TARDBP or C9ORF72 ALS mutations are dysfunctional despite maintaining viability. Nat Commun. 2015;6:5999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [128].Almeida S, Krishnan G, Rushe M, et al. Production of poly(GA) in C9ORF72 patient motor neurons derived from induced pluripotent stem cells. Acta Neuropathol. 2019;138:1099–1101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [129].Bursch F, Kalmbach N, Naujock M, et al. Altered calcium dynamics and glutamate receptor properties in iPSC-derived motor neurons from ALS patients with C9orf72, FUS, SOD1 or TDP43 mutations. Hum Mol Genet. 2019;28:2835–2850. [DOI] [PubMed] [Google Scholar]
- [130].Dafinca R, Barbagallo P, Farrimond L, et al. Impairment of mitochondrial calcium buffering links mutations in C9ORF72 and TARDBP in iPS-derived motor neurons from patients with ALS/FTD. Stem Cell Reports. 2020;14:892–908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [131].Tang X, Toro A, T G S, et al. Divergence, Convergence, and Therapeutic Implications: A Cell Biology Perspective of C9ORF72-ALS/FTD. Mol Neurodegener. 2020;15:34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [132].Ugolino J, Ji YJ, Conchina K, et al. Loss of C9orf72 enhances autophagic activity via deregulated mTOR and TFEB signaling. PLoS Genet. 2016;12:e1006443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [133].Atanasio A, Decman V, White D, et al. C9orf72 ablation causes immune dysregulation characterized by leukocyte expansion, autoantibody production, and glomerulonephropathy in mice. Sci Rep. 2016;6:23204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [134].Burberry A, Suzuki N, Wang JY, et al. Loss-of-function mutations in the C9ORF72 mouse ortholog cause fatal autoimmune disease. Sci Transl Med. 2016;8:347ra393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [135].Belzil VV, Katzman RB, Petrucelli L. ALS and FTD: an epigenetic perspective. Acta Neuropathol. 2016;132:487–502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [136].Barres BA, Barde Y. Neuronal and glial cell biology. Curr Opin Neurobiol. 2000;10:642–648. [DOI] [PubMed] [Google Scholar]
- [137].Jensen BK, Schuldi MH, McAvoy K, et al. Synaptic dysfunction induced by glycine-alanine dipeptides in C9orf72-ALS/FTD is rescued by SV2 replenishment. EMBO Mol Med. 2020;12:e10722. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [138].Atkinson RA, Fernandez-Martos CM, Atkin JD, et al. C9ORF72 expression and cellular localization over mouse development. Acta Neuropathol Commun. 2015;3:59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [139].Frick P, Sellier C, Mackenzie IR, et al. Novel antibodies reveal presynaptic localization of C9orf72 protein and reduced protein levels in C9orf72 mutation carriers. Acta Neuropathol Commun. 2018;6:72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [140].O’Rourke JG, Bogdanik L, Yanez A, et al. C9orf72 is required for proper macrophage and microglial function in mice. Science. 2016;351:1324–1329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [141].Burberry A, Wells MF, Limone F, et al. C9orf72 suppresses systemic and neural inflammation induced by gut bacteria. Nature. 2020;582:89–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [142].McCauley ME, O'Rourke JG, Yanez A, et al. C9orf72 in myeloid cells suppresses STING-induced inflammation. Nature. 2020;585:96–101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [143].Lan Y, Sullivan PM, Hu F. SMCR8 negatively regulates AKT and MTORC1 signaling to modulate lysosome biogenesis and tissue homeostasis. Autophagy. 2019;15:871–885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [144].Behrends C, Sowa ME, Gygi SP, et al. Network organization of the human autophagy system. Nature. 2010;466:68–76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [145].Wong PM, Puente C, Ganley IG, et al. ULK1 complex: sensing nutrient signals for autophagy activation. Autophagy. 2013;9:124–137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [146].Lin MG, Hurley JH. Structure and function of the ULK1 complex in autophagy. Curr Opin Cell Biol. 2016;39:61–68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [147].Ho WY, Tai YK, Chang JC, et al. The ALS-FTD-linked gene product, C9orf72, regulates neuronal morphogenesis via autophagy. Autophagy. 2019;15:827–842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [148].Amick J, Tharkeshwar AK, Amaya C, et al. WDR41 supports lysosomal response to changes in amino acid availability. Mol Biol Cell. 2018;29:2213–2227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [149].Amick J, Tharkeshwar AK, Talaia G, et al. PQLC2 recruits the C9orf72 complex to lysosomes in response to cationic amino acid starvation. J Cell Biol. 2020;219: DOI: 10.1083/jcb.201906076 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [150].Liang C, Shao Q, Zhang W, wet al. Smcr8 deficiency disrupts axonal transport-dependent lysosomal function and promotes axonal swellings and gain of toxicity in C9ALS/FTD mouse models. Hum Mol Genet. 2019;28:3940–3953. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [151].Amick J, Ferguson SM. C9orf72: at the intersection of lysosome cell biology and neurodegenerative disease. Traffic. 2017;18:267–276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [152].Lawrence RE, Fromm SA, Fu Y, et al. Structural mechanism of a Rag GTPase activation checkpoint by the lysosomal folliculin complex. Science. 2019;366:971–977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [153].Vigil D, Cherfils J, Rossman KL, et al. Ras superfamily GEFs and GAPs: validated and tractable targets for cancer therapy? Nat Rev Cancer. 2010;10:842–857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [154].Shaw RJ. mTOR signaling: RAG GTPases transmit the amino acid signal. Trends Biochem Sci. 2008;33:565–568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [155].Lamber EP, Siedenburg AC, Barr FA. Rab regulation by GEFs and GAPs during membrane traffic. Curr Opin Cell Biol. 2019;59:34–39. [DOI] [PubMed] [Google Scholar]
- [156].Homma Y, Hiragi S, Fukuda M. Rab family of small GTPases: an updated view on their regulation and functions. Febs J. 2020. DOI: 10.1111/febs.15453 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [157].Muller MP, Goody RS. Molecular control of Rab activity by GEFs, GAPs and GDI. Small GTPases. 2018;9:5–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [158].Kiral FR, Kohrs FE, Jin EJ, et al. Rab GTPases and membrane trafficking in neurodegeneration. Curr Biol. 2018;28:R471–R486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [159].Tsun ZY, Bar-Peled L, Chantranupong L, et al. The folliculin tumor suppressor is a GAP for the RagC/D GTPases that signal amino acid levels to mTORC1. Mol Cell. 2013;52:495–505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [160].Shen K, Huang RK, Brignole EJ, et al. Architecture of the human GATOR1 and GATOR1-Rag GTPases complexes. Nature. 2018;556:64–69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [161].Iyer S, Subramanian V, Acharya KR. C9orf72, a protein associated with amyotrophic lateral sclerosis (ALS) is a guanine nucleotide exchange factor. PeerJ. 2018;6:e5815. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [162].Sivadasan R, Hornburg D, Drepper C, et al. C9ORF72 interaction with cofilin modulates actin dynamics in motor neurons. Nat Neurosci. 2016;19:1610–1618. [DOI] [PubMed] [Google Scholar]
- [163].Chaineau M, Ioannou MS, McPherson PS. Rab35: gEFs, GAPs and effectors. Traffic. 2013;14:1109–1117. [DOI] [PubMed] [Google Scholar]
- [164].Kiontke S, Langemeyer L, Kuhlee A, et al. Architecture and mechanism of the late endosomal Rab7-like Ypt7 guanine nucleotide exchange factor complex Mon1-Ccz1. Nat Commun. 2017;8:14034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [165].Hutagalung AH, Novick PJ. Role of Rab GTPases in membrane traffic and cell physiology. Physiol Rev. 2011;91:119–149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [166].Petit CS, Roczniak-Ferguson A, Ferguson SM. Recruitment of folliculin to lysosomes supports the amino acid-dependent activation of Rag GTPases. J Cell Biol. 2013;202:1107–1122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [167].Nookala RK, Langemeyer L, Pacitto A, et al. Crystal structure of folliculin reveals a hidDENN function in genetically inherited renal cancer. Open Biol. 2012;2:120071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [168].De Franceschi N, Wild K, Schlacht A, et al. Longin and GAF domains: structural evolution and adaptation to the subcellular trafficking machinery. Traffic. 2014;15:104–121. [DOI] [PubMed] [Google Scholar]
- [169].Talaia G, Amick J, Ferguson SM. Receptor-like role for PQLC2 amino acid transporter in the lysosomal sensing of cationic amino acids. bioRxiv. 2020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [170].Aoe T, Cukierman E, Lee A, et al. The KDEL receptor, ERD2, regulates intracellular traffic by recruiting a GTPase-activating protein for ARF1. Embo J. 1997;16:7305–7316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [171].Garcia-Mata R, Szul T, Alvarez C, et al. ADP-ribosylation factor/COPI-dependent events at the endoplasmic reticulum-Golgi interface are regulated by the guanine nucleotide exchange factor GBF1. Mol Biol Cell. 2003;14:2250–2261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [172].Ge L, Melville D, Zhang M, et al. ER-Golgi intermediate compartment is a key membrane source for the LC3 lipidation step of autophagosome biogenesis. Elife. 2013;2:e00947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [173].Pang W, Hu F. Cellular and physiological functions of C9ORF72 and implications for ALS/FTD. J Neurochem. 2020. DOI: 10.1111/jnc.15255 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [174].Wang M, et al. Wang H, Tao Z, C9orf72 associates with inactive Rag GTPases and regulates mTORC1-mediated autophagosomal and lysosomal biogenesis. Aging Cell. 2020;19:e13126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [175].Schumann T, Konig J, Henke C, et al. Solute carrier transporters as potential targets for the treatment of metabolic disease. Pharmacol Rev. 2020;72:343–379. [DOI] [PubMed] [Google Scholar]
- [176].Hadley B, Litfin T, Day CJ, et al. Nucleotide sugar transporter SLC35 family structure and function. Comput Struct Biotechnol J. 2019;17:1123–1134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [177].Zhang Y, Zhang Y, Sun K, et al. SLC transporter in nutrient and metabolic sensing, regulation, and drug development. J Mol Cell Biol. 2019;11:1–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [178].Shinde SR, Maddika S. Post translational modifications of Rab GTPases. Small GTPases. 2018;9:49–56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [179].Salaun C, Greaves J, Chamberlain LH. The intracellular dynamic of protein palmitoylation. J Cell Biol. 2010;191:1229–1238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [180].Kong E, Peng S, Chandra G, et al. Dynamic palmitoylation links cytosol-membrane shuttling of acyl-protein thioesterase-1 and acyl-protein thioesterase-2 with that of proto-oncogene H-ras product and growth-associated protein-43. J Biol Chem. 2013;288:9112–9125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [181].Meinnel T, Dian C, Giglione C. Myristoylation, an ancient protein modification mirroring eukaryogenesis and evolution. Trends Biochem Sci. 2020;45:619–632. [DOI] [PubMed] [Google Scholar]
- [182].Schrodinger LLC The PyMOL molecular graphics system, version 1.8 (2015).
- [183].Chi X, Jin X, Chen Y, et al. Structural insights into the gating mechanism of human SLC26A9 mediated by its C-terminal sequence. Cell Discov. 2020;6:55. [DOI] [PMC free article] [PubMed] [Google Scholar]