

Abstract
Artificial metalloenzymes (ArMs), created by introducing synthetic cofactors into protein scaffolds, are an emerging class of biocatalyst for non-natural reactions. In vitro reconstitution of cofactors and proteins has been a limiting step in the directed evolution of ArMs because purification of individual host proteins is time-consuming. We describe the application of a platform to combine the P450 enzyme and the cofactor Ir(Me)MPIX in vivo, with coexpression P450 and the heme transporter encoded by the hug operon. We applied this platform to the development an ArMs catalyzing the insertion of the acyclic carbene from α-diazopropanoate esters (Me-EDA) into the N-H bonds of N-alkyl anilines. The mutants of the ArMs Ir(Me)CYP119 identified by an evolution campaign involving more than 4000 mutants are shown to catalyze the reaction of Me-EDA with N-methyl anilines to form chiral amino esters with high TON and good enantioselectivity, there by demonstrating that the directed evolution of ArMs can rival that of natural enzymes in vivo.
Keywords: Artificial metalloenzymes, Directed evolution, Whole cell, N-H insertion, P450
Graphical Abstract
Whole-cell, E. coli platform enables directed evolution campaigns with ArMs containing an abiotic Ir-porphyrin cofactor involving the generation of more than 4000 mutants to develop an enantioselective insertion of a simple acyclic carbene from a diazo ester into the N-H bond of N-methyl aniline that has evaded prior enzyme systems and formed racemic product with mutants of natural P450s

Introduction
Enzymes are well-established catalysts for the selective and environmentally benign synthesis of fine chemicals and complex drugs.[1] Although valuable, natural enzymes suffer from a limited scope of substrates and a limited set of transformations. Therefore, directed evolution and protein engineering has sought to expand the scope of unnatural substrates that react with natural enzymes and to achieve unnatural reactions for the construction of bioactive molecules. Examples range from the synthesis of sitagliptin catalyzed by an evolved transaminase[2] to the transfer of carbenes catalyzed by repurposed hemoproteins.[3]
The creation of artificial metalloenzymes (ArMs) possessing synthetic cofactors is a promising alternative strategy to achieve new reactivity with protein scaffolds. Strategies to create ArMs include the use of supramolecular assembly,[4] anchoring of a cofactor to unnatural amino acids,[5] and swapping native metals with noble metals.[6] Like that of native enzymes for unnatural reactions, high reactivity and selectivity for reactions catalyzed by ArMs require modifications to the protein scaffold. The identification of appropriate modifications by directed evolution is often a slower process than with native classes of enzymes, due to the additional step needed to introduce the abiotic cofactor and, in some cases, to remove the excess free cofactor.[7]
Although screening for catalytic activity and selectivity in cell lysates can be faster than screening purified proteins, the construction of ArMs in cell lysates remains challenging, due to non-specific binding and inhibition caused by cytosolic compounds, such as glutathione (Figure 1, a).[8] One could imagine that whole-cell platforms for the recombination of cofactors and proteins could be an alternative approach to accelerate the high-throughput screening of ArMs by eliminating the need for in vitro reconstitution steps. One way to overcome these hurdles is to transport the protein to the outer membrane or the periplasm of E. coli by tagging the proteins for transport from the cytoplasm and making the protein accessible to the cofactor and assembly in vivo (Figure 1, b).[9] In addition to these approaches, transferring the cofactor into the cytoplasm is the alternative strategy to realize the construction and reactions of ArMs in whole cells (Figure 1, c).[10] Recently, coexpression of transporters, such as ChuA and the Hug system, has been used to incorporate cofactors into heme proteins, resulting in a whole-cell catalyst for abiotic reactions and unnatural biosynthesis.[11] Chiral amines, one of the most important motifs in biomolecules and drugs, have been targets for catalytic processes for many years. Biocatalysis has become an approach for the enantioselective synthesis of chiral amines by transamination or reduction of imines.[12] In addition to these natural processes, repurposed hemoproteins and ArMs have generated chiral amines by nitrene insertion into C-H bond[13] and carbene insertions into N-H bonds.[14] Although high enantioselectivity has been achieved from carbene insertions into Si-H and B-H bonds catalyzed by engineered heme proteins, related enzymatic, enantioselective insertions of carbenes into N-H bonds is limited to specific, cyclic diazo compounds or primary arylamines.[15] This limitation results from the multi-step pathway that generates a ylide by the attack of the amine and the need for this ylide to undergo proton transfer when bound to the metal or after dissociation from the metal, but before dissociation from the enzyme binding site, if the process is to be enantioselective, design or evolution of an enzyme that catalyzes such enantioselective processes, particularly for alkyl-substituted amines, has been challenging.
Figure 1.

Approaches to assemble ArMs during a directed evolution campaign. (a) Screening in cell lysates with an additional step to remove nonspecific conjugation; (b) screening in vivo by cell surface display, and (c) in vivo assembly of Ir-CYP119 with a system to transport the cofactor into the cytoplasm.
We report the application of a whole-cell platform to the directed evolution of ArMs for the catalytic synthesis of chiral amines by enantioselective insertion of an acylic carbene into the N-H bond of N-alkyl arylamines. The whole-cell system, comprising an iridium-containing heme analog, a heme transporter system and coexpressed mutants of the host protein CYP119, was used directly as the catalyst to screen thousands of colonies without cell lysis or protein purification to identify a mutant that catalyzes the synthesis of N-methyl amino esters with good enantioselectivity and high turnover numbers (TON) by the reaction of carbene and amine classes that couple with low enantioselectivity when catalyzed by mutants of natural hemoproteins. The enzyme reacts with much higher turnover numbers than chemocatalytic systems to form the tertiary amine ester with comparable enantioselectivity.[16]
Results and Discussion
Enantioselective carbene insertion into the N-H bond of N-methyl anilines catalyzed by ArMs containing a synthetic Ir-porphyrin cofactor.
Previously, we reported insertions of nitrenes into C-H bonds to form chiral amines catalyzed by ArMs generated by reconstitution of mutants of CYP119, a P450 from the thermophile Sulfolobus solfataricus with the artificial cofactor Ir(Me)MPIX, containing the iridium-methyl fragment in mesoporphyrin IX.[17] With this class of enzyme (Ir-CYP119) we sought to assess the potential of forming chiral amines by carbene insertions into the N-H bonds of amines catalyzed by Ir-CYP119. Tetraarylporphrin complexes of the Ir-Me unit are known to insert carbenes into the N-H bonds of arylamines,[18] and the reaction of aniline with ethyl 2-diazopropanoate (Me-EDA) catalyzed by the free Ir(Me)MPIX cofactor formed the chiral amine in good yield and high TON. Encouraged by the high reactivity of the cofactor alone, we sought to use the combination of this cofactor and mutants of the host protein CYP119 to form the chiral amine enantioselectively, while maintaining the high reactivity of the Ir(Me)MPIX unit.
Following previously reported protocols,[19] a preliminary library containing 12 variants of Ir-CYP119 was generated and tested with a series of aniline derivatives. While the insertion process catalyzed by all 12 variants occurred in high yields with aniline, all of the reactions gave racemic product (Table 1, entry 1). However, the reaction of N-Me aniline catalyzed by the mutant CNH-0 (155F, 213G, 254L, 317G) formed the chiral amine with a measurable 40:60 er and acceptable yield (Table 1 entry 2). Aniline derivatives containing electron-withdrawing groups on the nitrogen gave little product (Table 1, entries 3–4). Because of this high reactivity and measurable selectivity for the reaction of N-methyl aniline and the lack of enantioselectivity from prior work on the insertions of carbenes into the N-H bonds of N-alkyl anilines, we sought to develop reactions with this class of arylamine.
Table 1.
Initial studies of carbene insertion into the N-H bond of N-Me aniline catalyzed by (Me)IrCYP119.a
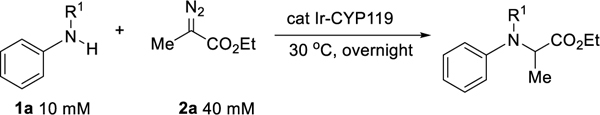
| |||||
|---|---|---|---|---|---|
|
| |||||
| entry | R1 | Catalyst | erb | Yieldb | TONc |
|
| |||||
| 1 | H | Purified ArM | 50:50 | 92% | 1840 |
| 2 | Me | Purified ArM | 40:60 | 56% | 1120 |
| 3 | Ac | Purified ArM | ND | <5% | -- |
| 4 | Ts | Purified ArM | ND | <5% | -- |
| 5 | Me | Cell lysis ArM | 50:50 | 54% | 1080 |
| 6d | Me | whole cell | 39:61 | 59% | 5867 |
| 7e | Me | whole cell | 45:55 | 23% | 2257 |
| 8f | Me | whole cell | ND | <5% | -- |
| 9g | Me | whole cell | ND | <5% | -- |
Reaction conditions: 2.5 μmol aniline derivatives, 10 μmol Me-EDA, 0.05 mol% Ir(Me)-MPIX with the CNH-0 mutant (155F, 213G, 254L, and 317G), 0.25 mL M9-N buffer, 30 °C, overnight.
The er and yield were determined by chiral HPLC with N,N-diethylaniline as the internal standard.
The TON was calculated by the number of substrate molecules converted to the product per iridium cofactor added to the cell culture.
The reactions were run in whole cells at an OD ~ 25 coexpressing the hug operon and CYP119 and 0.01 mol % Ir(Me)-MPIX.were
The reactions run in whole cells with only CYP119 expression.
The reactions run in whole cells with coexpressing the hug operon and a blank 2BT vector.
The reactions were run in whole cells coexpressing the hug operon and CYP119 but without the addition of Ir(Me)MPIX.
To increase the enantioselectivity of the carbene insertions into the N-H bond, we conducted directed evolution of the Ir-CYP119 system. While directed evolution of ArMs has been reported previously,[7] we sought to use a new approach to this process by conducting the evolution with our ArMs in whole E. coli cells. Our first work on the evolution of Ir-CYP119 enzymes was conducted with purified proteins. We later showed that screening of cell lysates was possible and would eliminate the time-consuming purification of CYP119,[20] but our initial studies on the insertion of the carbene from Me-EDA into the N-H bond of anilines catalyzed by cell lysates treated with the cofactor Ir(Me)MPIX occurred with almost no enantioselectivity, even for the reaction that occurred with measurable enantioselectivity when catalyzed by the purified protein (Table 1, entry 2 vs Table 1, entry 5). We presumed that this low selectivity resulted from the rapid background reactions catalyzed by unbound Ir(Me)MPIX. To minimize the formation of racemic products formed from the free cofactor, we sought to conduct this directed evolution with the holoenzyme assembled in vivo.
Recently, we demonstrated the efficient transport of an iridium-porphyrin complex into E. coli by a heterologous transport system to produce ArMs that form unnatural products by the combination of the reaction of the ArMs with natural enzymes in intact E. coli.[11] The heme transport system encoded by the hug operon from Plesiomonas shigelloides[21] led to the efficient assembly of the ArMs. Thus, to accelerate the directed evolution of ArMs for the insertion of carbenes into N-H bonds, we sought to generate mutants of ArMs containing the Ir(Me)MPIX cofactor in E. coli expressing this transport system. Indeed, insertion of the carbene from Me-EDA into the N-H bond of N-methyl aniline occurred in the presence of whole cells containing the CYP119 mutant CNH-0 and the hug operon to form the chiral amino ester product with similar selectivity and higher TON than the reactions catalyzed by the same mutant generated from purified protein (Table 1, entry 6)
Several control experiments were consistent with the reaction by the ArMs assembled in vivo by active transport of the cofactor. First, the reaction of N-Me aniline and Me-EDA catalyzed by the cell lacking the transporter occurred with lower selectivity and reactivity than that catalyzed by the cells containing the transporter (Table 1, entry 7). These data support the assertion that the products of the pHug operon facilitate the transport of the cofactor to enable assembly of the ArMs in vivo. Second, less than 5% of the product was formed from the reactions catalyzed by the cells lacking the gene for CYP119, indicating that this host protein is essential for reactivity. Finally, less than 5% of the product was formed from a reaction occurring without addition of the Me-Ir-MPIX cofactor, ruling out formation of the product by the combination of the CYP119 and a natural cellular component or component of the growth medium (Table 1, entries 8–9). Considering that the free cofactor catalyzes the reaction in pure buffer, we propose that the lack of formation of product by the cofactor without CYP119 results from poisoning of the cofactor by cellular components when it is unprotected by the CYP119 protein.
Directed evolution in whole cells of Ir-CYP119 for asymmetric N-H insertion into N-Me aniline.
Having demonstrated that the whole cells containing the transporter, host protein, and artificial cofactor catalyzed the reaction of Me-EDA with N-Me aniline with measurable enantioselectivity, the activity and selectivity were increased by directed evolution of the Ir-CYP119 in whole cells. The initial library was generated by combinatorial site-saturation mutagenesis of ten positions (69, 87, 208, 210, 152, 155, 254, 256, 282, 353) around the active site that significantly affected selectivity during our previous studies of carbene transfer reactions.[19] From several thousand colonies, 96 colonies were randomly chosen for sequencing. Between one and three of the ten positions were mutated in each colony, and mutations at all ten positions appeared among the 96 colonies. After screening of 900 colonies, 12 mutants were identified that reacted with comparable or higher selectivity than the parent mutant. After screening of 900 colonies, 12 mutants were identified that reacted with comparable or higher selectivity than the parent mutant. Comparing with the parent mutant, one or more of the six positions (152, 155, 208, 254, 256, 353) have been modified in these 12 mutants, which indicated these positions are hot spots from ten initial positions. Among these 12 mutants, CNH-1 (152L, 213G, 254A, and 317G) catalyzed insertion of the carbene unit into the N-H bond with the highest enantioselectivity (Scheme 1, run 1).
Scheme 1.

Directed evolution of carbene insertion into the N-H bond by screening of whole-cellsa,b
a) Reaction conditions: BL-21(DE) coexpressing the CYP119 mutants and hug operon, OD600 ~25, 0.0025 %mol Ir(Me)-MPIX, N-Me aniline (2.5 μmol), EDA (10 μmol), DMSO (10 μL), M9-N (250 μL), 30 °C, overnight. b) The ee was determined by chiral HPLC and was the average of three reactions; The TON was calculated by the number of substrate molecules converted to the product per iridium cofactor added to the cell culture and was the average of three reactions; ee was used instead of er in this scheme as the measure of enantioselectivity to show more clearly the experimental error in the selectivities.
Encouraged by this ability to screen the activity and selectivity of the ArMs in whole cells, we conducted a second round of evolution focused on the 152/153/155 positions, which are located in the F-G loop and are considered the entrance of the substrate channel.[22] Slightly higher enantioselectivity (36.1% ee) was observed for the reaction catalyzed by the new mutant CNH-2A (152S, 153N, 213G, 254A, and 317G) identified after conducting whole-cell reactions of 1080 colonies from the library generated by combinatorial site-saturation mutagenesis of three positions (Scheme 1, run 2A). These colonies were tested in less than two weeks, demonstrating that this whole-cell platform is a much faster system for conducting directed evolution than one based on purified proteins.
Following the approach of Focused Rational Iterative Site-specific Mutagenesis (FRISM),[22] a third round of evolution was conducted. A library was generated by combinatorial site-specific mutagenesis at the positions (208, 254, 256, and 353) that were found to positively affect the enantioselectivity during the first round of directed evolution but were not covered in run 2A. Instead of generating a saturation mutagenesis library at these four sites, we generated a library containing three amino acids of distinct size (A, L, F) at these four sites that were applied at this round of evolution. Screening 160 colonies of the library covering all four of the above positions showed that the mutant CNH-3A (152S, 153N, 208F, 213G, 254A, 256F, and 317G) catalyzes the insertion reaction with a further increased 39.3% ee (Scheme 1, run 3A). Based on this increase resulting from the installation of large amino acid residues at the 208 and 256 positions, mutants containing the other large amino acids (F/Y/W) at positions 208 and 256 were evaluated. However, none of these mutants reacted with higher enantioselectivity than that of the mutant CNH-3A (152N, 153S, 208F, 213G, 254A, 256F, and 317G). Revisiting the other positions (210, 254, and 353) by the FRISM strategy did not increase the enantioselectivity over that of the parent mutant. Thus, directed evolution along this path was terminated (Scheme 1, run 4A).
To create a new branch on the evolutionary tree, we conducted a round of evolution from a prior mutant CNH-1 (152L, 213G, 254A and 317G) with a library focused on positions 208, 210, 254, 256, and 353. Mutations at these positions increased enantioselectivity during the first round of directed evolution that was conducted with combinatorial site-specific mutagenesis. Evaluation of 540 colonies from this library led to a slightly higher ee (32.3% ee) when the reaction was catalyzed by the mutant CNH-2B (152L, 208F, 213G, 254A, 256F, and 317G) than with the starting mutant (Scheme 1, run 2B). Further exploration of mutants containing large amino acids at the 208 and 256 positions led to a more selective mutant CNH-3B (152L, 208F, 213G, 254A, 256Y, 317G) reacting with 39.6% ee (Scheme 1, run 3B). Finally, we found that an additional mutation of 254C (CNH-4B) further improved the ee to 42.5% (Scheme 1, run 4B) with over 20,000 TON in whole cells, based on the amount of iridium cofactor added to the cell culture (Table 2, entry1).
Table 2.
Evaluation of whole-cell reaction conditions for carbene insertion into the N-H bond of N-Me aniline.a,b
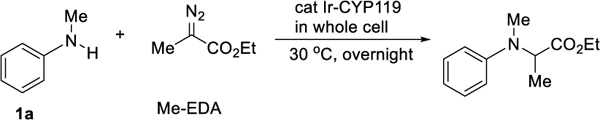
| |||||||
|---|---|---|---|---|---|---|---|
|
| |||||||
| entry | 1a (mM) | Me-EDA (mM) | reaction buffer | OD | Catalyst | erb | TONc |
|
| |||||||
| 1 | 10 | 40 | M9-N | 25 | Fresh cell | 71:29 | 21670 |
| 2 | 10 | 40 | M9-N | 25 | lyophilized cell | 75:25 | 20895 |
| 3 | 10 | 40 | M9-N | 50 | lyophilized cell | 78:22 | 17867 |
| 4 | 10 | 40 | NaPi | 50 | lyophilized cell | 81:19 | 14536 |
| 5 | 5 | 20 | NaPi | 50 | lyophilized cell | 87:13 | 3210 |
| 6d | 5 | 20 | NaPi | 50 | lyophilized cell | 91:9 | 8569 |
Reaction conditions: BL-21(DE) coexpressing the hug operon and the CYP119 mutant CNH-4B, 0.0025 mol % to 0.01 mol % Ir(Me)-MPIX based on the OD, 30 °C, 4 h.
The er was determined by chiral HPLC and was the average of three reactions.
The TON was calculated by the number of substrate molecules converted to the product per iridium cofactor added to the cell culture and was the average of three reactions.
The reaction was performed with the addition of Me-EDA in four batches over 4 h.
The obvious challenge confronting evolution to form mutants that catalyze this carbene insertion with substantial enantioselectivity and the similar selectivities from several lines of evolution suggest that the evolutionary landscape is rugged. We propose that this challenge arises from counteracting effects of amino acid residues during a multi-step process in which several steps and competing pathways can control enantioselectivity. The ability to achieve substantial enantioselectivity in such a system by generation and testing of more than 4000 colonies illustrates the value of generating and testing the ArMs in whole cells.
Having identified mutants that lead to substantial er, we sought to modify the reaction conditions to increase enantioselectivity further, and the next sections show that this mutant reacts under modified conditions with high enantioselectivity and turnover numbers.
Further increase in enantioselectivity and reactivity by varying reaction conitions
With a highly active and enantioselective mutant in hand, we investigated whether the enantioselectivity could be further improved by modifying the conditions under which the reaction is conducted. Considering the advantages of conducting reactions with lyophilized biocatalyst,[24] we conducted reactions in two different buffers with lyophilized cell pellets
The redissolved lyophilized cell pellets catalyzed the insertion reactions with higher er (75:25 vs 71:29) than those with fresh cells and with a similarly high TON (Table 2 entries 1–2) in M9-N buffer with the same OD. The increase in enantioselectivity from reactions catalyzed by lyophilized cells was attributed to increasing the permeability of the cells to the reagents, as well as reducing cellular components that might interfere with the reaction. The increased permeability to the reagents would increase the rate of the reaction catalyzed by the holoenzyme, thereby shortening the reaction time and reducing background reaction from free cofactor that could be released from the ArMs over the reaction time.
Changing the reaction buffer from the neutral M9-N (pH 7.4) to the slightly acidic NaPi buffer (pH 6) further increased the enantioselectivity, while maintaining high turnover numbers (Table 2, entries 4–5), most likely by precipitating the misfolded ArMs and reducing the background reactions. Finally, we found that the enantioselectivity further increased from 81:19 er to 87:13 er by reducing the concentration of substrates from 10 mM to 5 mM and the EDA from 40 mM to 20 mM. The higher enantioselectivity from the reactions with a lower concentration of Me-EDA suggests that the high concentration of Me-EDA destabilizes the ArMs, thereby generating the free cofactor that reacts to form the racemic product.
Although the er was higher for reactions with lower concentrations of Me-EDA, the turnover number was lower. Inspired by the increase in TON achieved by slow addition of the diazo compound during our previous studies on C-H insertion reactions catalyzed by Ir-CYP119,[20] we sought to increase the TON of the addition to the N-H bond by the addition of Me-EDA in batches to prevent decomposition of the diazo reagent and retain the concentration of Me-EDA that leads to high enantioselectivity. Consistent with this hypothesis, high reactivity (8569 TON) and high enantioselectivity (91: 9 er) were observed for reactions conducted by the addition of Me-EDA in four batches over 4 h. Given that the substituent of the ester in the diazo reagents has previously influenced enantioselectivity, [15a] we tested other diazo esters, but the highest er was achieved with the ethyl ester (Scheme S1).
After identifying mutants and reaction conditions that lead to high activity and enantioselectivity for this carbene transfer, we conducted the reactions of Me-EDA with a variety of N-Me aniline derivatives catalyzed by ArMs containing the Ir(Me)MPIX cofactor in E. coli. These reactions occurred with various aniline derivatives with good to moderate enantioselectivity under mild conditions. Similar to N-Me aniline (1a, Table 2, entry 6), para-methyl-substituted N-Me aniline (scheme3, 1b) reacted with Me-EDA to form the insertion product with good enantioselectivity (86:14 er) and high reactivity (5450 TON) with the same mutant (CHN-4B), and the p-methoxy-substituted N-Me aniline (1e) also formed the corresponding chiral amino ester with good selectivity (84:16 er) and high reactivity (2716 TON). Even the bulkier p-phenyl-substituted N-Me aniline (1i) also reacted to form the product of N-H insertion (3i) with good enantioselectivity (87:13 er) and high reactivity (2008 TON). However, the enantioselectivity and reactivity of o-methyl substituted N-Me aniline (1m) were lower than those with the m-methyl-substituted N-Me aniline (1j) and p-methyl-substituted N-Me aniline (1b), indicating the steric effects are more pronounced when the substituted group is closer to the reaction center. Moreover, 2-naphthylamine (1n), a substrate that reacted with low reactivity during previous studies of enzyme-catalyzed N-H insertion,[10] reacted readily in E. coli containing the ArMs with good TON (2400 TON) and substantial enantioselectivity (76:24 er). Slightly lower selectivity and reactivity were observed for the reaction of Me-EDA with more electron-deficient anilines. For example, the reaction of trifluoromethoxy-substituted 1f occurred with 69:31 er and 1428 TON, and reactions with m-fluoro and trifluoromethyl-substituted anilines (1k, 1l) reacted with lower enantioselectivity (1k). Nevertheless, the mild conditions enabled this system to react with good tolerance for functional groups, including a variety of halides (1c, 1d, 1g) and an ester (1h).
Conclusion
We have shown that a whole-cell E. coli platform enables directed evolution campaigns with ArMs containing an abiotic Ir-porphyrin cofactor. More than 4000 colonies were generated to develop an enantioselective insertion of an acylic carbene into the N-H bonds of N-methyl anilines, a scope of enantioselective reaction that has evaded prior enzymes generated by mutation of natural P450s. Our assembly of the mutants of ArMs for evolution was enabled by coexpression of the pHug transporter in commercial BL-21(DE) cells. Due to the mild conditions, this transformation accommodates a variety of functional groups. This method for assembling the mutants in whole cells during directed evolution enabled evaluation of much larger mutant libraries than had been possible by testing purified protein and led to products with enantioselectivities closer to those of purified proteins than to those from reactions conducted in cell lysates, which formed product from residual free cofactor. These results show that such whole-cell systems will be valuable for additional directed evolution campaigns aimed at new synthetic transformations.
Supplementary Material
Figure 2.

Structure of wild-type Fe-CYP119 (image prepared in Chimera from PDB 1IO7). The positions modified during the directed evolution are highlighted in green with red labels.
Scheme 2.

Scope of carbene insertion catalyzed by ArMs containing Me-Ir-MPIX in the whole cell.a,b,c
a) Reaction conditions: BL-21(DE) coexpressing the CYP119 CNH-4B and the hug operon, OD600 ~50, 0.01 %mol Ir(Me)-MPIX, N-Me aniline derivatives (5 μmol), EDA (20 μmol, addition in four batches over 4 h), DMSO (20 μL), NaPi (1.0 mL), 30 °C, overnight. b) The er was determined by chiral HPLC and was the average of three reactions. c) The TON was calculated by the number of substrate molecules converted to the product per iridium cofactor added to the cell culture and was the average of three reactions.
Acknowledgements
We thank the NIH Institutes of General Medical Sciences (GM-R35130387) for support of this work. Y. G. thanks a joint postdoc fellowship from Pharmaron and Shanghai Institute of Organic Chemistry (SIOC). Z. L thanks to the National Science Scholarship from the Singapore Agency for Science, Technology and Research.
Footnotes
Supporting information for this article is given via a link at the end of the document.
References
- [1].Devine PN, Howard RM, Kumar R, Thompson MP, Truppo MD, Turner NJ, Nat. Rev. Chem 2018, 2, 409–421 [Google Scholar]
- [2].Savile CK, Janey JM, Mundorff EC, Moore JC, Tam S, Jarvis WR, Colbeck JC, Krebber A, Fleitz FJ, Brands J, Devine PN, Huisman GW, Hughes GJ, Science 2010, 329, 305–309 [DOI] [PubMed] [Google Scholar]
- [3].Chen K, Arnold FH, Nat. Catal 2020, 3, 203–213. [Google Scholar]
- [4].a) Heinisch T, Ward TR, Acc. Chem. Res 2016, 49, 1711; [DOI] [PubMed] [Google Scholar]; b) Roelfes G, Acc. Chem. Res 2019, 52, 545–556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Lewis JC, Curr. Opin. Chem. Biol 2015, 25, 27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Natoli SN, Hartwig JF, Acc. Chem. Res 2019, 52, 326–335 [DOI] [PubMed] [Google Scholar]
- [7].Markel U, Sauer DF, Schiffels J, Okuda J, Schwaneberg U, Angew. Chem. Int. Ed 2019, 58, 4454–4464 [DOI] [PubMed] [Google Scholar]
- [8].Mallin H, Hestericová M, Reuter R, Ward TR, Nat. Protoc 2016, 11, 835–852. [DOI] [PubMed] [Google Scholar]
- [9].a) Heinisch T, Schwizer F, Garabedian B, Csibra E, Jeschek M, Vallapurackal J, Pinheiro VB, Marlière P, Panke S, Ward TR, Chem. Sci 2018, 9, 5383–5388; [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Jeschek M, Reuter R, Heinisch T, Trindler C, Klehr J, Panske S, T. R. Ward Nature 2016, 537, 661–665; [DOI] [PubMed] [Google Scholar]; c) Grimm AR, Sauer DF, Polen T, Zhu L, Hayashi T, Okuda J, Schwaneberg U, ACS Catal. 2018, 8, 2611–2614. [Google Scholar]
- [10].a) Winter MB, McLaurin EJ, Reece SY, Olea C, Nocera DG, Marletta MA, J. Am. Chem. Soc 2010, 132, 5582–5583; [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Lelyveld VS, Brustad E, Arnold FH, Jasanoff A, J. Am. Chem. Soc 2011, 133, 649–651; [DOI] [PMC free article] [PubMed] [Google Scholar]; c) Bordeaux M, Singh R, Fasan R, Bioorg. Med. Chem 2014, 22, 5697–5704; [DOI] [PMC free article] [PubMed] [Google Scholar]; d) Chordia S, Narasimhan S, Paioni AL, Baldus M, Roelfes G, Angew. Chem. Int. Ed 2021, 60, 5913–5920. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Huang J, Liu Z, Bloomer B, Clark D, Mukhopadhyay A, Keasling J, Hartwig J, Nat. Chem 2021, 10.1038/s41557-021-00801-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Ghislieri D, Turner NJ, Top. Catal 2014, 57, 284–300. [Google Scholar]
- [13].a) Hyster TK, Farwell CC, Buller AR, McIntosh JA, Arnold FH, J. Am. Chem. Soc 2014, 136, 15505–15508; [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Prier CK, Zhang RJK, Buller AR, Brinkmann-Chen S, Arnold FH, Nat. Chem 2017, 9, 629–634; [DOI] [PMC free article] [PubMed] [Google Scholar]; c) Yang, Cho I, Qi X, Liu P, Arnold FH, Nat. Chem 2019, 11, 987–993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].a) Wang ZJ, Peck NE, Renata H, Arnold FH, Chem. Sci 2014, 5, 598–601; [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Sreenilayam G, Fasan R, Chem. Commun 2015, 51, 1532–1534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].a) Steck V, Carminati DM, Johnson NR, Fasan R, ACS Catalysis 2020, 10, 10967–10977; [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Liu Z, Calvó-Tusell C, Zhou AZ, Kai C, Garcia-Borràs M, Arnold FH, Nat. Chem 2021, 10.1038/s41557-021-00794-z [DOI] [PubMed] [Google Scholar]
- [16].Zhu Y, Liu X, Dong S, Zhou Y, Li W, Lin L, Feng X, Angew. Chem. Int. Ed 2014, 53, 1636–1640. [DOI] [PubMed] [Google Scholar]
- [17].Dydio P, Key HM, Hayashi H, Clark DS, Hartwig JF, J. Am. Chem. Soc 2017, 139, 1750–1753. [DOI] [PubMed] [Google Scholar]
- [18].Anding BJ, Woo LK, Organometallics 2013, 32, 2599–2607. [Google Scholar]
- [19].Dydio P, Key HM, Nazarenko A, Rha JYE, Seyedkazemi V, Clark DS, Hartwig JF, Science 2016, 354, 102–106. [DOI] [PubMed] [Google Scholar]
- [20].Gu Y, Natoli SN, Liu Z, Clark DS, Hartwig JF, Angew. Chem. Int. Ed 2019, 58, 13954–13960. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Smith BJZ, Gutierrez P, Guerrero E, Brewer CJ, Henderson DP, Appl. Environ. Microbiol 2011, 77, 6703–6705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Basudhar D, Madrona Y, Kandel S, Lampe JN, Nishida CR, de Montellano PRO, J. Biol. Chem 2015, 290, 10000–10017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Li D, Wu Q, Reetz MT, Methods Enzymol. 2020, 643, 225–242. [DOI] [PubMed] [Google Scholar]
- [24].Montañez-Clemente I, Alvira E, Macias M, Ferrer A, Fonceca M, Rodriguez J, Rodriguez J, Gonzalez A, Barletta G, Biotechnol. Bioeng 2002, 78, 53–59. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.