

Abstract
Human PSCs hold tremendous potential for both basic biology and cell-based therapies for a wide variety of diseases. The ability to manipulate the genome of these cells using the CRISPR/Cas9 technology has expanded this potential by providing a valuable tool to engineer or correct disease-associated mutations. Because of the high efficiency by which CRISPR/Cas9 creates targeted double strand breaks, a major challenge has been the introduction of precise genetic modifications on one allele, without indel formation on the non-targeted allele. To overcome this obstacle, we describe the use of two oligonucleotide repair templates: one expressing the sequence change and the other maintaining the normal sequence. In addition, we have streamlined both the transfection and screening methodology to make this protocol efficient with small numbers of cells and limiting the amount of labor-intensive clone passaging. This protocol provides a technically simple approach for generating valuable tools to model human disease in stem cells.
BASIC PROTOCOL 1: Implementation of CRISPR based genome editing in human PSCs
BASIC PROTOCOL 2: Genetic modification of human PSCs using a double ODN CRISPR/Cas9 recombination system
Keywords: Human pluripotent stem cells (hPSCs), genome editing, homology directed repair, CRISPR/Cas9
Introduction
Pluripotent stem cells (PSCs) support the study of human biology and in vitro disease modeling. The ability to introduce and/or correct disease-associated mutations using the CRISPR/Cas9 system promotes greater tractability within these specialized cells (Brookhouser et. al., 2017; Hockemeyer & Jaenisch, 2016). However, engineering of highly specific alterations has been challenging, prompting the development of new methodologies to enhance the efficiency and ease of genome engineering in stem cells. A limiting factor in genome editing includes generating precise mono-allelic sequence modifications, without the introduction of nonspecific indels (Richardson et. al., 2016; Paquet et. al., 2016). To overcome this obstacle, we demonstrate the use of a double oligonucleotide (ODN) recombination system to introduce and/or correct mono-allelic genetic mutations. When using a single-stranded ODN (ssODN) template to generate a mutation, we found virtually all clones will harbor indel mutations in the non-targeted allele. To overcome this problem, we introduced two ssODNs, both of which contain silent mutations that block or prevent guide RNA (gRNA) recognition and re-cutting, but only one of which includes the desired nucleotide alteration. We show that this methodology results in introduction of the desired genetic alteration in one allele, without occurrence of indel generation in the other allele. Data collected from over ten unique gene editing experiments have demonstrated an average editing efficiency of 14%.
This unit begins with a description of gRNA design, cloning and efficiency testing in PSCs. The method utilizes transient transfection of plasmid DNA, and evaluation of gRNA cleavage efficiency (Basic Protocol 1). This is followed by a protocol for the introduction of precise heterozygous mutations by the recombination of two ssODNs (Basic Protocol 2). This method reduces the occurrence of indel formation in the second allele that often occurs when using a single ODN approach for homologous recombination.
NOTE: PSCs for CRISPR/Cas9 editing are cultured in human embryonic stem cell (hESC) media (see Reagents and Solutions) with irradiated mouse embryonic fibroblasts (MEFs) in a laminar-flow hood according to standard practices and maintained in a humidified incubator at 37°C, 5% CO2, 5% O2, and 90% N2 (Paluru, et. al., 2013).
BASIC PROTOCOL 1
Implementation of CRISPR based genome editing in human PSCs
This protocol outlines the design, cloning and efficiency testing of gRNAs required for CRISPR/Cas9 genome editing. Determination of guide cutting efficiency is performed in the target or a control stem cell line using a lipid-based transfection method.
Materials
Plasmids
pCas9_GFP (Addgene #44719)
gRNA_Cloning Vector (Addgene #41824)
Tissue Culture
ThermoFisher® Lipofectamine Stem Reagent (STEM00001)
TrypLE (ThermoFisher 12605036)
Y-27632 Dihydrochloride/Rock inhibitor (ROCKi) (Tocris 1254)
Falcon polystyrene 6 well tissue culture dishes (Corning 353046)
Falcon 5 mL polystyrene round-bottom tube with cell strainer-cap (Corning 352235)
Falcon 10-cm2 tissue culture dishes (Corning 353003)
Corning® Matrigel® Growth Factor Reduced (GFR) Basement Membrane Matrix (354230)
Iscove’s Modified Dulbecco’s Medium (IMDM) (Corning 12440053)
Murine Embryonic Fibroblasts (MEFs)- irradiated
Kits
Phusion High Fidelity DNA Polymerase (New England Biolabs M0530)
dNTPs (Roche 11969064001)
Gibson Assembly Kit (New England Biolabs E2611)
Purelink™ Quick Plasmid Miniprep Kit (Invitrogen K210011)
Nucleospin® Gel and PCR Clean-up Kit (Macherey-Nagel 740609)
Miscellaneous
Agarose (VWR N605)
AflII (New England Biolabs R0520)
Stellar™ competent cells (Takara 636763)
Kanamycin LB agar plates (50μg/ml Kanamycin)
Proteinase K Solution (Qiagen 19133)
Equipment
Orbital shaking incubator
Variable oxygen control CO2 incubator
Fluorescence-Activated Cell Sorter
Protocol steps with step annotations
Design of PCR Primers for Screening
-
Design forward and reverse PCR primers to amplify the region to be edited (Figure 1A).
The primers should asymmetrically flank the gRNA cleavage sites. The resulting amplicon can be ~300–1000 bp.
Optimize the PCR on control genomic DNA and confirm a clean amplicon by agarose gel electrophoresis.
A nested primer should also be designed for future sequencing of clones.
Figure 1. Guide RNA Design.
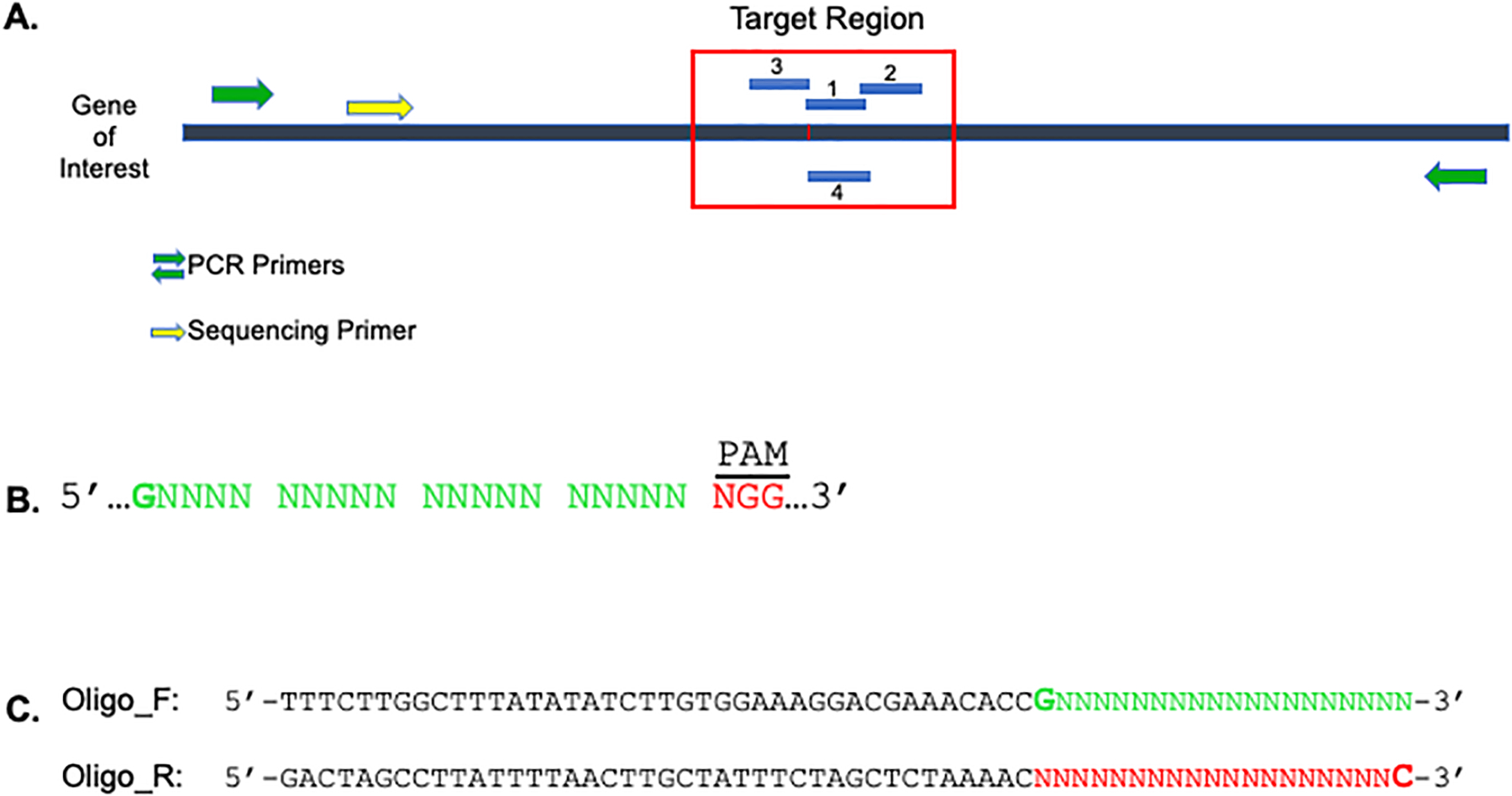
A. Identify gRNAs within ±20 bp of the target region (red box). gRNAs can be designed on either the sense or antisense strand. Design PCR primers to amplify ~300–100 bp flanking the region to be edited (green arrows). A nested sequencing primer should also be designed for future screening of clones (yellow arrow). B. 23 bp gRNA structure guideline with PAM at 3’ end. The 5’ most nucleotide can be introduced if the guanine does not occur in the sequence. C. Incorporation of gRNA sequence (minus the PAM site) into 60-mer ODNs.
Guide RNA Design
-
Identify two to three 23 bp 5’-G(N19)NGG-3’ guide sequences within 20 bp of the target site on either strand of DNA (Figure 1A & B).
If you are unable to find a G (guanine) at the first position (20th nucleotide preceding the PAM), it can be engineered into the gRNA (Figure 1B). However, the optimal condition is for this G to be within the target sequence.
-
Evaluate the gRNA sequences for genomic redundancy and off-target probability.
There are multiple resources that can be accessed for gRNA evaluation. We use the UCSC Genome Browser (https://genome.ucsc.edu/) and the CRISPOR website (http://crispor.tefor.net/).
Incorporate the 20 bp target sequence (excluding the PAM) into two 60-mer oligos as shown in Figure 1C (sequences are 5’ to 3’ and green and red are reverse compliments).
Order the two 60-mer oligos from IDT (www.idtdna.com) as 25 nmole DNA oligos (not ultramers) with standard desalting purification.
- Anneal the two oligos and generate a 100 bp dsDNA fragment using Phusion High Fidelity DNA polymerase.
- Resuspend forward and reverse oligos to a final concentration of 100 μM in water. Make a working stock of 10 μM.
-
Combine the following from working stocks:5 μl 10 μM forward oligo + 5 μl 10 μM reverse oligo
- Incubate at 95°C for 5 min.
- Cool at RT for 10 min.
- Add the following to each primer set (20 μl total volume):
- 0.4 μl 10 mM dNTPs
- 4 μl 5x Phusion HF buffer
- 0.2 μl Phusion polymerase
- 5.4 μl ddH2O
- Perform PCR amplification using the following parameters:
- 98°C × 30 sec
30 cycles: 98°C × 10 sec 55°C × 20 sec 72°C × 30 sec - 72°C × 5 min
- 4°C hold
Run the PCR product on a 1% agarose gel.
-
Excise a 100 bp band and purify with a gel extraction kit.
A nonspecific band at ~200 bp is sometimes observed.
Preparation of gRNA_Cloning Vector Plasmid
- Digest the gRNA_Cloning Vector with AflII:
- 10 μg gRNA_Cloning Vector
- 4 μl 10X CutSmart Buffer
- 24 μl water
- 1.5 μl AflII
Incubate at 37°C O/N.
Run digest on a 1% agarose gel and excise bands (expected band size is ~3519 bp).
Purify with a gel extraction kit.
Gibson Assembly of gRNA Vector
- Using the NEB Gibson Assembly Kit, set-up each reaction at a 1:5 ratio of AflII digested gRNA_Cloning Vector to 100 bp gel purified Phusion insert.
- 50–100 ng AflII digested vector
- 250–500 ng (5x) 100bp insert
- 10 μl 2x master mix
- ddH2O to 20 μl
Incubate at 50°C for 15 min.
Dilute Gibson Assembly reaction (20 μl) 1:3 in ddH2O and proceed to plasmid transformation.
Transformation of gRNA_Cloning Vector
Add 3 μl of the diluted Gibson Assembly reaction mixture to 50 μl of competent cells.
Incubate cells on ice for 20 minutes, heat shock at 42°C for 45 seconds, and return to ice for 2 minutes.
Add 900 μl of SOC media to each sample and shake at 37°C for 1 hour in an orbital shaking incubator.
Plate 500 μl of each sample on KanR LB plates and grow O/N at 37°C.
Pick 3–5 colonies per gRNA. Inoculate each colony in 4 mL LB and grow O/N at 37°C on an orbital shaking incubator.
Purify plasmid DNA using a miniprep plasmid isolation kit.
- Sequence each gRNA using the following primers:
- Forward: GTACAAAAAAGCAGGCTTTAAAGG
- Reverse: TGCCAACTTTGTACAAGAAAGCT
If the gRNA sequence is correct, continue with cell transfection. If the sequence is not found, cloning was unsuccessful, and additional bacterial colonies need to be screened.
Testing gRNA Cleavage Efficiency (Figure 2)
Figure 2. CRISPR-Cas9 Guide RNA Cleavage Efficiency.
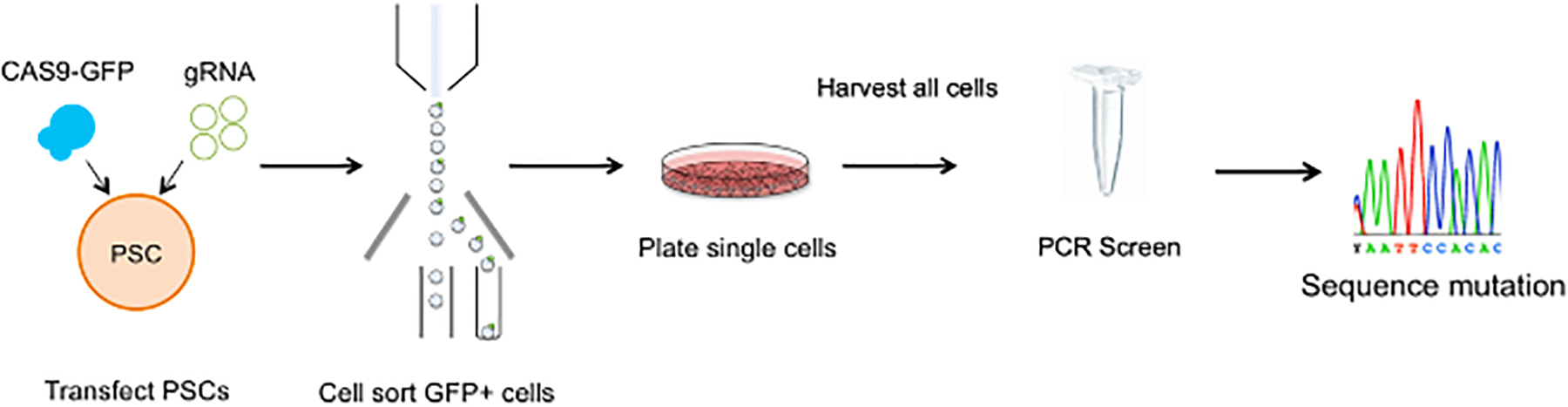
PSCs are transfected using a lipid mediated reagent and sorted for GFP 48 hours post-transfection. Single cells are plated into a single well and grown to confluency. The pooled cells are harvested and screened by PCR for gRNA cleavage confirmation. Sequence results are analyzed using a program to determine cutting efficiency.
-
Plate target cell line in a 6-well dish on irradiated MEFs to reach 80–85% confluency after an overnight incubation. Each gRNA is tested in duplicate requiring 2 wells of cells.
Each cell line will require optimization to achieve the desired plating density.
Change hESC medium (2.0 mL/well) at least 3 hours before transfection.
- For each transfection, add the following reagents to a 1.5 mL eppendorf tube:
- 50 μl base media (normal base media with no additions)
- 0.5 μg pCas9_GFP vector
- 0.5 μg guide RNA miniprep
- 3 μl Lipofectamine Stem - added last
Mix by pipetting and incubate at RT for 15 minutes.
Add dropwise to the cells and incubate for 48 hours, changing media 24 hours post transfection.
- Harvest cells for sorting after 48 hours.
- Remove MEFs with a 3 min RT TrypLE incubation.
- Wash cells 1× with IMDM medium and scrape into hESC medium + 10 μM ROCKi, combining duplicate wells.
- Pellet cells at 300×g for 3 min and resuspend in 0.5 mL hESC medium + 10 μM ROCKi.
- Filter into a 5 mL tube through a cell-strainer cap.
-
Using FACS, sort GFP positive cells.
The percent of GFP+ cells is typically between 1–6%.
Use hESC containing ROCKi and Pen/Strep for sorting and collection medium.
-
Transfer the sorted cells directly into 1 well of a 6 well dish (previously coated with 1:3 matrigel and 1×106 irradiated MEFs, see Reagents and Solutions) in growth medium containing ROCKi.
Do not pellet sorted cells before plating.
Change medium daily using hESC medium without ROCKi until colonies are well established (7–10 days).
Harvest pooled cells as previously described. Remove MEFs with a 3 minute TrypLE incubation, wash, and scrape cells into hESC.
-
Pellet cells in microfuge tubes at 10,000×g for 5 min and remove supernatant.
Pellets can be stored at −20°C to screen at a later time.
Clone Screening
-
Resuspend cell pellets in 50–100 μl of Proteinase K Buffer (see Reagents and Solutions).
Resuspension volume will need to be adjusted according to pellet size.
Incubate at 55°C for 1 hour, 95°C for 10 min.
Vortex vigorously and centrifuge at 10,000×g for 5 min.
Analyze 5 μl of the Proteinase K digest (dirty miniprep) in a PCR reaction using the primer set designed to flank the edited region. Include genomic DNA from the parental cell line as an unedited control.
Sequence PCR products using the nested sequencing primer previously designed.
-
Input control and edited sequencing results into the Synthego ICE program (or similar gene editing analysis program) to determine guide cutting efficiency and specificity.
Proceed to Basic Protocol 2 with the most efficient gRNA. We have found that a cleavage efficiency of >20% serves as a notional threshold for successful editing. gRNAs with lower cutting efficiency can be used if experimentally limited to this option.
BASIC PROTOCOL 2
Genetic modification of human PSCs using a double ODN CRISPR/Cas9 recombination system
This protocol is used to introduce precise mutations within a gene of interest and to allow efficient generation of a mutation in a single allele without generating an indel in the second allele. Using a prescreened gRNA (see Basic Protocol 1), two ssODNs are designed with and without the desired base alteration(s). Additional silent mutations are introduced into the ssODNs to prevent gRNA cutting and to streamline screening of clones.
Materials
Plasmids
pCas9_GFP (Addgene #44719)
pCAG-i53-bpA-EF1BFP (Addgene #111145)
Tissue Culture
ThermoFisher® Lipofectamine Stem Reagent (STEM00001)
TrypLE (ThermoFisher 12605036)
Y-27632 Dihydrochloride/Rock inhibitor (ROCKi) (Tocris 1254)
Falcon polystyrene 6 well tissue culture dishes (Corning 353046)
Falcon 10-cm2 tissue culture dishes (Corning 353003)
Corning® Matrigel® Growth Factor Reduced (GFR) Basement Membrane Matrix (354230)
Iscove’s Modified Dulbecco’s Medium (IMDM) (Corning 12440053)
Murine Embryonic Fibroblasts (MEFs) - irradiated
Kits
Nucleospin® Gel and PCR Clean-up Kit (Macherey-Nagel 740609)
Miscellaneous
Agarose (VWR N605)
Proteinase K Solution (Qiagen 19133)
Equipment
Variable oxygen control CO2 incubator
Fluorescence-Activated Cell Sorter
Protocol steps with step annotations
Oligonucleotide Design
Design 100 bp ssODNs centered around the most efficient gRNA sequence determined in protocol 1 (Figure 3A).
-
Introduce a silent mutation in the PAM sequence of the oligo, but if not possible, introduce 3–4 silent mutations within the gRNA sequence.
These mutations prevent gRNA “re-cleavage” of the recombined ODN.
As a screening strategy, introduce a unique restriction site within ~25 bp of the gRNA sequence using silent mutations. Targeted clones will be identified by restriction enzyme digestion.
-
Design one ODN with the desired base change(s) to create the mutation of interest and one ODN without the base change(s).
These changes should be no more than ~20 bp from the predicted CRISPR/Cas9 cut site, as recombination drops off considerably at greater distances (Paquet et. al., 2016).
Order the two ssODNs from IDT as 4 nmole Ultramer® DNA Oligos with standard desalting purification.
Resuspend in water to make 0.5–1.0 μg/μl stocks. Store stocks at −20°C.
Figure 3. Oligonucleotide Design and Recombination Screening.
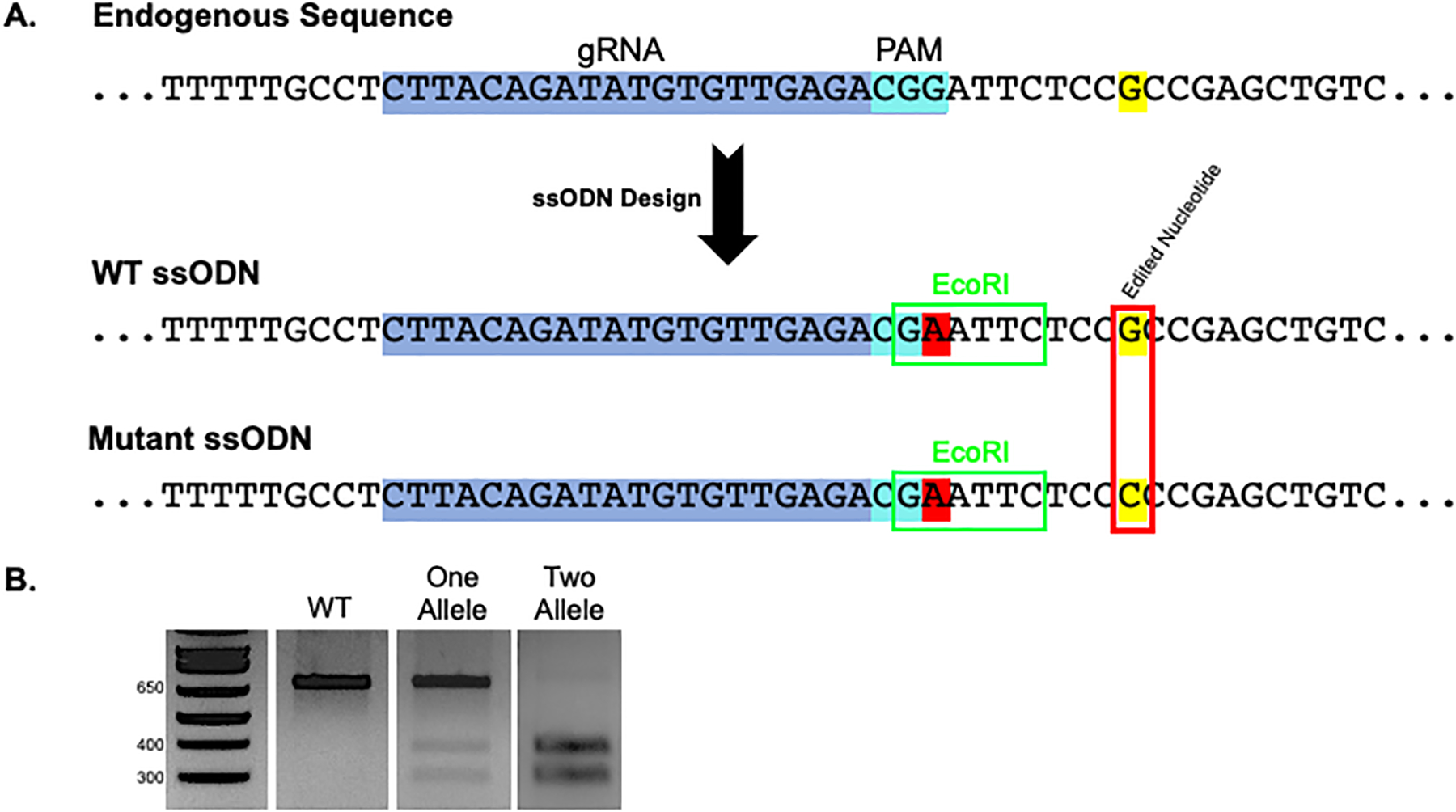
A. 100 bp ssODNs are designed flanking the optimal gRNA (blue/aqua shaded sequence). Silent/blocking mutation(s) are introduced in the PAM site or throughout the gRNA sequence to prevent subsequent cleavage after recombination (red shaded nucleotide). Introduction of a unique restriction site within the PCR amplicon allows for simplified screening of clones (green box). This example depicts an EcoRI site formed through introduction of a silent mutation in the PAM site. However, the restriction site does not need to be made within the gRNA sequence. Finally, wild-type (WT) and mutant ODNs are designed incorporating the endogenous sequence (WT) or a mutation of interest (mutant) (red box). B. Potential screening outcomes after PCR amplification and restriction digest include no recombination (WT), which shows an uncleaved band, one allele recombination, which shows both uncleaved and cleaved bands, and two allele recombination, which shows only cleaved product.
Transfection Setup
Plate the target cell line in a 6-well dish on irradiated MEFs to reach 80–85% confluency after an overnight incubation.
-
Change hESC medium at least 3 hours before transfection.
Additional bFGF (75–100 ng/ml) can be added to the media (1.5 mL/per well) to increase proliferation.
- For each transfection, add the following reagents to a 1.5 mL eppendorf tube:
- 50 μl base media (Opti-MEM or DMEM)
- 0.5 μg pCas9_GFP vector
- 0.5 μg guide RNA
- 0.5 μg targeting oligo (if using 2 oligos, use 0.25 μg each)
- 0.25 μg i53 vector
-
3 μl Lipofectamine Stem - added lastThe transcription factor 53 binding protein 1 (53BP1) hinders HDR mechanisms. Addition of i53 inhibits this effect, therefore promoting HDR (Canny et. al., 2018).
Mix by pipetting and incubate at RT for 15 minutes.
-
Add dropwise to the cells and incubate for 48 hours, changing media 24 hours post transfection.
The MEF feeder layer will also exhibit a low efficiency of transfection. This is to be expected and will not influence isolating edited clones as the MEFs are removed prior to sorting.
- Harvest cells for sorting after 48 hours.
- Remove MEFs with a 3 min RT TrypLE incubation.
- Rinse cells 1× with IMDM media, and scrape into hESC medium + 10 μM ROCKi, combining duplicate wells.
- Pellet cells at 300×g for 3 min and resuspend in 0.5 mL hESC medium + 10 μM ROCKi.
- Filter into a 5 mL tube through a cell-strainer cap.
-
Using FACs, sort GFP positive cells.
Use hESC + ROCKi and Pen/Strep for sorting and collection medium.
-
Transfer the sorted cells directly to a 10 cm2 dish containing hESC medium + ROCKi (coated with 1:3 matrigel, see Reagents and Solutions, and 1×106 irradiated MEFs).
Plate a maximum of 15k cells per 10 cm2 dish. A minimum of ~5k GFP+ cells appear to be required to achieve adequate clone formation.
- Change medium daily with hESC media and manually pick colonies after 10–15 days, when colonies are ~1 mm in diameter.
- Using a P200 pipette and microscope, carefully scrape a single clone and draw cells into the pipette.
- Disperse the cells by gently pipetting 3–4 times in a 96 well plate in the medium drawn up with the colony.
- Distribute:
- Half to a 24 or 48 well dish containing hESC medium + ROCKi (pre-coated with 1:3 matrigel and 1×106 irradiated MEFs).
-
Half to PCR strip tubes for screening.Isolate 24–48 clones per condition.
Clone Screening
-
Pellet cells in PCR strip tubes at 10,000×g for 5 min and remove supernatant.
Pellets can be stored at −20°C to screen at a later time.
Resuspend cells in 20 μl of Proteinase K Buffer (see Reagents and Solutions).
Incubate at 55°C for 1 hour, 95°C for 10 min.
Vortex vigorously and centrifuge at 10,000×g for 5 min.
Add 5 μl of the Proteinase K digest to a 30 μl PCR reaction using primers flanking the gene edited region.
Run 5 μl of the PCR reaction on a 1.5% agarose gel to ensure product formation and purify the remaining PCR product using a PCR clean-up kit.
-
Digest half of the PCR product using the restriction enzyme specific for the unique site designed in the ssODN and run on a gel to select clones with recombination events (Figure 3B).
An uncut band represents no recombination (WT). Evidence of both uncleaved and cleaved bands indicates compound heterozygous recombination (HTZ). In our experience, these are not desirable clones, as the uncut allele most likely contains an indel. Complete cleavage of the PCR product indicates recombination has occurred in both alleles (HMZ). Sequencing is required to determine which ssODN was used as a template.
-
Sequence clones using the nested sequencing primer previously designed.
Focus on HMZ clones due to the high probability that the HTZ clones will have an indel in the unrecombined allele.
Align sequences with the WT gene to ensure proper editing.
After confirmation, it is recommended to subclone the targeted clones to ensure a homogeneous population.
Expand cell stocks and confirm normal CNV and/or karyotype of clones.
REAGENTS AND SOLUTIONS
Proteinase K Buffer
50 mM Tris-HCl
15 mM ammonium sulfate pH 9.3
2.5 mM MgCl2
0.1% Tween 20
100 μg/mL Proteinase K
1:3 Matrigel
Corning® Matrigel® Growth Factor Reduced (GFR) Basement Membrane Matrix diluted 1:3 in IMDM medium. Keep all components on ice.
hESC Media
DMEM/F12
15% knock-out serum replacement
100 μM non-essential amino acids
1 mM sodium pyruvate
2 mM glutamine, 50 U/ml penicillin
mM β–mercaptoethanol (Sigma, St Louis, MO)
-
10 ng/ml human bFGF (R&D Systems)
NOTE: All incubations are performed in a humidified incubator at 37°C, 5% CO2, 5% O2, and 90% N2.
COMMENTARY
Background Information
Many genetic disorders have been studied by reprogramming cells from patients with a specific disease to iPSCs, but this approach is time consuming and severely limited by patient availability. Studies are also confounded by the genetic backgrounds of patients. These problems can be circumvented by introduction of the disease mutations in a control iPSC line (Anderson & Francis, 2018). The process can be completed in a shorter period of time and provides an isogenic control for subsequent studies. In order to introduce these mutations, the CRISPR/Cas9 system is increasingly preferred based on its high efficiency and ease of use compared with zinc-finger nucleases and transcription activator-like effector nucleases (TALENs) (Ding et. al., 2013; Hou et. al., 2013). When combined with a ssODN template repair that occurs via homologous recombination, the introduction of specific mutations can be readily achieved (Richardson et. al., 2017).
Critical Parameters
This protocol has been optimized for PSCs cultured on a feeder layer of irradiated MEFs. Although feeder-free growth conditions may result in significantly higher transfection efficiency, cell survival post-transfection and editing efficiency may be reduced compared to cells grown on MEFs. Transfection efficiency of PSCs grown on MEFs is often <5%, but we have found that a sufficient number of GFP+ cells can easily be generated after cell sorting.
We have also optimized this protocol using iPSCs grown in hypoxic culture conditions. Other labs have successfully implemented the use of normal culture conditions for CRISPR gene editing, and therefore it would be anticipated that normoxic conditions could be applied to this protocol (Giacalone et. al., 2018).
The design of optimal PCR primers for amplification of the edited sequence is essential for both gRNA efficiency testing and clone screening. Without a clean PCR product, both gRNA cleavage and restriction enzyme screening will be difficult to evaluate. In addition, the screening procedure used in this protocol allows for expedited screening so identification of possible targets prior to splitting of picked clones can occur. This method allows the investigator to save a significant amount of effort by only splitting and expanding clones that are positive in the initial screen. Some projects may not work with the dirty PCR prep and require expansion of all clones, and preparation of a high-quality genomic DNA preparation.
Location of gRNAs must reside within coding regions, as the silent/blocking mutations used to prevent recutting of edited alleles will have unknown effects in non-coding regions. This protocol could be used to target non-coding, single nucleotide polymorphisms with two caveats. First, the nucleotide change must be within the PAM sequence. Second, only homozygous changes can be made efficiently without indel formation (data not shown).
In this protocol, transfection of plasmid DNA is accomplished with ThermoFisher® Lipofectamine Stem reagent. This protocol can work with other lipid transfection reagents but will require further optimization. The use of lipid transfection allows the generation of sufficient numbers of transfected cells from two wells of a 6-well dish of PSCs. Lipid transfection is also less stressful on the cells compared to electroporation and is technically easier to perform. Delivery of the CRISPR/Cas9 components as a ribonucleoprotein (RNP) complex has shown numerous benefits, including increased editing efficiency, and reduced off-target effects due to transient expression (Zhang, et. Al. 2021). This method can be considered as a beneficial alternative for introducing the editing machinery but would require optimization of the protocol.
Defining off-target events incurred through CRISPR editing Our protocol recommends the utilization of bioinformatic programs to predict the off-target probability of a gRNA, which use the degree of similarity between the target DNA sequence and the gRNA sequence to predict potential off-target sites. More definitive evaluations of an edited line would be beneficial. Numerous technologies have been designed to detect off-target cleavage including whole genome sequencing (WGS) and next generation sequencing (NGS). Recent advances have shown that both cell-based and biochemical NGS methods including GUIDE-Seq, CIRCLE-SEQ, and DISCOVER-Seq provide in depth coverage to capture double stranded break events (Sullivan et. al., 2020; Chaudhari et. al., 2020). Employment of these methods would be advantageous in providing a well characterized research tool.
This protocol can also be amended to introduce deletions into a gene of interest. By designing two gRNAs flanking the region to be deleted, we have successfully deleted sequences up to ~10k bp. The cutting efficiency of each gRNA should be tested individually, then added to the transfection mixture at equal concentrations. Screening is accomplished using PCR primers flanking the deletion. With larger deletions, these primers will only create a product if the sequence has been removed. To confirm deletion or identify single allele deletions, a primer within the deleted region is designed and combined with the opposing flanking primer in addition to primer sets located completely within the deleted region.
Troubleshooting
For troubleshooting, please refer to Table 1.
Table 1.
Troubleshooting
| Technique | Problem | Possible Cause | Solution |
|---|---|---|---|
| Transformation of gRNA cloning vector | No gRNA sequence in vector | Inefficient cloning | Screen additional clones or repeat |
| Cleavage efficiency of Cas9 gRNA | Poor to undetectable cleavage | SNPs | Sequence target region in cells to be edited to confirm matching sequence |
| Refractory chromosomal region | Design guides in another region | ||
| Lethal mutation | None | ||
| ssODN Design | Cannot create unique restriction site with silent mutations | Limited sequence availability | A unique restriction site can be removed with silent mutations, but gRNA cleavage sometimes creates false positives when screening |
| Plasmid transfections | Poor cell survival | Toxic load of DNA | Decrease concentrations of plasmid/ssODN |
| Low efficiency | Cell density | Determine the cell number for 80–85% confluency | |
| Plasmid ratios incorrect | Test multiple plasmid concentrations to achieve optimal GFP expression | ||
| Lipofectamine Stem volume | Optimize volume needed for transfection | ||
| Clone screening | Heterogeneity of clones | Clone proximity too close upon manual isolation | Plate sorted cells at a higher dilution (no more than 15,000 sorted cells per 10 cm2 dish) |
| No PCR product or dirty PCR product | Proteinase K minipreps may not be adequately clean for PCR reaction | Prepare purified genomic DNA |
Understanding Results
Introduction of two ssODNs (WT and mutant) will result in three possible recombination outcomes (Figure 4).
Figure 4. Recombination Outcomes.
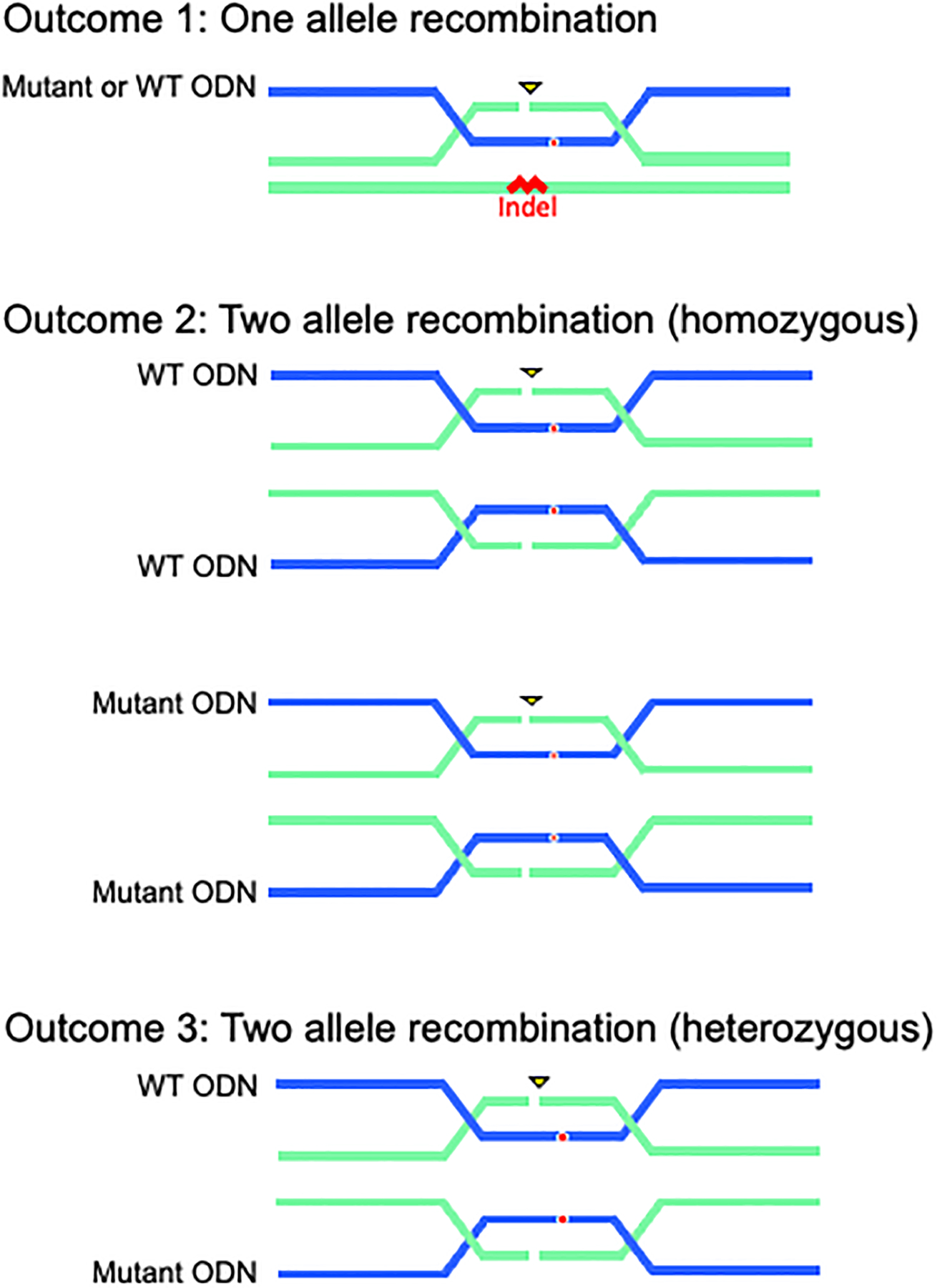
Potential outcomes include 1) single allele recombination (recombination in one allele and the presence or absence of indel formation in the second allele), 2) two allele recombination with either WT or MUT ODN (homozygous), or 3) two allele recombination with WT AND mutant ODN (heterozygous).
Outcome 1 shows homologous recombination with one ssODN. In our experience, when one allele recombines with an ODN, the second allele will harbor an indel. Either the mutant or WT ssODN may recombine with the one allele, which can be identified by TOPO PCR Cloning and sequence analysis.
Outcome 2 shows homologous recombination on both alleles with either the mutant or WT ssODNs.
Outcome 3 shows heterozygous recombination on both alleles with the mutant and WT ssODNs. One allele contains the desired base change, and the other allele contains the wild-type sequence. Both are identified by sequence analysis.
Using the two ssODN approach described in this protocol, we have found recombination efficiencies for Outcome 3 to be 0–21%. Figure 5 shows six representative editing experiments. Selected clones were karyotyped, and all were found to be normal. Significant differences in editing success are evident for each gene. Of note, low numbers of clones or an inability to identify gRNAs that cut may be due to chromatin structure and/or gene lethality. Addition of the pCAG-i53-bpA-EF1BFP plasmid significantly increases HDR occurrence (Figure 6). Concurrent transfections with or without the addition of the i53 plasmid were performed in three independent genes and iPSC lines. This plasmid is therefore recommended in all experiments utilizing ssODNs for HDR.
Figure 5. Recombination Efficiencies.
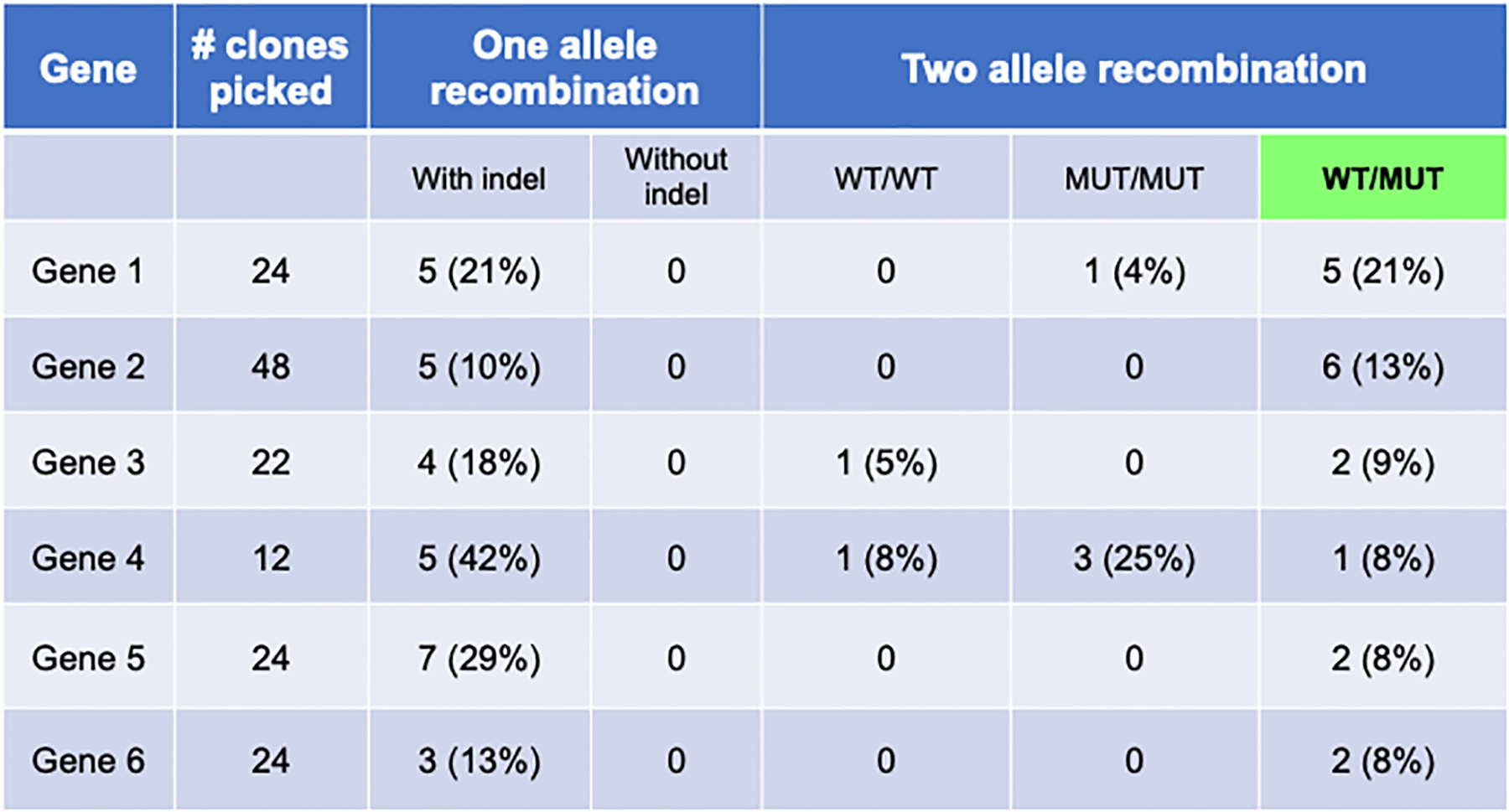
Table displays results from multiple genes edited with described protocol. When using one ssODN (one allele recombination), only one allele recombination with indel formation occurs. Introduction of two ssODN results in varying efficiencies of the three outcomes described in Figure 4.
Figure 6. HDR Efficiency is increased with the addition of 53BP1 inhibitory plasmid.
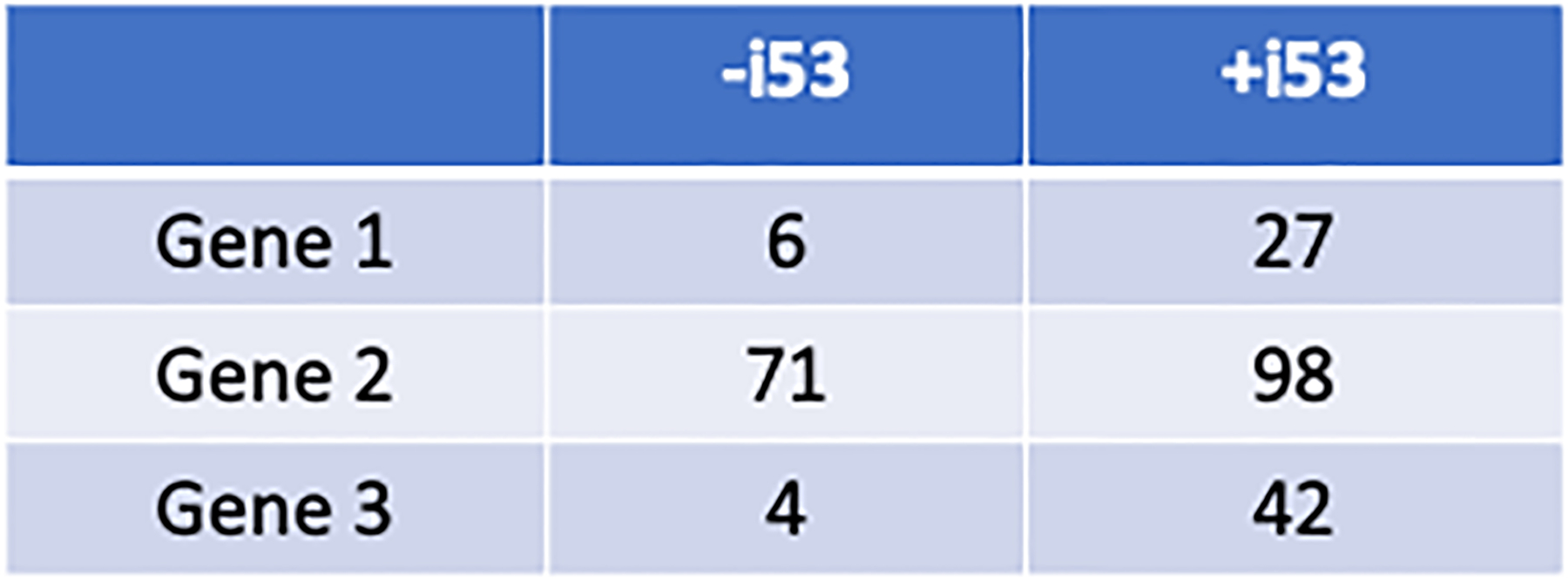
Recombination efficiency (%) in the absence and presence of the pCAG-i53-bpA-EF1BFP vector.
Time Considerations
The total amount of time for the described protocols largely depends on the success of gRNA cleavage. The following outlines an estimation of the time required for each task to be completed by a researcher familiar with PSC culture and molecular biology.
Design and cloning of gRNAs can take 4–5 days (not including time for shipment of ODNs). The gRNA efficiency testing of the protocol is time consuming, but an important required component of the assay. Sorted, GFP positive cells can take up to 2 weeks to be confluent enough for analysis. PCR screening of pooled cells will take 1–2 days. If an efficient gRNA is not identified in the first round of screening, additional gRNAs will need to be designed and cloned. This could delay the protocol by multiple weeks.
Once an optimal gRNA is identified, ssODN design can be accomplished in 1 day. Again, after a 48-hour transfection of gRNA and ODNs, clone expansion will take approximately 2 weeks. After manual isolation of clones, clone screening (PCR amplification and restriction enzyme digestion) will take 2–3 days. Potential edited clones can be sent for overnight sequencing, and upon confirmation of genome editing, cells can be expanded. It can be expected to take 2–3 weeks to expand and freeze stocks. Figure 7 depicts the protocol timeline.
Figure 7. Schematic representation of timeline.
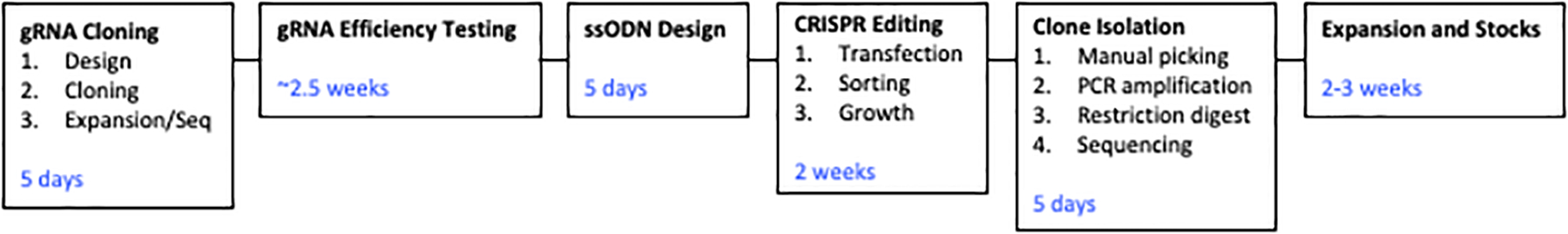
Outline of the major steps of the protocol and the approximate timing of each. Some steps incorporate ordering/shipping times.
Significance Statement.
Human pluripotent stem cells offer a valuable tool for the study and treatment of human disease. The ability to introduce and/or correct disease-associated mutations using the CRISPR/Cas9 system promotes greater tractability within these specialized cells. However, engineering of highly specific alterations has been challenging, prompting the development of new methodologies to enhance the efficiency and ease of genome engineering in stem cells. This report provides an updated version of an efficient and technically straightforward protocol for genetically manipulating these cells using CRISPR/Cas9 editing tools. Improvements to our protocol include a streamlined method for determining guide RNA cutting efficiency, and the addition of recombination enhancements to the transfection protocol.
ACKNOWLEDGMENTS
This research was supported by funding from U01HL099656 (PG and DLF) and U01HL134696 (PG and DLF) from the National Heart, Lung, and Blood Institute (NHLBI), National Institutes of Health.
Footnotes
CONFLICT OF INTEREST STATEMENT
Authors have no conflict of interest to declare.
DATA AVAILABILITY STATEMENT
The data that support the findings of this study are available from the corresponding author upon reasonable request.
Literature Cited
- Anderson RH & Francis KR (2018). Modeling rare diseases with induced pluripotent stem cell technology. Mol. And Cell. Probes. Article in Press 10.1016/j.mcp.2018.01.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brookhouser N, Raman S, Potts C, Brafman DA (2017). May I cut in? Gene Editing approaches in human induced pluripotent stem cells. Cells, 6(1):5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Canny MD, Moatti N, Wan LCK, Fradet-Turcotte A, Krasner D, Mateos-Gomez PA, Zimmermann M, Orthwein A, Juang YC, Zhang W, Noordermeer SM, Seclen E, Wilson MD, Vorobyov A, Munro M, Ernst A, Ng TF, Cho T, Cannon PM, Sidhu SS, Sicheri F, Durocher D (2018). Inhibition of 53BP1 favors homology-dependent DNA repair and increases CRISPR-Cas9 genome-editing efficiency. Nat Biotechnol, 36(1):95–102. doi: 10.1038/nbt.4021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chaudhari HG, Penterman J, Whitton HJ, Spencer SJ, Flanagan N, Lei Zhang MC, Huang E, Khedkar AS, Toomey JM, Shearer CA, Needham AW, Ho TW, Kulman JD, Cradick TJ, Kernytsky A Evaluation of Homology-Independent CRISPR-Cas9 Off-Target Assessment Methods. CRISPR J. 3(6):440–453. doi: 10.1089/crispr.2020.0053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ding Q, Regan SN, Xia Y, Oostrom LA, Cowan CA, and Mu-sunuru K (2013). Enhanced efficiency of human pluripotent stem cell genome editing through replacing TALENs with CRISPRs. Cell Stem Cell, 12, 393–394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Giacalone JC, Sharma TP, Burnight ER, Fingert JF,Mullins RF, Stone EM, & Tucker BA (2018). CRISPR-Cas9-based genome editing of human induced pluripotent stem cells. Current Protocols in Stem Cell Biology, 44,5B.7.1–5B.7.22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hockemeyer D & Jaenisch R (2016) Induced Pluripotent Stem Cells Meet Genome Editing. Cell Stem Cell 18:573–586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hou Z, Zhang Y, Propson NE, Howden SE, Chu LF, Sontheimer EJ, and Thomson JA (2013). Efficient genome engineering in human pluripotent stem cells using Cas9 from Neis-seria meningitidis. Proc. Natl. Acad. Sci 110(39): 15644–15649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paluru P, Hudock KM, Cheng X, Mills JA, Ying L, Galvao AM, Lu L, Tiyaboonchai A, Sim X, Sullivan SK, et al. (2013). The negative impact of wnt signaling on megakaryocyte and primitive erythroid progenitors derived from human embryonic stem cells. Stem Cell Res. 12, 441–451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paquet D, Kwart D, Chen A, Sproul A, Jacob S, Teo S, Olsen KM, Gregg A, Noggle S & Tessier-Lavigne M (2015). Efficient introduction of specific homozygous and heterozygous mutations using CRISPR/Cas9. Nature, 533:125. [DOI] [PubMed] [Google Scholar]
- Ran F, Hsu P, Wright J, and Agarwala V (2013). Genome engineering using the CRISPR-Cas9 system. Nat. Protoc 8, 2281–2308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Richardson CD, Ray GJ, DeWitt MA, Curie GL, Corn JE (2016). Enhancing homology-directed genome editing by catalytically active and inactive CRISPR-Cas9 using asymmetric donor DNA. Nat. Biotech 34:339–345. [DOI] [PubMed] [Google Scholar]
- Richardson CD, Kazane KR, Feng SJ, Zelin E, Bray NL, Schäfer AJ, Floor SN, Corn JE (2018) CRISPR-Cas9 genome editing in human cells occurs via the Fanconi Anemia pathway. Nat. Genet 50(8): 1132–1139. doi: 10.1038/s41588-018-0174-0. [DOI] [PubMed] [Google Scholar]
- Sullivan NT, Allen AG, Atkins AJ, Chung C-H, Dampier W, Nonnemacher MR, Wigdahl B (2020) Designing safer CRISPR/Cas9 therapeutics for HIV: Defining factors that regulate and technologies used to detect off-target editing. Front. Microbiol 11:1872. doi: 10.3389/fmicb.2020.01872. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang S, Shen J, Li D, Cheng Y 2021. Strategies in the delivery of Cas9 ribonucleoprotin for CRISPR/Cas9 genome editing. Theranostics. 11(2): 614–648. doi: 10.7150/thno.47007. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
The data that support the findings of this study are available from the corresponding author upon reasonable request.