

Graphical abstract
Keywords: miR-142 in AML, miR-96 in hearing loss, miR-184 in keratoconus, miR-204 in retinal dystrophy, miR-15/miR-16 in CLL, miR140 in skeletal dysplasia
Abstract
MicroRNAs (miRNAs) are small non-coding RNAs that posttranscriptionally regulate the expression of most genes. They are involved in regulating many physiological processes, and aberrations in the levels of different miRNAs play an important role in the development of many diseases, including autoimmune diseases, neuropsychiatric diseases, and cancers. Although miRNAs are being intensively studied and levels of many miRNAs are either specifically increased or decreased in particular diseases, very little is known about the genetic variations of miRNA genes and their impact on the functioning of miRNA genes and human diseases. To shed more light on the potential effects of genetic variants in miRNA genes, we review here representative examples of SNPs, mutations linked to Mendelian diseases, and cancer somatic mutations located in miRNA genes and discuss their potential effects on the expression of miRNA genes, i.e., the structure and processing of miRNA precursors, the levels of generated miRNAs, miRNA target recognition/silencing, and impact on human diseases.
1. Introduction
MicroRNAs (miRNAs) are small (∼21–23 nt) single-stranded noncoding RNAs whose canonical function is posttranscriptional regulation, usually downregulation, of the expression of their target genes. The specificity of target recognition is conditioned by the complementary interaction of miRNAs with their target sites, usually located in the 3′ untranslated region (3′UTR) of the targeted mRNAs. There are approximately 1900 and 567 miRNA precursors/genes annotated for humans in the most reputable miRNA databases, i.e., miRBase v.22.1 [1] and miRGeneDB v2.1, [2], [3], respectively. miRNA genes may be located within protein-coding genes or constitute independent transcriptional units (often annotated as miRNA host genes) and may encode either one or multiple miRNAs arranged in so-called miRNA clusters [4]. miRNA biogenesis is a multistep process described in detail elsewhere [5], [6]. Briefly, most miRNA genes are transcribed by RNA polymerase II. The most crucial part of the miRNA primary transcript (pri-miRNA) is a characteristic hairpin structure that upon cleavage by the microprocessor complex, in which the most important part is RNase DROSHA accompanied by DGCR8 and several other proteins, is released to the nucleoplasm as a secondary miRNA precursor (pre-miRNA). The pre-miRNA is exported to the cytoplasm, where it is further processed by the miRNA-induced silencing complex (miRISC) loading complex (RLC). The main component of the RLC is RNase DICER1, which cuts off the apical loop of the hairpin-shaped pre-mRNA, generating an ∼ 22-bp-long miRNA duplex, usually with 2 nucleotide-long 3′-end overhangs. Within the miRISC, the miRNA duplex is unwound, separating miRNA strands. One of the strands becomes the mature miRNA (miR-5p or miR-3p) and functions as a guide strand to recognize and silence target mRNAs, a process that is assisted by AGO and TNR6A proteins. It should be noted that the biogenesis of some miRNAs may be further regulated by different RNA-binding proteins and may be conducted by other alternative pathways [7], [8].
miRNAs regulate many important biological processes, e.g., the development, differentiation, and maintenance of homeostasis [9]. Many miRNAs have been identified to be upregulated or downregulated in different physiological and pathological conditions (diseases), and the role of many miRNAs in different human diseases, including various cancers, has been convincingly demonstrated [10].
Nonetheless, little attention has been given to the genetic variations in miRNA genes and their potential consequences for the functioning of miRNA genes, aberrations of miRNA-regulated processes, and human conditions, including diseases. Except for mutations in miRNA seed regions, which are crucial for target recognition, there is little knowledge or guidelines on how to interpret mutations located in other parts of miRNA genes. Are they all neutral variants or potentially deleterious mutations? In this review, we present the representative examples of single nucleotide polymorphisms (SNPs), germline mutations in Mendelian diseases, and cancer somatic mutations identified thus far in miRNA genes and discuss the potential consequences of genetic variants in these genes. We focused on the structural and functional consequences of genetic variants located in the different parts of miRNA genes, and their effect on miRNA genes expression, miRNA precursor structure, miRNA biogenesis, level of mature miRNA strands, and recognition of miRNA targets. We also discuss the implications of miRNA variants for human diseases.
2. Potential effects of genetic variants on miRNA biogenesis and function
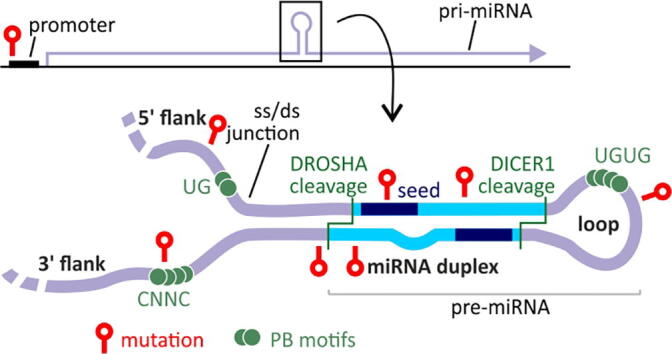
The particular sequence motifs and structural conformation of miRNA precursors are crucial for the proper biogenesis of miRNAs (Fig. 1A). Any aberrations caused by mutations or SNPs may lead to alterations at a particular step of miRNA biogenesis, resulting in changes in the miRNA level, precision of miRNA processing, 5p/3p miRNA balance, and specificity of miRNA target recognition (Fig. 1).
Fig. 1.

The potential effect of mutations on the functionality of miRNA genes (expression, precursor structure and processing, miRNA generation and target recognition). A) A schematic representation of the miRNA gene (above) and canonical miRNA precursor (below), with indicated miRNA precursor subregions and functional elements. B-G) Different effects of miRNA gene mutations. Positions of miRNA mutations are indicated as red lollipop symbols. The effect of the mutation on miRNA precursor structure (B), regulatory protein binding sites (C), DROSHA/DICER1 cleavage sites and precision of miRNA processing (D), 5p/3p strand balance (E), target recognition and creation of the new targets (F), and transcription of miRNA genes (G). (For interpretation of the references to color in this figure legend, the reader is referred to the web version of this article.)
The primary effect of each mutation in sequences coding for miRNA precursors, regardless of the subregion of the precursor in which the mutation occurs, is altered precursor structure and stability. This effect may be minor and may not change the general geometry of the precursor structure or be more profound and completely rearrange/destroy the structure. The effect of a mutation on the structure, among others, depends on the type of mutation, its localization, and the thermodynamic properties (stability) of the precursor. Generally, it may be expected that mutations/structural rearrangements in or in the direct vicinity of a miRNA hairpin structure (pre-miRNA) may have a more substantial effect on miRNA biogenesis and functioning of miRNA genes than mutations in other parts of the precursor (pri-miRNA) [11]. The mutations inducing larger structural rearrangements can make the precursor unrecognizable to the miRNA biogenesis enzymes and thus may stop miRNA biogenesis (Fig. 1B). Minor structural changes, such as new or removed bulges, internal loops, or other rearrangements inducing local changes in structure stability, may affect the precision or efficiency of miRNA precursor processing, mature miRNA selection and 5p/3p strand balance (Fig. 1B) [12]. Regardless of the impact on the structure, mutations in miRNA genes may disrupt or create new protein binding sites of RNA-binding proteins (RBPs), thus affecting the regulation of proper miRNA processing (Fig. 1C) [13]. Mutations in DROSHA or DICER1 cleavage sites may block or affect the precision of cleavage (Fig. 1D), which in turn may affect the balance between isomiRs or lead to the generation of new isomiRs [14], [15], [16]. Furthermore, variants in the miRNA duplex may change its stability, affecting the preferences of mature strand selection, and 5p/3p strand balance (Fig. 1E). The most obvious effects of mutations on miRNA functionality are due to mutations that occur in seed regions. A seed mutation in most cases will preclude proper recognition and silencing of its targets, but it may also lead to the recognition of novel targets (Fig. 1F) [17].
In addition to mutations in miRNA precursors, mutations in miRNA gene promoters and other regulatory sequences may affect the function of miRNA genes. Genetic variants in promoters may destroy, modify, or create new transcription factor-binding sites and thus may affect transcription efficiency and miRNA levels (Fig. 1G).
Mutations in the 3′UTRs of target genes and in genes that play a role in miRNAs biogenesis may also influence miRNAs functionality and biogenesis. However, in this review, we did not focus on these aspects, as they are well described in other publications [18], [19], [20], [21]. Still, other mechanisms that may have an impact on the expression of miRNA genes and function of miRNAs are epigenetic regulation of gene expression by promoters methylation [22], [23], methylation of miRNA-coding genomic regions [24], or histones modifications [25], as well as posttranscriptional modifications as A-to-I RNA editing [26], [27] or methylation of adenosine, guanosine or cytosine in miRNAs [28], but also these aspects are not the subject of this review.
Although most alterations in miRNA genes most likely act as loss-of-function mutations, some of these alterations may also be gain-of-function mutations. Such mutations could generate new miRNAs that recognize and downregulate new targets or create new competing endogenous RNAs (ceRNAs). New miRNAs may result from mutations in seed regions, mutations generating new isomiRs, or the preferential selection of a previously unused strand. Gain-of-function mutations may also result from the creation of new protein binding sites either in the promoter or in the miRNA precursor.
The examples presented above include those that may be directly deduced from the current knowledge about miRNA biology and certainly do not cover all the possibilities of how mutations may affect the functioning of the miRNA genes.
3. Seminal analyses of genetic variants in miRNA genes
The first global analysis of SNPs in miRNA genes showed that the density of SNPs within the precursor (hairpin-forming) sequence of miRNA genes is significantly lower than that in ∼ 300-nucleotide-long flanking regions [29]. This indicates that sequences coding for pre-miRNAs are crucial parts of miRNA genes in which genetic variation are not well-tolerated and confirms the functional importance and conservativeness of these sequences. This seminal result was subsequently confirmed in several later studies, which took advantage of a growing number of annotated SNPs and human miRNAs [30], [31], [32], [33]. In later studies that analyzed the SNP density in the subregions of miRNA precursors, it was shown that SNP density is almost equally decreased in the seed, loop, and miRNA duplex sequences. This finding indicates that all parts of pre-miRNA sequences (not only seeds) are similarly sensitive to genetic variants and suggests the substantial functional potential of all mutations in pre-miRNA sequences [31], [32], [33]. A similar conclusion about the conservative character of miRNA genes can also be drawn from the observation that the density of common copy number variants (CNVs) is significantly decreased in the regions of miRNA genes in comparison to the whole genome [30].
Despite the decreased density of SNPs in sequences coding for miRNA precursors, there are still numerous SNPs identified in these regions. The first SNPs in miRNA precursor-coding sequences were identified experimentally by sequencing 173 human pre-miRNA sequences (all known at the time of the analysis) in 96 samples from Japanese individuals [34]. The experiment allowed the identification of 10 SNPs located in 10 different pre-miRNA sequences. The identified SNPs were located in different subregions of pre-miRNAs, including mature miRNAs, passenger strands, apical loops, and directly adjacent (up to ∼ 10 nucleotides) flanking sequences. Among the identified SNPs was a substitution located in MIR30C2 in the 10th nucleotide of mature miR-30c-2-5p (n.16C>A; mutation annotation based on HGVS nomenclature, in reference to precursor transcript sequences from miRBase v.22.1) (Table 1). The mutation led to increased level of pre-miR-30c-2 and was predicted to affect the stability of miRNA:target pairing and proper target recognition.
Table 1.
Functionally tested genetic variants in miRNA genes.
| miRNA gene | HGVS; dbSNP ID | variant type | main miRNA | region (position)$ | free energy [kcal/mol]* | pri-miR level | pre-miR level | 5p-miR level | 3p-miR level | implicated disease | references |
|---|---|---|---|---|---|---|---|---|---|---|---|
| MIR15A/MIR16-1 cluster | wild-type | 5p | – | ||||||||
| n.*7C>A | germline mut | 3p flank | – | ↑ | ↓ | ↓ | – | CLL (cancer predisposition) | Calin 2005; Allegra 2011 | ||
| MIR30C2 | reference allele | 5p | –32,3 | ||||||||
| n.16C>A; rs1296517452 | SNP | miR-5p (10th nt) | −29,3 | – | ↑ | – | – | Iwai 2005 | |||
| MIR96 | wild-type | 5p | −48.40 | ||||||||
| n.13G>A | germline mut | 5p seed (4th nt) | −41.30 | – | no change | ↓ | no change | NSHL (Mendelian) | Mencia 2009; Solda 2012 | ||
| n.14C>A | germline mut | 5p seed (5th nt) | −41.80 | – | – | ↓ | – | NSHL (Mendelian) | Mencia 2009 | ||
| n.57T>C | germline mut | 3p seed (5th nt) | −44.60 | – | no change | ↓ | ↓ | NSHL (Mendelian) | Solda 2012 | ||
| n.57T>C, n.23A>G | compensatory muts | 3p seed (5th nt), miR-5p (15th nt) | −49.80 | – | no change | no change | no change | Solda 2012 | |||
| MIR125A | reference allele | 5p | −46.10 | ||||||||
| n.22G>T; rs12975333 | SNP | 5p seed (7th nt) | −40.00 | ↑ | ↓ | ↓ | – | Duan 2007 | |||
| MIR140 | wild-type | 3p | −52.00 | ||||||||
| n.24A>G | germline mut | 5p seed (1st nt) | −52.00 | – | – | ↑ | ↓ | skeletal dysplasia (Mendelian) | Grigelioniene 2019 | ||
| MIR142 | wild-type | 3p | −44.30 | ||||||||
| n.55A>G | somatic mut | 3p seed (3rd nt) | −44.00 | – | no change | ↓ | ↑ | AML, other blood cancers | Trissal 2018; Marshall 2020 | ||
| n.58G>C | somatic mut | 3p seed (6th nt) | −40.40 | – | no change | ↓ | no change | AML, other blood cancers | Trissal 2018; Marshall 2020 | ||
| n.57T>C | somatic mut | 3p seed (5th nt) | −40.40 | – | – | – | – | AML, other blood cancers | Marshall 2020 | ||
| n.59T>C | somatic mut | 3p seed (7th nt) | −46.70 | – | – | – | – | AML, other blood cancers | Kwanhian 2012 | ||
| n.59T>A | somatic mut | 3p seed (7th nt) | −40.20 | – | – | – | – | AML, other blood cancers | Kwanhian 2012 | ||
| MIR146A | reference allele | 5p | −37.50 | ||||||||
| n.60C>G; rs2910164 | SNP | 3p seed (3rd nt) | −40.30 | -/no change# | ↑/no change# | ↑/↓# | – | associated with PTC and other cancers, autoimmune and infectious diseases | Jazdzewski 2008/Shen 2008; Lofgren 2012; Zhang 2014 | ||
| MIR184 | wild-type | 3p | −35.50 | ||||||||
| n.57C>T | germline mut | 3p seed (4th nt) | −38.50 | – | – | – | – | keratoconus, EDICT (Mendelian) | Hudges 2011, Iliff 2012 | ||
| n.3A>G | germline mut | 5p flank | −34.70 | – | – | – | ↓ | keratoconus (Mendelian) | Lechter 2013 | ||
| n.8C>A | germline mut | 5p flank | –33.40 | – | – | – | ↓ | keratoconus (Mendelian) | Lechter 2013 | ||
| MIR204 | wild-type | 5p | −41.00 | ||||||||
| n.37C>T | germline mut | 5p seed (4th nt) | −38.30 | – | no change | no change | – | retinal dystrophy (Mendelian) | Conte 2015 | ||
| MIR499A | reference allele | 5p | −45.40 | ||||||||
| n.73A>G; rs3746444 | SNP | 3p seed (3rd nt) | −45.00 | ↑ | no change | ↓ | no change | associated with different cardiovascular diseases and cancers | Ding 2018 | ||
| MIR502 | reference allelle | 3p | −59.00 | ||||||||
| n.13C>G; rs782404826 | SNP | 5p flank | −53.30 | – | ↓ | ↓ | ↓ | Sun, 2009 | |||
| MIR510 | reference allele | 5p | −39.20 | ||||||||
| n.6G>A; rs1458999778 | SNP | 5p flank | −39.50 | – | ↑ | ↑ | ↑ | Sun, 2009 | |||
| MIR510 | reference allele | 5p | −39.20 | ||||||||
| n.48T>C rs1041712363 | SNP | 3p seed (3rd nt) | −35.50 | – | ↓ | ↓ | ↓ | Sun, 2009 | |||
| MIR890 | reference allele | 5p | −35.60 | ||||||||
| n.66G>C; rs138166791 | SNP | miR-3p (21st nt) | –32.20 | – | ↓ | ↓ | ↓ | Sun, 2009 | |||
| MIR892B | reference allele | 3p | −42.70 | ||||||||
| n.60T>C; rs146806052 | SNP | miR-3p (15th nt) | −38.10 | – | ↓ | ↓ | ↓ | Sun, 2009 | |||
| MIR934 | reference allele | 5p | −55.00 | ||||||||
| n.15T>G; rs1196000263 | SNP | miR-5p (1st nt) | −52.90 | – | – | ↓ | ↑ | Sun, 2009 | |||
| MIRLET7I | reference allele | 5p | – | ||||||||
| rs10877887 (T>C) | SNP | promoter | – | ↓ | – | – | – | associated with different cancers and other diseases | Liu 2017 | ||
| MIRLET7A1/MIRLET7F1/MIRLET7D cluster | reference allele | 5p | – | ||||||||
| rs13293512 (T>C) | SNP | promoter | – | ↓ | – | – | – | associated with different cancers and other diseases | Sima 2015; Dong 2018 |
$ According to miRNA sequences annotated in miRGeneDB; * free energy calculated for the predicted structure adopted by sequences of pre-miRNAs with 20nt flanks, as shown in Fig. 2; # depending on the study.
Subsequent computational analysis of SNPs annotated in dbSNP revealed 12 SNPs located in 12 different pre-miRNAs (annotated in miRBase 2007). Among the identified SNPs was a G>T substitution (rs12975333; n.22G>T) in MIR125A, located at the 7th nucleotide of the miR-125a-5p seed sequence (Table 1). In addition to affecting target recognition and reducing the effectiveness of miRNA-mediated translational suppression, it was shown in in vitro experiments that the alternative allele (T) introduces an additional mismatch to the precursor duplex and blocks precursor processing from pri-miRNA to pre-miRNA [35].
Another early study investigated the effects of 24 SNPs identified in schizophrenia and autism patients in miRNA genes linked to chromosome X [36]. The SNPs were located in different subregions of the miRNA precursors. Subsequent systematic in vitro analyses demonstrated that six of the SNPs which were analyzed in details, regardless of their location in miRNA precursors, affect miRNA biogenesis. One of the SNPs detected in MIR510 (n.6G>A located in 5p flank region), resulted in an increased level of the mature miRNA, four SNPs, i.e., in MIR502 (n.13C>G located in 5p flank region), MIR510 (n.48T>C located in 3rd nt in 3p seed sequence), MIR890 (n.66G>C in 21st nt in 3p strand), and MIR892B (n.60T>C in 15th nt in 3p strand) resulted in a decreased level of the mature miRNAs, and one SNP in MIR934 (n.15T>G in 1st nt in 5p strand) resulted in altered DROSHA/DICER1 cleavage sites and the generation of novel miRNA (isomiR) (summarized in Table 1).
The abovementioned studies were the first to demonstrate the effects of genetic variants on miRNA biogenesis and the function of miRNA genes. However, as an increasing number of genetic variants in miRNA genes are detected, e.g., the latest genome-wide study identified 1994 SNPs (identified in the 1000 Genomes Project samples) in 1025 (out of 1904 annotated in miRBase) miRNA precursors [33], future functional analyses of such variants are to be expected.
4. SNPs in miRNA genes associated with various diseases
Due to the function of miRNAs in the regulation of many biological processes and role in human diseases, numerous studies have been conducted that investigate the association between SNPs in miRNA genes and various human phenotypes and diseases, and numerous such associations have been reported [21], [37], [38]. However, it must be noted that most of these associations have been identified in small studies, were not replicated in independent cohorts, and are not significant at the genome-wide level; therefore, the results of such association studies should be interpreted with caution. Below, we describe several of the most convincing or most intensively investigated examples.
4.1. MIRLET7 family promoters
Let-7 was one of the first discovered microRNAs. It was originally detected in Caenorhabditis elegans [39] and was also found in metazoans [40]. In the human genome, there are 12 LET7 genes encoding 9 different mature miRNAs, which are detected at high levels during embryogenesis and in most tissues in adults [2], [41]. Let-7 family members play an important role during development and differentiation and act as tumor suppressors (commonly downregulated) in many types of cancers. Their functions have been discussed in many articles, including [41], [42].
There are two common SNPs in the promoters of the MIRLET7 genes. These SNPs are rs10877887 T>C in the promoter of MIRLET7I, with a minor allele frequency (MAF) of ∼ 40 %, and rs13293512 T>C in the promoter of the MIRLET7A1/MIRLET7F1/MIRLET7D cluster, with an MAF of 27 % [43], in which the minor alleles was suggested to decrease level of the corresponding miRNAs [44], [45], [46]. In silico analysis suggested that these SNPs may affect the affinity or binding sites for the predicted transcription factors myeloid zinc finger 1 (MZF1) and interferon regulatory factor 1 (IRF1) for rs10877887 and rs13293512, respectively. Moreover, rs10877887 is located in a CpG island, and thus, it may lead to the downregulation of MIRLET7I expression by epigenetic mechanisms [47]. Subsequent association studies showed that these SNPs are associated with the risk of numerous diseases, including different types of cancer. Both rs10877887 and rs13293512 were associated with a risk of intracranial aneurysm (IA) [45] and major depressive disorder (MDD) [48]. Moreover, the C allele of rs10877887 was associated with an increased risk of death in patients with hepatocellular carcinoma (HCC) [47] and an increased risk of cervical squamous cell carcinoma (CSCC) [46], whereas the C allele of rs13293512 was associated with a higher risk of breast cancer [49] and lower risk of ischemic stroke [50].
4.2. MIR146A
Most likely, the most intensively studied SNP in miRNA genes is rs2910164 (n.60C>G) in MIR146A. The SNP is located in the 3rd nucleotide of the miR-146a-3p seed sequence, and its frequency is highly variable among human populations. The alternative allele (G) frequency ranges from ∼ 75 % in European populations and ∼ 60 % in African populations to < 50 % in East-Asian populations [43]. miR-146-5p is commonly expressed in immune system cells [51] and in most tissues, including the thyroid [2], [52], [53], and its level is altered in many diseases and many types of cancers [54], [55].
The first association study of rs2910164 reported that the heterozygous (C/G) genotype was associated with a higher risk of papillary thyroid carcinoma (PTC; odds ratio (OR) = 1.6) [56]. In the same study, it was shown that the C allele reduces (by almost 2 times) the levels of pre-miR-146a and mature miR-146a-5p and consequently results in a less efficient downregulation of miR-146a-5p targets, including TRAF6, IRAK1, and PTC1 [56]. However, the effect of this SNP on the miRNA level was not consistent in other studies [57], [58], [59].
Subsequent studies showed that miR-146a-3p (considered before as a passenger strand) is also an active miRNA and that the SNP located in its seed sequence increases the number of miR-146a-3p targets, including many targets specific either to the G or C allele [60]. These findings suggest a gain-of-function effect of this SNP. The later mechanistic analysis of the SNP effect on miRNA biogenesis demonstrated that the C allele increases reversed positioning of the pri-miR-146a stem-loop in the microprocessor (DROSHA/DGCR8) complex, resulting in the generation of an improper (nonfunctioning) pre-miR-146a precursor [61]. This most likely results from structural changes (additional mismatch in the stem-loop) and the destabilization of the miRNA precursor structure induced by the C allele. It should be noted that not all further results were consistent with the observed association between rs2910164 and PTC; however, the discrepancies may result from different ethnicities of tested populations and different genetic models tested (not considering the heterozygote advantage/disadvantage model) (summarized in [62]).
Later studies of rs2910164 showed the association between the C allele (in the dominant model) and an increased risk of lung cancer [63], [64], [65], earlier onset of familial breast and ovarian cancers [57], and increased risk of sporadic breast cancer [66]. However, the C allele was also associated with a lower risk of cervical, bladder, liver, gastric, and oral cancer [65], [67] and a lower risk of death in patients with advanced colorectal cancer [68], but, these results were not always conclusive or confirmed in independent studies [69], [70].
The C allele of rs2910164 was also associated with other diseases, including higher susceptibility (risk) to multiple sclerosis in women [71] and leprosy [72] and lower risk of ankylosing spondylitis [73] and psoriasis [58].
4.3. MIR499A
Another SNP in the miRNA gene commonly associated with different diseases is rs3746444 (n.73A>G) in MIR499A, with an MAF in the global population of ∼ 20 % [43]. This SNP is located in the 3rd nucleotide of the miR-499a-3p seed sequence.
The main miRNA generated from MIR499A is miR-499a-5p. It plays a role in the maturation of cardiomyocytes and cardiac differentiation. It targets mostly genes involved in early cardiogenesis, and its level increases during this process [74], [75]. In a normal heart, it exerts antiapoptotic activity, and its level is often decreased in pathological conditions in the heart [76], [77]. On the other hand, it was reported that miR-499a-5p has proapoptotic and antiproliferative activity in cancer cells and is considered as a tumor suppressor [78], [79].
The G allele replaces the Watson-Crick base pair A:U with the wobble base pair G:U in the miRNA duplex and slightly destabilizes the structure of the miRNA precursor. In cells transfected with both allele variants, in cells with the G vs A allele, the level of the pri-miRNA was increased, pre-miRNA and miR-499a-3p levels did not change, and miR-499a-5p levels were significantly decreased [80]. These findings indicate that the main effect of the G allele (although located in the 3p seed sequence) is a decrease of miR-499a-5p, which is the more important functional strand [78], [80].
The G allele of rs3746444 was associated with increased susceptibility to inflammatory arthritis [81], a higher risk of asthma [82], diabetic polyneuropathy, cardiovascular autonomic neuropathy [83], and cardiovascular diseases but not with congenital heart disease and cerebrovascular diseases [84]. It was also associated with various cancer types, and the meta-analysis of data from 65 cancer association studies showed an association between the G allele and the increased global risk of cancer as well as the risk of individual cancers, such as cervical squamous cell carcinoma, hepatocellular carcinoma, lung cancer, and prostate cancer, but was not associated with AML, breast, bladder, colorectal, esophageal, gastric, and oral squamous cell cancers [85]. It must be noted, however, that most of the individual studies included in the meta-analysis were of a very low statistical power; therefore, these results should be interpreted with caution.
5. Mutations in miRNA genes in Mendelian and familial diseases
There are hundreds of well-characterized Mendelian disorders. Almost all of these diseases are caused by mutations in protein-coding genes. However, for the last several years, an increasing number of such diseases caused by mutations in genes coding for non-coding RNAs, including microRNA-coding genes, have been reported. The summary of basic characterization of all mutations described below is shown in Table 1.
5.1. MIR96 and non-syndromic hearing loss
The first discovered Mendelian disease attributed to mutations in a miRNA gene was non-syndromic hearing loss (NSHL) [86], a heterogeneous hereditary disease with an overall prevalence ranging from approximately 1 per 1000 in newborns to greater than 3 per 1000 in adolescents [87]. More than 120 genes/loci are attributed to this disease [88]. One of the NSHL loci, DFNA50, was mapped on 7q32; however, despite sequencing of many candidate protein-coding genes in the region, no causative mutation has been found [89]. Only a detailed genetic analysis of MIR96, also present in the locus, allowed the detection of two mutations segregating with the disease in two large Spanish families and demonstrated the dominant character of the mutations [86]. MIR96 is expressed in the cochlear in inner and outer hair cells and plays a role during their differentiation [90], [91]. It is also important in auditory hindbrain development [92].
The mutations found in the patients with NSHL are located at the 4th (n.13G>A) and 5th (n.14C>A) nucleotides of the seed sequence of the 5p mature miRNA (miR-96-5p). As both mutations were located in the seed sequence, it was assumed that the main effect of the mutations was the disruption of miRNA targets recognition. This effect of mutations was confirmed by the use of a luciferase reporter assay that showed the reduced silencing of the predicted wild-type miRNA targets by the mutant miRNAs. It was also shown that these two mutations result in mismatches (internal single nucleotide loops) in the miRNA duplex, which destabilize the precursor structure (Fig. 2A) and lead to a decrease in the level of miR-96-5p [86]. However, another mutation identified later in the Italian NSHL family was found in the opposite strand in the 6th nucleotide of the 3p seed region (n.57T>C). This mutation introduced a mismatch to the miRNA duplex, enlarging the existing internal loop and destabilizing the precursor structure (Fig. 2A). Subsequent functional analyses showed that the mutation result in decreased levels of both miR-96-5p and miR-96-3p but not the level of pre-miR-96. This finding implies that mutation-induced structural aberrations affect the precursor cleavage by DICER1. To further investigate the effect of the mutation on the structure of the miRNA precursor, in the opposite strand of the mutation, the additional compensating mutation (n.23A>G) was introduced, restoring the Watson-Crick pairing with the mutation residue and the original structure of the precursor (Fig. 2A). The double mutant showed similar levels of miR-96-5p and miR-96-3p and a similar level of miR-96-5p targets downregulation as that observed in the wild-type [93]. As MIR96, along with MIR182 and MIR183, form one polycistronic cluster, mutations in NSHL patients were also analyzed in these two genes; however, no additional mutations were found in MIR182 or MIR183 [86], [93].
Fig. 2.

The examples of the effect of the mutations on the predicted 2D and 3D miRNA precursor structures. A) The effect of the mutations in MIR96 causing NSHL. The modeled RNA sequences encompassed pre-miR-96 (based on miRGeneDB) together with 20-nucleotide-long upstream and downstream flanking sequences and the corresponding versions with the indicated mutations. In 2D structures, miRNA strands and seeds are indicated in blue and dark blue, respectively; mutations are indicated in red; the start of the reference miRBase sequence is indicated by a green arrowhead, and the entire miRBase sequence is indicated in bold. 2D structures were modeled with the use of mfold [160] and VARNA [161] software with default settings. 3D structures were generated in RNAComposer [162] using default settings and visualized in PyMOL. The structure of the wild-type precursor is indicated in black, and the mutants are indicated in green. The wild-type and mutant structures were superimposed using PyMOL. The effect of particular mutations on the level of the precursor, 5p, and 3p strands (based on the data from indicated references) is shown on the right, next to the structures. From the top are the following structures: wild-type, n.13G>A and n.14C>A mutations identified in the Spanish families [86], and the n.57T>C mutation identified in the Italian family, and n.57T>C together with the n.23A>G compensating mutation [93]. B) The effect of the selected somatic mutations in the MIR142 in blood cancers. From the top: wild type, n.58G>C, n.59T>C, and n.59T>A [150], [152]. The color and setup scheme as in A. (For interpretation of the references to color in this figure legend, the reader is referred to the web version of this article.)
The role of the MIR96 loss-of-function mutations in NSHL is consistent with the progressive hearing loss observed in the diminuendo mice with the induced miR-96-5p seed mutation, which resulted in the upregulation of several of the wild-type targets in the mutated mice. However, a parallel gain-of-function effect is also possible, as several genes in the mutated mice were downregulated in comparison to those in the wild-type animals [94]. Moreover, in heterozygous MIR96/MIR183 knockout mice, no serious alterations in hearing were observed (homozygotes were deaf), while in heterozygous diminuendo mice (with the seed mutation), hearing loss was observed. These findings further suggest a gain-of-function effect of this seed mutation [95].
5.2. MIR184 and eye diseases
Another miRNA gene mutation has been identified in MIR184 at the 4th nucleotide of the miR-184-3p seed sequence (n.57C>T) in two eye diseases. This mutation has been identified in a large Irish family affected by an autosomal-dominant form of severe keratoconus with early-onset cataract [96] and in a family with endothelial dystrophy, iris hypoplasia, congenital cataract, and stromal thinning (EDICT) syndrome, in which it clearly segregated with the disease phenotype [97]. Keratoconus is a heterogeneous disease characterized by progressive thinning and steeping of the cornea, with a prevalence widely ranging from 0.2 to 5000 per 100 000 people, depending on the diagnostic criteria or the ethnicity of the tested group [98]. Consistent with the pathogenesis of the diseases, miR-184-3p is the most abundant miRNA in the cornea, and it is expressed predominantly in the basal and suprabasal cells of the corneal epithelium and lens [99], [100]. It plays a role in the neovascularization of the cornea [101] and the proliferation and differentiation of epidermal cells [102]. Previous studies have shown that the crucial function of miR-184-3p is competition for the binding sites with miR-205-5p and disabling the miR-205-5p to downregulate two of its targets, INPPL1 and ITGB4, playing a role in the pathogenesis of eye diseases, i.e., in the proper functioning of corneal basal epithelial hemidesmosomes and in keratocytes apoptosis, respectively [96], [103]. This mutation stabilizes the structure of the miRNA precursor by reducing the internal loop size in miRNA duplex due to A:U base pairing instead of A:C mismatch. Functional in vitro experiments showed that the mutant miR-184-3p fails to block the miR-205-5p binding sites in its targets, thus enabling the downregulation of INPPL1 and ITGB4 by miR-205-5p [96]. Regardless of affecting the competition with miR-205-5p, the n.57C>T mutation may also affect the recognition/downregulation of the genuine miR-184-3p targets. For example, it was demonstrated in mouse and human keratinocytes that the mutant miR-184-3p failed to repress K15 and FIH1 and to induce Notch activation [102], demonstrating another loss-of-function effect of this mutation.
Another two mutations, n.3A>G and n.8C>A, in MIR184 were found in Australian and British families with keratoconus. Both mutations are located in the 5p flank region and result in a lowered level of miR-184-3p – by approximately 40 % for n.3A>G and almost complete reduction for n.8C>A. Both n.3A>G and n.8C>A mutations lead to the destabilization of the miRNA precursor structure, respectively, by changing the A:U to the wobble base pair G:U, and by introducing a mismatch in the miRNA duplex that enlarges the internal loop near the DROSHA cleavage site, and this may lead to a complete block of miRNA processing [104].
5.3. MIR204 and retinal dystrophy
Another ocular disease caused by a mutation in a miRNA gene is inherited retinal dystrophy (IRD). Linkage analysis in a large multigeneration British family and the subsequent sequencing of affected individuals revealed a mutation in MIR204 in the 4th position of the miR-204-5p seed sequence (n.37C>T). The mutation perfectly segregated within the family with this disease in an autosomal dominant manner [105]. IRD is a large group of heterogeneous disorders that manifest as the dysfunction or degeneration of photoreceptors, which leads to visual impairment or blindness [106]. It was shown in mice that miR-204-5p is detected at a higher level in the retinal pigment epithelium (RPE) and the ciliary body and at a lower level in the inner nuclear layer of the retina and in the choroid plexus in the brain [107]. miR-204-5p plays a crucial role in the development and proper function of the retina [108] and potentially has a neuroprotective effect on photoreceptor cells [109].
This mutation has a destabilizing effect on the miR-204 precursor structure. It replaces the Watson-Crick pair C:G with the wobble pair U:G in the miRNA duplex and does not cause any other structural aberrations. The in vitro experiments showed that this mutation does not affect the level of the precursor and miR-204-5p, which suggests that it does not affect miRNA biogenesis. However, in silico predictions revealed a much higher number of potential targets for the mutant than for the wild-type miR-204-5p. The RNA-seq analysis of cells transfected with plasmids coding for the wild-type or the mutated pre-miR-204 showed that the mutation both affects the downregulation of the genuine targets of the wild-type miR-204-5p and downregulates predicted targets of the mutant miR-204-5p. Subsequent injection of mutant miR-204-5p into medaka fish (Oryzias latipes) induced phenotypes consistent with the disease, including photoreceptor alterations with reduced numbers of both cones and rods as a result of increased apoptosis. These findings suggest the predominant gain-of-function effect of the mutation, although causative mutation-specific targets have not been indicated [105].
5.4. MIR140 and skeletal dysplasia
Skeletal dysplasia is a very heterogeneous group of diseases containing more than 400 different disorders [110], which affect mostly cartilage and bones [111]. The prevalence of these diseases at birth is approximately 1–1.6:5 000 [112], [113]. Skeletal dysplasia is an inherited genetic disease caused by mutations in numerous genes [110], including a recently identified mutation in MIR140 [114]. miR-140-3p is the most abundant microRNA in cartilage, and its level is approximately tenfold higher than miR-140-5p in mouse cartilage [115], [116], but both strands are functional. miR-140-3p regulates cartilage development and homeostasis. In knockout mice, a mild skeletal phenotype of the disease is observed [117].
The extensive sequence analysis of two families with skeletal dysplasia revealed the same heterozygous mutation, n.24A>G, in MIR140. In both families, the mutation occurred de novo, and in one family, it segregated with the disease (in the other family, it was not informative as the affected proband did not have offspring) [114]. This mutation is located at the 1st position of the miR-140-5p seed sequence. Mice with the mutation introduced by CRISPR/Cas technology (homozygotes and heterozygotes) developed several skeletal abnormalities consistent with the skeletal dysplasia symptoms in the analyzed patients. The level of miR-140-5p slightly increased in mice with the mutation in comparison to that in the wild-type mice, and the level of miR-140-3p decreased. The RNA-seq analysis revealed that genuine targets of the wild-type miR-140-5p were not efficiently silenced by the mutated miR-140-5p, whereas many new, predicted mutation-specific targets were effectively downregulated. Targets for the mutant miR-140-5p include many genes important for skeletal development and homeostasis, including Loxl3, Btg1, and Trps, as well as genes involved in the hypoxia response (e.g., Hif1a) or extracellular matrix genes (e.g., Col10a1). These data suggest that the n.24A>G mutation in MIR140 is both a loss- and gain-of-function mutation [114].
5.5. MIR17HG and Feingold syndrome type 2
Feingold syndrome is an autosomal dominant disease, manifested by microcephaly, learning disabilities, short stature, and brachymesophalangy, usually associated with mutations in the MYCN locus. The whole-genome comparative genome hybridization (CGA) analysis of 10 index patients with skeletal abnormalities consistent with Feingold syndrome but without mutation in MYCN revealed two different microdeletions (2.98 Mb and 165 kb long) in the 13q31-q32 region. Both deletions segregated with the disease in the patients’ families and the minimal overlapping region of the deletions encompassed MIR17HG [118], the gene coding miR-17–92 cluster known as OncomiR-1 due to its involvement in carcinogenesis and its oncogenic role in many types of cancers. In normal cells, miR-17–92 cluster is involved in lung, heart, skeletal, and immune system development [119]. Later, similar microdeletions from 165 kb to 17 Mb in 13q31-q32 were found in several other ethnically diverse patients/families with similar conditions, consistently named Feingold syndrome type 2 (FGLDS2) [120], [121]. Detailed functional studies showed that the deletions lead to a haploinsufficiency of MIR17HG and that no other gene is involved in the disease [118], [121].
On the other hand, microduplications in 13q31, overlapping MIR17HG were found in several patients with symptoms such as tall stature, macrocephaly, developmental delay, and skeletal abnormalities [122].
6. Cancer somatic mutations in miRNA genes
6.1. MIR15A and MIR16-1 and chronic lymphocytic leukemia
Chronic lymphocytic leukemia (CLL) is one of the most common types of leukemia, with a median diagnosis age of 70 years [123] and an incidence of 4.7 per 100 000 people per year in the USA [124]. Somatic deletions overlapping the 13q14 region was found in more than 50 % of patients with CLL [125], [126]. Further analysis showed that MIR15A and MIR16-1, encoded at that locus, are either deleted or otherwise downregulated in 68 % of CLL patients [127]. However, the levels of both miR-15a-5p and miR-16–1-5p are very high in normal CD5+ lymphocytes [128], indicating an important role of the miRNAs in the functioning of CD5+ B cells. It was later shown that independent of the deletion, the miRNAs can also be downregulated by other mechanisms, including silencing due to the activity of histone deacetylases [129]. The deletion of this region was also observed in other types of cancers, such as mantle cell lymphoma (MCL) [126], multiple myeloma, and prostate cancer [130].
Subsequent in vitro and in vivo functional studies confirmed the tumor suppressor character of these miRNAs, among others demonstrating their proapoptotic properties manifested by targeting and downregulating a key antiapoptotic gene, BCL2, which is frequently increased in many cancers, including CLL. Consequently, the somatic deletion of the MIR15A/MIR16-1 cluster is the main mechanism of BCL2 upregulation and thus decreased apoptosis in CLL [128]. Other oncogenes that are regulated by miR-15a-5p and miR-16–1-5p are MCL1, CCND1, and WNT3A [131]. Interestingly, the analysis of CLL patients (n = 75) for point mutations in the MIR15A/MIR16-1 region revealed a germline substitution in the 3p-flank region (7 nucleotides downstream) of MIR16-1 (n.*7C>A) in two unrelated CLL patients. In both patients, somatic deletion of the other MIR15A/MIR16-1 allele (loss-of-heterozygosity) and a substantial reduction in the miR-16-1-5p level were also observed. Subsequent functional analyses showed that the mutation results in an increased level of the primary precursor of miR-15a/miR-16-1, and decreased levels of pre-miR-15a and pre-miR-16-1 as well as both mature miRNAs [132]. These findings suggest that the mutation affects miRNA biogenesis at the DROSHA processing step [133].
The identification of MIR15A/MIR16-1 in a region frequently deleted in CLL prompted further analyses of somatic copy number alterations of miRNA genes in cancer. First, it was shown that miRNA genes are overrepresented in fragile sites and other regions frequently altered in cancer, which suggests that miRNA genes may be frequently affected by copy-number alterations in cancer [134]. Subsequently, it was demonstrated that many miRNA genes undergo very high and frequent somatic copy number alterations, either deletions or amplifications, in different cancer types [135], [136], [137], suggesting a more global role of somatic copy number variation in the regulation of miRNAs in cancer.
6.2. MIR142 and blood cancers
The recent pan-cancer analysis, based on an analysis of data of greater than 10 000 cancer samples generated within The Cancer Genome Atlas (TCGA) [138] and Pan-cancer Analysis of Whole Genome (PCAWG) [139] projects, revealed that the most frequently mutated miRNA gene in all cancer types is MIR142 [with the highest frequency in diffuse large B-cell lymphoma (DLBCL)] [140]. Altogether, over 50 somatic mutations in MIR142 have been identified in different studies thus far [summarized in [140], [141]]. Although the mutations are dispersed over the entire pre-miR-142 sequence (including the directly adjacent ∼ 20-nucleotide-long flanking sequences), the mutations are enriched in the miR-142-3p seed sequence. Furthermore, the mutations in the 4th and 7th nucleotides (n.56G>N and n.59T>N; N – different substitutions) of the miR-142-3p seed sequence are the most frequent mutations. The distribution of the somatic mutations over the entire pre-miR-142 sequence and heterogeneity of the seed mutations (different positions of the 3p seed are mutated with different substitutions) strongly suggest the loss-of-function character of the mutations and tumor suppressor character of MIR142. Consistent with the function of miR-142-3p (and to a lesser extent miR-142-5p) in hematopoiesis, immune homeostasis, and the immune response that is exhibited mostly in the blood [142] and the high level of these miRNAs in peripheral blood cells [143], somatic mutations in MIR142 occur predominantly (almost exclusively) in hematologic malignancies, including acute myeloid leukemia (AML; 0.4-3.5 % of samples) [140], [144], [145], chronic lymphocytic leukemia (CLL; 0.5–1.1 %) [141], [146], follicular lymphoma (FL; 9.5–25 %) [147], [148], [149], and DLBCL (12–27 %) [140], [147], [148], [150]. However, single somatic mutations have also been detected in solid tumors, such as breast cancer, endometrial cancer, bladder cancer and glioblastoma [140].
It was demonstrated in different functional analyses that both miR-142-5p and 3p regulate many important hematologic targets (summarized in [141]), including Rac Family Small GTPase 1 (RAC1), TNFRSF13C encoding B-cell activating factor receptor (BAFF-R), interleukin 6 (IL6), and CD274 encoding programmed death-ligand 1 (PD-L1), an important immune checkpoint molecule. In knockout mice, aberrant development of marginal zone B cells, peripheral B1 B cells and T cells, dendritic cells, immunodeficiency, and impaired erythropoiesis have been found [151], [152], [153].
Despite the detection of mutations in different parts of MIR142, functional tests have been performed only for a few mutations located in the 3p seed sequence. As expected, the different seed mutations had slightly different effects on miRNA biogenesis and levels [150], [152], [154] and as shown in the structural modeling performed by us, the different seed mutations have a different effect on the structure of miRNA precursor (Table 1, Fig. 2B). For example, in vitro overexpression experiments and analysis of patient samples showed that mutations n.55A>G and n.58G>C, located in the 3rd and 6th positions of the miR-142-3p seed, do not affect the level of pre-miR-142, have little or no effect on the level of miR-142-3p, and substantially reduce the level of miR-142-5p [152]. The decrease in the miR-142-5p level most likely results from the destabilization of the miRNA duplex at the 3p side of miR-142-5p and preferential loading of miR-142-3p into the RISC. Regardless of the effect of the mutations on the miRNA level, both n.55A>G and n.58G>C, as well as n.59T>C and n.59T>A located at the 7th nucleotide of the seed sequence, disabled the recognition and downregulation of the well-recognized targets of miR-142-3p, RAC1, TGFBR1, ADCY9, and ASHL1 [150], [152], confirming the loss-of-function character of the mutations. Subsequently, it was demonstrated in mouse models that inefficient downregulation of ASHL1 by the mutant miRNA results in increased levels of HOXA9 and A10 (positively regulated by ASHL1) and aberration in hematopoietic differentiation. This enhances myeloid differentiation and suppresses lymphoid lineages, ultimately leading to leukemic transformation and AML [152]. Independently, it was noticed, in a few AML patients, that MIR142 mutations co-occur with the hotspot mutations in IDH2 (R140), and it was demonstrated that the effects of MIR142 and IDH2 mutations synergize, leading to AML transformation [154].
It was also demonstrated that the novel miR-142-3p, resulting from the n.59T>C mutation may recognize and downregulate new mutation-specific targets, i.e., ZEB1 and ZEB2, playing a role in tumorigenesis. Thus, some of the mutations may also act as gain-of-function alterations [150].
6.3. Global analysis of cancer somatic mutations in miRNA genes
Thanks to the development of new sequencing technologies enabling whole-genome sequencing studies of large sets of cancer samples [155], [156], many more cancer-driving mutations in non-coding regions, including miRNA genes, may emerge in the near future. The abovementioned pan-cancer analysis cataloged and characterized 7110 somatic mutations in 1179 miRNA genes in TCGA samples and 1523 somatic mutations in 856 miRNA genes in PCAWG samples [140]. The mutations were almost equally distributed in all parts of the miRNA precursors, including the miRNA duplex (both within and outside of seed sequences), DICER1/DROSHA cleavage sites, apical loop, and 25 nucleotides long upstream and downstream flanking sequences. It was demonstrated, with the use of different computational analyses, that many of the mutations may affect miRNA precursor structure and stability or distract existing protein binding motifs and that the seed mutations severely affect the predicted miRNA targets [17], [140]. Although most of these mutations are most likely randomly occurring neutral variants, some of the miRNA genes were shown to be significantly overmutated across cancers or specific cancer types, and some were identified as potential cancer driver genes. However, it must be noted that future studies are needed to prove the functional potential of the identified mutations. Among these interesting examples (except the abovementioned MIR142) are (i) MIR205 predominantly mutated in melanoma, with a total of 17 mutations in pan-cancer, including hotspot mutation (n.35C>T) at the 1st nucleotide of miR-205-5p seed sequence, (ii) MIRLET7D with hotspot indels mutations in the upstream flanking sequence, (iii) MIR487B with 16 mutations and the hotspot mutation (n.54C>T) at the 3rd nucleotide of the miR-487b-3p seed sequence occurring in melanoma, and (iv) MIR122 with two mutations in the upstream 5p flanking sequence that were identified in liver cancer. All miRNAs encoded by these genes are well recognized as playing important roles in cancer. MIR122 has also been detected as an overmutated potential cancer driver in liver carcinoma in others genome wide studies utilizing different cancer driver identification strategies [156], [157]. It is worth mentioning that miR-122-5p is a highly liver-specific miRNA that plays many important regulatory roles in liver cells and is involved in the development of hepatocellular carcinoma [158].
7. Summary and outlook
Genetic variants located in miRNA genes lead to a wide range of changes in miRNA precursor structure and function by affecting the processing of the precursors (miRNA biogenesis) and the level of mature miRNAs, as well as by altering target recognition. To date, numerous SNPs associated with different diseases and disease-related conditions have been reported [21], [37], [38]. Moreover, mutations in miRNA genes, i.e., MIR96, MIR184, MIR204, and MIR140 and microdeletions of MIR17HG that have been implicated in hereditary disorders, and cancer germline and somatic mutations in MIR15A/MIR16-1 and MIR142 have been convincingly documented, strongly suggesting their disease- or cancer-driving potential. The examples cited in this article show that mutations in miRNA genes can affect miRNA genes in different ways, and while most mutations have a loss-of-function effect, some may also act as gain-of-function mutations.
We believe that the presented examples may help to interpret the consequences of these mutations and to better predict the effects of genetic variants that are identified in miRNA genes. However, we also have to admit that the available data are still very limited. Additionally, the ability to predict the effect of mutations in miRNA genes is far from that in protein-coding sequences, where the genetic code greatly helps to distinguish deleterious/functional mutations from those of neutral or of uncertain character. The most important limitations of analyses of mutations in miRNA genes are briefly discussed below.
First, only a small fraction of variants identified in miRNA genes are functionally tested or otherwise characterized. For example, hundreds of SNPs [33] or thousands of cancer somatic mutations [17], [140] have been identified in miRNA genes; however, only a few of them have been tested in any way. Second, functional testing is strongly biased toward analyses of mutations in seed sequences, which are considered to be the most crucial part of miRNA genes. Seed mutations have the most direct impact on target recognition/silencing, which is easier to demonstrate than the effects of mutations in other parts of miRNA genes. This ascertainment bias is also reflected in the examples presented in our study. However, seed mutations account for a small fraction of genetic variants in miRNA genes, i.e., ∼4% of SNPs [33] and ∼ 10 % of cancer somatic mutations [17], [140] identified in miRNA genes, and the higher functionality of seed sequences is not reflected in a higher conservation or substantially lower SNP density in seed sequences [33] compared to other parts of the miRNA hairpin. Third, most functional testing of miRNA gene mutations is performed with the use of simple in vitro tests based on very strong overexpression of artificial truncated sequences of miRNA genes and miRNA targets. Such analyses, although useful in primary screening, often may generate false-positive results. The effects of miRNA gene mutations are rarely tested in a natural sequence context and/or in patient samples. At least a partial solution for this limitation may be the generation of specific mutations with the use of CRISPR/Cas-based technologies. Fourth, there is still a lack of systematic functional analyses of sequence variants in miRNA genes. The only such study that analyze the effect of ∼ 20 SNPs was performed over 10 years ago in pre-next generation sequencing era [36]. Fifth, most of the association studies of SNPs in miRNA genes are strongly underpowered (performed in small panels of samples) and rarely replicated in independent studies. They rarely meet the high standards of genetic association studies. Last, there are many hereditary and sporadic genetic diseases in which the exact etiology and causative variants are not known [159]; however, mutations in the non-protein-coding regions, including miRNA genes, are still considered a last resort.
Despite all limitations listed above, we believe that the recent extension of genetic analyses toward the whole genome, instead of concentrating on protein-coding sequences, will allow identification and a better understanding of genetic variants in miRNA genes as well as in other non-protein-coding parts of the genome. Of course, such a shift, except for the increased sequencing power, will require systematic functional and genetic analyses of a much wider number of mutations and the development of new molecular, computational and statistical methods that allow much more robust and high-throughput characterization of variants in miRNA genes, convincingly distinguishing functional deleterious mutations from neutral variants.
CRediT authorship contribution statement
Magdalena Machowska: Conceptualization, Visualization, Resources, Writing - original draft, Writing - review & editing. Paulina Galka-Marciniak: Conceptualization, Funding acquisition, Writing - review & editing. Piotr Kozlowski: Conceptualization, Supervision, Funding acquisition, Writing - original draft, Writing - review & editing.
Declaration of Competing Interest
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
Acknowledgments
This work was supported by research grants from the National Science Centre Poland: 2016/22/A/NZ2/00184 and 2020/39/B/NZ5/01970 (to P.K.), and 2020/39/D/NZ2/03106 (to P.G-M.)
Contributor Information
Magdalena Machowska, Email: mmachowska@ibch.poznan.pl.
Paulina Galka-Marciniak, Email: pgalka@ibch.poznan.pl.
Piotr Kozlowski, Email: kozlowp@ibch.poznan.pl.
References
- 1.Kozomara A., Birgaoanu M., Griffiths-Jones S. miRBase: from microRNA sequences to function. Nucleic Acids Res. 2019;47:D155–D162. doi: 10.1093/nar/gky1141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Fromm B., Domanska D., Høye E., Ovchinnikov V., Kang W., Aparicio-Puerta E., et al. MirGeneDB 2.0: the metazoan microRNA complement. Nucleic Acids Res. 2020;48:D132–D141. doi: 10.1093/nar/gkz885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Fromm B., Høye E., Domanska D., Zhong X., Aparicio-Puerta E., Ovchinnikov V., et al. MirGeneDB 2.1: Toward a complete sampling of all major animal phyla. Nucleic Acids Res. 2022;50:D204–D210. doi: 10.1093/nar/gkab1101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Liu B., Shyr Y., Cai J., Liu Q. Interplay between miRNAs and host genes and their role in cancer. Brief Funct Genomics. 2019;18:255–266. doi: 10.1093/bfgp/elz002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Bartel D.P. Metazoan MicroRNAs. Cell. 2018;173:20–51. doi: 10.1016/j.cell.2018.03.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Lin S., Gregory R.I. MicroRNA biogenesis pathways in cancer. Nat Rev Cancer. 2015;15:321–333. doi: 10.1038/nrc3932. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Leitão A.L., Enguita F.J. A structural view of miRNA biogenesis and function. Non-Coding RNA. 2022;8 doi: 10.3390/ncrna8010010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Treiber T., Treiber N., Meister G. Regulation of microRNA biogenesis and its crosstalk with other cellular pathways. Nat Rev Mol Cell Biol. 2019;20:5–20. doi: 10.1038/s41580-018-0059-1. [DOI] [PubMed] [Google Scholar]
- 9.Vidigal J.A., Ventura A. The biological functions of miRNAs: lessons from in vivo studies. Trends Cell Biol. 2015;25:137–147. doi: 10.1016/j.tcb.2014.11.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Condrat C.E., Thompson D.C., Barbu M.G., Bugnar O.L., Boboc A., Cretoiu D., et al. miRNAs as biomarkers in disease: latest findings regarding their role in diagnosis and prognosis. Cells. 2020;9:276. doi: 10.3390/cells9020276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Rice G.M., Shivashankar V., Ma E.J., Baryza J.L., Nutiu R. Functional atlas of primary miRNA maturation by the microprocessor. Mol Cell. 2020;80:892–902.e4. doi: 10.1016/j.molcel.2020.10.028. [DOI] [PubMed] [Google Scholar]
- 12.Li S., Le T.N.Y., Nguyen T.D., Trinh T.A., Nguyen T.A. Bulges control pri-miRNA processing in a position and strand-dependent manner. RNA Biol. 2021;18:1716–1726. doi: 10.1080/15476286.2020.1868139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Auyeung V.C., Ulitsky I., McGeary S.E., Bartel D.P. Beyond secondary structure: primary-sequence determinants license Pri-miRNA hairpins for processing. Cell. 2013;152:844–858. doi: 10.1016/j.cell.2013.01.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Tomasello L., Distefano R., Nigita G., Croce C.M. The MicroRNA family gets wider: the isomirs classification and role. Front Cell Dev Biol. 2021;9:1–15. doi: 10.3389/fcell.2021.668648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Starega-Roslan J., Galka-Marciniak P., Krzyzosiak W.J. Nucleotide sequence of miRNA precursor contributes to cleavage site selection by Dicer. Nucleic Acids Res. 2015;43:10939–10951. doi: 10.1093/nar/gkv968. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Galka-Marciniak P., Olejniczak M., Starega-Roslan J., Szczesniak M.W., Makalowska I., Krzyzosiak W.J. siRNA release from pri-miRNA scaffolds is controlled by the sequence and structure of RNA. Biochim Biophys Acta – Gene Regul Mech. 2016;1859:639–649. doi: 10.1016/j.bbagrm.2016.02.014. [DOI] [PubMed] [Google Scholar]
- 17.Galka-Marciniak P, Urbanek-Trzeciak MO, Nawrocka PM, Dutkiewicz A, Giefing M, Lewandowska MA, et al. Somatic mutations in miRNA genes in lung cancer—potential functional consequences of non-coding sequence variants. Cancers (Basel) 2019;11. https://doi.org/10.3390/cancers11060793. [DOI] [PMC free article] [PubMed]
- 18.Guo Z., Shu Y., Zhou H., Zhang W. Identification of diagnostic and prognostic biomarkers for cancer: Focusing on genetic variations in microRNA regulatory pathways (Review) Mol Med Rep. 2016;13:1943–1952. doi: 10.3892/mmr.2016.4782. [DOI] [PubMed] [Google Scholar]
- 19.Galka-Marciniak P., Urbanek-Trzeciak M.O., Nawrocka P.M., Kozlowski P. A pan-cancer atlas of somatic mutations in miRNA biogenesis genes. Nucleic Acids Res. 2021;49:601–620. doi: 10.1093/nar/gkaa1223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Moszyńska A., Gebert M., Collawn J.F., Bartoszewski R. SNPs in microRNA target sites and their potential role in human disease. Open Biol. 2017;7 doi: 10.1098/rsob.170019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Chhichholiya Y., Suryan A.K., Suman P., Munshi A., Singh S. SNPs in miRNAs and target sequences: role in cancer and diabetes. Front Genet. 2021;12:1–24. doi: 10.3389/fgene.2021.793523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Baer C., Claus R., Plass C. Genome-wide epigenetic regulation of miRNAs in cancer. Cancer Res. 2013;73:473–477. doi: 10.1158/0008-5472.CAN-12-3731. [DOI] [PubMed] [Google Scholar]
- 23.Pilala K.M., Papadimitriou M.A., Panoutsopoulou K., Barbarigos P., Levis P., Kotronopoulos G., et al. Epigenetic regulation of MIR145 core promoter controls miR-143/145 cluster in bladder cancer progression and treatment outcome. Mol Ther – Nucleic Acids. 2022;30:311–322. doi: 10.1016/j.omtn.2022.10.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Glaich O., Parikh S., Bell R.E., Mekahel K., Donyo M., Leader Y., et al. DNA methylation directs microRNA biogenesis in mammalian cells. Nat Commun. 2019;10:1–11. doi: 10.1038/s41467-019-13527-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Pajares M.J., Alemany-cosme E., Goñi S., Bandres E., Palanca-Ballester C., Sandoval J. Epigenetic regulation of microRNAs in cancer: shortening the distance from bench to bedside. Int J Mol Sci. 2021;22 doi: 10.3390/ijms22147350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Nishikura K. A-to-I editing of coding and non-coding RNAs by ADARs. Nat Rev Mol Cell Biol. 2016;17:83–96. doi: 10.1038/nrm.2015.4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Michlewski G., Cáceres J.F. Post-transcriptional control of miRNA biogenesis. RNA. 2019;25:1–16. doi: 10.1261/rna.068692.118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Cheray M., Etcheverry A., Jacques C., Pacaud R., Bougras-Cartron G., Aubry M., et al. Cytosine methylation of mature microRNAs inhibits their functions and is associated with poor prognosis in glioblastoma multiforme. Mol Cancer. 2020;19:36. doi: 10.1186/s12943-020-01155-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Saunders M.A., Liang H., Li W.H. Human polymorphism at microRNAs and microRNA target sites. Proc Natl Acad Sci U S A. 2007;104:3300–3305. doi: 10.1073/pnas.0611347104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Marcinkowska M., Szymanski M., Krzyzosiak W.J., Kozlowski P. Copy number variation of microRNA genes in the human genome. BMC Genomics. 2011;12:183. doi: 10.1186/1471-2164-12-183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Han M., Zheng Y. Comprehensive analysis of single nucleotide polymorphisms in human MicroRNAs. PLoS One. 2013;8 doi: 10.1371/journal.pone.0078028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Torruella-Loran I., Laayouni H., Dobon B., Gallego A., Balcells I., Garcia-Ramallo E., et al. MicroRNA genetic variation: from population analysis to functional implications of three allele variants associated with cancer. Hum Mutat. 2016;37:1060–1073. doi: 10.1002/humu.23045. [DOI] [PubMed] [Google Scholar]
- 33.Villegas-Mirón P., Gallego A., Bertranpetit J., Laayouni H., Espinosa-Parrilla Y. Signatures of genetic variation in human microRNAs point to processes of positive selection and population-specific disease risks. Hum Genet. 2022 doi: 10.1007/s00439-021-02423-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Iwai N., Naraba H. Polymorphisms in human pre-miRNAs. Biochem Biophys Res Commun. 2005;331:1439–1444. doi: 10.1016/j.bbrc.2005.04.051. [DOI] [PubMed] [Google Scholar]
- 35.Duan R., Pak C.H., Jin P. Single nucleotide polymorphism associated with mature miR-125a alters the processing of pri-miRNA. Hum Mol Genet. 2007;16:1124–1131. doi: 10.1093/hmg/ddm062. [DOI] [PubMed] [Google Scholar]
- 36.Sun G., Yan J., Noltner K., Feng J., Li H., Sarkis D.A., et al. SNPs in human miRNA genes affect biogenesis and function. RNA. 2009;15:1640–1651. doi: 10.1261/rna.1560209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Slaby O., Bienertova-Vasku J., Svoboda M., Vyzula R. Genetic polymorphisms and microRNAs: new direction in molecular epidemiology of solid cancer. J Cell Mol Med. 2012;16:8–21. doi: 10.1111/j.1582-4934.2011.01359.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Park J.H., Jeong G.H., Lee K.S., Lee K.H., Suh J.S., Eisenhut M., et al. Genetic variations in MicroRNA genes and cancer risk: a field synopsis and meta-analysis. Eur J Clin Invest. 2020;50:1–15. doi: 10.1111/eci.13203. [DOI] [PubMed] [Google Scholar]
- 39.Reinhart B.J., Slack F.J., Basson M., Pasquienelll A.E., Bettlnger J.C., Rougvle A.E., et al. The 21-nucleotide let-7 RNA regulates developmental timing in Caenorhabditis elegans. Nature. 2000;403:901–906. doi: 10.1038/35002607. [DOI] [PubMed] [Google Scholar]
- 40.Pasquinelli A.E., McCoy A., Jiménez E., Saló E., Ruvkun G., Martindale M.Q., et al. Expression of the 22 nucleotide let-7 heterochronic RNA throughout the metazoa: a role in life history evolution? Evol Dev. 2003;5:372–378. doi: 10.1046/j.1525-142X.2003.03044.x. [DOI] [PubMed] [Google Scholar]
- 41.Lee H., Han S., Kwon C.S., Lee D. Biogenesis and regulation of the let-7 miRNAs and their functional implications. Protein Cell. 2016;7:100–113. doi: 10.1007/s13238-015-0212-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Roush S., Slack F.J. The let-7 family of microRNAs. Trends Cell Biol. 2008;18:505–516. doi: 10.1016/j.tcb.2008.07.007. [DOI] [PubMed] [Google Scholar]
- 43.dbSNP. https://www.ncbi.nlm.nih.gov/snp/ 2021.
- 44.Dong H.Y., Huang Z.C., Zhang H.L., Xiao Z.G., Liu Q. Rs13293512 polymorphism located in the promoter region of let-7 is associated with increased risk of radiation enteritis in colorectal cancer. J Cell Biochem. 2018;119:6535–6544. doi: 10.1002/jcb.26733. [DOI] [PubMed] [Google Scholar]
- 45.Sima X, Sun H, Zhou P, You C. A potential polymorphism in the promoter of let-7 is associated with an increased risk of Intracranial Aneurysm: A case-control study. Med (United States) 2015;94. https://doi.org/10.1097/MD.0000000000002267. [DOI] [PMC free article] [PubMed]
- 46.Liu J., Ni S. Association between genetic polymorphisms in the promoters of let-7 and risk of cervical squamous cell carcinoma. Gene. 2018;642:256–260. doi: 10.1016/j.gene.2017.11.038. [DOI] [PubMed] [Google Scholar]
- 47.Xie K., Liu J., Zhu L., Liu Y., Pan Y., Wen J., et al. A potentially functional polymorphism in the promoter region of let-7 family is associated with survival of hepatocellular carcinoma. Cancer Epidemiol. 2013;37:998–1002. doi: 10.1016/j.canep.2013.09.005. [DOI] [PubMed] [Google Scholar]
- 48.Liang Y., Zhao G., Sun R., Mao Y., Li G., Chen X., et al. Genetic variants in the promoters of let-7 family are associated with an increased risk of major depressive disorder. J Affect Disord. 2015;183:295–299. doi: 10.1016/j.jad.2015.04.035. [DOI] [PubMed] [Google Scholar]
- 49.Sun R., Gong J., Li J., Ruan Z., Yang X., Zheng Y., et al. A genetic variant rs13293512 in the promoter of let-7 is associated with an increased risk of breast cancer in Chinese women. Biosci Rep. 2019;39 doi: 10.1042/BSR20182079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Zhang L., Yang J., Xue Q., Yang D., Lu Y., Guang X., et al. An rs13293512 polymorphism in the promoter of let-7 is associated with a reduced risk of ischemic stroke. J Thromb Thrombolysis. 2016;42:610–615. doi: 10.1007/s11239-016-1400-1. [DOI] [PubMed] [Google Scholar]
- 51.Testa U., Pelosi E., Castelli G., Labbaye C. miR-146 and miR-155: Two key modulators of immune response and tumor development. Non-Coding RNA. 2017:3. doi: 10.3390/ncrna3030022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Iacona J.R., Lutz C.S. miR-146a-5p, Expression, regulation, and functions in cancer. WIREs RNA. 2019:10. doi: 10.1002/wrna.1533. [DOI] [PubMed] [Google Scholar]
- 53.Pignatti E., Vighi E., Magnani E., Kara E., Roncati L., Maiorana A., et al. Expression and clinicopathological role of miR146a in thyroid follicular carcinoma. Endocrine. 2019;64:575–583. doi: 10.1007/s12020-019-01845-9. [DOI] [PubMed] [Google Scholar]
- 54.Volinia S., Calin G.A., Liu C.G., Ambs S., Cimmino A., Petrocca F., et al. A microRNA expression signature of human solid tumors defines cancer gene targets. Proc Natl Acad Sci U S A. 2006;103:2257–2261. doi: 10.1073/pnas.0510565103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Li L., Chen X.P., Li Y.J. MicroRNA-146a and human disease. Scand J Immunol. 2010;71:227–231. doi: 10.1111/j.1365-3083.2010.02383.x. [DOI] [PubMed] [Google Scholar]
- 56.Jazdzewski K., Murray E.L., Franssila K., Jarzab B., Schoenberg D.R., de la Chapelle A. Common SNP in pre-miR-146a decreases mature miR expression and predisposes to papillary thyroid carcinoma. Proc Natl Acad Sci. 2008;105:7269–7274. doi: 10.1073/pnas.0802682105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Shen J., Ambrosone C.B., Dicioccio R.A., Odunsi K., Lele S.B., Zhao H. A functional polymorphism in the miR-146a gene and age of familial breast/ovarian cancer diagnosis. Carcinogenesis. 2008;29:1963–1966. doi: 10.1093/carcin/bgn172. [DOI] [PubMed] [Google Scholar]
- 58.Zhang W., Yi X., Guo S., Shi Q., Wei C., Li X., et al. A single-nucleotide polymorphism of miR-146a and psoriasis: an association and functional study. J Cell Mol Med. 2014;18:2225–2234. doi: 10.1111/jcmm.12359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Löfgren S.E., Frostegård J., Truedsson L., Pons-Estel B.A., D’Alfonso S., Witte T., et al. Genetic association of miRNA-146a with systemic lupus erythematosus in Europeans through decreased expression of the gene. Genes Immun. 2012;13:268–274. doi: 10.1038/gene.2011.84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Jazdzewski K., Liyanarachchi S., Swierniak M., Pachucki J., Ringel M.D., Jarzab B., et al. Polymorphic mature microRNAs from passenger strand of pre-miR-146a contribute to thyroid cancer. Proc Natl Acad Sci U S A. 2009;106:1502–1505. doi: 10.1073/pnas.0812591106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Le C.T., Nguyen T.L., Nguyen T.D., Nguyen T.A. Human disease-associated single nucleotide polymorphism changes the orientation of DROSHA on pri-mir-146a. RNA. 2020;26:1777–1786. doi: 10.1261/RNA.077487.120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Chen H., Zhang H., Liu Y., Chen Z., Gu J., Cui D., et al. MiR-146a rs2910164 Polymorphism and risk of papillary thyroid carcinoma: a meta-analysis. Genet Test Mol Biomarkers. 2018;22:674–679. doi: 10.1089/gtmb.2018.0038. [DOI] [PubMed] [Google Scholar]
- 63.Vinci S., Gelmini S., Pratesi N., Conti S., Malentacchi F., Simi L., et al. Genetic variants in miR-146a, miR-149, miR-196a2, miR-499 and their influence on relative expression in lung cancers. Clin Chem Lab Med. 2011;49:2073–2080. doi: 10.1515/CCLM.2011.708. [DOI] [PubMed] [Google Scholar]
- 64.Sodhi K.K., Bahl C., Singh N., Behera D., Sharma S. Functional genetic variants in pre-MIR-146a and 196a2 genes are associated with risk of lung cancer in North Indians. Futur Oncol. 2015;11:2159–2173. doi: 10.2217/fon.15.143. [DOI] [PubMed] [Google Scholar]
- 65.Nikolić Z.Z., Savić Pavićević D.L., Vučic N.L., Romac S.P., Brajušković G.N. Association between a Genetic Variant in the hsa-miR-146a Gene and Cancer Risk: An Updated Meta-Analysis. Public Health Genomics. 2015;18:283–298. doi: 10.1159/000438695. [DOI] [PubMed] [Google Scholar]
- 66.Brincas H.M., Augusto D.G., Mathias C., Cavalli I.J., de Lima R.S., Kuroda F., et al. A genetic variant in microrna-146a is associated with sporadic breast cancer in a southern brazilian population. Genet Mol Biol. 2019:42. doi: 10.1590/1678-4685-GMB-2019-0278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Yue C., Wang M., Ding B., Wang W., Fu S., Zhou D., et al. Polymorphism of the pre-miR-146a is associated with risk of cervical cancer in a Chinese population. Gynecol Oncol. 2011;122:33–37. doi: 10.1016/j.ygyno.2011.03.032. [DOI] [PubMed] [Google Scholar]
- 68.dos Santos J.S., Zunta G.L., Negrini A.B., Ribeiro M.S.G., Martinez C.A.R., Ribeiro M.L., et al. The association of a single-nucleotide variant in the microRNA-146a with advanced colorectal cancer prognosis. Tumor Biol. 2020;42:1–9. doi: 10.1177/1010428320923856. [DOI] [PubMed] [Google Scholar]
- 69.Morales S., De Mayo T., Gulppi F.A., Gonzalez-Hormazabal P., Carrasco V., Reyes J.M., et al. Genetic variants in pre-miR-146a, pre-miR-499, pre-miR-125a, pre-miR-605, and pri-miR-182 are associated with breast cancer susceptibility in a south American population. Genes (Basel) 2018;9:1–18. doi: 10.3390/genes9090427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Xia Z.G., Yin H.F., Long Y., Cheng L., Yu L.J., Guo W.J., et al. Genetic variant of miR-146a rs2910164 C > G and gastric cancer susceptibility. Oncotarget. 2016;7:34316–34321. doi: 10.18632/oncotarget.8814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Li Y., Du C., Wang W., Ma G., Cui L., Zhou H., et al. Genetic association of MiR-146a with multiple sclerosis susceptibility in the chinese population. Cell Physiol Biochem. 2015;35:281–291. doi: 10.1159/000369695. [DOI] [PubMed] [Google Scholar]
- 72.Cezar-de-Mello P.F.T., Toledo-Pinto T.G., Marques C.S., Arnez L.E.A., Cardoso C.C., Guerreiro L.T.A., et al. Pre-miR-146a (rs2910164 G>C) single nucleotide polymorphism is genetically and functionally associated with leprosy. PLoS Negl Trop Dis. 2014:8. doi: 10.1371/journal.pntd.0003099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Xu H.Y., Wang Z.Y., Chen J.F., Wang T.Y., Wang L.L., Tang L.L., et al. Association between ankylosing spondylitis and the miR-146a and miR-499 polymorphisms. PLoS One. 2015;10:1–9. doi: 10.1371/journal.pone.0122055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Wilson K.D., Hu S., Venkatasubrahmanyam S., Fu J.-D., Sun N., Abilez O.J., et al. Dynamic MicroRNA expression programs during cardiac differentiation of human embryonic stem cells. Circ Cardiovasc Genet. 2010;3:426–435. doi: 10.1161/CIRCGENETICS.109.934281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Li X., Wang J., Jia Z., Cui Q., Zhang C., Wang W., et al. MiR-499 regulates cell proliferation and apoptosis during late-stage cardiac differentiation via Sox6 and cyclin D1. PLoS One. 2013;8:1–14. doi: 10.1371/journal.pone.0074504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Wang J.X., Jiao J.Q., Li Q., Long B., Wang K., Liu J.P., et al. MiR-499 regulates mitochondrial dynamics by targeting calcineurin and dynamin-related protein-1. Nat Med. 2011;17:71–78. doi: 10.1038/nm.2282. [DOI] [PubMed] [Google Scholar]
- 77.Li Y., Lu J., Bao X., Wang X., Wu J., Li X., et al. MiR-499-5p protects cardiomyocytes against ischaemic injury via anti-apoptosis by targeting PDCD4. Oncotarget. 2016;7:35607–35617. doi: 10.18632/oncotarget.9597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Liu H., Ji F., Lu Y., Chen S.Y. MiR-499b-5p inhibits cervical cancer cell proliferation and induces apoptosis by targeting the Notch1 signaling pathway. Eur Rev Med Pharmacol Sci. 2021;25:6220–6231. doi: 10.26355/eurrev_202110_26992. [DOI] [PubMed] [Google Scholar]
- 79.Wang J., Li J., Chen L., Fan Z., Cheng J. MicroRNA-499 suppresses the growth of hepatocellular carcinoma by downregulating astrocyte elevated gene-1. Technol Cancer Res Treat. 2020;19:1–9. doi: 10.1177/1533033820920253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Ding W., Li M., Sun T., Han D., Guo X., Chen X., et al. A polymorphism rs3746444 within the pre-miR-499 alters the maturation of miR-499-5p and its antiapoptotic function. J Cell Mol Med. 2018;22:5418–5428. doi: 10.1111/jcmm.13813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Wang D., Pan G. Association of rs2910164 Polymorphism in miRNA-146 and rs3746444 Polymorphism in miRNA-499 with Inflammatory Arthritis: A meta-analysis. Biomed Res Int. 2019 doi: 10.1155/2019/7305750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Toraih E.A., Hussein M.H., Al Ageeli E., Riad E., AbdAllah N.B., Helal G.M., et al. Structure and functional impact of seed region variant in MIR-499 gene family in bronchial asthma. Respir Res. 2017:18. doi: 10.1186/s12931-017-0648-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Ciccacci C., Latini A., Greco C., Politi C., D’Amato C., Lauro D., et al. Association between a MIR499A polymorphism and diabetic neuropathy in type 2 diabetes. J Diabetes Complications. 2018;32:11–17. doi: 10.1016/j.jdiacomp.2017.10.011. [DOI] [PubMed] [Google Scholar]
- 84.Bastami M., Choupani J., Saadatian Z., Vahed S.Z., Mansoori Y., Daraei A., et al. MiRNA polymorphisms and risk of cardio-cerebrovascular diseases: a systematic review and meta-analysis. Int J Mol Sci. 2019:20. doi: 10.3390/ijms20020293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Yang X., Li X., Zhou B. A meta-analysis of miR-499 rs3746444 polymorphism for cancer risk of different systems: evidence from 65 case-control studies. Front Physiol. 2018;9:1–14. doi: 10.3389/fphys.2018.00737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Mencía A., Modamio-Høybjør S., Redshaw N., Morín M., Mayo-Merino F., Olavarrieta L., et al. Mutations in the seed region of human miR-96 are responsible for nonsyndromic progressive hearing loss. Nat Genet. 2009;41:609–613. doi: 10.1038/ng.355. [DOI] [PubMed] [Google Scholar]
- 87.Morton C.C., Nance W.E. Newborn hearing screening — A silent revolution. N Engl J Med. 2006;354:2151–2164. doi: 10.1056/NEJMra050700. [DOI] [PubMed] [Google Scholar]
- 88.Vona B., Doll J., Hofrichter M.A.H., Haaf T. Non-syndromic hearing loss: clinical and diagnostic challenges. Medizinische Genet. 2020;32:117–129. doi: 10.1515/medgen-2020-2022. [DOI] [Google Scholar]
- 89.Modamio-Høybjør S., Moreno-Pelayo M.A., Mencía A., del Castillo I., Chardenoux S., Morais D., et al. A novel locus for autosomal dominant nonsyndromic hearing loss, DFNA50, maps to chromosome 7q32 between the DFNB17 and DFNB13 deafness loci. J Med Genet. 2004;41:1–5. doi: 10.1136/jmg.2003.012500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Weston M.D., Pierce M.L., Rocha-Sanchez S., Beisel K.W., Soukup G.A. MicroRNA gene expression in the mouse inner ear. Brain Res. 2006;1111:95–104. doi: 10.1016/j.brainres.2006.07.006. [DOI] [PubMed] [Google Scholar]
- 91.Kuhn S., Johnson S.L., Furness D.N., Chen J., Ingham N., Hilton J.M., et al. miR-96 regulates the progression of differentiation in mammalian cochlear inner and outer hair cells. Proc Natl Acad Sci U S A. 2011;108:2355–2360. doi: 10.1073/pnas.1016646108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Schlüter T., Berger C., Rosengauer E., Fieth P., Krohs C., Ushakov K., et al. miR-96 is required for normal development of the auditory hindbrain. Hum Mol Genet. 2018;27:860–874. doi: 10.1093/hmg/ddy007. [DOI] [PubMed] [Google Scholar]
- 93.Soldà G., Robusto M., Primignani P., Castorina P., Benzoni E., Cesarani A., et al. A novel mutation within the MIR96 gene causes non-syndromic inherited hearing loss in an Italian family by altering pre-miRNA processing. Hum Mol Genet. 2012;21:577–585. doi: 10.1093/hmg/ddr493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Lewis M.A., Quint E., Glazier A.M., Fuchs H., De Angelis M.H., Langford C., et al. An ENU-induced mutation of miR-96 associated with progressive hearing loss in mice. Nat Genet. 2009;41:614–618. doi: 10.1038/ng.369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Lewis M.A., Di Domenico F., Ingham N.J., Prosser H.M., Steel K.P. Hearing impairment due to Mir183/96/182 mutations suggests both loss-of-function and gain-of-function effects. DMM. Dis Model Mech. 2021:14. doi: 10.1242/dmm.047225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Hughes A.E., Bradley D.T., Campbell M., Lechner J., Dash D.P., Simpson D.A., et al. Mutation altering the miR-184 seed region causes familial keratoconus with cataract. Am J Hum Genet. 2011;89:628–633. doi: 10.1016/j.ajhg.2011.09.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Iliff B.W., Amer Riazuddin S., Gottsch J.D. A single-base substitution in the seed region of miR-184 causes EDICT syndrome. Investig Ophthalmol Vis Sci. 2012;53:348–353. doi: 10.1167/iovs.11-8783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Santodomingo-Rubido J., Carracedo G., Suzaki A., Villa-Collar C., Vincent S.J., Wolffsohn J.S. Keratoconus: an updated review. Contact Lens Anterior Eye. 2022;45 doi: 10.1016/j.clae.2021.101559. [DOI] [PubMed] [Google Scholar]
- 99.Ryan D.G., Oliveira-Fernandes M., Lavker R.M. MicroRNAs of the mammalian eye display distinct and overlapping tissue specificity. Mol Vis. 2006;12:1175–1184. [PubMed] [Google Scholar]
- 100.Drewry M., Helwa I., Allingham R.R., Hauser M.A., Liu Y. miRNA profile in three different normal human ocular tissues by miRNA-seq. Investig Ophthalmol Vis Sci. 2016;57:3731–3739. doi: 10.1167/iovs.16-19155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Zong R., Zhou T., Lin Z., Bao X., Xiu Y., Chen Y., et al. Down-regulation of microRNA-184 is associated with corneal neovascularization. Investig Ophthalmol Vis Sci. 2016;57:1398–1407. doi: 10.1167/iovs.15-17417. [DOI] [PubMed] [Google Scholar]
- 102.Nagosa S., Leesch F., Putin D., Bhattacharya S., Altshuler A., Serror L., et al. microRNA-184 induces a commitment switch to epidermal differentiation. Stem Cell Rep. 2017;9:1991–2004. doi: 10.1016/j.stemcr.2017.10.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Yu J., Ryan D.G., Getsios S., Oliveira-Fernandes M., Fatima A., Lavker R.M. MicroRNA-184 antagonizes microRNA-205 to maintain SHIP2 levels in epithelia. Proc Natl Acad Sci U S A. 2008;105:19300–19305. doi: 10.1073/pnas.0803992105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Lechner J., Bae H.A., Guduric-Fuchs J., Rice A., Govindarajan G., Siddiqui S., et al. Mutational analysis of MIR184 in sporadic keratoconus and myopia. Investig Ophthalmol Vis Sci. 2013;54:5266–5272. doi: 10.1167/iovs.13-12035. [DOI] [PubMed] [Google Scholar]
- 105.Conte I., Hadfield K.D., Barbato S., Carrella S., Pizzo M., Bhat R.S., et al. MiR-204 is responsible for inherited retinal dystrophy associated with ocular coloboma. Proc Natl Acad Sci U S A. 2015;112:E3236–E3245. doi: 10.1073/pnas.1401464112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Georgiou M., Fujinami K., Michaelides M. Inherited retinal diseases: therapeutics, clinical trials and end points—A review. Clin Exp Ophthalmol. 2021;49:270–288. doi: 10.1111/ceo.13917. [DOI] [PubMed] [Google Scholar]
- 107.Deo M., Yu J.Y., Chung K.H., Tippens M., Turner D.L. Detection of mammalian microRNA expression by in situ hybridization with RNA oligonucleotides. Dev Dyn. 2006;235:2538–2548. doi: 10.1002/dvdy.20847. [DOI] [PubMed] [Google Scholar]
- 108.Bereimipour A., Najafi H., Mirsane E.S., Moradi S., Satarian L. Roles of miR-204 in retinal development and maintenance. Exp Cell Res. 2021;406 doi: 10.1016/j.yexcr.2021.112737. [DOI] [PubMed] [Google Scholar]
- 109.Karali M., Guadagnino I., Marrocco E., De Cegli R., Carissimo A., Pizzo M., et al. AAV-miR-204 protects from retinal degeneration by attenuation of microglia activation and photoreceptor cell Death. Mol Ther - Nucleic Acids. 2020;19:144–156. doi: 10.1016/j.omtn.2019.11.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Warman M.L., Cormier-Daire V., Hall C., Krakow D., Lachman R., Lemerrer M., et al. Nosology and classification of genetic skeletal disorders: 2010 revision. Am J Med Genet Part A. 2011;155:943–968. doi: 10.1002/ajmg.a.33909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Krakow D. Skeletal dysplasia. Paediatr Child Heal (United Kingdom) 2020;30:209–19. https://doi.org/10.1016/j.paed.2020.03.004.
- 112.Orioli I.M., Castilla E.E., Barbosa-Neto J.G. The birth prevalence rates for the skeletal dysplasias. J Med Genet. 1986;23:328–332. doi: 10.1136/jmg.23.4.328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Barbosa-Buck C.O., Orioli I.M., da Graça D.M., Lopez-Camelo J., Castilla E.E., Cavalcanti D.P. Clinical epidemiology of skeletal dysplasias in South America. Am J Med Genet Part A. 2012:158. doi: 10.1002/ajmg.a.35246. [DOI] [PubMed] [Google Scholar]
- 114.Grigelioniene G., Suzuki H.I., Taylan F., Mirzamohammadi F., Borochowitz Z.U., Ayturk U.M., et al. Gain-of-function mutation of microRNA-140 in human skeletal dysplasia. Nat Med. 2019;25:583–590. doi: 10.1038/s41591-019-0353-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Wienholds E, Kloosterman WP, Miska E, Alvarez-Saavedra E, Berezikov E, de Bruijn E, et al. MicroRNA expression in zebrafish embryonic development. Supporting Material. Science (80-) 2005;309:310–1. https://doi.org/10.1126/science.1114519. [DOI] [PubMed]
- 116.Crowe N., Swingler T.E., Le L.T.T., Barter M.J., Wheeler G., Pais H., et al. Detecting new microRNAs in human osteoarthritic chondrocytes identifies miR-3085 as a human, chondrocyte-selective, microRNA. Osteoarthr Cartil. 2016;24:534–543. doi: 10.1016/j.joca.2015.10.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Miyaki S., Sato T., Inoue A., Otsuki S., Ito Y., Yokoyama S., et al. MicroRNA-140 plays dual roles in both cartilage development and homeostasis. Genes Dev. 2010;24:1173–1185. doi: 10.1101/gad.1915510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.de Pontual L., Yao E., Callier P., Faivre L., Drouin V., Cariou S., et al. Germline deletion of the miR-17∼92 cluster causes skeletal and growth defects in humans. Nat Genet. 2011;43:1026–1030. doi: 10.1038/ng.915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Mogilyansky E., Rigoutsos I. The miR-17/92 cluster: a comprehensive update on its genomics, genetics, functions and increasingly important and numerous roles in health and disease. Cell Death Differ. 2013;20:1603–1614. doi: 10.1038/cdd.2013.125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Grote L.E., Repnikova E.A., Amudhavalli S.M. Expanding the phenotype of feingold syndrome-2. Am J Med Genet Part A. 2015;167:3219–3225. doi: 10.1002/ajmg.a.37368. [DOI] [PubMed] [Google Scholar]
- 121.Lei J., Han L., Huang Y., Long M., Zhao G., Yan S., et al. Feingold syndrome type 2 in a patient from China. Am J Med Genet Part A. 2021;185:2262–2266. doi: 10.1002/ajmg.a.62190. [DOI] [PubMed] [Google Scholar]
- 122.Siavrienė E., Preikšaitienė E., Maldžienė Ž., Mikštienė V., Rančelis T., Ambrozaitytė L., et al. A de novo 13q31.3 microduplication encompassing the miR-17 ∼ 92 cluster results in features mirroring those associated with Feingold syndrome 2. Gene. 2020;753 doi: 10.1016/j.gene.2020.144816. [DOI] [PubMed] [Google Scholar]
- 123.Hallek M., Al-Sawaf O. Chronic lymphocytic leukemia: 2022 update on diagnostic and therapeutic procedures. Am J Hematol. 2021;96:1679–1705. doi: 10.1002/ajh.26367. [DOI] [PubMed] [Google Scholar]
- 124.SEER SE and ERPNCI. Cancer Stat Facts: Leukemia — Chronic Lymphocytic Leukemia (CLL). 2021.
- 125.Döhner H., Stilgenbauer S., Benner A., Leupolt E., Kröber A., Bullinger L., et al. Genomic aberrations and survival in chronic lymphocytic leukemia. N Engl J Med. 2000;343:1910–1916. doi: 10.1056/nejm200012283432602. [DOI] [PubMed] [Google Scholar]
- 126.Stilgenbauer S., Nickolenko J., Wilhelm J., Wolf S., Weitz S., Döhner K., et al. Expressed sequences as candidates for a novel tumor suppressor gene at band 13q14 in B-cell chronic lymphocytic leukemia and mantle cell lymphoma. Oncogene. 1998;16:1891–1897. doi: 10.1038/sj.onc.1201764. [DOI] [PubMed] [Google Scholar]
- 127.Calin G.A., Dumitru C.D., Shimizu M., Bichi R., Zupo S., Noch E., et al. Frequent deletions and down-regulation of micro-RNA genes miR15 and miR16 at 13q14 in chronic lymphocytic leukemia. Proc Natl Acad Sci U S A. 2002;99:15524–15529. doi: 10.1073/pnas.242606799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Cimmino A., Calin G.A., Fabbri M., Iorio M.V., Ferracin M., Shimizu M., et al. miR-15 and miR-16 induce apoptosis by targeting BCL2. Proc Natl Acad Sci U S A. 2005;102:13944–13949. doi: 10.1073/pnas.0506654102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Sampath D., Liu C., Vasan K., Sulda M., Puduvalli V.K., Wierda W.G., et al. Histone deacetylases mediate the silencing of miR-15a, miR-16, and miR-29b in chronic lymphocytic leukemia. Blood. 2012;119:1162–1172. doi: 10.1182/blood-2011-05-351510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Dong J.T., Boyd J.C., Frierson H.F. Loss of heterozygosity at 13q14 and 13q21 in high grade, high stage prostate cancer. Prostate. 2001;49:166–171. doi: 10.1002/pros.1131. [DOI] [PubMed] [Google Scholar]
- 131.Aqeilan R.I., Calin G.A., Croce C.M. MiR-15a and miR-16-1 in cancer: discovery, function and future perspectives. Cell Death Differ. 2010;17:215–220. doi: 10.1038/cdd.2009.69. [DOI] [PubMed] [Google Scholar]
- 132.Calin G.A., Ferracin M., Cimmino A., Di Leva G., Shimizu M., Wojcik S.E., et al. A MicroRNA signature associated with prognosis and progression in chronic lymphocytic leukemia. N Engl J Med. 2005;353:1793–1801. doi: 10.1056/nejmoa050995. [DOI] [PubMed] [Google Scholar]
- 133.Allegra D., Mertens D. In-vivo quantification of primary microRNA processing by Drosha with a luciferase based system. Biochem Biophys Res Commun. 2011;406:501–505. doi: 10.1016/j.bbrc.2011.02.055. [DOI] [PubMed] [Google Scholar]
- 134.Calin G.A., Sevignani C., Dumitru C.D., Hyslop T., Noch E., Yendamuri S., et al. Human microRNA genes are frequently located at fragile sites and genomic regions involved in cancers. Proc Natl Acad Sci. 2004;101:2999–3004. doi: 10.1073/pnas.0307323101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Zhang L., Huang J., Yang N., Greshock J., Megraw M.S., Giannakakis A., et al. microRNAs exhibit high frequency genomic alterations in human cancer. Proc Natl Acad Sci. 2006;103:9136–9141. doi: 10.1073/pnas.0508889103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Czubak K., Lewandowska M.A., Klonowska K., Roszkowski K., Kowalewski J., Figlerowicz M., et al. High copy number variation of cancer-related microRNA genes and frequent amplification of DICER1 and DROSHA in lung cancer. Oncotarget. 2015;6:23399–23416. doi: 10.18632/oncotarget.4351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Vischioni C., Bove F., De Chiara M., Mandreoli F., Martoglia R., Pisi V., et al. miRNAs copy number variations repertoire as hallmark indicator of cancer species predisposition. Genes (Basel) 2022:13. doi: 10.3390/genes13061046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Weinstein J.N., Collisson E.A., Mills G.B., Shaw K.R.M., Ozenberger B.A., Ellrott K., et al. The cancer genome atlas pan-cancer analysis project. Nat Genet. 2013;45:1113–1120. doi: 10.1038/ng.2764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Campbell P.J., Getz G., Korbel J.O., Stuart J.M., Jennings J.L., Stein L.D., et al. Pan-cancer analysis of whole genomes. Nature. 2020;578:82–93. doi: 10.1038/s41586-020-1969-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Urbanek-Trzeciak M.O., Galka-Marciniak P., Nawrocka P.M., Kowal E., Szwec S., Giefing M., et al. Pan-cancer analysis of somatic mutations in miRNA genes. EBioMedicine. 2020;61 doi: 10.1016/j.ebiom.2020.103051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Galka-Marciniak P., Kanduła Z., Tire A., Wegorek W., Gwozdz-Bak K., Handschuh L., et al. Mutations in the miR-142 gene are not common in myeloproliferative neoplasms. Sci Rep. 2022:12. doi: 10.1038/s41598-022-15162-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Sharma S. Immunomodulation: a definitive role of microRNA-142. Dev Comp Immunol. 2017;77:150–156. doi: 10.1016/j.dci.2017.08.001. [DOI] [PubMed] [Google Scholar]
- 143.Merkerova M., Belickova M., Bruchova H. Differential expression of microRNAs in hematopoietic cell lineages. Eur J Haematol. 2008;81:304–310. doi: 10.1111/j.1600-0609.2008.01111.x. [DOI] [PubMed] [Google Scholar]
- 144.Thol F., Scherr M., Kirchner A., Shahswar R., Battmer K., Kade S., et al. Clinical and functional implications of microRNA mutations in a cohort of 935 patients with myelodysplastic syndromes and acute myeloid leukemia. Haematologica. 2015;100:e120–e124. doi: 10.3324/haematol.2014.120345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.The Cancer Genome Atlas Research Network Genomic and epigenomic landscapes of adult de novo acute myeloid leukemia. N Engl J Med. 2013;368:2059–2074. doi: 10.1056/NEJMoa1301689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Puente X.S., Beà S., Valdés-Mas R., Villamor N., Gutiérrez-Abril J., Martín-Subero J.I., et al. Non-coding recurrent mutations in chronic lymphocytic leukaemia. Nature. 2015;526:519–524. doi: 10.1038/nature14666. [DOI] [PubMed] [Google Scholar]
- 147.Morin R.D., Assouline S., Alcaide M., Mohajeri A., Johnston R.L., Chong L., et al. Genetic landscapes of relapsed and refractory diffuse large B-cell lymphomas. Clin Cancer Res. 2016;22:2290–2300. doi: 10.1158/1078-0432.CCR-15-2123. [DOI] [PubMed] [Google Scholar]
- 148.Hezaveh K., Kloetgen A., Bernhart S.H., Das M.K., Lenze D., Richter J., et al. Alterations of microRNA and microRNA-regulated messenger RNA expression in germinal center b-cell lymphomas determined by integrative sequencing analysis. Haematologica. 2016;101:1380–1389. doi: 10.3324/haematol.2016.143891. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.Bouska A., Zhang W., Gong Q., Iqbal J., Scuto A., Vose J., et al. Combined copy number and mutation analysis identifies oncogenic pathways associated with transformation of follicular lymphoma. Leukemia. 2017;31:83–91. doi: 10.1038/leu.2016.175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.Kwanhian W., Lenze D., Alles J., Motsch N., Barth S., Döll C., et al. MicroRNA-142 is mutated in about 20% of diffuse large B-cell lymphoma. Cancer Med. 2012;1:141–155. doi: 10.1002/cam4.29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151.Kramer N.J., Le W.W., Reyes E.Y., Kumar B., Chen C.C., Ramakrishna C., et al. Altered lymphopoiesis and immunodeficiency in miR-142 null mice. Blood. 2015;125:3720–3730. doi: 10.1182/blood-2014-10-603951. [DOI] [PubMed] [Google Scholar]
- 152.Trissal M.C., Wong T.N., Yao J.C., Ramaswamy R., Kuo I., Baty J., et al. MIR142 loss-of-function mutations derepress ASH1L to increase HOXA gene expression and promote leukemogenesis. Cancer Res. 2018;78:3510–3521. doi: 10.1158/0008-5472.CAN-17-3592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153.Mildner A., Chapnik E., Manor O., Yona S., Kim K.W., Aychek T., et al. Mononuclear phagocyte miRNome analysis identifies miR-142 as critical regulator of murine dendritic cell homeostasis. Blood. 2013;121:1016–1027. doi: 10.1182/blood-2012-07-445999. [DOI] [PubMed] [Google Scholar]
- 154.Marshall A., Kasturiarachchi J., Datta P., Guo Y., Deltcheva E., James C., et al. Mir142 loss unlocks IDH2R140-dependent leukemogenesis through antagonistic regulation of HOX genes. Sci Rep. 2020;10:19390. doi: 10.1038/s41598-020-76218-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155.Rheinbay E., Nielsen M.M., Abascal F., Wala J.A., Shapira O., Tiao G., et al. Analyses of non-coding somatic drivers in 2,658 cancer whole genomes. Nature. 2020;578:102–111. doi: 10.1038/s41586-020-1965-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156.Dietlein F, Wang AB, Fagre C, Tang A, Besselink NJM, Cuppen E, et al. Genome-wide analysis of somatic noncoding mutation patterns in cancer. Science (80-) 2022;376. https://doi.org/10.1126/science.abg5601. [DOI] [PMC free article] [PubMed]
- 157.Hornshøj H., Nielsen M.M., Sinnott-Armstrong N.A., Świtnicki M.P., Juul M., Madsen T., et al. Pan-cancer screen for mutations in non-coding elements with conservation and cancer specificity reveals correlations with expression and survival. Npj Genomic Med. 2018;3:1. doi: 10.1038/s41525-017-0040-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158.Chun K.H. Molecular Targets and Signaling Pathways of microRNA-122 in Hepatocellular Carcinoma. Pharmaceutics. 2022:14. doi: 10.3390/pharmaceutics14071380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159.Marwaha S., Knowles J.W., Ashley E.A. A guide for the diagnosis of rare and undiagnosed disease: beyond the exome. Genome Med. 2022;14:1–22. doi: 10.1186/s13073-022-01026-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160.Zuker M. Mfold web server for nucleic acid folding and hybridization prediction. Nucleic Acids Res. 2003;31:3406–3415. doi: 10.1093/nar/gkg595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 161.Darty K., Denise A., Ponty Y. VARNA: Interactive drawing and editing of the RNA secondary structure. Bioinformatics. 2009;25:1974–1975. doi: 10.1093/bioinformatics/btp250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 162.Popenda M., Szachniuk M., Antczak M., Purzycka K.J., Lukasiak P., Bartol N., et al. Automated 3D structure composition for large RNAs. Nucleic Acids Res. 2012;40:1–12. doi: 10.1093/nar/gks339. [DOI] [PMC free article] [PubMed] [Google Scholar]