

Summary
Arthritic diseases have attracted enormous scientific interest because of increased worldwide prevalence and represent a significant socioeconomic burden. Osteoarthritis (OA) is the most prevalent form of arthritis. It is a disorder of the diarthrodial joints, characterized by degeneration and loss of articular cartilage associated with adjacent subchondral bone changes. Chronic and unresolving inflammation has been identified as a critical factor driving joint degeneration and pain in OA. Despite numerous attempts at therapeutic intervention, no effective disease-modifying agents targeting OA inflammation are available to the patients. Inflammasomes are protein complexes known to play a critical role in the inflammatory pathology of several diseases, and their roles in OA pathogenesis have become evident over the last decade. In this sense, it is relevant to evaluate the vital role of inflammasomes as potential modulators of pathogenic features in OA. This review will provide an overview and perspectives on why understanding inflammasome activation is critical for identifying effective OA therapies. We elaborate on the contribution of extracellular mediators from the circulatory system and synovial fluid as well as intracellular activators within the synovial fibroblasts and articular chondrocytes toward invoking the inflammasome in OA. We further discuss the merits of emerging inflammasome targeting therapies and speculate on the potential strategies for inflammasome blockade for OA therapy.
Subject areas: Health sciences, Biological sciences, Physiology
Graphical abstract
Health sciences; Biological sciences; Physiology
Introduction
Arthritic diseases have attracted enormous scientific interest because of their increased worldwide prevalence. In particular, osteoarthritis (OA) displays the highest prevalence among arthritic diseases and represents an enormous economic cost to society, government, and health care systems.1 Globally the prevalence of OA has rapidly increased by 113.25% in nearly 3 decades. The number of cases doubled from 247.51 million in 1990 to 527.81 million cases in 2019.2 OA is considered a complex degenerative disorder of the diarthrodial joints, characterized by degradation and loss of articular cartilage with pathological changes in the adjacent subchondral bone.3 Traditionally, OA is considered a non-inflammatory disease. Age-related joint degeneration, because of stress from trauma or metabolic dysregulation was attributed as the causative factor for the development of this disease.4,5 However, increasing evidence has accumulated showing that low-grade inflammation is a critical factor driving OA pathogenesis.6 Advanced diagnostic techniques, such as ultrasound and magnetic resonance imaging (MRI) to detect synovitis in osteoarthritic joints, set the tone for a more comprehensive understanding of inflammation in OA.7,8,9,10 The presence of diverse inflammatory mediators in the synovial fluid of OA patients has strengthened the vital role of inflammation in OA pathogenesis.11 Certainly, cytokines and adipokines have been proposed as biomarkers for OA. These molecules actively participate in OA by driving articular cartilage degeneration, bone marrow lesions, and increased Western Ontario and McMaster (WOMAC) scores indicative of pain.12,13 Despite increasing evidence regarding proinflammatory mediators and their correlation with clinical and radiographical features, the exact pathogenic mechanisms leading to OA remain unclear.
The most prominent contribution of these inflammatory mediators in OA is to promote cartilage breakdown. Several studies have supported this hypothesis by demonstrating that IL-1β, IL-6, TNF-α, and IL-18 belong to the select group of proinflammatory cytokines that remain elevated both systemically and locally in OA.13 This group of cytokines participates not only in chronic inflammation but also in low-grade inflammation, also known as inflammaging, a critical process underlying OA.14 Cartilage breakdown is characterized by the release of cartilage matrix fragments, such as type II collagen and advanced glycation end products (AGEs). Once cleavage occurs, these components converge on local stimulation of chondrocytes, synovial fibroblasts, and immune cells.15,16,17 Eventually, local stimulation and activation of synovial and immune cells initiate the release of matrix metalloproteinases (MMPs) and damage-associated molecular patterns (DAMPs), generating a positive activation loop that enhances and promotes OA development. The accumulation of numerous DAMPs induces the activation of diverse inflammatory processes. Over the past few years, emerging evidence has indicated that inflammasome activation is one of the main pathogenic processes activated by DAMPs in OA.18,19,20,21,22
Inflammasomes are multi-protein complexes that play a critical role in the innate immune response. Activation of these complexes is mediated by the binding of DAMPs to pattern recognition receptors (PPRs), which is followed by the activation of cytosolic caspases.23 Together, they promote apoptosis while also driving the upregulation and activation of proinflammatory cytokines such as IL-1β and IL-18. Dysregulation of the inflammasome activity has been shown to be a major disruptor of tissue homeostasis leading to several inflammatory and metabolic diseases such as diabetes, obesity, atherosclerosis, chronic neurodegenerative diseases, and some cancers. The corresponding pathological mechanisms of these diseases have been extensively discussed in several review articles.24,25,26 In OA, NLRP3 and NLRP1 are important inflammasome components, which act as the triggers of pathogenic processes in macrophages.27,28 Because inflammation has been identified as a critical factor driving radiographic progression and pain in OA, the role of inflammasomes presents a promising strategy to target these molecules in distinct cell types, such as chondrocytes and fibroblast-like synoviocytes (FLS), which are key players in OA.27 Evaluation of inflammasome regulation might provide relevant evidence concerning the vital role of these multi-protein complexes as potential modulators of pathogenic features in OA. This review will therefore elaborate on the emerging roles of inflammasomes in OA and describe the diverse strategies targeting inflammasome-dependent mechanisms being developed as OA disease-modifying therapies.
Pathological roles of inflammasomes in arthritis and associated conditions
Inflammation and tissue damage are critical hallmarks of arthritic diseases (Figure 1). The role of inflammasomes in the most common arthritic diseases, for instance, rheumatoid arthritis (RA), Gout, and juvenile idiopathic arthritis (JIA) has been described in detail in several reviews pinpointing how understanding their pathogenic mechanisms can lead to the development of novel therapeutic options.29,30,31
Figure 1.

Schematic overview of the NLRP3 inflammasome complex
Inflammasomes are multi-protein complexes formed by the oligomerization of distinct domains. NLRP3 inflammasomes have an LRR domain at the C-terminus, a NACHT domain in the middle, and a PYD domain at the N-terminus. The PYD domain from NLRP3 interacts with the PYD domain from ASC, and CARD from ASC protein recruits pro-caspase-1 through interaction with its CARD domain. In the canonical inflammasome pathway, the NLRP3 is assembled after PAMPs or DAMPs are recognized by cytosolic PRRs or inflammasome sensors. This recognition leads to an increase in the transcriptional activity of the nuclear factor-κB (NF-κB), caspase-1-dependent cleave of GSDMD, and the release of proinflammatory cytokines, such as IL-1β and IL-18. NLRP3 inflammasome activation promotes inflammatory and pathogenic processes involved in the pathogenesis of arthritic diseases, for instance, osteoarthritis, rheumatoid arthritis, gout, and juvenile idiopathic arthritis. NLRP3, NOD-, LRR- and pyrin domain-containing protein 3; ASC, apoptosis-associated speck-like protein containing CARD; LRR, leucine-rich repeat; PYD, pyrin domain; CARD, caspase recruitment domain.
Spondyloarthropathies are a form of arthritic diseases that affect the spine in addition to joints. They are characterized by chronic pain, joint inflammation, and bone changes. Ankylosing spondylitis (AS) and psoriatic arthritis (PsA) are examples of spondyloarthropathies in which inflammasome activation has been implicated. In AS, decreased expression of TNFAIP3 and DEPTOR in peripheral blood monocytes correlated with NLRP3 inflammasome activation and high expression of IL1B and IL18.32 High mRNA expression of NLRP3 and IL1B in PBMC from AS patients compared with healthy blood donors was also reported by an independent study, supporting the role of inflammasomes in AS pathogenesis.33 High protein levels of NLRP3 inflammasome components and IL-1β production were observed in PBMC from AS mice.34 In addition, Guggino et al. provided interesting evidence about the critical role of the NLRP3, NLRC4, and AIM2 inflammasomes in the gut of both AS mice and AS patients.35 The overexpression of inflammasome components in PBMC from AS patients was additionally associated with increased serum levels of IL-1β and IL-18, which positively correlated with the AS Disease Activity Score (ASDAS).35 Regarding PsA, several single-nucleotide polymorphisms (SNP) in inflammasome and inflammasome-related genes have been reported. These SNPs are located in NLRP1, NLRP3, and CARD8 genes and were involved with PsA risk and the disease phenotype.36 CLIC1 is a chloride intracellular channel protein that functions as an activator of NLRP3 in macrophages.37 High CLIC1 expression in the skin from PsA was involved with increased angiogenesis and might suggest NLRP3 involvement in increased angiogenesis of PsA joints.38
The pathogenic processes involving inflammasome activation are not only restricted to arthritic diseases but also arthritic-associated conditions. One of the most common pathologies in inflammatory arthritis patients is osteoporosis, which causes bone loss and increases fracture risk (Jiang et al., 2021a). The global strategy for osteoporosis patients has focused on improving bone mineral density (BMD); however, specialized pharmacological approaches are still required.39 In this sense, NLRP3 inactivation has emerged as a determining process to ameliorate osteoporosis hallmarks. A study performed in the ovariectomized (OVX) mouse model, used for studying postmenopausal osteoporosis, indicated that OVX mice highly expressed NLRP3 inflammasome components in femoral bone, an effect that was successfully suppressed by melatonin treatment.40 In addition, repression of NLRP3 resulted in the restoration of osteogenic differentiation by upregulating Runx2 and osteocalcin.40
OA is a disease classified as a chronic low-grade inflammatory pathology (Figure 2). Increased expression of inflammatory mediators in the OA joint supports the induction of locally active non-resolving inflammatory processes.41,42 A role for inflammasome activation in non-resolving inflammation has started to emerge.43 It has been proposed that NLRP1 and NLRP3 containing inflammasomes not only promote the production of IL-18 and IL-1β from various cell types within the joints but also induce a unique form of cell death known as pyroptosis in the articular chondrocytes. In the following sections of this review, we will discuss in detail the discoveries made over the past decade that demonstrate a prominent role of inflammasome activation in driving the chronic inflammatory pathology of OA.
Figure 2.

Overview of non-resolving inflammatory processes in osteoarthritis
Several active processes drive inflammation in OA. For instance, cellular stress, tissue damage, and trauma are critical processes commonly involved with the inflammation onset. These processes produce molecules associated with damage (DAMPs), pathogens (PAMPs), and lifestyle (LAMPs), which are recognized by the innate immune system through PRRs. This recognition synergizes the activation of the immune system in the vascular, molecular, and cellular environment to provide the appropriate defense conditions. However, in inflammatory diseases such as OA, homeostatic processes fail and the resolution model is not achieved, thus, the inflammation is perpetuated. In this scenario, the proinflammatory cytokine profile increases; MMPs, ADAMTs, and calcium crystals are produced. Moreover, the role of NLRP3 in maintaining and promoting inflammation has been described as critical in OA pathogenesis due to the regulation of IL-1β, which activates chondrocytes and FLS. Both cell subsets actively participate in mediating pathogenic processes converging in ECM degradation, cartilage breakdown, fibrosis, synovitis, and production of inflammatory mediators. In addition, macrophages display pathogenic features after TLR-4 activation, promoting NLRP3-dependent responses, and nuclear translocation of NF-κB and IL-1β/IL-18 production. Abbreviations: PRRs, Pattern Recognition Receptors; DAMPs, Damage-associated molecular patterns; PAMPs, Pathogen-associated molecular patterns; LAMPs, Lifestyle-associated molecular patterns; MMP, Matrix metalloproteinases; ADAMTs, Disintegrin and metalloproteinase with thrombospondin motifs; NLRP3, NLR family pyrin domain containing 3; ECM, Extracellular matrix; AGEs, Advanced glycation end products; BCP, Basic calcium phosphatase; CPPD, Calcium pyrophosphatase dihydrate; HIF-1α, Hypoxia-inducible factor- 1α; HO-1, heme oxygenase-1; Nrf2, nuclear factor erythroid 2 related factor 2; FLS, Fibroblast-like synoviocytes; Mθ, macrophage.
Pathological mechanisms driving inflammasome activation in OA
Extracellular factors drive inflammasome activation in OA
Numerous systemic and local soluble factors have been reported to be involved in driving inflammasome activation in arthritic diseases, which happens in two steps.44,45 The first step requires a primary signal to activate intracellular cascades to transcribe and translate the inflammasome factor NLRP3 and the proinflammatory cytokines pro-IL-1β and pro-IL-18. Signal one is mediated through PAMPs (Pathogen-associated molecular patterns) or DAMPs binding to their specific PRRs. Consequently, a second step is triggered resulting in inflammasome assembly, caspase-1 activation, and finally, the release of the active form of IL-1β and IL-18 to promote inflammatory and pathogenic mechanisms.
In OA, the primary signal is predominantly constituted by TLRs agonists.44,45,46 The second signal for NLRP3 inflammasome activation can be mediated via several factors. One of these factors is hydroxyapatite (HA), a type of calcium phosphatase crystal found in synovial fluid samples from OA patients.18 In addition, basic calcium phosphatase (BCP) and calcium pyrophosphatase dihydrate (CPPD) crystals, commonly found in cartilage, synovial fluid, and menisci of OA patients, activate the inflammasome.19 The mechanism of action for these calcium crystals is NLRP3-dependent and independent; however, IL-1β production and aggravation of joint degeneration appear to take place after NLRP3-dependent calcium crystal activation (Figure 3). Purine metabolites have been proposed as an inflammasome activator in OA. It is well known that chronic local and systemic production of free fatty acids, ATP, and purine are critical factors that trigger inflammasome-dependent inflammation in OA from the stress of obesity-associated metainflammation, type 2 diabetes mellitus (T2DM) and metabolic syndrome (MetS).47
Figure 3.

Multiple extracellular factors activate the NLRP3 inflammasome in OA patients
The main reported promoters of NLRP3 inflammasome activation include calcium-containing crystals, i.e., CPPD, BCP, and hydroxyapatite. Moreover, purinergic activators of the NLRP3 are ATP and adenosine. These promoters have been involved in the high expression of NLRP3 inflammasome components such as NLRP3, ASC, active caspase-1, and the production of IL-1β and IL-18 in cartilage and synovium from OA patients. Abbreviations: BCP, Basic calcium phosphatase; CPPD, Calcium pyrophosphatase dihydrate; ATP, Adenosine triphosphate; NLRP3, NLR family pyrin domain containing 3; ASC, Apoptosis-associated speck-like protein containing a CARD.
Controversial has been the systemic role of inflammasomes in OA. Although different studies have reported increased expression of inflammasome components in the cartilage and synovium, these findings differ in blood cell studies. In this sense, NLRP3 and IL-1β were evaluated in two cohorts of erosive (EHOA) and non-erosive (NEHOA) hand OA patients. However, the study was unable to demonstrate increased secretion of IL-1β in serum samples from hand OA (HOA) patients when compared with control subjects.20 In addition, PBMC from HOA patients exerted a decreased gene expression of both IL1B and NLRP3. Of interest, the NLRP3/beta-actin ratio significantly increased in NEHOA compared with EHOA and the control group.20 On the contrary, an independent study reported high soluble levels of ASC (apoptosis-associated speck-like protein containing a CARD), caspase-1, and IL-1β in OA patients compared with healthy blood donors; however, its consequence on OA phenotype was not discussed.48 Soluble factors associated with inflammasome activation have also been observed locally. Synovial fluid from temporomandibular OA patients showed high concentrations of NLRP3, caspase-1, IL-1β, and IL-18, suggesting these molecules initiate degenerative processes in situ.49 High NLRP3 expression was also reported in synovium from OA patients,21 and LPS-primed OA FLS were able to highly express pro-IL-1β22; however, whether this expression is NLRP3-dependent is yet to be elucidated.
Inflammasome activation driven by chondrocytes in OA
Cartilage breakdown is the primary pathogenic process observed in OA. The breakdown is mediated by several mechanisms, for instance, overexpression of MMP and ADAMTS (A disintegrin and metalloproteinase with thrombospondin motifs) that promote sustained degradation of ECM (extracellular matrix) components, increased apoptosis of articular chondrocytes and cell senescence.
Inflammasome factors, in particular NLRP3, have been extensively investigated in the OA articular chondrocytes (Figure 4); Two independent studies have reported that deficiency of NLRP3−/− did not prevent cartilage damage in two different animal models for OA,50,51 even after performing biomechanical load stimulation and inflammatory stress in chondrocytes.50 Low-grade mechanical loading in anterior cruciate ligament transection (ACLT)-induced OA model reduced NLRP3, caspase-1, and IL-1β in chondrocytes.52 In fact, low-grade mechanical loading had a significant effect in ameliorating disease hallmarks, for instance, cartilage degeneration, reducing OARSI (Osteoarthritis Research Society International) score and abnormal bone remodeling, and finally, decreasing the expression of joint catabolic factors (ADAMTS5, MMP3, MMP13).52 In another study, increased expression of NLRP3 and caspase-1 with increased expression of IL-1β and IL-18 was detected in cartilage from the ACLT- and the monosodium iodoacetate-induced OA models, strongly supporting a role for inflammasome activation in joint degeneration.53,54 Similarly, LPS-primed chondrocytes significantly produced and released IL-1β and IL-18 but also activated pyroptosis, an inflammatory type of lytic cell death, via the NLRP3-caspase-1-GSDMD axis.54 Also, supporting a role for inflammasome in papain-induced experimental OA model, overexpression of the NLRP3-ASC-caspase-1-IL-1β/IL-18 axis in the chondrocytes was associated with cartilage degradation and NF-κB activation.55 Taken together, these studies show that the downregulation of NLRP3-inflammasome is either inconsequential or protective in articular cartilage. However, its upregulation is detrimental to joint homeostasis, making it a highly attractive therapeutic target.
Figure 4.

Mechanisms of NLRP3 activation in OA chondrocytes
Chondrocytes have become active players driving NLRP3 inflammasome-dependent mechanisms. Stimulation of these cells with LPS or calcium-containing crystals, which are critical activators of the NF-κB signaling pathway favors the transcription of NLRP3, ASC, CASP1, IL1B, and IL18. Moreover, IL-1β can also activate several pathogenic processes, for instance, NF-κB activation and overexpression of miRNAs that function as activators of NLRP3. The miR-30b-3p directly induces the activation of NLRP3 and binds to SIRT1 mRNA, which is involved in chondrocyte apoptosis induction. The miR-244-5p act by blocking the inhibitory effect of the long-non-coding RNA SNHG7, an inhibitor of NLRP3. Adenosine is another NLRP3 inductor after binding with A2AR, A2BR, and A3R. Similarly, ATP can activate the NLRP3 inflammasome by interacting with P2X7R and P2X4R. Complex mechanisms have also been described for NLRP3 activation, i.e., the stimulation of chondrocytes with H2O2 caused upregulation of USP7. USP7 is a ubiquitin protease capable of deubiquitinating NOX4, thus, preventing NOX4 proteasomal degradation. NOX4 promotes the release of ROS, a process that leads to oxidative stress and converges in NLRP3 activation. An additional mechanism for NLRP3 activation is mediated by the upregulation of Ctbp in cartilage from OA patients, where Ctbp proteins act as transcriptional co-activators of the AP1 transcription factor. The active complex Ctbp-p300-AP1 specifically binds to the promoter region of the NLRP3 gene promoting its transcription in OA chondrocytes. On the other hand, NRF2 functions as a transcriptional repressor of NLRP3 and NF-κB in an HO-1-dependent mechanism; however, in OA several molecules such as AGEs inhibit the effect of NRF2 and contribute to the promotion and maintenance of the inflammatory process. Abbreviations: IL-1β, Interleukin-1 beta; IL-1R, Interleukin-1 receptor; LPS, Lipopolysaccharide; TLR, Toll-like receptors; CPPD, Calcium pyrophosphatase dihydrate; ATP, Adenosine triphosphate; NLRP3, NLR family pyrin domain containing 3; A2AR, Adenosine A2A receptor; A2BR, Adenosine A2B receptor; A3R, Adenosine A3 receptor; P2X7R, P2X purinoceptor 7; P2X4R, P2X purinoceptor 4; USP7, Ubiquitin specific protease 7; NOX4, NADPH oxidase 4; MyD88, Myeloid differentiation primary response 88; IRAK1/4, Interleukin 1 receptor-associated kinase 1/4; TRAF6, TNF receptor-associated factor 6; SIRT, Sirtuin 1; FoxO3a, Forkhead box class O 3a; NF-κB, Nuclear factor kappa-light-chain-enhancer of activated B cells; CASP1, Caspase-1; ASC, Apoptosis-associated speck-like protein containing a CARD; GSDMD, Gasdermin D; ROS, Reactive oxygen species; AGEs, Advanced glycation end products; Ctbp, C-terminal-binding proteins; p300, E1A binding protein p300; AP1, Activator protein 1 complex; NRF2, Nuclear factor erythroid 2–related factor 2; HO-1, Heme oxygenase-1.
The mechanisms underlying inflammasome activation in OA chondrocytes have also been reported. Increased levels of C-terminal-binding proteins (CtBPs; CtBP1 and CtBP2) in the cartilage of OA patients was put forth as one such mechanism. The CtBPs directly interact with p300 and subsequently bind with AP1 subunits.56 The CtBP-p300-AP1 transactivation complex interacts with the NLRP3 promoter to activate its transcription and subsequently, cleavage of caspase-1 and increased expression of IL-1β in chondrocytes occurs. This study also reported that increased expression of CtBPs in OA chondrocytes is mediated by the downregulation of DNA methyltransferases (DNMT1 and DNMT3), which causes the demethylation of CTBP1 and CTBP2 genes.56
The role of long non-coding RNAs (lncRNA) and micro-RNAs have also been implicated in the regulation of inflammasome activity in OA. In this regard, the small nuclear RNA host gene 7 (SNHG7) has been reported as a critical downregulator of NLRP3 inflammasome activation, which is downregulated in OA cartilage.57 The mechanism mediating SNHG7 downregulation is presumably mediated by the overexpression of miR-214-5p, which to some extent can partially inhibit the cartilage-protecting effects of SNHG7 in IL-1β-stimulated chondrocytes.57 The miR-30b-3p is another interesting microRNA proposed as a critical driver of inflammation in chondrocytes. The miR-30b-3p was found to be upregulated in joint tissues from OA patients and in chondrocytes obtained from DMM rats.58 miR-30b-3p likely targets SIRT1 in chondrocytes and promotes apoptosis and activation of NRLP3 inflammasome components via inhibition of the chondroprotective SIRT1/FoxO3 axis.58
Nuclear factor erythroid 2-related factor 2 (NRF2), a transcription factor member of the leucine zipper family, is yet another regulator of NLRP3 inflammasome activation in articular chondrocytes.59,60 A balance between NRLP3 and NRF2 activity in the chondrocytes was found to regulate OA pathogenesis; for instance, NRF2 was found to be downregulated after stimulation with AGEs in the SW1353 chondrocyte cell line, thereby resulting in the upregulation of NLRP3 and ASC, with the secretion of IL-1β and IL-18.59 ROS production and oxidative stress downstream of NADPH oxidase 4 (NOX4) are also known to regulate NLRP3 levels in chondrocytes.59 Oxidative stress upregulates ubiquitin-specific protease (USP7) causing the deubiquitination of NOX4. As a result, NOX4 escapes from the proteasomal degradation enhancing ROS production and NLRP3 and caspase-1 activation.61 This model of oxidative stress-induced OA was characterized by IL-1β and IL-18 secretion, GSDMD-dependent pyroptosis of chondrocytes, and MMP production.61 The increased expression of USP7 and NOX4 in human OA cartilage further supports that the USP7-NOX4-ROS-NLRP3 axis may be relevant to joint degeneration in OA patients.
Activated fibroblast-like synoviocytes mediate NLRP3 activation in OA
Tissue fibrosis that occurs in OA shows an increase in the production of ECM and angiogenesis, leading the synovial tissue to a process of chronic inflammation that has repercussions on surrounding tissues.62,63 In addition, the synovium components allow the joints' nutrition, lubrication, and structure; therefore, the synovium becomes a relevant tissue in OA.64 Fibroblast-like synoviocytes (FLS) are a key player in inflammation and joint destruction because of their activation and release of proinflammatory markers. Several studies have focused on centralizing the role that inflammasomes play directly in the joint and how their activation or inhibition can affect FLS homeostasis.65
One of the main cytokines responsible for FLS activation is IL-1β, which is regulated by NLRP3, and studies from NLRP3−/− FLS stimulated with LPS confirmed the involvement of NLRP3-dependent IL-1β and IL-18 production by FLS.64 Increased hypoxia levels mediate HIF-1α activation in OA and other inflammatory pathologies66; this process has been directly related to the activation of NLPR3 and aggravation of synovitis and fibrosis in OA pathology.63,67 Simultaneously, immunohistochemical analysis of synovium from OA patients and murine model showed higher expression of NLRP3, ASC, NRF2, and heme oxygenase-1 (HO-1) in the synovium. Although the relationship of these molecules in the activation of NLRP3 seems inconclusive, it was determined that ROS could induce their activation once stimulated with LPS in vivo. Even though induction by LPS is considered a mechanism of inflammatory OA; this study also suggests that beyond activation, NLRP3 is vital to trigger pathogenic OA hallmarks.64 Cultures of human FLS isolated from knee OA (KOA) have shown a role for LPS and ATP as molecules capable of activating NLRP3. In addition, increased mRNA levels of NLRP3, ASC, CASP1, and increased soluble levels of IL-1β and IL-18 suggest that LPS and ATP signaling converge in inflammasome-mediated activation mechanisms.68
Even though the expression of NLRP3 and the release of IL-1β and IL-18 in the synovium have been confirmed, the mechanisms by which they promote inflammation have not been related to cartilage degradation and MMP production in OA; therefore, the role of FLS expressing NLRP3, and cartilage degradation remain controversial.64,69 Functional studies in FLS have supported this controversy by reporting that the inflammasome assembly was unsuccessful in active synoviocytes.67 The synovium not only expresses NLRP3, but also NLRP1, NLRP6, NLRP10, NLRP12, and NLRP14, whereas NLRP3 and NLRP1 are the established mechanisms for the activation of pyroptosis.43,65,67 A different NLRP complex may likely be involved in regulating the inflammasome pathways in OA FLS.
Caspase activation mediates inflammation and pyroptosis in OA
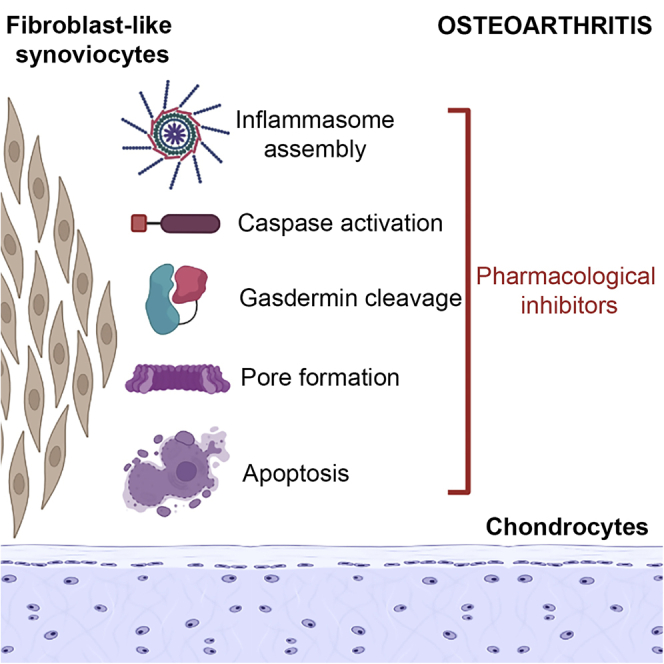
Pyroptosis is a type of highly inflammatory lytic programmed cell death prompted by inflammasomes. This form of cell death has characteristics such as DNA fragmentation, chromatin agglutination, caspase-1 activation, and flattening of the cytoplasm due to plasma membrane leakage.70,71 Pyroptosis is mediated by pore formation (1–2 μm diameter) because of the polymerization of the Gasdermin family members, allowing mature IL-1β and IL-18, followed by the release of cell content and osmotic lysis and release of proinflammatory cytokines.71 Inflammasome-mediated pyroptosis can occur via the canonical or the non-canonical mode of activation (Figure 5). The canonical inflammasome pathway primarily involves caspase-1-dependent cleavage of Gasdermin D (GSDMD) whereas the non-canonical pathway is primarily driven by the activation of caspase 4/5 (caspase 11 in mouse).72 In OA pyroptosis was shown to occur in the perichondrium, triggered by the activation of different caspases where inflammasomes act as platforms for caspase activation.28 Danger signals are present in joints of OA patients, such as high mobility box 1 (HMGB1), uric acid, ATP, S100 proteins, heat shock proteins (HSPs), and hyaluronan fragments, triggering the immune system, promoting pyroptosis, sustaining inflammation, and creating a chronic low-grade inflammation.14,28 In OA pathogenesis, DAMPs accumulation is a driver of proinflammatory signals that promote pyroptosis and production of MMP-1, MMP-2, and MMP-13 and promote cartilage degeneration and damaged chondrocytes associated with OA morphological alterations consistent with pyroptosis.28,73 Several Gasdermin family members were also implicated in OA pathology. GSDMD level is upregulated in OA synovium and its knockout provided protection from cartilage degeneration and synovitis in a surgically induced post-traumatic OA mouse model.26 Similarly, Gasdermin E (GSDME) level is reported to be upregulated in the synovial macrophages and synovial fibroblasts of OA and RA synovium and its deficiency mitigated synovitis in mouse models of RA.74,75 In addition, we speculated a role for Gasdermin C (GSDMC) in OA as its upstream regulator, caspase-6, 76 was shown to be upregulated in chondrocytes exposed to mechanical stress in the ACLT rat osteoarthritis model.77
Figure 5.

Pyroptotic pathways related to OA
The canonical pyroptotic pathway involves the activation of caspase-1 activity leading to the cleavage of GSDMD through the activation of nod-like receptor (NLR) proteins. GSDMD translocases to the plasma membrane and form pores that converge in cellular membrane rupturing. Caspase-8 is activated by TNF-α derived from macrophages and activates GSDMD and GSDMC, resulting in pyroptosis. In the non-canonical pyroptotic pathway, caspase-3/6 activation triggers pyroptosis by cleaving GSDMC. Moreover, p-Stat3 drives PD-L1 translocation, promoting the expression of GSDMC. Caspase-3 cleaves GSDME. Pyroptosis in the non-canonical pathway is induced by caspases-4/5/11 that activate GSDMD and is mediated by Pannexin-1 causing the release of cellular ATP and inducing pyroptotic cell death by the ion channel P2X7 receptor. Finally, the potential therapeutic agents for targeting pyroptosis in OA animal models are shown within the green boxes. Abbreviations: P2X7R, P2X purinoceptor 7; ATP, Adenosine triphosphate; GSDMD, Gasdermin D; GSDME, Gasdermin E; GSDMC, Gasdermin C.
Another class of molecules associated with OA pathogenesis are micro-RNAs, which have been associated with anti-inflammatory properties, specifically, miRNA-146a and miRNA-140-5p, via RNA silencing and transcriptional regulation of gene expression.78 The miR-146a and miR-146b counteract with proinflammatory molecules, increase chondrocytes proliferation, and reduce cell death by targeting the NF-κB pathway. In addition, miR-140-5p was shown to alleviate OA cartilage injury by repressing chondrocyte pyroptosis via the CTSB/NLRP3 axis.78,79
Targeting inflammasome-dependent mechanisms in OA
The development of disease-modifying OA drugs (DMOADs) has become the most important challenge worldwide. Several non-steroidal anti-inflammatory drugs are currently used to ameliorate OA symptoms; however, these drugs only slightly relieve pain and are ineffective in preventing disease progression. In the following sections, we will discuss studies on various natural and synthetic compounds that inhibited inflammasome-dependent pathways in OA.
Emerging strategies for inhibiting inflammasome activation by extracellular factors
The role of calcium crystals in activating inflammasome-dependent mechanisms promoting OA severity and its inhibition might represent an attractive therapeutic strategy for OA patients. In this regard, Phosphocitrate (PC) has been proposed as an inhibitor of calcium-containing crystals, and several studies have supported its therapeutic activity in ameliorating OA hallmarks.19,80,81 Another study revealed that PC decreased the expression of genes involved in proliferation, angiogenesis, inflammatory response, and pain in OA FLS.82 Considering the reported evidence about the effect of PC in OA, a new small molecule has been reported as a PC analog: Carolinas Molecule-01 (CM-01) shares structural similarities with PC and was orally administered to OA-prone Hartley guinea pigs.83 The results suggested that CM-01 treatment protects from cartilage degeneration, osteophyte formation, and meniscal calcification; thus, preventing the development of OA, which leaves open a window for a new orally administered OA drug. The mechanism of action for PC on different tissues in aged animals needs further investigation.
Extracellular ATP levels are also known to modulate inflammasomes in OA. For instance, the purinergic receptors P2X7 and P2X4, which are primarily activated by extracellular ATP, can initiate NLRP1 and NLRP3-dependent inflammatory responses in OA.65,84,85,86 AZD9056 is a potent P2X7 receptor antagonist which has shown significant efficacy by downregulating the expression of P2X7, modulating the activation of the NF-κB signaling pathway, and proinflammatory mediators in rats with monoiodoacetic acid-induced (MIA) OA.87 Another study implemented the same MIA model to test in vivo the P2X7R antagonist, A740003, reporting comparable results. Moreover, A740003 was effective in downregulating the expression of NLRP3, cleaved caspase-1, IL-1β, and finally, decreasing the percentage of pyroptotic cells.84 In the same line of evidence, 5-BDBD and A-438079, two potent antagonists of P2X4 and P2X7, respectively, were evaluated in urothelial cells, showing significant inhibition of caspase-1 activation88; however, the role of these modulators requires more investigation within the OA pathology.
Hyaluronic acid (HA) is another important modulator of inflammation in OA pathology; however, its role in inhibiting inflammasome components is yet to be described. In this regard, a recent study reported that intra-articular administration of HA in temporomandibular joint OA patients significantly inhibited the secretion of NLRP3, IL-1β, and IL-18 in FLS.49 Curcumin has also been evaluated in the DMM OA model, demonstrating a significant reduction of OA score and inflammatory markers. Although the authors suggested that the curcumin mechanism of action targets the NLRP3-Caspase-1-IL-1β axis, those results were observed in THP-1 cells and further investigation in OA primary cells and preclinical models must be performed to corroborate these findings.89
Emerging treatment strategies for inflammasome inhibition in OA chondrocytes
The mechanisms mediating inflammasome activation differ between cell types within the osteoarthritic joint; for this reason, the inflammasome inhibitory mechanisms might be slightly different and dependent on the studied cell type, such as chondrocytes (Figure 6). For instance, Sinomenine, the major bioactive compound of the traditional herb Sinomenium acutum, has been proposed as a natural compound with anti-inflammatory effects in OA chondrocytes.53 The suggested mechanism of action depends on the upregulation of miR-223-3p, which has a binding site for the NLRP3 gene. miR-223-3p was found to be downregulated in IL-1β-stimulated chondrocytes, which was reverted by Sinomenine treatment. Sinomenine also inhibited the protein levels of NLRP3, ASC, caspase-1, IL-1β, and IL-18.53 In the same line of evidence, Icariin, an extract of the herb Epimedium spp., has also shown anti-inflammatory effects in OA. The effect of Icariin was evaluated in LPS-primed chondrocytes where NLRP3 expression and caspase-1 activity were suppressed.54 Another natural compound with anti-inflammatory properties is ursolic acid (UA). UA is a pentacyclic triterpenoid found in the peel of several fruits and medicinal plants and has shown promising results as a therapeutic agent in OA.90 The precise mechanism of action for UA is yet to be elucidated; however, it has been proposed that UA treatment can significantly decrease the protein levels of NLRP3, ASC, and cleaved caspase-1 in rat chondrocytes stimulated with TNF-α.90 In addition, Licochalcone A (Lico A), a flavonoid extracted from the roots of Glycyrrhiza spp., has demonstrated optimistic results modulating NLRP3 inflammasome-associated processes in OA. The proposed mechanism suggests an upregulation of NRF2 and HO-1, both considered regulators of inflammatory mechanisms.91 Lico A can additionally suppress the inflammatory response in LPS-primed chondrocytes and protect mice from cartilage degeneration in the ACLT-induced OA model.91 Similarly, Cardamonin, a biological compound extracted from Alpinia katsumadai, has been evaluated in IL-1β-stimulated chondrocytes exerting anti-inflammatory properties. Cardamonin is capable of suppressing NLRP3 inflammasome signaling and upregulating the expression of NRF2.60 These findings provide reasonable evidence for a regulatory mechanism between NRF2 and NLRP3 in chondrocytes in vitro; nevertheless, in vivo models are required to corroborate those results. Baicalein, a flavonoid found in Scutellaria spp., also showed promising results in vivo OA models by downregulating NLRP3 and caspase-1 and reducing chondrocyte damage.92 Although the suggested mechanism of action for Baicalein indicated an association with the regulation of oxidative stress markers, the current evidence was unable to demonstrate the specific mechanism. Further research must be performed to clarify this concern. Similarly, Quercetin has exerted important anti-inflammatory and antioxidant properties. Decreased OARSI score, soluble IL-1β, and IL-18, with decreased cartilage expression of NLRP3 and caspase-3, have been reported after Quercetin administration in the ACLT-induced OA model.93 These findings were further corroborated in vitro by stimulating chondrocytes with IL-1β. The mechanism of action for Quercetin was suggested to inhibit IRAK1-dependent IL-1β stimulation; thus, causing inhibition of NLRP3 activation, suppression of cell apoptosis, ROS production, and IL-1β and IL-18 secretion.93
Figure 6.

Targeting NLRP3 inflammasomes in OA chondrocytes
Natural and synthetic compounds have been proposed as mediators of NLRP3-dependent mechanisms in OA. Unspecific inhibitors include molecules that do not directly inhibit known inflammasome drivers or components but have a significant indirect effect downregulating the expression of NLRP3 components and critical OA hallmarks in vivo and in vitro. Inhibitors of NLRP3 activators target upstream molecules involved in NLRP3 activation, regulation, or expression. Some of these inhibitors also upregulate the expression of NRF2, an important repressor of NLRP3 expression. Specific inhibitors targeting NLRP3 include small molecules that bind to the NLRP3 NATCH motive and inhibit its structural conformation change toward an active form. Sinomenine is a natural compound that upregulates the expression of the miR-223-3p, a microRNA that directly targets the NLRP3 mRNA blocking its translation toward a functional protein.
Synthetic compounds have equally exerted modulatory properties in OA. Dexmedetomidine, which acts as a highly selective α2-adrenergic agonist with sedative and analgesic properties, has also been associated with pain relief and cartilage protection in OA. The reported mechanism of action suggests that dexmedetomidine might target inflammasome-dependent mechanisms in the cartilage of OA rats by significantly decreasing the expression of NLRP3, ASC, and cleaved caspase-1.55Of interest, histamine receptor 1 (H1R) expression and activation have also been detected in chondrocytes after stimulation with AGEs, and loratadine, a synthetic blocker for H1R, has emerged as a modulator of inflammasomes in OA.59 AGEs-H1R interaction induced the activation of intracellular signaling cascades that converge in NLRP3 activation. Loratadine treatment seems to participate in modulating inflammasome activation in two simultaneous processes: (1) Interfering with AGEs signaling and (2) facilitating nuclear translocation of NRF2, which, as previously mentioned, can act as a repressor of NLRP3 transcription.59 In previous sections of this review, we discussed the two-step mechanism for inflammasome activation, which requires one signal mediated by PRRs. In this sense, TLR4 belongs to those critical PRRs that trigger NLRP3 inflammasome signaling. Amitriptyline is employed as an antidepressant drug that has been repurposed as a pain modulator and has also exerted exciting results by blocking TLR4-LPS interactions in OA chondrocytes.92 Amitriptyline could downregulate NLRP3 expression and IL-1β secretion in OA chondrocytes in an inhibitory mechanism that seems to be TLR4-dependent.92 Repurposing available drugs represent a promising strategy to encourage researchers to follow new lines of evidence focused on ameliorating signs and symptoms in OA patients.
Earlier in this review, we explored the activation of the USP7-NOX4-ROS-NLRP3 axis as a critical driver of inflammation in OA. In this regard, an interesting study reported that inhibition of different components of this axis had a potential therapeutic effect in OA chondrocytes. Apocynin, a ROS inhibitor, along with the molecules P22077 and GLX351322, which are USP7 and NOX4 inhibitors, respectively, were able to suppress the NLRP3 inflammasome-dependent mechanisms.61 Chondrocyte pyroptosis and expression of MMP-1 and MMP-3 were successfully targeted after inhibition of those three axis components,61which supports that targeting oxidative stress can indirectly suppress the inflammasome-dependent mechanism in OA.
Small synthetic molecules have emerged as potential DMOADs. CY-09 is a small molecule that interferes with the ATP-binding motif of the NLRP3 NACHT domain; thus, representing a specific inhibitor for this inflammasome component. CY-09 was evaluated in TNF-α-stimulated chondrocytes and in the DMM OA model showing that the NLRP3 inflammasome and GSDMD-mediated pyroptosis were effectively targeted both in vivo and in vitro.94 MCC950 is another small molecule described as a specific inhibitor of NLRP3 because of its binding to the Walter B motif at the NLRP3 NACHT domain. MCC950 has shown meaningful results in IL-1β-treated chondrocytes by suppressing NLRP3 and cleaved caspase-1, a mechanism that is dependent on the activation of the Nrf2/HO-1/NQO2 axis.95 Those findings were further corroborated by using the DMM model. MCC950 has also exerted its therapeutic effect by decreasing the expression of several catabolic factors and suppressing ROS production in chondrocytes.95 Some other small molecules have demonstrated an effective indirect inhibition of NLRP3, for instance, the alpha 7 nicotinic acetylcholine receptor (α7nAChR) agonist, PNU-282987. This α7nAChR agonist has been tested in the MIA-induced arthritis model, causing decreased expression of MMP-1 and MMP-13. Of interest, several NLRP3 inflammasome components were also inhibited in the cartilage of MIA rats, findings that were additionally corroborated in IL-1β-treated chondrocytes.96 The indirect mechanism by which α7nAChR protects from OA progression is presumably through inhibition of IκBα degradation, which prevents NF-κB phosphorylation, thus indirectly targeting NLRP3 inflammasome activation and, finally, conferring chondroprotection.96
Emerging treatment strategies for inflammasome inhibition in OA FLS
An emerging area of interest relates to distinct natural molecules that can block the activation of NLRP3 in OA FLS (Table 1). For instance, casticin was a molecule capable of reducing the effect of LPS in FLS by decreasing the expression of caspase-1.62 Vanillic acid was also able to decrease IL-18 and IL-1β expression,97 a result further confirmed by studying the effect of this molecule in combination with LPS in an experimental model of OA.98 Furthermore, Chrysin, a flavonoid found in several plants, could inhibit the NLRP3-dependent production of IL-18 and IL-1β, and suppressed the expression of NLRP3 and caspase-1.99 In the same line of evidence, agnuside, a chemical compound isolated from Vitex negundo L., has exerted inhibitory properties by decreasing the expression of NLRP3.63
Table 1.
Potential therapeutic agents targeting NLRP3-dependent mechanisms in OA FLS
| Therapeutic agent | Experimental model | Relevant data | Reference |
|---|---|---|---|
| Estromal cell-derived factor-1 (SDF-1) | Human FLS | SDF-1 was found to increase the proliferation of OA FLS. SDF-1 downregulated the gene and protein expression levels of NLRP3 inflammasome, GSDMD, and IL-1β | Wang et al.100 |
| Murine model of collagenase-induced OA | SDF-1 treatment delays pathological phenotypes. Intra-articular injection of 120 ng/kg SDF-1 twice a week reverses high expression of NLRP3, ASC, caspase-1, and protein markers of pyroptosis | ||
| Agnuside (extract of Vitex negundo L. (Verbenaceae)) | Rat FLS | Agnuside reverses NLRP3 expression | Zhang et al.63 |
| Monoiodoacetic acid (MIA)-induced KOA in rats | Agnuside relieves the state of hypoxia in KOA rats and reduces mRNA and protein levels of NLRP3 in KOA rats | ||
| Casticin | MIA-induced KOA in rats | Upregulated NLRP3 inflammasome complex in MIA group were reduced by use of casticin significantly | Li et al.62 |
| Rat FLS | The increase of NLRP3, ASC, pro-caspase-1 expression was reduced by casticin | ||
| Vanillic Acid | MIA-induced KOA in rats | The mRNA and protein expressions of caspase-1, NLRP3, and ASC showed a reduction after vanillic acid treatment. The use of vanillic acid promotes less inflammation. | Ma et al.98 |
| Rat FLS | The mRNA and protein expressions of IL-18 and IL-1β were decreased in the vanillic acid treatment group | ||
| Chrysin (Scutellariae Radix) | MIA-induced KOA in rats | MIA modeling resulted in increased NLRP3, caspase-1 and ASC mRNA expression in synovial tissue compared with the normal group, but chrysin reverses this effect. The high IL-1β and IL-18 expression levels in serum from KOA rats were significantly suppressed by chrysin. | Liao et al.99 |
| Rat FLS | High NLRP3, caspase-1 and ASC mRNA expression is suppressed by chrysin. The expression of the proinflammatory factors IL-1β and IL-18 was reduced in the cell supernatant upon treatment with Chrysin | ||
| Estradiol (E2) | Human FLS | NLRP3 mRNA and protein expression were significantly suppressed by E2. The cleaved caspase-1, the production of IL-1b and IL-18 were also significantly decreased by E2 treatment, whereas ASC and pro-caspase-1 was not significantly changed | Shi et al.68 |
| KMU-1170 (a derivative of indolin-2-one) | Human FLS | KMU-1170 pretreatment inhibited LPS-induced phosphorylation of IKKα/β and NF-κB p65 associated with proinflammatory cytokines (IL-1β, TNF-α, and IL-6) production | Baek et al.22 |
Synthetic molecules have also been studied as important inflammasome inhibitors. KMU-1170, a novel multi-protein kinase inhibitor, has shown the ability to suppress the activation of the NLRP3 inflammasome signaling pathway in human osteoarthritic FLS.22 Another study reported that estradiol significantly suppresses IL-18, IL-1β, and caspase-1 production and downregulated the expression of NLRP3 at the mRNA and protein levels, whereas pro-caspase-1 production was not significantly changed.68 Moreover, the exogenous stromal cell-derived factor-1 (SDF-1) downregulated the gene expression and protein levels of NLRP3 inflammasome.100 Limitations in knowledge regarding the role of inflammasome-dependent mechanisms in OA FLS are a critical issue that must be taken into consideration in future studies.
Other potential future directions for inflammasome targeting in OA
In addition to the research focusing on non-specific inhibitors of inflammasomes, inhibition of NLPR3 activators, and specific molecules targeting NLRP3 inflammasomes, other research directions related to chondrocytes and FLS inflammasome-dependent activation in OA should be considered. In this regard, adenosine deaminase (ADA) could represent a promising therapeutic modulator in OA because of its effect promoting the degradation of adenosine into inosine. Of interest, adenosine degradation promoted decreased NLRP3 inflammasome activation and reduction of IL-1β and ATP levels.101
Targeting pyroptosis in OA has been proposed in in vitro experimental studies, where chondrocytes were activated by LPS and ATP, and treatment with disulfiram and glycyrrhizin acid resulted in suppression of the inflammatory response and reduction of pyroptosis.102 Another interesting finding was that treatment with Icariin also suppressed GSDMD expression, and chondrocyte pyroptosis, and protected against cartilage degeneration in the MIA rat OA model.54 Other molecules; for instance, morroniside and loganin were assessed in a mouse model of OA, and the authors found that those two iridoid glycosides extracted from Cornus officinalis were protective against cartilage degradation by reducing chondrocyte pyroptosis via inhibition of NF-κB signaling.103,104 Furthermore, the effect of the combined use of the Hedgehog signaling inhibitor, GANT-61, and indomethacin was reported in experimental OA mice. GANT-61 and indomethacin had a synergistic effect on OA via mitigation of chondrocytes pyroptosis and reduction of IL-1β, IL-18, TNF-α, IL-2, and IL-6 expression.105 In the same line of evidence, Lico A, mitigated the progression of OA in a mouse model, attenuated LPS-induced chondrocytes pyroptosis, and reduced the expression of NLRP3, GSDMD, caspase-1, IL-1β, and IL-18. The authors concluded that Lico A may have a therapeutic potential in OA.91 Another potential agent for the treatment of OA that has been recently described is Quercetin. This molecule was tested in different models of OA Wistar rats, showing that quercetin can reduce LPS-induced apoptosis and ECM degradation by inhibiting NLRP3-mediated pyroptosis.106
The role of GSDMD in mediating IL-1β secretion is well known. Several reports have pointed out that loss or inhibition of GSDMD results in attenuated IL-1β and IL-18 release. For instance, in autoimmune diseases, the GSDMD inhibition attenuated synovitis and cartilage degeneration.107 In addition, necrosulfonamide, an inhibitor of pyroptotic cell death, was investigated and proposed as a therapy in animal models of inflammatory diseases and resulted in the molecular regulation of inflammatory response by promoting GSDMD inhibition.108 Other studies have reported inhibition of pyroptosis by blocking GSDMD using FDA-approved drugs such as disulfiram (used to treat alcohol addiction)109 and dimethyl fumarate.110 Overall, these findings provide relevant evidence that points out the inhibition of GSDMD as a potential anti-inflammatory strategy in pyroptosis-driven diseases such as OA.
Metainflammation is the loss of metabolic homeostasis that involves a dysregulation of the immune system, and this process is associated with low-level inflammation.111 Metainflammation is characterized by increased levels of catabolic mediators and proinflammatory cytokines, for instance, TNF-α, IL-6, and IL-1β.112 Those factors contribute to chondrocyte dysfunction and aggravate OA progression.47 Thus, evaluating the effect of distinct metainflammation inhibitors might represent a promising strategy in OA. From this perspective, the evaluation of Levornidazole, a nitroimidazole derivative, has been demonstrated to block NLRP3 activation through the reduction of ROS generation.113 Moreover, studies evaluating the effect of resveratrol, a natural polyphenol, reported that this molecule can cause the inadequate assembly of ASC on the mitochondria114 and inhibit NF-κB-dependent responses.115 Therefore, resveratrol might function as a potential anti-inflammatory and antioxidant molecule, which may be used to attenuate the metainflammation observed in OA. In addition, melatonin, an endogenous indoleamine, has exerted immunomodulatory properties, and apparently, this molecule can influence energy metabolism. Melatonin has shown inhibitory properties on the expression of NLRP3 inflammasome-associated genes in adipose tissue and inhibited NF-κB phosphorylation, a key factor in metainflammation.116 Remarkably, another study showed that certain components, such as parthenolide and vinylsulfones, could inhibit the activation of caspase-1 and NF-κB. Regarding synthetic molecules targeting inflammasome components, Bay 11–7082, displayed interesting results and might be considered a potential inhibitor of NLRP3.117 Lastly, γ-tocotrienol, an analog of vitamin E, could also be considered a metainflammation inhibitor because this molecule can attenuate adipose tissue inflammation and insulin resistance in diet-induced obesity. The γ-tocotrienol molecule was able to repress inflammasome activation, caspase-1 cleavage, and IL-1β secretion, exerting anti-inflammatory and anti-pyroptotic properties.118 Taken together, these findings from non-joint tissues have improved our understanding of the potential role of inflammasome in OA.
Conclusion
Over the last decade, the role of inflammasomes driving chronic inflammation and pathogenic hallmarks in OA has become increasingly evident. Several in vitro and preclinical studies have suggested that the activation of NLRP3 inflammasomes in tissue-resident cells, such as chondrocytes and FLS, plays a crucial role in OA. An abundance of the research discussed in this review, suggests that targeting the inflammasome-dependent mechanisms could slow down OA progression, but whether these approaches could reverse the pathology of fully established disease in humans remains to be established. Several studies were also limited by an independent focus on either chondrocytes or FLS rather than considering the joint as a functional unit with different cell/tissue types. Moreover, primary FLS and chondrocytes in culture are known to lack subtype diversity that is observed under in vivo conditions.119,120,121,122 Therefore, profiling the gene expression of various inflammasome components from published single-cell RNA-seq datasets from human OA patient synovium and cartilage will be very informative. Finally, OA is a complex disease that can be categorized into multiple disease subtypes depending on the age, and mechanical, metabolic, inflammatory, and genetic characteristics of the patient. Hence, conducting translational research to identify potential responders and non-responders of the inflammasome regulators coupled with the development of effective delivery strategies for the identified bioactive compounds will be needed to effectively exploit the inflammasome pathway for OA therapy.
Acknowledgments
We thank the support from the National Council of Science and Technology (CONACYT-Mexico) grant (800977) assigned to SR-P (CVU: 660472) and Startup funds from the Department of Orthopedics, Emory University School of Medicine assigned to PB. We would also like to thank Heather Boldt, Director of the English Language Support Program and Global Engagement at Emory University for her critical review during the manuscript preparation. The figures were created with BioRender.com.
Author contributions
S.R-P. and P.B. developed the idea. S.R-P and I.V.R-P. accomplished the review framework. S.R-P., I.V.R-P., D.E.M-F., and L.A.H-P. wrote the first draft of the manuscript. S.R-P. and P.B. supervised manuscript writing and performed the final editing. All authors listed have made a substantial, direct, and intellectual contribution to the work and approved it for publication.
Declaration of interests
All authors declare no conflict of interest.
Contributor Information
Sergio Ramirez-Perez, Email: sdrami2@emory.edu.
Pallavi Bhattaram, Email: pallavi.bhattaram@emory.edu.
References
- 1.Gabriel S.E., Michaud K. Epidemiological studies in incidence, prevalence, mortality, and comorbidity of the rheumatic diseases. Arthritis Res. Ther. 2009;11:229. doi: 10.1186/ar2669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Long H., Liu Q., Yin H., Wang K., Diao N., Zhang Y., Lin J., Guo A. Prevalence trends of site-specific osteoarthritis from 1990 to 2019: findings from the global burden of disease study 2019. Arthritis Rheumatol. 2022;74:1172–1183. doi: 10.1002/art.42089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Martel-Pelletier J., Barr A.J., Cicuttini F.M., Conaghan P.G., Cooper C., Goldring M.B., Goldring S.R., Jones G., Teichtahl A.J., Pelletier J.P. Osteoarthritis. Nat. Rev. Dis. Primers. 2016;2:16072. doi: 10.1038/nrdp.2016.72. [DOI] [PubMed] [Google Scholar]
- 4.Chen D., Shen J., Zhao W., Wang T., Han L., Hamilton J.L., Im H.J. Osteoarthritis: toward a comprehensive understanding of pathological mechanism. Bone Res. 2017;5:16044. doi: 10.1038/boneres.2016.44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Courties A., Gualillo O., Berenbaum F., Sellam J. Metabolic stress-induced joint inflammation and osteoarthritis. Osteoarthritis Cartilage. 2015;23:1955–1965. doi: 10.1016/j.joca.2015.05.016. [DOI] [PubMed] [Google Scholar]
- 6.Terkawi M.A., Ebata T., Yokota S., Takahashi D., Endo T., Matsumae G., Shimizu T., Kadoya K., Iwasaki N. Low-grade inflammation in the pathogenesis of osteoarthritis: cellular and molecular mechanisms and strategies for future therapeutic intervention. Biomedicines. 2022;10 doi: 10.3390/biomedicines10051109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Kortekaas M.C., Kwok W.Y., Reijnierse M., Huizinga T.W., Kloppenburg M. In erosive hand osteoarthritis more inflammatory signs on ultrasound are found than in the rest of hand osteoarthritis. Ann. Rheum. Dis. 2013;72:930–934. doi: 10.1136/annrheumdis-2012-201458. [DOI] [PubMed] [Google Scholar]
- 8.Mancarella L., Addimanda O., Pelotti P., Pignotti E., Pulsatelli L., Meliconi R. Ultrasound detected inflammation is associated with the development of new bone erosions in hand osteoarthritis: a longitudinal study over 3.9 years. Osteoarthritis Cartilage. 2015;23:1925–1932. doi: 10.1016/j.joca.2015.06.004. [DOI] [PubMed] [Google Scholar]
- 9.Haugen I.K., Slatkowsky-Christensen B., Bøyesen P., Sesseng S., van der Heijde D., Kvien T.K. MRI findings predict radiographic progression and development of erosions in hand osteoarthritis. Ann. Rheum. Dis. 2016;75:117–123. doi: 10.1136/annrheumdis-2014-205949. [DOI] [PubMed] [Google Scholar]
- 10.Baker K., Grainger A., Niu J., Clancy M., Guermazi A., Crema M., Hughes L., Buckwalter J., Wooley A., Nevitt M., Felson D.T. Relation of synovitis to knee pain using contrast-enhanced MRIs. Ann. Rheum. Dis. 2010;69:1779–1783. doi: 10.1136/ard.2009.121426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Boehme K.A., Rolauffs B. Onset and progression of human osteoarthritis-can growth factors, inflammatory cytokines, or differential miRNA expression concomitantly induce proliferation, ECM degradation, and inflammation in articular cartilage? Int. J. Mol. Sci. 2018;19 doi: 10.3390/ijms19082282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Zhu J., Ruan G., Cen H., Meng T., Zheng S., Wang Y., Li B., Zhu Z., Han W., Winzenberg T., et al. Association of serum levels of inflammatory markers and adipokines with joint symptoms and structures in participants with knee osteoarthritis. Rheumatology. 2022;61:1044–1052. doi: 10.1093/rheumatology/keab479. [DOI] [PubMed] [Google Scholar]
- 13.Zhu Z., Otahal P., Wang B., Jin X., Laslett L.L., Wluka A.E., Antony B., Han W., Wang X., Winzenberg T., et al. Cross-sectional and longitudinal associations between serum inflammatory cytokines and knee bone marrow lesions in patients with knee osteoarthritis. Osteoarthritis Cartilage. 2017;25:499–505. doi: 10.1016/j.joca.2016.10.024. [DOI] [PubMed] [Google Scholar]
- 14.Millerand M., Berenbaum F., Jacques C. Danger signals and inflammaging in osteoarthritis. Clin. Exp. Rheumatol. 2019;37:48–56. [PubMed] [Google Scholar]
- 15.Steenvoorden M.M., Huizinga T.W., Verzijl N., Bank R.A., Ronday H.K., Luning H.A., Lafeber F.P., Toes R.E., DeGroot J. Activation of receptor for advanced glycation end products in osteoarthritis leads to increased stimulation of chondrocytes and synoviocytes. Arthritis Rheum. 2006;54:253–263. doi: 10.1002/art.21523. [DOI] [PubMed] [Google Scholar]
- 16.Sofat N., Ejindu V., Heron C., Harrison A., Koushesh S., Assi L., Kuttapitiya A., Whitley G.S., Howe F.A. Biomarkers in painful symptomatic knee OA demonstrate that MRI assessed joint damage and type II collagen degradation products are linked to disease progression. Front. Neurosci. 2019;13:1016. doi: 10.3389/fnins.2019.01016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Garnero P., Peterfy C., Zaim S., Schoenharting M. Bone marrow abnormalities on magnetic resonance imaging are associated with type II collagen degradation in knee osteoarthritis: a three-month longitudinal study. Arthritis Rheum. 2005;52:2822–2829. doi: 10.1002/art.21366. [DOI] [PubMed] [Google Scholar]
- 18.Jin C., Frayssinet P., Pelker R., Cwirka D., Hu B., Vignery A., Eisenbarth S.C., Flavell R.A. NLRP3 inflammasome plays a critical role in the pathogenesis of hydroxyapatite-associated arthropathy. Proc. Natl. Acad. Sci. USA. 2011;108:14867–14872. doi: 10.1073/pnas.1111101108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Conway R., McCarthy G.M. Calcium-containing crystals and osteoarthritis: an unhealthy alliance. Curr. Rheumatol. Rep. 2018;20:13. doi: 10.1007/s11926-018-0721-9. [DOI] [PubMed] [Google Scholar]
- 20.Fioravanti A., Tenti S., McAllister M., Chemaly M., Eakin A., McLaughlin J., Bjourson A.J., Frati E., McGilligan V., Cheleschi S., Gibson D.S. Exploring the involvement of NLRP3 and IL-1β in osteoarthritis of the hand: results from a pilot study. Mediators Inflamm. 2019;2019:2363460. doi: 10.1155/2019/2363460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Liu Y., Wei W., Wang Y., Wan C., Bai Y., Sun X., Ma J., Zheng F. TNF-α/calreticulin dual signaling induced NLRP3 inflammasome activation associated with HuR nucleocytoplasmic shuttling in rheumatoid arthritis. Inflamm. Res. 2019;68:597–611. doi: 10.1007/s00011-019-01244-w. [DOI] [PubMed] [Google Scholar]
- 22.Baek H.S., Hong V.S., Kim S.H., Lee J., Kim S. KMU-1170, a novel multi-protein kinase inhibitor, suppresses inflammatory signal transduction in THP-1 cells and human osteoarthritic fibroblast-like synoviocytes by suppressing activation of NF-κB and NLRP3 inflammasome signaling pathway. Int. J. Mol. Sci. 2021;22 doi: 10.3390/ijms22031194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Newton K., Dixit V.M., Kayagaki N. Dying cells fan the flames of inflammation. Science. 2021;374:1076–1080. doi: 10.1126/science.abi5934. [DOI] [PubMed] [Google Scholar]
- 24.Holbrook J.A., Jarosz-Griffiths H.H., Caseley E., Lara-Reyna S., Poulter J.A., Williams-Gray C.H., Peckham D., McDermott M.F. Neurodegenerative disease and the NLRP3 inflammasome. Front. Pharmacol. 2021;12:643254. doi: 10.3389/fphar.2021.643254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Sharma B.R., Kanneganti T.D. NLRP3 inflammasome in cancer and metabolic diseases. Nat. Immunol. 2021;22:550–559. doi: 10.1038/s41590-021-00886-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Rathinam V.A., Fitzgerald K.A. Inflammasome complexes: emerging mechanisms and effector functions. Cell. 2016;165:792–800. doi: 10.1016/j.cell.2016.03.046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Sanchez-Lopez E., Coras R., Torres A., Lane N.E., Guma M. Synovial inflammation in osteoarthritis progression. Nat. Rev. Rheumatol. 2022 doi: 10.1038/s41584-022-00749-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.An S., Hu H., Li Y., Hu Y. Pyroptosis plays a role in osteoarthritis. Aging Dis. 2020;11:1146–1157. doi: 10.14336/ad.2019.1127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Spel L., Martinon F. Inflammasomes contributing to inflammation in arthritis. Immunol. Rev. 2020;294:48–62. doi: 10.1111/imr.12839. [DOI] [PubMed] [Google Scholar]
- 30.Kong R., Sun L., Li H., Wang D. The role of NLRP3 inflammasome in the pathogenesis of rheumatic disease. Autoimmunity. 2022;55:1–7. doi: 10.1080/08916934.2021.1995860. [DOI] [PubMed] [Google Scholar]
- 31.Jiang Q., Wang X., Huang E., Wang Q., Wen C., Yang G., Lu L., Cui D. Inflammasome and its therapeutic targeting in rheumatoid arthritis. Front. Immunol. 2021;12:816839. doi: 10.3389/fimmu.2021.816839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Zhai Y., Lin P., Feng Z., Lu H., Han Q., Chen J., Zhang Y., He Q., Nan G., Luo X., et al. TNFAIP3-DEPTOR complex regulates inflammasome secretion through autophagy in ankylosing spondylitis monocytes. Autophagy. 2018;14:1629–1643. doi: 10.1080/15548627.2018.1458804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Kim S.K., Cho Y.J., Choe J.Y. NLRP3 inflammasomes and NLRP3 inflammasome-derived proinflammatory cytokines in peripheral blood mononuclear cells of patients with ankylosing spondylitis. Clin. Chim. Acta. 2018;486:269–274. doi: 10.1016/j.cca.2018.08.022. [DOI] [PubMed] [Google Scholar]
- 34.Tian Z.G., Yao M., Chen J. Micheliolide alleviates ankylosing spondylitis (AS) by suppressing the activation of the NLRP3 inflammasome and maintaining the balance of Th1/Th2 via regulating the NF-κB signaling pathway. Ann. Transl. Med. 2020;8:991. doi: 10.21037/atm-20-4987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Guggino G., Mauro D., Rizzo A., Alessandro R., Raimondo S., Bergot A.S., Rahman M.A., Ellis J.J., Milling S., Lories R., et al. Inflammasome activation in ankylosing spondylitis is associated with gut dysbiosis. Arthritis Rheumatol. 2021;73:1189–1199. doi: 10.1002/art.41644. [DOI] [PubMed] [Google Scholar]
- 36.Juneblad K., Kastbom A., Johansson L., Rantapää-Dahlqvist S., Söderkvist P., Alenius G.M. Association between inflammasome-related polymorphisms and psoriatic arthritis. Scand. J. Rheumatol. 2021;50:206–212. doi: 10.1080/03009742.2020.1834611. [DOI] [PubMed] [Google Scholar]
- 37.Domingo-Fernández R., Coll R.C., Kearney J., Breit S., O'Neill L.A.J. The intracellular chloride channel proteins CLIC1 and CLIC4 induce IL-1β transcription and activate the NLRP3 inflammasome. J. Biol. Chem. 2017;292:12077–12087. doi: 10.1074/jbc.M117.797126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Bordean L., Chis M., Raica M., Cotoi O.S., Ceausu A.R., Avram C., Cimpean A.M. CLIC1 expression in skin biopsies from patients with rheumatoid and psoriatic arthritis as a potential tool to predict therapy response. In Vivo. 2021;35:2559–2567. doi: 10.21873/invivo.12538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Kanis J.A., McCloskey E.V., Johansson H., Oden A. Approaches to the targeting of treatment for osteoporosis. Nat. Rev. Rheumatol. 2009;5:425–431. doi: 10.1038/nrrheum.2009.139. [DOI] [PubMed] [Google Scholar]
- 40.Xu L., Zhang L., Wang Z., Li C., Li S., Li L., Fan Q., Zheng L. Melatonin suppresses estrogen deficiency-induced osteoporosis and promotes osteoblastogenesis by inactivating the NLRP3 inflammasome. Calcif. Tissue Int. 2018;103:400–410. doi: 10.1007/s00223-018-0428-y. [DOI] [PubMed] [Google Scholar]
- 41.Rosenberg J.H., Rai V., Dilisio M.F., Sekundiak T.D., Agrawal D.K. Increased expression of damage-associated molecular patterns (DAMPs) in osteoarthritis of human knee joint compared to hip joint. Mol. Cell. Biochem. 2017;436:59–69. doi: 10.1007/s11010-017-3078-x. [DOI] [PubMed] [Google Scholar]
- 42.Koh S.M., Chan C.K., Teo S.H., Singh S., Merican A., Ng W.M., Abbas A., Kamarul T. Elevated plasma and synovial fluid interleukin-8 and interleukin-18 may be associated with the pathogenesis of knee osteoarthritis. Knee. 2020;27:26–35. doi: 10.1016/j.knee.2019.10.028. [DOI] [PubMed] [Google Scholar]
- 43.Zhao L.R., Xing R.L., Wang P.M., Zhang N.S., Yin S.J., Li X.C., Zhang L. NLRP1 and NLRP3 inflammasomes mediate LPS/ATP-induced pyroptosis in knee osteoarthritis. Mol. Med. Rep. 2018;17:5463–5469. doi: 10.3892/mmr.2018.8520. [DOI] [PubMed] [Google Scholar]
- 44.McAllister M.J., Chemaly M., Eakin A.J., Gibson D.S., McGilligan V.E. NLRP3 as a potentially novel biomarker for the management of osteoarthritis. Osteoarthritis Cartilage. 2018;26:612–619. doi: 10.1016/j.joca.2018.02.901. [DOI] [PubMed] [Google Scholar]
- 45.Mullen L.M., Chamberlain G., Sacre S. Pattern recognition receptors as potential therapeutic targets in inflammatory rheumatic disease. Arthritis Res. Ther. 2015;17:122. doi: 10.1186/s13075-015-0645-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Huang Z., Kraus V.B. Does lipopolysaccharide-mediated inflammation have a role in OA? Nat. Rev. Rheumatol. 2016;12:123–129. doi: 10.1038/nrrheum.2015.158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Gratal P., Lamuedra A., Medina J.P., Bermejo-Álvarez I., Largo R., Herrero-Beaumont G., Mediero A. Purinergic system signaling in metainflammation-associated osteoarthritis. Front. Med. 2020;7:506. doi: 10.3389/fmed.2020.00506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Chen Y., Fujuan Q., Chen E., Yu B., Zuo F., Yuan Y., Zhao X., Xiao C. Expression of AIM2 in rheumatoid arthritis and its role on fibroblast-like synoviocytes. Mediators Inflamm. 2020;2020:1693730. doi: 10.1155/2020/1693730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Jia M., Lv Y., Xu Y., Gong Z. A comparative analysis of NLRP3-related inflammatory mediators in synovial fluid in temporomandibular joint osteoarthritis and internal derangement. BMC Musculoskelet Disord. 2021;22:229. doi: 10.1186/s12891-021-04092-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Bougault C., Gosset M., Houard X., Salvat C., Godmann L., Pap T., Jacques C., Berenbaum F. Stress-induced cartilage degradation does not depend on the NLRP3 inflammasome in human osteoarthritis and mouse models. Arthritis Rheum. 2012;64:3972–3981. doi: 10.1002/art.34678. [DOI] [PubMed] [Google Scholar]
- 51.Nasi S., Ea H.K., So A., Busso N. Revisiting the role of interleukin-1 pathway in osteoarthritis: interleukin-1α and -1β, and NLRP3 inflammasome are not involved in the pathological features of the murine menisectomy model of osteoarthritis. Front. Pharmacol. 2017;8:282. doi: 10.3389/fphar.2017.00282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.He Z., Nie P., Lu J., Ling Y., Guo J., Zhang B., Hu J., Liao J., Gu J., Dai B., Feng Z. Less mechanical loading attenuates osteoarthritis by reducing cartilage degeneration, subchondral bone remodelling, secondary inflammation, and activation of NLRP3 inflammasome. Bone Joint Res. 2020;9:731–741. doi: 10.1302/2046-3758.910.Bjr-2019-0368.R2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Dong H.C., Li P.N., Chen C.J., Xu X., Zhang H., Liu G., Zheng L.J., Li P. Sinomenine attenuates cartilage degeneration by regulating miR-223-3p/NLRP3 inflammasome signaling. Inflammation. 2019;42:1265–1275. doi: 10.1007/s10753-019-00986-3. [DOI] [PubMed] [Google Scholar]
- 54.Zu Y., Mu Y., Li Q., Zhang S.T., Yan H.J. Icariin alleviates osteoarthritis by inhibiting NLRP3-mediated pyroptosis. J. Orthop. Surg. Res. 2019;14:307. doi: 10.1186/s13018-019-1307-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Cheng F., Yan F.F., Liu Y.P., Cong Y., Sun K.F., He X.M. Dexmedetomidine inhibits the NF-κB pathway and NLRP3 inflammasome to attenuate papain-induced osteoarthritis in rats. Pharm. Biol. 2019;57:649–659. doi: 10.1080/13880209.2019.1651874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Sun X., Xiao L., Chen J., Chen X., Chen X., Yao S., Li H., Zhao G., Ma J. DNA methylation is involved in the pathogenesis of osteoarthritis by regulating CtBP expression and CtBP-mediated signaling. Int. J. Biol. Sci. 2020;16:994–1009. doi: 10.7150/ijbs.39945. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Xu J., Pei Y., Lu J., Liang X., Li Y., Wang J., Zhang Y. LncRNA SNHG7 alleviates IL-1β-induced osteoarthritis by inhibiting miR-214-5p-mediated PPARGC1B signaling pathways. Int. Immunopharmacol. 2021;90:107150. doi: 10.1016/j.intimp.2020.107150. [DOI] [PubMed] [Google Scholar]
- 58.Xu H., Zhang J., Shi X., Li X., Zheng C. NF-κB inducible miR-30b-5p aggravates joint pain and loss of articular cartilage via targeting SIRT1-FoxO3a-mediated NLRP3 inflammasome. Aging (Albany NY) 2021;13:20774–20792. doi: 10.18632/aging.203466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Gao F., Zhang S. Loratadine alleviates advanced glycation end product-induced activation of NLRP3 inflammasome in human chondrocytes. Drug Des. Devel Ther. 2020;14:2899–2908. doi: 10.2147/dddt.S243512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Jiang J., Cai M. Cardamonin inhibited IL-1β induced injury by inhibition of NLRP3 inflammasome via activating nrf2/NQO-1 signaling pathway in chondrocyte. J. Microbiol. Biotechnol. 2021;31:794–802. doi: 10.4014/jmb.2103.03057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Liu G., Liu Q., Yan B., Zhu Z., Xu Y. USP7 inhibition alleviates H(2)O(2)-induced injury in chondrocytes via inhibiting NOX4/NLRP3 pathway. Front. Pharmacol. 2020;11:617270. doi: 10.3389/fphar.2020.617270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Li X., Mei W., Huang Z., Zhang L., Zhang L., Xu B., Shi X., Xiao Y., Ma Z., Liao T., et al. Casticin suppresses monoiodoacetic acid-induced knee osteoarthritis through inhibiting HIF-1α/NLRP3 inflammasome signaling. Int. Immunopharmacol. 2020;86:106745. doi: 10.1016/j.intimp.2020.106745. [DOI] [PubMed] [Google Scholar]
- 63.Zhang L., Li X., Zhang H., Huang Z., Zhang N., Zhang L., Xing R., Wang P. Agnuside alleviates synovitis and fibrosis in knee osteoarthritis through the inhibition of HIF-1α and NLRP3 inflammasome. Mediators Inflamm. 2021;2021:5534614. doi: 10.1155/2021/5534614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Chen Z., Zhong H., Wei J., Lin S., Zong Z., Gong F., Huang X., Sun J., Li P., Lin H., et al. Inhibition of Nrf2/HO-1 signaling leads to increased activation of the NLRP3 inflammasome in osteoarthritis. Arthritis Res. Ther. 2019;21:300. doi: 10.1186/s13075-019-2085-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Fan C., Zhao X., Guo X., Cao X., Cai J. P2X4 promotes interleukin-1β production in osteoarthritis via NLRP1. Mol. Med. Rep. 2014;9:340–344. doi: 10.3892/mmr.2013.1748. [DOI] [PubMed] [Google Scholar]
- 66.Anderson J.R., Chokesuwattanaskul S., Phelan M.M., Welting T.J.M., Lian L.Y., Peffers M.J., Wright H.L. (1)H NMR metabolomics identifies underlying inflammatory pathology in osteoarthritis and rheumatoid arthritis synovial joints. J. Proteome Res. 2018;17:3780–3790. doi: 10.1021/acs.jproteome.8b00455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Kolly L., Busso N., Palmer G., Talabot-Ayer D., Chobaz V., So A. Expression and function of the NALP3 inflammasome in rheumatoid synovium. Immunology. 2010;129:178–185. doi: 10.1111/j.1365-2567.2009.03174.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Shi J., Zhao W., Ying H., Zhang Y., Du J., Chen S., Li J., Shen B. Estradiol inhibits NLRP3 inflammasome in fibroblast-like synoviocytes activated by lipopolysaccharide and adenosine triphosphate. Int J. Rheum Dis. 2018;21:2002–2010. doi: 10.1111/1756-185x.13198. [DOI] [PubMed] [Google Scholar]
- 69.Ea H.K., Chobaz V., Nguyen C., Nasi S., van Lent P., Daudon M., Dessombz A., Bazin D., McCarthy G., Jolles-Haeberli B., et al. Pathogenic role of basic calcium phosphate crystals in destructive arthropathies. PLoS One. 2013;8:e57352. doi: 10.1371/journal.pone.0057352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Cookson B.T., Brennan M.A. Pro-inflammatory programmed cell death. Trends Microbiol. 2001;9:113–114. doi: 10.1016/s0966-842x(00)01936-3. [DOI] [PubMed] [Google Scholar]
- 71.Yu P., Zhang X., Liu N., Tang L., Peng C., Chen X. Pyroptosis: mechanisms and diseases. Signal Transduct. Target Ther. 2021;6:128. doi: 10.1038/s41392-021-00507-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Deets K.A., Vance R.E. Inflammasomes and adaptive immune responses. Nat. Immunol. 2021;22:412–422. doi: 10.1038/s41590-021-00869-6. [DOI] [PubMed] [Google Scholar]
- 73.Lambert C., Zappia J., Sanchez C., Florin A., Dubuc J.E., Henrotin Y. The damage-associated molecular patterns (DAMPs) as potential targets to treat osteoarthritis: perspectives from a review of the literature. Front. Med. 2020;7:607186. doi: 10.3389/fmed.2020.607186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Wu T., Zhang X.P., Zhang Q., Zou Y.Y., Ma J.D., Chen L.F., Zou Y.W., Xue J.M., Ma R.F., Chen Z., Dai L. Gasdermin-E mediated pyroptosis-A novel mechanism regulating migration, invasion and release of inflammatory cytokines in rheumatoid arthritis fibroblast-like synoviocytes. Front. Cell Dev. Biol. 2021;9:810635. doi: 10.3389/fcell.2021.810635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Zhai Z., Yang F., Xu W., Han J., Luo G., Li Y., Zhuang J., Jie H., Li X., Shi X., et al. Attenuation of rheumatoid arthritis through the inhibition of tumor necrosis factor-induced caspase 3/gasdermin E-mediated pyroptosis. Arthritis Rheumatol. 2022;74:427–440. doi: 10.1002/art.41963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Hou J., Zhao R., Xia W., Chang C.W., You Y., Hsu J.M., Nie L., Chen Y., Wang Y.C., Liu C., et al. PD-L1-mediated gasdermin C expression switches apoptosis to pyroptosis in cancer cells and facilitates tumour necrosis. Nat. Cell Biol. 2020;22:1264–1275. doi: 10.1038/s41556-020-0575-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Xu B., Xing R., Huang Z., Yin S., Li X., Zhang L., Ding L., Wang P. Excessive mechanical stress induces chondrocyte apoptosis through TRPV4 in an anterior cruciate ligament-transected rat osteoarthritis model. Life Sci. 2019;228:158–166. doi: 10.1016/j.lfs.2019.05.003. [DOI] [PubMed] [Google Scholar]
- 78.Barreto G., Manninen M., K K.E. Osteoarthritis and toll-like receptors: when innate immunity meets chondrocyte apoptosis. Biology. 2020;9 doi: 10.3390/biology9040065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Zhang L., Qiu J., Shi J., Liu S., Zou H. MicroRNA-140-5p represses chondrocyte pyroptosis and relieves cartilage injury in osteoarthritis by inhibiting cathepsin B/Nod-like receptor protein 3. Bioengineered. 2021;12:9949–9964. doi: 10.1080/21655979.2021.1985342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Cheung H.S., Sallis J.D., Demadis K.D., Wierzbicki A. Phosphocitrate blocks calcification-induced articular joint degeneration in a Guinea pig model. Arthritis Rheum. 2006;54:2452–2461. doi: 10.1002/art.22017. [DOI] [PubMed] [Google Scholar]
- 81.Sun Y., Kiraly A.J., Cox M., Mauerhan D.R., Hanley E.N., Jr. The role of inhibition by phosphocitrate and its analogue in chondrocyte differentiation and subchondral bone advance in Hartley Guinea pigs. Exp. Ther. Med. 2018;15:3320–3328. doi: 10.3892/etm.2018.5846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Sun Y., Mauerhan D.R., Franklin A.M., Norton J., Hanley E.N., Jr., Gruber H.E. Phosphocitrate is potentially a disease-modifying drug for noncrystal-associated osteoarthritis. BioMed Res. Int. 2013;2013:326267. doi: 10.1155/2013/326267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Sun Y., Kiraly A.J., Sun A.R., Cox M., Mauerhan D.R., Hanley E.N., Jr. Effects of a phosphocitrate analogue on osteophyte, subchondral bone advance, and bone marrow lesions in Hartley Guinea pigs. Bone Joint Res. 2018;7:157–165. doi: 10.1302/2046-3758.72.Bjr-2017-0253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Li Z., Huang Z., Zhang H., Lu J., Tian Y., Wei Y., Yang Y., Bai L. P2X7 receptor induces pyroptotic inflammation and cartilage degradation in osteoarthritis via NF-κB/NLRP3 crosstalk. Oxid. Med. Cell. Longev. 2021;2021:8868361. doi: 10.1155/2021/8868361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Li Z., Huang Z., Zhang H., Lu J., Tian Y., Piao S., Lin Z., Bai L. Moderate-intensity exercise alleviates pyroptosis by promoting autophagy in osteoarthritis via the P2X7/AMPK/mTOR axis. Cell Death Discov. 2021;7:346. doi: 10.1038/s41420-021-00746-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Li Z., Huang Z., Bai L. The P2X7 receptor in osteoarthritis. Front. Cell Dev. Biol. 2021;9:628330. doi: 10.3389/fcell.2021.628330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Hu H., Yang B., Li Y., Zhang S., Li Z. Blocking of the P2X7 receptor inhibits the activation of the MMP-13 and NF-κB pathways in the cartilage tissue of rats with osteoarthritis. Int. J. Mol. Med. 2016;38:1922–1932. doi: 10.3892/ijmm.2016.2770. [DOI] [PubMed] [Google Scholar]
- 88.Dunton C.L., Purves J.T., Hughes F.M., Jr., Jin H., Nagatomi J. Elevated hydrostatic pressure stimulates ATP release which mediates activation of the NLRP3 inflammasome via P2X(4) in rat urothelial cells. Int. Urol. Nephrol. 2018;50:1607–1617. doi: 10.1007/s11255-018-1948-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Sun Y., Liu W., Zhang H., Li H., Liu J., Zhang F., Jiang T., Jiang S. Curcumin prevents osteoarthritis by inhibiting the activation of inflammasome NLRP3. J. Interferon Cytokine Res. 2017;37:449–455. doi: 10.1089/jir.2017.0069. [DOI] [PubMed] [Google Scholar]
- 90.Wang C., Gao Y., Zhang Z., Chen C., Chi Q., Xu K., Yang L. Ursolic acid protects chondrocytes, exhibits anti-inflammatory properties via regulation of the NF-κB/NLRP3 inflammasome pathway and ameliorates osteoarthritis. Biomed. Pharmacother. 2020;130:110568. doi: 10.1016/j.biopha.2020.110568. [DOI] [PubMed] [Google Scholar]
- 91.Yan Z., Qi W., Zhan J., Lin Z., Lin J., Xue X., Pan X., Zhou Y. Activating Nrf2 signalling alleviates osteoarthritis development by inhibiting inflammasome activation. J. Cell Mol. Med. 2020;24:13046–13057. doi: 10.1111/jcmm.15905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Franco-Trepat E., Alonso-Pérez A., Guillán-Fresco M., Jorge-Mora A., Crespo-Golmar A., López-Fagúndez M., Pazos-Pérez A., Gualillo O., Bravo S.B., Bahamonde R.G. Amitriptyline blocks innate immune responses mediated by toll-like receptor 4 and IL-1 receptor: preclinical and clinical evidence in osteoarthritis and gout. Br. J. Pharmacol. 2022;179:270–286. doi: 10.1111/bph.15707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Li W., Wang Y., Tang Y., Lu H., Qi Y., Li G., He H., Lu F., Yang Y., Sun H. Quercetin alleviates osteoarthritis progression in rats by suppressing inflammation and apoptosis via inhibition of IRAK1/NLRP3 signaling. J. Inflamm. Res. 2021;14:3393–3403. doi: 10.2147/jir.S311924. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Zhang Y., Lin Z., Chen D., He Y. CY-09 attenuates the progression of osteoarthritis via inhibiting NLRP3 inflammasome-mediated pyroptosis. Biochem. Biophys. Res. Commun. 2021;553:119–125. doi: 10.1016/j.bbrc.2021.03.055. [DOI] [PubMed] [Google Scholar]
- 95.Ni B., Pei W., Qu Y., Zhang R., Chu X., Wang Y., Huang X., You H. MCC950, the NLRP3 inhibitor, protects against cartilage degradation in a mouse model of osteoarthritis. Oxid. Med. Cell. Longev. 2021;2021:4139048. doi: 10.1155/2021/4139048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Zhu X., Dai S., Xia B., Gong J., Ma B. Activation of the alpha 7 nicotinic acetylcholine receptor mitigates osteoarthritis progression by inhibiting NF-κB/NLRP3 inflammasome activation and enhancing autophagy. PLoS One. 2021;16:e0256507. doi: 10.1371/journal.pone.0256507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Starobova H., Nadar E.I., Vetter I. The NLRP3 inflammasome: role and therapeutic potential in pain treatment. Front. Physiol. 2020;11:1016. doi: 10.3389/fphys.2020.01016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Ma Z., Huang Z., Zhang L., Li X., Xu B., Xiao Y., Shi X., Zhang H., Liao T., Wang P. Vanillic acid reduces pain-related behavior in knee osteoarthritis rats through the inhibition of NLRP3 inflammasome-related synovitis. Front. Pharmacol. 2020;11:599022. doi: 10.3389/fphar.2020.599022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Liao T., Ding L., Wu P., Zhang L., Li X., Xu B., Zhang H., Ma Z., Xiao Y., Wang P. Chrysin attenuates the NLRP3 inflammasome cascade to reduce synovitis and pain in KOA rats. Drug Des. Devel Ther. 2020;14:3015–3027. doi: 10.2147/dddt.S261216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Wang S., Mobasheri A., Zhang Y., Wang Y., Dai T., Zhang Z. Exogenous stromal cell-derived factor-1 (SDF-1) suppresses the NLRP3 inflammasome and inhibits pyroptosis in synoviocytes from osteoarthritic joints via activation of the AMPK signaling pathway. Inflammopharmacology. 2021;29:695–704. doi: 10.1007/s10787-021-00814-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Baron L., Gombault A., Fanny M., Villeret B., Savigny F., Guillou N., Panek C., Le Bert M., Lagente V., Rassendren F., et al. The NLRP3 inflammasome is activated by nanoparticles through ATP, ADP and adenosine. Cell Death Dis. 2015;6:e1629. doi: 10.1038/cddis.2014.576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Li C., Li L., Lan T. Co-treatment with disulfiram and glycyrrhizic acid suppresses the inflammatory response of chondrocytes. J. Orthop. Surg. Res. 2021;16:132. doi: 10.1186/s13018-021-02262-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Hu J., Zhou J., Wu J., Chen Q., Du W., Fu F., Yu H., Yao S., Jin H., Tong P., et al. Loganin ameliorates cartilage degeneration and osteoarthritis development in an osteoarthritis mouse model through inhibition of NF-κB activity and pyroptosis in chondrocytes. J. Ethnopharmacol. 2020;247:112261. doi: 10.1016/j.jep.2019.112261. [DOI] [PubMed] [Google Scholar]
- 104.Yu H., Yao S., Zhou C., Fu F., Luo H., Du W., Jin H., Tong P., Chen D., Wu C., Ruan H. Morroniside attenuates apoptosis and pyroptosis of chondrocytes and ameliorates osteoarthritic development by inhibiting NF-κB signaling. J. Ethnopharmacol. 2021;266:113447. doi: 10.1016/j.jep.2020.113447. [DOI] [PubMed] [Google Scholar]
- 105.Liu Q., Wu Z., Hu D., Zhang L., Wang L., Liu G. Low dose of indomethacin and Hedgehog signaling inhibitor administration synergistically attenuates cartilage damage in osteoarthritis by controlling chondrocytes pyroptosis. Gene. 2019;712:143959. doi: 10.1016/j.gene.2019.143959. [DOI] [PubMed] [Google Scholar]
- 106.Wang Q., Ying L., Wei B., Ji Y., Xu Y. Effects of quercetin on apoptosis and extracellular matrix degradation of chondrocytes induced by oxidative stress-mediated pyroptosis. J. Biochem. Mol. Toxicol. 2022;36:e22951. doi: 10.1002/jbt.22951. [DOI] [PubMed] [Google Scholar]
- 107.Yang T., Sun K., Wang C., Swarnkar G., Quan S., Kress D., Xiao J., Alippe Y., Zheng H., Brophy R.H., et al. Gasdermin D deficiency attenuates arthritis induced by traumatic injury but not autoantibody-assembled immune complexes. Arthritis Res. Ther. 2021;23:286. doi: 10.1186/s13075-021-02668-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Jiang K., Tu Z., Chen K., Xu Y., Chen F., Xu S., Shi T., Qian J., Shen L., Hwa J., et al. Gasdermin D inhibition confers antineutrophil-mediated cardioprotection in acute myocardial infarction. J. Clin. Invest. 2022;132 doi: 10.1172/jci151268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Hu J.J., Liu X., Xia S., Zhang Z., Zhang Y., Zhao J., Ruan J., Luo X., Lou X., Bai Y., et al. FDA-approved disulfiram inhibits pyroptosis by blocking gasdermin D pore formation. Nat. Immunol. 2020;21:736–745. doi: 10.1038/s41590-020-0669-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Humphries F., Shmuel-Galia L., Ketelut-Carneiro N., Li S., Wang B., Nemmara V.V., Wilson R., Jiang Z., Khalighinejad F., Muneeruddin K., et al. Succination inactivates gasdermin D and blocks pyroptosis. Science. 2020;369:1633–1637. doi: 10.1126/science.abb9818. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Gregor M.F., Hotamisligil G.S. Inflammatory mechanisms in obesity. Annu. Rev. Immunol. 2011;29:415–445. doi: 10.1146/annurev-immunol-031210-101322. [DOI] [PubMed] [Google Scholar]
- 112.Aravindhan V., Madhumitha H. Metainflammation in diabetic coronary artery disease: emerging role of innate and adaptive immune responses. J. Diabetes Res. 2016;2016:6264149. doi: 10.1155/2016/6264149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Wang X., Wang S., Hu C., Chen W., Shen Y., Wu X., Sun Y., Xu Q. A new pharmacological effect of levornidazole: inhibition of NLRP3 inflammasome activation. Biochem. Pharmacol. 2015;97:178–188. doi: 10.1016/j.bcp.2015.06.030. [DOI] [PubMed] [Google Scholar]
- 114.Misawa T., Saitoh T., Kozaki T., Park S., Takahama M., Akira S. Resveratrol inhibits the acetylated α-tubulin-mediated assembly of the NLRP3-inflammasome. Int. Immunol. 2015;27:425–434. doi: 10.1093/intimm/dxv018. [DOI] [PubMed] [Google Scholar]
- 115.Wang S., Moustaid-Moussa N., Chen L., Mo H., Shastri A., Su R., Bapat P., Kwun I., Shen C.L. Novel insights of dietary polyphenols and obesity. J. Nutr. Biochem. 2014;25:1–18. doi: 10.1016/j.jnutbio.2013.09.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Favero G., Franceschetti L., Bonomini F., Rodella L.F., Rezzani R. Melatonin as an anti-inflammatory agent modulating inflammasome activation. Int J. Endocrinol. 2017;2017:1835195. doi: 10.1155/2017/1835195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Juliana C., Fernandes-Alnemri T., Wu J., Datta P., Solorzano L., Yu J.W., Meng R., Quong A.A., Latz E., Scott C.P., Alnemri E.S. Anti-inflammatory compounds parthenolide and Bay 11-7082 are direct inhibitors of the inflammasome. J. Biol. Chem. 2010;285:9792–9802. doi: 10.1074/jbc.M109.082305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Kim Y., Wang W., Okla M., Kang I., Moreau R., Chung S. Suppression of NLRP3 inflammasome by γ-tocotrienol ameliorates type 2 diabetes. J. Lipid Res. 2016;57:66–76. doi: 10.1194/jlr.M062828. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Sebastian A., McCool J.L., Hum N.R., Murugesh D.K., Wilson S.P., Christiansen B.A., Loots G.G. Single-cell RNA-seq reveals transcriptomic heterogeneity and post-traumatic osteoarthritis-associated early molecular changes in mouse articular chondrocytes. Cells. 2021;10 doi: 10.3390/cells10061462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Huang Z.Y., Luo Z.Y., Cai Y.R., Chou C.H., Yao M.L., Pei F.X., Kraus V.B., Zhou Z.K. Single cell transcriptomics in human osteoarthritis synovium and in silico deconvoluted bulk RNA sequencing. Osteoarthritis Cartilage. 2022;30:475–480. doi: 10.1016/j.joca.2021.12.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Wei K., Korsunsky I., Marshall J.L., Gao A., Watts G.F.M., Major T., Croft A.P., Watts J., Blazar P.E., Lange J.K., et al. Notch signalling drives synovial fibroblast identity and arthritis pathology. Nature. 2020;582:259–264. doi: 10.1038/s41586-020-2222-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Zhang F., Wei K., Slowikowski K., Fonseka C.Y., Rao D.A., Kelly S., Goodman S.M., Tabechian D., Hughes L.B., Salomon-Escoto K., et al. Defining inflammatory cell states in rheumatoid arthritis joint synovial tissues by integrating single-cell transcriptomics and mass cytometry. Nat. Immunol. 2019;20:928–942. doi: 10.1038/s41590-019-0378-1. [DOI] [PMC free article] [PubMed] [Google Scholar]