

Summary
Growth differentiation factor 15 (GDF15) is a stress-induced secreted protein whose circulating levels are increased in the context of obesity. Recombinant GDF15 reduces body weight and improves glycemia in obese models, which is largely attributed to the central action of GDF15 to suppress feeding and reduce body weight. Despite these advances in knowledge, the tissue-specific sites of GDF15 production during obesity are unknown, and the effects of modulating circulating GDF15 levels on insulin sensitivity have not been evaluated directly. Here, we demonstrate that hepatocyte Gdf15 expression is sufficient for changes in circulating levels of GDF15 during obesity and that restoring Gdf15 expression specifically in hepatocytes of Gdf15 knockout mice results in marked improvements in hyperinsulinemia, hepatic insulin sensitivity, and to a lesser extent peripheral insulin sensitivity. These data support that liver hepatocytes are the primary source of circulating GDF15 in obesity.
Subject areas: Cellular physiology, Molecular genetics, Diabetology
Graphical abstract
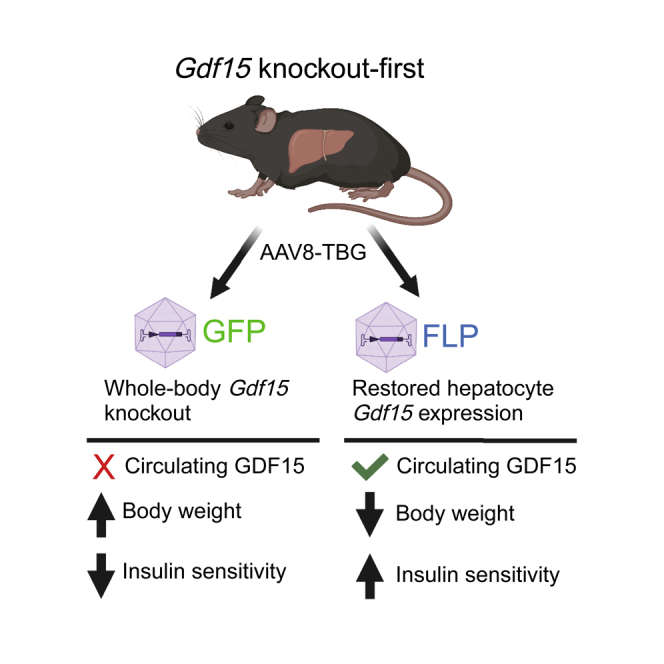
Highlights
-
•
Hepatocytes are the primary source of circulating GDF15 during obesity
-
•
Hepatocyte Gdf15 is sufficient to reverse high-fat diet-induced weight gain
-
•
Restoring hepatocyte Gdf15 expression in Gdf15 knockout mice improves liver insulin sensitivity
Cellular physiology; Molecular genetics; Diabetology
Introduction
Growth differentiation factor 15 (GDF15) is a distant member of the transforming growth factor β superfamily and is a stress-induced cytokine.1,2 Circulating levels of GDF15 are increased in a range of pathological states, including obesity,3 heart and kidney failure,4,5,6,7 interstitial lung disease,8 nonalcoholic steatohepatitis (NASH) and chronic liver disease,9,10 and in patients with cancer.11,12,13 Although GDF15 is firmly established as a biomarker of multiple disease states, whether it contributes to disease progression or plays a role in mitigating disease remains under debate. Indeed, GDF15 has been attributed pleiotropic and often conflicting roles in regulating cancer progression,14 energy metabolism,15,16 cellular migration,17,18 and inflammation.19,20 The recent identification of GDF15’s receptor, GDNF family receptor α-like (GFRAL), and its co-receptor, RET, casts many of these previous findings into new light.16,21,22,23 The fact that GFRAL expression is restricted to the hindbrain, where it signals to suppress feeding,24 suggests many of the proposed actions of GDF15 may be indirect and a consequence of altered feeding or body weight, or alternatively due to centrally mediated signals to the periphery.20 A potential exception to that rule, however, was recently reported, where the anti-inflammatory drug colchicine was shown to suppress myeloid cell activation via a GFRAL-independent mechanism that required intact hepatocyte expression of Gdf15, implicating an unidentified GDF15-induced hepatokine or GDF15 cleavage product.25 These novel observations and unresolved questions regarding the direct and indirect effects of GDF15 demonstrate the need for further study and more refined genetic approaches to delineate tissue-specific sites of GDF15 production and action in the context of multiple disease states.
With regards to obesity and its comorbidities, particularly insulin resistance and type 2 diabetes, evidence that endogenous GDF15 may constrain body weight gain and insulin resistance comes from studies in mice lacking Gdf15, as well as studies where circulating GDF15 levels are modulated through a variety of approaches. Gdf15 knockout (KO) mice fed high-fat diet (HFD) eat more, gain more weight, and develop more severe glucose and insulin intolerance.26 In contrast, elevating circulating GDF15 levels in obese models through overexpression or treatment with recombinant GDF15 protein reduces feeding and body weight and improves glucose homeostasis.5,16,27,28 Critically, the tissue-specific site(s) of GDF15 production during obesity are not known. Gdf15 mRNA is induced in the liver and adipose tissue in rodent models of obesity, but not in skeletal muscle, heart, or kidney.28,29 Human data addressing this question are limited, but in a single study comparing lean and obese women, there was no difference in adipose tissue Gdf15 expression.3 With regards to the liver, transcriptomic profiling and immunohistochemistry of samples from patients with moderate nonalcoholic fatty liver disease (NAFLD) and advanced NASH, both of which strongly associate with obesity, demonstrated increased expression of Gdf15 in hepatocytes, epithelial, and immune cells of the liver from NASH samples.30 Together, these observations suggest that in rodents, liver or adipose tissue cell types may contribute to changes in circulating GDF15, whereas in humans, multiple cell types within the liver may be the primary source. We therefore applied a definitive genetic approach to determine the hepatocyte-specific contribution of Gdf15 to changes in circulating GDF15 and effects on obesity and insulin sensitivity. To do so, we utilized a novel Gdf15 KO-first strain of mice where Gdf15 expression can be restored at its endogenous locus in a cell-type-specific fashion via expression of Flp recombinase (FLP), combined with adeno-associated viral (AAV)-mediated delivery of FLP to hepatocytes of adult mice.
Results
Hepatocyte Gdf15 expression is sufficient for obesity-associated increases in plasma GDF15
To determine the hepatocyte-specific contribution of Gdf15 to changes in circulating GDF15 during diet-induced obesity, we acquired a KO-first allele of Gdf15 from the Knockout Mouse Project (KOMP) and further manipulated this allele as shown in Figure 1A. In this unique strain, expression of Gdf15 is prevented by a β-galactosidase and neomycin selection (lacZ-neo) cassette that is knocked into the first intron of Gdf15. The lacZ-neo cassette is flanked by FLP recognition target (FRT) sites and can be removed by expression of Flp recombinase. Once removed, Gdf15 is expressed by the endogenous promoter. Ten-week-old Gdf15 KO-first male mice received tail vein injections of AAV of serotype 8 (AAV8) encoding either Gfp or Flp under control of the hepatocyte-specific thyroxine-binding globulin promoter (AAV8-TBG-Gfp or KO + GFP; AAV8-TBG-Flp or KO + FLP). Mice were maintained on a control low-fat diet (LFD; 10% kcal fat) for four weeks prior to initiating a HFD (60% kcal fat) for 12 weeks. In parallel, age-matched C57BL6 mice (wild-type [WT]) received tail vein injections of saline and were maintained on LFD (WT LFD) or HFD (WT HFD) for the same duration (see study timeline Figure 1B). As previously reported,28,29 HFD feeding increased Gdf15 mRNA in the liver and white and brown adipose tissue in WT mice (Figures 1C–1E), while there was no effect of diet on Gdf15 expression in the heart or skeletal muscle (Figures 1F and 1G). There was also a significant increase in kidney Gdf15 expression in WT HFD compared with LFD mice (Figure 1H), which was not observed previously.29 Differences in the effects of HFD feeding on kidney Gdf15 may be due to differences in diets used in the control (standard chow29 versus purified 10% kcal fat here) or experimental groups (45%29 versus 60% kcal fat here). FLP-mediated deletion of the lacZ-neo cassette in hepatocytes of Gdf15 KO mice restored liver Gdf15 expression to levels comparable with WT HFD mice (Figure 1C) without affecting Gdf15 deletion in other tissues (Figures 1D–1H). In situ hybridization studies using RNAscope with custom probes specific to exon 2 of Gdf15 confirmed hepatocyte-specific expression of Gdf15 in the KO + FLP group and no expression in KO + GFP mice (Figure 1I). We further tested the efficiency and specificity of AAV8-TBG-mediated gene delivery in our hands using a membrane-targeted tandem dimer Tomato (mT)/green fluorescent protein (mG) Cre-reporter strain that expresses mT basally and mG in the presence of Cre.31 Two weeks after tail vein injection with AAV8-TBG-Cre, mG fluorescence was detected in essentially 100% of hepatocytes in liver sections from mT/mG mice and not in any other liver cell types (Figure 1J), consistent with past studies establishing hepatocyte-specific delivery using AAV8-TBG.32 Under these conditions, circulating GDF15 was significantly increased in WT mice in response to HFD and undetectable in KO + GFP mice (Figure 1K). Re-expression of endogenous Gdf15 specifically in hepatocytes of Gdf15 KO mice restored plasma GDF15 to levels seen in WT mice fed HFD (Figure 1K), demonstrating that hepatocyte Gdf15 expression is sufficient for obesity-associated increases in circulating GDF15.
Figure 1.

Hepatocyte Gdf15 expression is sufficient for high-fat diet-induced increases in plasma GDF15
(A) Schematic of Gdf15 knockout-first strain and hepatocyte-specific Gdf15 re-expression model.
(B) Study group and timeline overview.
(C–H) Gdf15 mRNA levels relative to Actb expressed as fold-change relative to WT LFD for liver (C), white adipose tissue (D), brown adipose tissue (E), skeletal muscle (F), heart (G) and kidney (H).
(I) RNA in situ hybridization for Gdf15 exon two of liver samples from HFD-fed KO + GFP and KO + FLP mice.
(J) Photomicrographs of frozen liver sections from RosamTmG/mTmG mice after AAV8-TBG-CRE dosing.
(K) Plasma GDF15 levels. Data are the mean ± SEM for n = 6–9/group for (C, I and K), n = 4/group for (D–H). Scale bar in (I) and (J) represents 100 microns. Data analyzed by 1-way ANOVA with multiple comparison testing. ∗p < 0.05, ∗∗p < 0.01, ∗∗∗p < 0.001, ∗∗∗∗p < 0.0001.
Restoring endogenous expression of Gdf15 in hepatocytes reduces weight gain and reverses hyperinsulinemia
Prior to AAV injection, there was no difference in body weight between KO + GFP and KO + FLP groups, and two weeks after injection plasma GDF15 levels were significantly different and approached 60 pg/mL in the KO + FLP group (Figures S1A and S1B). Four weeks after AAV dosing, just prior to initiating HFD, body weight and composition between the two groups were not different, although there was a slight trend toward reduced lean mass in the KO + FLP group (p = 0.09; Figures S1C–S1E). HFD feeding significantly increased body weight in WT mice, and KO + GFP mice weighed significantly more than WT HFD mice (Figure 2A), as previously reported.26 KO + GFP mice also weighed significantly more than KO + FLP mice, and there was no difference in body weight when comparing WT HFD and KO + FLP mice (Figure 2A). The differences in body weight between LFD and HFD WT mice were primarily due to significant changes in fat and not lean mass, whereas KO + GFP mice weighed more than HFD WT mice due to significant increases in both fat and lean mass (Figures 2B and 2C). Surprisingly, the differences in body weight between KO + GFP and KO + FLP mice were due to significant changes in lean but not fat mass, where lean mass was on average 3.7 g less (p < 0.01; Figure 2C) and fat mass was 1.6 g less (p = 0.21; Figure 2B). Next, we measured liver triglyceride levels and detected a significant 4-fold increase in WT HFD compared with LFD WT mice and a significant 2-fold increase in KO + GFP compared with HFD WT (Figure 2D), consistent with previous reports indicating Gdf15 deficiency exacerbates diet-induced NAFLD.9 Steatosis was marginally reduced in KO + FLP compared with KO + GFP mice (20% decrease; p = 0.17 Figure 2D). Plasma triglycerides were significantly increased in WT HFD and KO + GFP compared with WT LFD and significantly reduced in KO + FLP mice compared with WT HFD but not KO + FLP (Figure 2E). We found no differences in nonesterified fatty acids (NEFAs) in KO + GFP or KO + FLP mice compared with one another or other groups (Figure 2F), whereas HFD WT mice had significantly reduced NEFAs compared with LFD WT (Figure 2F). Plasma insulin levels were significantly increased in KO + GFP mice compared with HFD WT and LFD WT mice (Figure 2G). This hyperinsulinemia in the absence of GDF15 was completely reversed in the KO + FLP mice whose plasma insulin levels were significantly less compared with KO + GFP mice (Figure 2G).
Figure 2.

Hepatocyte-specific restoration of Gdf15 expression reduces body weight and reverses hyperinsulinemia in Gdf15 knockout-first mice
(A) Body weight after LFD (WT; 10% kcal fat) and HFD (WT, KO + GFP, KO + FLP; 60% kcal fat) feeding.
(B and C) Fat and lean mass measured by 1H-NMR.
(D) Liver triglyceride levels expressed as mg per g liver.
(E–G) Plasma levels of triglycerides (E), nonesterified fatty acids (NEFA; F) and insulin (G). Data are the mean ± SEM for n = 6–9/group. Data analyzed by 1-way ANOVA with multiple comparison testing. ∗p < 0.05, ∗∗p < 0.01, ∗∗∗p < 0.001, ∗∗∗∗p < 0.0001.
Glucose intolerance and insulin resistance are reversed by hepatic Gdf15 expression
To better understand the differences in body weight and composition between KO + GFP and KO + FLP groups, we studied mice in metabolic cages two weeks after initiating the HFD such that body weight and composition were similar between groups (Figures S2A–S2C). We detected no differences in total activity, feeding, energy expenditure, or respiratory exchange ratio between groups (Figures S2D–S2G). We reassessed feeding after ten weeks of HFD feeding when differences in body weight were present. Mice were individually housed for 12 days, and food was weighed and replaced twice daily (8a.m. and 5p.m.). Feeding was reduced by 8% in KO + FLP mice when expressed on a per mouse basis (Figure 3A; p < 0.05). There were no differences in feeding when expressed per kg body weight or lean mass (Figures 3B and 3C). Next, we performed glucose tolerance tests after a 6-h morning fast. Fasting plasma glucose levels were significantly reduced and glucose tolerance was markedly improved in KO + FLP mice, as indicated by significantly reduced plasma glucose levels at several time points during the 120-min study and area under curve for the change in plasma glucose levels (Figures 3D–3F). Given the improved hyperinsulinemia and glucose tolerance in KO + FLP mice, and to ascertain tissue-specific contributions to changes in whole-body glucose homeostasis, we assessed insulin sensitivity by hyperinsulinemic euglycemic clamps. Clamps were performed after an overnight fast to reduce the influence of altered feeding on basal insulin levels and hepatic glycogen content between groups. Plasma glucose levels were matched at approximately 200 mg/dL during the hyperinsulinemic infusion (Figure 3G upper panel & 3H), and the glucose infusion rate (GIR) required to maintain euglycemia was approximately 5-fold greater in KO + FLP compared with KO + GFP mice (Figure 3G lower panel and 3I), demonstrating significantly improved whole-body insulin sensitivity in the KO + FLP mice. The 15.1 mg x kg−1 x min−1 improvement in whole-body insulin sensitivity (Figure 3I) was accounted for by a 4.4 mg × kg−1 × min−1 increase in whole-body glucose uptake and a 10.7 mg × kg−1 × min−1 increase in the suppression of endogenous or hepatic glucose production (EGP) in the KO + FLP mice (Figures 3J and 3K). These data indicated that most of the improvement in insulin sensitivity in the KO + FLP mice was due to changes in the liver compared with peripheral insulin action (70 versus 30%, respectively). Consistent with improved hyperinsulinemia in the ad libitum fed state (Figure 2G), fasting plasma insulin levels prior to the clamp were significantly reduced in KO + FLP mice (Figure 3L). And despite receiving matched insulin infusion rates during the clamp, plasma insulin levels were significantly lower in KO + FLP mice (Figure 3L), suggesting enhanced hepatic insulin clearance.
Figure 3.

Hepatocyte-specific restoration of GDF15 improves glucose homeostasis and insulin sensitivity in Gdf15 knockout-first mice
(A) Feeding per mouse based on change in food weight measured twice daily over 12 days for individually housed mice.
(B and C) Feeding data in A normalized per kg body weight (B) or lean mass (C).
(D) Plasma glucose levels measured after a 6 h morning fast.
(E) Plasma glucose levels during glucose tolerance test using 1 g/kg glucose dose.
(F) Area under the curve (AUC) for glucose in E.
(G) Plasma glucose levels (upper panel) and glucose infusion rate (GIR; lower panel) during hyperinsulinemic euglycemic clamp.
(H) Average plasma glucose levels during steady state or last 40 min of clamp.
(I) GIR during steady state.
(J) Whole-body glucose disposal during steady state.
(K) Endogenous glucose production (EGP) during steady state.
(L) Basal and clamped plasma insulin levels. Data are the mean ± SEM for n = 8–9 for (A–F) and n = 7–11 for (G–L). Data analyzed by Student’s t test or 2-way RM ANOVA. ∗p < 0.05, ∗∗p < 0.01, p < 0.001, ∗∗∗∗p < 0.0001.
Discussion
Using an in vivo genetic deletion and conditional, tissue-specific re-expression approach, we find that hepatocyte Gdf15 is sufficient for changes in circulating GDF15 and subsequent effects on constrained body weight and improved insulin sensitivity in an experimental model of obesity. Given past reports regarding changes in Gdf15 expression that suggested liver or adipose tissue as potential sites for increased circulating GDF15 in obesity,3,28,29,30 our data strongly support the conclusion that hepatocytes are the primary source of GDF15. There was, however, a nonsignificant 30% reduction in plasma GDF15 in the KO + FLP mice compared with HFD WT mice (Figure 1K), suggesting adipose tissue also contributes, albeit to a lesser extent, to circulating GDF15 in mice. During review of our manuscript, a report describing a Gdf15 liver-specific KO line was published that arrived at this same conclusion.33 Deletion of Gdf15 in liver hepatocytes reduced circulating GDF15 by 50% in HFD-fed mice and resulted in increased body weight, increased liver fat content, and impaired glucose and insulin tolerance. Furthermore, using a variety of approaches, including measuring gene expression from specific cell populations within the stromal vascular or adipocyte fractions of adipose tissue, RNAscope for Gdf15 mRNA in adipose tissue, and myeloid cell-specific Gdf15 KO mice and WT mice with Gdf15 KO bone marrow transplants, the authors demonstrated that changes in adipose tissue Gdf15 expression during obesity are likely due to changes in expression within immune cells and not adipocytes.33 Despite the modest difference in plasma GDF15 in the KO + FLP mice compared with WT HFD, the subsequent effects of restoring plasma GDF15 on feeding (Figure 3A), body weight and composition (Figures 2A–2C), and insulin sensitivity (Figure 3I), demonstrate that hepatocyte Gdf15 is sufficient for the physiological effects of GDF15.
The observation that restoring circulating GDF15 reduced increases in lean mass more than increases in fat mass (Figures 2B and 2C) came as a surprise based on the assumption that modest reductions in feeding (Figure 3A; ∼8%) would affect adiposity. Indeed, modest increases in feeding (8%) in GDF15 KO mice resulted in increased weight gain due to increased adiposity,26 and reductions in feeding in transgenic mice overexpressing Gdf15 resulted in reduced body weight due to reduced fat mass.27 Interestingly, differences in body weight in HFD-fed liver-specific Gdf15 KO mice could not be attributed to changes in adipose tissue weight or increased feeding, suggesting the increased liver weight or potentially changes in lean mass, which were not reported, may have contributed.33 Thus, while a reduction in feeding was consistent with lower body weight gain in our studies, the greater effect on lean mass and observations made in the liver-specific Gdf15 KO mice suggest feeding alone did not account for changes in body composition. One possibility is that the marked hyperinsulinemia (Figure 2G) in the KO + GFP mice, alongside more severe hepatic, as opposed to peripheral insulin resistance (Figures 3J and 3K), promoted greater increases in lean mass and to a lesser extent fat mass. Indeed, baseline differences in insulin dynamics and resistance were recently shown to affect changes in body composition following weight loss.34 Although not directly comparable to our model, these observations may provide insight into the unique changes in body composition observed here and suggest that these may relate to the pattern of improved insulin sensitivity.
Past studies addressing the mechanism by which GDF15 reduces body weight suggested increased energy expenditure, as opposed to reduced feeding, as a primary mechanism.15,35 Differences in body weight during metabolic cage studies in these reports, however, make interpretation of the normalized data difficult.36 More recently, identification of GFRAL and pair-feeding studies argued strongly that reduced feeding is the primary mechanism by which GDF15 reduces body weight.16,21 Our metabolic cage data in body weight-matched mice detected no differences in energy expenditure, activity, or feeding (Figures S2D–S2G), whereas manually performed feeding studies demonstrated reduced feeding in KO + FLP mice (Figure 2A), consistent with the conclusion that GDF15 reduces body weight via effects on feeding. The differences in feeding data in our studies performed at two and ten weeks after starting HFD may result from the time-dependent effects of HFD on circulating GDF15 levels, where significant increases in plasma GDF15 were reported to occur at eight but not four weeks of HFD.29 Thus, while we know that at two weeks post-AAV treatment plasma GDF15 levels were increased in KO + FLP mice (Figure S1B), circulating levels at two weeks post-HFD may have been insufficient to suppress feeding, whereas at ten weeks levels were likely high enough to produce this well-established effect of GDF15.
Past studies evaluating the effects of modulating GDF15 levels on glucose homeostasis in obese models relied solely on glucose tolerance tests, typically without concurrent measures of plasma insulin, limiting conclusions regarding insulin sensitivity and tissue-specific effects. These studies consistently report that GDF15 improves glucose tolerance,16,22,28 while the absence of GDF15 in KO models impairs glucose tolerance or increases fasting plasma glucose and insulin levels.9,26 Here, we addressed these limitations by reporting the first hyperinsulinemic euglycemic clamp in a GDF15-null model (Figure 3). We found that restoring circulating GDF15 markedly improved insulin sensitivity primarily due to enhanced suppression of endogenous (hepatic) glucose production by insulin (Figures 3I–3K). The pronounced effect of hepatocyte-Gdf15 expression on hepatic insulin sensitivity was also evident due to an apparent increase in insulin clearance (Figure 3L). Interestingly, despite improvements in hepatic insulin sensitivity, liver triglyceride levels, which are positively associated with insulin resistance in most settings, were not significantly reduced in the KO + FLP mice (Figure 2D). Liver triglyceride levels were, however, 20% less in the KO + FLP mice (p = 0.16 compared with KO + GFP), suggesting reductions in specific lipid species directly implicated in the pathogenesis of insulin resistance, such as ceramides or diacylglycerols,37,38 may have contributed to the phenotype. While it is tempting to speculate that restored expression of hepatocyte Gdf15 produced local effects with regards to insulin responsiveness, the fact that body weight was reduced in our model at the time of the clamp precludes any conclusions regarding GFRAL or feeding-independent effects. Future studies of this nature in body weight-matched or GFRAL-null animals will be of great interest.
Limitations of the study
One of the limitations of our study is the lack of mechanistic understanding for the reported change in body composition, specifically the more pronounced effect of restoring hepatocyte Gdf15 on lean as opposed to fat mass. In addition, although our main conclusion is that the hepatocyte is the primary source of circulating GDF15 during obesity, we cannot completely rule out the role of other cell types due to the 30% difference in circulating GDF15 levels that persisted when comparing HFD WT and KO + FLP mice. Although work by Patel and colleagues suggests that adipocytes do not contribute to circulating GDF15 levels during obesity,33 definitive adipose-specific Gdf15 KO studies have not yet been performed. An additional limitation to our studies was that assessments of glucose homeostasis and insulin sensitivity were only made in KO + GFP and KO + FLP groups. We focused our attention on these models because comparisons of glucose homeostasis in WT and Gdf15 KO were reported previously, and we expected generalized effects on insulin sensitivity secondary to changes in body weight. Given the more pronounced effects of restored hepatic Gdf15 on liver insulin sensitivity, inclusion of WT HFD mice would have allowed for a more complete interpretation of this unique phenotype. Also, our experimental design limited, in some ways, comparison to past studies regarding the effects of GDF15 on obesity, which are primarily intervention studies, as opposed to prevention studies, like ours. However, our studies were designed to test the question of hepatocyte sufficiency with regards to changes in plasma GDF15 during obesity, and future studies using this model are already underway to address questions regarding obesity intervention and body weight-independent effects of hepatocyte Gdf15 expression.
STAR★Methods
Key resources table
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Bacterial and virus strains | ||
| AAV8-TBG-GFP | Vector Biolabs | VB1728 |
| AAV8-TBG-FLP | Vector Biolabs | VB1724 |
| Chemicals, peptides, and recombinant proteins | ||
| 3-3H-glucose | Perkin Elmer | NET331C001MC |
| Insulin | Novolin-R, Novo Nordisk | NDC 0169-1833-11 |
| Dextrose | Sigma Aldrich | D9434 |
| Critical commercial assays | ||
| Qiagen RNeasy kit | Qiagen | 74104 |
| Quantitect Reverse Transcriptase Kit | Qiagen | 205313 |
| PowerUp SYBR Green Master Mix | Thermo Fisher | A25778 |
| GDF15 ELISA | R&D systems | MGD150 |
| RNAscope 2.5 High Definition-BROWN assay | Bio-Techne | 322300 |
| Hematoxylin QS | Vector laboratories | H-3404-100 |
| Total Cholesterol Kit, Wako Diagnostics | Fisher Scientific | NC9138103 |
| NEFA HR(2) solvent B, Wako Diagnostics | Fisher Scientific | NC9517311 |
| NEFA HR(2) solvent A, Wako Diagnostics | Fisher Scientific | NC9517309 |
| NEFA HR(2) Color reagent B, Wako Diagnostics | Fisher Scientific | NC9517310 |
| NEFA HR(2) Color reagent A, Wako Diagnostics | Fisher Scientific | NC9517308 |
| Triglyceride Infinity Reagent | Thermo Fisher | TR22421 |
| STELLUX Chemiluminescent Rodent Insulin ELISA | ALPCO Diagnostics | 80-INSMR-CH10 |
| Experimental models: Organisms/strains | ||
| Mouse: C57BL6/J mice (wildtype) | Jackson Laboratory | 000664 |
| Mouse sperm: Gdf15 knockout-first C57BL/6N-Gdf15tm1a(KOMP)Wtsi/H | Knock Out Mouse Project (KOMP) | EMMA Strain ID 06354 |
| Mouse: RosamTmG; B6.129(Cg)- Gt(ROSA)26Sortm4 (ACTB-tdTomato,−EGFP)Luo/J | Jackson Laboratory | 007676 |
| Mouse: C57BL6/J diet-induced obese mice | Jackson Laboratory | 380050 |
| Oligonucleotides | ||
| Gdf15 primers Predesigned and validated by IDT | Integrated DNA Technologies (IDT) | Mm.PT.58.13112185 |
| Actb; FWD GCAGCTCCTTCGTTGCCGGT; REV TACAGCCCGGGGAGCATCGT); Designed using IDT online software | IDT | N/A |
| Custom RNAscope probe targeting Gdf15 exon 2 | Bio-Techne | 300040 |
| Software and algorithms | ||
| Prism | GraphPad Software | Version 9.3.1 |
| Other | ||
| Mouse Diet: Regular chow diet | ProLab IsoPro | RMH 3000 |
| Mouse Diet: control low-fat diet (LFD) | Research Diets | D12450J |
| Mouse Diet: experimental high-fat diet (HFD) | Research Diets | D12492 |
Resource availability
Lead contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the lead contact, Michael J. Jurczak (jurczakm@pitt.edu).
Materials availability
The unique reagents and materials presented within this manuscript will be made available upon request.
Experimental model details
Animal
All animal studies were approved by the Institutional Animal Care and Use Committee at the University of Pittsburgh. Mice were housed at 22 ± 2 degrees Celsius at the University of Pittsburgh on a 12 h light/12 h dark cycle from 7am to 7pm, and provided ad libitum access to food and water except where noted for specific experimental endpoints. Eight-week-old C57BL6/J male mice were purchased from Jackson Labs (stock 000664) and allowed to recover two weeks prior to initiating studies described below. Gdf15 knockout-first (C57BL/6N-Gdf15tm1a(KOMP)Wtsi/H; EMMA Strain ID 06354) sperm was obtained from the Knock Out Mouse Project (KOMP) and used to fertilize C57BL6/J ovum prior to implanting into pseudopregnant wild-type C57BL6/J female mice at the University of Pittsburgh Mouse Embryo Services Core. Gdf15 knockout-first mice were backcrossed to the C57BL6/J strain a minimum of three times prior to study. RosamTmG mice were purchased from Jackson Labs (B6.129(Cg)-Gt(ROSA)26Sortm4(ACTB-tdTomato,-EGFP)Luo/J; stock 007676). Ten-week-old Gdf15 knockout-first mice received 1 × 1012 genomic copies (GC) per mouse of AAV8-TBG-GFP or AAV8-TBG-FLP by tail vein injection (Vector Biolabs; stocks VB1728 and VB1724. Ten-week-old male WT mice received volume-matched tail vein injections of saline. After tail vein injection, regular chow diets (ProLab IsoPro RMH 3000) were replaced with control low-fat diet (LFD, Research Diets, D12450J) for all mice. After four weeks of control LFD, half of the WT mice and all the Gdf15 knockout-first mice (GFP and FLP) were transitioned to experimental high-fat diet (HFD, Research Diets, D12492). Mice were maintained on diets for an additional 12 weeks. A second cohort of Gdf15 knockout-first mice received AAV8-TBG-GFP or FLP, as before, and underwent the same dietary protocol before undergoing hyperinsulinemic euglycemic clamps, as described below.
Method details
Gdf15 expression and circulating GDF15 levels
RNA was extracted using the Qiagen RNeasy kit per the manufacturer’s instructions. RNA was used to generate cDNA using the Quantitect Reverse Transcriptase Kit, followed by quantitative PCR (QPCR) using the Applied Biosystems QuantStudio3 System and PowerUp SYBR Green Master Mix. Primers were purchased from Integrated DNA Technologies (IDT) and either designed using IDT online software or were predesigned and validated by IDT. Sequences for designed primers were as follows: Actb (FWD GCAGCTCCTTCGTTGCCGGT; REV TACAGCCCGGGGAGCATCGT). Sequences for predesigned primers for Gdf15 are available upon purchase (IDT assay ID: MmPT.58.13112185). Primer efficiency was calculated from a four-point standard curve and used to calculate Gdf15 expression relative to Actb. Final data was expressed as fold-change relative to WT LFD on a per-tissue basis. Circulating GDF15 levels were measured using commercially available ELISA kits according to the manufacture’s protocol (R&D systems; MGD150).
RNA in situ hybridization and imaging
Because the first exon of Gdf15 is predicted to be expressed in Gdf15 knock-out first mice, we requested a custom RNAscope probe design that targeted the second exon of Gdf15 (Mm-Gdf15-O2; targeting c.764-1534 of NM_011819.3). This custom probe was used in conjunction with the RNAscope 2.5 High Definition-BROWN assay to stain formalin-fixed paraffin embedded liver sections according to the manufacture’s recommendations. Slides were counterstained with hematoxylin QS (Vector laboratories). For analysis of RosamTmG mice, livers were fixed in 4% PFA for two hours prior to cryoprotection in 30% sucrose in PBS overnight. The following morning, livers were embedded in OCT and sectioned. Sections were washed in PBS, stained with DAPI, and imaged immediately. All images were captured with a Nikon Eclipse Ni-E upright microscope equipped with both ORCA-FLASH 4.0 and DS-Fi3 cameras for widefield and brightfield imaging, respectively.
Plasma biochemistry and liver triglyceride measure
Whole blood was collected by cardiac puncture during dissection and transferred to heparin coated tubes for centrifugation to obtain plasma. Plasma non-esterified fatty acid and cholesterol levels were measured using kits from Wako Diagnostics according to the manufacturer’s instructions. Plasma triglyceride levels were measured using the Infinity Triglyceride Reagent (Thermo Fisher TR22421). Plasma insulin levels were measured using a Stellux Chemiluminescent Rodent Insulin ELISA (Alpco 80-INSMR-CH01). Blood glucose levels during the glucose tolerance tests were measured using a Bayer Contour Next EZ glucometer. Plasma glucose levels during the hyperinsulinemic euglycemic clamp were measured by the glucose oxidase method using the Analox GM9 Glucose Analyzer.
Glucose homeostasis and insulin resistance
Assessments of glucose homeostasis were performed according to recommendations from the Mouse Metabolic Phenotyping Consortium.39 Glucose tolerance tests were performed after a 6 h morning fast. Mice received a 1 g/kg dose of glucose by intraperitoneal injection and blood samples were collected at set time points by tail massage to determine plasma glucose levels. Hyperinsulinemic euglycemic clamps were performed as previously described with minor modifications.40 Five days prior to study, an indwelling catheter was surgically placed in the right jugular vein and passed subcutaneously such that it could be concealed in the scapular region on the dorsal side of the mouse. Mice were fasted overnight prior to study. Fasted and insulin-stimulated rates of glucose turnover were measured by isotope dilution using a primed/continuous infusion of 3-3H-glucose (Perkin Elmer NET331C001MC; prime = 0.7 μCi/kg over 3 min; 0.05 μCi/min fasted, 0.1 μCi/min clamp). Insulin was provided as a primed/continuous infusion (Novolin-R, Novo Nordisk; prime = 30 mU/kg over 3 min; continuous = 4.5 mU × kg−1 × min−1). Plasma glucose levels were checked every 10 min during the hyperinsulinemic infusion by tail vein message to collect blood and euglycemia was matched between groups using a variable infusion of 20% dextrose (Sigma Aldrich D9434).
Metabolic cage studies and body composition
Mice were housed in the Sable Systems Promethion Multi-plexed Mouse Metabolic Cage System for 72 h in a home-cage setting during which activity, feeding, drinking, energy expenditure by indirect calorimetry and respiratory exchange ratio were recorded. The first 24 h of housing was considered acclimation and the subsequent 48 h was used for data analysis. Body composition (fat and lean mass) was measured by EchoMRI.
Quantification and statistical analysis
Data shown are the mean ± the standard error of the mean. Data were compared by 1-way ANOVA followed by multiple comparison testing using Tukey’s correction for multiple comparison testing. Data were also analyzed by Student’s t-test and 2-way ANOVA where appropriate and noted. A p-value less than 0.05 was considered significant. Data was analyzed using GraphPad Prism version 9.3.1.
Acknowledgments
Studies were supported by funds provided by the University of Pittsburgh (MJJ), the Pittsburgh Foundation (MR2020 109502), and National Institutes of Health grant R01HL135062 (JKA) and R01DK114012 (MJJ), as well as through support from the Center for Metabolism and Mitochondrial Medicine.
Author contributions
M.J.J. and J.K.A. conceived the study. B.X., A.M., A.M.V., A.A.S., F.M.B., I.S., B.C., H.B., F.M.S., N.D., J.K.A. and M.J.J. performed experiments. B.X., I.S., J.K.A., and M.J.J. analyzed data and interpreted results. B.X., A.M., F.M.S., C.P.O., J.K.A., and M.J.J. designed and planned the experiments. A.M., J.C., J.K.A., and M.J.J. wrote the manuscript. All authors contributed to the preparation of manuscript and approved the final version.
Declaration of interests
The authors declare no competing interests.
Inclusion and diversity
We support inclusive, diverse, and equitable conduct of research.
Published: December 22, 2022
Footnotes
Supplemental information can be found online at https://doi.org/10.1016/j.isci.2022.105569.
Contributor Information
Jonathan K. Alder, Email: jalder@pitt.edu.
Michael J. Jurczak, Email: jurczakm@pitt.edu.
Supplemental information
Data and code availability
-
•
All data reported in this paper will be shared by the lead contact upon request.
-
•
This paper does not report original code.
-
•
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.
References
- 1.Bootcov M.R., Bauskin A.R., Valenzuela S.M., Moore A.G., Bansal M., He X.Y., Zhang H.P., Donnellan M., Mahler S., Pryor K., et al. MIC-1, a novel macrophage inhibitory cytokine, is a divergent member of the TGF-beta superfamily. Proc. Natl. Acad. Sci. USA. 1997;94:11514–11519. doi: 10.1073/pnas.94.21.11514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Lockhart S.M., Saudek V., O’Rahilly S. GDF15: a hormone conveying somatic distress to the brain. Endocr. Rev. 2020;41:bnaa007. doi: 10.1210/endrev/bnaa007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Dostálová I., Roubícek T., Bártlová M., Mráz M., Lacinová Z., Haluzíková D., Kaválková P., Matoulek M., Kasalicky M., Haluzík M. Increased serum concentrations of macrophage inhibitory cytokine-1 in patients with obesity and type 2 diabetes mellitus: the influence of very low calorie diet. Eur. J. Endocrinol. 2009;161:397–404. doi: 10.1530/EJE-09-0417. [DOI] [PubMed] [Google Scholar]
- 4.Kempf T., von Haehling S., Peter T., Allhoff T., Cicoira M., Doehner W., Ponikowski P., Filippatos G.S., Rozentryt P., Drexler H., et al. Prognostic utility of growth differentiation factor-15 in patients with chronic heart failure. J. Am. Coll. Cardiol. 2007;50:1054–1060. doi: 10.1016/j.jacc.2007.04.091. [DOI] [PubMed] [Google Scholar]
- 5.Johnen H., Lin S., Kuffner T., Brown D.A., Tsai V.W.-W., Bauskin A.R., Wu L., Pankhurst G., Jiang L., Junankar S., et al. Tumor-induced anorexia and weight loss are mediated by the TGF-β superfamily cytokine MIC-1. Nat. Med. 2007;13:1333–1340. doi: 10.1038/nm1677. [DOI] [PubMed] [Google Scholar]
- 6.Stiermaier T., Adams V., Just M., Blazek S., Desch S., Schuler G., Thiele H., Eitel I. Growth differentiation factor-15 in Takotsubo cardiomyopathy: diagnostic and prognostic value. Int. J. Cardiol. 2014;173:424–429. doi: 10.1016/j.ijcard.2014.03.014. [DOI] [PubMed] [Google Scholar]
- 7.Breit S.N., Carrero J.J., Tsai V.W.-W., Yagoutifam N., Luo W., Kuffner T., Bauskin A.R., Wu L., Jiang L., Barany P., et al. Macrophage inhibitory cytokine-1 (MIC-1/GDF15) and mortality in end-stage renal disease. Nephrol. Dial. Transplant. 2012;27:70–75. doi: 10.1093/ndt/gfr575. [DOI] [PubMed] [Google Scholar]
- 8.Zhang Y., Jiang M., Nouraie M., Roth M.G., Tabib T., Winters S., Chen X., Sembrat J., Chu Y., Cardenes N., et al. GDF15 is an epithelial-derived biomarker of idiopathic pulmonary fibrosis. Am. J. Physiol. Lung Cell Mol. Physiol. 2019;317:L510–L521. doi: 10.1152/ajplung.00062.2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Kim K.H., Kim S.H., Han D.H., Jo Y.S., Lee Y.-H., Lee M.-S. Growth differentiation factor 15 ameliorates nonalcoholic steatohepatitis and related metabolic disorders in mice. Sci. Rep. 2018;8:6789. doi: 10.1038/s41598-018-25098-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Lee E.S., Kim S.H., Kim H.J., Kim K.H., Lee B.S., Ku B.J. Growth differentiation factor 15 predicts chronic liver disease severity. Gut Liver. 2017;11:276–282. doi: 10.5009/gnl16049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Koopmann J., Buckhaults P., Brown D.A., Zahurak M.L., Sato N., Fukushima N., Sokoll L.J., Chan D.W., Yeo C.J., Hruban R.H., et al. Serum macrophage inhibitory cytokine 1 as a marker of pancreatic and other periampullary cancers. Clin. Cancer Res. 2004;10:2386–2392. doi: 10.1158/1078-0432.CCR-03-0165. [DOI] [PubMed] [Google Scholar]
- 12.Lerner L., Gyuris J., Nicoletti R., Gifford J., Krieger B., Jatoi A. Growth differentiating factor-15 (GDF-15): a potential biomarker and therapeutic target for cancer-associated weight loss. Oncol. Lett. 2016;12:4219–4223. doi: 10.3892/ol.2016.5183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Buckhaults P., Rago C., St Croix B., Romans K.E., Saha S., Zhang L., Vogelstein B., Kinzler K.W. Secreted and cell surface genes expressed in benign and malignant colorectal Tumors1. Cancer Res. 2001;61:6996–7001. [PubMed] [Google Scholar]
- 14.Emmerson P.J., Duffin K.L., Chintharlapalli S., Wu X. GDF15 and growth control. Front. Physiol. 2018;9:1712. doi: 10.3389/fphys.2018.01712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Chrysovergis K., Wang X., Kosak J., Lee S.-H., Kim J.S., Foley J.F., Travlos G., Singh S., Baek S.J., Eling T.E. NAG-1/GDF-15 prevents obesity by increasing thermogenesis, lipolysis and oxidative metabolism. Int. J. Obes. 2014;38:1555–1564. doi: 10.1038/ijo.2014.27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Mullican S.E., Lin-Schmidt X., Chin C.-N., Chavez J.A., Furman J.L., Armstrong A.A., Beck S.C., South V.J., Dinh T.Q., Cash-Mason T.D., et al. GFRAL is the receptor for GDF15 and the ligand promotes weight loss in mice and nonhuman primates. Nat. Med. 2017;23:1150–1157. doi: 10.1038/nm.4392. [DOI] [PubMed] [Google Scholar]
- 17.Kalli M., Minia A., Pliaka V., Fotis C., Alexopoulos L.G., Stylianopoulos T. Solid stress-induced migration is mediated by GDF15 through Akt pathway activation in pancreatic cancer cells. Sci. Rep. 2019;9:978. doi: 10.1038/s41598-018-37425-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Codó P., Weller M., Kaulich K., Schraivogel D., Silginer M., Reifenberger G., Meister G., Roth P. Control of glioma cell migration and invasiveness by GDF-15. Oncotarget. 2016;7:7732–7746. doi: 10.18632/oncotarget.6816. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Abulizi P., Loganathan N., Zhao D., Mele T., Zhang Y., Zwiep T., Liu K., Zheng X. Growth differentiation factor-15 deficiency augments inflammatory response and exacerbates septic heart and renal injury induced by lipopolysaccharide. Sci. Rep. 2017;7:1037. doi: 10.1038/s41598-017-00902-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Luan H.H., Wang A., Hilliard B.K., Carvalho F., Rosen C.E., Ahasic A.M., Herzog E.L., Kang I., Pisani M.A., Yu S., et al. GDF15 is an inflammation-induced central mediator of tissue tolerance. Cell. 2019;178:1231–1244.e11. doi: 10.1016/j.cell.2019.07.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Yang L., Chang C.-C., Sun Z., Madsen D., Zhu H., Padkjær S.B., Wu X., Huang T., Hultman K., Paulsen S.J., et al. GFRAL is the receptor for GDF15 and is required for the anti-obesity effects of the ligand. Nat. Med. 2017;23:1158–1166. doi: 10.1038/nm.4394. [DOI] [PubMed] [Google Scholar]
- 22.Emmerson P.J., Wang F., Du Y., Liu Q., Pickard R.T., Gonciarz M.D., Coskun T., Hamang M.J., Sindelar D.K., Ballman K.K., et al. The metabolic effects of GDF15 are mediated by the orphan receptor GFRAL. Nat. Med. 2017;23:1215–1219. doi: 10.1038/nm.4393. [DOI] [PubMed] [Google Scholar]
- 23.Hsu J.-Y., Crawley S., Chen M., Ayupova D.A., Lindhout D.A., Higbee J., Kutach A., Joo W., Gao Z., Fu D., et al. Non-homeostatic body weight regulation through a brainstem-restricted receptor for GDF15. Nature. 2017;550:255–259. doi: 10.1038/nature24042. [DOI] [PubMed] [Google Scholar]
- 24.Wang D., Day E.A., Townsend L.K., Djordjevic D., Jørgensen S.B., Steinberg G.R. GDF15: emerging biology and therapeutic applications for obesity and cardiometabolic disease. Nat. Rev. Endocrinol. 2021;17:592–607. doi: 10.1038/s41574-021-00529-7. [DOI] [PubMed] [Google Scholar]
- 25.Weng J.-H., Koch P.D., Luan H.H., Tu H.-C., Shimada K., Ngan I., Ventura R., Jiang R., Mitchison T.J. Colchicine acts selectively in the liver to induce hepatokines that inhibit myeloid cell activation. Nat. Metab. 2021;3:513–522. doi: 10.1038/s42255-021-00366-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Tran T., Yang J., Gardner J., Xiong Y. GDF15 deficiency promotes high fat diet-induced obesity in mice. PLoS One. 2018;13:e0201584. doi: 10.1371/journal.pone.0201584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Macia L., Tsai V.W.-W., Nguyen A.D., Johnen H., Kuffner T., Shi Y.-C., Lin S., Herzog H., Brown D.A., Breit S.N., et al. Macrophage inhibitory cytokine 1 (MIC-1/GDF15) decreases food intake, body weight and improves glucose tolerance in mice on normal & obesogenic diets. PLoS One. 2012;7:e34868. doi: 10.1371/journal.pone.0034868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Xiong Y., Walker K., Min X., Hale C., Tran T., Komorowski R., Yang J., Davda J., Nuanmanee N., Kemp D., et al. Long-acting MIC-1/GDF15 molecules to treat obesity: evidence from mice to monkeys. Sci. Transl. Med. 2017;9:eaan8732. doi: 10.1126/scitranslmed.aan8732. [DOI] [PubMed] [Google Scholar]
- 29.Patel S., Alvarez-Guaita A., Melvin A., Rimmington D., Dattilo A., Miedzybrodzka E.L., Cimino I., Maurin A.-C., Roberts G.P., Meek C.L., et al. GDF15 provides an endocrine signal of nutritional stress in mice and humans. Cell Metab. 2019;29:707–718.e8. doi: 10.1016/j.cmet.2018.12.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Govaere O., Cockell S., Tiniakos D., Queen R., Younes R., Vacca M., Alexander L., Ravaioli F., Palmer J., Petta S., et al. Transcriptomic profiling across the nonalcoholic fatty liver disease spectrum reveals gene signatures for steatohepatitis and fibrosis. Sci. Transl. Med. 2020;12:eaba4448. doi: 10.1126/scitranslmed.aba4448. [DOI] [PubMed] [Google Scholar]
- 31.Muzumdar M.D., Tasic B., Miyamichi K., Li L., Luo L. A global double-fluorescent Cre reporter mouse. Genesis. 2007;45:593–605. doi: 10.1002/dvg.20335. [DOI] [PubMed] [Google Scholar]
- 32.Russell J.O., Lu W.-Y., Okabe H., Abrams M., Oertel M., Poddar M., Singh S., Forbes S.J., Monga S.P. Hepatocyte-specific β-catenin deletion during severe liver injury provokes cholangiocytes to differentiate into hepatocytes. Hepatology. 2019;69:742–759. doi: 10.1002/hep.30270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Patel S., Haider A., Alvarez-Guaita A., Bidault G., El-Sayed Moustafa J.S., Guiu-Jurado E., Tadross J.A., Warner J., Harrison J., Virtue S., et al. Combined genetic deletion of GDF15 and FGF21 has modest effects on body weight, hepatic steatosis and insulin resistance in high fat fed mice. Mol. Metab. 2022;65:101589. doi: 10.1016/j.molmet.2022.101589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Wong J.M.W., Yu S., Ma C., Mehta T., Dickinson S.L., Allison D.B., Heymsfield S.B., Ebbeling C.B., Ludwig D.S. Stimulated insulin secretion predicts changes in body composition following weight loss in adults with high BMI. J. Nutr. 2022;152:655–662. doi: 10.1093/jn/nxab315. [DOI] [PubMed] [Google Scholar]
- 35.Chung H.K., Ryu D., Kim K.S., Chang J.Y., Kim Y.K., Yi H.-S., Kang S.G., Choi M.J., Lee S.E., Jung S.-B., et al. Growth differentiation factor 15 is a myomitokine governing systemic energy homeostasis. J. Cell Biol. 2017;216:149–165. doi: 10.1083/jcb.201607110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Kaiyala K.J., Schwartz M.W. Toward a more complete (and less controversial) understanding of energy expenditure and its role in obesity pathogenesis. Diabetes. 2011;60:17–23. doi: 10.2337/db10-0909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Samuel V.T., Petersen K.F., Shulman G.I. Lipid-induced insulin resistance: unravelling the mechanism. Lancet. 2010;375:2267–2277. doi: 10.1016/S0140-6736(10)60408-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Summers S.A. Sphingolipids and insulin resistance: the five Ws. Curr. Opin. Lipidol. 2010;21:128–135. doi: 10.1097/MOL.0b013e3283373b66. [DOI] [PubMed] [Google Scholar]
- 39.Ayala J.E., Samuel V.T., Morton G.J., Obici S., Croniger C.M., Shulman G.I., Wasserman D.H., McGuinness O.P., NIH Mouse Metabolic Phenotyping Center Consortium Standard operating procedures for describing and performing metabolic tests of glucose homeostasis in mice. Dis. Model. Mech. 2010;3:525–534. doi: 10.1242/dmm.006239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Edmunds L.R., Xie B., Mills A.M., Huckestein B.R., Undamatla R., Murali A., Pangburn M.M., Martin J., Sipula I., Kaufman B.A., et al. Liver-specific Prkn knockout mice are more susceptible to diet-induced hepatic steatosis and insulin resistance. Mol. Metab. 2020;41:101051. doi: 10.1016/j.molmet.2020.101051. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
-
•
All data reported in this paper will be shared by the lead contact upon request.
-
•
This paper does not report original code.
-
•
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.