

Abstract
Many drugs, or their antecedents, were discovered through observation of their effects on normal or disease physiology. For the past generation, this approach was largely supplanted by the powerful but reductionist approach of modulating specific molecular targets of interest. Modern phenotypic drug discovery (PDD) combines the original concept with modern tools and strategies, and has re-emerged over the past decade to systematically pursue drug discovery based on therapeutic effects in realistic disease models. Here, we discuss recent successes, as well as consider ongoing challenges and approaches to address them. We also explore how innovation in this area may fuel the next generation of successful projects.
Introduction
Historically, new medicines were discovered through observation of their therapeutic effect on disease phenotypes either directly in humans as part of traditional medicine or in models of disease. With the advent of the molecular biology revolution in the 1980s and the sequencing of the human genome in 2001, the focus shifted to specific molecular targets. Since 2011, however, Phenotypic Drug Discovery (PDD) has experienced a major resurgence following the surprising observation that a majority in first in class drugs were discovered empirically without a drug target hypothesis between 1999 and 2008.1 The modern version of this legacy strategy is defined by its focus on the modulation of a disease phenotype or biomarker rather than a pre-specified target to provide a therapeutic benefit.2 Ten years in, PDD is maturing as a field, serving as an accepted discovery modality in both academia and the pharmaceutical industry as opposed to a transient fad. This continued interest is rooted in notable successes in the past decade, including ivacaftor and lumicaftor for cystic fibrosis, risdiplam and branaplam for spinal muscular atrophy (SMA), SEP-363856 for schizophrenia, KAF156 for malaria and crisaborole for atopic dermatitis.
This is not to say that PDD approaches are a magic bullet to address issues with pharmaceutical industry productivity; the pros/cons of phenotypic screening need to be carefully balanced against molecular approaches for validated targets.3 While PDD has been successful, many historical examples used highly complex disease systems (in vivo models and even humans) rather than cell-based screens and/or were the result of serendipitous discoveries (Figure 1),4 and complex models are only now regaining prominence. Drug repurposing provides a compelling example of this state of affairs, with on the one hand well known examples of repurposed drugs based on serendipitous clinical observations (e.g. sildenafil, minoxidil, thalidomide, amantadine) and on the other hand a lack of approved repurposed drugs stemming from pre-planned screens of clinical compound collections.5 This raises the critical question of how best to prospectively approach the discovery of novel drugs using phenotypic screening.
Figure 1:
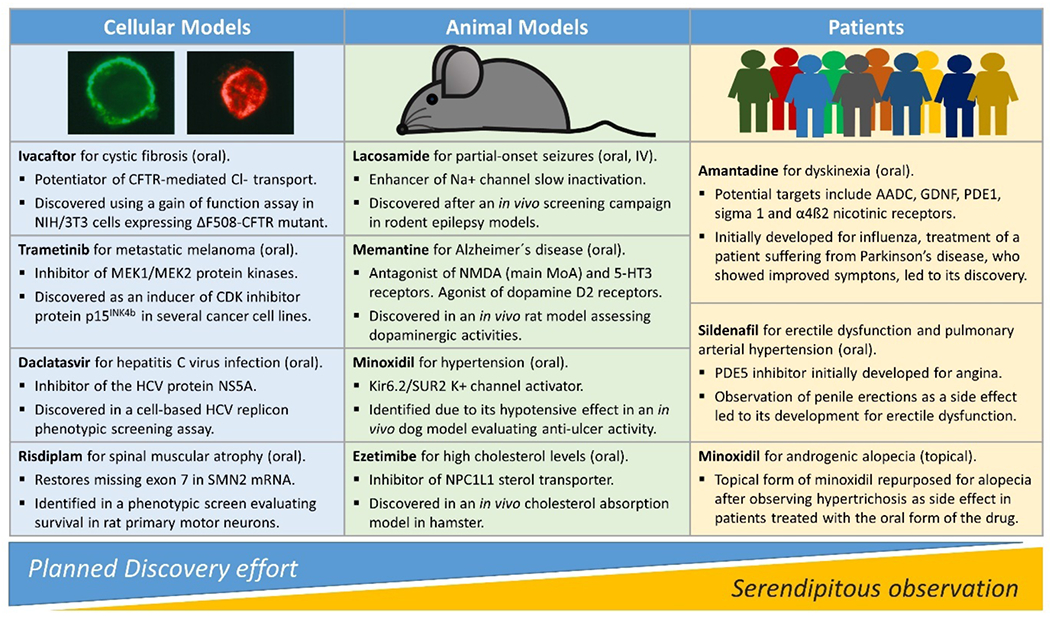
Modern PDD strikes a balance between planned discovery and serendipity. Approved drugs are listed based on the original phenotypic assay which first connected the compound series or the drug itself to the disease. Notably, while all discoveries from cellular screens represented the outcome of planned efforts, it was unexpected clinical side effects in patients which led to compound repurposing. References: ivacaftor,6,7 , daclatasvir,8 risdiplam,9 trametinib,10 lacosamide,11 memantine,12 minoxidil,13,14 ezetimibe,15 amantadine,16 sildenafil.17
In addition to a renewed appreciation for the complexities of physiology and pharmacology, PDD challenges our assumptions in terms of what is druggable with unusual targets and mechanisms of action (MoA), including polypharmacology, and what is a drug with unexpected compound properties. While important hurdles remain in terms of target identification, a helpful step for safety derisking and the mapping of a clinical path, exciting opportunities are emerging for the application of functional genomics, machine learning/artificial intelligence and improved disease models.
Given the major differences between PDD and target-based drug discovery (TDD) and the new technologies that can now be brought to bear on phenotypic programs, the field is evolving at a rapid pace, with a need to establish and share best practices across industry and academia.3,18–21 Although technical and cultural hurdles remain, here, we discuss how the renewed utilization of PDD has started to change the manner in which we conceptualize drug discovery and has proven to be an important testing ground for technical innovations in the life sciences. This perspective highlights the authors’ collective thoughts on how PDD has influenced concepts related to drug discovery and ends with a discussion of the challenges ahead for maximizing the effectiveness of PDD.
Drug discovery concepts recently shaped by PDD
Expansion of “druggable” target space
The main driver for PDD stems from the disproportionate number of first-in-class medicines derived from this approach.1 In contrast to TDD, which is based on an established causal relationship between a molecular target and a disease state, PDD relies on chemical interrogation of a disease-relevant biological system in a molecular-target-agnostic fashion. This empirical, biology-first strategy provides tool molecules to link therapeutic biology to previously unknown signaling pathways, molecular mechanisms and drug targets, as highlighted in the following examples.
Hepatitis C is a liver disease caused by the hepatitis C virus (HCV), which infects 3% of the population and is estimated to cause 300,000 deaths worldwide each year.22 In the past decade, the treatment of HCV has been revolutionized by the development of combinations of orally available direct-acting antivirals (DAAs) that inhibit HCV replication, and clear the virus in >90% of infected patients. Modulators of the HCV protein NS5A such as daclatasvir are a key component of these DAA combinations. The importance of NS5A, which is essential for HCV replication but has no known enzymatic activity, as well as its small-molecule modulators, were initially discovered using a HCV replicon phenotypic screen.23
Cystic fibrosis (CF) is a progressive and frequently fatal genetic disease caused by various mutations in the CF transmembrane conductance regulator (CFTR) gene that decrease CFTR function or interrupt CFTR intracellular folding and plasma membrane insertion.24 Target-agnostic compound screens using cell lines expressing wild-type or disease-associated CFTR variants identified compound classes that improved CFTR channel gating properties (potentiators such as ivacaftor), as well as compounds with an unexpected mechanism of action: enhancing the folding and plasma membrane insertion of CFTR (correctors such as tezacaftor and elexacaftor).6,7 Notably, a combination of elexacaftor, tezacaftor and ivacaftor was approved in 2019 which addresses 90% of the CF patient population.25
Inspired by observations that thalidomide effectively treated leprosy, modulated multiple anti-inflammatory cytokines, inhibited angiogenesis and showed activity in multiple myeloma,26 the optimized analogue lenalidomide gained FDA approval for several blood cancer indications and has been highly successful (sales > $12 billion in 2020).27–29 Significantly, the unprecedented molecular target and MoA of lenalidomide were only elucidated several years post-approval. Lenalidomide binds to the E3 ubiquitin ligase Cereblon and redirects its substrate selectivity to promote the ubiquitination and subsequent degradation of target proteins including the transcription factors IKZF1 and IKZF3.30 Furthermore, this novel MoA is now being intensively explored in the development of further targeted protein degraders, dubbed ‘bifunctional molecular glues’.31
Type 1 SMA is a rare neuromuscular disease with 95% mortality by 18 months of age. SMA is caused by loss-of-function mutations in the SMN1 gene, which encodes a protein known as survival of motor neuron (SMN) that is involved in the formation and maintenance of neuromuscular junctions. Humans also have a very closely related SMN2 gene, but a mutation that affects its splicing leads to exclusion of exon 7 and the production of an unstable shorter SMN variant. Phenotypic screens by two research groups independently identified small molecules that modulate SMN2 pre-mRNA splicing and increase levels of full-length SMN protein.32,33 Both compounds work by engaging two sites at the SMN2 exon 7 and stabilizing the U1 snRNP complex, 32,34,35 an unprecedented drug target and MoA. One such compound, risdiplam, was approved by the FDA in 2020 as the first oral disease-modifying therapy for SMA.
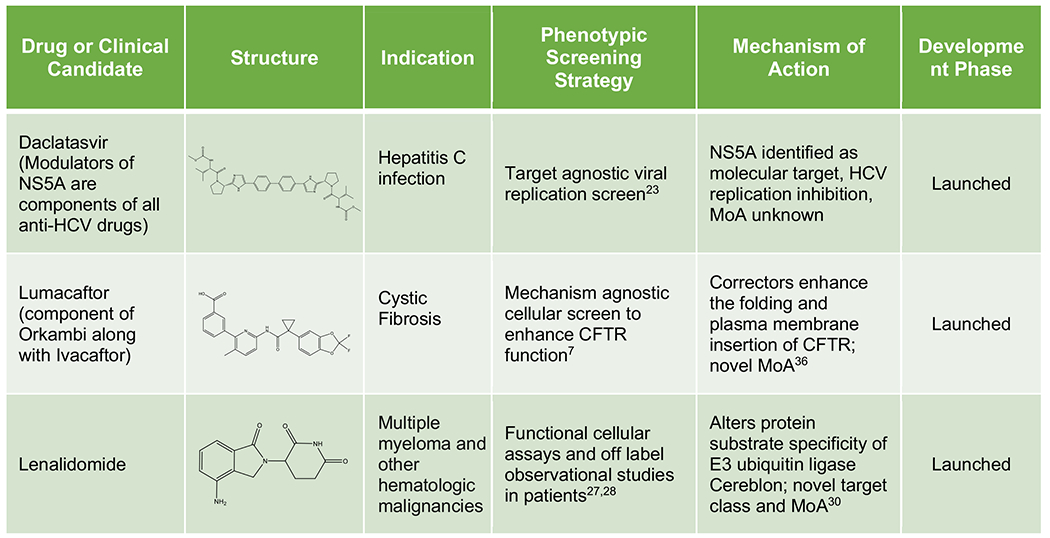
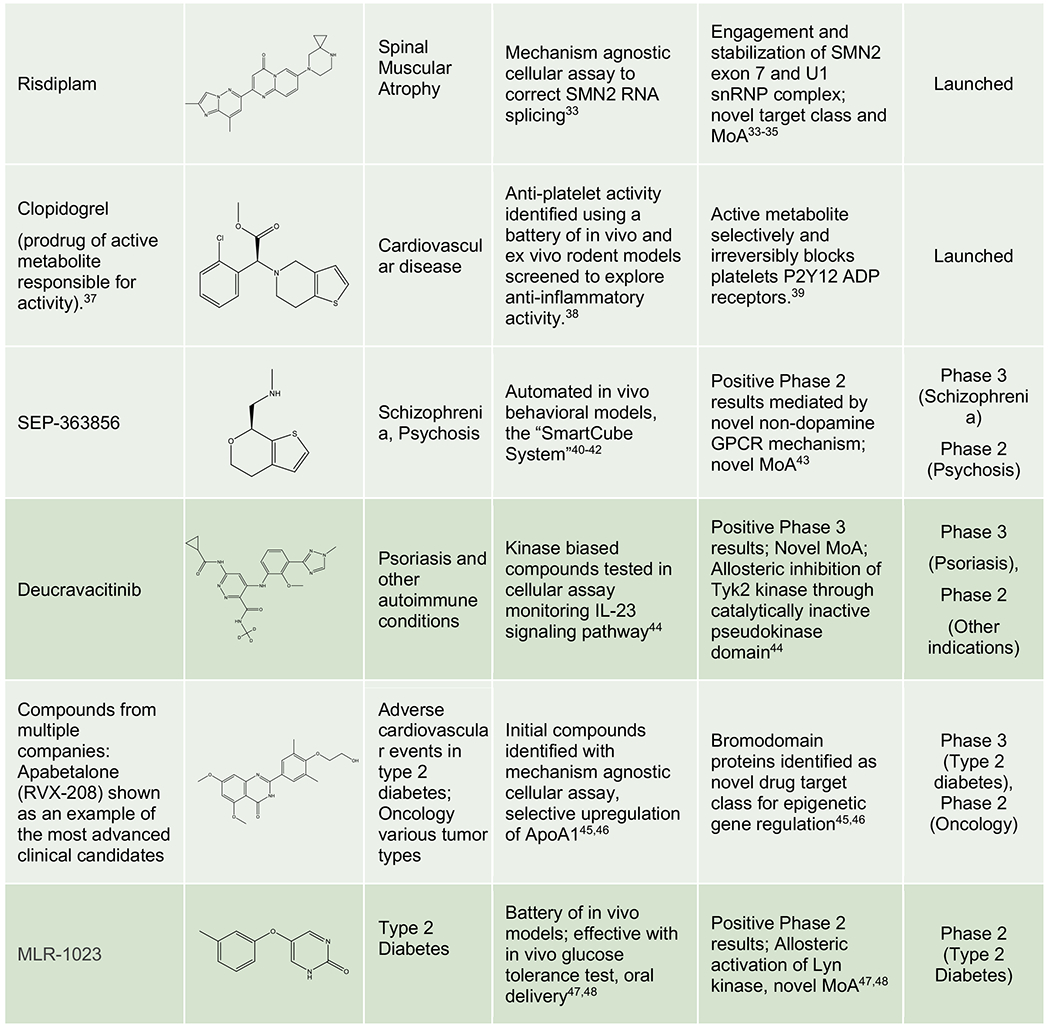
Table 1 presents further recent examples of approved or clinical-stage compounds originating from phenotypic screens, including those where the affected cellular processes are well defined but the specific entity which binds the compound is a multi-component “cellular machine”, a poorly defined molecular target. Taken together, these examples demonstrate how phenotypic strategies have expanded the “druggable target space” to include unexpected cellular processes (pre-mRNA splicing, target protein folding, trafficking, translation, and degradation), novel MoAs for traditional target classes (pseudo-kinase domain inhibition, allosteric kinase activation, masked covalent warhead), and revealed new classes of drug targets (e.g. bromodomains). They suggest that phenotypic strategies should be considered when no attractive target is known to modulate the pathway or disease phenotype of interest and/or the project goal is to obtain a first-in-class drug with a differentiated MoA.
Table 1:
Phenotypic Origins of Approved Drugs and Clinical Phase Molecules
|
|

|
Polypharmacology reexamined
With no restrictions in the available chemical and biological space other than those defined by the compound library and the disease model systems, phenotypic screening offers the opportunity to identify molecules engaging multiple targets in what is otherwise known as polypharmacology.51,52 In this scenario, the intended effect of a compound depends upon a combination of targets (on-targets); however, these are not necessarily its full target signature that may include targets not required for activity (off-targets).
In the quest for ever more selective drugs, polypharmacology has been traditionally associated with poorly optimized compounds prone to potential side effects due to the difficulty in tracking all the biological functions represented by off-targets. However at therapeutically relevant concentrations most, if not all, approved drugs are known to interact with multiple targets that often underlie side effects but can also contribute to clinical efficacy.53–55 In fact, the simultaneous low potency modulation of several targets to achieve efficacy “by synergy” has been suggested as a strategy to minimize side effects.56 One classic TDD example of unintended polypharmacology is imatinib, the first rationally designed kinase inhibitor approved by the FDA for the treatment of chronic myeloid leukemia’s (CML) and other cancers, and currently in clinical development for recent-onset type I diabetes.57 Initially regarded as an inhibitor of CML’s BCR-ABL fusion protein,58 imatinib also exhibits activity towards c-KIT and PDGFR receptor tyrosine kinases, among other targets, which are believed to contribute to its activity in several types of cancer.59,60
Multi-target approaches based on drug combinations, i.e. post-hoc polypharmacology by design, are well-accepted strategies for antiviral and oncology indications for which resistance to treatment can develop when only a single target is engaged.61–64 Polypharmacological drugs are also commonly used to treat central nervous system and heart diseases for which single-target-based approaches have shown limited success and classical in vivo phenotypic models have long been used for drug discovery.40,65,66 Generally speaking, multi-target drugs may be a better match for complex, polygenic diseases with multiple underlying mechanisms often involving interactions with immune or nervous system components.
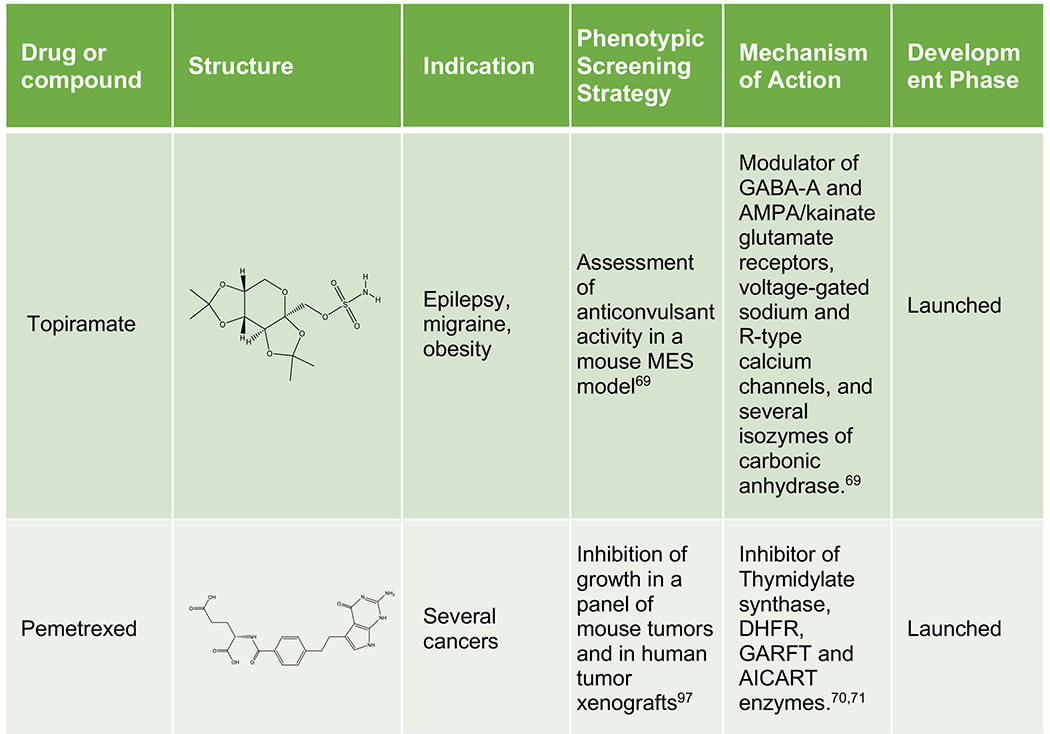
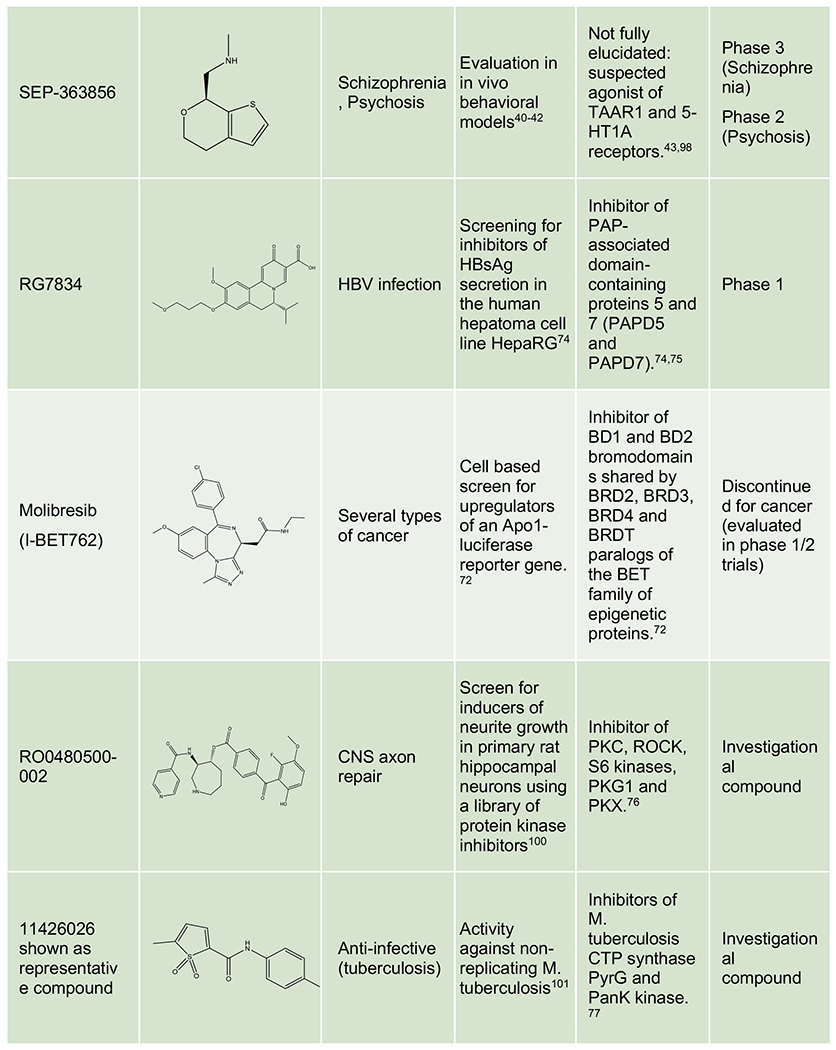
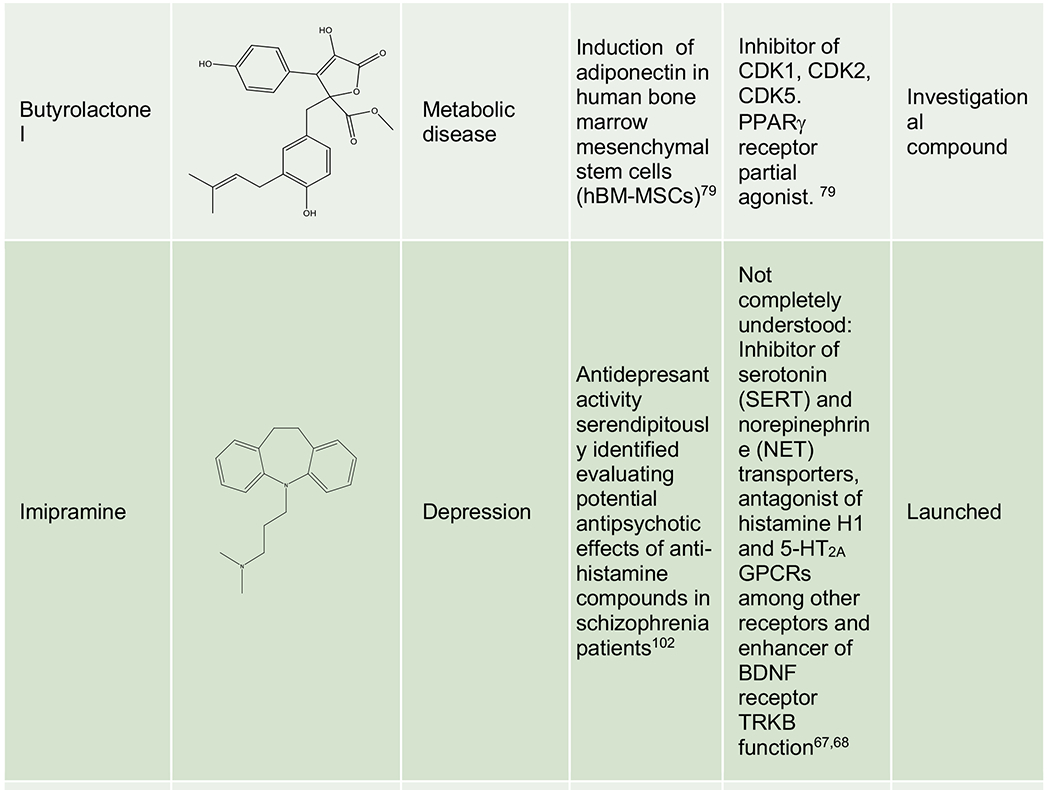
Phenotypic approaches have provided a number of drugs and candidate molecules that revealed on-target polypharmacology upon MoA identification. Representative examples include imipramine, a tricyclic antidepressant discovered in the 1950s with a prominent polypharmacological signature modulating some key CNS monoamine transporters and receptors,67,68 topiramate, a neurostabilizer for epilepsy and migraine that engages a selection of neuronal receptors, ion channels and enzymes,69 pemetrexed, a folate antimetabolite approved for mesothelioma and NSCLC that inhibits a combination of enzymes involved in folate metabolism and nucleotide synthesis,70,71 molibresib, a first-generation BET inhibitor tested in phase 1/2 clinical trials for several cancers,72,73 RG7834 evaluated for HBV infection,74,75 SEP-363856, a clinical candidate for schizophrenia identified in an in vivo phenotypic screen43 and new investigational compounds for CNS pathologies,76 infectious diseases,77 cancer78 and metabolic disorders (Table 2).79 Unsurprisingly, polypharmacology often occurs among proteins from the same family, sharing common structural domains or similar substrates or ligands (e.g. kinases, aminergic GPCRs, and BET domain proteins).
Table 2.
Representative examples of polypharmacology among marketed drugs, clinical candidates and investigational compounds. Generic name, code name or identifier from the original reference is indicated for each compound. For investigational compounds representative examples have been selected from the corresponding referenced publications.
|
|
|
Drug combinations, engineered multi-targeted drugs and multi-specific antibodies represent simplified polypharmacology scenarios, typically combining two moieties with selectivity for each of the individual targets.80–82 Engineering more complex polypharmacology into a single entity while balancing all other properties required by a drug candidate is a complex process that remains a daunting challenge, in spite of recent advances in molecular docking, computational technologies and artificial intelligence (AI).76,82–84
Phenotypic screening offers the possibility to identify hits with novel, unbiased polypharmacology signatures only limited by the target landscape of the model system and the number of activities potentially contained in each of the scaffolds available in the compound library.85,86 An important practical consideration is the use of a gain-of-signal phenotype to help focus efforts on productive polypharmacology rather than cellular stress or cytotoxicity through targets unrelated to the biology of interest.21,76 A first snapshot of polypharmacology signatures can be obtained using conventional selectivity panels.87,88 These signatures can serve as starting points for a reverse engineering SAR-based process aimed at identifying and optimizing their on /off-targets balance, mapping correlations between activity in the phenotypic assay and in each of the individual targets.76 An example of a systematic approach to map off-targets and increase selectivity for a phenotypic endpoint resulting from polypharmacology is provided by Tear et al, using hits from a phenotypic HTS screen to identify Trypanosoma brucei inhibitors.89 Phenotypic endpoints integrate the contributions of polypharmacoloy on-targets in a single read out, while reduction of the off-targets footprint can be explored with support from SAR and, when available, reference compounds selective for the targets identified.90
On-target(s) identification efforts may be more productive after some compound optimization is undertaken to reduce the off-targets signature of initial hit compounds and its potential confounding effects. Additional support from chemical and functional genomics tools at more advanced stages may also be required. Even if on-targets cannot be unequivocally identified, insight into the MoA and potential safety risks of candidate molecules can be obtained from the powerful phenotypic and molecular profiling platforms currently available using reference drugs as benchmarks.91–94 Polypharmacology is unlikely to be limited to the proteome. In our experience, phenotypic screening using a chemical library containing highly selective legacy compounds from target-based drug discovery programs can provide hits (e.g. kinase inhibitors) whose phenotypic activity does not seem to rely in their bona fide targets alone.90 This could be due to subtle polypharmacology resulting from a combination of lower potency activities or even involve interactions with non-protein targets like RNAs,95 not detected in conventional protein-only selectivity panels but certainly potential targets of phenotypic screening hits.96
From a drug discovery perspective, phenotypic screening-derived polypharmacology can take advantage of increasingly robust phenotypic models combined with advanced profiling, omics and computational technologies to evolve from serendipity to a SAR-based reverse engineering approach that can minimize potential safety risks while preserving and eventually optimizing phenotypic activity to increase the chances of clinical success.
Drug “likeness” revisited: PDD successes with low molecular weight compounds
Modern phenotypic screening may also help expand the range of molecular properties displayed by new drugs. Specifically, the molecular weight of drugs has increased significantly over the past few decades in conjunction with the advent of TDD.103 Historically, smaller molecules were discovered using phenotypic approaches, often using animal models (ibuprofen104 MW 206, minoxidil13 MW 209, memantine12 MW 179) (Figure 1). These molecules were well within the broadly accepted criteria for fragments, such as MW<300. Contemporary examples include the discoveries of lacosamide (MW 250) using a model of epilepsy11 and SEP-363856 (MW 183) for schizophrenia using a battery of CNS models,42,43,98 along with the repurposing of MLR-1023 (MW 202) towards type 2 diabetes and NASH after testing in a diversified suite of in vivo models,48,105 and of dymethyl fumarate (MW 144) for multiple sclerosis (Figure 1).106
The above examples raise a critical question. Why would fragment-sized molecules that would nowadays be looked at as weak hits in need of a large amount of potency optimization in the context of TDD programs be active in in vivo disease models and lead to similarly sized drugs after optimization? We hypothesize that several reasons might account for this unexpected pattern. First, fragment-sized molecules are known to deliver a higher fraction of valid hits when screened against specific targets due to their small size and larger number of orientations able to fit within a binding site.107 As noted previously, polypharmacology is a frequent feature of PDD drugs and may also contribute here (e.g. SEP-363856 interacts with TAAR1 and 5-HT1A receptors).43,98 Finally, smaller molecules have documented advantages in terms of intestinal permeability, operating through paracellular spaces, while they present fewer options to metabolizing enzymes due to their smaller size.108 While drug dosages and the corresponding exposures for many of these fragment-sized drugs may be high by the standards of larger molecules, they may still display high ligand efficiencies for their target(s) which allow them to be dosed safely.109 In other words, the same high compound concentration does not carry the same safety implications for molecules with MW of 200 vs 500 in terms of exposure.
An opportunity therefore exists to purposefully exploit this ability of phenotypic screening to tap into a chemical and pharmacology space poorly covered by TDD programs.110 The recent discoveries of SEP-363856, MLR-1023 and lacosamide suggest that phenotypic screening using in vivo animal models can still be successful. Such success will depend heavily on the clinical relevance of the model, a controversial topic but one still worthy of exploration as will be discussed later on. Looking forward, a second option to consider would be to adapt this concept of screening fewer and smaller molecules in complex, multi-cellular organoid assays. Fragment libraries are known to cover a large pharmacophore space despite numbering only in the thousands.107 Testing libraries of fragments or molecules in between fragments and standard compounds may potentially allow the coverage of a significant pharmacology space even with low-throughput, complex 3D assays.110
A related concept is the use of covalent fragments - a subset of fragments containing reactive moieties which can establish chemical bonds with proteins - to identify both novel targets and reveal chemistry starting points. Used in cell-based phenotypic assays, they are garnering recognition for their ability to access a greater slice of biology through the discovery of binding sites which may not be available to reversible compounds. Importantly, covalent fragments benefit from the strengths of both covalent targeting (sustained target engagement) and fragment-based screening (wide coverage of ligandable pockets with a small library).111 For example, profiling of covalent fragments in primary human T cells revealed inhibitors of T cell activation operating by distinct mechanisms including the direct functional perturbation and/or degradation of proteins.112 Notably, modulated targets included both previously liganded or unliganded proteins.
Target identification and project progression – a maturing discussion
Target identification for an active compound series is broadly accepted as being desirable to help derisk the project from a safety and clinical translation perspective. Whether it is absolutely necessary for compounds prior to entering the clinic has been the source of contentious debate over the years.3 This discussion is now maturing. Target identification is usually viewed as leading to a simple binary outcome: either the target is identified or it is not (Figure 3A). Here, we wish to offer a different framework for discussing this important topic. First, it is important to keep in mind that target identification is only a means to an end, with that end being to obtain decision-making information for compound series on the path to and in the clinic. Target identification therefore represents only one option to meet this goal. Furthermore, identifying the molecular target does not equate to understanding the MoA of a compound series and may even be misleading in terms of program decisions. For example, the documentation of NS5A being the target of HCV drug daclatasvir did not rationalize its notable sub-nM cellular potency nor did it explain how efficacy can be obtained with a compound to protein stoichiometry lower than 1 to 1,000.8 Similarly, the rational response to the identification of the ribosome as the target of a PCSK9 secretion inhibitor should have been to terminate the program as it raised the specter of significant safety liabilities through broad protein synthesis inhibition. However, further investigations including proteomics profiling revealed that its molecular MoA provided unexpectedly specific inhibition of the translation of the PCSK9 mRNA transcript due to the formation of a trimeric complex between the ribosome, the compound and the nascent PCSK9 polypeptide.49,50 Another class of examples includes compounds exhibiting a unique phenotypic effect in their class due to degradation of the target (e.g. fulvestrant with ER)113 or modification of its protein binding partners (e.g. DNMDP with PDE3A)114.
Figure 3.
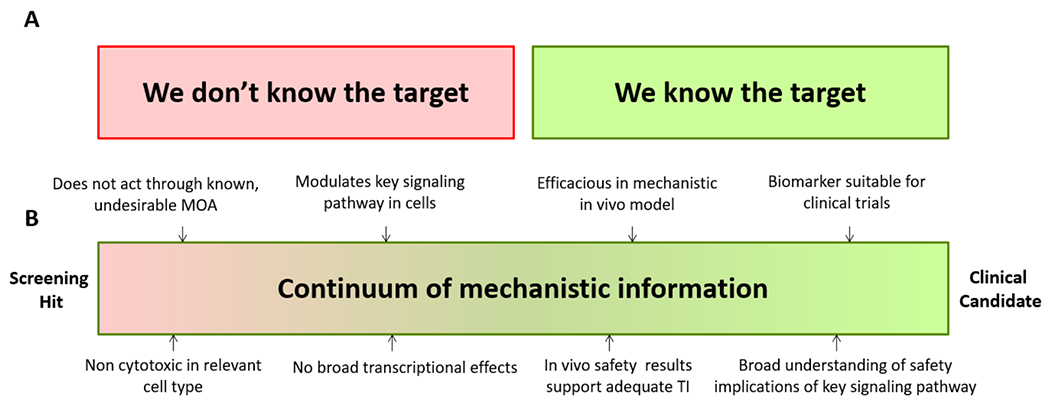
Target identification is sometimes perceived as necessary and having a simple binary outcome: the target is identified or it is not (Panel A, legacy thinking). We suggest instead that a continuum of information can be accessed which may address the true end goal of target identification, helping obtain sufficient confidence in safety and translation to support progression into the clinic (Panel B, emerging thinking). TI: Therapeutic index.
As an alternative to target identification, we suggest that much actionable knowledge about the MoA can be acquired empirically (Figure 3B). Practically, cellular and in vivo assays can provide unbiased compound assessment with readouts relevant to efficacy and safety while mechanistic studies, now often employing omics approaches, may reveal information related to the MoA such as the specific biological pathways impacted by the compound.115 Rather than the binary switch of identifying – or not - the target, a continuum of information can be obtained about a compound series during the course of a project which may lead to its progression to the clinic based on the accumulated confidence in safety and translation.
Target identification and mechanism-of-action profiling strategies
While finding direct binders to a small-molecule screening hit has the promise of delivering “a/the” target, it is not the complete picture when trying to deconvolute a phenotypic screen.116–118 Affinity (or photo-affinity) enrichment combined with chemo-proteomics methods119–123 or, more recently, Cellular Thermal Shift Assay (CETSA)124 to identify the protein (or proteins) that bind to the small molecule hit are a way to understand mechanism of action (e.g. P2X4 as the target of autophagy inhibitor Indophagolin).125 But this may not be sufficient, and as mentioned above for PCSK9 could even be misleading.
Developments in RNAi, and CRISPR-Cas9, have opened the ability to screen whole genome libraries, allowing gain of function and loss of function studies to be performed with high specificity.126 Genetic perturbations, in combination with compound treatment, provide further mechanistic understanding of MoA and may lead to identification of the molecular target itself (e.g. NAMPT as the target of anti-leukemia agent STF-118804 and DHODH as the target of antiviral GSK983).127–130 One large-scale effort is the Cancer Dependency Map that aims to systematically identify genetic dependencies and small-molecule sensitivities using massively-parallel compound screens in molecularly-barcoded cell lines using the PRISM method.131,132
More recently, molecular profiling methodologies that provide comprehensive information about biological changes resulting from a chemical perturbation have taken a prominent role in the follow up of phenotypic hits.21 Examples of such large-scale profiling efforts can be categorized into measurements of gene expression, cell morphology or biomarker activity. The Connectivity Map has gene expression profiles of test and annotated compounds that can be used for signature similarity mapping.133 An extension from that is the Library of Integrated Network-Based Cellular Signatures (LINCS),91,134 an NIH Common Fund program that catalogs changes in cell lines in response to chemical, genetic, and disease perturbations. Cell Painting uses morphological profiling: quantitative data are extracted from microscopy images of cells to identify biologically relevant similarities and differences among samples.135–138 The BioMap panel profiles primary cell systems upon treatment (chemical or biological), reading out biomarker activities that are increased or decreased in comparison to vehicle control.92,139,140 A key advantage provided by these technologies is the growing feasibility of testing numerous compounds (e.g. a list of top hits) rather than having to focus on a strictly limited number of compounds as with legacy proteomics strategies. Much of the value of these platforms is derived from the comparison of a hit’s biological signature to a databank of signatures obtained with reference, annotated compounds. It may reveal a match or help construct hypotheses regarding its MoA. For example, Tapinaroff was matched to an AHR agonist using the BioMap panel, with its own AHR agonism validated in follow up studies.141 Practically, molecular profiling is now becoming integrated in screening funnels and used in determining which hits and series will see further investment and investigation.21
These large-scale profiling methodologies, while higher in throughput and focused on pathway-level information, rely heavily on known compounds with similar phenotypes in the reference databases. Given that the purpose behind phenotypic screening is to discover new mechanisms of action, it may take time to build the knowledge base for these methods to live up to the expectation of a look-up table. However, many phenotypic screens are now guided by specific mechanistic information (mechanism-informed PDD as coined by Moffat et al).142 This strategy provides a key biological framework to place into context the data generating hypotheses for the MoA of the phenotypic hits based on the tools described above.
Clinical development considerations for PDD-derived compounds
In the absence of target information, progressing a PDD-derived preclinical drug candidate into the clinic poses several challenges to development teams. Simply phrased, target identity provides valuable information both for derisking safety concerns and for predicting and monitoring efficacy. This section will present specific strategies and examples to address these hurdles.
A ‘chain of translatability’ — a molecular-level association between mechanisms which drive the original phenotypic assay, subsequent preclinical disease models, and is an inherent component of the human disease — is critical for a PDD program to succeed in the clinic.3 For example, the HBV antiviral agent RG7834 lowers the secretion of non-infectious membranous particles containing the tolerogenic viral S antigen in vitro and therefore captures an essential (and prognostic) component of the disease in humans.74,75,143,144 The programs for branaplam and risdiplam for spinal muscular atrophy (SMA) also provide a strong example of this concept. As noted above, SMA is caused by loss-of-function mutations in the SMN1 gene, and both efforts originated from high-throughput phenotypic screening programs aimed at producing functional SMN to compensate by modulating the splicing of the almost identical SMN2 gene to include exon 7 (this exon is normally absent, resulting in an unstable Δ7 protein).9,32,33,145 These programs screened for a disease surrogate biomarker (inclusion of exon 7 in SMN2 mRNA) of clear clinical relevance, as the subsequent clinical studies demonstrated. For instance, in a phase I healthy male volunteer single escalating dose study, treatment with risdiplam resulted in the intended shift in SMN2 splicing towards full-length SMN2 mRNA, which further translated into a medically meaningful benefit in SMA patients in the SUNFISH (NCT02908685) pivotal clinical trial.146 To summarize, clinical development may be feasible in the absence of an identified target but with a clear biomarker to monitor in humans based on a strong molecular MoA understanding.
On the safety front, target information, and the accompanying knowledge about its physiological expression pattern and role, is often used to focus attention on potential safety signals. These may be investigated early in the life of a TDD program to either derisk or terminate it rapidly. Regulatory guidelines do not require target information for safety evaluation though. Instead, they provide a list of required toxicology studies, to guide the choice of acceptable compound doses for human testing. Safety derisking was the topic of a workshop at the 2019 Keystone symposium on PDD with multiple themes and strategies emerging from these discussions.
First, safety considerations need to be incorporated as early as hit triage and validation in phenotypic programs. Effective methods include cytotoxicity counterscreens as well as molecular profiling to remove hits acting through frequent hitter and other undesirable MoAs.21 Once the hit list has been trimmed to a few hits of interest, the use of active–inactive compound pairs has proven helpful to either increase or decrease confidence in a given compound series and its cognate target/MoA. These molecular tools can enable the identification of specific biomarkers and signatures of the MoA of interest while also allowing the assessment of adverse reactions that are not directly related to the compound mechanism of action (analogous to “off target” adverse effects in target-based drug discovery). This approach was followed to gain confidence in the MoA of the original PCSK9 secretion inhibition hit, R-IMPP.49 As often with screening hits, this hit was rather weak and promiscuous, raising concerns about further investment. The fact that its enantiomer, S-IMPP, was observed to be similarly promiscuous yet was inactive in the PCSK9 secretion assay suggested the R-IMPP series acted through a specific molecular target rather than through broad cell stress or injury (Figure 4A). Conversely, this strategy was used to justify the termination of a series of CFTR correctors being developed for cystic fibrosis.147 Here, following the observation of severe in vivo toxicity following chronic dosing of a lead molecule, a closely structurally related but inactive analogue was similarly tested in vivo. The inactive compound was well tolerated with similar exposure levels, suggesting that toxicity was more likely MoA-related rather than compound-related (Figure 4B).
Figure 4.
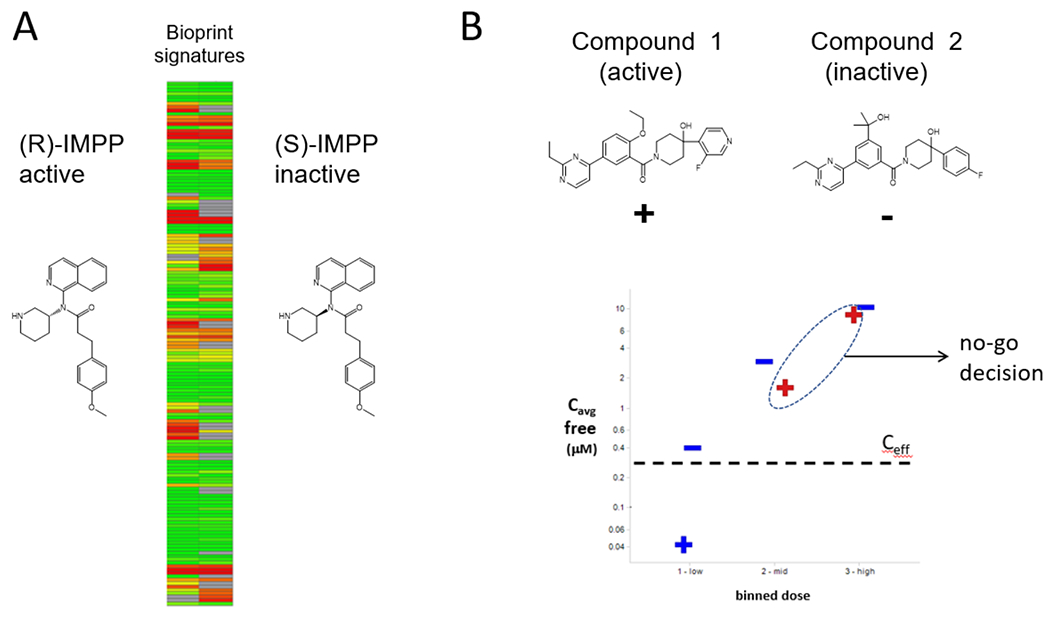
Utility of active / inactive compound pairs to address safety questions for compound series with unknown target(s) or MoA(s). A) Profiles in the BioPrint pharmacology panel148 of PCSK9 secretion inhibitor (R)-IMPP and its inactive analogue (S)-IMPP. Figure adapted with permission from ref 49 B) In vivo toxicology results following multi day dosing of of cystic fibrosis lead Compound 1 (+) and its inactive analogue Compound 2 (−). Blue coloring indicates the compound was tolerated while red lettering indicates it was not. Cave: average in vivo concentration, Ceff: predicted in vivo effective concentration for Compound 1.
Additionally, researchers can access more complex phenotypic characterizations and map compound-specific profiles to reference collections in zebrafish149 or in human primary cell-based disease systems such as Biomap, validated with fingerprints generated using clinically approved drugs.150,151 While these systems do not fully recapitulate the range of safety issues which may be observed in humans, they still provide an opportunity to detect some multiorgan liabilities while allowing the testing of significantly larger number of compounds or series. Finally, in vivo toxicology studies, which include testing of multiple compound doses in two separate animal species, constitute a key step and the subsequent determination of a no-observed-adverse-effect level (NOAEL) guides the choice of compound doses for human testing. Here, ensuring that the two species chosen display modulation of a disease biomarker or biological signature associated with the series MoA is important to maximize the value of these studies prior to entering the clinic.
Drug candidates derived from PDD can and have transitioned to the clinic in the absence of knowledge of the molecular target.1 These include for example, 1) Lenalidomide (2005 approval for multiple myeloma) with its MoA being elucidated in 2014,30 2) Lacosamide (2008 approval for epilepsy) with information with its likely complex MoA still under investigation,11,152,153 and 3) RG7834 which recently entered phase I clinical trials while target identification efforts had yet to succeed.74,75,154 Howeverin the absence of target information, it is necessary to identify surrogate disease biomarkers that translate effectively to human patients. Similarly, specific strategies can help evaluate safety risks for a given series and the associated MoA. An additional consideration is the unmet medical need and clinical landscape for a given indication as an absence of well validated targets may provide further impetus to progress a compound into the clinic in the absence of target information.3
Looking Forward
Surrogate Phenotypes for phenotypic screens
At times, phenotypic screening can present a conundrum. It is obviously valuable for diseases that do not have well validated therapeutic targets and which may be correspondingly less well understood or characterized. However, disease knowledge is essential to design a phenotypic assay with a relevant in vitro or in vivo biological system, stimulus and readout and establish the required Chain of Translation.3,18 In much the same way they are being used to define the MoA of a small molecule, high-dimensional profiles such as gene expression and cellular morphology could be used to define a surrogate disease phenotype as the readout of the phenotypic assay.155 Instead of a chemical compound, the perturbagen in this case is the disease itself with the screen aimed at reverting the system from diseased to healthy state.
Gene expression profiling has been used to define disease states, such as those caused by genomic alterations in cancer. For example, high-throughput mRNA profiles were used to cluster alleles found in lung adenocarcinoma based on their functional impact, a precursor to therapeutic strategy for variants of previously unknown significance.156 As indicated previously, the LINCS program is designed to create a network-based understanding of biology by cataloging changes in gene expression that occur in response to a perturbagen or disease state.134 A goal of the program is to develop a computational framework to discover therapies on the basis of restoring perturbed pathways and networks to their normal, healthy states. A recent study used high-throughput drug screening combined with in silico analyses of existing transcriptomic datasets to identify a compound capable of reversing pulmonary arterial hypertension (PAH) in vivo.157 The authors note that their studies could be further improved by generating LINCS gene expression signatures using vascular cells rather than cancer cells. The promise of these methods, however, is still in its infancy, as shown by elegant work done by Alvarez et al.158 Here, the authors combined gene expression profiling and several computational algorithms to define master regulator proteins, for gastroenteropancreatic neuroendocrine tumors (GEP-NETs) and then conducted transcriptome analysis of GEP-NET-derived cells, perturbed with a library of 107 compounds. Conceptually the method showed that compounds capable of inverting the coordinated activity of tumor-checkpoint master regulators can effectively destabilize tumor cell state. However, validation of two drugs predicted to induce patient-specific master regulator collapse was inconclusive. The authors provide an in-depth discussion of the reasons why that could be.
As discussed previously, cell morphological phenotypes, including shape, size, intensity, and texture of cellular compartments have been shown to change in response to perturbation - be it a small molecule or disease associated alleles. Already, the LINCS portal has incorporated such data from Cell Painting136 and the Drug Repurposing Hub159 reporting Cell Painting data for 1,571 compounds (92% of them mapped to a human protein target or assigned a mechanism-of-action label).135 These data could be used to define phenotypes reversed by drugs of known MoA.
Additional methods to computationally compare and visualize drug and disease gene expression profiles are used to define reversal of disease phenotypes. A ranking system, the Reverse Gene Expression Score (RGES) provided a systematic way to connect disease gene expression with drug-induced expression profiles.160 Integrating data from TCGA (https://tcga-data.nci.nih.gov/), LINCS,91,134 ChEMBL161 and CCLE162 allowed the researchers to show that drugs with efficacy in cancer cells had enhanced potency to reverse disease gene expression compared to ineffective drugs.160
Analogous to gene set enrichment analysis (GSEA), Nassiri et al developed a cell morphology enrichment analysis to assess the association between transcriptomic alterations and changes in cell morphology underscoring that the interdependence between transcription and cell morphology can be linked to disease state, in this case looking at cell morphological changes in a human bone osteosarcoma cell line.163
The repositories of high dimensional profiling datasets mentioned in this section already exist in the public domain. For drug discovery, the challenge of how to best use these data to define surrogate disease phenotypes that can be reversed as an indication of therapeutic efficacy remains as one of the next big hurdles for the field. Some pharmaceutical companies have started to use this approach as a screening platform. Starting in 2013, Recursion Pharmaceuticals may be the first industrial effort to use a surrogate disease signature via cell painting to screen for reversion of a surrogate disease signature. Their main focus is on drug repurposing aimed at rare monogenic disorders.164
Artificial intelligence and PDD
The application of artificial intelligence is well accepted in multiple domains of drug discovery and development (for a comprehensive review see 165) including drug design,166 protein folding,167 chemistry,168 in silico toxicity prediction,169 and drug repurposing.170 The exponential pace of application of this methodology relies on the ability of machine learning (ML) algorithms to identify patterns and learn from their association with certain parameters such as potency, selectivity, etc.
A query of the published literature on PubMed using the keywords “deep learning”, “artificial intelligence” and “phenotypic” or “drug discovery” results into two rather different class of papers: 1) classifiers applied to a large collection of compounds or chemical structures and 2) classifiers applied to phenotypic assay derived features. The vast majority of the published works belong to the first category (graph-based or deep learning-based with or without transfer learning from different training datasets) to a large collection of compounds or chemical structures with associated pharmacology data generated in previous screens. The classifier is not applied therefore to phenotype-derived features but to chemical structures in order to come up with potential novel scaffolds that in turn can become substrate for experimental validation. A prototypical example is the DeepMalaria study, where a graph-based model was trained on antiplasmodial hits from a GSK dataset to predict Plasmodium falciparum growth inhibition (and mammalian cell cytotoxicity) aiding in the rational selection of scaffolds as input for further investigation.171 To overcome the difficulty of low training data, transfer learning was used from an unrelated dataset. Those molecules were then subsequently validated in a phenotypic assay. Though the AI classifier is deployed on chemical structures prior to the phenotypic effort, that approach can still contribute to PDD as demonstrated by the identification of potent candidate antimalarial agents.171
Conversely, beyond the AI hype, efforts to deploy the classifier on the phenotype-derived features are still rare even though this is clearly a tantalizing application of this technology capable of revealing patterns hidden in what is apparently chaos to a casual observer. PDD relies by definition on phenotypic pattern changes to identify and optimize molecules with little or no knowledge of the biological target or MoA.172 Characterization of drug-induced perturbations at the cellular level (e.g., Cell Painting136) has shown that subcellular feature metrics can be used to cluster and classify compound and gene perturbations.138,173,174 ML is particularly useful in instances when the feature space is not well defined, and could therefore be remarkably enabling to PDD. Leveraging a large cell painting data set (126,779 morphological profiles induced by 30,616 compounds) for instance,138 Hofmarcher et al demonstrated that convolutional neural networks operating on raw images are able to extract morphological changes of cells from images, outperforming a traditional image-processing pipeline based on segmenting cells and subsequent feature extraction.175 Interestingly, application of different dyes to cells may not be necessary as other authors have shown that even bright field images can be used to train an algorithm that can discriminate specific phenotypes.177
There are also other opportunities for the integration of ML with PDD. For example, phenotypic screening and ML can be combined to extract target information from promiscuous compound collections such as unselective kinase inhibitors. In this area, studies showed that, even while using promiscuous kinase inhibitors, it was possible to deconvolute the kinase dependencies of active molecules and identify the kinase combinations whose inhibition delivered the desired outcomes of increased neurite outgrowth or breast cancer cell death.76,178
Two studies highlight the transformative potential of the application of ML to PDD with bacterial phenotypic fingerprinting and the repurposing of high-throughput images.94,176 Though others have deployed deep learning to antibiotics discovery,179 Zoffmann et al 94 using a combined high-content imaging and genomic approach in conjunction with a ML-powered dataset analysis effectively narrowed down, compared and predicted compound MoAs. In that study, application of ML could therefore define, in the feature space, compound ‘archetypes’, enabling chemists to proceed in their compound optimization efforts while constantly monitoring how such modifications affected the MoA of analogues — a major difference with traditional PDD. Janssen researchers brought this concept to the next level, integrating the high-throughput imaging, normally used to read out a handful of morphological features documenting a single biological process, with ML to establish a proof of concept that images from a given cellular assay can support activity prediction across a spectrum of biological assays.176 Specifically, they were able to predict compound activity against two different targets using cellular morphology information extracted from an unrelated imaging screen, increasing hit rates by >50 fold.
Further development of physiological and disease-relevant assay systems
Whether a phenotypic drug discovery project succeeds or fails depends on the inherent strength of the “chain of translatability”, discussed above, that links the primary phenotypic assay at the outset to patient efficacy at the end, often with an animal disease model in between.3 An analysis of the biopharmaceutical industry by Scannell and Bosley suggests that the decline in R&D efficiency may be caused by the progressive exhaustion of the most predictive disease models, and that the rate of creation of new disease relevant models may be a major constraint on R&D productivity.180 Notably, the authors also conclude that small increases in the predictive validity or translatability of disease models can offset large differences (i.e. orders of magnitude) in assay throughput.180 Taken together, the continued development of realistic disease-relevant assays with validated clinical translatability is of critical importance to future PDD efforts.
Fortunately, advances in diverse disciplines such as stem cell biology, functional genomics, bioengineering/microfabrication, and instrumentation/data analytics have converged to provide an expansive experimental pallet to develop potential disease relevant assay systems. Technology advances include but are not limited to: platform approaches to model organisms,181 high capacity in vivo mammalian pharmacology,40,182 the use of high fidelity Cas-9 methods to modulate gene regulation/structure,183,184 access to novel cellular systems such as primary cells, patient derived cells and induced pluripotent stem cell (iPSC)-derived cells,18,185–187 the use of mono or co-culture188–190 systems in 2D or 3D cell formats,191,192 and integration of microfabrication/bioengineering advances to provide micropatterned cell culture surfaces193, 3D matrices/microfluidic systems187, and organ on a chip194–197 capabilities.
The complexity and number of experimental variables pertinent to the design of disease relevant biological models are significant.18,185,186,198,199 Unfortunately, recapitulating all aspects of the relevant patient biology in a model is at best an aspirational goal. More realistically, research usually focuses on reproducing specific disease features deemed to be essential for model value. Even so, these are usually complex systems, and therefore require significant development, optimization and validation efforts.18,185,186 As with molecular target validation,200 disease models should be considered translational only after concordance is established between discovery and clinical phase data. This represents a high hurdle which is obtained only late in a project’s lifecycle.
Although relevant cell types and culture conditions are necessary to develop physiologically relevant models, their use alone is not sufficient to guarantee disease relevance.199 Critically, the chain of translatability of a cellular system should be benchmarked against multiple aspects of the human clinical condition such as morphology, multi-omics characterization, and pharmacological responses. The recent development of an in vitro model of non-alcoholic fatty liver disease and non-alcoholic steatohepatitis (NASH) illustrates this key point.196,201 This model uses 3D co-culture of primary human hepatocytes, Kupffer cells, and hepatic stellate cells which display disease-relevant tissue morphology, biomarker expression/secretion, transcriptional profiles and responsiveness to obeticholic acid, an advanced clinical compound which improves the histological features of NASH but has yet to secure FDA approval.196,201 Similarly, oncology models utilizing 3D organoids can be frequently derived from patient biopsies. Patient-derived organoids (PDOs) retain aspects of the original patient tumor such as histopathology, biomarker protein expression, and genomic features (copy number variations and mutational landscapes).202–205 Significantly, PDOs derived from gastrointestinal and pancreatic cancers show diverse ex vivo responses to standards of care but closely match the clinical chemotherapeutic responses of specific patients 203,204 and ex vivo responses of rectal cancer PDOs to chemoradiation treatment correlate with the clinical responses noted in individual patient tumors.202,205
In the era of big data, integration of real-world patient records from large populations (e.g. UK Biobank, FinnGen) and their omics data may also be used to help both build and validate model systems. Utilizing an in vitro phenotypic screen monitoring alpha-synuclein gene expression as a Parkinson’s disease (PD) model, Mittal et al identified βB2-adrenergic receptor modulators in a drug repurposing screen. Significantly, the in vitro model was subsequently validated by analysis of 4 million patient records which correlated the use of a β2AR agonist or antagonist with a reduced or increased risk of developing PD, respectively.206 Alternatively, the Tumor Profiler Study seeks to deliver patient specific treatment recommendations by integration of a patient’s real world clinical data with high-resolution multi-omics profiles and ex vivo drug response of the patient tumor within a clinically relevant turnaround time.207
Animal models of human disease are an important component of preclinical drug discovery. While nowadays they are used mostly to validate modulation of specific molecular targets, models utilizing mammals were a mainstay as primary phenotypic assays in drug discovery until the late 20th century and led to the discovery of therapeutic agents for indications such as epilepsy, gastric ulcers, hypertension, inflammation and pain.11,13,69,104,105 With the advent of the 21st century, this approach to drug discovery was all but ruled out at larger pharmaceutical companies.105
As the need for phenotypic assays with high translational potential becomes ever clearer, perhaps it is time to reconsider the wholesale abandonment of in vivo models as first line phenotypic screening systems. While whole organism models may be necessary to more fully recreate the disease state for certain complex or multi-organ indications poorly described by in vitro systems, even those including native cells and 3D architecture, the development of valid in vivo translational models is certainly not trivial however.180
Importantly, in vivo disease models have recognized issues with human disease translation due to factors including methodology, systematic data review, and critical disease specific disparities.208–210 Lack of species translation constitutes another significant hurdle as exemplified by the failure in clinical trials of flavonoid DMXAA despite promising efficacy in preclinical models. Following the identification of its target, the disconnect was traced to the selective activation of mouse STING over its human ortholog.211 Overall, ethical, cost, translatability and throughput considerations combine to place a high bar on the development and use of suitable in vivo models.
Efforts to enhance their translational value include the development of a “Mouse Hospital” and co-clinical trial concept, where in vivo preclinical mouse models and early clinical studies are closely aligned for in vivo testing of drugs in order to mitigate translational issues.212,213 Similarly, comparative expression profiles of genetic mouse models of Huntington’s disease (HD) correspond well with patient profiles, particularly for mRNAs that are decreased in HD striatum214 and thus increase the predictive validity of HD mouse models.
Advances in disease models may also encompass aspects of data acquisition and analysis rather than improvements in the model organism per se. SEP-363856 is a novel psychotropic agent with a mechanism of action which is independent of D2 and 5-HT2A modulation employed by legacy agents such as chlorpromazine. It was identified by SmartCube phenotypic screening — an automated system to capture digital video of various domains of mouse behavior followed by algorithmic data reduction to approximately 2,000 features and supervised learning to derive drug class signatures or behavioral barcodes based on mice treated with compounds validated for specific therapeutic indications and marketed drugs.40,41,215 The resulting drug class/behavioral signatures are high dimensional/high content and are likely to capture drug-induced aspects of behavior which may not be apparent in conventional manual, stand-alone mouse models.
While screening throughput is a significant hurdle in such in vivo systems, it is incorrectly thought to be an insurmountable barrier. Historical success stories document that hits could be identified as part of smaller, often hypothesis-based and pharmacophore-informed in vivo phenotypic screens.11,104 As discussed earlier, the success observed with very small molecules in legacy and recent screens may be rationalized by several key features collectively leading to increased odds of success.42,108 Finally, the in vivo profiling of 1,000 analogs which led to the discovery and development of SEP-363856 indicates that a reasonable throughput may still be obtained.43,215 Alternatively, CRISPR sgRNA technology using Cas9 mice enables genetic screens to identify pathways and nodes modulating some disease phenotypes of interest, especially in the field of oncology.216,217 Models based on lower order organisms such as Zebrafish also offer an opportunity for higher throughput but at the cost of being potentially further removed translationally.218
The inherent multi-disciplinary nature of disease model enablement and the iterative process to optimize and correlate discovery and therapeutic endpoints for these assets presents a major hurdle to the biopharmaceutical research community. Notably, much of the expertise in these diverse disciplines resides in academia whereas the end users and experience to identify/prioritize disease indications for model development to address unmet medical needs reside largely in the biopharmaceutical sector. Additional barriers to the development of new disease model systems include uncertainty and time constraints imposed by grant support of academic research along with lower appetite for fundamental research with long- or uncertaintime horizons in the for-profit sector. The development of physiological and disease relevant models may thus benefit from a non-profit, pre-competitive research organization,185,186 similar to consortia developing probe compounds to pre-competitive molecular targets.219–222
Concluding remarks
PDD has demonstrated its potential by identifying drugs, targets and MoAs that in many cases would have been impossible to discover using target-based approaches.1 3,4,5 223 This strategy offers a path to novel therapeutics when molecular information about disease pathophysiology is lacking, providing access to the untapped “dark biological matter” represented by the proteome and any other biomolecule and cellular process underlying disease.4,6 The choice of compound library and the clinical translatability of the phenotypic model are essential for the success of PDD. 5,7,8
Key aspects highlighted here include the discovery of new MoA only accessible to PDD, the need for phenotypic models that can better recapitulate the pathophysiology of complex diseases (for example, integrating immune or nervous system components), the opportunities offered by polypharmacology and the advantages of using libraries with smaller-than-conventional molecules. Increased uptake of bioactivity profiling and MoA characterization approaches applied to efficacy / safety assessment and the increasingly powerful computational technologies have become essential to extract the full potential of PDD.
The current challenge is how to rationally combine these key aspects to prospectively “industrialize” phenotypic drug discovery. Given the exponential growth in this area, we are confident that increased application of ML (and deep learning in particular) will contribute to the effective implementation of PDD (Figure 5). Our vision is that industrialization of phenotypic drug discovery coupled with the extensive experience accumulated after more than a decade of “modern” PDD practice will contribute to a more productive drug discovery process that seamlessly integrates target and phenotype-based approaches.223
Figure 5. Schematic overview of an industrialized phenotypic drug discovery process.
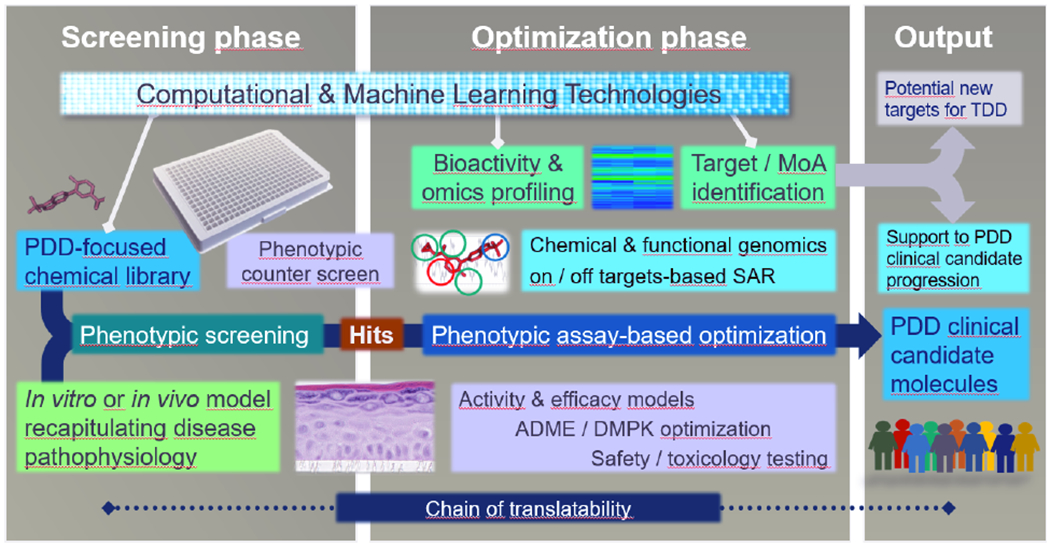
A chemical library designed for phenotypic screening (e.g. with smaller MW compounds) and a disease-relevant in vitro or in vivo model system capable of providing sufficient throughput, are combined in a phenotypic screening campaign to identify hits whose optimization, characterization and progression to clinical phases can take advantage of the current plethora of omics, profiling and computational approaches (including machine learning). Target or MoA information is used to support the progress of clinical PDD candidates. One additional possibility, if targets are identified, is their potential use as starting points for new TDD programs. Chain of translatability is shown to represent the molecular association between the mechanisms driving the phenotypic assay, the preclinical disease models, and human disease. Counter screen refers to an assay aimed at verifying the selectivity of the hit molecules versus other unintended phenotypic endpoints. SAR: structure-activity relationship, MoA: mechanism of action.
Figure 2.
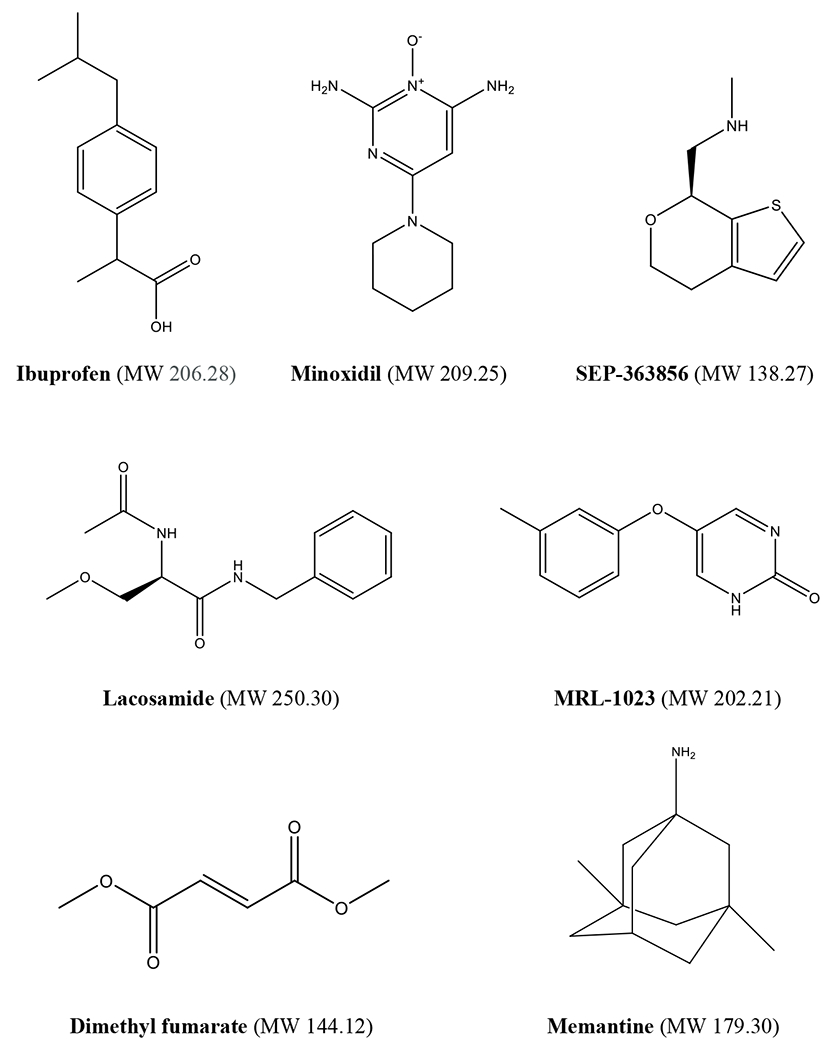
Low molecular weight clinical candidates and drugs derived from phenotypic approaches
Contributor Information
Fabien Vincent, Pfizer.
Arsenio Nueda, Almirall.
Jonathan Lee, PDD4Patients, ex-Eli Lilly.
Monica Schenone, Pfizer, ex-Broad Institute.
Marco Prunotto, Roche + University of Geneva.
Mark Mercola, Stanford.
References
- 1.Swinney DC & Anthony J How were new medicines discovered? Nat Rev Drug Discov 10, 507–519, doi: 10.1038/nrd3480 (2011). [DOI] [PubMed] [Google Scholar]
- 2.Lee JA & Berg EL Neoclassic drug discovery: the case for lead generation using phenotypic and functional approaches. J Biomol Screen 18, 1143–1155, doi: 10.1177/1087057113506118 (2013). [DOI] [PubMed] [Google Scholar]
- 3.Moffat JG, Vincent F, Lee JA, Eder J & Prunotto M Opportunities and challenges in phenotypic drug discovery: an industry perspective. Nat Rev Drug Discov 16, 531–543, doi: 10.1038/nrd.2017.111 (2017). [DOI] [PubMed] [Google Scholar]
- 4.Eder J, Sedrani R & Wiesmann C The discovery of first-in-class drugs: origins and evolution. Nat Rev Drug Discov 13, 577–587, doi: 10.1038/nrd4336 (2014). [DOI] [PubMed] [Google Scholar]
- 5.Edwards A What Are the Odds of Finding a COVID-19 Drug from a Lab Repurposing Screen? J. Chem. Inf. Model, doi: 10.1021/acs.jcim.0c00861 (2020). [DOI] [PubMed] [Google Scholar]
- 6.Van Goor F et al. Rescue of CF airway epithelial cell function in vitro by a CFTR potentiator, VX-770. Proc Natl Acad Sci U S A 106, 18825–18830, doi: 10.1073/pnas.0904709106 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Van Goor F et al. Correction of the F508del-CFTR protein processing defect in vitro by the investigational drug VX-809. Proc Natl Acad Sci U S A 108, 18843–18848, doi: 10.1073/pnas.1105787108 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Belema M & Meanwell NA Discovery of daclatasvir, a pan-genotypic hepatitis C virus NS5A replication complex inhibitor with potent clinical effect. J Med Chem 57, 5057–5071, doi: 10.1021/jm500335h (2014). [DOI] [PubMed] [Google Scholar]
- 9.Ratni H et al. Discovery of Risdiplam, a Selective Survival of Motor Neuron-2 ( SMN2) Gene Splicing Modifier for the Treatment of Spinal Muscular Atrophy (SMA). J Med Chem 61, 6501–6517, doi: 10.1021/acs.jmedchem.8b00741 (2018). [DOI] [PubMed] [Google Scholar]
- 10.Yoshida T et al. Identification and characterization of a novel chemotype MEK inhibitor able to alter the phosphorylation state of MEK1/2. Oncotarget 3, 1533–1545, doi: 10.18632/oncotarget.747 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Choi D, Stables JP & Kohn H Synthesis and anticonvulsant activities of N-Benzyl-2-acetamidopropionamide derivatives. J Med Chem 39, 1907–1916, doi: 10.1021/jm9508705 (1996). [DOI] [PubMed] [Google Scholar]
- 12.Alam S, Lingenfelter KS, Bender AM & Lindsley CW Classics in Chemical Neuroscience: Memantine. ACS Chem Neurosci 8, 1823–1829, doi: 10.1021/acschemneuro.7b00270 (2017). [DOI] [PubMed] [Google Scholar]
- 13.Martin D Guinter Kahn, Inventor of Baldness Remedy, Dies at 80. New York Times, A21 (2014). [Google Scholar]
- 14.Rossi A et al. Minoxidil use in dermatology, side effects and recent patents. Recent Pat Inflamm Allergy Drug Discov 6, 130–136, doi: 10.2174/187221312800166859 (2012). [DOI] [PubMed] [Google Scholar]
- 15.Clader JW The discovery of ezetimibe: a view from outside the receptor. J Med Chem 47, 1–9, doi: 10.1021/jm030283g (2004). [DOI] [PubMed] [Google Scholar]
- 16.Schwab RS, England AC Jr., Poskanzer DC & Young RR Amantadine in the treatment of Parkinson’s disease. JAMA 208, 1168–1170 (1969). [PubMed] [Google Scholar]
- 17.Ghofrani HA, Osterloh IH & Grimminger F Sildenafil: from angina to erectile dysfunction to pulmonary hypertension and beyond. Nat Rev Drug Discov 5, 689–702, doi: 10.1038/nrd2030 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Vincent F et al. Developing predictive assays: the phenotypic screening “rule of 3”. Sci Transl Med 7, 293ps215, doi: 10.1126/scitranslmed.aab1201 (2015). [DOI] [PubMed] [Google Scholar]
- 19.Haasen D et al. How Phenotypic Screening Influenced Drug Discovery: Lessons from Five Years of Practice. Assay Drug Dev Technol 15, 239–246, doi: 10.1089/adt.2017.796 (2017). [DOI] [PubMed] [Google Scholar]
- 20.Comess KM et al. Emerging Approaches for the Identification of Protein Targets of Small Molecules - A Practitioners’ Perspective. J Med Chem 61, 8504–8535, doi: 10.1021/acs.jmedchem.7b01921 (2018). [DOI] [PubMed] [Google Scholar]
- 21.Vincent F et al. Hit Triage and Validation in Phenotypic Screening: Considerations and Strategies. Cell Chem Biol 27, 1332–1346, doi: 10.1016/j.chembiol.2020.08.009 (2020). [DOI] [PubMed] [Google Scholar]
- 22.Zajac M et al. Hepatitis C - New drugs and treatment prospects. Eur J Med Chem 165, 225–249, doi: 10.1016/j.ejmech.2019.01.025 (2019). [DOI] [PubMed] [Google Scholar]
- 23.Lemm JA et al. Identification of hepatitis C virus NS5A inhibitors. J Virol 84, 482–491, doi: 10.1128/JVI.01360-09 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Boyle MP & De Boeck K A new era in the treatment of cystic fibrosis: correction of the underlying CFTR defect. Lancet Respir Med 1, 158–163, doi: 10.1016/S2213-2600(12)70057-7 (2013). [DOI] [PubMed] [Google Scholar]
- 25.Middleton PG et al. Elexacaftor-Tezacaftor-Ivacaftor for Cystic Fibrosis with a Single Phe508del Allele. N Engl J Med 381, 1809–1819, doi: 10.1056/NEJMoa1908639 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Singhal S et al. Antitumor activity of thalidomide in refractory multiple myeloma. N Engl J Med 341, 1565–1571, doi: 10.1056/NEJM199911183412102 (1999). [DOI] [PubMed] [Google Scholar]
- 27.Millrine D & Kishimoto T A Brighter Side to Thalidomide: Its Potential Use in Immunological Disorders. Trends Mol Med 23, 348–361, doi: 10.1016/j.molmed.2017.02.006 (2017). [DOI] [PubMed] [Google Scholar]
- 28.Lindner S & Kronke J The molecular mechanism of thalidomide analogs in hematologic malignancies. J Mol Med (Berl) 94, 1327–1334, doi: 10.1007/s00109-016-1450-z (2016). [DOI] [PubMed] [Google Scholar]
- 29.Urquhart L Top companies and drugs by sales in 2020. Nature Reviews Drug Discovery 20, 253 (2021). [DOI] [PubMed] [Google Scholar]
- 30.Lu G et al. The myeloma drug lenalidomide promotes the cereblon-dependent destruction of Ikaros proteins. Science 343, 305–309, doi: 10.1126/science.1244917 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Schreiber SL The Rise of Molecular Glues. Cell 184, 3–9, doi: 10.1016/j.cell.2020.12.020 (2021). [DOI] [PubMed] [Google Scholar]
- 32.Palacino J et al. SMN2 splice modulators enhance U1-pre-mRNA association and rescue SMA mice. Nat Chem Biol 11, 511–517, doi: 10.1038/nchembio.1837 (2015). [DOI] [PubMed] [Google Scholar]
- 33.Naryshkin NA et al. Motor neuron disease. SMN2 splicing modifiers improve motor function and longevity in mice with spinal muscular atrophy. Science 345, 688–693, doi: 10.1126/science.1250127 (2014). [DOI] [PubMed] [Google Scholar]
- 34.Sivaramakrishnan M et al. Binding to SMN2 pre-mRNA-protein complex elicits specificity for small molecule splicing modifiers. Nat Commun 8, 1476, doi: 10.1038/s41467-017-01559-4 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Campagne S et al. Structural basis of a small molecule targeting RNA for a specific splicing correction. Nat Chem Biol 15, 1191–1198, doi: 10.1038/s41589-019-0384-5 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Van Goor F et al. Rescue of DeltaF508-CFTR trafficking and gating in human cystic fibrosis airway primary cultures by small molecules. Am J Physiol Lung Cell Mol Physiol 290, L1117–1130, doi: 10.1152/ajplung.00169.2005 (2006). [DOI] [PubMed] [Google Scholar]
- 37.Savi P et al. Identification and biological activity of the active metabolite of clopidogrel. Thromb Haemost 84, 891–896 (2000). [PubMed] [Google Scholar]
- 38.Maffrand JP The story of clopidogrel and its predecessor, ticlopidine: Could these major antiplatelet and antithrombotic drugs be discovered and developed today? Comptes Rendus Chimie 15, 737–743, doi: 10.1016/j.crci.2012.05.006 (2012). [DOI] [Google Scholar]
- 39.Savi P et al. P2y(12), a new platelet ADP receptor, target of clopidogrel. Biochem Biophys Res Commun 283, 379–383, doi: 10.1006/bbrc.2001.4816 (2001). [DOI] [PubMed] [Google Scholar]
- 40.Alexandrov V, Brunner D, Hanania T & Leahy E High-throughput analysis of behavior for drug discovery. Eur J Pharmacol 750, 82–89, doi: 10.1016/j.ejphar.2014.11.047 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Roberds SL, Filippov I, Alexandrov V, Hanania T & Brunner D Rapid, computer vision-enabled murine screening system identifies neuropharmacological potential of two new mechanisms. Front Neurosci 5, 103, doi: 10.3389/fnins.2011.00103 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Liming Shao UCC, Kevin Fang Q, Powell Noel A., Campbell John E., Jones Philip G., Hanania Taleen, Alexandrov Vadim, Morganstern Irene, Sabath Emily, Zhong Hua M., Large Thomas H., Spear Kerry L.. In vivo phenotypic drug discovery: applying a behavioral assay to the discovery and optimization of novel antipsychotic agents Medicinal Chemistry Communications 7, 1093–1101 (2016). [Google Scholar]
- 43.Dedic N et al. SEP-363856, a Novel Psychotropic Agent with a Unique, Non-D2 Receptor Mechanism of Action. J Pharmacol Exp Ther 371, 1–14, doi: 10.1124/jpet.119.260281 (2019). [DOI] [PubMed] [Google Scholar]
- 44.Tokarski JS et al. Tyrosine Kinase 2-mediated Signal Transduction in T Lymphocytes Is Blocked by Pharmacological Stabilization of Its Pseudokinase Domain. J Biol Chem 290, 11061–11074, doi: 10.1074/jbc.M114.619502 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Nicodeme E et al. Suppression of inflammation by a synthetic histone mimic. Nature 468, 1119–1123, doi: 10.1038/nature09589 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Chung CW et al. Discovery and characterization of small molecule inhibitors of the BET family bromodomains. J Med Chem 54, 3827–3838, doi: 10.1021/jm200108t (2011). [DOI] [PubMed] [Google Scholar]
- 47.Ochman AR, Lipinski CA, Handler JA, Reaume AG & Saporito MS The Lyn kinase activator MLR-1023 is a novel insulin receptor potentiator that elicits a rapid-onset and durable improvement in glucose homeostasis in animal models of type 2 diabetes. J Pharmacol Exp Ther 342, 23–32, doi: 10.1124/jpet.112.192187 (2012). [DOI] [PubMed] [Google Scholar]
- 48.Saporito MS, Ochman AR, Lipinski CA, Handler JA & Reaume AG MLR-1023 is a potent and selective allosteric activator of Lyn kinase in vitro that improves glucose tolerance in vivo. J Pharmacol Exp Ther 342, 15–22, doi: 10.1124/jpet.112.192096 (2012). [DOI] [PubMed] [Google Scholar]
- 49.Petersen DN et al. A Small-Molecule Anti-secretagogue of PCSK9 Targets the 80S Ribosome to Inhibit PCSK9 Protein Translation. Cell Chem Biol 23, 1362–1371, doi: 10.1016/j.chembiol.2016.08.016 (2016). [DOI] [PubMed] [Google Scholar]
- 50.Lintner NG et al. Selective stalling of human translation through small-molecule engagement of the ribosome nascent chain. PLoS Biol 15, e2001882, doi: 10.1371/journal.pbio.2001882 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Hopkins AL Network pharmacology: the next paradigm in drug discovery. Nat Chem Biol 4, 682–690, doi: 10.1038/nchembio.118 (2008). [DOI] [PubMed] [Google Scholar]
- 52.Reddy AS & Zhang S Polypharmacology: drug discovery for the future. Expert Rev Clin Pharmacol 6, 41–47, doi: 10.1586/ecp.12.74 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Keiser MJ et al. Predicting new molecular targets for known drugs. Nature 462, 175–181, doi: 10.1038/nature08506 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Mestres J, Gregori-Puigjane E, Valverde S & Sole RV The topology of drug-target interaction networks: implicit dependence on drug properties and target families. Mol Biosyst 5, 1051–1057, doi: 10.1039/b905821b (2009). [DOI] [PubMed] [Google Scholar]
- 55.Lin A et al. Off-target toxicity is a common mechanism of action of cancer drugs undergoing clinical trials. Sci Transl Med 11, doi: 10.1126/scitranslmed.aaw8412 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Lotsch J & Geisslinger G Low-dose drug combinations along molecular pathways could maximize therapeutic effectiveness while minimizing collateral adverse effects. Drug Discov Today 16, 1001–1006, doi: 10.1016/j.drudis.2011.10.003 (2011). [DOI] [PubMed] [Google Scholar]
- 57.Gitelman SE et al. Imatinib therapy for patients with recent-onset type 1 diabetes: a multicentre, randomised, double-blind, placebo-controlled, phase 2 trial. Lancet Diabetes Endocrinol 9, 502–514, doi: 10.1016/S2213-8587(21)00139-X (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Capdeville R, Buchdunger E, Zimmermann J & Matter A Glivec (STI571, imatinib), a rationally developed, targeted anticancer drug. Nat Rev Drug Discov 1, 493–502, doi: 10.1038/nrd839 (2002). [DOI] [PubMed] [Google Scholar]
- 59.Wong S et al. Sole BCR-ABL inhibition is insufficient to eliminate all myeloproliferative disorder cell populations. Proc Natl Acad Sci U S A 101, 17456–17461, doi: 10.1073/pnas.0407061101 (2004). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Cohen P, Cross D & Janne PA Kinase drug discovery 20 years after imatinib: progress and future directions. Nat Rev Drug Discov 20, 551–569, doi: 10.1038/s41573-021-00195-4 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Crystal AS et al. Patient-derived models of acquired resistance can identify effective drug combinations for cancer. Science 346, 1480–1486, doi: 10.1126/science.1254721 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Ianevski A et al. Identification and Tracking of Antiviral Drug Combinations. Viruses 12, doi: 10.3390/v12101178 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.van Hasselt JGC & Iyengar R Systems Pharmacology: Defining the Interactions of Drug Combinations. Annu Rev Pharmacol Toxicol 59, 21–40, doi: 10.1146/annurev-pharmtox-010818-021511 (2019). [DOI] [PubMed] [Google Scholar]
- 64.Morphy R Selectively nonselective kinase inhibition: striking the right balance. J Med Chem 53, 1413–1437, doi: 10.1021/jm901132v (2010). [DOI] [PubMed] [Google Scholar]
- 65.Roth BL, Sheffler DJ & Kroeze WK Magic shotguns versus magic bullets: selectively nonselective drugs for mood disorders and schizophrenia. Nat Rev Drug Discov 3, 353–359, doi: 10.1038/nrd1346 (2004). [DOI] [PubMed] [Google Scholar]
- 66.Rusinova R, Koeppe RE 2nd & Andersen OS A general mechanism for drug promiscuity: Studies with amiodarone and other antiarrhythmics. J Gen Physiol 146, 463–475, doi: 10.1085/jgp.201511470 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Gillman PK Tricyclic antidepressant pharmacology and therapeutic drug interactions updated. Br J Pharmacol 151, 737–748, doi: 10.1038/sj.bjp.0707253 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Casarotto PC et al. Antidepressant drugs act by directly binding to TRKB neurotrophin receptors. Cell 184, 1299–1313 e1219, doi: 10.1016/j.cell.2021.01.034 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Maryanoff B Phenotypic Assessment and the Discovery of Topiramate. ACS Medicinal Chemistry Letters 7, 662–665, doi: 10.1021/acsmedchemlett.6b00176 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Taylor EC et al. A dideazatetrahydrofolate analogue lacking a chiral center at C-6, N-[4-[2-(2-amino-3,4-dihydro-4-oxo-7H-pyrrolo[2,3-d]pyrimidin-5- yl)ethyl]benzoyl]-L-glutamic acid, is an inhibitor of thymidylate synthase. J Med Chem 35, 4450–4454, doi: 10.1021/jm00101a023 (1992). [DOI] [PubMed] [Google Scholar]
- 71.Mendelsohn LG et al. Enzyme inhibition, polyglutamation, and the effect of LY231514 (MTA) on purine biosynthesis. Semin Oncol 26, 42–47 (1999). [PubMed] [Google Scholar]
- 72.Mirguet O et al. Discovery of epigenetic regulator I-BET762: lead optimization to afford a clinical candidate inhibitor of the BET bromodomains. J Med Chem 56, 7501–7515, doi: 10.1021/jm401088k (2013). [DOI] [PubMed] [Google Scholar]
- 73.Piha-Paul SA et al. Phase 1 Study of Molibresib (GSK525762), a Bromodomain and Extra-Terminal Domain Protein Inhibitor, in NUT Carcinoma and Other Solid Tumors. JNCI Cancer Spectr 4, pkz093, doi: 10.1093/jncics/pkz093 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Han X et al. Discovery of RG7834: The First-in-Class Selective and Orally Available Small Molecule Hepatitis B Virus Expression Inhibitor with Novel Mechanism of Action. J Med Chem, doi: 10.1021/acs.jmedchem.8b01245 (2018). [DOI] [PubMed] [Google Scholar]
- 75.Mueller H et al. A novel orally available small molecule that inhibits hepatitis B virus expression. J Hepatol 68, 412–420, doi: 10.1016/j.jhep.2017.10.014 (2018). [DOI] [PubMed] [Google Scholar]
- 76.Al-Ali H et al. Rational Polypharmacology: Systematically Identifying and Engaging Multiple Drug Targets To Promote Axon Growth. ACS Chem Biol 10, 1939–1951, doi: 10.1021/acschembio.5b00289 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Chiarelli LR et al. A multitarget approach to drug discovery inhibiting Mycobacterium tuberculosis PyrG and PanK. Sci Rep 8, 3187, doi: 10.1038/s41598-018-21614-4 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Sumi NJ et al. Divergent Polypharmacology-Driven Cellular Activity of Structurally Similar Multi-Kinase Inhibitors through Cumulative Effects on Individual Targets. Cell Chem Biol 26, 1240–1252 e1211, doi: 10.1016/j.chembiol.2019.06.003 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Ahn S et al. Cyclin-Dependent Kinase 5 Inhibitor Butyrolactone I Elicits a Partial Agonist Activity of Peroxisome Proliferator-Activated Receptor gamma. Biomolecules 10, doi: 10.3390/biom10020275 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Sun D et al. Dual-target kinase drug design: Current strategies and future directions in cancer therapy. Eur J Med Chem 188, 112025, doi: 10.1016/j.ejmech.2019.112025 (2020). [DOI] [PubMed] [Google Scholar]
- 81.Labrijn AF, Janmaat ML, Reichert JM & Parren P Bispecific antibodies: a mechanistic review of the pipeline. Nat Rev Drug Discov 18, 585–608, doi: 10.1038/s41573-019-0028-1 (2019). [DOI] [PubMed] [Google Scholar]
- 82.Proschak E, Stark H & Merk D Polypharmacology by Design: A Medicinal Chemist’s Perspective on Multitargeting Compounds. J Med Chem 62, 420–444, doi: 10.1021/acs.jmedchem.8b00760 (2019). [DOI] [PubMed] [Google Scholar]
- 83.Besnard J et al. Automated design of ligands to polypharmacological profiles. Nature 492, 215–220, doi: 10.1038/nature11691 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Da C et al. Data-Driven Construction of Antitumor Agents with Controlled Polypharmacology. J Am Chem Soc 141, 15700–15709, doi: 10.1021/jacs.9b08660 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Sweis RF Target (In)Validation: A Critical, Sometimes Unheralded, Role of Modern Medicinal Chemistry. ACS Med Chem Lett 6, 618–621, doi: 10.1021/acsmedchemlett.5b00183 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Monteleone S, Fuchs JE & Liedl KR Molecular Connectivity Predefines Polypharmacology: Aliphatic Rings, Chirality, and sp(3) Centers Enhance Target Selectivity. Front Pharmacol 8, 552, doi: 10.3389/fphar.2017.00552 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Bendels S et al. Safety screening in early drug discovery: An optimized assay panel. J Pharmacol Toxicol Methods 99, 106609, doi: 10.1016/j.vascn.2019.106609 (2019). [DOI] [PubMed] [Google Scholar]
- 88.Bowes J et al. Reducing safety-related drug attrition: the use of in vitro pharmacological profiling. Nat Rev Drug Discov 11, 909–922, doi: 10.1038/nrd3845 (2012). [DOI] [PubMed] [Google Scholar]
- 89.Tear WF et al. Selectivity and Physicochemical Optimization of Repurposed Pyrazolo[1,5-b]pyridazines for the Treatment of Human African Trypanosomiasis. J Med Chem 63, 756–783, doi: 10.1021/acs.jmedchem.9b01741 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Orellana A et al. Application of a phenotypic drug discovery strategy to identify biological and chemical starting points for inhibition of TSLP production in lung epithelial cells. PLoS One 13, e0189247, doi: 10.1371/journal.pone.0189247 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Subramanian A et al. A Next Generation Connectivity Map: L1000 Platform and the First 1,000,000 Profiles. Cell 171, 1437–1452 e1417, doi: 10.1016/j.cell.2017.10.049 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Berg EL Phenotypic chemical biology for predicting safety and efficacy. Drug Discov Today Technol 23, 53–60, doi: 10.1016/j.ddtec.2017.01.001 (2017). [DOI] [PubMed] [Google Scholar]
- 93.Drawnel FM et al. Molecular Phenotyping Combines Molecular Information, Biological Relevance, and Patient Data to Improve Productivity of Early Drug Discovery. Cell Chem Biol 24, 624–634 e623, doi: 10.1016/j.chembiol.2017.03.016 (2017). [DOI] [PubMed] [Google Scholar]
- 94.Zoffmann S et al. Machine learning-powered antibiotics phenotypic drug discovery. Sci Rep 9, 5013, doi: 10.1038/s41598-019-39387-9 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Connelly CM, Moon MH & Schneekloth JS Jr. The Emerging Role of RNA as a Therapeutic Target for Small Molecules. Cell Chem Biol 23, 1077–1090, doi: 10.1016/j.chembiol.2016.05.021 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Warner KD, Hajdin CE & Weeks KM Principles for targeting RNA with drug-like small molecules. Nat Rev Drug Discov 17, 547–558, doi: 10.1038/nrd.2018.93 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Taylor EC The Discovery and Synthesis of Alimta. Chemistry International -- Newsmagazine for IUPAC 33, 4–9, doi: 10.1515/ci.2011.33.5.4 (2011). [DOI] [Google Scholar]
- 98.Koblan KS et al. A Non-D2-Receptor-Binding Drug for the Treatment of Schizophrenia. N Engl J Med 382, 1497–1506, doi: 10.1056/NEJMoa1911772 (2020). [DOI] [PubMed] [Google Scholar]
- 99.Qian F et al. Inhibition of tumor cell growth, invasion, and metastasis by EXEL-2880 (XL880, GSK1363089), a novel inhibitor of HGF and VEGF receptor tyrosine kinases. Cancer Res 69, 8009–8016, doi: 10.1158/0008-5472.CAN-08-4889 (2009). [DOI] [PubMed] [Google Scholar]
- 100.Al-Ali H, Schurer SC, Lemmon VP & Bixby JL Chemical interrogation of the neuronal kinome using a primary cell-based screening assay. ACS Chem Biol 8, 1027–1036, doi: 10.1021/cb300584e (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Mori G et al. Thiophenecarboxamide Derivatives Activated by EthA Kill Mycobacterium tuberculosis by Inhibiting the CTP Synthetase PyrG. Chem Biol 22, 917–927, doi: 10.1016/j.chembiol.2015.05.016 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Brown WA & Rosdolsky M The clinical discovery of imipramine. Am J Psychiatry 172, 426–429, doi: 10.1176/appi.ajp.2015.14101336 (2015). [DOI] [PubMed] [Google Scholar]
- 103.Shultz MD Two Decades under the Influence of the Rule of Five and the Changing Properties of Approved Oral Drugs. J Med Chem 62, 1701–1714, doi: 10.1021/acs.jmedchem.8b00686 (2019). [DOI] [PubMed] [Google Scholar]
- 104.Rainsford KD in Ibuprofen: Discovery, Development and Therapeutics (ed Rainsford Kim D.) Ch. 1, 1–21 (Wiley-Blackwell; |, 2015). [Google Scholar]
- 105.Lipinski CA & Reaume AG High throughput in vivo phenotypic screening for drug repurposing: Discovery of MLR-1023 a novel insulin sensitizer and novel Lyn kinase activator with clinical proof of concept. Bioorg Med Chem 28, 115425, doi: 10.1016/j.bmc.2020.115425 (2020). [DOI] [PubMed] [Google Scholar]
- 106.Faissner S & Gold R Oral Therapies for Multiple Sclerosis. Cold Spring Harb Perspect Med 9, doi: 10.1101/cshperspect.a032011 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Jhoti H, Williams G, Rees DC & Murray CW The ‘rule of three’ for fragment-based drug discovery: where are we now? Nat Rev Drug Discov 12, 644–645, doi: 10.1038/nrd3926-c1 (2013). [DOI] [PubMed] [Google Scholar]
- 108.Raymer B & Bhattacharya SK Lead-like Drugs: A Perspective. J Med Chem 61, 10375–10384, doi: 10.1021/acs.jmedchem.8b00407 (2018). [DOI] [PubMed] [Google Scholar]
- 109.Hopkins AL, Keseru GM, Leeson PD, Rees DC & Reynolds CH The role of ligand efficiency metrics in drug discovery. Nat Rev Drug Discov 13, 105–121, doi: 10.1038/nrd4163 (2014). [DOI] [PubMed] [Google Scholar]
- 110.Ayotte Y et al. Fragment-Based Phenotypic Lead Discovery To Identify New Drug Seeds That Target Infectious Diseases. ACS Chem Biol 16, 2158–2163, doi: 10.1021/acschembio.1c00657 (2021). [DOI] [PubMed] [Google Scholar]
- 111.Lu Wenchao, K. M, Tinghu Zhang, Jianwei Che, Patricelli Matthew P., Jones Lyn H., Chouchaniae Edward T., Nathanael S. Gray Fragment-based covalent ligand discovery. RSC Chemical Biology, doi: 10.1039/D0CB00222D (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Parker CG et al. Ligand and Target Discovery by Fragment-Based Screening in Human Cells. Cell 168, 527–541 e529, doi: 10.1016/j.cell.2016.12.029 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Wijayaratne AL & McDonnell DP The human estrogen receptor-alpha is a ubiquitinated protein whose stability is affected differentially by agonists, antagonists, and selective estrogen receptor modulators. J Biol Chem 276, 35684–35692, doi: 10.1074/jbc.M101097200 (2001). [DOI] [PubMed] [Google Scholar]
- 114.de Waal L et al. Identification of cancer-cytotoxic modulators of PDE3A by predictive chemogenomics. Nat Chem Biol 12, 102–108, doi: 10.1038/nchembio.1984 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Hughes RE, Elliott RJR, Dawson JC & Carragher NO High-content phenotypic and pathway profiling to advance drug discovery in diseases of unmet need. Cell Chem Biol 28, 338–355, doi: 10.1016/j.chembiol.2021.02.015 (2021). [DOI] [PubMed] [Google Scholar]
- 116.Schenone M, Dancik V, Wagner BK & Clemons PA Target identification and mechanism of action in chemical biology and drug discovery. Nat Chem Biol 9, 232–240, doi: 10.1038/nchembio.1199 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Terstappen GC, Schlupen C, Raggiaschi R & Gaviraghi G Target deconvolution strategies in drug discovery. Nat Rev Drug Discov 6, 891–903, doi: 10.1038/nrd2410 (2007). [DOI] [PubMed] [Google Scholar]
- 118.Kosaka T et al. Identification of molecular target of AMP-activated protein kinase activator by affinity purification and mass spectrometry. Anal Chem 77, 2050–2055, doi: 10.1021/ac0484631 (2005). [DOI] [PubMed] [Google Scholar]
- 119.Ong SE et al. Identifying the proteins to which small-molecule probes and drugs bind in cells. Proc Natl Acad Sci U S A 106, 4617–4622, doi: 10.1073/pnas.0900191106 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Harding MW, Galat A, Uehling DE & Schreiber SL A receptor for the immunosuppressant FK506 is a cis-trans peptidyl-prolyl isomerase. Nature 341, 758–760, doi: 10.1038/341758a0 (1989). [DOI] [PubMed] [Google Scholar]
- 121.Seneviratne U et al. Photoaffinity Labeling and Quantitative Chemical Proteomics Identify LXRbeta as the Functional Target of Enhancers of Astrocytic apoE. Cell Chem Biol 28, 148–157 e147, doi: 10.1016/j.chembiol.2020.09.002 (2021). [DOI] [PubMed] [Google Scholar]
- 122.Huang Z et al. Global Portrait of Protein Targets of Metabolites of the Neurotoxic Compound BIA 10-2474. ACS Chem Biol 14, 192–197, doi: 10.1021/acschembio.8b01097 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Wang ZH et al. C/EBPbeta regulates delta-secretase expression and mediates pathogenesis in mouse models of Alzheimer’s disease. Nat Commun 9, 1784, doi: 10.1038/s41467-018-04120-z (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Martinez Molina D et al. Monitoring drug target engagement in cells and tissues using the cellular thermal shift assay. Science 341, 84–87, doi: 10.1126/science.1233606 (2013). [DOI] [PubMed] [Google Scholar]
- 125.Carnero Corrales MA et al. Thermal proteome profiling identifies the membrane-bound purinergic receptor P2X4 as a target of the autophagy inhibitor indophagolin. Cell Chem Biol 28, 1750–1757 e1755, doi: 10.1016/j.chembiol.2021.02.017 (2021). [DOI] [PubMed] [Google Scholar]
- 126.Schmidt R et al. CRISPR activation and interference screens decode stimulation responses in primary human T cells. Science 375, eabj4008, doi: 10.1126/science.abj4008 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Neggers JE et al. Target identification of small molecules using large-scale CRISPR-Cas mutagenesis scanning of essential genes. Nat Commun 9, 502, doi: 10.1038/s41467-017-02349-8 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Deans RM et al. Parallel shRNA and CRISPR-Cas9 screens enable antiviral drug target identification. Nat Chem Biol 12, 361–366, doi: 10.1038/nchembio.2050 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Matheny CJ et al. Next-generation NAMPT inhibitors identified by sequential high-throughput phenotypic chemical and functional genomic screens. Chem Biol 20, 1352–1363, doi: 10.1016/j.chembiol.2013.09.014 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Cheng J et al. Small-molecule probe reveals a kinase cascade that links stress signaling to TCF/LEF and Wnt responsiveness. Cell Chem Biol, doi: 10.1016/j.chembiol.2021.01.001 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Tsherniak A et al. Defining a Cancer Dependency Map. Cell 170, 564–576 e516, doi: 10.1016/j.cell.2017.06.010 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Yu C et al. High-throughput identification of genotype-specific cancer vulnerabilities in mixtures of barcoded tumor cell lines. Nat Biotechnol 34, 419–423, doi: 10.1038/nbt.3460 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Lamb J et al. The Connectivity Map: using gene-expression signatures to connect small molecules, genes, and disease. Science 313, 1929–1935, doi: 10.1126/science.1132939 (2006). [DOI] [PubMed] [Google Scholar]
- 134.Keenan AB et al. The Library of Integrated Network-Based Cellular Signatures NIH Program: System-Level Cataloging of Human Cells Response to Perturbations. Cell Syst 6, 13–24, doi: 10.1016/j.cels.2017.11.001 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Gustafsdottir SM et al. Multiplex cytological profiling assay to measure diverse cellular states. PLoS One 8, e80999, doi: 10.1371/journal.pone.0080999 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Bray MA et al. Cell Painting, a high-content image-based assay for morphological profiling using multiplexed fluorescent dyes. Nat Protoc 11, 1757–1774, doi: 10.1038/nprot.2016.105 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Pahl A & Sievers S. The Cell Painting Assay as a Screening Tool for the Discovery of Bioactivities in New Chemical Matter. Methods Mol Biol 1888, 115–126, doi: 10.1007/978-1-4939-8891-4_6 (2019). [DOI] [PubMed] [Google Scholar]
- 138.Bray MA et al. A dataset of images and morphological profiles of 30 000 small-molecule treatments using the Cell Painting assay. Gigascience 6, 1–5, doi: 10.1093/gigascience/giw014 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Kunkel EJ et al. Rapid structure-activity and selectivity analysis of kinase inhibitors by BioMAP analysis in complex human primary cell-based models. Assay Drug Dev Technol 2, 431–441, doi: 10.1089/adt.2004.2.431 (2004). [DOI] [PubMed] [Google Scholar]
- 140.Kunkel EJ et al. An integrative biology approach for analysis of drug action in models of human vascular inflammation. FASEB J 18, 1279–1281, doi: 10.1096/fj.04-1538fje (2004). [DOI] [PubMed] [Google Scholar]
- 141.Smith SH et al. Tapinarof Is a Natural AhR Agonist that Resolves Skin Inflammation in Mice and Humans. J Invest Dermatol 137, 2110–2119, doi: 10.1016/j.jid.2017.05.004 (2017). [DOI] [PubMed] [Google Scholar]
- 142.Moffat JG, Rudolph J & Bailey D. Phenotypic screening in cancer drug discovery - past, present and future. Nat Rev Drug Discov 13, 588–602, doi: 10.1038/nrd4366 (2014). [DOI] [PubMed] [Google Scholar]
- 143.Kwon H & Lok AS Hepatitis B therapy. Nat Rev Gastroenterol Hepatol 8, 275–284, doi: 10.1038/nrgastro.2011.33 (2011). [DOI] [PubMed] [Google Scholar]
- 144.Zhu D et al. Clearing Persistent Extracellular Antigen of Hepatitis B Virus: An Immunomodulatory Strategy To Reverse Tolerance for an Effective Therapeutic Vaccination. J Immunol 196, 3079–3087, doi: 10.4049/jimmunol.1502061 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Cheung AK et al. Discovery of Small Molecule Splicing Modulators of Survival Motor Neuron-2 (SMN2) for the Treatment of Spinal Muscular Atrophy (SMA). J Med Chem 61, 11021–11036, doi: 10.1021/acs.jmedchem.8b01291 (2018). [DOI] [PubMed] [Google Scholar]
- 146.Sturm S et al. A phase 1 healthy male volunteer single escalating dose study of the pharmacokinetics and pharmacodynamics of risdiplam (RG7916, RO7034067), a SMN2 splicing modifier. Br J Clin Pharmacol 85, 181–193, doi: 10.1111/bcp.13786 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Vincent F in Phenotypic Drug Discovery: Recent Advances and Insights from Chemical and Systems Biology (Keystone Symposium).
- 148.Krejsa CM et al. Predicting ADME properties and side effects: the BioPrint approach. Curr Opin Drug Discov Devel 6, 470–480 (2003). [PubMed] [Google Scholar]
- 149.Cassar S et al. Use of Zebrafish in Drug Discovery Toxicology. Chem Res Toxicol 33, 95–118, doi: 10.1021/acs.chemrestox.9b00335 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.Shah F et al. Mechanisms of Skin Toxicity Associated with Metabotropic Glutamate Receptor 5 Negative Allosteric Modulators. Cell Chem Biol 24, 858–869 e855, doi: 10.1016/j.chembiol.2017.06.003 (2017). [DOI] [PubMed] [Google Scholar]
- 151.Kleinstreuer NC et al. Phenotypic screening of the ToxCast chemical library to classify toxic and therapeutic mechanisms. Nat Biotechnol 32, 583–591, doi: 10.1038/nbt.2914 (2014). [DOI] [PubMed] [Google Scholar]
- 152.Rogawski MA, Tofighy A, White HS, Matagne A & Wolff C Current understanding of the mechanism of action of the antiepileptic drug lacosamide. Epilepsy Res 110, 189–205, doi: 10.1016/j.eplepsyres.2014.11.021 (2015). [DOI] [PubMed] [Google Scholar]
- 153.Labau JIR et al. Lacosamide Inhibition of NaV1.7 Channels Depends on its Interaction With the Voltage Sensor Domain and the Channel Pore. Front Pharmacol 12, 791740, doi: 10.3389/fphar.2021.791740 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154.Javanbakht H. in Phenotypic Drug Discovery: Recent Advances and Insights from Chemical and Systems Biolog. (ed Keystone Symposia). [Google Scholar]
- 155.Pandika M. Mining Gene Expression Data for Drug Discovery. ACS Cent Sci 4, 944–947, doi: 10.1021/acscentsci.8b00529 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156.Berger AH et al. High-throughput Phenotyping of Lung Cancer Somatic Mutations. Cancer Cell 30, 214–228, doi: 10.1016/j.ccell.2016.06.022 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157.Gu M. et al. iPSC-endothelial cell phenotypic drug screening and in silico analyses identify tyrphostin-AG1296 for pulmonary arterial hypertension. Sci Transl Med 13, doi: 10.1126/scitranslmed.aba6480 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158.Alvarez MJ et al. A precision oncology approach to the pharmacological targeting of mechanistic dependencies in neuroendocrine tumors. Nat Genet 50, 979–989, doi: 10.1038/s41588-018-0138-4 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159.Corsello SM et al. The Drug Repurposing Hub: a next-generation drug library and information resource. Nat Med 23, 405–408, doi: 10.1038/nm.4306 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160.Chen B. et al. Reversal of cancer gene expression correlates with drug efficacy and reveals therapeutic targets. Nat Commun 8, 16022, doi: 10.1038/ncomms16022 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 161.Mendez D. et al. ChEMBL: towards direct deposition of bioassay data. Nucleic Acids Res 47, D930–D940, doi: 10.1093/nar/gky1075 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 162.Barretina J. et al. The Cancer Cell Line Encyclopedia enables predictive modelling of anticancer drug sensitivity. Nature 483, 603–607, doi: 10.1038/nature11003 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 163.Nassiri I & McCall MN Systematic exploration of cell morphological phenotypes associated with a transcriptomic query. Nucleic Acids Res 46, e116, doi: 10.1093/nar/gky626 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 164.Pharmaceuticals, R (ed Securities and Exchange Commission; ) (2021). [Google Scholar]
- 165.Vamathevan J. et al. Applications of machine learning in drug discovery and development. Nat Rev Drug Discov 18, 463–477, doi: 10.1038/s41573-019-0024-5 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 166.Schneider P. et al. Rethinking drug design in the artificial intelligence era. Nat Rev Drug Discov 19, 353–364, doi: 10.1038/s41573-019-0050-3 (2020). [DOI] [PubMed] [Google Scholar]
- 167.Callaway E. ‘It will change everything’: DeepMind’s AI makes gigantic leap in solving protein structures. Nature 588, 203–204, doi: 10.1038/d41586-020-03348-4 (2020). [DOI] [PubMed] [Google Scholar]
- 168.Cova T & Pais A Deep Learning for Deep Chemistry: Optimizing the Prediction of Chemical Patterns. Front Chem 7, 809, doi: 10.3389/fchem.2019.00809 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 169.Idakwo G et al. A review on machine learning methods for in silico toxicity prediction. J Environ Sci Health C Environ Carcinog Ecotoxicol Rev 36, 169–191, doi: 10.1080/10590501.2018.1537118 (2018). [DOI] [PubMed] [Google Scholar]
- 170.Issa NT, Stathias V, Schurer S & Dakshanamurthy S Machine and deep learning approaches for cancer drug repurposing. Semin Cancer Biol, doi: 10.1016/j.semcancer.2019.12.011 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 171.Keshavarzi Arshadi A, Salem M, Collins J, Yuan JS & Chakrabarti D DeepMalaria: Artificial Intelligence Driven Discovery of Potent Antiplasmodials. Front Pharmacol 10, 1526, doi: 10.3389/fphar.2019.01526 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 172.Chandrasekaran SN, Ceulemans H, Boyd JD & Carpenter AE Image-based profiling for drug discovery: due for a machine-learning upgrade? Nat Rev Drug Discov 20, 145–159, doi: 10.1038/s41573-020-00117-w (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 173.Rohban MH et al. Systematic morphological profiling of human gene and allele function via Cell Painting. Elife 6, doi: 10.7554/eLife.24060 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 174.Caicedo JC, Singh S & Carpenter AE Applications in image-based profiling of perturbations. Curr Opin Biotechnol 39, 134–142, doi: 10.1016/j.copbio.2016.04.003 (2016). [DOI] [PubMed] [Google Scholar]
- 175.Hofmarcher M, Rumetshofer E, Clevert DA, Hochreiter S & Klambauer G Accurate Prediction of Biological Assays with High-Throughput Microscopy Images and Convolutional Networks. J Chem Inf Model 59, 1163–1171, doi: 10.1021/acs.jcim.8b00670 (2019). [DOI] [PubMed] [Google Scholar]
- 176.Simm J et al. Repurposing High-Throughput Image Assays Enables Biological Activity Prediction for Drug Discovery. Cell Chem Biol 25, 611–618 e613, doi: 10.1016/j.chembiol.2018.01.015 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 177.O’Duibhir E et al. Machine Learning Enables Live Label-Free Phenotypic Screening in Three Dimensions. Assay Drug Dev Technol 16, 51–63, doi: 10.1089/adt.2017.819 (2018). [DOI] [PubMed] [Google Scholar]
- 178.Gautam P, Jaiswal A, Aittokallio T, Al-Ali H & Wennerberg K Phenotypic Screening Combined with Machine Learning for Efficient Identification of Breast Cancer-Selective Therapeutic Targets. Cell Chem Biol, doi: 10.1016/j.chembiol.2019.03.011 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 179.Stokes JM et al. A Deep Learning Approach to Antibiotic Discovery. Cell 181, 475–483, doi: 10.1016/j.cell.2020.04.001 (2020). [DOI] [PubMed] [Google Scholar]
- 180.Scannell JW & Bosley J When Quality Beats Quantity: Decision Theory, Drug Discovery, and the Reproducibility Crisis. PLoS One 11, e0147215, doi: 10.1371/journal.pone.0147215 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 181.Lam PY & Peterson RT Developing zebrafish disease models for in vivo small molecule screens. Curr Opin Chem Biol 50, 37–44, doi: 10.1016/j.cbpa.2019.02.005 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 182.Ciallella JR & Reaume AG In vivo phenotypic screening: clinical proof of concept for a drug repositioning approach. Drug Discov Today Technol 23, 45–52, doi: 10.1016/j.ddtec.2017.04.001 (2017). [DOI] [PubMed] [Google Scholar]
- 183.Anzalone AV et al. Search-and-replace genome editing without double-strand breaks or donor DNA. Nature 576, 149–157, doi: 10.1038/s41586-019-1711-4 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 184.le Sage C, Lawo S & Cross BCS CRISPR: A Screener’s Guide. SLAS Discov 25, 233–240, doi: 10.1177/2472555219883621 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 185.Friese A et al. The Convergence of Stem Cell Technologies and Phenotypic Drug Discovery. Cell Chem Biol 26, 1050–1066, doi: 10.1016/j.chembiol.2019.05.007 (2019). [DOI] [PubMed] [Google Scholar]
- 186.Horvath P et al. Screening out irrelevant cell-based models of disease. Nat Rev Drug Discov 15, 751–769, doi: 10.1038/nrd.2016.175 (2016). [DOI] [PubMed] [Google Scholar]
- 187.Benam KH et al. Engineered in vitro disease models. Annu Rev Pathol 10, 195–262, doi: 10.1146/annurev-pathol-012414-040418 (2015). [DOI] [PubMed] [Google Scholar]
- 188.E LB, Hsu YC & Lee JA Consideration of the cellular microenvironment: physiologically relevant co-culture systems in drug discovery. Adv Drug Deliv Rev 69-70, 190–204, doi: 10.1016/j.addr.2014.01.013 (2014). [DOI] [PubMed] [Google Scholar]
- 189.Hetheridge C, Mavria G & Mellor H Uses of the in vitro endothelial-fibroblast organotypic co-culture assay in angiogenesis research. Biochem Soc Trans 39, 1597–1600, doi: 10.1042/BST20110738 (2011). [DOI] [PubMed] [Google Scholar]
- 190.Thelu A, Catoire S & Kerdine-Romer S Immune-competent in vitro co-culture models as an approach for skin sensitisation assessment. Toxicol In Vitro 62, 104691, doi: 10.1016/j.tiv.2019.104691 (2020). [DOI] [PubMed] [Google Scholar]
- 191.Carragher N et al. Concerns, challenges and promises of high-content analysis of 3D cellular models. Nat Rev Drug Discov 17, 606, doi: 10.1038/nrd.2018.99 (2018). [DOI] [PubMed] [Google Scholar]
- 192.Kelm JM, Lal-Nag M, Sittampalam GS & Ferrer M Translational in vitro research: integrating 3D drug discovery and development processes into the drug development pipeline. Drug Discov Today 24, 26–30, doi: 10.1016/j.drudis.2018.07.007 (2019). [DOI] [PubMed] [Google Scholar]
- 193.Thery M Micropatterning as a tool to decipher cell morphogenesis and functions. J Cell Sci 123, 4201–4213, doi: 10.1242/jcs.075150 (2010). [DOI] [PubMed] [Google Scholar]
- 194.Jalili-Firoozinezhad S et al. A complex human gut microbiome cultured in an anaerobic intestine-on-a-chip. Nat Biomed Eng 3, 520–531, doi: 10.1038/s41551-019-0397-0 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 195.Maoz BM et al. A linked organ-on-chip model of the human neurovascular unit reveals the metabolic coupling of endothelial and neuronal cells. Nat Biotechnol 36, 865–874, doi: 10.1038/nbt.4226 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 196.Kostrzewski T et al. A Microphysiological System for Studying Nonalcoholic Steatohepatitis. Hepatol Commun 4, 77–91, doi: 10.1002/hep4.1450 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 197.Vunjak-Novakovic G, Ronaldson-Bouchard K & Radisic M Organs-on-a-chip models for biological research. Cell 184, 4597–4611, doi: 10.1016/j.cell.2021.08.005 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 198.Abbott RD & Kaplan DL Strategies for improving the physiological relevance of human engineered tissues. Trends Biotechnol 33, 401–407, doi: 10.1016/j.tibtech.2015.04.003 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 199.Ainslie GR et al. Microphysiological lung models to evaluate the safety of new pharmaceutical modalities: a biopharmaceutical perspective. Lab Chip 19, 3152–3161, doi: 10.1039/c9lc00492k (2019). [DOI] [PubMed] [Google Scholar]
- 200.Williams M Target validation. Curr Opin Pharmacol 3, 571–577, doi: 10.1016/j.coph.2003.06.001 (2003). [DOI] [PubMed] [Google Scholar]
- 201.Kostrzewski T et al. Modelling human liver fibrosis in the context of non-alcoholic steatohepatitis using a microphysiological system. Commun Biol 4, 1080, doi: 10.1038/s42003-021-02616-x (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 202.Ganesh K et al. A rectal cancer organoid platform to study individual responses to chemoradiation. Nat Med 25, 1607–1614, doi: 10.1038/s41591-019-0584-2 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 203.Tiriac H et al. Organoid Profiling Identifies Common Responders to Chemotherapy in Pancreatic Cancer. Cancer Discov 8, 1112–1129, doi: 10.1158/2159-8290.CD-18-0349 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 204.Vlachogiannis G et al. Patient-derived organoids model treatment response of metastatic gastrointestinal cancers. Science 359, 920–926, doi: 10.1126/science.aao2774 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 205.Yao Y et al. Patient-Derived Organoids Predict Chemoradiation Responses of Locally Advanced Rectal Cancer. Cell Stem Cell 26, 17–26 e16, doi: 10.1016/j.stem.2019.10.010 (2020). [DOI] [PubMed] [Google Scholar]
- 206.Mittal S et al. beta2-Adrenoreceptor is a regulator of the alpha-synuclein gene driving risk of Parkinson’s disease. Science 357, 891–898, doi: 10.1126/science.aaf3934 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 207.Irmisch A et al. The Tumor Profiler Study: integrated, multi-omic, functional tumor profiling for clinical decision support. Cancer Cell 39, 288–293, doi: 10.1016/j.ccell.2021.01.004 (2021). [DOI] [PubMed] [Google Scholar]
- 208.Bolker JA Animal Models in Translational Research: Rosetta Stone or Stumbling Block? Bioessays 39, doi: 10.1002/bies.201700089 (2017). [DOI] [PubMed] [Google Scholar]
- 209.Hooijmans CR & Ritskes-Hoitinga M Progress in using systematic reviews of animal studies to improve translational research. PLoS Med 10, e1001482, doi: 10.1371/journal.pmed.1001482 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 210.van der Worp HB et al. Can animal models of disease reliably inform human studies? PLoS Med 7, e1000245 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 211.Kim S et al. Anticancer flavonoids are mouse-selective STING agonists. ACS Chem Biol 8, 1396–1401, doi: 10.1021/cb400264n (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 212.Clohessy JG & Pandolfi PP Mouse hospital and co-clinical trial project--from bench to bedside. Nat Rev Clin Oncol 12, 491–498, doi: 10.1038/nrclinonc.2015.62 (2015). [DOI] [PubMed] [Google Scholar]
- 213.Clohessy JG & Pandolfi PP The Mouse Hospital and Its Integration in Ultra-Precision Approaches to Cancer Care. Front Oncol 8, 340, doi: 10.3389/fonc.2018.00340 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 214.Kuhn A et al. Mutant huntingtin’s effects on striatal gene expression in mice recapitulate changes observed in human Huntington’s disease brain and do not differ with mutant huntingtin length or wild-type huntingtin dosage. Hum Mol Genet 16, 1845–1861, doi: 10.1093/hmg/ddm133 (2007). [DOI] [PubMed] [Google Scholar]
- 215.Shao L et al. In vivo phenotypic drug discovery: applying a behavioral assay to the discovery and optimization of novel antipsychotic agents. MedChemCommun 7, 1093–1101 (2016). [Google Scholar]
- 216.Manguso RT et al. In vivo CRISPR screening identifies Ptpn2 as a cancer immunotherapy target. Nature 547, 413–418, doi: 10.1038/nature23270 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 217.Kuhn MS, Antonio J; Platt, Randall J Moving from in vitro to in vivo CRISPR screens. Gene and Genome Editing 2, doi: 10.1016/j.ggedit.2021.100008 (2021). [DOI] [Google Scholar]
- 218.Patton EE, Zon LI & Langenau DM Zebrafish disease models in drug discovery: from preclinical modelling to clinical trials. Nat Rev Drug Discov 20, 611–628, doi: 10.1038/s41573-021-00210-8 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 219.Rodgers G et al. Glimmers in illuminating the druggable genome. Nat Rev Drug Discov 17, 301–302, doi: 10.1038/nrd.2017.252 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 220.Carter AJ et al. Target 2035: probing the human proteome. Drug Discov Today 24, 2111–2115, doi: 10.1016/j.drudis.2019.06.020 (2019). [DOI] [PubMed] [Google Scholar]
- 221.Muller S et al. Donated chemical probes for open science. Elife 7, doi: 10.7554/eLife.34311 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 222.Arrowsmith CH et al. The promise and peril of chemical probes. Nat Chem Biol 11, 536–541, doi: 10.1038/nchembio.1867 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 223.Swinney DC & Lee JA Recent advances in phenotypic drug discovery. F1000Res 9, doi: 10.12688/f1000research.25813.1 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]