

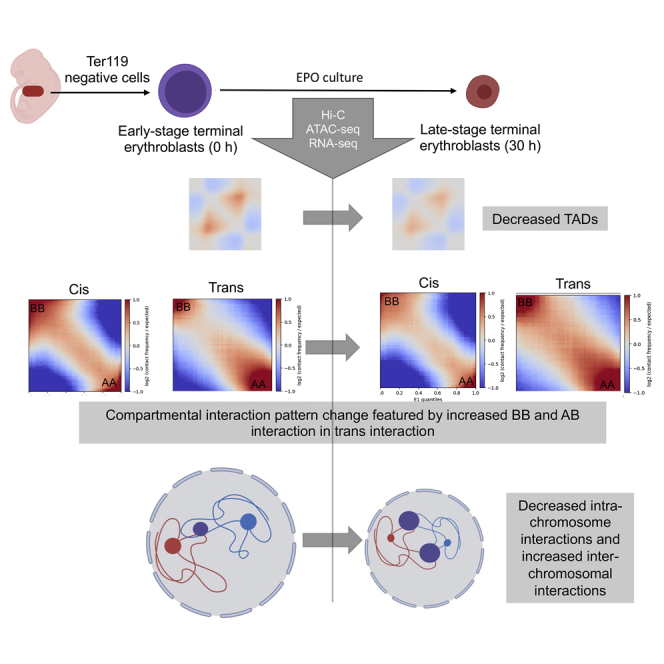
Summary
Mammalian terminal erythropoiesis involves chromatin and nuclear condensation followed by enucleation. Late-stage erythroblasts undergo caspase-mediated nuclear opening that is important for nuclear condensation through partial histone release. It remains unknown the dynamic changes of three-dimensional (3D) genomic organization during terminal erythropoiesis. Here, we used Hi-C to determine the chromatin structural change during primary mouse erythroblast terminal differentiation. We also performed RNA-sequencing and ATAC-sequencing under the same experimental setting to further reveal the genome accessibility and gene expression changes during this process. We found that late-stage terminal erythropoiesis involves global loss of topologically associating domains and establishment of inter-chromosomal interactions of the heterochromatin regions, which are associated with globally increased chromatin accessibility and upregulation of erythroid-related genes.
Subject areas: Cell biology, Structural biology
Graphical abstract
Highlights
-
•
3D genomic organization changes during mouse terminal erythropoiesis are revealed
-
•
Late-stage terminal erythropoiesis involves global loss of TADs
-
•
Decreased intra- and increased inter-chromosomal interactions during erythropoiesis
Cell biology; Structural biology
Introduction
Under constant replenishment, the development of mammalian erythrocytes can be broadly divided into two stages, commitment of hematopoietic stem and progenitor cells to erythroid lineage and terminal differentiation of erythroblasts to enucleated red blood cells. During late stages of terminal erythropoiesis, erythroblasts undergo a series of unique changes including nuclear condensation, enucleation, and loss of major organelles.1,2,3,4,5,6
Besides terminal erythropoiesis, nuclear and chromatin condensation also occurs during apoptosis. Apoptotic mechanisms including caspase activation are known to play an important role in erythroid nuclear condensation.7,8 We recently reported that late-stage erythroblasts undergo caspase-mediated transient nuclear opening that mediates partial histone release to the cytoplasm and nuclear condensation.7,9,10,11,12,13 Mechanistically, inactivation of caspases inhibits caspase-mediated lamin B cleavage and compromises nuclear condensation and enucleation. Loss of caspase-3 or ectopic expression of a caspase-3 non-cleavable lamin B mutant also inhibits nuclear opening and condensation. Same as mouse erythroblasts, disruption of caspase functions in human CD34+ cells leads to compromised nuclear opening and histone release.10
Histone cytoplasmic release during terminal erythropoiesis was also reported to be mediated by exportin 7, a highly erythroid specific exportin. Loss of exportin 7 inhibits histone and DNA release, as well as nuclear condensation.11 Moreover, a recent report showed that loss of Wdr, a gene essential for lamin B degradation, could induce anemia in zebrafish and disrupt erythroid nuclear condensation in mouse.12 Together, these studies demonstrate that histone release is essential in nuclear condensation and enucleation. However, the chromatin structural change during histone release and nuclear condensation remains unclear.
To address this question, we used a murine fetal liver primary erythroid culture system to investigate the changes in the three-dimensional (3D) genomic landscape during terminal erythropoiesis. Hi-C, ATAC-seq and RNA-seq experiments were performed to integrate the study of chromatin structural changes during terminal erythropoiesis. We found that terminal erythropoiesis involves compaction and establishment of inter-chromosomal interactions of the heterochromatin regions, which is associated with globally increased accessibility and upregulation of erythroid-related genes.
Results
Loss of topologically associating domains during mouse terminal erythropoiesis
To study the 3D chromatin architectural changes during terminal erythropoiesis, we used a well-established murine primary erythroid in vitro differentiation system in which Ter119 negative fetal liver erythroid precursors from E13.5 fetus were cultured in erythropoietin (EPO)-containing medium for 48 h.14 These cells undergo gradual chromatin and nuclear condensation, as well as 4 to 5 rounds of nuclear opening formation with associated histone cytoplasmic release, before the start of enucleation at around 30 h in culture9 (Figure 1A).
Figure 1.

Loss of topologically associating domains during mouse terminal erythropoiesis
(A) Schematic overview of the mouse primary fetal liver culture system and overall experimental design.
(B) Hi-C contact matrices of chromosome 2 (35-85Mb) at 100 kb resolution in freshly isolated cells (0 h) and cells cultured in EPO containing medium for 30 h.
(C) Measurement of Hi-C genomic contact distance in cells from B.
(D) Number of TADs identified in the indicated cells.
(E) The distribution of TAD length in the indicated cells.
(F) The insulation scores of TADs identified in the indicated cells.
(G) Aggregated Hi-C interaction frequencies and the neighboring regions (+/− 0.5 TAD length) in the indicated cells.
(H) Representative Hi-C maps on regions chr3:41-51Mb and chr4:74-83Mb with disrupted chromatin domains in cells at 30 h group.
Our previous study demonstrated that mouse fetal liver erythroid cells cultured at 30 h exhibit dramatic chromatin condensation compared to the cells at the beginning of culture.15 Thus, to compare the dynamic change of chromatin 3D structures during mouse terminal erythropoiesis, we performed Hi-C with cells at 0 and 30 h in culture. By comparing the difference of Hi-C contact map, we observed increased contacts in 30 h group at a higher resolution on certain chromosome regions (Figure 1B) and no detectable changes in others (data not shown), compared with 0 h group. Further quantitative analysis in the whole genome level revealed that cells at 30 h group show detectable shift of contact distance with less long-range (>10 million base pairs) interactions compared with those at 0 h group. Short (<1 million base pairs) and medium-range (1-10 million base pairs) interactions are indiscernible (Figure 1C).
To further investigate the chromatin structural change, we performed a Topologically Associating Domains (TADs) analysis. We found that TADs are decreased in the late stage of erythroid terminal differentiation (Figure 1D). The TAD lengths are predominantly less than 2 Mb in both groups (Figure 1E). The average profile of insulation score on TADs demonstrated weakened boundaries in late-stage cells (30 h) compared with those at 0 h (Figure 1F). We further aggregated the Hi-C contact map on the TADs and the surrounding regions. We observed decreased interactions within the TADs in 30 h group cells compared to cells at 0 h (Figure 1G). We also observed multiple regions with disrupted TADs in cells at 30 h group, exemplified at chr3:41-51Mb and chr4:74-83Mb regions (Figure 1H).
Increased inter-chromosomal interactions during erythroid chromatin condensation
Chromosome can be arranged into A and B compartments on a large scale in Hi-C analyses. A and B compartments are associated with open/active and closed/inactive chromatins, respectively. To further characterize the DNA structure during terminal erythropoiesis, we investigated the A/B compartment and compartment switches in these cells. Comparing cells at 0 versus 30 h, 2.73% of B compartments in cells at 0 h switch to A compartment at 30 h whereas 2.24% A switch to B. The rest of the compartments remain unchanged (Figure 2A).
Figure 2.

Increased inter-chromosomal interactions during erythroid chromatin condensation
(A) A/B compartment switches in cells at 30 h compared with cells at 0 h. AA: A to A switch, BA: B to A switch, AB: A to B switch, BB: B to B switch.
(B) Cis (upper panels) and trans (lower panels) saddle plot analyses for Hi-C A/B compartment interactions in cells at 0 and 30 h. The analysis is based on A/B compartment defined in the 0 h condition.
(C) Quantification of B.
To compare the genome-wide compartment interactions, we performed a saddle plots analysis. We determined and fixed genes’ compartment identity at 0 h and applied for all group analysis for better picturing of the dynamic compartmental changes during development. We then generated saddle plots for cis and trans interactions, representing relative intra-chromosomal and inter-chromosomal interactions, respectively (Figure 2B). The analysis demonstrates that the interaction patterns change significantly during development. The cis interactions show a slight decrease in AA, BB and AB interactions in cells at 30 h whereas the trans interactions increase in BB and AB interactions in these cells (Figure 2C). These results indicate increased inter-chromosomal interactions during chromatin condensation.
Changes of chromatin accessibility during terminal erythropoiesis
We next performed ATAC-seq analyses on these cells to determine the chromatin accessibility changes during terminal erythropoiesis. Cells from the same experimental conditions as above were used (0 and 30 h). In total, we identified 17,941 peaks, among which 1,518 peaks show differential accessibility (>2-fold change, adjusted pvalue < 0.00001). Compared to cells at 0 h, 394 peaks were upregulated and 1,124 were downregulated in cells at 30 h group (Figure 3A).
Figure 3.

Changes of chromatin accessibility during terminal erythropoiesis
(A) Volcano plot of open peaks detected by ATAC-seq comparing cells at 30 vs 0 h. Red dots: >2-fold change, adjusted pvalue <0.00001; Green dots: >2-fold change, adjusted pvalue >0.00001; Blue dots: <2-fold change, adjusted pvalue <0.00001; Gray dot: <2-fold change, adjusted pvalue >0.00001.
(B) K-means clustering of open chromatin regions of cells at 0 and 30 h. Experiments were performed in duplicate (r).
(C) Quantification of peak density in cluster 9, 10, 11, and 12 in B.
We then grouped peaks in ATAC-seq into 12 clusters using seqMINER. Among these clusters, cluster 9 and 10 show decreased peak reads at 30 h, suggesting reduced accessibility (Figures 3B and3C). Of interest, Gene Ontology (GO) analysis reveals that many genes in these clusters are involved in immune responses (Figure S1). Cluster 11 and 12 show increased accessibility at 30 h (Figures 3B and3C). GO analysis shows that genes in these two clusters are closely related to terminal erythropoiesis including porphyrin metabolism and biosynthesis, heme metabolism and biosynthesis, erythrocyte differentiation, iron hemostasis, etc. (Figure S1).
Gene expression changes during terminal erythropoiesis
Under the same experimental settings, we then performed RNA-sequencing analyses to further determine the changes in gene expression during terminal erythropoiesis. In total, 23,564 genes were identified, among which 3,076 genes were found differentially expressed between 0 and 30 h groups (fold change>4, p<0.01). Among the differentially expressed genes, 813 genes were upregulated in cells at 30 h compared to those at 0 h, whereas 2,263 were downregulated (Figure 4A). We then generated a heatmap with most significantly differentially expressed genes. As expected, we observed that erythroid-related genes are among those that are most significantly upregulated (Figure 4B). GO enrichment analysis showed that the upregulated genes are closely related to erythropoiesis, as well as cell cycle and autophagy that are also well known to be involved in terminal erythropoiesis (Figure S2).
Figure 4.

Gene expression changes during terminal erythropoiesis
(A) Volcano plot of RNA-seq data comparing cells at 30 h to those at 0 h.
(B) Heatmap analysis of RNA-seq data between cells at 30 and 0 h groups in triplicate. The most significantly differentially expressed genes are listed on the right.
Cross dataset analyses reveal diverse ways of gene expression regulation during terminal erythropoiesis
The A and B compartments in Hi-C are generally related to active and inactive areas in chromatin, respectively. Chromatin accessibility is also positively associated with genes’ expression and activity. To test whether these are applicable during terminal erythropoiesis, we cross-analyzed datasets in Hi-C, ATAT-seq, and RNA-seq. We identified 3,787 genes switched from compartment B to A, which represents increased gene activity. When compared to the RNA-seq data, 121 genes switched from B to A showed increased expression, whereas 612 genes with significantly increased expression did not show compartment change in PCA analysis (Fold change>4, p<0.01). Among these 121 genes, we identified 25 genes with increased chromatin accessibility in ATAC-seq (Figure 5A). Notably, most of the genes in these datasets do not overlap, which indicates that gene expression is regulated in diverse ways during terminal erythropoiesis.
Figure 5.

Cross analyses of Hi-C, ATAC-seq, and RNA-seq
(A) Green: genes switched from compartment B to A in Hi-C; Pink: Genes with increased peaks in ATAC-seq; Blue: Genes with increased expression in RNA-seq. Representative genome browser view of genes with increased expression and chromatin accessibility is on the right.
(B) Gene Ontology analysis of the 25 overlapped genes in three datasets.
GO analysis shows that these 25 genes are closely related to terminal erythropoiesis (Figure 5B). We reviewed literature and listed related studies on these genes in Table 1. Within these 25 genes, Kel, Rhag and Abcg2 are well known red blood cell markers. Genes like Arl4a, Hpf1, Snca, and Trim10 have been reported to be involved in terminal erythropoiesis. The roles of other genes in erythropoiesis remain unclear.
Table 1.
Genes that are upregulated during terminal erythropoiesis with increased chromatin accessibility and compartment switch from B to A
| OFFICIAL GENE SYMBOL | Gene Name | Alias | Function | Reference |
|---|---|---|---|---|
| 1110034G24Rik | RIKEN cDNA 1110034G24 gene(1110034G24Rik) | Shld1 | ||
| Abcg2 | ATP-binding cassette, sub-family G (WHITE), member 2(Abcg2) | ABC15, ABCP, BCRP, BCRP1, BMDP, CD338, CDw338, EST157481, GOUT1, MRX, MXR, MXR1, UAQTL1, MXR-1 | Abcg2 null mice show similar phenotype as erythropoietic protoporphyria | Jonker et al.16 |
| Arl4a | ADP-ribosylation factor-like 4A(Arl4a) | Knockdown of arl4a in Zebrafish induces reduced definitive hematopoiesis by interfering with Notch Receptor maturation | Guo et al.17 | |
| Cdc42ep3 | CDC42 effector protein (Rho GTPase binding) 3(Cdc42ep3) | BORG2, CEP3, UB1 | ||
| Cpeb4 | cytoplasmic polyadenylation element binding protein 4(Cpeb4) | KIAA1673 | Knockdown of Cpeb4 causes inhibition of terminal erythropoiesis | Hu et al.18 |
| Gda | guanine deaminase(Gda) | GAH, Cytoplasmic PSD-95 Interactor, P51-Nedasin, EC 3.5.4.3 | ||
| Ghitm | growth hormone inducible transmembraneprotein(Ghitm) | TMBIM5, DERP2, HSPC282, PTD010, My021 | ||
| Hace1 | HECT domain and ankyrin repeat containing, E3 ubiquitin protein ligase 1(Hace1) | KIAA1320, EC 2.3.2.26, EC 6.3.2, SPPRS | ||
| Hpf1 | histone PARylation factor 1(Hpf1) | C4orf27, FLJ20534, Chromosome 4 Open Reading Frame 27, UPF0609 Protein C4orf27 | Modulation of PARP2, which sustains erythropoiesis | Farres et al.19 Kurgina et al.20 |
| Ifit1bl1 | interferon induced protein with tetratricpeptide repeats 1B like 1(Ifit1bl1) | IFNAl1, G10P1, IFI56, GARG-16, IFI-56K | ||
| Kel | Kell blood group(Kel) | CD238, ECE3 | Endopeptidase that cleaves big endothelin-3. | Lee et al.21 |
| Lrrc1 | leucine rich repeat containing 1(Lrrc1) | LANO, DJ523E19.1 | ||
| Mllt3 | myeloid/lymphoid or mixed-lineage leukemia; translocated to, 3(Mllt3) | YEATS3, AF9 | Regulate Erythrocyte/Megakaryocyte fate decision | Pina et al.22 |
| Ncoa7 | nuclear receptor coactivator 7(Ncoa7) | ERAP140, DJ187J11.3. TLDC4 | ||
| Pkhd1l1 | polycystic kidney and hepatic disease 1-like 1(Pkhd1l1) | Fibrocystin-L | ||
| Pla2g16 | phospholipase A2, group XVI(Pla2g16) | HREV107-3, HREV107, AdPLA, PLAAT-3 | ||
| Rhag | Rhesus blood group-associated A glycoprotein(Rhag) | SLC42A1, CD241, RH50A | Amonia transport | Stewart et al.23Genetet et al.24 |
| Scai | suppressor of cancer cell invasion(Scai) | NET40, FLJ36664, C9orf126 | ||
| Sec61g | SEC61, gamma subunit(Sec61g) | SSS1 | ||
| Smc2os | structural maintenance of chromosomes 2, opposite strand(Smc2os) | |||
| Snca | synuclein, alpha(Snca) | NACP, PARK1. PD1, PARK4 | Regulate oxidative stress | Renella et al.25 |
| Trim10 | tripartite motif-containing 10(Trim10) | RFB30, HERF1, RNF9 | regulating splicing of exon16 in 4.1R and required for terminal erythropoiesis | Blaybel et al.26 |
| Tspan8 | tetraspanin 8(Tspan8) | CO-029, TM4SF3 | Zhao et al.27 | |
| Zfand6 | zinc finger, AN1-type domain 6(Zfand6) | AWP1, ZFAND5B, ZA20D3 | ||
| Zfp760 | zinc finger protein 760(Zfp760) |
Discussion
In this study, we report for the first time the genomewide chromatin 3D architectural changes during murine terminal erythroid differentiation. Our study reveals decreased long-range chromosome interactions and TADs during terminal erythropoiesis. Similar to the decreased TADs in the late-stage murine terminal erythroblasts, it was reported that chicken mature erythrocytes lack typical TADs found in mammals and chicken embryonic fibroblasts.28 A more recent study comparing erythrocytes 3D genome organization across various vertebrates further showed lack of TADs in vertebrate erythrocytes, including late-stage erythroblasts in mouse.29 Because chicken mature erythrocytes also undergo nuclear condensation despite retaining their nuclei, these findings suggest that the terminal erythropoiesis is evolutionally conserved among vertebrates. It is possible that immature chicken erythroblasts in the bone marrow could have typical TADs as those found in the murine erythroblasts and gradually decrease. It would be interesting to explore from the perspective of comparative biology.
Besides compartment switching, we also found increased trans interactions in BB and AB compartments during development, suggesting increased inter-chromosomal interactions. Overall, our study reveals a dynamic process during chromatin condensation in terminal erythropoiesis. In this process, a small portion of genome DNA switches between active and inactive states. Chromatin penetrates the compartmental boundary and builds new interactions, which is featured by decreased intra-chromosomal and increased inter-chromosomal interactions (Figure 6).
Figure 6.

Schematic overview of chromatin 3D structural changes during terminal erythropoiesis
Blue and red strings represent independent chromosomes. Blue and red dots represent intra-chromosomal interactions. Purple dots represent inter-chromosomal interactions. During differentiation, chromosomal interactions within the same chromosome decrease, whereas the interactions between chromosomes increase.
Another key feature in terminal erythropoiesis is nuclear opening that is mediated by partial histone release and facilitates nuclear condensation.9,30,31 This process occurs more frequently during the polychromatic to orthochromatic stage. We previously reported that caspases play critical roles in this process by cleaving lamin B to generate transient nuclear openings for histone release. Histone cytoplasmic release during terminal erythropoiesis was also reported to be mediated by exportin 7, a highly erythroid-specific exportin. Loss of exportin 7 inhibits histone and DNA release, as well as nuclear condensation.11 Moreover, a recent report showed that loss of Wdr, a gene essential for lamin B degradation, could induce anemia in zebrafish and disrupt erythroid nuclear condensation in mouse.12 Together, these studies demonstrate that histone release is essential in nuclear condensation and enucleation. It would be interesting to investigate whether inhibition or downregulation of these proteins would affect chromatin 3D structural changes. In this respect, erythroid specific and inducible knockout models will be essential to reveal the detailed mechanisms.
Limitations of the study
The current study used cultured mouse fetal liver cells. The culture artifact could be a potential concern. In addition, the rapid differentiation process of mouse fetal liver cells within 48 h makes it difficult to dissect the chromatin reconstruction at each stage of terminal erythropoiesis.
STAR★Methods
Key resources table
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| APC Rat Anti-Mouse TER-119 | BD | Cat# 557909; RRID:AB_398635 |
| FITC Rat Anti-Mouse CD71 | BD | Cat# 567260; RRID:AB_2916522 |
| Deposited data | ||
| HiC-seq data for erythrocyte differentiation | This paper | GSE184974 |
| ATAC-seq and RNA-seq data for erythrocyte differentiation | This paper | GSE185470 |
| Experimental models: Organisms/strains | ||
| C57BL/6j mice | Jax Lab | strain #: 000,664 |
| Software and algorithms | ||
| runHiC python package | Wang32 | https://pypi.org/project/runHiC/ |
| cooltools | Venev et al.33 | https://github.com/open2c/cooltools |
| cworld matrix2insulation and insulation2tads | NA | https://github.com/dekkerlab/cworld-dekker |
| juicebox | NA | https://github.com/aidenlab/Juicebox |
| MACS2 | Zhang et al.34 | NA |
| seqMINER | Ye et al.35 | NA |
| bedtools | Quinlan et al.36 | NA |
| TopHat2 | Kim et al.37 | NA |
Resource availability
Lead contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the lead contact, Peng Ji (peng-ji@fsm.northwestern.edu).
Materials availability
This study did not generate new unique reagents.
Experimental model and subject details
E13.5 fetal liver cells were acquired by breeding male and female C57BL/6J mice purchased from the Jackson laboratory. The mice were maintained at animal facility in center for comparative medicine, Feinberg School of Medicine at Northwestern University. After 1 night of breeding (day 1), female mice were separated and observed for 2 weeks. At day 14 after breeding, the pregnant female mice were sacrificed, and fetal livers were then retrieved. The fetal livers were then homogenized by repeated pipetting and filtered through 40 μm cell strainer to obtain single cells. The filtered cells were incubated with biotin-antimouse Ter119 antibody for 15 min and washed with PBS. After washing, the cells were resuspended and Ter119 positive cells were pulled down with streptavidin magnetic beads. The remained Ter119 negative cells were then used for the following experiments.
The Ter119 negative cells were either processed for sequencing directly or cultured in erythropoietin (EPO)-containing medium (15% FBS, 1% BSA, 10μg/ml Insulin, 200μg/ml Holo-transferrin, 10−4M β-Mercaptoethanol, 1% Penicillin-Streptomycin, 2mM Glutamine, 2U/ml EPO in IMDM) for up to 48 h. The cells were harvested at 30 h for the follow up studies.15,38
Method details
Mice
C57BL/6J mice were purchased from the Jackson laboratory (strain #: 000,664). Both male and females were used. All animal studies were performed in accordance with the Guidelines for the Care and Use of Laboratory Animals and were approved by the Institutional Animal Care and Use Committees at Northwestern University.
Cell culture
Purification of mouse fetal liver erythroblast precursors and in vitro culture were performed as previously described.31 Briefly, fetal liver erythroid precursors were isolated from the E13.5 fetal liver by depletion of Ter119 positive cells using a biotinylated anti-Ter119 antibody. The Ter119 negative cells were then incubated in EPO-containing medium (15% FBS, 1% BSA, 10μg/ml Insulin, 200 μg/mL Holo-transferrin, 10−4M β-Mercaptoethanol, 1% Penicillin-Streptomycin, 2mM Glutamine, 2U/ml EPO in IMDM) for differentiation and enucleation for up to 48 h. The cells were harvested at 30 h for further analysis.
Cell surface marker staining
Cells were washed in PBS and resuspended in 100 μL PBS with a concentration of 1 million cells/ml. 0.5 μg APC-Ter119 antibody (BD, Cat# 557909) and FITC-CD71(BD, Cat# 567260) were added to the cell suspension. In turn, Hoechst 33,342 was added to the cell suspension to a final concentration of 20 μg/mL. The Cells were incubated in dark under room temperature for 10 min and washed with PBS. The washed cells were then resuspended with PBS containing 1 μg/mL propidium iodide to exclude dead cells. The resuspended cells will then be analyzed by flow cytometry within 1 h.
Total RNA extraction
Cells were harvested and washed for 3 times with PBS. Qiagen RNeasy kit was used to extract total RNA. Briefly, cells were lysed and homogenized in lysis buffer. Ethanol was then added to the lysate followed by centrifugation through silica membrane. After 2 times wash, RNAs were eluted by elution buffer and stocked for further analysis.
Hi-C data analysis
The adapters of the raw reads were trimmed and mapped against the mm10 reference genome using the runHiC python package (https://pypi.org/project/runHiC/). The contact matrix was binned at multiple resolutions: 5, 10, 25, 40, 50, 100, 250, 500kb, 1, 2.5, 5 and 10Mb using the run-cool2multirescool script from the 4DN consortium. The ICE normalization was performed for each resolution. For A/B compartment analysis, we first identified A/B compartment at 100kb resolution using the cooltools (https://github.com/open2c/cooltools) call-compartment function. To quantify the A and B compartment interactions, we used the cooltools compute-saddle function. The cworld matrix2insulation and insulation2tads (https://github.com/dekkerlab/cworld-dekker) were used for insulations scores calculation and TAD identification. We also generated the multi-resolution.hic files using the juicer tools for the convenience of example Hi-C matrices visualization in juicebox (https://github.com/aidenlab/Juicebox). The accession number for Hi-C is GSE184974.
RNA-seq analysis
RNA-seq reads were aligned to the mouse genome (NCBI37/mm10) using TopHat2.37 Expression of each gene was calculated from the resulting alignment bam file by HTSeq-count.39 Differentially expressed genes were determined using R package DESeq240 with 2-fold change or 4-fold change as cutoff. Gene ontology analysis for changed genes was performed using DAVID.41
ATAC-seq analysis
Paired-end ATAC-seq reads were aligned to the mouse genome (NCBI37/mm10) using Bowtie242 with option ‘‘--very-sensitive-local’’. Mitochondrial reads were excluded from downstream analysis. Peak calling was performed on each individual sample by MACS234 with parameter ‘‘-BAMPE’’. Peaks from different ATAC-seq samples were merged using bedtools.36 Merged peaks were used as genome coordinate for K-means clustering which was performed using seqMINER.35 10 million randomly sampled mapped reads from each sample were used as input. Signals were calculated in a 1 kb region (±500 bp) surrounding the center of the peak with 20 nt bin. Peaks assignment to the genes and subsequent ontology analysis were done with GREAT.43 The accession number for ATAC-seq and RNA-seq is GSE185470.
Quantification and statistical analysis
Results are expressed as mean ± SEM unless otherwise indicated. Statistical comparisons between two groups were performed with a two-tailed unpaired Student’s t test using GraphPad Prism version 6.0 software. p <0.05 was considered to be significant.
Acknowledgments
We thank Dr. Ching Man Wai’s technical support during this study. This work was supported by National Institute of Diabetes and Digestive and Kidney Disease (NIDDK) grant R01-DK124220 (P.J.), National Heart, Lung, and Blood Institute (NHLBI) grant R01-HL148012 (P.J.), and R01-HL150729 (P.J.). P.J. is a scholar of the Leukemia and Lymphoma Society.
Author contributions
H.B., Y.H., J.W., A.M., D.H., Y.L., H.B., X.H., K.R., and E.L. performed the experiments and interpreted data. Y.H. performed the Hi-C experiment. J.W., Z.X., and D.W. analyzed the Hi-C data. H.B., Y.L., F.Y., and P.J. designed the experiments, interpreted data, and edited the manuscript. H.B. and P.J. wrote the manuscript.
Declaration of interests
The authors declare no competing interests.
Published: December 22, 2022
Footnotes
Supplemental information can be found online at https://doi.org/10.1016/j.isci.2022.105554.
Contributor Information
Feng Yue, Email: yue@northwestern.edu.
Peng Ji, Email: peng-ji@fsm.northwestern.edu.
Supplemental information
Data and code availability
-
•
All data were deposited at NCBI GEO database. The accession number for Hi-C is GEO: GSE184974. The accession number for ATAC-seq and RNA-seq data is GEO: GSE185470.
-
•
This paper does not report original code.
-
•
Any additional information required to reanalyze the data reported in this paper is available from the lead contactupon request.
References
- 1.Migliaccio A.R. Erythroblast enucleation. Haematologica. 2010;95:1985–1988. doi: 10.3324/haematol.2010.033225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Ji P., Murata-Hori M., Lodish H.F. Formation of mammalian erythrocytes: chromatin condensation and enucleation. Trends Cell Biol. 2011;21:409–415. doi: 10.1016/j.tcb.2011.04.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Mei Y., Liu Y., Ji P. Understanding terminal erythropoiesis: an update on chromatin condensation, enucleation, and reticulocyte maturation. Blood Rev. 2021;46:100740. doi: 10.1016/j.blre.2020.100740. [DOI] [PubMed] [Google Scholar]
- 4.Hattangadi S.M., Wong P., Zhang L., Flygare J., Lodish H.F. From stem cell to red cell: regulation of erythropoiesis at multiple levels by multiple proteins, RNAs, and chromatin modifications. Blood. 2011;118:6258–6268. doi: 10.1182/blood-2011-07-356006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Popova E.Y., Krauss S.W., Short S.A., Lee G., Villalobos J., Etzell J., Koury M.J., Ney P.A., Chasis J.A., Grigoryev S.A. Chromatin condensation in terminally differentiating mouse erythroblasts does not involve special architectural proteins but depends on histone deacetylation. Chromosome Res. 2009;17:47–64. doi: 10.1007/s10577-008-9005-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Jayapal S.R., Lee K.L., Ji P., Kaldis P., Lim B., Lodish H.F. Down-regulation of Myc is essential for terminal erythroid maturation. J. Biol. Chem. 2010;285:40252–40265. doi: 10.1074/jbc.M110.181073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Zermati Y., Garrido C., Amsellem S., Fishelson S., Bouscary D., Valensi F., Varet B., Solary E., Hermine O. Caspase activation is required for terminal erythroid differentiation. J. Exp. Med. 2001;193:247–254. doi: 10.1084/jem.193.2.247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Carlile G.W., Smith D.H., Wiedmann M. Caspase-3 has a nonapoptotic function in erythroid maturation. Blood. 2004;103:4310–4316. doi: 10.1182/blood-2003-09-3362. [DOI] [PubMed] [Google Scholar]
- 9.Zhao B., Mei Y., Schipma M.J., Roth E.W., Bleher R., Rappoport J.Z., Wickrema A., Yang J., Ji P. Nuclear condensation during mouse erythropoiesis requires caspase-3-mediated nuclear opening. Dev. Cell. 2016;36:498–510. doi: 10.1016/j.devcel.2016.02.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Zhao B., Liu H., Mei Y., Liu Y., Han X., Yang J., Wickrema A., Ji P. Disruption of erythroid nuclear opening and histone release in myelodysplastic syndromes. Cancer Med. 2019;8:1169–1174. doi: 10.1002/cam4.1969. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Hattangadi S.M., Martinez-Morilla S., Patterson H.C., Shi J., Burke K., Avila-Figueroa A., Venkatesan S., Wang J., Paulsen K., Gorlich D., et al. Histones to the cytosol: exportin 7 is essential for normal terminal erythroid nuclear maturation. Blood. 2014;124:1931–1940. doi: 10.1182/blood-2013-11-537761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Zhen R., Moo C., Zhao Z., Chen M., Feng H., Zheng X., Zhang L., Shi J., Chen C. Wdr26 regulates nuclear condensation in developing erythroblasts. Blood. 2020;135:208–219. doi: 10.1182/blood.2019002165. [DOI] [PubMed] [Google Scholar]
- 13.Zhao B., Yang J., Ji P. Chromatin condensation during terminal erythropoiesis. Nucleus. 2016;7:425–429. doi: 10.1080/19491034.2016.1226717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Zhang J., Socolovsky M., Gross A.W., Lodish H.F. Role of Ras signaling in erythroid differentiation of mouse fetal liver cells: functional analysis by a flow cytometry-based novel culture system. Blood. 2003;102:3938–3946. doi: 10.1182/blood-2003-05-1479. [DOI] [PubMed] [Google Scholar]
- 15.Ji P., Jayapal S.R., Lodish H.F. Enucleation of cultured mouse fetal erythroblasts requires Rac GTPases and mDia2. Nat. Cell Biol. 2008;10:314–321. doi: 10.1038/ncb1693. [DOI] [PubMed] [Google Scholar]
- 16.Jonker J.W., Buitelaar M., Wagenaar E., Van Der Valk M.A., Scheffer G.L., Scheper R.J., Plosch T., Kuipers F., Elferink R.P., Rosing H., et al. The breast cancer resistance protein protects against a major chlorophyll-derived dietary phototoxin and protoporphyria. Proc. Natl. Acad. Sci. USA. 2002;99:15649–15654. doi: 10.1073/pnas.202607599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Guo Y., Cheng B.Y.L., Wang D., Ma A.C.H., He B.L., Man T.K., Cheung M.P.L., Shi X., Ng N.K.L., Leung A.Y.H. Function of Arl4aa in the initiation of hematopoiesis in zebrafish by maintaining golgi complex integrity in hemogenic endothelium. Stem Cell Rep. 2020;14:575–589. doi: 10.1016/j.stemcr.2020.02.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Hu W., Yuan B., Lodish H.F. Cpeb4-mediated translational regulatory circuitry controls terminal erythroid differentiation. Dev. Cell. 2014;30:660–672. doi: 10.1016/j.devcel.2014.07.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Farres J., Llacuna L., Martin-Caballero J., Martinez C., Lozano J.J., Ampurdanes C., Lopez-Contreras A.J., Florensa L., Navarro J., Ottina E., et al. PARP-2 sustains erythropoiesis in mice by limiting replicative stress in erythroid progenitors. Cell Death Differ. 2015;22:1144–1157. doi: 10.1038/cdd.2014.202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Kurgina T.A., Moor N.A., Kutuzov M.M., Naumenko K.N., Ukraintsev A.A., Lavrik O.I. Dual function of HPF1 in the modulation of PARP1 and PARP2 activities. Commun. Biol. 2021;4:1259. doi: 10.1038/s42003-021-02780-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Lee S., Lin M., Mele A., Cao Y., Farmar J., Russo D., Redman C. Proteolytic processing of big endothelin-3 by the kell blood group protein. Blood. 1999;94:1440–1450. [PubMed] [Google Scholar]
- 22.Pina C., May G., Soneji S., Hong D., Enver T. MLLT3 regulates early human erythroid and megakaryocytic cell fate. Cell Stem Cell. 2008;2:264–273. doi: 10.1016/j.stem.2008.01.013. [DOI] [PubMed] [Google Scholar]
- 23.Stewart A.K., Shmukler B.E., Vandorpe D.H., Rivera A., Heneghan J.F., Li X., Hsu A., Karpatkin M., O'Neill A.F., Bauer D.E., et al. Loss-of-function and gain-of-function phenotypes of stomatocytosis mutant RhAG F65S. Am. J. Physiol. Cell Physiol. 2011;301:C1325–C1343. doi: 10.1152/ajpcell.00054.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Genetet S., Ripoche P., Picot J., Bigot S., Delaunay J., Armari-Alla C., Colin Y., Mouro-Chanteloup I. Human RhAG ammonia channel is impaired by the Phe65Ser mutation in overhydrated stomatocytic red cells. Am. J. Physiol. Cell Physiol. 2012;302:C419–C428. doi: 10.1152/ajpcell.00092.2011. [DOI] [PubMed] [Google Scholar]
- 25.Renella R., Schlehe J.S., Selkoe D.J., Williams D.A., LaVoie M.J. Genetic deletion of the GATA1-regulated protein alpha-synuclein reduces oxidative stress and nitric oxide synthase levels in mature erythrocytes. Am. J. Hematol. 2014;89:974–977. doi: 10.1002/ajh.23796. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Blaybel R., Theoleyre O., Douablin A., Baklouti F. Downregulation of the Spi-1/PU.1 oncogene induces the expression of TRIM10/HERF1, a key factor required for terminal erythroid cell differentiation and survival. Cell Res. 2008;18:834–845. doi: 10.1038/cr.2008.68. [DOI] [PubMed] [Google Scholar]
- 27.Zhao K., Wang Z., Hackert T., Pitzer C., Zoller M. Tspan8 and Tspan8/CD151 knockout mice unravel the contribution of tumor and host exosomes to tumor progression. J. Exp. Clin. Cancer Res. 2018;37:312. doi: 10.1186/s13046-018-0961-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Fishman V., Battulin N., Nuriddinov M., Maslova A., Zlotina A., Strunov A., Chervyakova D., Korablev A., Serov O., Krasikova A. 3D organization of chicken genome demonstrates evolutionary conservation of topologically associated domains and highlights unique architecture of erythrocytes' chromatin. Nucleic Acids Res. 2019;47:648–665. doi: 10.1093/nar/gky1103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Ryzhkova A., Taskina A., Khabarova A., Fishman V., Battulin N. Erythrocytes 3D genome organization in vertebrates. Sci. Rep. 2021;11:4414. doi: 10.1038/s41598-021-83903-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Zhao B., Keerthivasan G., Mei Y., Yang J., McElherne J., Wong P., Doench J.G., Feng G., Root D.E., Ji P. Targeted shRNA screening identified critical roles of pleckstrin-2 in erythropoiesis. Haematologica. 2014;99:1157–1167. doi: 10.3324/haematol.2014.105809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Zhao B., Mei Y., Yang J., Ji P. Mouse fetal liver culture system to dissect target gene functions at the early and late stages of terminal erythropoiesis. J. Vis. Exp. 2014;9:e51894. doi: 10.3791/51894. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Wang X. 2016. runHiC: A User-Friendly Hi-C Data Processing Software Based on Hiclib (Zenodo) [Google Scholar]
- 33.Venev S., Abdennur N., Goloborodko A., Flyamer I., Fudenberg G., Nuebler J., Galitsyna A., Akgol B., Abraham S., Kerpedjiev P., Imakaev M. 2021. open2c/cooltools: v0.4.1 (Zenodo) [Google Scholar]
- 34.Zhang Y., Liu T., Meyer C.A., Eeckhoute J., Johnson D.S., Bernstein B.E., Nusbaum C., Myers R.M., Brown M., Li W., Liu X.S. Model-based analysis of ChIP-seq (MACS) Genome Biol. 2008;9:R137. doi: 10.1186/gb-2008-9-9-r137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Ye T., Krebs A.R., Choukrallah M.A., Keime C., Plewniak F., Davidson I., Tora L. seqMINER: an integrated ChIP-seq data interpretation platform. Nucleic Acids Res. 2011;39:e35. doi: 10.1093/nar/gkq1287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Quinlan A.R., Hall I.M. BEDTools: a flexible suite of utilities for comparing genomic features. Bioinformatics. 2010;26:841–842. doi: 10.1093/bioinformatics/btq033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Kim D., Pertea G., Trapnell C., Pimentel H., Kelley R., Salzberg S.L. TopHat2: accurate alignment of transcriptomes in the presence of insertions, deletions and gene fusions. Genome Biol. 2013;14:R36. doi: 10.1186/gb-2013-14-4-r36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Liu Y., Mei Y., Han X., Korobova F.V., Prado M.A., Yang J., Peng Z., Paulo J.A., Gygi S.P., Finley D., Ji P. Membrane skeleton modulates erythroid proteome remodeling and organelle clearance. Blood. 2021;137:398–409. doi: 10.1182/blood.2020006673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Anders S., Pyl P.T., Huber W. HTSeq--a Python framework to work with high-throughput sequencing data. Bioinformatics. 2015;31:166–169. doi: 10.1093/bioinformatics/btu638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Love M.I., Huber W., Anders S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014;15:550. doi: 10.1186/s13059-014-0550-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Huang da W., Sherman B.T., Lempicki R.A. Systematic and integrative analysis of large gene lists using DAVID bioinformatics resources. Nat. Protoc. 2009;4:44–57. doi: 10.1038/nprot.2008.211. [DOI] [PubMed] [Google Scholar]
- 42.Langmead B., Salzberg S.L. Fast gapped-read alignment with Bowtie 2. Nat. Methods. 2012;9:357–359. doi: 10.1038/nmeth.1923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.McLean C.Y., Bristor D., Hiller M., Clarke S.L., Schaar B.T., Lowe C.B., Wenger A.M., Bejerano G. GREAT improves functional interpretation of cis-regulatory regions. Nat. Biotechnol. 2010;28:495–501. doi: 10.1038/nbt.1630. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
-
•
All data were deposited at NCBI GEO database. The accession number for Hi-C is GEO: GSE184974. The accession number for ATAC-seq and RNA-seq data is GEO: GSE185470.
-
•
This paper does not report original code.
-
•
Any additional information required to reanalyze the data reported in this paper is available from the lead contactupon request.