

Summary
Background
ABL-class gene fusions other than BCR-ABL1 have been detected in ~3% of children with newly diagnosed acute lymphoblastic leukemia (ALL) and may be targeted by tyrosine kinase inhibitors (TKIs). This international study establishes the baseline characteristics and outcome of ABL-class ALL patients in the pre-TKI era.
Methods
Patients’ characteristics and outcome of 122 children (1–18 years) with ABL-class B-cell precursor ALL were retrospectively collected through the Ponte di Legno consortium. Patients were enrolled in pediatric trials between 2000 and 2018 and were not exposed to TKIs during their first-line protocols. Event-free (EFS) and overall survival (OS) were determined by Kaplan-Meier methodology, and the cumulative incidence of relapse (CIR) and treatment-related mortality by a competing risk model.
Findings
Outcome of all ABL-class cases at 5 years was 31.0% (standard deviation (SD) 4.6) for CIR, 59.1% (95%CI 50.5–69.1) for EFS and 76.1% (95%CI 68.6–84.5) for OS. ABL-class patients displayed a high frequency of poor prednisone response (28 of 57 patients, 49%) and IKZF1 deletions (36 of 59 patients, 61%), but both features lacked prognostic value. MRD-levels at the end of induction therapy (EOI) were very high (≥1×10−2) in 66% of ABL-class cases (61 of 93 patients), and most prevalent detected in ABL2 (6 of 7 patients, 86%) and PDGFRB-fusion (43 of 49 patients, 88%) cases. MRD-EOI ≥1×10−2 was predictive of an unfavorable outcome among ABL-class patients (HREFS 3.33, 95%CI 1.46–7.56; p=0.0039. The 5-year EFS was 80% (95%CI 58.7–100) for CSF1R (n=10), 68.6% (95%CI 54.5–86.3) for ABL1 (n=40), 52.9% (95%CI 41.5–67.5) for PDGFRB (n=64), and 37.5% (95%CI 15.3–91.7) for ABL2 (n=8) fusion cases (p=0.059). Sixty-nine percent of relapses (25 of 36) occurred within 3 years after diagnosis. The 5-years CIR of patients who received hematopoietic stem cell transplantation (n=41; 17.8% SD 6.2) was lower compared to the non-transplanted group (n=43; 45.1% SD 8.4; p=0.013), but EFS and OS did not differ between the two groups.
Interpretation
Children with ABL-class B-ALL have a poor outcome on therapies without TKIs despite the use of high-risk chemotherapy regimens and frequent transplantation in first remission. This paper provides baseline outcome for evaluating the potential benefit of upfront TKI usage in ABL-class patients.
Keywords: ABL-class, tyrosine kinase inhibitors, ALL, clinical outcome, children
Introduction
Gene expression profiling studies have identified over 20 genetic subtypes of B-cell precursor acute lymphoblastic leukemia (B-ALL) in children. One particular group of interest because of a high risk of relapse is BCR-ABL1-like or Philadelphia chromosome (Ph)-like B-ALL. This group, first described in 2009, whilst negative for the BCR-ABL1 fusion has a gene expression profile that mirrors that of BCR-ABL1 positive ALL.1,2 This BCR-ABL1-like/-Ph-like group is characterized by a high frequency of lesions involving ABL-class genes (12–18% of cases) as well as JAK-pathway genes (JAK2, EPOR), chemokine receptors (CRLF2) and/or MEK-ERK pathway genes.3–5 The ABL-class group of genomic alterations consists mainly of in-frame fusions that join ABL1, ABL2, CSF1R and PDGFRB, among others, to genes normally expressed during B-cell development. The resulting chimeric proteins have profound tyrosine kinase activity in cells in which the ABL-class genes are usually not abundantly expressed, resulting in the activation of pathways involved in the survival and proliferation of immature lymphoid cells.6,7
The addition of tyrosine kinase inhibitors (TKIs) such as imatinib and dasatinib to chemotherapy-based therapies has significantly improved the outcome for children with newly diagnosed BCR-ABL1 positive leukemia.8–12 Similar to BCR-ABL1-positive ALL, ABL-class ALL is associated with high risk (HR) features of older age and higher white blood cell count at diagnosis and high minimal residual disease levels at the end of induction therapy (MRD EOI).5,13 ABL-class fusions are found in much higher frequency (4%) in National Cancer Institute (NCI) high risk (HR) patients compared with 0.2% of standard risk patients.14,15 Imatinib and dasatinib both have significant activity in pre-clinical models of ABL-class fusions which mirrors the results observed in BCR-ABL1 positive pre-clinical models.3,6,7 Given the molecular similarities to BCR-ABL1-positive ALL, there are strong grounds for assessing the potential benefit of TKIs in ABL-class patients. Several anecdotal reports have described excellent responses to additional TKI therapy but lacked information regarding long-term outcome.6,16,17 Two recent reports highlight the potential efficacy of TKIs in patients but these are still hampered by small patient numbers and late introduction of TKI therapy.13,18 The ABL-class cohort therefore remains a heterogenous group of patients with unverified baseline characteristics and outcome for each of the ABL1, ABL2, CSF1R and PDGFRB fusion types separately.
The Ponte di Legno group consists of >20 established ALL study groups worldwide and was initiated to address outcome questions in rare subsets of newly diagnosed pediatric ALL patients for which individual study groups have only a limited number of cases. We undertook a retrospective study to investigate the clinical outcome of newly diagnosed ABL-class patients treated on first-line trials without TKIs. The results described in this paper will serve as reference to interpret the potential benefit of adding TKIs in the front-line treatment of children with ABL-class ALL.
Methods
Study design and participants
Newly diagnosed pediatric B-ALL cases (1–18 years of age) were retrospectively included in this study based on the presence of an ABL-class fusion, enrollment in a pediatric trial between 2000 and 2018, and no exposure to TKIs during their first-line protocol. Patient and outcome characteristics were collected from study groups belonging to the Ponte di Legno consortium using a standard case report form. In accordance with the declaration of Helsinki, written informed consent was obtained from parents or guardians, and the institutional review boards approved the use of patient data for research purposes. Patients were enrolled in pediatric trials between 3 October 2000 and 28 August 2018 and were not exposed to TKIs during their first-line protocols. Patients were categorized by NCI risk criteria into standard risk (SR) and high risk (HR; age ≥10 year and/or WBC ≥50×109/L at diagnosis). Minimal residual disease (MRD) testing was performed using IG/TCR PCR and/or flow-MRD depending on the protocol guidelines per study group. Results of these MRD monitoring methods are comparable according to previously published results.19 Data of both detection methods were merged in this study defining 1% by flow-MRD as reflecting a value equivalent to 1×10−2 by IG/TCR MRD. In most cases MRD values were measured at the end of induction (EOI), although less commonly data were also collected at the end of consolidation (EOC). The EOI and EOC MRD time points are taken roughly 29–35 days and 77–80 days after start treatment, respectively, with minor differences between protocols. Response to a therapeutic window with 7 days of prednisone and one dose of intrathecal methotrexate was collected for BFM-based trials. A poor prednisone response (PPR) was defined by persistence of ≥1,000 blasts per μl peripheral blood on day 8 of treatment. Patients received risk-stratified treatment based on the criteria set by each treatment protocol. In the present study, patients treated with either standard, medium, or non-high risk arms of individual protocols were collectively assigned to the non-high risk (non-HR) treatment group and only patients receiving high risk therapy were assigned to the HR treatment group.
Procedures
The ABL-class cohort was defined by fusion genes involving ABL1, ABL2, CSF1R and PDGFRB gene loci. ABL-class cases were identified by the diagnostic and research laboratories of participating study groups. Patients were retrospectively tested, often as part of research interests to characterize the poor prognostic subset of Ph-like/BCR-ABL1-like ALL. B-ALL cases negative for prognostically relevant genetic lesions (BCR-ABL1, KMT2A-rearranged, ETV6-RUNX1, TCF3-PBX1, high hyperdiploidy) were subjected to total RNA sequencing, reverse transcriptase-PCR or fluorescence in situ hybridization (FISH) often prompted by cytogenetic or karyotypic evidence for abnormal chromosomal regions affecting 1q25 (ABL2), 5q13–34 (CSF1R and PDGFRB)20 and 9q34 (ABL1), or by the gene expression signatures used to discover the Ph-like/BCR-ABL1-like cases.1,3,4,21 Identification of ABL-class cases was dependent on sample availability and individual study groups’ decisions to perform additional analyses. In the appendix p 1–2 the detection methods are described which were used by the Dutch Childhood Oncology Group (DCOG) as an example. Some cases included in this study have been presented in publications about new fusion gene discovery (including CRLF2, EPOR and JAK-fusions) in BCR-ABL1-like/-Ph-like patients5,6,20,22 and in a publication about outcome in a single protocol.13
In 59 (48.4%) of 122 cases, the presence of Ikaros (IKZF1) deletions (intragenic or fully deleted) was assessed by the multiplex ligation-dependent probe amplification (SALSA MLPA P202, MRC Holland). In a limited number of cases other B-cell development genes, including PAX5 and CDKN2A/2B deletions were also analysed using the SALSA MLPA P335 assay. Data on PAR1 and ERG status had limited availability by some study groups and IKZF1-plus status could be assessed according to Stanulla et al.23 Instead, we compiled a derivative IKZF1-“plus” group of cases having a deletion in IKZF1 with concomitant deletions in PAX5 and/or CDKN2A/2B, which largely (>85%) overlaps with the previously reported IKZF1-plus group.23,24
Outcomes
Complete remission (CR) was defined as <5% leukemic cells in the bone marrow and recovery of normal hematopoiesis, absence of peripheral blood leukemic cells and no evidence of disease at any other site. Early death was defined as death in induction prior to CR. Treatment related mortality (TRM) was defined as any death in first CR. Non-responders represented cases who failed to achieve CR after two courses of chemotherapy. Relapse was defined by disease recurrence after initial CR. The time between diagnosis and start of treatment is typically between 0 and 2 days.
Statistical analysis
The Pearson χ2 and Kruskal-Wallis test were used to compare age, white blood cell counts and MRD levels between the four ABL-class fusion types. A competing risk model with relapse and death was employed to estimate the cumulative incidence of relapse (CIR) and the cumulative incidence of treatment-related mortality (TRM) from first diagnosis for cases who reached CR. The Gray’s test was used to compare cumulative incidences between ABL-class patients. Kaplan-Meier (KM) methodology was used to estimate event-free survival (EFS) and overall survival (OS). EFS was defined as time from diagnosis to first event. Events were defined as non-response, early death, relapse, second malignancy and death in first remission. OS was estimated from diagnosis to date of death by any cause. Individuals without an event were censored at the last date of contact. EFS and OS KM-curves between ABL-class patients were statistically compared using the log-rank test and the 5-year survival percentage and standard error (SE) are given. To quantify the effect of risk factors on EFS, a Cox proportional hazard regression model was used to estimate the hazard ratio (HR) and the 95% confidence interval (CI). The effect of hematopoietic stem cell transplantation (HSCT) was investigated by a landmark approach to avoid immortal time bias. A waiting time to HSCT of 6 months was taken as landmark, and outcome events were only considered if occurring after the landmark. Analyses have been performed with SPSS version 25. All analyses concerning the competing risks model were performed in R software environment (version 3.2.2) with cmprsk package version 2.2–7. All plots for EFS, OS and CIR start at t=diagnosis.
Role of the funding source
The funder of the study had no role in study design, data collection, data analysis, data interpretation, or writing of the report. The corresponding author had full access to all of the data and the final responsibility to submit for publication.
Results
Newly diagnosed pediatric B-ALL patients (1–18 years of age) with ABL-class fusions enrolled in pediatric trials between 2000 and 2018 were eligible for this study, if they were not exposed to TKIs during their first-line protocols. Outcome characteristics of 122 patients were collected from 14 international study groups participating in the Ponte di Legno group (Figure 1). The breakdown per study group including NCI risk group is provided in appendix p 3. Data of 33 BFM/AIEOP patients was included in a prior single protocol study.13 Two-thirds of the 122 patients (84) were diagnosed before 2010, from 3 October 2000 till 20 November 2009; the remaining 38 were diagnosed from 22 February 2000 till 28 August 2018. Since most study groups only selectively screened for ABL-class fusions, the frequency of ABL-class fusion types (see appendix p 6) may not represent a population-based distribution of lesions. The vast majority of ABL-class fusions involve PDGFRB (64 patients, 52%) and ABL1 (40 patients, 33%), with a minority of cases having fusions involving CSF1R (10 patients, 8%) and ABL2 (8 patients, 7%). Fourteen different fusion partner genes were found, and their frequencies per ABL fusion type are outlined in appendix p 4 and 6. EBF1 was the predominant partner for PDGFRB (50/64 cases), ZMIZ1 for ABL1 (16/40), SSBP2 for CSF1R (8/10), and RCSD1 and ZH3HAV1 for ABL2 (each 4/8).
Figure 1: Cohort overview.
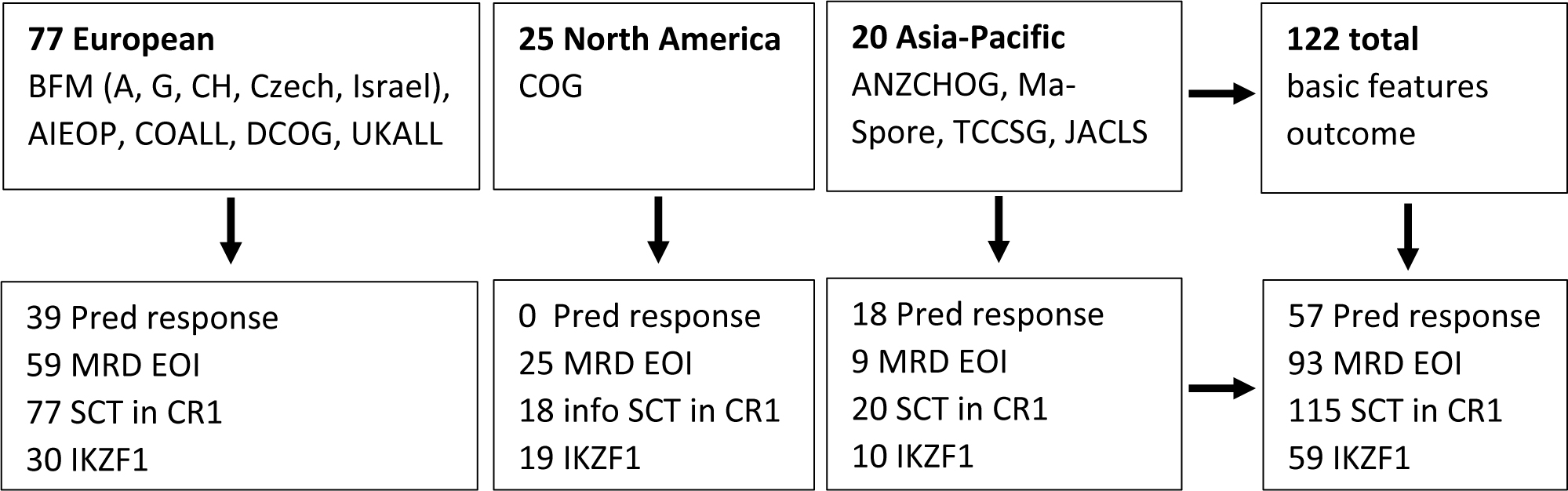
The diagram shows the distribution of pre-TKI ABL class patients collected by the different study groups and the number of patients with information on prednisone response, end of induction minimal residual disease, stem cell transplant in first complete remission and IKZF1 deletion.
The ABL-class fusions were most often seen in patients classified as NCI high risk (93 of 121 patients, 77%), and were skewed towards NCI high risk for all four ABL-class fusion types. The mean age at diagnosis was 9.7±5.1 year and the mean presenting white blood cell count (WBC) was 97.9±114.8×109/L. Age and WBC varied between ABL-class types (Pearson χ2, p=0.0066 and p=0.079, respectively), with the highest means for ABL2 fusion cases (Table 1; appendix p 7). Central nervous system involvement (CNS3) was detected at diagnosis in 4 of the 116 cases (3.4%) for which these data were available, including 1 CSF1R and 3 PDGFRB fusion cases. Testicular involvement was not observed in 44 males with available data. The male:female ratio was 1.7:1 and did not vary between ABL-class types (p=0.86; Table 1). In 36 (61%) of 59 cases tested for deletion status of IKZF1, PAX5, and CDKN2A/B, an IKZF1 deletion was detected. In 13 (22%) of 59 patients only an IKZF1 deletion was found and in 23 (39%) an IKZF1 deletion plus a PAX5 and/or a CDKN2A/B deletion. The frequency of IKZF1 deletion was not significantly associated with certain ABL-class fusion types (Table 1). Neither an IKZF1 deletion nor the IKZF1-“plus” genotype was associated with an unfavorable outcome compared to patients with wildtype IKZF1 (appendix p 8).
Table 1:
Patients characteristics and risk factors
| Risk Factor | Category | ABL-class | ABL1 | ABL2 | CSF1R | PDGFRB | Pearson χ2 |
|---|---|---|---|---|---|---|---|
| N (%) | N | N | N | N | |||
| 122 | 40 | 8 | 10 | 64 | |||
| Sex | Male | 76 (62%) | 25 | 4 | 7 | 40 | |
| Female | 46 (38%) | 15 | 4 | 3 | 24 | 0.86* | |
| Age at diagnosis | mean±sd (years) | 9.7±5.1 | 7.1±5.3 | 14±4.3 | 10.4±4.5 | 10.7±4.5 | |
| 1–9 yr | 54 (44%) | 26 | 1 | 3 | 24 | ||
| 10–18 yr | 68 (56%) | 14 | 7 | 7 | 40 | 0.0066* | |
| WBC at diagnosis | mean±sd (counts ×10e9/L) | 97.9±114.8 | 99.1±110.1 | 142.4±71.1 | 65.5±100.4 | 96.7±124 | |
| <50 | 62 (51%) | 21 | 0 | 7 | 34 | ||
| 50–100 | 18 (15%) | 4 | 2 | 1 | 11 | ||
| ≥100 | 41 (34%) | 14 | 6 | 2 | 19 | 0.079* | |
| NCI risk | SR | 28 (23%) | 14 | 0 | 2 | 12 | |
| HR | 93 (77%) | 25 | 8 | 8 | 52 | 0.081* | |
| CNS involvement | no | 112 (97%) | 38 | 8 | 9 | 57 | |
| yes | 4 (3%) | 0 | 0 | 1 | 3 | 0.34* | |
| Testis involvement | no | 44 (100%) | 15 | 3 | 4 | 22 | |
| yes | 0 (0%) | 0 | 0 | 0 | 0 | N/A | |
| Ikaros status | IKZF1 wildtype | 23(39%) | 4 | 2 | 2 | 15 | |
| IKZF1 deleted | 36 (61%) | 12 | 2 | 5 | 17 | 0.44* | |
| of which only IKZF1 deleted | 13 (22%) | 5 | 0 | 2 | 6 | ||
| or IKZF1 and PAX5 and/or CDKN2A/2B deleted | 23 (39%) | 7 | 2 | 3 | 11 | 0.72** | |
Percentage of cases per category out of total number of cases with registered information is given in parentheses.
Pearson χ2 p-values are estimates because the number of cases is less than 5 for some variables.
IKZF1 “plus” versus IKZF1 only.
N/A, Pearson χ2 not applicable since no patients with testis involvement were reported. Number of patients with missing data: WBC 1; NCI risk 1; CNS involvement 6; testis involvement 32.
The response to prednisone as a single systemic agent was assessed in 57 ABL-class patients, of whom 28 (49%) had a poor response (PPR; Table 1). The frequency of PPR varied between <10% for CSF1R (0 out of 4 patients) and ABL1 fusion cases (1 out of 14 patients) and ≥60% for ABL2 (3 out of 5 patients) and PDGFRB fusion cases (24 out of 34 patients; 71%, p=0.00015). A PPR in ABL-class cases was not predictive of an unfavorable clinical outcome (EFS p=0.35; appendix p 9). Non-HR and HR treatment was given to 28 (23%) and 93 (77%) of 121 ABL-class cases, respectively (Table 2). In the total cohort there was no significant difference in the EFS of non-HR (5-year 70.7%, 95%CI 54.3–92.1) and HR cases (5-year 55.4%, 95%CI 45.9–67.1; p=0.22; appendix p 10). Most PDGFRB fusion cases were NCI HR (52 of 64 patients, 81%) and had very high MRD levels at the end of induction. The few PDGFRB fusion cases who received non-HR treatment had a poor outcome; 6 out of 7 non-HR treated patients experienced an event compared to 24 out of 57 HR treated PDGFRB-fusion cases (appendix p 10; 5-year EFS of non-HR 28.6% (95%CI 8.9–92.2) and HR cases 56.5% (95%CI 44.5–71.8), p=0.032).
Table 2:
Risk stratification and response of children with ABL-class positive B-ALL
| Response criteria | Category | ABL-class | ABL1 | ABL2 | CSF1R | PDGFRB | Pearson χ2 | ||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| N (%) | N | N | N | N | |||||||
| 122 | 40 | 8 | 10 | 64 | |||||||
| Prednisone window response | PGR | 29 (51%) | 13 | 2 | 4 | 10 | |||||
| PPR | 28 (49%) | 1 | 3 | 0 | 24 | 0.00015* | |||||
| Treatment arm | non-HR | 28 (23%) | 16 | 1 | 4 | 7 | |||||
| HR | 93 (77%) | 23 | 7 | 6 | 57 | 0.0023* | |||||
| MRD EOI | negative | 5 (5%) | 3 | 0 | 1 | 1 | |||||
| <10−4, including positive not quantifiable | 11 (12%) | 8 | 0 | 3 | 0 | ||||||
| 10−4 to 10−2 | 16 (17%) | 9 | 1 | 1 | 5 | ||||||
| ≥10−2 | 61 (66%) | 9 | 6 | 3 | 43 | <0.0001* | |||||
| Type of events | no event, in CR | 72 (59%) | 27 | 3 | 8 | 34 | |||||
| total number of events | 50 (41%) | 13 | 5 | 2 | 30 | N/A** | |||||
| early death | 3 (2.5%) | 1 | 0 | 0 | 2 | ||||||
| non-responder | 3 (2.5%) | 0 | 0 | 0 | 3 | ||||||
| relapse | 35 (29%) | 12 | 2 | 2 | 19 | ||||||
| 2nd malignancy | 2 (1.5%) | 0 | 0 | 0 | 2 | ||||||
| death in 1st CR | 7 (6%) | 0 | 3 | 0 | 4 | ||||||
| HSCT in HR treated cases | no | 43 (51%) | 13 | 1 | 4 | 25 | |||||
| (landmark 6 months) | in CR | 25 (67%) | 8 | 0 | 3 | 14 | |||||
| relapse | 18 (33%) | 5 | 1 | 1 | 11 | ||||||
| yes | 41 (49%) | 8 | 2 | 2 | 29 | N/A** | |||||
| in 2nd CR | 25 (61%) | 4 | 1 | 1 | 19 | ||||||
| total number of events after HSCT | 16 (39%) | 4 | 1 | 1 | 10 | ||||||
| relapse | 7 (17%) | 1 | 1 | 1 | 4 | ||||||
| 2nd malignancy | 2 (5%) | 0 | 0 | 0 | 2 | ||||||
| treatment-related mortality | 7 (17%) | 3 | 0 | 0 | 4 | ||||||
PPR, prednisone poor response, defined by ≥1,000 blasts per μl of peripheral blood at day 8 of a therapeutic window with prednisone; PGR, prednisone good response, defined by <1,000 blasts per μl of peripheral blood at day 8. Percentage of cases in each category out of the total number of cases with registered information is given in parentheses. One exception: HSCT variable: the total number of patients in 2nd CR and those suffering from any event was set to 100%. Three non-responders (all PDGFRB fusion positive cases) also received HSCT, one relapsed, one suffered from treatment-related mortality and one achieved a 2nd CR. These 3 patients were included in the HSCT outcome analysis for whom time from landmark at 6 months to relapse, to death and last contact was used, respectively.
Pearson χ2 p-values are estimates because the number of cases is less than 5 for some variables.
N/A, Pearson χ2 not applicable since time-related occurrence of events. Number of patients with missing or no data: Prednisone window response 65; treatment arm 1; MRD EOI 29; HSCT 7.
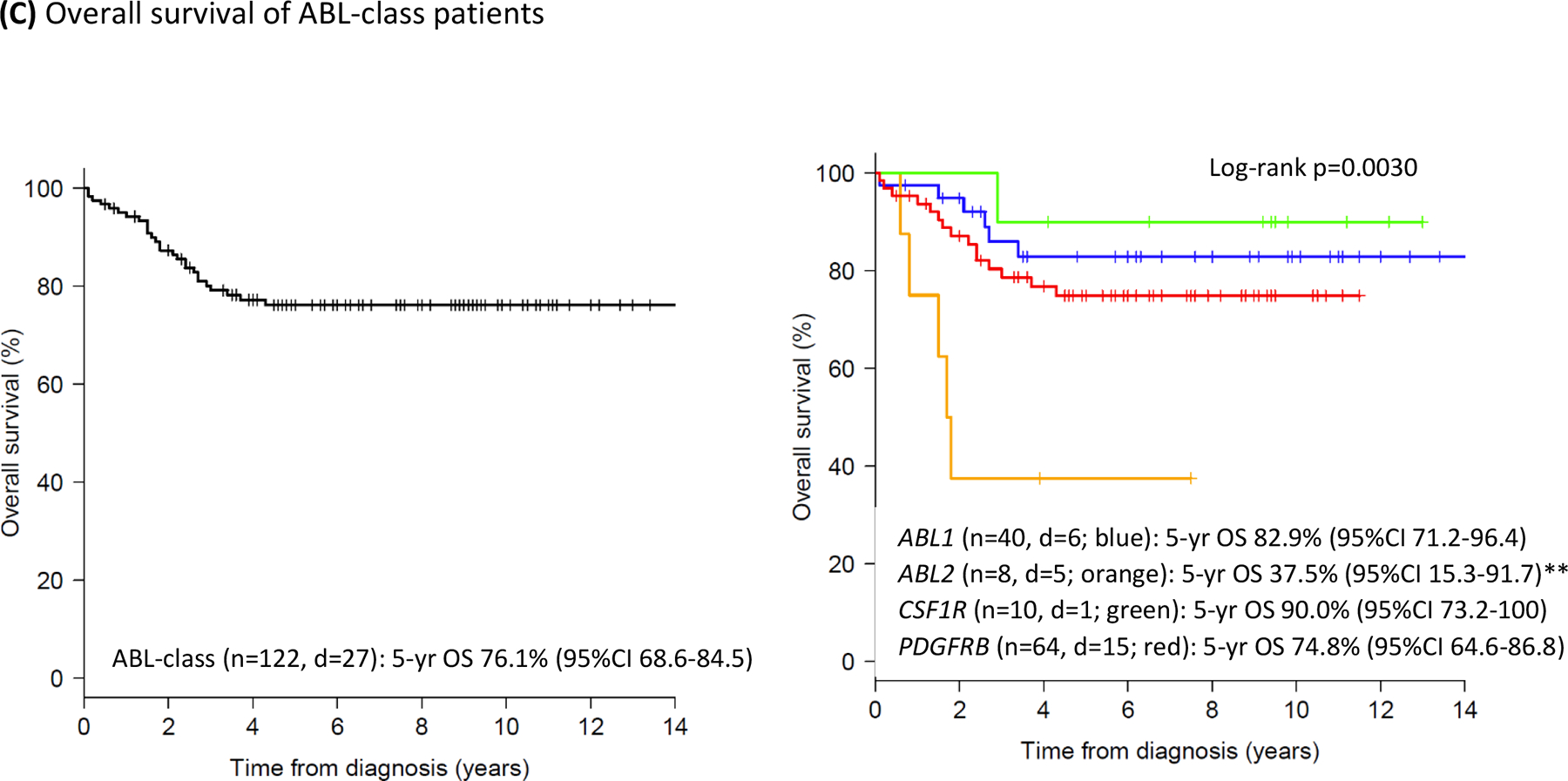
The 5-year EFS and OS of the total group of ABL-class patients was 59.1% (95%CI 50.5–69.1) and 76.1% (95%CI 68.6–84.5), respectively with median follow up of 6.7 years (interquartile range 3.9–9.4) for those without an event. Death in induction (early death) occurred in 3 (2.5%) and death in complete remission occurred in 7 (6%) patients. Two and a half percent (3 patients) were non-responders at the end of consolidation treatment (Table 2). These patients continued on protocol and reached CR; one remained in CR, one relapsed, and one patient died of HSCT-related toxicity. The 5-year cumulative incidence of relapse (CIR) of the total group was 31.0% (SD 4.6) (Figure 2A). In 25 (69%) of 36 cases relapse occurred within 3 years of diagnosis. Most relapses were found in the bone marrow (29 of 35; 83%; for 1 relapse location was not reported). Extramedullary relapse (mostly CNS) was seen either alone or combined with medullary relapse in 11 (31%) of the relapsed cases. Twenty of the 36 relapsed patients remained alive in second CR. Outcomes varied between ABL-class fusions. The 5-year EFS was 37.5% (95%CI 15.3–91.7) for ABL2, 52.9% (95%CI 41.5–67.5) for PDGFRB, 68.6% (95%CI 54.5–86.3) for ABL1 and 80.0% (95%CI 58.7–100) for CSF1R fusion cases (Figure 2B; p=0.059). The corresponding 5-year CIR was 25.0% (SD 16.9) for ABL2, 29.6% (SD 8.2) for ABL1, 34.3% (SD 6.5) for PDGFRB and 20.0% (SD 13.4) for CSF1R fusion cases (Figure 2A; p=0.82). The 5-year OS varied between 37.5% (95%CI 15.3–91.7) for ABL2 and ≥75% for the other fusion types (Figure 2C; p=0.0030). The highest Hazard Ratio was observed for ABL2-fusion cases compared to the remaining ABL-class cases (HROS 4.90, 95% CI 1.84–13.03, p=0.0015).
Figure 2: Outcome characteristics of children with ABL-class B-ALL in the pre-TKI era.
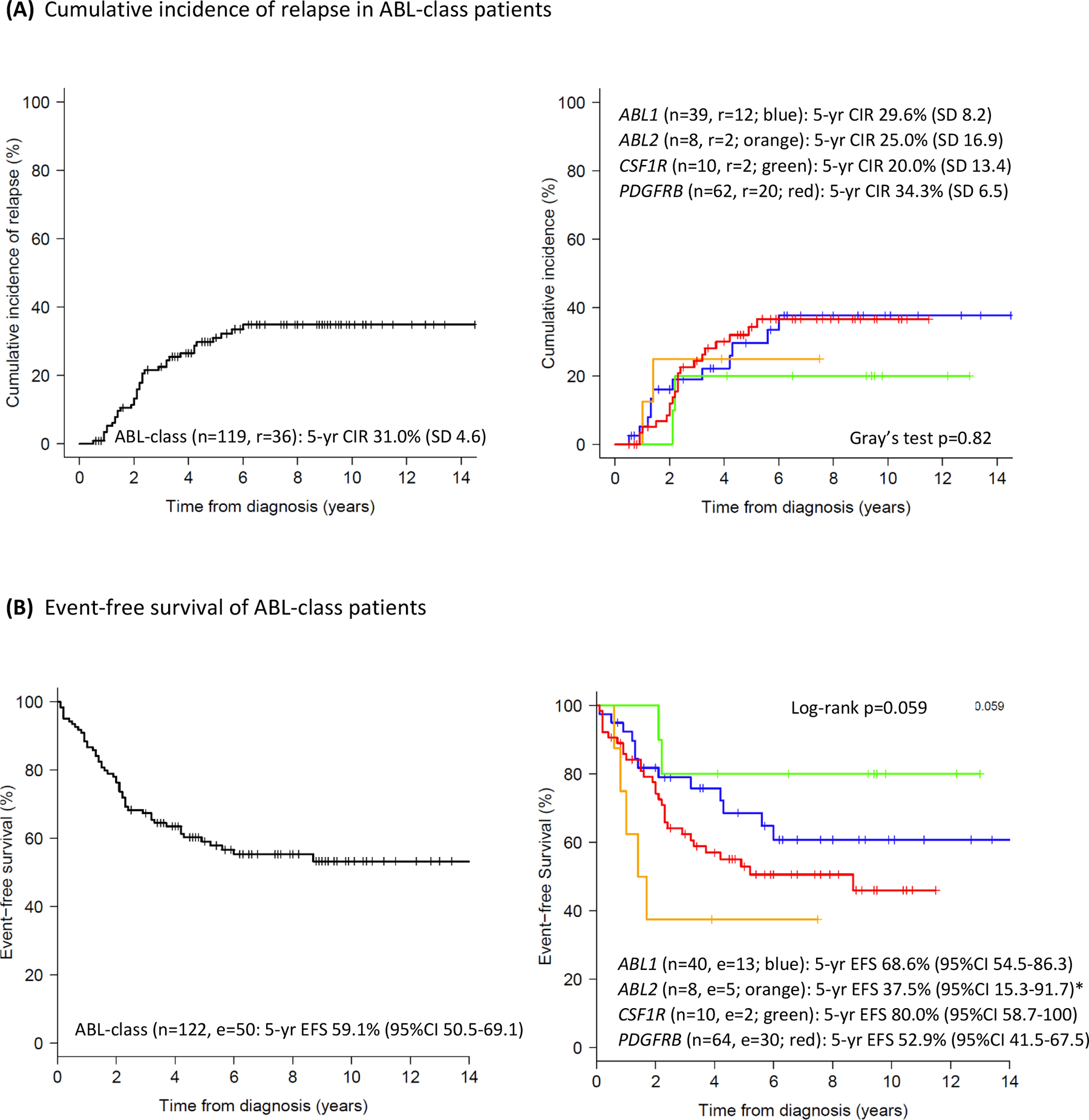
(A) CIR curve of 119 ABL-class patients, excluding 3 early death cases (left panel) and per ABL fusion type (right panel), Gray’s test p=0.82.
(B) EFS curve of 122 ABL-class patients (left panel) and per ABL fusion type (right panel), log-rank p=0.059. *ABL2-fusion versus remaining ABL-class: HR 2.40, 95%CI 0.95–6.10, Cox p-value 0.064.
(C) OS curve of 122 ABL-class patients (left panel) and per ABL fusion type (right panel), log-rank p=0.0030. **ABL2-fusion versus remaining ABL-class: HR 4.90, 95%CI 1.84–13.03, Cox p-value 0.0015.
Color code ABL fusions: ABL1, blue; ABL2, orange; CSF1R, green; PDGFRB, red; n=number of patients in the analysis, r=relapse, e=event, d=death.
MRD levels at the end of induction (EOI) were reported in 93 out of 122 cases. The EOI MRD levels were positive in 88 (95%) cases, ≥1×10−4 in 77 (83%), and ≥1×10−2 in 61 (66%) of the ABL-class cases, with notable variation between fusions (Table 2, Figure 3A). ABL1 fusion patients had the most favorable MRD EOI response, i.e. 20 (69%) of 29 patients were <1×10−2 although only 11 (38%) were <1×10−4. The EOI MRD levels of ≥1×10−2 were significantly associated with ABL2 (6 of 7 patients, 86%) and PDGFRB (43 of 49 patients, 88%) fusions (Kruskal Wallis, p<0.0001). Levels of MRD were not affected by the different 14 fusion partners of the ABL-class genes (appendix p 11). From the 41 cases with available MRD data both at EOI and at end of consolidation (EOC), 7 (17%) had negative or non-quantifiable MRD at EOI and 16 (39%) at EOC (appendix p 12; Paired test p<0.0001). Because of the refractory nature of ABL-class ALL, we used a threshold of 1×10−2 to compare outcome. ABL-class patients with MRD EOI ≥1×10−2 had an unfavorable 5-year EFS of 44.7% (95%CI 33.2–60.4) compared to 81.9% (95%CI 68.7–97.7) for those patients with MRD levels <1×10−2 (Figure 3B, p=0.0023) which, in multivariate Cox models, appeared independent from NCI risk group and treatment arm (appendix p 5). The 5-year CIR corresponding to MRD EOI levels <1×10−2 and ≥1×10−2 were 18.1% (SD 7.5) and 41.1% (SD 7.0) respectively (Gray’s test p=0.10; Figure 3C).
Figure 3: Minimal residual disease levels at the end of induction therapy in children with ABL-class B-ALL.
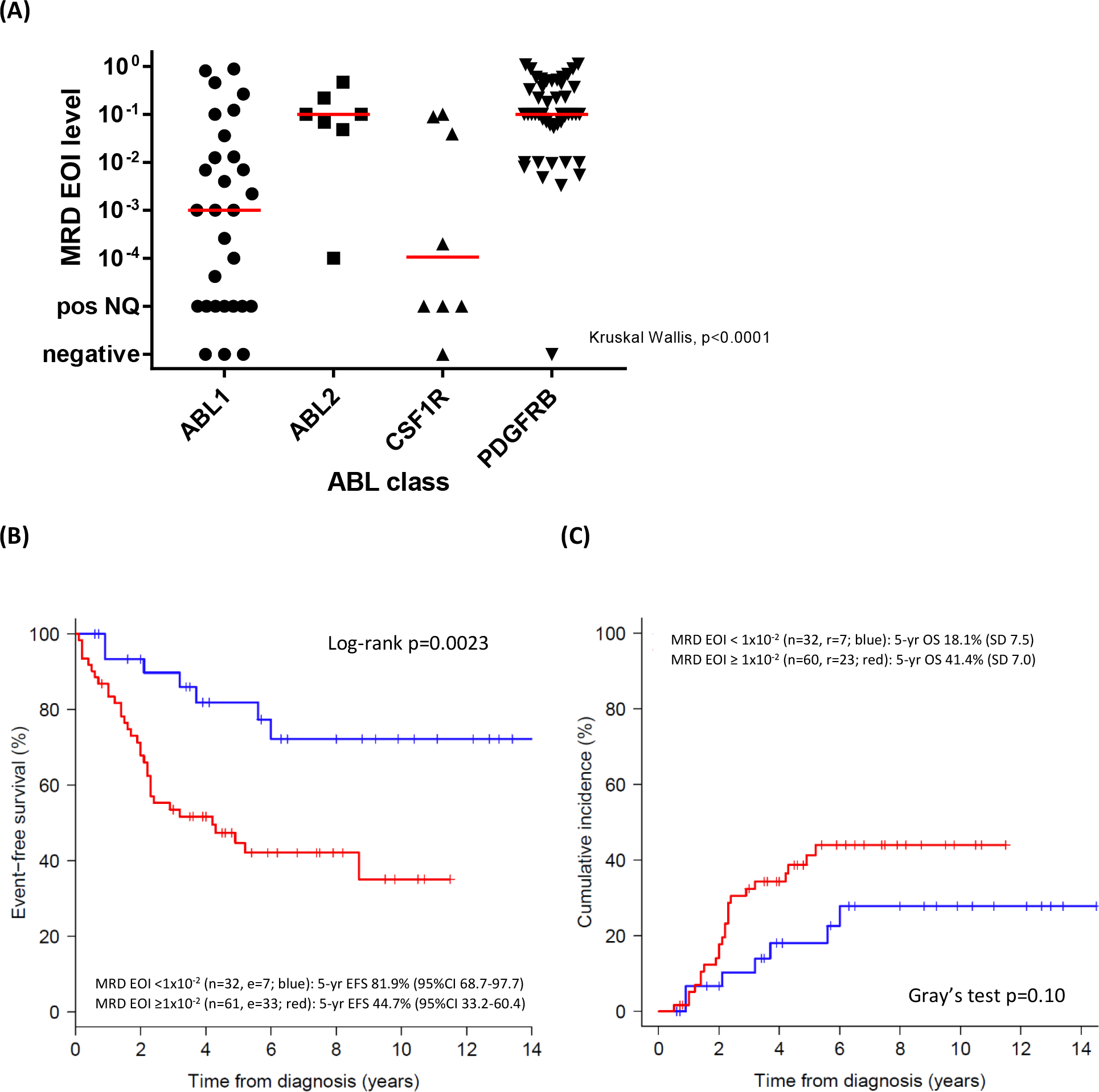
(A) Absolute MRD levels at the end of induction (EOI) per ABL-class type. The red line indicates the median value per ABL-class type. Kruskal Wallis p<0.0001. ABL1 and CSF1R MRD EOI levels are lower than those of ABL2 (p=0.044 and p=0.041, respectively) and PDGFRB cases (p<0.0001 and p=0.0010, respectively).
(B) Event-free survival of ABL-class cases according to MRD EOI levels. MRD EOI <1×10−2, n= 32, blue line, 5-year EFS 81.9% (95%CI 68.7–97.7) and MRD EOI ≥1×10−2, n=61, red line, 5-year EFS 44.7% (95%CI 33.2–60.4), log-rank P=0.0023. Cox proportional HR 3.34, 95% CI 1.47–7.60, p=0.0039.
(C) Cumulative incidence of relapse according to MRD EOI levels. MRD EOI levels <1×10−2, n=32, blue, 5-year CIR 18.1% (SD 7.5) and MRD EOI levels ≥1×10−2, n=60, red, 5-year CIR 41.1% (SD 7.0), Gray’s test p=0.10.
In total, 2 non-HR and 41 HR cases received a hematopoietic stem cell transplantation (HSCT) in first CR out of 115 cases for whom the HSCT status was reported. The median time to transplant was 6.7 months (interquartile range 5.8–7.9). The clinical outcome of 41 HR patients who received HSCT was compared to that of 43 HR patients without HSCT who survived at least 6 months from diagnosis without any event (landmark analysis). Sixteen events occurred after HSCT (7 relapses, 7 deaths in second CR and 2 second malignancies). The transplant-related mortality was high (7/41), and 6/7 of transplant-related deaths occurred in patients transplanted before 2010. In the group without HSCT, 18 events occurred, all relapses. The relapses and death in second CR (indicative of HSCT/treatment-related mortality) mainly occurred in the first 2 years following HSCT, after which the CIR and EFS curves stabilized. In contrast, relapses occurred up to 5-year after the landmark in the non-transplanted group (appendix p 13). The estimated CIR significantly differed between patients with HSCT (5-year 17.8%, SD 6.2) versus those without HSCT (5-year 45.1%, SD 8.4; P=0.013), but EFS and OS estimates did not significantly differ between the two groups (appendix p 13).
Discussion
This Ponte di Legno group study shows that the ABL-class subset of B-ALL, especially those with ABL2 and PDGFRB fusions, are characterized by very high risk features, a high frequency of poor prednisone response (PPR), high MRD EOI levels and an unfavorable long-term outcomes. The 5-year estimates in this pre-TKI era are 31% CIR, 59% EFS and 76% OS. These outcome data are highly inferior compared to that of other children with newly diagnosed B-ALL treated with contemporary treatment protocols, i.e. <8% CIR, >85% EFS and >90% OS.25,26 Our study also shows that the outcome varies between the four different types of ABL-class fusions with ABL2 and PDFGRB fusion cases having the most unfavorable baseline characteristics of older age, high WBC and high MRD EOI levels.
ABL-class patients are characterized by older age and high WBC at diagnosis and 75% are classified as NCI-HR compared to 30–35% of the general pediatric ALL population.26,27 The percentage of CNS involvement in ABL-class patients (3.4%) is low and comparable to the frequency seen in NCI HR cases of a large reference cohort (2.4%).28 In contrast, ABL-class patients more frequently have a PPR compared to reference cohorts of newly diagnosed ALL (49% versus <10%, respectively).25,26 Sixty-six percent of ABL-class patients have a very high and prognostically unfavorable MRD EOI level of ≥1×10−2 compared to less than 10% in reference cohorts.25,26 Only 39% had negative/non-quantifiable MRD at the end of consolidation therapy, which is much lower than in other subsets of B-ALL, e.g. 77% in the AIEOP-BFM ALL 2000 trial.26 Positive MRD at this late time-point is associated with a high risk of relapse and often used as an indication for HSCT. Considering the strong predictive role of MRD, a significant reduction in MRD levels in ABL-class patients during the first months of therapy is an important aim to reduce the relapse risk and to reduce the intensity of treatment (e.g. by avoiding HSCT) and its associated complications and mortality.
The frequency of IKZF1 deletions in ABL-class patients was high (61%) and comparable to the frequency in BCR-ABL1 positive ALL (75%), both being much higher than the 15% in BCR-ABL1 negative pediatric ALL.2,4,29 We found that an IKZF1 deletion with or without additional deletions in PAX5, CDKN2A or CDKN2B did not have additive prognostic value in contrast to the findings in non-ABL-class patients.23 Hence, treatment should not be modified on the basis of an additional IKZF1 deletion in ABL-class patients.
While it is possible that higher risk patients were selected to undergo HSCT in first CR, we found that overall survival for ABL-class cases treated without TKI therapy was very similar with chemotherapy alone or HSCT in first CR. The reduced number of relapses following HSCT was counterbalanced by the number of treatment-related deaths. Similar results were observed in a recent study analyzing both TKI and non-TKI treated ABL-class patients enrolled in recent AIEOP-BFM trials.13 In a landmark analysis (taking into account a 6 months waiting time to transplant), we noticed that relapses occurred over a longer timeframe of 5.5 years from diagnosis in the non-transplanted group, whereas the transplanted group suffered more often from early relapses (within 2.5 years of diagnosis). The occurrence of relapses early after HSCT is a known observation in ALL and indicates the failure of the conditioning regimen and the intended immune control by the engrafted immune cells. Early relapse rate in the transplanted group was not linked to a specific type of ABL-class fusion.
The data collected on ABL-class patients in this Ponte di Legno study was limited to the first-line treatment, up to the occurrence of the first event. The 122 patients were treated on >20 treatment protocols (1–22 patients each) between 2000 and 2018, which did not allow separate outcome analyses per protocol. MRD-guided risk stratification started to be used from 2000 onwards but was not implemented in all protocols in the same way. While for all but 29 patients MRD at the end of induction was available, only 41 patients also had MRD evaluated at the end of consolidation. Over the study period, incremental improvement in the overall survival of pediatric B-ALL was achieved, for example in the Netherlands from 86% (2000–04) to 91% (2005–09) and 93% (2010–15).30 Two-thirds of the ABL-class patients were diagnosed before 2010, suggesting that there was a decline of recruited patients possibly due to the increased first-line use of TKIs in recent years. The transplant-related mortality was high, and 6/7 of transplant-related deaths occurred in patients transplanted before 2010. Further analysis of possible reasons for HSCT failure was limited by the fact that HSCT details including conditioning, donor type, stem cell source and pre-HSCT MRD were not available. The current study was not designed to evaluate the effect of HSCT in ABL class patients, and the number of patients was too low to draw conclusions on the effectiveness of HSCT in CR1 as an effective salvage. Similarly, no data were collected on the use of TKIs or immunotherapies as second line treatment in our study. Given the time period of this study, it is very unlikely that immunotherapies were used in first line therapy of these protocols.
The signaling pathways activated by ABL-class fusions strongly suggest that ABL-class patients may benefit from the addition of TKIs to combination chemotherapy. Preclinical studies conducted in vitro (Ba/F3 and Arf−/− cell lines), ex vivo (patients’ cells) and in vivo (mouse models), all provide evidence that leukemic cells harboring ABL-class fusions are sensitive to different TKIs, including first (imatinib), second (e.g. dasatinib, bosutinib) and third generation (e.g. ponatinib) TKIs. TKIs were also efficacious in several case studies of children largely with refractory or relapsed ALL (appendix p 14 and 15, and references herein). Furthermore, the FRALLE group recently reported that all 8 MRD EOI positive ABL-class patients who received TKI achieved and remained in first complete remission for a prolonged time.18 Similar, the AIEOP-BFM group recently reported that TKIs applied at different stages of therapy resulted in only one relapse among 13 children with ABL-class leukemia.13 Together, these studies illustrate that TKIs can be beneficial to ABL-class patients. However, evidence for TKI efficacy in CSF1R fusion positive patients is lacking and limited to preclinical studies showing some sensitivity of CSF1R-fusion positive cells to TKIs.8,31
Targeting the ABL-class lesions by TKIs may be as effective as their use in children with BCR-ABL1 positive ALL.8,9,11,32 Most promising results have been obtained by giving TKIs continuously over a longer period in BCR-ABL1-positive ALL.9,11 Given the baseline 5-year EFS of 59% observed in the present study including the use of alloSCT in a significant proportion of patients, TKI addition to upfront therapies may increase the long-term outcome also for ABL-class cases.
In conclusion, this Ponte di Legno study shows that without TKI the outcome of children with ABL-class B-cell precursor ALL is highly unfavorable compared to non-ABL-class ALL patients. The availability of TKIs that have established to be safe and effective when combined with chemotherapy in BCR-ABL1 positive patients will fast track the use of TKI-containing combination therapies for ABL-class patients. This paper establishes the outcome standard to which these TKI-containing therapies will be compared.
Supplementary Material
Research in context.
Evidence before this study
In the last decade, it has become clear that ABL-class gene fusions other than BCR-ABL1 are detected in ~3% of children with acute lymphoblastic leukemia (ALL). Preclinical studies suggest that leukemic cells carrying ABL-class fusions can be targeted successfully by tyrosine kinase inhibitors (TKIs). The addition of TKIs to the therapy of BCR-ABL1-positive ALL has significantly improved the outcome but it is unknown whether this holds true for ALL with other ABL-class fusions. Moreover, the ABL-class fusion group is heterogeneous and includes patients with ABL1, ABL2, CSF1R and PDGFRB fusion types. The outcome for patients with these subtypes of ALL is not known because their occurrences are rare. A systematic search was not performed.
Added value of this study
This study was undertaken by the Ponte di Legno group. This group consists of >20 established ALL study groups worldwide and was initiated to address outcome questions in rare subsets of newly diagnosed pediatric ALL patients. We investigated the characteristics and outcome of children with ABL-class positive ALL treated on recent first-line trials without TKIs.
Implications of all the available evidence
The results described in this paper will serve as reference to interpret the potential benefit of adding TKIs in the front-line treatment of children with ABL-class positive ALL.
Acknowledgments
This study was financially supported by the Oncode institute (MLdB), Pediatric Cancer Foundation Rotterdam (MLdB, RP), Dutch Cancer Society (MLdB), the Kika foundation (MLDB, JMB), the Deutsche Krebshilfe (GC, MS), Blood Cancer UK (AVM) and AIRC grants (AIRC 2017 20564; CRUK/AIRC/FC AECC 22791 and AIRC 5 per mille 21147; AB); Cancer Australia APP1128727 (RS). This study was also supported by NCI grant R35 CA197695 (CGM), NIH grants U10 CA98543 and U10 CA180886 (COG Chair’s grants), U10 CA98413 and U10 CA180899 (COG Statistics and Data Center grants), U24 CA114766 and U24-CA196173 (COG Specimen Banking), St Baldrick’s Foundation funding. SPH is the Jeffrey E. Perelman Distinguished Chair in Pediatrics at The Children’s Hospital of Philadelphia. MLL is the Benioff Chair of Children’s Health and the Deborah and Arthur Ablin Endowed Chair for Pediatric Molecular Oncology at Benioff Children’s Hospital. Ponte di Legno working group and all affiliated study group members contributing to this study are acknowledged. Diagnostic and research laboratories linked to Ponte di Legno working group members are acknowledged for ABL-class testing of patients. In particular, Aurélie van Kleef-Boeree (DCOG), Udo zur Stadt (COALL), Gianni Cazzaniga (AIEOP), Andishe Attarbaschi (A-BFM), Marketa Zaliova (C-BFM), Sarah Elitzur (Israel-BFM), Claire Schwab and member laboratories of the UKCCG (UK-ALL) and Deborah White (ANZCHOG) are acknowledged for coordinating ABL-class testing in their study groups. All data centers and data managers associated with the Ponte di Legno group are acknowledged for providing highly accurate clinical data linked to these patients.
Funding
This study was financially supported by the Oncode institute (MLdB), Pediatric Cancer Foundation Rotterdam (MLdB, RP), Dutch Cancer Society (MLdB), the Kika foundation (MLDB, JMB), the Deutsche Krebshilfe (GC, MS), Blood Cancer UK (AVM) and AIRC grants (AIRC 2017 20564; CRUK/AIRC/FC AECC 22791 and AIRC 5 per mille 21147; AB); Cancer Australia APP1128727 (RS). This study was also supported by NCI grant R35 CA197695 (CGM), NIH grants U10 CA98543 and U10 CA180886 (COG Chair’s grants), U10 CA98413 and U10 CA180899 (COG Statistics and Data Center grants), U24 CA114766 and U24-CA196173 (COG Specimen Banking), St Baldrick’s Foundation funding.
Footnotes
Declaration of interest
MLdB, AVM, HAdGK, JMB, MF, GE, TI, AY, RS, LDP, NK, KGR, AV, AA, MZ, SE, GCazzaniga, AB, MLL and RP declare no competing interests. GCario reports personal fees from Jazz Pharmaceuticals and Novartis outside the submitted work. SPH reports personal fees from Novartis, Amgen, and other from Amgen, outside the submitted work. CGM reports personal fees from Illumina and grants from Loxo Oncology during the conduct of the study, and grants from Abbvie and Pfizer and personal fees from Amgen outside the submitted work. MS reports grants from SHIRE, JazzPharma, Servier, SigmaTau, Amgen, and Novartis during the conduct of the study, and personal fees from SHIRE, Servier, and JazzPharma outside the submitted work.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Contributor Information
Monique L den Boer, Dutch Childhood Oncology Group (DCOG), Princess Máxima Center for Pediatric Oncology, Utrecht, The Netherlands.
Gunnar Cario, AIEOP/BFM-Germany, University Hospital Schleswig-Holstein, Kiel, Germany.
Anthony V Moorman, UK-ALL study group, Leukaemia Research Cytogenetics Group, Wolfson Childhood Cancer Research Centre, Translational and Clinical Research Institute, Newcastle University, Newcastle upon Tyne, UK.
Hester A de Groot-Kruseman, Dutch Childhood Oncology Group (DCOG), Princess Máxima Center for Pediatric Oncology, Utrecht, The Netherlands.
Judith M Boer, Dutch Childhood Oncology Group (DCOG), Princess Máxima Center for Pediatric Oncology, Utrecht, The Netherlands.
Marta Fiocco, Dutch Childhood Oncology Group (DCOG), Princess Máxima Center for Pediatric Oncology, Utrecht, The Netherlands; Institute of Mathematics, Leiden University, Leiden, The Netherlands.
Gabriele Escherich, COALL study group, University Medical Center Hamburg Eppendorf, Germany.
Toshihiko Imamura, JACLS study group, Department of Pediatrics, Graduate School of Medical Science, Kyoto, Japan.
Allen Yeoh, Ma-Spore study group, Khoo Teck Puat - National University Children’s Medical Institute, Yong Loo Lin School of Medicine, National University of Singapore, Singapore, Singapore.
Rosemary Sutton, ANZCHOG study group, Children’s Cancer Institute, University of New South Wales and Cancer Centre for Children-Children’s Hospital at Westmead, Sydney, New South Wales, Australia.
Luciano Dalla-Pozza, ANZCHOG study group, Children’s Cancer Institute, University of New South Wales and Cancer Centre for Children-Children’s Hospital at Westmead, Sydney, New South Wales, Australia.
Nobutaka Kiyokawa, TCCSG study group, Department of Pediatric Hematology and Oncology Research, National Research Institute for Child and Development, Tokyo, Japan.
Martin Schrappe, AIEOP/BFM-Germany, University Hospital Schleswig-Holstein, Kiel, Germany.
Kathryn G Roberts, SJCRH study group; St Jude Children’s Research Hospital, Memphis, TN USA.
Charles G Mullighan, SJCRH study group; St Jude Children’s Research Hospital, Memphis, TN USA.
Stephen P Hunger, COG study group, Monrovia, CA, USA; Department of Pediatrics and the Center for Childhood Cancer Research, Children’s Hospital of Philadelphia and the Perelman School of Medicine at the University of Pennsylvania, PA, USA.
Ajay Vora, Department of Haematology, Great Ormond Street Hospital, London, UK.
Andishe Attarbaschi, AIEOP-BFM Austria, St. Anna Kinderspital, Vienna, Austria, USA.
Marketa Zaliova, AIEOP-BFM Czech Republic, University Hospital Motol, Prague, Czech Republic.
Sara Elitzur, AIEOP-BFM Israel, Schneider Children’s Medical Center of Israel, Petach Tikvah, Israel.
Giovanni Cazzaniga, AIEOP-BFM Italy, Universtà di Milano-Bicocca, S. Gerardo Hospital, Monza, Italy.
Andrea Biondi, AIEOP-BFM Italy, Universtà di Milano-Bicocca, S. Gerardo Hospital, Monza, Italy.
Mignon L Loh, COG study group, Monrovia, CA, USA; Department of Pediatrics, Benioff Children’s Hospital and the Helen Diller Family Comprehensive Cancer Center, University of California, San Francisco, San Francisco, CA, USA.
Rob Pieters, Dutch Childhood Oncology Group (DCOG), Princess Máxima Center for Pediatric Oncology, Utrecht, The Netherlands.
Data sharing
Requests to receive de-identified study data can be submitted to the corresponding author and should include a description of the research question.
References
- 1.Den Boer ML, van Slegtenhorst M, De Menezes RX, et al. A subtype of childhood acute lymphoblastic leukaemia with poor treatment outcome: a genome-wide classification study. Lancet Oncol 2009; 10(2): 125–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Mullighan CG, Su X, Zhang J, et al. Deletion of IKZF1 and prognosis in acute lymphoblastic leukemia. N Engl J Med 2009; 360(5): 470–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Roberts KG, Morin RD, Zhang J, et al. Genetic alterations activating kinase and cytokine receptor signaling in high-risk acute lymphoblastic leukemia. Cancer Cell 2012; 22(2): 153–66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.van der Veer A, Waanders E, Pieters R, et al. Independent prognostic value of BCR-ABL1-like signature and IKZF1 deletion, but not high CRLF2 expression, in children with B-cell precursor ALL. Blood 2013; 122(15): 2622–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Boer JM, Steeghs EM, Marchante JR, et al. Tyrosine kinase fusion genes in pediatric BCR-ABL1-like acute lymphoblastic leukemia. Oncotarget 2017; 8(3): 4618–28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Roberts KG, Li Y, Payne-Turner D, et al. Targetable kinase-activating lesions in Ph-like acute lymphoblastic leukemia. N Engl J Med 2014; 371(11): 1005–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Roberts KG, Yang YL, Payne-Turner D, et al. Oncogenic role and therapeutic targeting of ABL-class and JAK-STAT activating kinase alterations in Ph-like ALL. Blood Adv 2017; 1(20): 1657–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Schultz KR, Carroll A, Heerema NA, et al. Long-term follow-up of imatinib in pediatric Philadelphia chromosome-positive acute lymphoblastic leukemia: Children’s Oncology Group study AALL0031. Leukemia 2014; 28(7): 1467–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Slayton WB, Schultz KR, Kairalla JA, et al. Dasatinib Plus Intensive Chemotherapy in Children, Adolescents, and Young Adults With Philadelphia Chromosome-Positive Acute Lymphoblastic Leukemia: Results of Children’s Oncology Group Trial AALL0622. J Clin Oncol 2018; 36(22): 2306–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Biondi A, Schrappe M, De Lorenzo P, et al. Imatinib after induction for treatment of children and adolescents with Philadelphia-chromosome-positive acute lymphoblastic leukaemia (EsPhALL): a randomised, open-label, intergroup study. Lancet Oncol 2012; 13(9): 936–45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Biondi A, Gandemer V, De Lorenzo P, et al. Imatinib treatment of paediatric Philadelphia chromosome-positive acute lymphoblastic leukaemia (EsPhALL2010): a prospective, intergroup, open-label, single-arm clinical trial. Lancet Haematol 2018; 5(12): e641–e52. [DOI] [PubMed] [Google Scholar]
- 12.Shen S, Chen X, Cai J, et al. Effect of Dasatinib vs Imatinib in the Treatment of Pediatric Philadelphia Chromosome-Positive Acute Lymphoblastic Leukemia: A Randomized Clinical Trial. JAMA Oncol 2020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Cario G, Leoni V, Conter V, et al. Relapses and treatment-related events contributed equally to poor prognosis in children with ABL-class fusion positive B-cell acute lymphoblastic leukemia treated according to AIEOP-BFM protocols. Haematologica 2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Roberts KG, Reshmi SC, Harvey RC, et al. Genomic and outcome analyses of Ph-like ALL in NCI standard-risk patients: a report from the Children’s Oncology Group. Blood 2018; 132(8): 815–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Tran TH, Harris MH, Nguyen JV, et al. Prognostic impact of kinase-activating fusions and IKZF1 deletions in pediatric high-risk B-lineage acute lymphoblastic leukemia. Blood Adv 2018; 2(5): 529–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Lengline E, Beldjord K, Dombret H, Soulier J, Boissel N, Clappier E. Successful tyrosine kinase inhibitor therapy in a refractory B-cell precursor acute lymphoblastic leukemia with EBF1-PDGFRB fusion. Haematologica 2013; 98(11): e146–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Weston BW, Hayden MA, Roberts KG, et al. Tyrosine kinase inhibitor therapy induces remission in a patient with refractory EBF1-PDGFRB-positive acute lymphoblastic leukemia. J Clin Oncol 2013; 31(25): e413–6. [DOI] [PubMed] [Google Scholar]
- 18.Tanasi I, Ba I, Sirvent N, et al. Efficacy of tyrosine kinase inhibitors in Ph-like acute lymphoblastic leukemia harboring ABL-class rearrangements. Blood 2019; 134(16): 1351–5. [DOI] [PubMed] [Google Scholar]
- 19.Neale GA, Coustan-Smith E, Stow P, et al. Comparative analysis of flow cytometry and polymerase chain reaction for the detection of minimal residual disease in childhood acute lymphoblastic leukemia. Leukemia 2004; 18(5): 934–8. [DOI] [PubMed] [Google Scholar]
- 20.Schwab C, Ryan SL, Chilton L, et al. EBF1-PDGFRB fusion in pediatric B-cell precursor acute lymphoblastic leukemia (BCP-ALL): genetic profile and clinical implications. Blood 2016; 127(18): 2214–8. [DOI] [PubMed] [Google Scholar]
- 21.Reshmi SC, Harvey RC, Roberts KG, et al. Targetable kinase gene fusions in high-risk B-ALL: a study from the Children’s Oncology Group. Blood 2017; 129(25): 3352–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Imamura T, Kiyokawa N, Kato M, et al. Characterization of pediatric Philadelphia-negative B-cell precursor acute lymphoblastic leukemia with kinase fusions in Japan. Blood Cancer J 2016; 6: e419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Stanulla M, Dagdan E, Zaliova M, et al. IKZF1(plus) Defines a New Minimal Residual Disease-Dependent Very-Poor Prognostic Profile in Pediatric B-Cell Precursor Acute Lymphoblastic Leukemia. J Clin Oncol 2018; 36(12): 1240–9. [DOI] [PubMed] [Google Scholar]
- 24.Hamadeh L, Enshaei A, Schwab C, et al. Validation of the United Kingdom copy-number alteration classifier in 3239 children with B-cell precursor ALL. Blood Adv 2019; 3(2): 148–57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Pieters R, de Groot-Kruseman H, Van der Velden V, et al. Successful Therapy Reduction and Intensification for Childhood Acute Lymphoblastic Leukemia Based on Minimal Residual Disease Monitoring: Study ALL10 From the Dutch Childhood Oncology Group. J Clin Oncol 2016; 34(22): 2591–601. [DOI] [PubMed] [Google Scholar]
- 26.Conter V, Bartram CR, Valsecchi MG, et al. Molecular response to treatment redefines all prognostic factors in children and adolescents with B-cell precursor acute lymphoblastic leukemia: results in 3184 patients of the AIEOP-BFM ALL 2000 study. Blood 2010; 115(16): 3206–14. [DOI] [PubMed] [Google Scholar]
- 27.Hunger SP, Mullighan CG. Redefining ALL classification: toward detecting high-risk ALL and implementing precision medicine. Blood 2015; 125(26): 3977–87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Winick N, Devidas M, Chen S, et al. Impact of Initial CSF Findings on Outcome Among Patients With National Cancer Institute Standard- and High-Risk B-Cell Acute Lymphoblastic Leukemia: A Report From the Children’s Oncology Group. J Clin Oncol 2017; 35(22): 2527–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.van der Veer A, Zaliova M, Mottadelli F, et al. IKZF1 status as a prognostic feature in BCR-ABL1-positive childhood ALL. Blood 2014; 123(11): 1691–8. [DOI] [PubMed] [Google Scholar]
- 30.Reedijk AMJ, Coebergh JWW, de Groot-Kruseman HA, et al. Progress against childhood and adolescent acute lymphoblastic leukaemia in the Netherlands, 1990–2015. Leukemia 2020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Lilljebjorn H, Agerstam H, Orsmark-Pietras C, et al. RNA-seq identifies clinically relevant fusion genes in leukemia including a novel MEF2D/CSF1R fusion responsive to imatinib. Leukemia 2014; 28(4): 977–9. [DOI] [PubMed] [Google Scholar]
- 32.Biondi A, Cario G, De Lorenzo P, et al. Long-term follow up of pediatric Philadelphia positive acute lymphoblastic leukemia treated with the EsPhALL2004 study: high white blood cell count at diagnosis is the strongest prognostic factor. Haematologica 2019; 104(1): e13–e6. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Requests to receive de-identified study data can be submitted to the corresponding author and should include a description of the research question.