

Abstract
Objectives
Neurodevelopmental disorders (NDDs) are a group of conditions that are clinically and etiologically heterogeneous. Biallelic variants in ACBD6 were previously reported in 7 patients with NDDs. Unfortunately, their clinical information remains very limited with descriptions of only their neurologic and craniofacial features. The purpose of this report is to expand the clinical phenotype of the ACBD6-associated NDDs.
Methods
We identified 2 Thai siblings with NDDs. Clinical and radiologic features of the proband were described. The affected siblings and parents underwent whole-exome sequencing and PCR-Sanger sequencing.
Results
Clinical manifestations that have never been previously reported include morbid obesity, pancytopenia with severe infections, diabetes mellitus, cirrhosis, and renal failure, leading to deaths in their early 30s. Molecular studies identified a novel homozygous 1 base-pair duplication (c.360dup; p.Leu121Thrfs*27) in the ACBD6 gene.
Discussion
This study reported 1 novel single base-pair duplication, expanding the mutational spectrum, and described the clinical features establishing the entity of ACBD6-associated NDDs.
Neurodevelopmental disorders (NDDs) are a diverse group of conditions that include intellectual disability, global developmental delay, autism spectrum disorder, and attention-deficit/hyperactivity disorder.1 Next-generation sequencing approaches have resulted in the identification of an increasing number of NDD-associated genes.2 Previous studies reported ACBD6 as a gene associated with NDD,3,4 but no causal relationship was definitely established.
The acyl-coenzyme A binding domain-containing protein 6 (ACBD6), encoded by the ACBD6 gene (OMIM *616352), is a modular protein with an acyl-coenzyme A binding domain at its N terminus and 2 ankyrin motifs at its C terminus. ACBD6 binds long-chain acyl CoAs with a strong preference for unsaturated, C18:1-CoA and C20:4-CoA, and oversaturated, C16:0-CoA, acyl species.5
Variants in the ACBD6 gene have been previously reported in 7 patients with NDDs without the details of their phenotype.3,4,6 Here, we report 2 Thai siblings who harbor a novel homozygous pathogenic variant c.360dup (p.Leu121Thrfs*27) in ACBD6. Their clinical and radiologic features are described.
Methods
Patients
Two siblings affected with NDDs and their healthy parents were studied (Figure 1C).
Figure 1. Facial Features of the Siblings With ACBD6-Associated Syndrome.

(A and B) Patient II-1 and II-2 were obese with epicanthal folds, narrow palpebral fissures, flat nasal bridge, hypoplastic ala nasi, and no ear lobes. (C) The pedigree shows 2 affected siblings and their parents. The horizontal bars above individual symbols represent persons who were examined.
Standard Protocol Approvals, Registrations, and Patient Consents
All family members have consented to genomic studies. This study was approved by the Institutional Review Board of the Faculty of Medicine, Chulalongkorn University.
Genetic Findings
The genomic DNA of the 2 affected and their parents (Figure 1) was prepared from peripheral leukocytes for Illumina sequencing library. Subsequently, it was enriched for the desired target using the Illumina Exome Enrichment protocol. The captured libraries were sequenced using the Illumina HiSeq 2000 Sequencer. The reads were mapped against University of California Santa Cruz genome browser hg19 using Burrows-Wheeler Aligner software. The single nucleotide variants and Indels were detected by Sequence Alignment/Map tools. The Variant call format files were analyzed using BaseSpace Variant Interpreter software (Illumina).
To confirm the presence of the identified variant, we performed PCR and Sanger sequencing of all family members using a forward primer 5′-CCTGGCCAGCAATTATTTTA-3′ and a reverse primer 5′-GGGGAGGACGATTAGGAAAA-3′. The PCR products were sent for direct sequencing in both directions at Macrogen, Inc. (Seoul, Korea). The sequence was analyzed using Sequencher (version 4.2; Gene Codes Corporation, Ann Arbor, MI).
Data Availability
All datasets generated during the current study are not publicly available but are available from the corresponding author on reasonable request.
Results
Clinical Features
Patient 1, the proband, was a 25-year-old Thai woman with NDDs (Figure 1A). She was born at term to nonconsanguineous parents. She had delayed development since infancy, and at 25 years of age, she was minimally verbal, walked slowly, and followed simple instructions with a Global Assessment of Functioning score of 41–50 (on a scale of 0–100). She had been overweight since childhood. Her weight was 101 kg, height 156 cm, and BMI 41.5 kg/m2. She had pancytopenia with hemoglobin of 8.6 g/dL (12.0–15.0 g/dL), white cell counts 2.9 × 103/μL (4.5–11.0 × 103/μL), and platelet counts 59 × 103/μL (150–450 × 103/μL), which contributed to recurrent infections requiring repeated hospitalization and IV antibiotics. Specific examples included a soft-tissue infection of the perineum at 19 years of age, an abscess in the right breast at 20 years of age, and pneumonia with an accompanying pleural effusion at 26 years of age (eFigure 1, A and B, links.lww.com/NXG/A567). At 21 years of age, she was found to have diabetes mellitus type 2 with a fasting blood glucose level of 233 mg/dL (70–99 mg/dL) and HbA1C of 10.6% (4.0%–6.0%). She subsequently developed diabetic retinopathy, dyslipidemia, and hypertension with her blood pressure of 160/96 mm Hg. At 26 years of age, liver cirrhosis with splenomegaly was found (eFigure 1, E and F, links.lww.com/NXG/A567). At 29 years of age, the patient developed paraplegia and was found to have a T5-T8 epidural abscess (Figure 3, C and D). CT of the thoracic spine demonstrated intravertebral disc herniation and spondylosis of the lower thoracic spine (eFigures 1C and D, links.lww.com/NXG/A567), whereas CT of the brain revealed diffusely mild brain atrophy and cranial vault thickening (Figure 3, A and B). At 31 years of age, she developed stage 5 chronic renal disease with blood urea nitrogen of 180 mg/dL (7–20 mg/dL), suffered acute respiratory failure, and died.
Figure 3. Radiologic Findings of the Patient II-1.
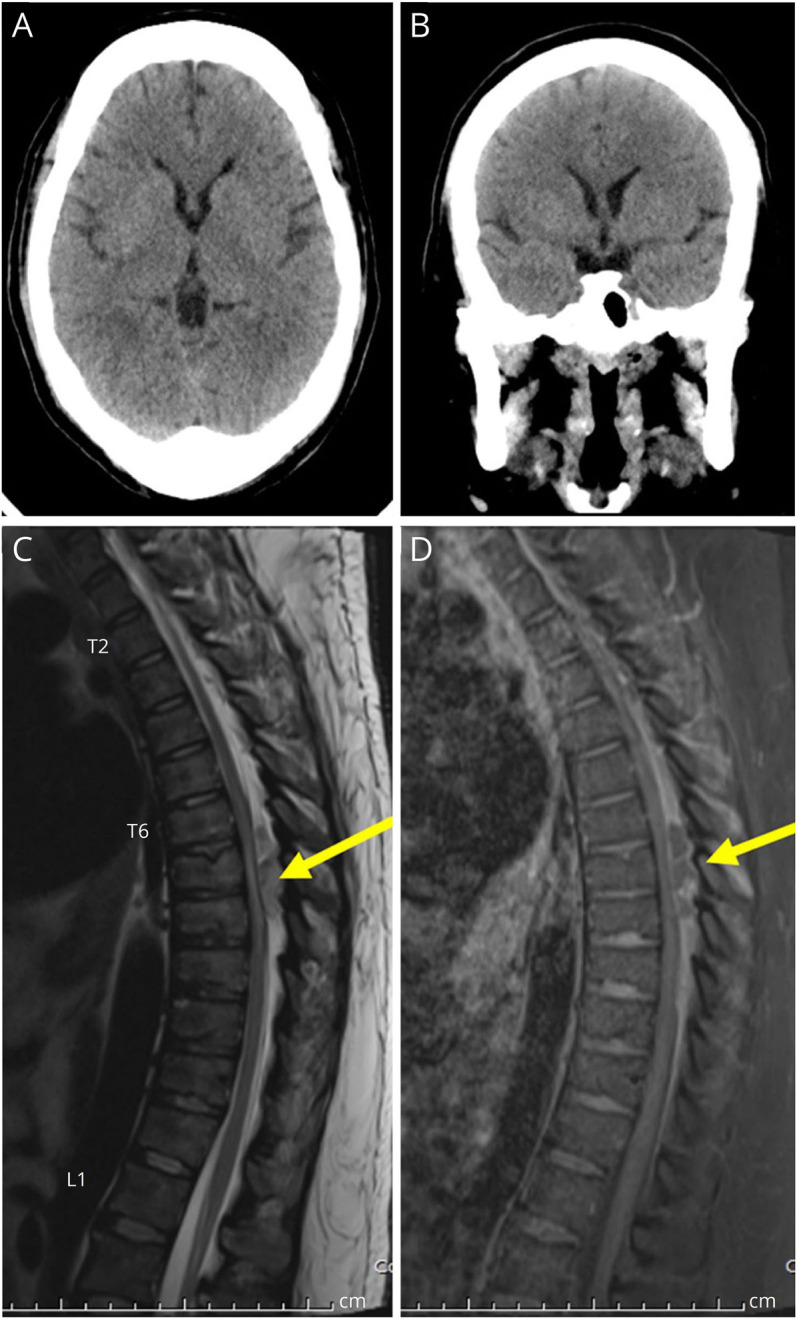
(A and B) CT scan of the brain shows diffuse brain atrophy, disproportionate with the patient's age. There is the diffuse and thickened cranial vault. (C and D) MRI of the thoracic spine: sagittal T2-weighted image and sagittal gadolinium-enhanced T1-weighted image with fat suppression show an oval shape, 0.8 × 3.8 cm (anteroposterior × vertical diameter) in size, peripheral enhancing hyperintense on T2-weighted lesion in the posterior epidural space of T6-8 levels, suggesting an epidural abscess. This posterior epidural lesion causes severe narrowing of the thecal sac with spinal cord compression. There is hyperintense spinal cord signal intensity on T2-weighted images involving T7-T8 levels, suggesting compressive myelopathy.
Patient 2 was a younger brother of Patient 1 (Figure 1B). According to his mother, he had similar clinical course to patient 1 including severely delayed development and morbid obesity. However, his mother did not have him engaged with medical care. Thus, no detailed diagnostic data are available. At 30 years of age, he developed a poor appetite with accompanying constipation, aspiration, restlessness, and seizure before dying at home.
Genetic Findings
Whole-exome sequencing (WES) of the 4 family members yielded 12.6–14.7 Gb per exome, with the mean depths of target regions of 46.6–54.5X per exome. With the assumption of an autosomal recessive model, only 1 candidate pathogenic variant was identified. It was a homozygous 1 base-pair duplication, c.360dup (p.Leu121Thrfs*27), in ACBD6 (NM_032360.4) found in both patients (Figure 2A). Their father and mother were heterozygous for this variant. Sanger sequencing confirmed the findings (Figure 2B). The variant has never been previously reported and subsequently submitted to the ClinVar database (ncbi.nlm.nih.gov/clinvar/) (VCV001177532).
Figure 2. Molecular Findings of the Siblings With ACBD6-Associated Syndrome.

(A and B) Findings of 1 base-pair deletion by whole-exome sequencing and Sanger sequencing. BAM files and electropherograms of the father, the mother, 2 siblings, and a control in panels 1–5, respectively. The 2 patients were homozygous, whereas the father and the mother were heterozygous for the c.360dup (p.Leu121Thrfs*27) in ACBD6.
Discussion
Although there have been 7 patients from 4 families reported in the literature with ACBD6 mutations, their clinical information is very limited (Table 1). In this manuscript, we report 2 siblings with a homozygous ACBD6 mutation. Their clinical features and courses leading to death in their early 30s were described.
Table 1.
Reported Clinical Features and Genetic Variants of the Patients With Mutations in ACBD6 in the Literature

WES revealed that the 2 siblings were homozygous for a novel c.360dup located in the acyl-CoA binding domain of ACBD6. This variant is predicted to result in a frameshift leading to the premature stop codon (p.Leu121Thrfs*27). The human ACBD6 (282 amino acids) consists of the acyl-CoA binding domain (amino acids 1–129), the linker (amino acids 130–178), and the 2 ankyrin-repeat motifs (amino acids 177–282).6 This frameshift precludes the generation of all functional domains. Nonetheless, it is likely to lead to nonsense mRNA mediated decay. Consequently, loss-of-function mutations in ACBD6 lead to the N-myristoylation deficiency.6 The variant is absent in publicly available databases including National Heart, Lung, and Blood Institute7, gnomAD8, ClinVar9, and Thai reference exome (T-REx) variant database.
From the previously reported cases of ACBD6 mutations,3,4,6 only neurologic symptoms and craniofacial features were described. Notably, our report describes clinical features of patients with ACBD6 mutations in more details and up to their deaths in their 30s. Particularly, the patient (II-1) has a diagnosis of diabetes, cirrhosis, renal failure, and hypertension. Her brother was reportedly to have similar symptoms. Earlobes are present bilaterally in the mother, whereas absent in the father. In addition, bilateral epicanthal folds are present in both parents. These 2 features present in the 2 affected siblings therefore may be the phenotypic expression inherited from their parents.
The wide range of affected organs and tissues are consistent with the fact that ACBD6 is expressed in several tissues, including the brain, kidney, liver, thymus, thyroid, adrenal gland, lung, small intestine, stomach, spleen, heart, skeletal muscle, salivary gland, and trachea (source: RNA sequencing of total RNA from 20 human tissues ncbi.nlm.nih.gov/gene/84320?report=expression&bioproject=PRJNA280600).
In conclusion, our report delineates the detailed clinical features and suggests a natural course of affected individuals with biallelic loss-of-function variants in ACBD6.
Acknowledgment
The authors thank the patients and their family for participating in this study.
Appendix. Authors
Study Funding
This study was supported by the Health Systems Research Institute (65-040).
Disclosure
The authors report no disclosures relevant to the manuscript. Full disclosure form information provided by the authors is available with the full text of this article at Neurology.org/NG.
References
- 1.Hanly C, Shah H, Au PYB, Murias K. Description of neurodevelopmental phenotypes associated with 10 genetic neurodevelopmental disorders: a scoping review. Clin Genet. 2021;99(3):335-346. [DOI] [PubMed] [Google Scholar]
- 2.Vickers RR, Gibson JS. A review of the genomic Analysis of children presenting with developmental delay/intellectual disability and associated dysmorphic features. Cureus. 2019;11(1):e3873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Kochinke K, Zweier C, Nijhof B, et al. Systematic phenomics Analysis deconvolutes genes mutated in intellectual disability into biologically coherent modules. Am J Hum Genet. 2016;98(1):149-164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Najmabadi H, Hu H, Garshasbi M, et al. Deep sequencing reveals 50 novel genes for recessive cognitive disorders. Nature. 2011;478(7367):57-63. [DOI] [PubMed] [Google Scholar]
- 5.Soupene E, Serikov V, Kuypers FA. Characterization of an acyl-coenzyme A binding protein predominantly expressed in human primitive progenitor cells. J Lipid Res. 2008;49(5):1103-1112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Soupene E, Schatz UA, Rudnik-Schoneborn S, Kuypers FA. Correction: requirement of the acyl-CoA carrier ACBD6 in myristoylation of proteins: activation by ligand binding and protein interaction. PLoS One. 2022;17(4):e0267678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.NHLBI Exome Sequencing Project (ESP)Exome Variant Server. Accessed November 22, 2022. evs.gs.washington.edu.
- 8.gnomAD: Genome Aggregation Database. Accessed November 22, 2022. gnomad.broadinstitute.org
- 9.ClinVar. Accessed November 22, 2022. ncbi.nlm.nih.gov/clinvar/
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
All datasets generated during the current study are not publicly available but are available from the corresponding author on reasonable request.