

Abstract
Concerns over the potential use of variola virus as a biological weapon have prompted new interest in the development of small molecule therapeutics to prevent and treat smallpox infection. Since smallpox is no longer endemic, human clinical trials designed to link antiviral efficacy to clinical outcome have been supplanted by antiviral efficacy evaluations in animal models of orthopoxvirus disease. This poses a unique challenge for drug development; how can animal efficacy data with a surrogate virus be used to establish clinical correlates predictive of human disease outcome? This review will examine the properties of selected animal models that are being used to evaluate poxvirus antivrial drug candidates, and discuss how data from these models can be used to link drug efficacy to clinical correlates of human disease.
Keywords: animal model, antiviral, biodefense, inflammatory disease, orthopoxvirus, pathogenesis, regulatory, smallpox, systemic spread
The ‘animal efficacy’ rule (US FDA regulation 21 Code of Federal Regulations [CFR] 314 Subpart I) is designed to evaluate new drug products when human efficacy studies are not ethical or feasible. This rule was developed to facilitate the development of drugs for the prevention and treatment of diseases caused by pathogens considered to be potential bioweapon threats. Many of these pathogens are no longer endemic or exist in isolated areas where outbreaks affect small numbers of people, thereby reducing the feasibility of conducting large-scale human trials to measure drug efficacy. Thus, regulatory approval of new drug products must rely not only on safety data in humans, but also on animal efficacy evaluations that provide data predictive of human disease outcome.
Satisfying the animal efficacy rule will be challenging, especially in the area of smallpox antiviral drug development, since variola virus is no longer endemic in the human population, access to variola virus is highly restricted, and animal models of orthopoxvirus disease require the use of surrogate viruses to cause infection and disease. Moreover, current molecular markers and techniques used to describe human disease had not been developed prior to smallpox eradication. Thus, linking animal model data to human disease poses many challenges. This review will provide an overview of poxvirus pathogenesis in humans and other animal hosts. In addition, a description of several animal models currently in use to evaluate antipoxvirus drugs will be discussed. The goal of this review is to define disease parameters in these model systems that can be used to link antiviral efficacy to clinical outcome in humans.
Poxvirus replication cycle
Variola virus is the etiological agent of smallpox and belongs to the family Orthopoxviridae that includes vaccinia (smallpox vaccine strain), monkeypox, cowpox and camelpox viruses [1]. Orthopoxviruses are large double-stranded DNA viruses that replicate exclusively in the cytoplasm of infected cells. There are four types of infectious virus particles produced during productive infection; intracellular mature virus (IMV), intracellular enveloped virus (IEV), cell-associated enveloped virus (CEV) and extracellular enveloped virus (EEV). The intracellular and extracellular forms of virus are thought to play unique roles in orthopoxvirus pathogenesis [1,2].
IMV particles assemble from crescent-shaped membranes in virus factory areas of the cytoplasm. Particle formation requires a series of temporally regulated proteolytic cleavage events of viral core proteins that result in condensation of the viral core [3]. The core particles are enveloped by intracellular membranes to form IMV particles. Once formed, these particles remain inside the cell and are released upon cell lysis. IMV particles are stable to the environment and are thought to play a role in host transmission [2].
Approximately 10% of the total infectious particles produced during infection are wrapped in virus-modified membranes derived from post-trans Golgi or endosomal membrane systems to form IEV particles [4]. Once formed, IEV particles travel to the cell surface in a microtubule-dependent fashion where the outer membrane of the IEV containing vesicle fuses with the plasma membrane to release CEV that remain associated with the cell surface. Approximately 1% of CEV particles are released into the extracellular space as EEV particles by a process involving motile actin tail formation [5,6].
The extracellular virus particles, EEV and CEV, are responsible for efficient cell-to-cell spread and long-range dissemination of the virus in the host [4,7,8]. Virus variants containing defects in genes required for the production of extracellular virus particles produce small plaques in vitro and are attenuated for virus spread in vivo[9–11]. Virus strains that produce higher proportions of extracellular virus particles as part of the infection cycle are more virulent in mice [7]. Passive immunization with antibodies directed against IMV particles are less protective than antibodies directed against whole virus, suggesting that neutralizing antibodies to EEV antigens play a significant role in disease prevention [12]. Finally, inhibitors of extracellular virus formation or more specifically, production of EEV particles, protect animals from lethal orthopoxvirus infection [13–15]. These observations suggest that extracellular virus particles play an important role in virus spread and orthopoxvirus disease progression.
At least seven virus-specific gene products are required for synthesis of extracellular virus particles. These gene products participate in the wrapping of IMV particles (B5R and F13L), transport of particles to the cell surface (F12L), actin tail formation (A33R, A34R and A36R) and release of particles from the cell (A33R, A34R and B5R). EEV particles have a higher specific infectivity compared with IMV particles and are more resistant to complement-mediated neutralization [16,17]. Complement resistance is the result of incorporation of host complement control proteins, CD46, CD55 and CD59, into the outer membrane of the virus [17]. These properties of extracellular virus particles facilitate long-range spread of the virus in the host.
Natural history of human orthopoxvirus infections
The natural history of orthopoxvirus infections can be divided into two stages, localized replication at the site of infection and systemic spread. Many factors influence the course of disease progression, including the amount of virus entering the host, route of entry and host response to infection. Although variola virus is no longer endemic, the natural history of variola infection and clinical manifestations of smallpox has been well documented [18].
Orthopoxvirus infection of humans produces a spectrum of diseases that range from severe disseminated lesional disease characteristic of the most common type of variola virus infection to localized lesional infection caused by vaccinia virus. Four major types of clinical disease are associated with variola virus infection and are defined by the morphology of the virus-specific lesion and severity of disease symptoms. Ordinary smallpox is characterized by raised pustular skin lesions that can be confluent or discrete. Variola sine eruptione is characterized by fever without rash and requires serological analysis to confirm diagnosis. Flat-type smallpox is characterized by confluent flat pustules, and hemorrhagic-type smallpox is characterized by widespread hemorrhages in the skin and mucous membranes. Both flat- and hemorrhagic-type smallpox are usually fatal with 97% mortality in diagnosed cases [17,19].
Three other species of orthopoxviruses have been found to infect humans and cause disease. Monkeypox virus, which is likely a disease of rodents, can cause a generalized infection in humans resembling a milder version of smallpox. Monkeypox virus is poorly transmissible from person to person, and outbreaks result in small numbers of people contracting disease. Vaccinia virus is a laboratory vaccine strain with no known natural reservoir that causes localized infection and generates protective immunity against variola virus. Cowpox virus, which is thought to be carried by rodents, can be transmitted to humans by contact with infected animals. Cowpox virus infection produces a self-limiting, localized infection similar to vaccinia virus infection [18]. Disease severity in all cases is influenced by host immune status. Individuals suffering from certain skin disorders or who are immunocompromised suffer more severe infection [20].
Other species of orthopoxviruses that are genetically related to variola virus such as camelpox and ectromelia viruses have not been found to infect humans. The genetic basis for susceptibility of poxviruses is not well understood, but is thought to be related to acquisition and adaptive evolution of host-response modifier genes [21–23]. These genes are often found to be virulence factors that downregulate the host immune response and thereby facilitate systemic virus spread. Phylogenetic analysis of poxvirus genomes has identified a number of gene families undergoing positive selection, many of which are candidate host-response modifier genes. These genes tend to localize towards the ends of the viral genome [21,23,24]. Gene families which are evolutionarily more stable encode proteins required for basic virus replication functions and are located in the central region of the genome [23]. Thus, inhibitors that target conserved replication functions developed against one species of orthopoxvirus often work equally well to inhibit replication of multiple orthopoxvirus species in cell culture [14,25].
A pair-wise genomic comparison of variola virus with selected orthopoxviruses demonstrated a number of genes that differ from variola virus at the nucleotide level. The ends of the viral genome appear to have the greatest level of divergence (Figure 1). While the function of most of these genes is unknown, several genes can be classified as potential host-response modifiers. An example of genes found in ectromelia virus that are not found in variola virus is shown in Table 1. Thus, even within Orthopoxviridae, there are species-specific differences in host-response modifier genes. While orthopoxviruses exhibit a broad host range in cell culture, the susceptibility in animals (including humans) is likely to be related to the acquisition and adaptive evolution of host-response modifier genes that provide a selective advantage for virus replication in a particular animal host.
Figure 1. Gene synteny between variola virus and selected species of orthopoxviruses.
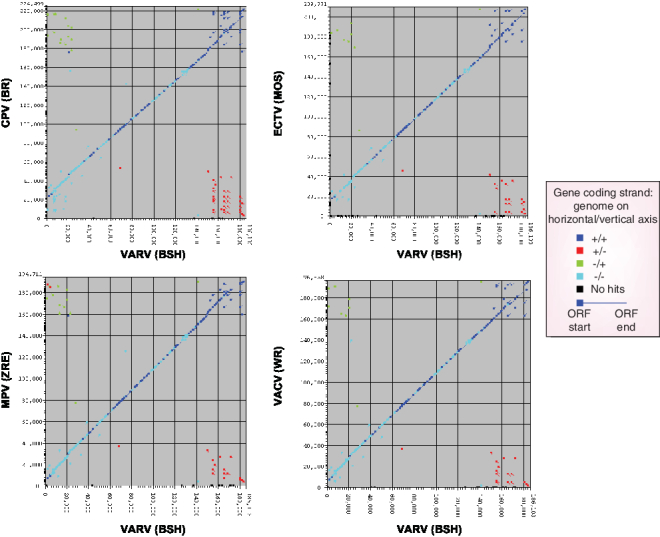
A pairwise nucleotide comparison of VARV (BSH) with CPV (BR); ECTV (MOS); MPV (ZRE); VACV (WR). Paralogs with similarity >90% fall on the diagonal axis while paralogs with lower homology are plotted away from the diagonal axis. Genes that are not found in variola virus are plotted on the y-axis and genes that found in variola virus but not in the test genome are plotted on the x-axis. The E-value cutoff was set at 0.01 and the percentage identify cutoff was set at 0% to show the maximum degree of similarity between genomes. The majority of genes that are plotted away from the diagonal shift to the x- and y-axes when the E-value cutoff is set to 1×10-25 and percent identify cutoff is set to 40%. The direction of the ORF is shown. In general, the similarity between genes on the diagonal falls from ˜99% in the middle of the genome to ˜90–95% at the termini. The sequence data and tools for this analysis can be found at [102].
CPV (BR); Cowpox virus strain Red; ECTV (MOS); Ectromelia virus strain Moscow; MPV (ZRE); Monkeypox virus strain Zaire 96-|-16; ORF; Open reading frame; VACV (WR); Vaccinia virus strain Western Reserve; VARV (BSH); Variola virus strain Bangladesh; +: Forward reading frame; −: Backward reading frame; +/−: Forward gene in query aligns with a backward gne in target.
Table 1. Ectromelia virus genes not found in variola virus.
| Open reading frame | Start* | Finish* | Function | Ref. |
|---|---|---|---|---|
| EVM004 | 10,494 | 9673 | Unknown | |
| EVM006 | 12,824 | 12,597 | Unknown | |
| EVM007 | 13,077 | 12,766 | Unknown | |
| EVM009 | 14,351 | 14,016 | CD30, cytokine inhibition | [56] |
| EVM010 | 16,733 | 14,442 | Unknown | |
| EVM014 | 22,782 | 22,603 | Unknown | |
| EVM024 | 37,765 | 36,932 | Unknown | |
| EVM047 | 60,995 | 61,492 | Unknown | |
| EVM117.5 | 133,000 | 132,839 | Phospholipase D-like protein | [57] |
| EVM137 | 152,897 | 153,427 | Virulence factor | [58] |
| EVM153 | 173,249 | 174,760 | Unknown | |
| EVM157 | 178,451 | 178,996 | Tumor necrosis factor binding protein C-terminal | [21] |
| EVM159 | 180,883 | 181,146 | Unknown | |
| EVM163 | 183,858 | 184,844 | Interleukin-1β binding protein | [21] |
*Start and finish refer to the starting nucleotide position and ending nucleotide position in the open reading frame, respectively. EVM: Ectromelia virus strain Moscow (AF012825).
Host response to systemic infection mediates disease progression
The host response to variola infection and the ability of the virus to counteract the host response, determines the severity of smallpox by limiting virus replication and spread during the early stages of infection and contributing to the immunopathology associated with late-stage infection [18,26–28]. From studies with ectromelia virus, immune cells encounter invading virus in the oral mucosa and express growth factors, chemokines, cytokines and interferon which trigger host immune-effector cells to migrate to the site of infection [29]. The virus counteracts this by producing a variety of host-response modifier proteins that downregulate host inflammatory signals [21,30]. Studies conducted with ectromelia and vaccinia virus have shown that viral mutants lacking functional interleukin-18 binding protein are attenuated for replication in mice [31,32]. Vaccinia virus mutants that lack functional soluble tumor necrosis factor receptor encoded by the cytokine response modifier E (CrmE) gene or chemokine-binding protein are less virulent in mice [33,34]. Moreover, patients who are immunocompromised fail to mount an efficient host response to infection and exhibit higher mortality rates and more severe disease [18]. Likewise, people with certain types of immune-related skin ailments suffer higher rates of complications resulting from vaccination with the vaccinia virus vaccine [20]. Thus, the ability of the virus to spread in the host and cause systemic disease is regulated by the robustness of the initial host response to viral infection and the effectiveness of viral immune evasion strategies.
While the host response to infection is important for restricting virus spread, it may also contribute to late-stage disease [35]. In animals, orthopoxvirus infection causes the release of cytokines, chemokines and other mediators of inflammation into the bloodstream that result in vascular dysfunction, coagulopathy and multiorgan failure [29,34,36,37]. Moreover, in humans, the spectrum of disease from fatal hemorrhagic type to self-limiting lesional disease can be associated with the degree of inflammatory response to systemic viral infection. Autopsy results from smallpox patients show dilation of the capillaries and endothelial swelling in the capillary walls, leukocyte mobilization and disseminated intravascular coagulation, consistent with pathology resulting from an inflammatory response to infection [18,26–28].
In light of these observations, smallpox can be thought of as an acute inflammatory disease where the host response to systemic infection stimulates immune cells to release proinflammatory cytokines causing severe disease and death. It is tempting to speculate that virus-mediated inhibition of the host response may exacerbate this condition and cause a build up of activated immune cells which must elicit a stronger cytokine response to overcome virus-specific immune evasion strategies.
Variola infection of humans
The life-cycle of the variola virus resembles that of other orthopoxviruses in which infection of the natural human host results in virus replication at the periphery followed by systemic spread. The spread of variola in humans has been inferred from animal studies, especially those conducted in mice infected with ectromelia virus [18]. The variola virus is thought to enter the respiratory tract via aerosolized droplets, seeding mucous membranes and passing rapidly into local lymph nodes. Based on animal studies, the virus replicates in the local lymph tissue to produce a primary viremia. The virus then travels to the spleen, liver and reticulo–endothelial system where replication in these organs produces a secondary viremia. If this mechanism of intense viral multiplication occurs in humans, it is remarkable in that it is accompanied by a clinical latency in which physical symptoms of infection are absent. In animal models of orthopoxvirus disease, virus replication stimulates the production of proinflammatory cytokines that can be detected in the blood [19,35]. While techniques for cytokine profiling were not available during the time when smallpox was endemic, hematological analysis of patients suffering from normal forms of smallpox found that thrombocytopenia was relatively common with more severe hematological abnormalities found in hemorrhagic forms of disease. Thrombocytopenia associated with variola virus infection was attributed to disseminated intravascular coagulation, a clinical condition that could be related to excessive inflammatory cytokine secretion [27,28]. While induction of cytokines in response to variola virus infection likely occurs in humans, physical symptoms of an inflammatory response are not evident early in the infection process [21,30].
Clinical latency ends with the rapid onset of severe headache, backache and fever, termed the prodromal phase. The prodromal phase correlates with a secondary viremia in which infectious virus can be detected in the mucous membranes of the mouth and pharynx. The virus invades the capillary epithelium of the dermal layer in skin, perivascular cells and epidermis where replication results in necrosis and the formation of a rash. The spleen, lymph nodes, liver, bone marrow, kidneys and other viscera may contain large quantities of virus based upon data from animal studies. Replication in the epidermis may be enhanced by secretion of virus-specific growth factors that bind to cellular receptors on keratinocytes and stimulate growth [38]. Histological studies of skin lesions from smallpox patients have documented the changes in skin leading to lesion formation [18]. The earliest changes detected were a dilation of the capillaries in the papillary layer of the dermis followed by swelling of the endothelial cells in the wall of blood vessels [18,26].
Infection results in 30% mortality with the cause of death attributed to toxemia, associated with immune complexes and hypotension. Toxemia is a poorly defined clinical condition thought to be caused by an excessive inflammatory immune response similar to septicemia associated with systemic bacterial infections. Examination of dermal layers of blood vessels from autopsy patients infected with variola virus show extensive leakage of the endothelial layer consistent with the presence of high levels of proinflammatory cytokines [18,26].
Treatment of smallpox
The most effective therapy for the treatment and prevention of smallpox is vaccination with vaccinia virus [18]. While vaccinia virus is closely related to variola virus, it is much less virulent causing localized infection in normal healthy adults rather than systemic disease. Localized infection generates a robust immune response that results in protective immunity. The immunogenicity of vaccinia virus forms the basis for the current smallpox vaccine which has been used to eradicate endemic smallpox [19]. The route of inoculation also contributes to the reduced virulence of vaccinia virus. Virus entering through the skin as opposed to the oral mucosa produces a localized infection that is cleared by the immune system. While intentional dermal infection or intranasal inoculation with variola virus (variolization) also produces a localized infection of the skin, systemic spread occurs more frequently with virus-induced lesions forming away from the site of inoculation, and fatal smallpox occurring in approximately 1% of patients. Variolization had been used for many years prior to the development of the vaccinia virus-based vaccine [18].
Replication of vaccinia virus is similar to variola virus except that infection remains localized and virus spread is uncommon in normal immunocompetent people. Vaccinia virus establishes productive infection in basilar epithelium following inoculation with multiple stabs from a bifurcated needle to establish a local cellular reaction. By 6–8 days post inoculation, a grayish–white pustule develops that is 1–2 cm in diameter, usually having central umbilication. Central crusting begins and spreads peripherally over a period of 3–5 days. Local edema and a hard crust remain until the third week post inoculation [18]. The host response to infection leads to the development of protective immunity against variola virus.
Smallpox antiviral drug candidates
Currently, there are no FDA-approved drugs to treat smallpox. A number of compounds that are in early preclinical development have been tested for antiorthopoxvirus activity [25]. In general, two classes of compounds have been found to be active in treating orthopoxvirus infection; those that directly inhibit virus replication and those that target systemic spread. Cidofovir (Vistide®), a broad-spectrum DNA polymerase inhibitor, is an example of the first compound class. Cidofovir which is currently licensed for the treatment of cytomegalovirus retinitis in AIDS patients is also active against orthopoxvirus replication in vitro and in vivo[25,39]. The compound targets the viral DNA polymerase to directly inhibit virus replication [40]. Orally bioavailable prodrugs have been developed to overcome the need for intravenous administration [41]. The second class of compounds is represented by those that target systemic spread. Compounds such as Gleevec, CI-1033 and ST-246 inhibit systemic spread without directly inhibiting virus replication [13–15]. Gleevec and CI-1033 inhibit host Abl-family tyrosine kinases and Erb-1 family of kinases, respectively to prevent formation of EEV particles [13,15]. ST-246 targets the poxvirus F13L protein, which is required for the formation of CEV and EEV particles [14]. While these compounds fail to inhibit IMV particle production in vitro, they have been shown to protect mice from lethal orthopoxvirus infection and/or reduce disease. Thus, inhibitors of poxvirus replication that block replication and/or systemic spread in animal models of orthopoxvirus disease are likely to be effective in reducing disease severity in humans.
Animal models of orthopoxvirus disease
The specter of bioterrorism has led to renewed interest in developing therapies to treat and prevent smallpox. Animal models of orthopoxvirus disease that are predictive for human disease outcome are an important component of the drug and vaccine development process. A number of animal models of orthopoxvirus disease have been developed to evaluate antipoxvirus compounds [42]. While these models are useful for evaluating antiviral activity of compounds in animals, each model by itself fails to capture all aspects of human disease and, therefore can not be predictive of clinical outcome. Thus, multiple models of orthopoxvirus infection will be required to evaluate antiviral efficacy of poxvirus inhibitor compounds.
The most relevant animal models of orthopoxvirus infection involve the use of host-adapted virus and require replication at the periphery followed by systemic virus spread in a manner similar to variola virus infection of humans. These models are appealing because virus replication in target tissue and systemic spread is determined by the host response to infection. Some animal models of orthopoxvirus infection have been developed using nonhost-adapted orthopoxviruses to establish disease. These models require inoculation of animals with large quantities of virus sometimes delivered by unnatural routes. Delivering large amounts of virus by unnatural routes alters pathogenesis by allowing virus access to different tissues and stimulating a non-natural host response. The term ‘host adapted’ is used in a broad sense and is defined as genetic alterations that result in increased virulence for a specific host. These changes may include evolutionary adaptations such as acquisition of host-response modifier functions such as those found in ectromelia virus for example, or unknown changes that result in increased virulence, such as those found in rabbitpox virus, which is genetically very similar to vaccinia virus but more pathogenic in rabbits [43]. To better understand the pathogenesis of orthopoxvirus infection in animals and humans, the natural history of poxvirus infection in several host species is described.
Intranasal vaccinia virus/cowpox virus infection of mice
Vaccinia virus mouse models have been developed to measure the efficacy of antipoxvirus compounds [39,44]. Similar models have also been developed using cowpoxvirus [36,39]. The pathogenesis of infection is dictated by the route of entry and models using intracranial, intravenous or intranasal inoculation have been described [44–46]. Intranasal inoculation of mice with vaccinia or cowpox virus produces local replication and systemic disease providing a system capable of assessing the antiviral activity of compounds that inhibit multiple steps in the virus life-cycle. To establish infection, mice are inoculated with 104–106 plaque-forming units (PFU) of vaccinia virus or cowpox virus in a small volume (20 µl) to each nare. Although establishment of lethal infection requires a large inoculating dose of virus, the infection starts locally in the nasal tissue and lungs before spreading systemically [39,44]. The virus replicates in the reticulo–endothelial system and can be found in the liver, spleen, lung and kidney [39]. By day 4 post inoculation, mice begin losing weight and become lethargic. Mice continue to lose weight and their general appearance declines with most animals exhibiting signs of severe disease such as ruffled fur, hunched posture and unsteady gate. By day 8 post inoculation, mice are moribund and have lost as much as 30% of their initial body weight.
Mortality is the primary end-point in this model with 100% of mice succumbing to infection by day 10 post inoculation. Disease progression can be monitored by measuring the change in weight during the course of infection. The change in weight correlates with systemic replication of virus in mice and is a noninvasive method of determining disease severity. The percentage weight change is useful for determining disease severity when treatment protects mice from lethal infection. Thus, the efficacy of compounds that prevent mortality but do not completely inhibit virus replication and all aspects of disease progression can be assessed in this model. To quantify the level of virus spread, animals must be sacrificed at selected time points post infection and virus titers measured in the liver, spleen, lung, kidney and other organs. Antiviral efficacy is measured by the decreased mortality, inhibition of virus-induced weight loss and reduction in viral titers in the liver, spleen, lung, kidney and other tissues.
Ectromelia virus infection of mice
Ectromelia virus is a laboratory pathogen of mice that has been used as a model system to characterize disease pathogenesis of orthopoxvirus infections [18,29,47,48]. The pathogenesis of ectromelia virus disease closely resembles human smallpox, with distinct stages of localized replication, systemic virus spread and lesion formation, however, the time course of infection and disease progression is much shorter (Figure 2)[47].
Figure 2. A comparison of the pathogenesis of orthopovirus infection.
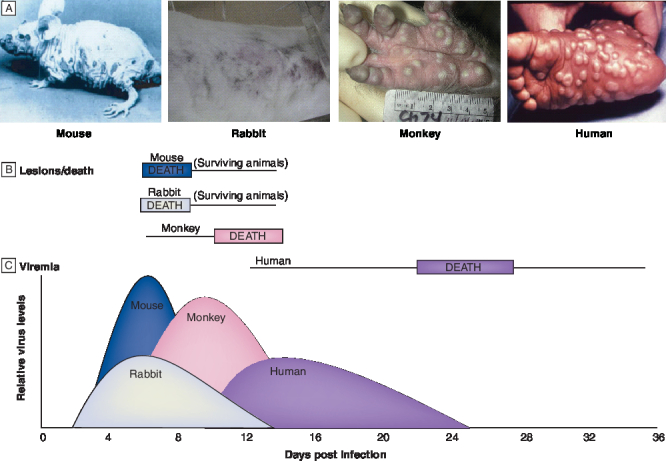
(A) Lesion formation caused by footpad inoculation of a naturally hairless mouse with 10 PFU of ectromella virus (ECTV) at day 6 post infection (courtesy of Zentralinstitut for Versuchstiere. Federal Republic of Germany) [47], intranasal inoculation of a rabbit with 150 PFU of rabbitpox virus (RPV) at day 6 post infection (reprinted with permission from Chaed Roy, US Army Research Institute for Infectious Disease [USAMRIID]), intravenous inoculation of a rhesus macaque with 106 PFU of monkeypox virus (MPV) at day 10 post infection (reprinted with permission from Jay Hooper, USAMRIID) [53], and variola virus (VARV) infection of a human at day 22 post infection [101]. (B) Diagram showing time of death (rectangles) and duration of lesion formation (lines) for orthopovirus infections of a mouse, rabbit, monkey or human. (C) Diagram showing the time of appearance of viremia detected in spleen (rabbit, mouse) or in throat swabs (monkeys, humans) and quantified by plaque assay (RPV, ECTV, MPV) in cell culture of chorioallantoic membranes (VARV). The height of the peaks represents relative levels of viral titers and was modified from previously published data [18,47,52,53].
Ectromelia virus enters through abrasions in the skin and replicates in local lymphoid cells [29]. Virus can be detected in these tissues by immunofluorescence within a few hours after inoculation [49–51]. The virus multiplies in the lymphatic endothelial cells, macrophages and lymphocytes within the node over a period of 2–4 days [29]. Following this latency period, the virus spreads through the lymph and enters the bloodstream to cause a primary viremia. The virus is rapidly removed by macrophages lining the sinusoids of the liver, spleen and bone marrow [49]. Infection of the parenchymal cells of liver and lymphoid cells of the spleen produces high virus titers that are released into the bloodstream to cause a secondary viremia [50,51]. In highly susceptible animals, replication in the liver and spleen produces focal necrotic lesions, acute hepatitis and multiorgan failure. In mice that are less susceptible to infection, a rash develops following the secondary viremia [47]. The rash is caused by virus replication in the perivascular cells, dermal endothelial cells and epidermis (Figure 2).
A lethal ectromelia virus mouse model has been established to evaluate the efficacy of antiviral drugs [41,48]. In susceptible mice as little as 1 PFU of ectromelia virus causes lethal infection [41]. Mice are inoculated with ectromelia virus either by footpad scarification, which is similar to the natural route of infection, or intranasal delivery [47]. The virus multiplies in the lymphatic endothelial cells, macrophages and lymphocytes within the regional node over a period of 2–4 days. By day 4 post inoculation, animals appear ill with hunched posture, ruffled coat and increased respiration. Viral replication in the liver and spleen and other internal organs cause death in the infected animal between days 6 and 10 post inoculation [47].
The rate of disease progression is similar to the intranasal vaccinia virus model with mortality occurring in 100% of mice by day 10 post inoculation [41]. Disease progression can be monitored by measuring the change in weight during the course of infection. To quantify the level of virus spread, animals are sacrificed at selected time points post infection, and virus titers measured in the liver, spleen, lung, kidney and other organs [14,41]. Detecting virus in these tissues is an excellent measure of virus spread since viral inoculums contain 50–100 PFU, and the virus is likely to be detectable only after significant levels of replication and spread. Like the intranasal vaccinia virus model, antiviral efficacy is measured by decreased mortality, inhibition of virus-induced weight loss and reduction in viral titers in the liver, spleen and other tissues.
Rabbitpox virus infection of rabbits
Rabbitpox virus is highly adapted to replicate in rabbits and as little as 15 PFU can establish productive infection in most rabbit strains [52]. Rabbits are inoculated with rabbitpox virus in the footpad by intradermal injection or the nasal cavity by aerosol spray. Virus replicates in the mucosa or local lymph tissue to produce a primary viremia which lasts 2–4 days [52]. The virus spreads through the lymph to the blood, ultimately seeding the liver, spleen and other internal organs. Virus replication at these sites often results in multiorgan failure and fatal disease [18]. Rabbits that survive infection of internal organs develop a secondary viremia where high levels of circulating virus seed the endothelial cells that line the dermal blood vessels to produce a rash on the skin that appears by day 6 post inoculation (Figure 2). Vertical transmission of the virus to susceptible rabbits has been observed allowing for the potential assessment of drug effects on virus spread within a population [52].
To establish infection, rabbits are inoculated with 100 PFU of rabbitpox virus by intranasal administration through aerosol delivery to the respiratory tract. By day 6 post inoculation animals develop fever, listlessness and purulent discharges from the eyes and nose [52]. A rash develops between day 6 and 8 post inoculation; however, skin lesions range from a few scattered lesions to confluency. Most animals die without developing a rash and death is accompanied by a fall in body temperature to below normal levels [18,52]. Thus, quantifying lesion number or severity of the rash is a subjective measure of systemic virus spread and may not be possible in all cases. Since rabbits can tolerate more frequent and larger volume blood draws, blood chemistries can be measured to correlate changes in hematological status with disease progression. To quantify the level of virus spread, animals are sacrificed at selected time points post infection and virus titers measured in the liver, spleen, lung, kidney and other organs. Antiviral efficacy is assessed by measuring decreased mortality, reduced virus titers in organs and improvement of hematological status. Histological examination of tissue from sacrificed animals further defines the effects of antiviral compounds on viral pathogenesis.
Monkeypox virus infection of nonhuman primates
Primate models of orthopoxvirus disease have been developed to evaluate efficacy of new smallpox vaccines [53]. These models are also being considered for the evaluation of antiorthopoxvirus drugs [54]. Administration of 107 PFU of monkeypox virus to nonhuman primates delivered by intravenous or intratracheal injection produces a lethal infection that reproduces the lesional disease characteristic of smallpox and monkeypox in humans (Figure 2). Typically, animals die between days 10 and 14 post infection with over 750 poxvirus lesions [53].
Monkeypox virus infection of nonhuman primates causes fever, decreased oxygen saturation, elevated heart rate and other changes in vital signs that can be measured by telemetry (J Huggins, pers. comm.). Moreover, changes in blood chemistries can be monitored over time since repeated blood draws do not compromise animal health. At selected time points post infection, skin lesions can be quantified in anesthetized animals and disease severity scores calculated based upon lesion number. In addition, virus yields can be measured in serum and whole blood samples and from throat swabs taken from anesthetized animals to quantify the level of viremia [53]. Quantifying lesion numbers and virus yields provide noninvasive methods for evaluating systemic virus spread. Viral titers and histopathology can be measured in selected organs and tissues from sacrificed animals at different time points post infection to determine the degree of virus spread and extent of disease. Thus, there are multiple secondary end-points in this model system that can be used to assess antiviral efficacy. One of the major drawbacks of delivering large amounts of infectious virus directly to the bloodstream by intravenous administration is that this route bypasses the mucosa and produces a lesional disease that is characteristic of post secondary viremia.
Variola virus infection of nonhuman primates
Variola virus can infect nonhuman primates to produce a disease resembling human smallpox; however, lethal infection requires administration of large quantities of virus delivered by intravenous injection [55]. A recent study has shown that intravenous delivery of 109 PFU of the Harper strain of variola virus produced a significant lesional disease, with more than 250 lesions in one animal, and 100% mortality in all three animals between day 4 and 10 post inoculation. Intravenous infection with 108 PFU produced significantly lower mortality rates (33%) [55]. Thus, high-dose virus delivered by intravenous injection is required to establish disease. Intravenous injection establishes instant viremia bypassing the normal requirement for replication at the periphery and systemic spread. Since the natural history of disease progression is dramatically altered, it is unclear how antiviral efficacy would correlate with human disease outcome in this model.
In summary, animal models of orthopoxvirus infection that use host-adapted viruses to establish disease are more likely to be predictive of human disease outcome than infection models that require administration of large quantities of virus delivered by unnatural routes. While small animal models (mice and rabbits) are useful for assessing antiviral efficacy, mortality in these systems is due to multiorgan failure caused by virus replication in vital organs and does not appear to be related to an excessive inflammatory immune response to systemic infection as in human smallpox. In addition, animals die prior to the formation of lesions that are characteristic of smallpox disease eliminating the possibility of using this noninvasive measure of clinical disease severity. A comparison of the pathogenesis of orthopoxvirus infection of mice, rabbits, nonhuman primates and humans is presented in Figure 2.
Linking animal efficacy to human disease
The molecular pathogenesis of human smallpox is poorly defined since modern techniques such as cytokine profiling and quantitative virology were unavailable prior to eradication. Thus, surrogate markers predictive of human disease outcome have not been well established. From historical accounts, the clinical course of smallpox resembles that of ectromelia virus infection of mice, or rabbitpox virus infection of rabbits, where acute systemic infection results from inoculation with a small amount of virus at the periphery [18]. While it is recognized that no single model is likely to be predictive of human disease outcome, especially now that smallpox is no longer endemic and disease markers cannot be qualified, certain disease models that focus on infection with host-adapted viruses will be more informative in assessing antiviral activity. These models allow evaluation of antiviral activity in a coevolved system (like human smallpox) and measurement of many aspects of viral pathogenesis. Disease models that require high-dose virus administered by unnatural routes may artificially alter pathogenesis by inducing different arms of the immune system or by introducing virus to tissues and cell types not normally associated with viral infection.
Animal models where infection is established with a low dose of virus and where pathogenesis is defined based upon the appearance of disease markers such as skin lesions or inflammatory cytokines, and not strictly on the levels of viral replication in the animal would be most desirable for linking antiviral efficacy to human disease.
Disease markers that define orthopoxvirus infections
Skin pocks
The appearance of pox lesions are the hallmark of smallpox. These lesions are easily quantifiable and require local replication and systemic viral spread for their formation. The appearance of pox lesions in humans is well characterized [18]. Smallpox lesions are predictive of clinical outcome and disease grading scales based upon lesion number, have provided clinicians with a noninvasive method for assessing disease severity.
Unfortunately, lesion formation is not the primary disease symptom in the most common models of orthopoxvirus pathogenesis. In the ectromelia virus mouse model, susceptible mice die without developing lesional disease. Moreover, in less susceptible mouse strains, lesions are not well-defined pox, but appear as a generalized rash, which is difficult to quantify [18,47]. This situation also occurs in the rabbitpox model of orthopoxvirus disease where most rabbits succumb to infection before developing quantifiable skin lesions [18,52]. In animals that develop rash, the onset of skin lesions occur after the peak of viremia, suggesting that animals must survive replication of the virus in the internal organs in order for the virus to seed endothelial cells in the capillaries of the skin. Mouse models have been developed that emphasize lesion formation [45]. Tail vein injection of vaccinia virus into normal mice results in the appearance of quantifiable, pox-like lesions that resolve over the course of several weeks [14,45]. While this model is useful for measuring virus replication and pox-like lesion formation in an animal, the route of viral entry, which bypasses normal routes of systemic spread, and ensuing nonlethal disease, make lesion formation in this model less predictive of human disease outcome. Lesion formation in the monkeypox model suffers from similar drawbacks. Lesion formation requires administration of large quantities of virus delivered by intravenous injection [53]. In addition, both the mouse tail vein model and the monkeypox nonhuman primate model use nonhost-adapted virus to establish disease.
Proinflammatory cytokines
Smallpox is an inflammatory disease and the cytokine response to infection would likely be predictive of disease outcome. However, methods for assessing cytokine levels were not available during the time when smallpox was endemic, thus no direct correlation between cytokine levels during smallpox outbreaks and clinical severity of disease can be made. Moreover, in animal models of orthopoxvirus disease, mortality is not caused by an excessive inflammatory immune response to systemic infection, but correlates more closely with virus replication in vital organs [18,36]. While the cytokine response is an important marker of the host response to infection it may not be possible to use this disease marker to predict clinical outcome in humans.
Systemic virus spread
Smallpox is a systemic disease with the appearance of generalized pox lesions and lethal consequences of virus infection being directly related to the degree of virus spread [18,35]. In animal models of orthopoxvirus disease, the degree of systemic spread can be quantified by measuring viral titers in the liver, spleen and other internal organs. While there is little information on the levels of virus replication in humans during natural infection, the virus has been cultivated from oropharangeal secretions of smallpox patients during the prodromal phase of infection and at beginning stages of lesion formation [18]. In addition, autopsy results from patients who died within several days after the first sign of rash showed evidence of necrotic lesion formation in the reticulo–endothelial system consistent with virus replication in internal organs [26]. Thus, measuring viral titers in the internal organs of animals as a measure of systemic spread will likely correlate most closely with human clinical outcome. A summary of disease markers that define pathogenesis of orthopoxvirus infection in different animal models is presented in Table 2.
Table 2. Factors that influence pathogenesis of orthopoxvirus infections.
| Animal | Virus | Route | Inoculum (PFU) | Lesions | Systemic disease | Death (days) | Ref. |
|---|---|---|---|---|---|---|---|
| Mouse | VV | IN | 105 | - | + | 6–8 | [14,39] |
| ID | 105 | 1 | - | - | [59] ¶ | ||
| IV | 104 | 20–50 | + | - | [14,60] | ||
| ECTV | IN | 1–10 | - | + | 6–8§ | [14,41] | |
| ID | 1 | - | + | 6–8§ | [47] | ||
| Rabbit | RPV | IN | 1–15 | Rash | + | 10–14 | [52] |
| Monkey | MPV | IT | 107 | >100 | + | 15–19 | [61] |
| IV | 107 | >100 | + | 6–16 | [53] | ||
| VARV | IN | 108.5 | Mild | - | - | [62] | |
| IV | 109 | >250 | + | 4–10 | [55] | ||
| Human | VV | ID | 105 | 1 | - | - | [63] |
| VARV | ID | Scab material | 1–50 | + | - | [18] | |
| MPV | IN | Unknown | >100 | + | 20–30* | [18] | |
| VARV | IN | 1 | >100 | + | 20–30‡ | [18] |
*Mortality observed in approximately 10–15% of patients.
‡Mortality observed in approximately 30% of patients.
§In highly susceptible BALB/c mouse strains.
¶Study performed in a hairless immunocompetent mouse. ECTVL Ectromelia virus; ID: Intradermal; IN: Intranasal; IT: Intratracheal; IV: Intravenous; MPV: Monkeypox virus; PFU: Plaque-forming units; RVP: Rabbitpox virus; VARV: Variola virus; VV: Vaccinia virus.
Conclusion
Development of antiviral drugs for treatment of diseases where human clinical trials that measure compound efficacy are either unethical or are not feasible poses a challenge for drug development. The animal efficacy rule has been developed to provide data to support regulatory approval of drugs for these indications. The design of animal models that can be used to link antiviral efficacy data to human disease outcome will be challenging since the pathogenesis in these model systems is often caused by different mechanisms. Animal models that measure viral pathogenesis using systems that retain coevolved virus host interactions are likely to be most informative.
Expert commentary
Development of therapeutics in which human efficacy evaluations are not feasible will require the use of animal models to assess therapeutic efficacy. This requirement will likely be necessary to facilitate regulatory approval of therapeutics for biodefense targets where disease is no longer endemic or the numbers of patients contracting disease is not large enough to conduct statistically robust efficacy evaluations. To satisfy this requirement a link must be established between therapeutic efficacy in animal models of disease to clinical outcome in humans. For development of antiviral compounds to treat smallpox infection, no single model recapitulates all aspects of human disease. To satisfy the animal efficacy rule, a combination of animal models that mimic specific aspects of orthopoxvirus disease will be required.
Models that require small amounts of virus delivered through mucosal routes to establish infection are most desirable for modeling smallpox infection. Infection by this route requires local replication and systemic virus spread to cause disease, thereby reproducing the critical aspects of human smallpox. Moreover, host-adapted virus species such as ectromelia virus in mice and rabbitpox virus in rabbits which have acquired unique strategies through positive adaptive evolution of host-response modifier genes, allow for virus spread in the host in a manner similar to variola virus in humans. Lesional disease models that involve tail vein inoculation of mice with vaccinia virus, or intravenous delivery of large quantities of monkeypox virus to nonhuman primates are limited in that viremia and disease is induced by intravenous injection bypassing normal routes of systemic spread. In addition, delivery of virus by this route may elicit a different host response relative to virus that spreads systemically following localized replication at the site of inoculation. Lesional disease models are important in that they mimic pock formation characteristic of smallpox disease, but would be less useful for assessing efficacy of therapeutics that prevent systemic infection.
To link antiviral efficacy to human disease, the pharmacokinetic (PK) and pharmacodynamic (PD) variables that track with drug efficacy must be identified in multiple animal species. To establish a PK–PD link, exposure–response relationships are constructed from sequential measurements of the microbiological end-point (reduction in lesion formation, cytokine response, virus yield from organs or other disease markers) as a function of plasma drug concentration. From these analyses, the PD variable(s) that track with drug efficacy can be determined. This information can be used to establish dosing regimens based upon human drug exposure in the absence of viral infection to determine the dose of a compound that will likely provide therapeutic benefit. Establishing the PK–PD link in several animal models using multiple disease markers to define antiviral efficacy will provide data that has the potential of being predictive of human clinical outcome.
Five-year view
Future research will focus on standardizing existing animal models of orthopoxvirus disease with the ultimate goal towards compliance with Good Laboratory Practice (GLP) guidelines. GLP compliance will provide regulatory agencies consistent and validated data to assess the quality of therapeutics for biodefense indications. Regulatory agencies will need to come to agreement as to which features of the current disease models will act as surrogate markers of human disease and most likely to be predictive of outcome of human smallpox. This information will provide the framework for development of biodefense-specific therapeutics. Development of new animal models using host-adapted virus strains will be important for assessing efficacy of biodefense therapeutics since infection and disease are established under conditions that more closely resemble human infection. An emphasis on models that can be conducted safely under Biosafety Level 2 conditions will provide broader access to resources for companies and facilitate biodefense drug development.
Acknowledgements
We would like to thank Chelsea Byrd for critical review of the manuscript. In addition, we would like to thank Mark Buller, Chad Roy and Jay Hooper for supplying images of poxvirus lesions shown in Figure 2.
-
•
Regulatory approval of therapeutics, where human clinical trials are not possible or are unethical, requires the use of animal models of human disease for assessment of efficacy.
-
•
Therapeutic efficacy in animal models must be predictive of human disease outcome.
-
•
For smallpox antiviral drug development, no animal models exist that have been proven to be predictive of clinical outcome.
-
•
Models that require low-dose virus in the inoculum to establish disease will be most informative since they require localized virus replication and systemic spread which mirrors critical features of human smallpox.
-
•
Multiple animal models in different animal species will be required to establish clear dose–response relationships and establish pharmacokinetic variables that track with antiviral efficacy.
-
•
Human dosing regimens most likely to provide clinical benefit can be designed based on the pharmacokinetic–pharmacodynamic link established in animals and human pharmacokinetic data.
References
Papers of special note have been highlighted as: • of interest •• of considerable interest
- 1.Moss B. Poxviridae and their replication. In: Fields Virology. Knipe DM, Howley PM (Eds), Raven Press, Ltd., NY, USA, 2849–2884 (2001). [Google Scholar]; • An excellent review of poxvirus molecular virology.
- 2.Smith GL, Vanderplasschen A, Law M. The formation and function of extracellular enveloped vaccinia virus. J. Gen. Virol. 83(Pt 12), 2915–2931 (2002). [DOI] [PubMed] [Google Scholar]; • An excellent review of the role of extracellular virus in orthopoxvirus replication.
- 3.Byrd CM, Bolken TC, Hruby DE. The vaccinia virus I7L gene product is the core protein proteinase. J. Virol. 76(17), 8973–8976 (2002). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Blasco R, Moss B. Extracellular vaccinia virus formation and cell-to-cell virus transmission are prevented by deletion of the gene encoding the 37,000-Dalton outer envelope protein. J. Virol. 65(11), 5910–5920 (1991). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Cudmore S, Cossart P, Griffiths G, Way M. Actin-based motility of vaccinia virus. Nature 378(6557), 636–638 (1995). [DOI] [PubMed] [Google Scholar]
- 6.Roper RL, Wolffe EJ, Weisberg A, Moss B. The envelope protein encoded by the A33R gene is required for formation of actin-containing microvilli and efficient cell-to-cell spread of vaccinia virus. J. Virol. 72(5), 4192–4204 (1998). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Payne LG. Significance of extracellular enveloped virus in the in vitro and in vivo dissemination of vaccinia. J. Gen. Virol. 50(1), 89–100 (1980). [DOI] [PubMed] [Google Scholar]
- 8.Payne LG, Kristensson K. Extracellular release of enveloped vaccinia virus from mouse nasal epithelial cells in vivo. J. Gen. Virol. 66(Pt 3), 643–646 (1985). [DOI] [PubMed] [Google Scholar]
- 9.McIntosh AA, Smith GL. Vaccinia virus glycoprotein A34R is required for infectivity of extracellular enveloped virus. J. Virol. 70(1), 272–281 (1996). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Stern RJ, Thompson JP, Moyer RW. Attenuation of B5R mutants of rabbitpox virus in vivo is related to impaired growth and not an enhanced host inflammatory response. Virology 233(1), 118–129 (1997). [DOI] [PubMed] [Google Scholar]
- 11.Zhang WH, Wilcock D, Smith GL. Vaccinia virus F12L protein is required for actin tail formation, normal plaque size, and virulence. J. Virol. 74(24), 11654–11662 (2000). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Boulter EA, Zwartouw HT, Titmuss DH, Maber HB. The nature of the immune state produced by inactivated vaccinia virus in rabbits. Am. J. Epidemiol. 94(6), 612–620 (1971). [DOI] [PubMed] [Google Scholar]
- 13.Reeves PM, Bommarius B, Lebeis S et al. Disabling poxvirus pathogenesis by inhibition of Abl-family tyrosine kinases. Nature Med. 11(7), 731–739 (2005). [DOI] [PubMed] [Google Scholar]
- 14.Yang G, Pevear DC, Davies MH et al. An orally bioavailable antipoxvirus compound (ST-246) inhibits extracellular virus formation and protects mice from lethal orthopoxvirus challenge. J. Virol. 79(20), 13139–13149 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Yang H, Kim SK, Kim M et al. Antiviral chemotherapy facilitates control of poxvirus infections through inhibition of cellular signal transduction. J. Clin. Invest. 115(2), 379–387 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Vanderplasschen A, Hollinshead M, Smith GL. Antibodies against vaccinia virus do not neutralize extracellular enveloped virus but prevent virus release from infected cells and comet formation. J. Gen. Virol. 78(Pt 8), 2041–2048 (1997). [DOI] [PubMed] [Google Scholar]
- 17.Vanderplasschen A, Mathew E, Hollinshead M, Sim RB, Smith GL. Extracellular enveloped vaccinia virus is resistant to complement because of incorporation of host complement control proteins into its envelope. Proc. Natl Acad. Sci. USA 95(13), 7544–7549 (1998). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Fenner F, Henderson DA, Arita I, Jazek Z, Ladnyi ID. Smallpox and its eradication. WHO, Geneva, Switzerland, 1–1421 (1988). [Google Scholar]; •• Classic treatise of the smallpox eradication effort that includes a comprehensive review of the pathogenesis, natural history and epidemiology of variola virus infection.
- 19.Breman JG, Henderson DA. Diagnosis and management of smallpox. N. Engl. J. Med. 346(17), 1300–1308 (2002). [DOI] [PubMed] [Google Scholar]; • An outstanding overview of the natural history and clinical manifestations of variola virus infection.
- 20.Bray M. Pathogenesis and potential antiviral therapy of complications of smallpox vaccination. Antiviral Res. 58(2), 101–114 (2003). [DOI] [PubMed] [Google Scholar]
- 21.Chen N, Buller RM, Wall EM, Upton C. Analysis of host response modifier ORFs of ectromelia virus, the causative agent of mousepox. Virus Res. 66(2), 155–173 (2000). [DOI] [PubMed] [Google Scholar]; • Identification and functional charaterization of host-response modifier genes in ectromelia virus.
- 22.Gubser C, Hue S, Kellam P, Smith GL. Poxvirus genomes: a phylogenetic analysis. J. Gen. Virol. 85(Pt 1), 105–117 (2004). [DOI] [PubMed] [Google Scholar]
- 23.McLysaght A, Baldi PF, Gaut BS. Extensive gene gain associated with adaptive evolution of poxviruses. Proc. Natl Acad. Sci. USA 100(26), 15655–15660 (2003). [DOI] [PMC free article] [PubMed] [Google Scholar]; • A bioinformatic approach was used to evaluate poxvirus genome evolution to identify gene families undergoing positive selection.
- 24.Massung RF, Esposito JJ, Liu LI et al. Potential virulence determinants in terminal regions of variola smallpox virus genome. Nature 366(6457), 748–751 (1993). [DOI] [PubMed] [Google Scholar]
- 25.Baker RO, Bray M, Huggins JW. Potential antiviral therapeutics for smallpox, monkeypox and other orthopoxvirus infections. Antiviral Res. 57(1–2), 13–23 (2003). [DOI] [PMC free article] [PubMed] [Google Scholar]; • A review of compounds that inhibit orthopoxvirus replication in vitro.
- 26.Bras G. The morbid anatomy of smallpox. Doc. Med. Geogr. Trop. 4(4), 303–351 (1952). [PubMed] [Google Scholar]; • A comprehensive analysis of smallpox pathogenesis using data from autopsy patients.
- 27.McKenzie PJ, Githens JH, Harwood ME, Roberts JF, Rao AR, Kempe CH. Haemorrhagic smallpox. 2. Specific bleeding and coagulation studies. Bull. World Health Organ. 33(6), 773–782 (1965). [PMC free article] [PubMed] [Google Scholar]
- 28.Roberts JF, Coffee G, Creel SM et al. Haemorrhagic smallpox. I. Preliminary haematological studies. Bull. World Health Organ. 33(5), 607–613 (1965). [PMC free article] [PubMed] [Google Scholar]
- 29.Esteban DJ, Buller RM. Ectromelia virus: the causative agent of mousepox. J. Gen. Virol. 86(Pt 10), 2645–2659 (2005). [DOI] [PubMed] [Google Scholar]; • Outstanding review of the pathogenesis of ectromelia virus infection.
- 30.Johnston JB, McFadden G. Poxvirus immunomodulatory strategies: current perspectives. J. Virol. 77(11), 6093 (2003). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Born TL, Morrison LA, Esteban DJ et al. A poxvirus protein that binds to and inactivates IL-18, and inhibits NK cell response. J. Immunol. 164(6), 3246–3254 (2000). [DOI] [PubMed] [Google Scholar]
- 32.Reading PC, Smith GL. Vaccinia virus interleukin-18-binding protein promotes virulence by reducing gamma interferon production and natural killer and T-cell activity. J. Virol. 77(18), 9960–9968 (2003). [DOI] [PMC free article] [PubMed] [Google Scholar]; • An example of the role of host-response modifier genes in pathogenesis of orthopoxvirus infection.
- 33.Reading PC, Khanna A, Smith GL. Vaccinia virus CrmE encodes a soluble and cell surface tumor necrosis factor receptor that contributes to virus virulence. Virology 292(2), 285–298 (2002). [DOI] [PubMed] [Google Scholar]
- 34.Reading PC, Symons JA, Smith GL. A soluble chemokine-binding protein from vaccinia virus reduces virus virulence and the inflammatory response to infection. J. Immunol. 170(3), 1435–1442 (2003). [DOI] [PubMed] [Google Scholar]
- 35.Bray M, Buller M. Looking back at smallpox. Clin. Infect. Dis. 38(6), 882–889 (2004). [DOI] [PubMed] [Google Scholar]; • An excellent review of smallpox relating historical data to our present day understanding of viral pathogenesis.
- 36.Martinez MJ, Bray MP, Huggins JW. A mouse model of aerosol-transmitted orthopoxviral disease: morphology of experimental aerosol-transmitted orthopoxviral disease in a cowpox virus-BALB/c mouse system. Arch. Pathol. Lab. Med. 124(3), 362–377 (2000). [DOI] [PubMed] [Google Scholar]; • The pathogenesis of cowpox virus infection in mice.
- 37.Zaucha GM, Jahrling PB, Geisbert TW, Swearengen JR, Hensley L. The pathology of experimental aerosolized monkeypox virus infection in cynomolgus monkeys (Macaca fascicularis). Lab. Invest. 81(12), 1581–1600 (2001). [DOI] [PMC free article] [PubMed] [Google Scholar]; • Histopathology of monkeypox infection of nonhuman primates.
- 38.Kim M, Yang H, Kim SK et al. Biochemical and functional analysis of smallpox growth factor (SPGF) and anti-SPGF monoclonal antibodies. J. Biol. Chem. 279(24), 25838–25848 (2004). [DOI] [PubMed] [Google Scholar]
- 39.Quenelle DC, Collins DJ, Kern ER. Efficacy of multiple- or single-dose cidofovir against vaccinia and cowpox virus infections in mice. Antimicrob. Agents Chemother. 47(10), 3275–3280 (2003). [DOI] [PMC free article] [PubMed] [Google Scholar]; • Antiviral efficacy in lethal vaccinia and cowpox virus mouse models.
- 40.Magee WC, Hostetler KY, Evans DH. Mechanism of inhibition of vaccinia virus DNA polymerase by cidofovir diphosphate. Antimicrob. Agents Chemother. 49(8), 3153–3162 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Buller RM, Owens G, Schriewer J, Melman L, Beadle JR, Hostetler KY. Efficacy of oral active ether lipid analogs of cidofovir in a lethal mousepox model. Virology 318(2), 474–481 (2004). [DOI] [PubMed] [Google Scholar]; • Antiviral efficacy in a lethal ectromelia virus mouse model.
- 42.Smee DF, Sidwell RW. A review of compounds exhibiting anti-orthopoxvirus activity in animal models. Antiviral Res. 57(1–2), 41–52 (2003). [DOI] [PMC free article] [PubMed] [Google Scholar]; • A comprehensive review of animal models used to evaluate antiorthopoxvirus compounds.
- 43.Li G, Chen N, Roper RL et al. Complete coding sequences of the rabbitpox virus genome. J. Gen. Virol. 86(Pt 11), 2969–2977 (2005). [DOI] [PubMed] [Google Scholar]
- 44.Nelson JB. The behavior of pox viruses in the respiratory tract. J. Exp. Med. 68, 401–412 (1938). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.De Clercq E, De Somer P. Effect of interferon, polyacrylin acid, and polymethacrylic acid on tail lesions on mice infected with vaccinia virus. Appl. Microbiol. 16(9), 1314–1319 (1968). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Thompson RL, Price M, Minton SA Jr, Falco EA, Hitchings GH. Protection of mice against the vaccinia virus by the administration of phenoxythiouracils. J. Immunol. 67(6), 483–491 (1951). [PubMed] [Google Scholar]
- 47.Fenner F, Buller RML. Mousepox. In: Viral Pathogenesis. Nathanson N (Ed.), Lippincott-Raven Publishers, PA, USA, 535–553 (1997). [Google Scholar]
- 48.Schriewer J, Buller RM, Owens G. Mouse models for studying orthopoxvirus respiratory infections. Methods Mol. Biol. 269, 289–308 (2004). [DOI] [PubMed] [Google Scholar]
- 49.Mims CA. The response of mice to large intravenous injections of ectromelia virus. I. The fate of injected virus. Br. J. Exp. Pathol. 40, 533–542 (1959). [PMC free article] [PubMed] [Google Scholar]
- 50.Mims CA. The response of mice to large intravenous injections of ectromelia virus. II. The growth of virus in the liver. Br. J. Exp. Pathol. 40, 543–550 (1959). [PMC free article] [PubMed] [Google Scholar]
- 51.Mims CA. Aspects of the pathogenesis of virus diseases. Bacteriol. Rev. 28, 30–71 (1964). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Westwood JC, Boulter EA, Bowen ET, Maber HB. Experimental respiratory infection with poxviruses. I. Clinical virological and epidemiological studies. Br. J. Exp. Pathol. 47(5), 453–465 (1966). [PMC free article] [PubMed] [Google Scholar]
- 53.Hooper JW, Thompson E, Wilhelmsen C et al. Smallpox DNA vaccine protects nonhuman primates against lethal monkeypox. J. Virol. 78(9), 4433–4443 (2004). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Huggins JW, Smee DF, Martinez M, Bray M. Cidofovir (HPMPC) treatment of monkeypox. Antiviral Res. 37, A37 (1998). [Google Scholar]
- 55.Jahrling PB, Hensley LE, Martinez MJ et al. From the cover: exploring the potential of variola virus infection of cynomolgus macaques as a model for human smallpox. Proc. Natl Acad. Sci. USA 101(42), 15196–15200 (2004). [DOI] [PMC free article] [PubMed] [Google Scholar]; • Variola virus infection of nonhuman primates.
- 56.Saraiva M, Smith P, Fallon PG, Alcami A. Inhibition of type 1 cytokine-mediated inflammation by a soluble CD30 homologue encoded by ectromelia (mousepox) virus. J. Exp. Med. 196(6), 829–839 (2002). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Afonso CL, Tulman ER, Lu Z et al. The genome of camelpox virus. Virology 295(1), 1–9 (2002). [DOI] [PubMed] [Google Scholar]
- 58.Roper RL. Characterization of the vaccinia virus A35R protein and its role in virulence. J. Virol. 80(1), 306–313 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Neyts J, Leyssen P, Verbeken E, De CE. Efficacy of cidofovir in a murine model of disseminated progressive vaccinia. Antimicrob. Agents Chemother. 48(6), 2267–2273 (2004). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Neyts J, Verbeken E, De CE. Effect of 5-iodo-2´-deoxyuridine on vaccinia virus (orthopoxvirus) infections in mice. Antimicrob. Agents Chemother. 46(9), 2842–2847 (2002). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Stittelaar KJ, van AG, Kondova I et al. Modified vaccinia virus Ankara protects macaques against respiratory challenge with monkeypox virus. J. Virol. 79(12), 7845–7851 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.LeDuc JW, Jahrling PB. Strengthening national preparedness for smallpox: an update. Emerg. Infect. Dis. 7(1), 155–157 (2001). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Frey SE, Couch RB, Tacket CO et al. Clinical responses to undiluted and diluted smallpox vaccine. N. Engl. J. Med. 346(17), 1265–1274 (2002). [DOI] [PubMed] [Google Scholar]
Websites
- 101.WHO. Smallpox slide set, 2005 www.who.int/emc/diseases/smallpox/ slideset/
- 102.PBR shared orthologs syntenic plot www.poxvirus.org/synteny.asp