

Abstract
Peptide ligases are versatile enzymes that can be utilized for precise protein conjugation for bioengineering applications. Hyperactive peptide asparaginyl ligases (PALs), such as butelase-1, belong to a small class of enzymes from cyclotide-producing plants that can perform site-specific, rapid ligation reactions after a target peptide asparagine/aspartic acid (Asx) residue binds to the active site of the ligase. How PALs specifically recognize their polypeptide substrates has remained elusive, especially at the prime binding side of the enzyme. Here we report crystal structures that capture VyPAL2, a catalytically efficient PAL from Viola yedoensis, in an activated state, with and without a bound substrate. The bound structure shows one ligase with the N-terminal polypeptide tail from another ligase molecule trapped at its active site, revealing how Asx inserts in the enzyme’s S1 pocket and why a hydrophobic residue is required at the P2′ position. Besides illustrating the anchoring role played by P1 and P2′ residues, these results uncover a role for the Gatekeeper residue at the surface of the S2 pocket in shifting the nonprime portion of the substrate and, as a result, the activity toward ligation or hydrolysis. These results suggest a picture for proenzyme maturation in the vacuole and will inform the rational design of peptide ligases with tailored specificities.
Solving the structure of a peptide ligase in an activated state with and without its substrate will help inform the rational design of specific peptide ligases as bioengineering tools.
IN A NUTSHELL.
Background: Proteases, protein scissors that cut proteins into smaller pieces, can be found almost everywhere in the natural world, including in plants. However, protein ligases, which stitch small pieces of proteins together, are much harder to find. Yet protein ligases such as peptide asparaginyl ligases (PALs) are very valuable, because they have many applications in biomedical research, such as conjugating proteins with a label for imaging, or fusing an antibody with a toxin to specifically kill cancer cells in patients.
Question: How do PALs recognize their peptide substrates? We studied VyPAL2, a PAL from the medicinal plant Viola yedoensis with very strong ligase activity at neutral pH. We also asked how VyPAL2 can cut itself into two pieces at acidic pH, so that it becomes active.
Findings: We expressed the VyPAL2 protein in insect cells and obtained crystals of the active protein. Comparing our crystal structures with published protease structures, we found a backbone shift induced by a specific residue that acts as an important activity determinant. We also found that the catalytic cysteine, which was reported to be the key residue for activation, is not necessary for maturation. Instead, a catalytic histidine plays a key role and we came up with a scheme to account for auto-activation.
Next steps: The results described here will help engineer ligases with altered specificity that can be used in biotechnology and precision medicine.
Introduction
Peptide ligases, such as sortase A (Schneewind et al., 1992), constitute attractive tools for protein conjugation (the attachment of a small molecule to a protein) (Mao et al., 2004), protein semi-synthesis (the assembly of a complete protein from its separate fragments) (Noike et al., 2015; Cao et al., 2016), peptide cyclization (Antos et al., 2009), or live cell labeling (Bi et al., 2017). Ligases extracted from plants could play a crucial role in the active field of molecular epigenetics, where precision tools are needed to produce target proteins, such as exquisitely modified histone proteins (Bagert and Muir, 2021). Following the discovery of cyclotide sequences in graminaceous crop plants (Mulvenna et al., 2006) and the realization that these cyclic polypeptides, which have medicinal and agricultural applications, depend on plant asparaginyl endopeptidases (AEPs) for their maturation (Mylne et al., 2012), a quest to clone the genes encoding AEPs was started. In 2014, a peptide ligase specific for asparagine (Asn; see Supplemental Table S1 for a list of abbreviations used throughout the manuscript) was successfully isolated from the cyclotide-producing plant blue pea (Clitoria ternatea) and named butelase-1 (Nguyen et al., 2014). Compared to bacterial sortase A, butelase-1 is a hyperactive peptide asparaginyl ligase (PAL) with a reported catalytic efficiency of 1,314,000 M−1s−1 that has proven useful for many applications such as for protein or peptide macrocyclization or the enzymatic engineering of live bacterial cell surfaces (Cao et al., 2015, 2016; Nguyen et al., 2015a, 2015b, 2016a, 2016b; Bi et al., 2017). Subsequently, several PALs including OaAEP1b (Harris et al., 2015), HeAEP3 (Jackson et al., 2018), and VyPAL2 (Hemu et al., 2019) were identified from other cyclotide-producing plants: Oldenlandia affinis, Hybanthus enneaspermus, and Viola yedoensis, respectively.
AEPs (or legumains; Kembhavi et al., 1993) belong to the subfamily C13 of cysteine protease. Legumains such as butelase-2 (Serra et al., 2016), OaAEP2 (Serra et al., 2016), and HaAEP1 from sunflower (Helianthus annuus) (Haywood et al., 2018) are predominantly endowed with protease activity at neutral pH, with a low level of peptide ligase activity (Dall and Brandstetter, 2013; Zhao et al., 2014; Haywood et al., 2018; Zauner et al., 2018a; Dall et al., 2020, 2021). A third group of enzymes, including CeAEP from jack bean (Canavalia ensiformis) (Bernath-Levin et al., 2015), PxAEP3 from Petunia x hybrida (Jackson et al., 2018), AtLEGγ from Arabidopsis thaliana (Zauner et al., 2018a), and VyAEP1 from V. yedoensis (Hemu et al., 2019) catalyze (at near neutral pH) the formation of both ligation and hydrolytic products from peptide substrates carrying proper amino-acid recognition sequences. In contrast to “bi-functional” or “predominant” AEPs, butelase-1 (Nguyen et al., 2014), OaAEP1b (Harris et al., 2015), OaAEP3-5 (Harris et al., 2019), and VyPAL2 (Hemu et al., 2019) catalyze the formation of ligation products, with no hydrolytic product observed.
In plants, all these enzymes (with either predominantly Asx peptide ligase, endoprotease, or a hybrid of protease/ligase activity) are expressed as inactive zymogens. The expressed protein contains a vacuole-targeted signal peptide, an N-terminal pro-domain, a catalytic core domain, and a C-terminal cap domain that covers the active site of the folded proenzyme (Kuroyanagi et al., 2002). Auto-activation is performed in vivo in the acidic vacuolar compartment of the plant, leading to the conversion of the proenzyme into its mature active form via proteolysis of the immature polypeptide chain, presumably in trans (Nguyen et al., 2014). Similarly, in vitro activation of the recombinant proenzyme is usually performed in the laboratory at pH values ranging from 4 to 5. Under these acidic conditions, the zymogen undergoes autolytic activation, leading to the cleavage of both the pro- and cap domains at the N- and C-termini of the catalytic core, respectively (Harris et al., 2015; Yang et al., 2017; Jackson et al., 2018; Hemu et al., 2019) and the release of mature active enzyme. Auto-activation thus appears as a one-off capacity of these enzymes to catalyze a hydrolysis reaction, regardless of the predominant activity of the mature form (protease or ligase), raising questions about the catalytic mechanism underlying auto-activation.
AEPs and PALs share a high level of structural similarity, suggesting that their enzymatic activity is controlled by subtle structural differences near the catalytic center (Hemu et al., 2019). AEPs and PALs have a conserved catalytic triad formed by a cysteine and histidine (lining the S1 active site pocket) and an Asn. The S1 pocket houses Asx (either Asn or Asp) as the P1 residue following the nomenclature proposed for the polypeptide substrate by (Schechter and Berger, 1967). Mechanisms for ligation and hydrolysis were previously described (Hemu et al., 2019, 2020) (see Figure 1A).
Figure 1.

Mechanism of ligation by PALs, and amino-acid composition of the S2 and S1′ pockets. A, Two steps are necessary for the reaction. First, the substrate peptide binds to the active site, with the P1 Asx bound into the S1 pocket. The catalytic cysteine performs a nucleophilic attack that breaks the peptide bond between the P1 and P1′ residues; as a result, a thioester intermediate is formed. The second step is the resolution of the intermediate by an incoming nucleophile (a water molecule in the case of hydrolysis and the amine group of a peptide in the case of a ligation reaction). Substrate P4–P1 and P1′–P2′ binding occurs via interactions with specific residues in the S1–S4 and S1′–S2′ pockets. Nucleophilic attack by the catalytic cysteine on the P1 residue carbonyl carbon atom leads to the formation of the acyl-enzyme intermediate. Nucleophilic attack by a water molecule is inhibited to favour nucleophilic attack by the amine group of the incoming peptide. B, Nature of residues in the S2 (LAD 1 region) and S1′ (LAD2 region) pockets characteristic of ligases and proteases. The numbers above the sequence alignment refer to residue numbers in VyPAL2. Residues in the Gatekeeper position are shaded in gray. Abbreviations for amino acids are as follows: A: Ala, C: Cys, D: Asp, E: Glu, F: Phe, G: Gly, H: His, I: Ile, K: Lys, L: Leu, M: Met, N: Asn, P: Pro, Q: Gln, R: Arg, S; Ser, Snn: Asi (Aspartimide), T: Thr, V: Val, W: Trp, Y: Tyr.
Recent structure–function studies identified important determinants of peptide bond formation, which were named “marker of ligase activity” (Jackson et al., 2018) and “Ligase activity determinants” (LADs) (Hemu et al., 2019). Local structural features immediately surrounding the S1 Asx-binding site were proposed to control (1) the conformation of the bound peptide substrate; (2) the kinetics of peptide departure at the enzyme nonprime site; and (3) the access to the S-acyl enzyme intermediate of either water molecules (leading to hydrolysis of the peptide substrate) or an incoming nucleophile (leading to ligation) at the prime side. Rules were proposed (Hemu et al., 2019) that were largely validated via the successful conversion of butelase-2, a pure protease, into a peptide ligase by solely mutating the LAD1 (S2 pocket) and LAD2 (near the S1′ pocket) residues, while mutations at other non-LAD positions had only a limited impact on activity (Hemu et al., 2020).
Of special functional importance is the “Gatekeeper,” the central residue of the LAD1 tripeptide motif. The importance of this residue was discovered via a site-directed mutagenesis study of OaAEP1b, a weakly active PAL isolated from O. affinis. The Gatekeeper, which is Cys247 in the wild-type (WT) OaAEP1b enzyme, plays a key role in controlling both the directionality and the kinetics of the reaction (Yang et al., 2017). A single mutation of Cys247 to Ala was sufficient to increase the kcat/Km value for ligation by over 150-fold compared to the WT OaAEP1b enzyme. Likewise, the cognate substitution of Gatekeeper Gly252 of butelase-2 into Val or Ile yielded butelase-2 mutants with a markedly increased ligase activity (Hemu et al., 2020) (Figure 1B). Thus, the presence of a Gly as Gatekeeper in the amino-acid sequence is indicative of a predominantly protease activity for the mature enzyme, whilst residues with aliphatic side chains at this position favor ligase activity, as is the case for butelase-1 (Val237) or VyPAL2 (Ile244) (Hemu et al., 2019, 2020).
Despite the publication of the crystal structures of several plant AEP and PALs, either in their pro-enzyme or active forms (Dall and Brandstetter, 2013; Zhao et al., 2014; Yang et al., 2017; Haywood et al., 2018; Zauner et al., 2018a; Hemu et al., 2019; James et al., 2019), our understanding of the mechanistic and structural importance of the LAD1 and LAD2 residues and of the Gatekeeper remains incomplete. Here we sought to identify structural determinants that account for the substrate recognition and ligase activity of VyPAL2, a very efficient PAL discovered via mining the transcriptome of V. yedoensis that can be conveniently expressed in recombinant form Hemu et al. (2019). We obtained crystal structures following acid-induced auto-activation, including one structure where residues from the N-terminal propeptide of one ligase molecule are bound to the active site of the neighboring molecule in the crystal lattice. This structure gives a complete atomic view of substrate binding, including at the prime side of the enzyme. Together with supporting biochemical and enzymatic data, we uncovered key structural features associated with specific substrate recognition. We propose a testable mechanism for peptide ligase activity in which the Gatekeeper plays a crucial role, which agrees with kinetics observations. Moreover, our analysis of the auto-activation of VyPAL2 proenzymes bearing mutations at their His and Cys catalytic residues pointed to alternative routes used for their maturation process and for catalytic activity.
Results
Ligase activity and crystal structure of the activated enzyme
We expressed, purified, and activated VyPAL2 as previously reported (Hemu et al., 2019). To conform to the LAD1 of butelase-1 (Figure 1B), which has the highest ligase activity documented so far (Nguyen et al., 2014; Hemu et al., 2019, 2020), we expressed, purified, and activated a single mutant of the VyPAL2 proenzyme with the mutation Ile244Val at the Gatekeeper position (VyPAL2-I244V) in a similar manner. Briefly, activation was carried out by incubating the proenzyme at pH 4.2 for 2 h at 37°C, followed by size-exclusive chromatography to purify the core catalytic domain (Figure 2A; Supplemental Figure S1). Upon activation, for both VyPAL2 and VyPAL2-I244V, a time-dependent decrease in the amount of the 50 kDa pro-enzyme was observed by sodium dodecyl sulfate–poly acrylamide gel electrophoresis (SDS–PAGE). This was accompanied by the appearance of a ∼37 kDa band corresponding to the active form. Activation was completed after 2 h of incubation (Figure 2, A and B).
Figure 2.

Expression, activation, ligase activity, and crystal structure of VyPAL2-I244V. A, Schematic sequence representation of VyPAL2-I244V. The respective VyPAL2 genes encoding complete amino-acid sequences were cloned into the expression vector, with the signal peptide substituted by a hexa-His-tag for affinity purification (see “Materials and methods”). Catalytic residues and the aspartimide moiety (Snn171) next to the catalytic His172 are indicated in brown, N-linked glycosylation sites are indicated in cyan, and domain boundaries (as deduced from a previous LC–MS/MS analysis) are indicated in black (please also see SI of Hemu et al. (2019)). Proteolytic cleavage sites are indicated with arrows above the N- and C-terminal sequences. The products from low pH activation of the proenzyme were analyzed using (B) SDS–PAGE with Coomassie blue staining. Incubation of VyPAL2 for 2 h led to the production of a fully mature enzyme. C, Analysis of ligase activity using a Förster Resonance Energy Transfer assay. Red squares (VyPAL2-I244V) and blue points (VyPAL2) represent mean values and error bars denote standard deviations. D, Comparison between the crystal structures of the VyPAL2 pro-enzyme monomer (right, PDB access code: 6IDV (Hemu et al., 2019)) and the activated VyPAL2 protein (left, this work). The proteins are displayed with α-helices as ribbons and β-strands as arrows. The color-code used for each of the three domains of the proenzymes is used throughout the manuscript (core domain: green, cap domain: wheat (light yellow), linker region connecting the catalytic core and cap domain: orange).
Next, we determined the peptide ligase activity of the WT VyPAL2 enzyme and VyPAL2-I244V using a fluorometric assay, allowing real-time monitoring of the formation of a ligated peptide product (Figure 2C). Two model peptide substrates with the amino-acid sequences PIE(EDANS)YNAL and GIK(DABSYL)SIP, containing the “Asn–Ala–Leu” tripeptide and ‘Gly–Ile” dipeptide ligase recognition motifs at their C- and N-termini, respectively, were fluorescently labeled. Upon ligation, the fluorescent signal of the EDANS moiety of the first peptide becomes quenched by the DABSYL moiety of the second peptide, whilst the Ala–Leu dipeptide was released (Figure 2C). We performed ligation reactions of VyPAL2 and VyPAL2-I244V at 37°C for 2 min to determine the initial velocity at pH 6.5. The Vmax value of VyPAL2-I244V (53.76 RFU/s) increased by approximately two-fold compared to the WT VyPAL2 enzyme (28.68 RFU/s), while the Km values were comparable, with Km = 9.351 μM for the WT enzyme and 7.455 μM for VyPAL2-I244V (Figure 2C).
We then concentrated the purified auto-activated VyPAL2 and VyPAl2-I244V proteins to 6.7 mg mL−1 and 4.5 mg mL−1, respectively, prior to crystallization. The crystals were mounted on a cryo-loop and flash frozen in liquid nitrogen. We collected X-ray diffraction intensities extending to 2.3 and 1.59 Å resolution for VyPAL2 and VyPAL2-I244V, respectively, and determined the structures of their active forms by molecular replacement using the core domain of the VyPAL2 proenzyme structure as a search probe (Hemu et al., 2019) (Supplemental Table S2). Two essentially identical molecules are present in the asymmetric unit. The small interface between these two molecules (679 Å2 and 473 Å2 for VyPAL2 and VyPAL2-I244V, respectively, as determined by the PISA server, http://www.ebi.ac.uk/pdbe/prot_int/pistart.html) rules out the formation of a functional dimer in solution. This agrees with the observed elution volume in size exclusion chromatography (SEC) corresponding to a monomer with an apparent molecular weight of ∼40 kDa.
The structure of the active enzyme comprising residues Ile51 to Glu331 adopts the conserved core domain architecture also seen in legumains: a central β-sheet containing six β-strands surrounded by five large and four smaller α-helices (Figure 2D; Supplemental Figure S2). Compared to the equivalent core domain in the context of the immature proenzyme VyPAL2 (Hemu et al., 2019), no major structural change occurs upon maturation, as indicated by a local root mean square deviation (r.m.s.d.) (http://superpose.wishartlab.com/) of 0.3 Å for 276 superimposed α-carbon (Figure 2D; Supplemental Figure S3). Hence, activation appears to have little impact on the structure of the core domain, besides cap removal, leading to a complete exposure of the catalytic site to the solvent and to diffusing protein substrates (Figure 2D). The structures of VyPAL2 and VyPAL2-I224V are highly similar, with a r.m.s.d. of 0.41 Å (Supplemental Figures S3 and S4). Further comparisons with other active PALs such as Arabidopsis AtLEGγ (Zauner et al., 2018b) (PDB access code: 5OBT), sunflower HaAEP1 (Haywood et al., 2018) (PDB code: 6AZT), or butelase-1 (James et al., 2019) (PDB code: 6DHI) return an average r.m.s.d. value of 1.21 Å (Supplemental Table S3), confirming that no major change of conformation occurs when the enzymes undergo maturation.
Crystallization of an enzyme–substrate complex
To trap a complex with a peptide substrate, we generated a VyPAL2 mutant by mutating the catalytic Cys214 to alanine. This mutant is catalytically inactive, as it lost the ability to form an acyl-enzyme intermediate with the substrate. Remarkably, this mutant undergoes rapid maturation at acidic pH, and its core domain could be purified using a procedure akin to that of the WT enzyme (Figure 3A). Moreover, the auto-activation of VyPAL2-C214A appears to be even faster than that of VyPAL2-I244V, as the full processing of the proenzyme was observed after only 60 min versus 2 h for VyPAL2-I244V (Figures 2, B and 3, A; Supplemental Figure S4). As expected, VyPAL2-C214A showed highly reduced activity in our peptide ligation assay (Figure 2C), supporting a crucial role for Cys214 in the ligation and cyclization reactions but not maturation.
Figure 3.

Expression, maturation, and crystal structures of VyPAL2-C214A. A, left: Schematic representations of VyPAL2-C214A sequences. Before activation (upper diagram), following acid-induced activation and crystallization (middle and lower diagrams, respectively). Residues visible in the electron density map are displayed in bold, while residues immediately at the N-terminus likely to be cleaved are in plain font. B, Schematic diagram of crystal packing showing how VyPAL2-C214A molecules are stacked in the monoclinic C2 unit cell. Molecules related by a unit-cell translation along b are displayed as ribbons of the same color, whilst molecules related by the two-fold axis along the same axis are displayed in green and pink (middle). The N-terminal tail of each molecule inserts in the molecule stacked underneath. C, The N-terminal peptide bound to the enzyme. The molecular surface of the peptide ligase is displayed in gray. Overlaid is a Fourier difference map displayed as a blue mesh with coefficients 2Fo–Fc, with phases from the refined model, contoured at a level of 1σ over the mean.
Following maturation at acidic pH and purification of the activated form, well-diffracting crystals of the core domain of VyPAL2-C214A were obtained under two conditions (see “Materials and methods”). We collected X-ray diffraction data at resolutions of 1.9 and 1.8 Å, respectively, for two crystal forms of VyPAL2-C214A (labeled as “Form I” and “Form II”) (Supplemental Table S2).
Form I crystals grew after 3–5 days and contained one monomer per asymmetric unit. Molecules were packed in staggered rows along the unique b axis (Figure 3B). Residues Ser44 to Val329 of the protein could be traced with confidence in the electron density map, including residues Asn43–Asp49 at the N-terminal cleavable proenzyme region, which were in well-defined electron density (Figure 3C). In particular, the scissile (cleavable) P1–P1′ (Schechter and Berger, 1967) bond Asp49–Ser50 was neither cleaved during the 1-h low pH activation step nor during the following purification and crystallization steps (over 3–5 days). As anticipated, this cleavable N-terminal tail appears flexible and projects away from the enzyme catalytic core. In the crystal lattice, this tail inserts into the active site of the neighboring molecule related to the first by the crystallographic dyad along the b axis (Figure 3, B and C). Thus, in the crystal lattice, the N-terminal region (Asn43–Asp49) of a molecule occupies the active site of the molecule located underneath. Of note, Asp49 is deeply buried in the S1 pocket, while residues Asp47, Asp48, Ser50, and Ile51 occupy the S3, S2, S1′, and S2′ pockets, respectively (Figure 4). These observations agree with previous liquid chromatography mass spectrometry (LC–MS/MS) results, demonstrating that the peptide bond C-terminal to Asp49 of VyPAL2 constitutes one of the several possible N-terminal maturation sites of the proenzyme (Hemu et al., 2019). Hence, the N-terminal peptide Asn43–Ile51 bound to the active site of VyPAL2-C214A likely represents an enzyme–substrate complex trapped during N-terminal maturation.
Figure 4.
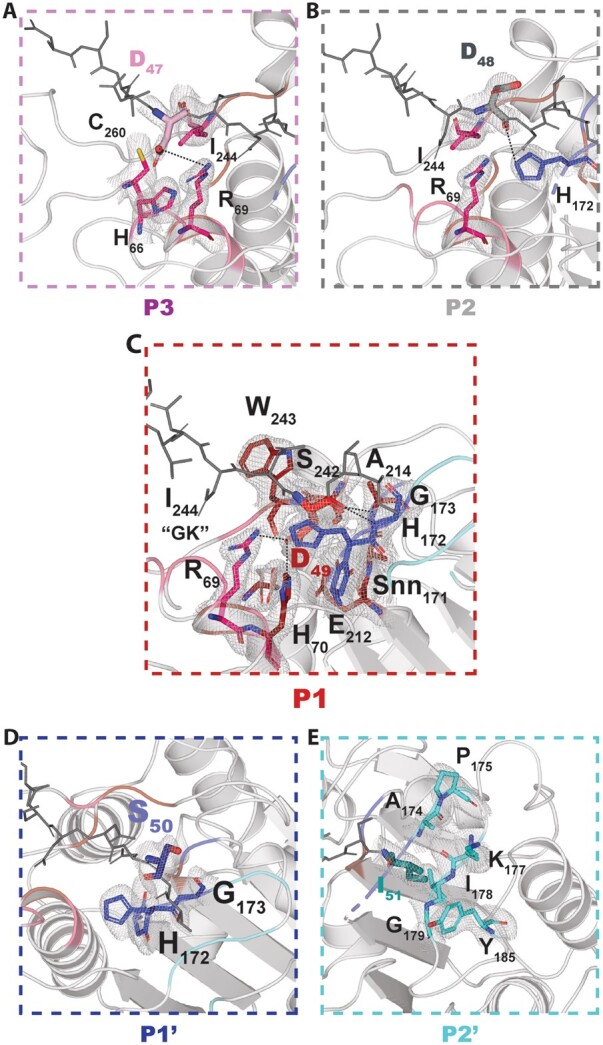
Detailed views of the ligase specificity pockets. Magnified views of the S3–S2′ specificity pockets of VyPAL2. For each pocket, the cognate peptide substrate (sticks) is colored in wheat (A, P3), gray (B, P2), red (C, P1) blue (D, P1′), and cyan (E, P2′), while the rest of the peptide is in gray. Residues from the specificity pockets are colored in magenta (A, S3), red (B, S2), red (C, S1) blue (D, S1′), and cyan (E, S2′) and are shown as sticks and labeled. Polar interactions between the substrate residues and residues lining the specificity pockets (hydrogen bonds or salt bridges) are depicted as dashed lines. Overlaid is a Fourier difference map displayed as a blue mesh with coefficients 2Fo–Fc, with phases from the refined model, contoured at a level of 1σ over the mean.
In contrast, Form II crystals only grew after a 2-month incubation (Supplemental Table S1; Supplemental Figure S2). In this crystal form, the N-terminal region (Asn43–Asp49) could not be observed, suggesting that the N-terminal region is slowly cleaved before crystallization.
Analysis of the enzyme–substrate complex
Form I crystals offer a unique opportunity to analyze substrate binding for an activated PAL and to compare it to the unbound auto-activated structure. The bound N-terminal peptide sits in a shallow groove at the surface of the ligase. Intermolecular interactions between the ligase and its substrate appear to be primarily mediated by P1 residue Asp49 and Ile51 (P2′), which act as two anchoring points (Figure 4, A and B). The monomer can be superimposed with the activated VyPAL2-I244V structure with an r.m.s.d. 0.41 Å. Thus, binding of the Asn43–Ile51 N-terminal substrate is not accompanied by significant conformational changes (compare left and right parts in Figure 5A). Asp49 is bound in the S1 pocket, as one would expect for the isosteric enzyme’s P1 substrate Asn. At the acidic pH of 4.5 used to obtain form I crystals, the carboxylic sidechains of Asp49 as well as Glu212 and Asp263 present in the S1 pocket are likely to be protonated, favoring binding, while binding would be disfavored at neutral pH (Zhang et al., 2021). Since the N-terminal Asn43–Ile51 region represents a catalytically functional substrate positioned in the active site, we used this experimental structure to model an active enzyme–substrate complex by mutating in silico a Cys back at position 214. The Cys rotamer was chosen based on the catalytic cysteine from the activated WT protein structure as well as the inhibitor-bound AtLEGγ cysteine residue (Dall et al., 2020). This allowed us to precisely define the enzyme-binding pockets at both the prime and nonprime positions S3–S2′ (Figures 4 and 5).
Figure 5.

Peptide-binding groove and substrate recognition by VyPAL2. A, The left part shows the peptide binding groove of the ligase, as derived from the crystal structure of VyPAL2 (Supplemental Table S2). The right part is derived from the structure of VyPAL2-C214A (Supplemental Table S2, form I) bound to the peptide displayed in magenta. B, Residues lining the substrate binding pockets are depicted as sticks. The VyPAL2 core domain is represented as a gray surface. The S1 pocket is red, the S3 pocket is magenta, the S1′ pocket is blue, the S2′ pocket is cyan. C, The active site residues His172 and C214 (mutated here to Ala) are on opposite sides of the peptide substrate, and adding back a sulfhydryl group at Ala214 to the model shows that the distance from the scissile carbon is consistent with an inline attack of the carbon atom of the carbonyl group as displayed.
The overall interface between the substrate and the protein is 435 Å2, and the total binding energy is −11.7 kcal mol−1 (according to the PISA server http://www.ebi.ac.uk/pdbe/prot_int/pistart.html). P1 and P2′ residues constitute the main anchors to maintain the substrate with the largest buried surface areas (112 Å2 and 128 Å2, respectively). One face of the S1 pocket is lined with negatively charged residues Asp263 and Glu212, while positively charged residues are found on the other side: Arg69 and His70. At neutral pH, this asymmetric charge distribution dictates tight binding of Asn by providing complementary H-bonds to both its oxygen and nitrogen sidechain atoms. In the S2′ pocket, Tyr185 and Lys177 establish van der Waals interactions with the P2′ residue. The floor of this pocket is formed by the backbone atoms of Ile178 and Gly179. This is consistent with the previous specificity studies, suggesting a higher affinity for hydrophobic P2′ residues (Hemu et al., 2019). Finally, P1′, P2, and P3 residues have smaller buried surface areas of 72 Å2, 83 Å2, and 72 Å2, respectively, accounting for a relaxed specificity of the substrate at these positions.
In summary, the present structure represents a possible model for the complex between active enzyme and substrate before acyl-enzyme formation. As a caveat, it remains to be seen if the same substrate reorientation will be observed with respect to the CMK-inhibited AtLEGγ complex, when the WT VyPAL2 active site (e.g. not the VyPAL2-C214A inactive mutant) is intact and when the true acyl-enzyme intermediate forms. The ligation reaction catalyzed by VyPAL2 can be divided into two steps: first, a nucleophilic attack of the catalytic cysteine on the carbonyl of the P1 residue leads to acyl-enzyme formation, breaking the peptide bond between the P1 and P1′ residues. Second, the incoming N-terminal amine performs a nucleophilic attack on the acyl-enzyme intermediate releasing the product from the catalytic cysteine. When we replaced Ala214 with a cysteine residue in silico, we found that the P1–P1′ scissile amide bond lies between the Asp49 and the Ser50 residues and that the Asp49 carbonyl moiety is located at 2.5 Å from the Sγ atom of the catalytic Cys214 (Figure 5C). This distance is consistent with the distance (2.5 Å) separating the active site Cys Sγ and the inhibitor carbonyl carbon in the AtLEGγ complex structure (Dall et al., 2020), as well as the results of density functional theory-based quantum mechanics/molecular mechanics studies on legumains prior to nucleophilic attack (2.9 Å) (Elsässer et al., 2017). This distance is thus compatible with a conformation of the enzyme adopted before nucleophilic attack and leading to acyl-enzyme intermediate formation, further suggesting that the present structure represents an enzyme–substrate complex.
The “Gatekeeper” residue induces a shift in the substrate position at the nonprime site
It was previously observed that mutation of a Cys residue (Cys247) near the active site of OaAEP1b (PDB access code: 5H0I) to larger amino acids (Thr, Met, Val, Leu, and Ile), reduced the catalytic efficiency of ligation, while mutations to smaller residues such as Ala resulted in an over 100-fold improved ligation efficiency. Moreover, mutation of this “Gatekeeper” residue to Gly resulted in an increased amount of hydrolysis product, suggesting that residues at this site play a major role in modulating enzyme function (Yang et al., 2017). Interestingly, molecular dynamics (MD) simulation suggested that a shift in the position of the N-terminal (nonprime) portion of the substrate occurs due to steric hindrance introduced by a Gatekeeper residue bulging at the surface of the S2 substrate-binding pocket (Hemu et al., 2019). The experimental structure of the ligase–peptide complex reported here confirms and extends this hypothesis: while there is excellent substrate overlap at the P1 position between VyPAL2-C214A form I (this work) and AtLEGγ covalently bound to Ac-YVAD-cmk (PDB access code: 5OBT), a repositioning of residues P2–P3 of the substrate occurs in the presence of a protruding sidechain at the Gatekeeper position (Figure 6; Supplemental Figure S5): in the presence of Ile244 as the Gatekeeper, P2–P3 residues are pushed away by ∼4 Å from the surface of the ligase active site compared to the position adopted when the Gatekeeper is a Gly, allowing the peptide substrate to come closer to the enzyme surface. As a result, a hydrogen bond forms between the backbone carbonyl oxygen of the substrate P2 residue (Asp48) and the imidazole ring of catalytic His172 (Figure 6A). The formation of this hydrogen bond is possible in the orientation of the histidine imidazole ring adopted in the experimental structure, with the Nε atom oriented toward the substrate. Conversely, reorientation of the histidine sidechain toward the catalytic cysteine, as seen in AEP structures, enables the activation of an attacking water molecule (Figure 6B; Supplemental Figure S5).
Figure 6.

Active site residues and the role of the Gatekeeper position in substrate positioning. A, Structure of the enzyme–substrate complex simulated based on the experimental VyPAL2-C214A structure (Supplemental Table S2, form I). Ala214 was replaced with a cysteine. The enzyme core is shown as ribbons and colored in slate. The catalytic Cys214 and His172 as well as the Ile244 Gatekeeper residue are shown as sticks. Residues Asn67 and Arg69 putatively involved in the auto-activation mechanism are shown as sticks with distances from the His172 imidazole ring indicated (see text). The hydrogens of His172 are also shown. The P4–P2′ (NDDDSI) substrate is shown as sticks, colored in magenta, and the 2Fo–Fc electronic density map contoured at 1σ is colored in light blue. B, Structure of the enzyme–inhibitor complex of AtLEGg (PDB code: 5OBT). The enzyme core is shown as ribbons and colored in light gray. The catalytic residues Cys219 and His172 as well as the Gly249 Gatekeeper residue are shown as sticks. The YVAD-CMK covalent inhibitor is shown as sticks, with residues labeled P4–P1. The proposed nucleophilic water molecule (shown as a red sphere) is located at a distance of 2.9 Å from His172 in the axis of the thioester bond between the Cys219 and P1 Asp–CMK residues. For reference, the position of the VyPAL2-C214A substrate superimposed onto the AtLEGγ bound structure is represented as lines and colored in magenta.
In summary, the presence of a Gly residue at the Gatekeeper position is conducive to the positioning of the catalytic His such that it can activate an incoming water molecule for hydrolysis, while the presence of a bulkier residue such as Ile leads to a reorientation of the same histidine, which is detrimental to hydrolysis and thus favors ligation.
A histidine residue of VyPAL2 is required for auto-activation
PALs and AEPs are naturally expressed as proenzymes that require activation (Supplemental Movie S1). The activation steps consist of a series of sequential hydrolysis events targeting an N-terminal pro-peptide of ∼50 residues and the C-terminal cap domain (∼150 residues). We previously identified several activation sites present in VyPAL2 (Hemu et al., 2019). Auto-activation of AEPs and PALs represents a proteolysis reaction regardless of the main activity (protease or ligase) of the mature enzyme. It is commonly thought that the auto-activation mechanism involves the nucleophilic attack of the catalytic cysteine onto the peptide carbonyl at Asn and Asp residues located in the N-terminal and linker regions of the proenzyme. In light of this concept, the ability of VyPAL2-C214A to efficiently auto-activate at acidic pH was rather surprising, as it lacks the catalytic cysteine able to perform the nucleophilic attack. To further explore the auto-activation mechanism, we generated a single mutant of VyPAL2 in which its catalytic histidine was mutated to alanine. The VyPAL2-H172A single mutant was unable to auto-activate (Figure 7B), as no reduction in the amount of the proenzyme occurred during the assay. This result suggests that Cys214 is not involved in nucleophilic attack and points to an alternative route for VyPAL2 auto-activation, where His172 is the catalytic residue required for proenzyme activation.
Figure 7.

His172 is the catalytic residue necessary for proenzyme VyPAL2 activation. A, Schematic sequence representations of VyPAL2-H172A and VyPAL2-N67A/C214A. B, The products from low pH processing of the VyPAL2-H172A and VyPAL2-N67A/C214A proenzymes were analyzed using SDS–PAGE with Coomassie blue staining. Incubation of VyPAL2-H172A for up to 2 h did not lead to any processed enzyme. C, Proposed mechanism of the processing of VyPAL2 that is Cys214 independent and where His172 is the main catalytic residue. This mechanism involves the formation of an acyl-imidazole intermediate, which is highly unstable and rapidly resolved by the attack of a nucleophilic water molecule.
Inspection of the active site revealed that the distance between the guanidinium group of Arg69 and the Nε group of the His172 imidazole ring is 3.6 Å (Figure 6A). Thus, the Arg69 sidechain is well positioned to attract electrons from the imidazole ring of His172, lowering its pKa compared to the value of 6.6 ± 1 it has as a free amino acid (Grimsley et al., 2009). In this scheme, Asn67 assists His172 by acting as a proton acceptor. The Asn67 residue, the third member of the cysteine protease catalytic triad, is thought to orient the catalytic histidine. The imidazole nitrogen of His172 acts as a nucleophile to attack and cleave the P1–P1′ scissile bond, forming an acyl-imidazole intermediate. As protons are continuously taken up and given back to the surrounding solvent near the catalytic site, once formed, the acyl-imidazole intermediate could be quickly hydrolyzed to complete the activation process (Figure 7C). In this mechanism, Asn67 would play an essential role by accepting the proton from His172 and thus helping to activate it for subsequent nucleophilic attack. To test this hypothesis, we generated a VyPAL2-C214A/N67A double mutant (the VyPAL2-N67A single mutant precipitates upon activation). While VyPAL2-C214A could be easily activated, under the same activation conditions, VyPAL2-C214A/N67A lost most of its activation ability (Figure 7B), supporting the proposed activation mechanism.
Discussion
Proteases and ligases share high amino-acid sequence and structural identity. PALs could have divergently evolved from AEPs to catalyze transpeptidation or ligation reactions required to produce cyclic peptides in plants as a mechanism for defense against pathogens. At the molecular level, the nature of this evolution was recently clarified with the characterization of the Gatekeeper residue and LADs concepts (Yang et al., 2017; Hemu et al., 2019, 2020). In this concept, only a few specific positions in the substrate-binding site are sufficient to convert an AEP into a PAL. Here, we uncovered a possible molecular mechanism accounting for the primary role of the Gatekeeper residue in determining the direction of the enzyme activity.
Ligation or hydrolysis reactions appear to proceed through two shared initial steps: (1) substrate binding and nucleophilic attack by the Cys sulfhydryl moiety on the P1 carbonyl (Figure 1A) leads to the formation of an acyl-enzyme intermediate and cleavage of the P1–P1′ peptide bond and (2) nucleophilic attack onto this acyl-enzyme intermediate breaks the transient thioester bond formed between the catalytic cysteine and the P1 Asx residue. The main difference between ligation and hydrolysis derives from the selection of the incoming nucleophilic group to resolve the acyl-enzyme intermediate: either a water molecule for proteolysis or an amine from an incoming peptide for ligation. This proposed overall scheme concurs with the elegant hydrogen–deuterium exchange measurements that demonstrated the absence of an isotopic shift in a kalata B1 cyclotide peptide produced by a peptide cyclization reaction, hence ruling out the role of a water molecule in the ligation reaction carried out by OaAEP1b (Harris et al., 2015) and favoring the scheme where direct aminolysis and transpeptidation are performed by an incoming amine (Hemu et al., 2019).
PALs and AEPs share almost identical structures; subtle differences at their LAD1 and LAD2 regions play a key role in determining the direction of the reaction (Hemu et al., 2019). The LAD2 region, located at the primed side of the peptide-binding site, alters the enzyme’s preference: nonhydrophobic residues at LAD2 favor the presence of water molecules, while bulky residues tend to exclude an incoming peptide. For instance, the Y168A mutation targeting LAD2 was sufficient to endow the VyPAL3 and VcAEP proteases with significant peptide cyclase activity. Conversely, a small exposed hydrophobic residue such as alanine at this position will favor ligation, as its hydrophobicity would decrease the local affinity for water molecules from the solvent, while its small size can easily accommodate an incoming peptide (Figure 5; Supplemental Figure S6). Here, as seen in form I complex, VyPAL2 possesses Ala174–Pro175–Gly176 at the LAD2 region, which maintain the local secondary structure of the protein (a β-turn) and have the required size and hydrophobicity to bind Ile51 as P2′ of an incoming peptide.
Another key determinant of activity lies in the Gatekeeper residue centered at LAD1 on the nonprime side of the binding site. In the VyPAL2-C214A form I complex, the Gatekeeper (Ile244) and the catalytic His172 are located at opposite sides of the peptide-binding groove (Supplemental Movie S2). The aliphatic sidechain of Ile244 lies at a van der Waals contact distance of 3.3 Å from the α-carbon residue P2 (Asp48) of the peptide ligand (Figures 4 and 5). A comparison with enzymes having a Gly residue as the Gatekeeper, such as AtLEGγ, covalently bound to Ac-YVAD-cmk, reveals that the corresponding P2–P3 residues are displaced by ∼4 Å from the ligase active site (Figure 6; Supplemental Figure S5). As a result, the displacement induced by an aliphatic sidechain as the Gatekeeper allows a hydrogen bond to form between the carbonyl oxygen of the substrate P2 residue (Asp48) and the imidazole ring of catalytic His172 (Figure 6). In contrast, when the Gatekeeper is a glycine, the corresponding distance is too far to establish a hydrogen bond (Figure 6B). Accordingly, this hydrogen bond is not observed in other active form protease structures. Thus, the present orientation of the imidazole ring of His172, constrained by this polar interaction with the peptide, seems incompatible with a role in activating an incoming water molecule for nucleophilic attack and appears to be a unique feature of a ligase. Conversely, in AEPs, the catalytic histidine can act as a base to assist in the activation of the water molecule that is positioned above Gly173 in VyPAL2 (corresponding to Gly178 in the case of AtLEGγ) for the nucleophilic attack of the thioester bond. We propose that this subtle change in the peptide substrate conformation induced by the sidechain of the Gatekeeper explains why PALs favor a nucleophilic attack by the amine group of an incoming peptide, and why at a more acidic pH, the ligation reaction becomes less favored due to protonation of the incoming amine group.
At neutral pH values, the cap domain strongly associates with the core domain in the zymogen form due to electrostatic interactions between the two domains, preventing auto-proteolytic activation. Conversely, at acidic pH, the interaction between both domains is loosened, exposing the catalytic surface of the core domain, which can then cleave (in trans) peptide bonds located in the linker region (Zhao et al., 2014) (Supplemental Movie S1). Our results show that auto-activation can be performed without the presence of the catalytic Cys, while the catalytic His is necessary for this process (Figures 3, A and 7, A), suggesting that auto-activation and peptide hydrolysis/ligation use different molecular mechanisms, although they share the same active site. In this respect, it should be noted that this auto-activation activity occurs at a much slower rate compared to the catalytic activity of the mature enzyme (compare Figure 1, B and C, for instance). Based on our results, we propose a mechanism for the auto-activation of VyPAL2-C214A where at acidic pH, the imidazole of the catalytic histidine residue acts as both an acid and a base.
For a folded protein, the local chemical environment surrounding a His can drastically affect the pKa value of its sidechain imidazole group, which can adopt values ranging from 2.4 to 9.2, as surveyed by Grimsley et al. (2009). It is thus possible that, in the presence of the positively charged Arg69 and the binding of an incoming peptide substrate to the active site, the pKa of the His172 side chain becomes much lower than its usual pKa as a free amino acid. In this case, the imidazole group will remain in its neutral (unprotonated) form and can thus act as a nucleophile. Upon binding of one of the activation motifs present in the proenzyme, Nδ of His172 in the VyPAL2-C214A mutant would serve as a nucleophile to attack the carbonyl carbon of the scissile peptide bond, forming an acyl-imidazole intermediate. The amide hydrogens of Gly173 and Ala214 form hydrogen bonds with the carbonyl oxygen of the scissile peptide bond. The same hydrogen bonds can still form with the oxyanion for its stabilization. This is crucial for the formation of the acyl-enzyme intermediate, since the first step of this mechanism has the highest activation energy and hence constitutes the rate-determining step.
The acyl-imidazole intermediate is very unstable and undergoes rapid hydrolysis, leading to the release of the core and cap domains. In turn, we propose that the role of Asn67 is to weaken the H–Nε bond of His172 such that the proton can be more easily dissipated to bulk solvent, which is necessary in order for His172 imidazole to serve as a nucleophile to attack an Asx–peptide bond in the proenzyme during the auto-activation process. Although Asn residues are not typically considered to be ionizable or act as a general base, it is possible that proximity would account for the weak catalytic activity observed. In the structure, the oxygen atom of the Asn67 sidechain amide is perfectly positioned to form a strong hydrogen bond with H–Nε of the His172 sidechain imidazole, facilitating the abstraction of this proton. Without this proton transfer system (as in the case of the Asn67Ala mutant), the peptide bond cleavage ability of His172 will be greatly reduced, which is fully consistent with the experimental data showing that VyPAL2-N67A/C214A cannot be auto-activated (Figure 7B). Finally, we note that this proposed activation mechanism provides an elegant answer to the conundrum faced by “pure” asparaginyl ligases, which must retain a level of hydrolysis activity required for their activation.
In summary, we found that the N-terminus of VyPAL2-C214A containing the Asp49–Ser50–Ile51 sequence constitutes a P1P1′P2′ substrate proteolytic motif, allowing us to perform a detailed structural analysis of the active site pockets of the enzyme that are used for both protease and ligase activities. In this respect, peptide asparaginyl ligases (PALs) appear to be opportunistic catalysts that have evolved from asparaginyl endoproteases by recycling essentially the same binding site, with the introduction of only a few key residues near the conserved S1 pocket. A slight distortion in the conformation of the substrate peptide appears to be sufficient to endow the same catalytic site with a novel activity. This dual usage of the specificity pockets is also reflected in the way we can interpret the present form I crystal structure: the complex can be seen as either a pro-enzyme–substrate complex or a ligase–product complex. In the former, the proenzyme is caught in the act of cleaving in trans the N-terminal tail of a companion molecule for maturation, whilst in the latter, the ligase has just performed a protein–peptide ligation reaction using Asp as the P1 residue.
Materials and methods
Protein expression and auto-activation
The expression and purification of VyPAL2 and VyPAL2 mutant proteins from V. yedoensis (Ile244Val, Cys214Ala, His172Ala, Asn67Ala, and Cys214Ala) were carried out as previously described (Hemu et al., 2019). A list of primers used to generate the mutations is given in Supplemental Table S4. Briefly after mutagenesis, the genes encoding VyPAL and its mutants were expressed in Sf9 insect cells using the Bac-to-Bac protocol (Invitrogen, Waltham, MA, USA). Protein purification was performed in three steps, with IMAC affinity purification followed by ion exchange and SEC (with 1× PBS, pH 7.4, 1-mM DTT). The protein was then concentrated and stored at 4°C.
Following gel filtration, proenzymes or their mutants were concentrated to 2 mg mL−1. The optimal activation time was estimated following a time-course analysis. Zymogens were activated at 37°C in 50-mM citrate, 100-mM NaCl in a 1:1 volume ratio (v/v), with the addition of 0.5-mM N-Laurocryosine, 1-mM DTT, final pH 4.2. The samples were checked using SDS–PAGE to determine the optimal activation time. After scaled-up proenzyme activation, the activation mixture was subjected to SEC purification with buffer containing 20-mM MES, pH 6.5, 0.1-M NaCl, 1-mM DTT. Fractions containing the active form protein were pooled and concentrated for further use.
Crystallization, data collection, and structure determination
The active enzymes were concentrated to 4–9 mg mL−1 and screened against the JCSG-plus HT-96 (Molecular Dimensions) screening kit at a protein: mother liquid volume ratio of 1:1, 1:2, and 2:1 (v/v). Crystals were obtained for the active form of VyPAL2 (6.7 mg mL−1): 0.1-M sodium acetate, pH 4, 0.2-M lithium sulfate, 30% (w/v) PEG 8000; VyPAL2-I244V (10.4 mg mL−1): 0.1-m Bis–Tris pH = 7.5, 25% (w/v) PEG 3350; VyPAL2-C214A (Form I, 4.93 mg mL−1): 0.2-M Lithium sulfate, 0.1-M sodium acetate, 30% (w/v) PEG 8000 pH = 4.6; and VyPAL2-C214A (Form II, 9.14 mg mL−1): 0.2-M potassium formate, 20% (w/v) PEG 3350. All crystals were obtained at a protein:mother liquid volume ratio of 1:1. Crystals suitable for X-ray data collection were frozen in liquid nitrogen, placed in 20% (v/v) glycerol, and shipped to synchrotrons for data collection. Data were integrated using XDS (Kabsch, 2010) and the structures were solved using 6IDV as a search probe in Phaser (CCP4) (Winn et al., 2011). Manual corrections of the structures were performed using the Coot program for molecular graphics (Emsley et al., 2010) and refined using Buster TNT (GlobalPhasing Ltd.) (Smart et al., 2012). Processing, refinement statistics, and PDB access codes are presented in Supplemental Table S2.
Kinetic studies
Substrate peptides were synthesized by Bio Basic and consisted of the N-terminal substrate PIE(EDANS)YNAL and the C-terminal substrate GIK(DABSYL)SIP. Ligation assays were performed in a 100-µL reaction mixture containing 60 µL of MES buffer (20-mM MES, pH 6.5, 0.1-M NaCl, 1-mM DTT) and 20 µL of serial dilutions (in MES buffer) of a substrate mixture containing both N- and C-terminal substrates at a ratio (EDANS: DABSYL) of 1:3. The final concentrations of EDANS substrate were 50, 25, 12.5, 6.25, 3.125, 1.5625, and 0.78125 µM. The enzyme was added immediately before injection into the injector of a Tecan 10M Spark microplate reader; 20 µL of the enzyme at 100 nM was added to the reaction to give a final concentration of 20 nM. The reactions were run at 37°C for 2 min and measured at 5-s intervals to calculate the initial rates during the linear portion of the progress curve. All samples were added to a Thermo Fisher Scientific-Nunclon 96 Flat Black plate, and fluorescence was measured using a Tecan 10M Spark microplate reader with an excitation wavelength of 336 nm and an emission wavelength of 490 nm.
Statistical analysis
In Figure 2C, measurements for each concentration were performed as triplicate. The initial velocity of the reaction was determined using the Magellan software provided by the manufacturer. The statistical analysis for the X-ray crystallographic studies presented in this study is summarized in Supplemental Table S2.
Accession numbers
The Genbank accession number for the amino-acid sequence of PAL 2 (VyPAL2) belonging to enzyme family EC 3.4.22.34, is QCW05335.1. The coordinates and structure factors of the following crystals structures have been deposited in the PDB at www.rcsb.org with the following accession numbers: VyPAL2 active form, 7F5Q; VyPAL2-I244V, 7F5J; VyPAL2-C214A form I, 7F5P; VyPAL2-C214A form II, 7FA0. Please also refer to Supplemental Table S2. The plasmids to express VyPAL2, VyPAL2-I244V, and VyPAL2-C214A are available from the authors.
Supplemental data
The following materials are available in the online version of this article.
Supplemental Figure S1. SEC profiles of WT VyPAL2 and I244V.
Supplemental Figure S2. Comparison of the active structures of VyPAL2 and its mutants.
Supplemental Figure S3. Comparison of the active structures of VyPAL2 and the VyPAL2 proenzyme structure.
Supplemental Figure S4. Comparison of the activation kinetics between VyPAL2 and VyPAL2-C214A.
Supplemental Figure S5. Effects of the LAD1 “Gatekeeper” residue on the molecular surface of VyPAL2.
Supplemental Figure S6. Effects of the presence of a large bulky residue in the LAD2 region.
Supplemental Table S1. List of abbreviations.
Supplemental Table S2. Data collection and refinement statistics.
Supplemental Table S3. R.M.S.D. values of VyPAL2 with other plant AEPs (PDBeFold).
Supplemental Table S4. Mutations and primers.
Supplemental Movie S1. Transactivation of the VyPAL2 proenzyme by a VyPAL2 active enzyme.
Supplemental Movie S2. A hypothesis for the role of the Gatekeeper residue.
Supplementary Material
Acknowledgments
We thank scientists and beamline staff at the following locations for their expert assistance: (1) Spring-8 Synchrotron where the VyPAL2 active enzyme structure was collected, (2) Swiss Light Source (SLS) where VyPAL2-C214A form II was collected, and (3) MX2 (Australian Synchrotron) where VyPAL2-C214A form I and VyPAL2-I244V datasets were collected.
Funding
This research was supported by Academic Research Grant Tier 3 (MOE2016-T3-1-003) from Singapore Ministry of Education (MOE) to the J.L., J.P.T., and C.F.L. laboratories.
Conflict of interest statement. None declared.
Contributor Information
Side Hu, School of Biological Sciences, Nanyang Technological University, Singapore City, 637551, Singapore; NTU Institute of Structural Biology, Nanyang Technological University, Singapore City, 636921, Singapore.
Abbas El Sahili, School of Biological Sciences, Nanyang Technological University, Singapore City, 637551, Singapore; NTU Institute of Structural Biology, Nanyang Technological University, Singapore City, 636921, Singapore.
Srujana Kishore, School of Biological Sciences, Nanyang Technological University, Singapore City, 637551, Singapore; NTU Institute of Structural Biology, Nanyang Technological University, Singapore City, 636921, Singapore.
Yee Hwa Wong, School of Biological Sciences, Nanyang Technological University, Singapore City, 637551, Singapore; NTU Institute of Structural Biology, Nanyang Technological University, Singapore City, 636921, Singapore.
Xinya Hemu, School of Biological Sciences, Nanyang Technological University, Singapore City, 637551, Singapore.
Boon Chong Goh, NTU Institute of Structural Biology, Nanyang Technological University, Singapore City, 636921, Singapore; Antimicrobial Resistance Interdisciplinary Research Group, Singapore-MIT Alliance for Research and Technology Centre, Singapore City, 138602, Singapore.
Sang Zhipei, School of Biological Sciences, Nanyang Technological University, Singapore City, 637551, Singapore.
Zhen Wang, School of Biological Sciences, Nanyang Technological University, Singapore City, 637551, Singapore.
James P Tam, School of Biological Sciences, Nanyang Technological University, Singapore City, 637551, Singapore.
Chuan-Fa Liu, School of Biological Sciences, Nanyang Technological University, Singapore City, 637551, Singapore.
Julien Lescar, School of Biological Sciences, Nanyang Technological University, Singapore City, 637551, Singapore; NTU Institute of Structural Biology, Nanyang Technological University, Singapore City, 636921, Singapore.
A.E.S. and J.L. designed the research. S.H., A.E.S., X.H., and B.C.G. performed the research. S.K., Y.H.W., Z.S., and Z.W. contributed reagents. S.H., A.E.S., J.P.T., C.F.L., and J.L. analyzed the data. A.E.S., C.F.L., and J.L. wrote the paper.
The authors responsible for distribution of materials integral to the findings presented in this article in accordance with the policy described in the Instructions for Authors (https://academic.oup.com/plcell) are: Julien Lescar (julien@ntu.edu.sg) and Chuan Fa Liu (cfliu@ntu.edu.sg).
References
- Antos JM, Popp MWL, Ernst R, Chew GL, Spooner E, Ploegh HL (2009) A straight path to circular proteins. J Biol Chem 284: 16028–16036 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bagert JD, Muir TW (2021) Molecular epigenetics: chemical biology tools come of age. Ann Rev Biochem 90: 287–320 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bernath-Levin K, Nelson C, Elliott AG, Jayasena AS, Millar AH, Craik DJ, Mylne JS (2015) Peptide macrocyclization by a bifunctional endoprotease. Chem Biol 22: 571–582 [DOI] [PubMed] [Google Scholar]
- Bi X, Yin J, Nguyen GKT, Rao C, Halim NBA, Hemu X, Tam JP, Liu CF (2017) Enzymatic engineering of live bacterial cell surfaces using butelase 1. Angew Chem Int Ed 56: 7822–7825 [DOI] [PubMed] [Google Scholar]
- Cao Y, Nguyen GKT, Chuah S, Tam JP, Liu CF (2016) Butelase-mediated ligation as an efficient bioconjugation method for the synthesis of peptide dendrimers. Bioconjugate Chem 27: 2592–2596 [DOI] [PubMed] [Google Scholar]
- Cao Y, Nguyen GKT, Tam JP, Liu CF (2015) Butelase-mediated synthesis of protein thioesters and its application for tandem chemoenzymatic ligation. Chem Commun 51: 17289–17292 [DOI] [PubMed] [Google Scholar]
- Dall E, Brandstetter H (2013) Mechanistic and structural studies on legumain explain its zymogenicity, distinct activation pathways, and regulation. Proc Natl Acad Sci USA 110: 10940–10945 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dall E, Stanojlovic V, Demir F, Briza P, Dahms SO, Huesgen PF, Cabrele C, Brandstetter H (2021) The peptide ligase activity of human legumain depends on fold stabilization and balanced substrate affinities. ACS Catalysis 11: 11885–11896 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dall E, Zauner FB, Soh WT, Demir F, Dahms SO, Cabrele C, Huesgen PF, Brandstetter H (2020) Structural and functional studies of Arabidopsis thaliana legumain beta reveal isoform specific mechanisms of activation and substrate recognition. J Biol Chem 295: 13047–13064 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elsässer B, Zauner FB, Messner J, Soh WT, Dall E, Brandstetter H (2017) Distinct roles of catalytic cysteine and histidine in the protease and ligase mechanisms of human legumain as revealed by DFT-based QM/MM simulations. ACS Catal 7: 5585–5593 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Emsley P, Lohkamp B, Scott WG, Cowtan K (2010) Features and development of coot. Acta Crystallogr D Biol Crystallogr 66: 486–501 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grimsley GR, Scholtz JM, Pace CN (2009) A summary of the measured pK values of the ionizable groups in folded proteins. Protein Sci 18: 247–251 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harris KS, Guarino RF, Dissanayake RS, Quimbar P, McCorkelle OC, Poon S, Kaas Q, Durek T, Gilding EK, Jackson MA, Craik DJ, et al. (2019) A suite of kinetically superior AEP ligases can cyclise an intrinsically disordered protein. Sci Rep 9: 10820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harris KS, Durek T, Kaas Q, Poth AG, Gilding EK, Conlan BF, Saska I, Daly NL, Van Der Weerden NL, Craik DJ, Anderson MA (2015) Efficient backbone cyclization of linear peptides by a recombinant asparaginyl endopeptidase. Nat Commun 6: 1–10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haywood J, Schmidberger JW, James AM, Nonis SG, Sukhoverkov KV, Elias M, Bond CS, Mylne JS (2018) Structural basis of ribosomal peptide macrocyclization in plants. Elife 7: 1–22 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hemu X, El Sahili A, Hu S, Wong K, Chen Y, Wong YH, Zhang X, Serra A, Goh BC, Darwis DA, et al. (2019) Structural determinants for peptide-bond formation by asparaginyl ligases. Proc Natl Acad Sci USA 116: 11737–11746 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hemu X, el Sahili A, Hu S, Zhang X, Serra A, Goh BC, Darwis DA, Chen MW, Sze SK, Liu CF, Lescar J, Tam JP (2020) Turning an asparaginyl endopeptidase into a peptide ligase. ACS Catal 10: 8825–8834 [Google Scholar]
- Jackson MA, Gilding EK, Harris KS, Kaas Q, Poon S, Shafee T, Yap K, Jia H, Guarino R, Chan LY, et al. (2018) Molecular basis for the production of cyclic peptides by plant asparaginyl endopeptidases. Nat Commun 9: 1–12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- James AM, Haywood J, Leroux J, Ignasiak K, Elliott AG, Schmidberger JW, Fisher MF, Nonis SG, Fenske R, Bond CS, et al. (2019) The macrocyclizing protease butelase 1 remains autocatalytic and reveals the structural basis for ligase activity. Plant J 98: 988–999 [DOI] [PubMed] [Google Scholar]
- Kabsch W (2010) XDS. Acta Crystallogr D Biol Crystallogr 66: 125–132 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kembhavi AA, Buttle DJ, Knight CG, Barrett AJ (1993) Two cysteine endoproteases of legume seeds - purification and characterisation by use of specific fluorometric assays. Arch Biochem Biophys 303: 208–213 [DOI] [PubMed] [Google Scholar]
- Kuroyanagi M, Nishimura M, Hara-Nishimura I (2002) Activation of Arabidopsis Vacuolar processing enzyme by self-catalytic removal of an auto-inhibitory domain of the c-terminal propeptide. Plant Cell Physiol 43: 143–151 [DOI] [PubMed] [Google Scholar]
- Mao H, Hart SA, Schink A, Pollok BA (2004) Sortase-mediated protein ligation: a new method for protein engineering. J Am Chem Soc 126: 2670–2671 [DOI] [PubMed] [Google Scholar]
- Mulvenna JP, Mylne JS, Bharathi R, Burton RA, Shirley NJ, Fincher GB, Anderson MA, Craik DJ (2006) Discovery of cyclotide-like protein sequences in graminaceous crop plants: Ancestral precursors of circular proteins? Plant Cell 18: 2134–2144 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mylne JS, Chan LY, Chanson AH, Daly NL, Schaefer H, Bailey TL, Nguyencong P, Cascales L, Craik DJ (2012) Cyclic peptides arising by evolutionary parallelism via asparaginyl-endopeptidase-mediated biosynthesis. Plant Cell 24: 2765–2778 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nguyen GKT, Cao Y, Wang W, Liu CF, Tam JP (2015a) Site-specific N-terminal labeling of peptides and proteins using butelase 1 and thiodepsipeptide. Angew Chem Int Ed 54: 15694–15698 [DOI] [PubMed] [Google Scholar]
- Nguyen GKT, Hemu X, Quek JP, Tam JP (2016a) Butelase-mediated macrocyclization of d-amino-acid-containing peptides. Angew Chem Int Ed 55: 12802–12806 [DOI] [PubMed] [Google Scholar]
- Nguyen GKT, Kam A, Loo S, Jansson AE, Pan LX, Tam JP (2015b) Butelase 1: a versatile ligase for peptide and protein macrocyclization. J Am Chem Soc 137: 15398–15401 [DOI] [PubMed] [Google Scholar]
- Nguyen GKT, Qiu Y, Cao Y, Hemu X, Liu CF, Tam JP (2016b) Butelase-mediated cyclization and ligation of peptides and proteins. Nat Protocol 11: 1977–1988 [DOI] [PubMed] [Google Scholar]
- Nguyen GKT, Wang S, Qiu Y, Hemu X, Lian Y, Tam JP (2014) Butelase 1 is an Asx-specific ligase enabling peptide macrocyclization and synthesis. Nat Chem Biol 10: 732–738 [DOI] [PubMed] [Google Scholar]
- Noike M, Matsui T, Ooya K, Sasaki I, Ohtaki S, Hamano Y, Maruyama C, Ishikawa J, Satoh Y, Ito H, et al. (2015) A peptide ligase and the ribosome cooperate to synthesize the peptide pheganomycin. Nat Chem Biol 11: 71–76 [DOI] [PubMed] [Google Scholar]
- Schechter I, Berger A (1967) On the size of the active site in proteases. I. Papain. Biochem Biophys Res Commun 27: 157–162 [DOI] [PubMed] [Google Scholar]
- Schneewind O, Model P, Fischetti VA (1992) Sorting of protein a to the staphylococcal cell wall. Cell 70: 267–281 [DOI] [PubMed] [Google Scholar]
- Serra A, Hemu X, Nguyen GKT, Nguyen NTK, Sze SK, Tam JP (2016) A high-throughput peptidomic strategy to decipher the molecular diversity of cyclic cysteine-rich peptides. Sci Rep 6: 23005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smart OS, Womack TO, Flensburg C, Keller P, Paciorek W, Sharff A, Vonrhein C, Bricogne G (2012) Exploiting structure similarity in refinement: automated NCS and target-structure restraints in BUSTER. Acta Crystallogr D Biol Crystallogr 68: 368–380 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Winn MD, Ballard CC, Cowtan KD, Dodson EJ, Emsley P, Evans PR, Keegan RM, Krissinel EB, Leslie AGW, McCoy A, et al. (2011) Overview of the CCP4 suite and current developments. Acta Crystallogr D Biol Crystallogr 67: 235–242 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang R, Wong YH, Nguyen GKT, Tam JP, Lescar J, Wu B (2017) Engineering a catalytically efficient recombinant protein ligase. J Am Chem Soc 139: 5351–5358 [DOI] [PubMed] [Google Scholar]
- Zauner FB, Dall E, Regl C, Grassi L, Huber CG, Cabrele C, Brandstetter H (2018a) Crystal structure of plant legumain reveals a unique two-chain state with pH-dependent activity regulation. Plant Cell 30: 686–699 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zauner FB, Elsässer B, Dall E, Cabrele C, Brandstetter H (2018b) Structural analyses of Arabidopsis thaliana legumain reveal differential recognition and processing of proteolysis and ligation substrates. J Biol Chem 293: 8934–8946 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang D, Wang Z, Hu S, Balamkundu S, To J, Zhang X, Lescar J, Tam JP, Liu CF (2021) PH-controlled protein orthogonal ligation using asparaginyl peptide ligases. J Am Chem Soc 143: 8704–8712 [DOI] [PubMed] [Google Scholar]
- Zhao L, Hua T, Crowley C, Ru H, Ni X, Shaw N, Jiao L, Ding W, Qu L, Hung LW, et al. (2014) Structural analysis of asparaginyl endopeptidase reveals the activation mechanism and a reversible intermediate maturation stage. Cell Res 24: 344–358 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.