

Abstract
NOTCH1 is a well-established lineage specifier for T cells and among the most frequently mutated genes throughout all subclasses of T cell acute lymphoblastic leukemia (T-ALL). How oncogenic NOTCH1 signaling launches a leukemia-prone chromatin landscape during T-ALL initiation is unknown. Here we demonstrate an essential role for the high-mobility-group transcription factor Tcf1 in orchestrating chromatin accessibility and topology, allowing aberrant Notch1 signaling to convey its oncogenic function. Although essential, Tcf1 is not sufficient to initiate leukemia. The formation of a leukemia-prone epigenetic landscape at the distal Notch1-regulated Myc enhancer, which is fundamental to this disease, is Tcf1-dependent and occurs within the earliest progenitor stage even before cells adopt a T lymphocyte or leukemic fate. Moreover, we discovered a unique evolutionarily conserved Tcf1-regulated enhancer element in the distal Myc-enhancer, which is important for the transition of preleukemic cells to full-blown disease.
Key Points
-
•
Tcf1 shapes a Notch1-induced T-ALL-prone chromatin landscape in early hematopoietic progenitors.
-
•
Tcf1 orchestrates chromatin accessibility within the Notch1-regulated Myc super-enhancer.
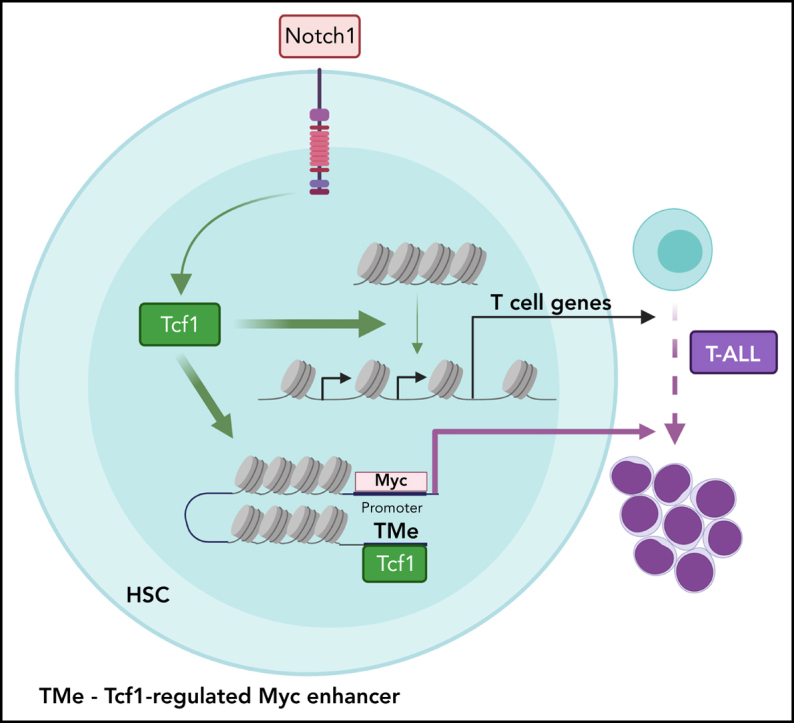
Introduction
Signaling events governing cell identity, differentiation, and proliferation converge on spatial folding of chromatin, eliciting programs essential for all cellular functions.1 Thus, the adoption of the appropriate genome architecture is imperative for development and tissue homeostasis. Dysregulation of chromatin topology, as a consequence of genomic alterations or mutations affecting chromatin regulatory proteins, has functionally been linked to cancer.2, 3, 4, 5 Nevertheless, how lineage-specifying transcription factors establish a cancer-permissive chromatin landscape is not well understood. The Notch signaling pathway regulates many cell fate decisions during development and homeostasis and is dysregulated in cancer.6, 7, 8 In the hematopoietic system, Notch1 is an essential specifier of the T cell lineage9, 10 and is one of the most frequently mutated genes in T cell acute lymphoblastic leukemia (T-ALL).11, 12 Activating NOTCH1 mutations occur in many subclasses of T-ALL, and the growth of these cancers often remains NOTCH1-dependent.13, 14 Aberrant NOTCH signaling has been associated with the regulation of enhancers in T-ALL,15 in particular with activation of the proto-oncogene MYC through a distal enhancer located ∼1.5 Mb downstream of its promoter.16, 17 Mechanisms by which aberrant Notch establishes a leukemia-prone chromatin landscape are currently unknown.
Tcf1 harbors a histone deacetylase domain18 and has been shown to be an important epigenetic regulator during multiple stages of T cell development (TCD).19, 20 In T-ALL, canonical Wnt signaling, mediated through β-catenin/Tcf1, has been shown to be active in a subpopulation of cells enriched for leukemia-initiating cells. Importantly, genetic inactivation of β-catenin reduced leukemia initiation frequency in Notch1-driven T-ALL.21
Here we assessed the function of the Notch1 target gene Tcf7, which encodes for Tcf1,22, 23 and the canonical Wnt mediator β-catenin for their coordinate role with oncogenic Notch1 to induce T-ALL. In this context, the transcriptome and the epigenome were analyzed using ATAC-seq, ChIP-seq, and in situ Hi-C. Analysis of hematopoietic progenitors allowed us to gain novel insights into the formation of chromatin topology at T-ALL initiation. Finally, we assessed the leukemia-prone landscape of the Notch1-regulated Myc enhancer region for conserved regulatory elements.
Methods
Mice
Gt(ROSA)26Sortm1(Notch1)Dam/J (N1IClox/lox), Notch1lox/lox, B6.129-Ctnnb1tm2Kem/KnwJ (β-cateninlox/lox), and Mx1Cre mouse lines have been described.9, 24, 25, 26 Additional information on compound lines, the conditional Tcf7 and the TMe mouse lines, are described in supplemental Information.
Flow cytometry and cell sorting
LSKs were defined as CD45.2+ lineage-negative CD117+Sca1+eGFP+. For ATAC-seq, RNA-seq, ChIP-seq, and Hi-C analyses, oncogenic Notch1-expressing LSKs were sorted using a FACSAria (Becton Dickinson) or MoFlo Astrios EQ (Beckman Coulter). Oncogenic NOTCH1-expressing human cord blood (CB) cells used for ATAC-seq were defined as CD45+CD34-CD38+CD5-CD7+eGFP+ and sorted using a FACSAria. Sorting and analysis strategies are outlined in supplemental Information. The purity of sorted subsets was >97%. Flow-cytometric data were acquired on a Gallios (Beckman Coulter) and analyzed using FlowJo v10.7.0. Primary and secondary antibodies are listed in supplemental Table 2.
Ethics statement
All animal work was carried out in accordance with Swiss national guidelines. This study (VD1099) was reviewed and approved by the cantonal veterinary service.
Results
Tcf7 is essential for Notch1-mediated T-ALL induction
We generated a conditional gene-targeted mouse line for the Tcf7 gene to investigate the function of Tcf1 in the context of Notch1-driven T-ALL. Due to the importance of Notch19 and Tcf122, 27 in T cell fate specification, we analyzed 6- to 8-week-old age-matched Tcf7Δ/Δ Mx1Cre and Notch1Δ/Δ Mx1Cre animals. As expected, gene inactivation of either Notch1 or Tcf7 resulted in impaired TCD (supplemental Figure 1A-H), confirming the validity of the conditional Tcf7 loss-of-function (LoF) alleles. Interestingly, Tcf7Δ/Δ Mx1Cre mice did not develop T cell lymphoma within the investigated time frame as previously reported for Tcf7−/− and Tcf7Δ/Δ Vav-Cre animals,28, 29 which may be explained by the different developmental stages at which Tcf7 is inactivated.
Confirming the importance of both genes in TCD, we assessed the function of Tcf1 in the context of Notch1-driven T-ALL. The R26 N1IClox/+Mx1Cre genetic Notch1 gain-of-function (GoF) mouse model was used to induce leukemogenesis26 in bone marrow (BM) chimeras. Mx1Cre-mediated recombination results in the expression of a dominant active form of Notch1 (N1IC) linked to an eGFP reporter driven from the Rosa26 locus.26 N1IClox/+Tcf7lox/lox Mx1Cre compound animals were used to interrogate LoF of Tcf7 in oncogenic Notch1-driven T-ALL (Figure 1A). Activation of the Mx1Cre recombinase drove the inactivation of Tcf7, leading to loss of Tcf1 protein (supplemental Figure 1I).
Figure 1.

Initiation of Notch1-driven T-ALL is Tcf7-dependent. (A) Schematic representation of BM chimeras transplanted with the indicated genotypes: R26 N1IClox/+ or R26 N1IClox/+ Tcf7lox/lox (Controls, gray), R26 N1IClox/+Mx1Cre (N1IC, red), or R26 N1IClox/+ Tcf7lox/loxMx1Cre (N1IC Tcf7lox/lox, blue) and treatment schedule. (B) Kaplan-Meier survival plot of chimeras after last poly(I:C) injection. N1IC mice (n = 8), N1IC Tcf7Δ/Δ (n = 10), and Controls (n = 7) followed for 199 days. Log-rank (Mantel-Cox) test, ****P value < .0001. (C-D) Phenotypic flow cytometric analysis of transplanted (CD45.2+) and induced (eGFP+ for N1IC and N1IC Tcf7Δ/Δ) BM cells from Controls (n = 4), N1IC (n = 6), and N1IC Tcf7Δ/Δ (n = 5). Quantification of absolute numbers is shown for T cells, erythroid cells, myeloid cells, and B cells. Data are represented as mean ± standard error of the mean (SEM). Unpaired t-test, *P value < .05; **P value < .01; ***P value < .001; ****P value < .0001. (E) Schematic representation of BM chimeras with Controls (gray, n = 3), R26 N1IClox/lox Mx1Cre (N1ICΔ/Δ, red, n = 18), R26 N1IClox/lox Tcf7lox/lox Mx1Cre (N1ICΔ/Δ Tcf7Δ/Δ, blue, n = 15), and R26 N1IClox/lox β-cateninlox/lox Mx1Cre (N1ICΔ/Δ β-cateninΔ/Δ, light green, n = 9) mice. (F) Kaplan-Meier survival analysis of transplanted mice after last poly(I:C) injection and followed for 115 days. Log-rank (Mantel-Cox) test, ***P value < .001; ****P value < .0001. (G) Phenotypic flow cytometric analysis of transplanted (CD45.2+) and induced (eGFP+ for N1ICΔ/Δ, N1ICΔ/Δ Tcf7Δ/Δ, and N1ICΔ/Δ β-cateninΔ/Δ) BM. Panels on the left depict representative plots of CD4 and CD8 T cells with quantification of absolute numbers on the right. Data are represented as mean ± SEM. Unpaired t-test, ****P value < .0001.
Expression of N1IC resulted in T-ALL development (Figure 1B). Flow cytometric analysis of N1IC GoF chimeric BM cells revealed an accumulation of CD4+CD8+ double-positive (DP) and CD8+ single-positive leukemic cells (Figure 1C), with concomitant loss of hematopoietic lineages including erythroblasts, B cells, and myeloid cells (Figure 1D).
Surprisingly, none of the N1IC Tcf7Δ/Δ BM chimeras developed disease (Figure 1B). Importantly, abrogation of T-ALL in N1IC Tcf7Δ/Δ BM chimeras was not a mere consequence of loss of the transplant (supplemental Figure 1J). However, erythroblasts, myeloid cells, immature B220+IgM- B cell progenitors, and to a somewhat lesser extent, mature B220+IgM+ B cells were efficiently suppressed (Figure 1D). To determine a potential role of β-catenin-mediated canonical Wnt signaling in Notch1-driven T-ALL, we performed β-catenin LoF studies in an oncogenic Notch1 GoF background, in which N1IC is expressed from both Rosa26 alleles. Again, no disease was observed in N1ICΔ/Δ Tcf7Δ/Δ chimeras, whereas all N1ICΔ/Δ β-cateninΔ/Δ BM chimeras succumbed to T-ALL, albeit with a short-term kinetic delay compared with N1ICΔ/Δ chimeras (Figure 1E-G; supplemental Figure 1I).
These results demonstrate that Tcf1 exerts essential, β-catenin-independent functions in Notch1-driven T-ALL initiation, while suppression of other hematopoietic lineages imposed by forced Notch signaling appears to be largely Tcf1-independent.
Oncogenic Notch1 requires Tcf1 to induce a T cell-specific gene expression program in early hematopoietic progenitors
Mx1Cre-mediated recombinase activity has been previously demonstrated in all blood lineages, including hematopoietic stem cells.30Mx1Cre-mediated N1IC expression occurs in hematopoietic progenitors, including LSKs within the different chimeric cohorts. As Tcf7 gene inactivation profoundly affects N1IC-driven T-ALL induction, we aimed to investigate early changes during disease onset. Thus, we performed RNA-seq on sorted LSK BM progenitors from Controls (N1IClox/+ Tcf7lox/lox), Tcf7Δ/Δ, N1IC, and N1IC Tcf7Δ/Δ chimeras 72 hours postMx1Cre-mediated recombination (Figure 2A). Analysis of differential gene expression revealed no major differences between Controls and Tcf7Δ/Δ LSKs, indicating that ablation of Tcf7 alone had no major impact on gene expression (Figure 2B; supplemental Figure 2A). In contrast, the expression of leukemogenic N1IC led to profound reprogramming of the cellular transcriptome. We identified 414 and 423 genes significantly down- and upregulated, respectively, in N1IC-expressing LSKs compared with Controls (Figure 2B; supplemental Figure 2A). Among the 423 upregulated genes, 119 were Tcf1-dependent as they were lost in the LSK population of N1IC Tcf7Δ/Δ chimeras. Interestingly, analysis of the gene ontology biological processes of the Tcf7-dependent upregulated genes in LSKs derived from N1IC chimeras revealed an association with lymphocyte differentiation/activation and included T cell activation and differentiation (Figure 2B-C; supplemental Figure 2B). Quantitative analysis confirmed the upregulation of T cell-specific transcripts, including Tcf7, Il2rα, Gata3, and CD3ε in N1IC-expressing LSKs. In contrast, N1IC was unable to upregulate T cell-specific genes in the absence of Tcf1 (Figure 2D), albeit that Notch target genes such as Notch1 itself and Hes1 were upregulated by N1IC, independently of Tcf1 (Figure 2E-F).
Figure 2.

Tcf7 regulates the expression of genetic T cell signature in BM progenitors in response to oncogenic Notch1. (A) Experimental setup: induced CD45.2+ BM cells from Controls (black, n = 3), Tcf7Δ/Δ (light blue, n = 3), N1IC (red, n = 3), or N1IC Tcf7Δ/Δ (blue, n = 3) mice were FACS purified for lineage-, cKit+ (CD117+), and Sca1+ BM progenitors (LSK) for RNA-seq analysis. Representative flow cytometric plots are shown. (B) Heatmap depicting regulated genes in N1IC vs Controls (FDR <0.05, −1.5 > FC > 2) from gene ontology (GO) T cell activation collection, shown for all experimental groups. (C) Enrichment of biological pathways from GO biological process (GOBP) collection in genes with induced expression by N1IC and Tcf7 from RNA-seq on LSK cells. Top 20 pathways are shown. P values were calculated with Fisher's exact test. (D) Expression of investigated genes measured as TPM (transcripts per kilobase million) with induced expression by N1IC and Tcf7 from RNA-seq on LSK cells. Barplots from left to right of each graph: Controls, Tcf7Δ/Δ, N1IC, and N1IC Tcf7Δ/Δ. Data are represented as mean ± standard error of the mean (SEM). One-way ANOVA, *P value < .05; **P value < .01; ***P value < .001. (E) Enrichment of biological pathways from GOBP collection in genes with induced expression by N1IC and independent of Tcf7 from RNA-seq on LSK cells. Top 20 pathways are shown. P values were calculated with Fisher's exact test. (F) Expression of investigated genes measured as TPM with induced expression by N1IC and independent of Tcf7 from RNA-seq on LSK cells. Barplots from left to right of each graph: Controls, Tcf7Δ/Δ, N1IC, and N1IC Tcf7Δ/Δ. Data are represented as mean ± SEM. One-way ANOVA, **P value < .01; ***P value < .001.
Next, we tested whether Tcf7 GoF in Notch1-proficient BM progenitors would be sufficient to induce ectopic TCD and/or T-ALL. Thus, we performed Tcf7 GoF experiments using Tcf7 IRES eGFP-expressing BM chimeras. Analysis of Tcf7 GoF chimeras 13 weeks posttransplantation revealed neither ectopic TCD nor T-ALL induction compared with Controls or retrovirally-driven N1IC BM chimeras analyzed at endpoint (supplemental Figure 2C-F).
Altogether, this demonstrates that Tcf1 is essential for N1IC to elicit a T cell-specific gene expression program. However, Tcf1 alone in vivo is not sufficient to induce a T cell program with leukemic self-renewal activity even in a Notch1-proficient background.
Tcf1 regulates chromatin accessibility in N1IC-expressing LSKs
As Tcf1 has been associated with epigenetic regulation of TCD,18, 20 we asked whether Tcf1 is necessary to modulate chromatin accessibility enabling forced N1IC to permit a leukemia-specific program in hematopoietic progenitors. Thus, we performed ATAC-seq on LSKs derived from Controls, Tcf7Δ/Δ, N1IC, and N1IC Tcf7Δ/Δ chimeras (Figure 3A). Differential accessibility analysis showed pronounced modulation of the epigenome in response to N1IC and Tcf1 (Figure 3B; supplemental Figure 3A). Genes in proximity to N1IC-induced Tcf1-dependent chromatin modulation were predominantly associated with T cell differentiation, proliferation, and activation (Figure 3C; supplemental Figure 3B). Indeed, N1IC-driven chromatin accessibility at the promoter and putative enhancer regions of T cell-specific genes, including Ptcrα and Cd3ε, were Tcf1-dependent (Figure 3D).
Figure 3.

Notch1 and Tcf1 epigenetically establish T-lineage specification in early BM progenitors. (A) Induced CD45.2+ BM cells from Controls (black, n = 3), Tcf7Δ/Δ (light blue, n = 3), N1IC (red, n = 3), or N1IC Tcf7Δ/Δ (blue, n = 3) mice were FACS purified for lineage-, cKit+ (CD117), and Sca1+ BM progenitors (LSK) for ATAC-seq analysis. Characteristic flow cytometric plots are shown. (B) Heatmap depicting all regulated genomic loci in comparison N1IC vs Controls and then used as a reference and compared with N1IC Tcf7Δ/Δ (FDR < 0.01) for ATAC-seq called and centered peaks, shown for all experimental groups. Color scale for centered values is shown below the heatmap. (C) Enrichment of biological pathways from gene ontology biological process (GOBP) collection in genes with induced proximal accessibility by N1IC and Tcf7. Top 20 pathways are shown from ontologies with a fold enrichment >2 and FDR <0.05. P values were calculated with Fisher's exact test. (D) Integrative genomics viewer chromatin accessibility profiles for all experimental groups are shown at the promoter of Ptcrα and for Cd3ε. Tracks were group-scaled. Schematic representation of genetic loci is depicted below the profiles. (E-F) Footprint analysis for transcription factors binding regulated by (E) N1IC vs Controls and (F) N1IC vs N1IC Tcf7Δ/Δ.
Lineage determination of early hematopoietic progenitors is governed by specific master transcription factors (TFs) and their controlled binding to regulatory chromatin loci.31, 32 To assess whether such regulation in T-ALL initiation is dependent on N1IC- and Tcf1-mediated chromatin modulation, we performed TF footprint and TF binding sites (BSs) analysis of gained and lost ATAC-seq peaks. The top differential accessible BSs identified were Runx, Tcf1, Lef1, and RBPJ in both N1IC-expressing vs Controls and N1IC vs N1IC Tcf7Δ/Δ settings. These findings confirmed that chromatin loci are open for these TFs in a Notch1- and Tcf1-regulated manner. Gain of Gata TF BSs as a consequence of Tcf7 deficiency in N1IC vs N1IC Tcf7Δ/Δ footprint analysis indicates that in an N1IC GoF context, their repression is Tcf1-dependent. In contrast, Cebp and Pax5 TF BSs correlate with negative differential binding scores independent of Tcf7 (Figure 3E-F; supplemental Figure 3C-D).
These results suggest that forced N1IC expression in LSKs modulates a chromatin landscape that allows induction of T cell-specific genes, and this process appears to be Tcf1-dependent. Contrarily, oncogenic N1IC simultaneously closes chromatin loci that would be permissive for induction of alternative cell fates such as myeloid, B- or erythroid-cell lineages. These processes are regulated by Tcf7-dependent and -independent mechanisms.
Dynamic large-scale genomic interactions promoting leukemogenesis rely on Notch1 and Tcf1
Using chromatin accessibility analysis, we identified an early involvement of Tcf1 and Notch1 in the events of T-ALL commitment from hematopoietic progenitors. It is not clear whether 3D genome folding is equally affected, hence impacting cell fate decisions in oncogenic processes.1 We thus performed in situ Hi-C, allowing for spatial profiling of all genomic loci at once33 on LSKs derived from N1IC, N1IC Tcf7Δ/Δ, and control chimeras (Figure 4A). Data resolution of 2.5 kb34 and improved quality over recently published in situ Hi-C on hematopoietic stem cells35 allowed us to proceed confidently with the analysis of topology (Figure 4B; supplemental Figure 4A-D).
Figure 4.


Notch1 and Tcf1 regulate 3D organization of chromatin compartments and domains. (A) Induced CD45.2+ BM cells from Controls (black, n = 2), N1IC (red, n = 2), or N1IC Tcf7Δ/Δ (blue, n = 2) mice were FACS purified for lineage-, cKit+ (CD117), and Sca1+ BM progenitors (LSK) and processed for in situ Hi-C analysis. Characteristic flow cytometric plots are shown. (B) Juicebox-generated contact matrices from chromosome 15: whole chromosome, at 250 kb resolution (far left); 50 kb resolution (middle left); chromatin domains at 10 kb resolution (middle right); chromatin loops (blue squares) at highest resolution of 2.5 kb (far right) shown for Controls. The 1D regions corresponding to a contact matrix are indicated in the diagrams below and right. The intensity of each pixel represents the normalized number of contacts between a pair of loci. Maximum intensity: 1350, 293, 20, 3 (from left to right). (C-D) Chromatin subcompartment switching between (C) Controls vs N1IC and (D) N1IC Tcf7Δ/Δ vs N1IC (left panels). X-axis indicates the number and direction of subcompartments switching: stable (0), toward active (+), and toward inactive (−). Association with gene expression differences (FDR < 0.1) for genes within dynamic compartments is shown in right panels. Unpaired Wilcoxon test, ****P value < .0001. (E) IGV CCCTC-Binding Factor (CTCF), chromatin accessibility, and H3K27ac profiles for all experimental groups shown for Tspan9. Tracks were group-scaled. Schematic representation of genetic loci is depicted below the profiles. Compartment tracks identified by Calder are shown at the bottom for all experimental conditions. (F) Quantitative comparison of identified TAD boundary changes differential, nondifferential, and shifted (nonoverlapping) in comparison N1IC vs Controls and N1IC vs N1IC Tcf7Δ/Δ. (G) Proportion of TAD boundary changes classified into 5 categories for all comparisons as indicated. (H) Schematic depiction of Notch1- and Tcf1-dependent TAD regulating the expression of Grap2 gene. Hi-C matrix at 10 kb resolution is shown on top, TopDom and TADCompare analyzed TAD with differential boundary is shown below (highlighted in red) together with boundary score visualization. IGV profiles for CTCF and ATAC-seq are shown for N1IC LSKs. Tracks were group-scaled. Representation of genetic loci is depicted below the tracks.
Genomic compartment A regions contain open, active chromatin, while B compartments are correlated with heterochromatin.36, 37 While large genomic regions of A and B compartments remain relatively stable, fine-grained subcompartments analysis can reveal subtle compartment changes.33, 38 Direct analysis of subcompartment switching between N1IC vs Controls, N1IC Tcf7Δ/Δ vs N1IC, and N1IC Tcf7Δ/Δ vs Controls LSKs revealed high genome stability (65% to 70% stability). However, the proportion of the subcompartment changes, irrespectively of genotypes and comparison, varied between 12% and 15% (Figure 4C-D; supplemental Figure 4E). Interestingly, subcompartment rank switching correlated with transcription changes of differentially expressed genes after integration of our RNA-seq data comparing N1IC vs Controls and N1IC Tcf7Δ/Δ vs Controls. No significant correlations were obtained when comparing N1IC Tcf7Δ/Δ vs N1IC (Figure 4C-D; supplemental Figure 4E). A specific example of a Notch1-induced and Tcf7-dependent gene, whose expression was directly correlated with subcompartment switching (N1IC vs Controls and N1IC Tcf7Δ/Δ vs N1IC), is Tspan939 (Figure 4E; supplemental Figure 4F).
We next identified topologically associating domains (TADs) from contact matrices at 10 kb resolution. Since boundaries of TADs are known to be enriched for CTCF,33, 40 we performed CTCF analysis using ChIP-seq on sorted LSKs from Controls, N1IC, and N1IC Tcf7Δ/Δ. Enrichment analysis at both TAD anchors identified oriented binding of CTCF at over 37% of chromatin domains (supplemental Figure 5A). Thus, only these domains were taken into consideration for subsequent analyses. To quantify modulations of TADs between conditions, we performed an analysis of TAD boundary changes. Forty-five TAD boundaries were increasing with overexpression of N1IC. These were found to be Tcf1-dependent as their score decreased in N1IC Tcf7Δ/Δ LSKs (Figure 4F). On the contrary, 52 TAD boundaries had a decreased score and were Tcf1-dependent in N1IC LSKs (Figure 4F). The majority of TAD boundaries were classified as nondifferential, and only around 2.5% to 3% were categorized as increased in strength in different genetic comparisons (Figure 4F-G). Genes proximal to dynamic TAD boundaries revealed no genome-wide significant correlation between gene expression and TAD boundary changes (supplemental Figure 5B). Grap2 has been identified as an example among differentially expressed genes due to Notch1- and Tcf1-dependent changes in TAD boundaries (Figure 4H; supplemental Figure 5C-E).41, 42
Our analysis revealed fine-tuned coordination by N1IC and Tcf7 cooperatively orchestrate distinctive higher-order genomic folding during leukemic fate programming.
Establishment of EPIs in T-ALL initiation partly depends on Tcf1
After assessing the effects of Notch1 and Tcf1 on high-order genome organization, we focused on 3D chromatin loop interactions established in N1IC and N1IC Tcf7Δ/Δ. Local chromatin interactions are of importance when connecting regulatory regions (RRs), such as promoters with distal enhancers, leading to modulated gene expression.43 Identified interactions were established and compared between Controls, N1IC, and N1IC Tcf7Δ/Δ genotypes to determine both genotype-specific and shared enhancer-promoter interactions (EPIs) (Figure 5A).
Figure 5.


Chromatin looping and activation status of regulatory elements are Notch1- and Tcf1-dependent. (A) Three-way quantitative comparison of identified chromatin loops for condition-specific and shared loops for Controls, N1IC, and N1IC Tcf7Δ/Δ. (B) Overrepresentation analysis (ORA) for N1IC-specific loop-associated genes for phenotype catalog. Top 10 pathways are shown. P values were calculated with Fisher's exact test. (C) Association of gene expression differences (adjusted P value ≤ .05) for genes within dynamic condition-specific loops (from panel A). Left and right panels show log2 fold-change for condition-specific loop-associated genes from RNA-seq analysis. (D) Induced CD45.2+ BM cells from Controls (black, n = 3), N1IC (red, n = 3), or N1IC Tcf7Δ/Δ (blue, n = 3) mice were FACS purified for lineage-, cKit+ (CD117), and Sca1+ BM progenitors (LSK) for histone mark ChIP-seq analysis with H3K27ac antibody. (E) H3K27ac ChIP-seq signal at the non-TSS loop anchor. Shown at 5 kb windows around the center of a peak or loop anchor. Enrichment for H3K27ac is scaled. Quantification of the global signal for non-TSS loop anchors is depicted on the right. Data are shown from left to right for Controls, N1IC, and N1IC Tcf7Δ/Δ. (F) Gene-annotated scatterplot for differential EPI (x-axis) for differentially expressed genes (log2FC, y-axis) in comparison N1IC vs Controls. Dots for genes with differential EPI >1 are shown in red. (G) ORA for genes with differential EPI in comparison N1IC vs Controls for gene ontology biological processes (GOBP) catalog. Top 20 pathways are shown. P values were calculated with Fisher's exact test. (H) Expression of Bcl11b measured as transcripts per million (TPM) from RNA-seq on LSK cells. Data are represented as mean ± standard error of the mean. One-way ANOVA, ***P value < .001. (I) Representation of 5 kb interacting regions between the indicated genomic coordinates color-coded based on their EPI strength value (top). Identified enhancer (gray line) and promoter (yellow line) interaction is enlarged in the top left corner. Integrative genomics viewer CTCF, chromatin accessibility, and H3K27ac profiles are shown for all experimental conditions (bottom). Tracks were group-scaled. Schematic representation of genetic loci is depicted below the profiles.
Since not all identified chromatin loops connect genes with potential RRs, we focused on those with confirmed proximity to transcription start sites (TSS) at one of the anchors. Investigation of chromatin loops with differential expression dependent on both Notch1 and Tcf1 revealed genes from leukemic ontology associated with T-ALL, such as Pdgfrb (Figure 5B; supplemental Figure 6A).6 Although the investigation of Notch1-dependent but Tcf1-independent chromatin loops in N1IC Tcf7Δ/Δ identified leukemic ontology as being highly enriched, the affected genes cannot be considered T-ALL-related, rather they have been described in myeloid malignancies (supplemental Figure 6B-C).44 Moreover, analysis of data for genome-wide correlation within dynamic condition-specific chromatin loops revealed no significant differences (Figure 5C).
Regulation of gene expression in T-ALL by NOTCH1 has been linked to distal enhancers, where NOTCH1 occupancy correlates with increased levels of H3K27ac.15 Thus, LSKs from Controls, N1IC, and N1IC Tcf7Δ/Δ chimeras were isolated for genome-wide H3K27acChIP-seq analysis (Figure 5D). We investigated whether identified chromatin loops were connecting active RRs in Notch1- or Tcf1-dependency by profiling H3K27ac at non-TSS loop anchors. Interestingly, the highest genome-wide acetylation levels of connected chromatin loci were detected in N1IC-specific loops, highlighting the importance of Tcf1 in such distal activation downstream of Notch1 signaling (Figure 5E). Subsequently, all genes controlled by Notch1 and Tcf1 were examined for the involvement of distal regulatory elements. We performed unbiased genome-wide analysis for Notch1- and Tcf1-dependent EPIs correlated with differential gene expression. We identified up to 239 differential EPIs, of which 147 correlated with a respective increase or decrease in gene expression (Figure 5F; supplemental Figure 6D-F). Dynamic EPIs were analyzed with respect to dynamic subcompartments. Increased EPI strength positively correlated with increasing compartment ranks across analyzed comparisons (supplemental Figure 6G-I). Direct comparison of differential EPI between experimental chimeras revealed that ontologies related to lymphocyte and T cell activation were significantly enriched in N1IC (Figure 5G). Expression of Bcl11b45 and Gpr17446 are 2 examples of genes regulated by differential EPI being Notch- and Tcf1-dependent (Figure 5H-I; supplemental Figure 6J-K).
Taken together, 3D chromatin interactions in T-ALL-prone LSKs are regulated by Tcf1 in order to calibrate the transcriptional output of genes during Notch1-mediated transformation processes.
A novel Tcf1-regulated Myc-enhancer region is essential for Notch1-driven T-ALL progression
A conserved Notch1 MYC enhancer (NMe) located 1.5 Mb downstream of the MYC promoter drives transformation and T-ALL.16 Notch1-Rbpj BSs are located in a broad super-enhancer region that directly interacts with the MYC promoter via long-range chromatin looping.17 Genetic deletion of the NMe impairs T-ALL development.16 We investigated whether the NMe and Myc gene/promoters are regulated in a Tcf7-dependent manner during the early initiation phase of T-ALL in N1IC-expressing LSKs. While chromatin accessibility at the Myc promoter was not significantly different, accessibility at the NMe site was Tcf7-dependent (Figure 6A; supplemental Table 3). However, in N1IC-expressing LSKs, gene expression and enhancer-promoter looping of Myc itself were not differentially upregulated at this early stage (supplemental Figure 7A-B).
Figure 6.


Tcf1 exerts a crucial regulatory function within distal Myc enhancers. (A) Integrative genomics viewer (IGV) profiles for Controls, N1IC, and N1IC Tcf7Δ/Δ from ATAC-seq analysis performed on sorted murine LSKs for Myc promoter (left panel), NMe (red), and TMe (green) (right panel). Tracks were group-scaled. Schematic representation of genetic loci is depicted below the profiles. (B) TMe evolutionary conservation tree (left panel) and predicted ultraconserved transcription factor binding motifs in the TMe sequence (right panel). PhastCons conservation scores are indicated above the sites (score >0.5). (C) Schematic representation of lentiviral (LV) overexpression experiment using CD34+ human CB cells transduced with LV overexpressing NOTCH1 (red, n = 2). Cells were used for ATAC-seq analysis. ATAC-seq (GSM4743251 and GSM4743252) data sets from CD34+ human CB cells were used as Controls. (D) Enrichment of biological pathways from GO biological process collection in genes with induced proximal accessibility by human NOTCH1 and Controls CD34+ human CB cells. Top 10 pathways are shown from ontologies with an FDR ≤ 0.01. P values were calculated with Fisher's exact test. (E) TOBIAS footprint analysis NOTCH1 vs Controls. Pathway enrichment analysis from KEGG pathway catalog regulated by TCF1-confirmed footprint. Top 5 pathways are shown with corresponding FDR. The panel below represents IGV chromatin accessibility profiles for both experimental groups shown for IL2RΑ. Tracks were group-scaled; scaling is shown in the top left corner. Schematic representation of genetic loci is depicted below the profiles together with footprint analysis at TCF1-binding motif. (F) IGV chromatin accessibility profiles for TMe in human NOTCH1 and Controls CD34+ human CB cells. Tracks were group-scaled. (G) Flow cytometric-based analysis of intracellular TCF1 levels in NOTCH1 (red) and Controls (gray) CD34+ human CB cells.
Unexpectedly, analysis of chromatin accessibility revealed a prominent regulatory site 14 kb downstream of the NMe regulated by Notch1 in a Tcf1-dependent manner. We named this newly discovered region TMe (Tcf1-regulated Myc enhancer) (Figure 6A; supplemental Table 3). Attributing functional relevance to this novel TMe, we hypothesized that cis-acting elements within this accessible chromatin region would be conserved between different species, similarly to the NMe.47 Phylogenic footprint analysis of TMe across vertebrates revealed multiple highly conserved regulatory elements (Figure 6B). The high conservation of this cluster of TF BSs in placental mammals is indicative of a functional role for the TMe region. Hence, we assessed the ability of forced NOTCH1 expression to establish an oncogenic chromatin landscape in human hematopoietic progenitor cells. We used a system that models NOTCH1-mediated T-ALL in vitro through lentiviral transduction of human CB cells.48 Specifically, oncogenic NOTCH1 was expressed in CD34+ cells and analyzed by ATAC-seq (Figure 6C). Gene Ontology analysis revealed that NOTCH1-regulated chromatin modulation is associated with immune response-activating cell surface receptor signaling, T cell receptor (TCR) signaling, leukocyte activation, and others (Figure 6D). Taking advantage of footprint analysis, we identified TCF1 as a regulator of ontologies such as TCR signaling and pathways in cancer. Moreover, forced NOTCH1 expression induced chromatin accessibility in T cell genes, such as IL2RA, and, although statistically not significant, accessibility of the TMe region increased and resulted in TCF1 expression (Figure 6E-G; supplemental Table 4) in CD34+ cells. TMe is also accessible in NOTCH1-dependent DND-41 T-ALL cells (supplemental Figure 7C). This indicates that not only are DNA sequences conserved between mammals but that Notch-mediated chromatin accessibility gain is also conserved between mice and humans. Furthermore, ChIP and reverse ChIP analysis using murine and human T-ALL cell lines indicated that Tcf1 binds to both NMe and TMe (Figure 7A; supplemental Figure 7D-E).
Figure 7.

TMe enhancer site is essential for T-ALL initiation. (A) ChIP-qPCR analysis for Tcf1 binding in murine T-ALL (mT-ALL, n = 3), wild type LSKs (Neg Ctrl, n = 3) and IgG Controls (n = 2) at NMe (each left bar, red) and TMe (each right bar, green). Data are represented as mean ± standard error of the mean (SEM). (B) Genomic localization of TMe in mm10, depicting the strategy for deletion of genomic element. Integrative genomics viewer profiles for N1IC (top, red, n = 3) and N1IC TMe−/− (bottom, green, n = 3) showing chromatin accessibility from ATAC-seq analysis on sorted LSKs. Tracks were group-scaled, scaling is shown in the top left corner. (C) Schematic representation of BM chimeras R26 N1IClox/+Mx1Cre (N1IC), R26 N1IClox/+ TMe+/−Mx1Cre (N1IC TMe+/−), and R26 N1IClox/+ TMe−/−Mx1Cre (N1IC TMe−/−) mice. (D) Kaplan-Meier survival analysis of transplanted mice after last poly(I:C) injection. N1IC mice (red, n = 8), N1IC TMe+/− (orange, n = 10), and N1IC TMe−/− (green, n = 11) were followed for 341 days post poly(I:C) injection. Log-rank (Mantel-Cox) test, *P value < .05; ***P value < .001; ****P value < .0001. (E) Relative percentages of CD45.2+ transplanted cells in peripheral blood of N1IC mice (red, n = 12), N1IC TMe+/− (orange, n = 11), and N1IC TMe−/− (green, n = 12), post poly(I:C) injection. Timepoints are indicated below the graph. Data are represented as mean ± SEM. (F) Flow cytometric-based phenotypic analysis of transplanted (CD45.2+) and induced (eGFP+) BM cells. Plots depict representative profiles from N1IC mouse T-ALL midstage progression (red), N1IC TMe+/− at the endpoint (orange), and N1IC TMe−/− at the endpoint (green). (G) Total protein analysis by western blot for Myc, Tcf1, and β-actin on FACS purified T cells from BM of experimental groups: N1IC CD8+ (n = 2), N1IC DP (CD4+CD8+, n = 2), N1IC TMe+/− DP (CD4+CD8+, n = 4), and N1IC TMe−/− DP (CD4+CD8+, n = 2), as indicated. (H) Total protein analysis by western blot for Myc and β-actin on FACS purified T cell blasts from BM of experimental groups: N1IC CD8+ (n = 3) and N1IC TMe+/− CD8+ (n = 3), as indicated. (I) 3C-qPCR analysis for interactions between Myc promoter and NMe on FACS purified T cells from BM of experimental groups: N1IC CD8+ (n = 3), N1IC DP (n = 3), N1IC TMe−/− DP (n = 3). Bacterial artificial chromosome (BAC) clones spanning the region of interest were used as negative control. Data are represented as mean ± SEM. Unpaired t-test, *P value < .05.
We thus hypothesized that Tcf1 orchestrates Notch1-induced chromatin organization in the distal Myc-enhancer through binding to NMe and/or TMe. To test the putative function of the TMe region in the context of Notch1-driven T-ALL, we generated CRISPR-targeted mice for TMe and established R26 N1IClox/+ TMe+/−Mx1Cre and R26 N1IClox/+ TMe−/−Mx1Cre compound lines. TMe−/− mice are fertile, viable, and exhibit normal hematopoietic development (supplemental Figure 8). The consequences of TMe deletion in the context of Notch-driven T-ALL were assessed in BM chimeras. The effect of TMe genomic deletion on epigenetic features was addressed by ATAC-seq on LSKs of N1IC and N1IC TMe−/− mice. As expected, chromatin accessibility was readily detectable for both NMe and TMe regions in N1IC-derived LSKs, while only the NMe region retained chromatin accessibility in N1IC TMe−/− chimeras (Figure 7B).
Strikingly, analysis of leukemogenesis revealed that none of the N1IC TMe−/−chimeras developed T-ALL and only 40% of N1IC TMe+/− succumbed, while chimerism was stable over time on all genetic backgrounds (Figure 7C-E). Flow cytometric analysis of BM cells at midstage disease of N1IC and endpoint of N1IC TMe+/− and N1IC TMe−/− chimeras revealed similar proportions of ectopic CD4+CD8+ DP preleukemic cells in all compound chimeras (Figure 7F). While preleukemic cells in N1IC BM chimeras progressed from a Myc-negative DP stage to Myc-expressing CD8+ leukemic cells, surprisingly efficient progression to aggressive leukemia is impaired in the majority of N1IC TMe+/− and in all N1IC TMe−/− chimeras (Figure 7F-G). Accordingly, comparable Myc expression was observed between N1IC and N1IC TMe+/− chimeras that developed T-ALL (Figure 7H). Moreover, 3C analysis showed that NMe interacts with the Myc promoter in sorted Myc-expressing N1IC CD8+ T-ALL cells. Interaction is reduced in Myc-negative N1IC DP and even more so in N1IC TMe−/− DP cells (Figure 7G,I), raising the possibility that TMe is important for efficient Myc promoter-enhancer interaction during disease progression.
Discussion
We discovered an essential function of Tcf1 in orchestrating chromatin accessibility allowing aberrant Notch1 signaling to execute its tumorigenic function in T-ALL. Tcf7 has been shown to be a direct target gene of Notch1 in TCD22, 23 and implicated in the regulation of TCD.19, 20 This function of Tcf1 has been shown to be independent of canonical Wnt signaling.49, 50, 51 Similarly, our results show that β-catenin is dispensable in Notch1-induced T-ALL. Nonetheless, β-catenin deficiency resulted in a delayed T-ALL progression, which is in agreement with previous observations that canonical Wnt signaling is active in a subpopulation of leukemia-initiating cells.21
β-catenin-independent functions of Tcf1 during TCD have been linked to its ability to function as an epigenetic regulator. Tcf1 with Lef1 establish the CD8 T cell-lineage by repressing CD4-lineage-associated genes via histone deacetylase activity.18 Overexpression of Tcf1 in fibroblasts or progenitors in vitro can regulate T cell-specific genes.20, 22, 23 Thus, we assessed whether Tcf1 in vivo is sufficient to mimic oncogenic Notch1. Retroviral overexpression of Tcf1 in hematopoietic progenitors was insufficient to induce ectopic TCD or T-ALL in vivo, even in a Notch1-proficient background. In N1IC-expressing LSKs, however, ex vivo epigenetic and gene expression profiling revealed that Tcf1 is essential to modulate chromatin accessibility and allows Notch1-driven expression of T cell-specific and growth-promoting genes. Without Tcf1, the chromatin structure of promoters and enhancers of T cell-specific genes such as CD3ε, Ptcrα, and the distal Myc enhancer are not accessible despite oncogenic Notch expression. Thus, Notch1-induced, Tcf1-dependent regulation of chromatin accessibility appears to be an early key event in T-ALL induction. This Tcf1-specific function is in agreement with the previously established role of Tcf1 during normal TCD.20, 22
Lineage specification requires not only the expression of distinct genes but also simultaneous repression of other lineage programs.52 Our ATAC-seq footprint analyses reveal that forced N1IC expression in LSKs induces repression of master regulators of other blood lineages such as Cebps, Pax5, and Gatas. Thus, Tcf1 contributes to proper lineage specification independently of the gene expression of such master regulators.
Regulation of leukemogenesis in BM progenitors is governed by chromatin accessibility of regulatory elements proximal to genes, as assessed by ATAC-seq, as well as by distal chromatin interactions established through EPIs. Genome-wide identification of the 3D landscape revealed fine-tuned control of T-ALL-associated genes. Dynamic chromatin subcompartments, TADs, and loops directly affect the genes reported in clinical T-ALL.39, 53 However, we found that dynamics of genome folding are much more subtle than changes observed during cellular differentiation.54 Importantly, Tcf1 is essential for such an organization of chromatin topology and activation of specific regulatory elements in Notch1-initiated leukemogenesis.
NOTCH1 has been shown to regulate the MYC oncogene through the NMe17 in T-ALL. Although Myc is not differentially expressed in Controls, N1IC and N1IC Tcf7Δ/Δ LSKs, our epigenetic analyses revealed that accessibility of NMe is already established early in N1IC-expressing LSKs in a Tcf1-dependent manner. In the absence of Tcf1, forced N1IC-expression cannot render NMe accessible. Thus, the chromatin landscape is already shaped for future events when cells express appropriate TFs and/or additional chromatin regulators. We identified an additional evolutionarily conserved TMe region 14 kb downstream of NMe and tested its functional importance. We hypothesized that TMe is important to establish chromatin accessibility of NMe. However, genomic deletion of TMe revealed that NMe accessibility could still be established in LSKs of N1IC TMe−/− chimeras. ChIP-qPCR analysis identified direct regulation of NMe by bound Tcf1. Whereas NMe-deficient mice develop marked thymic atrophy characterized by severe reduction of DP thymocytes,16 TMe−/− mice revealed that TMe is dispensable for physiological TCD. Strikingly, in T-ALL initiation, none of the N1IC TMe−/− chimeras developed T-ALL and only 40% of N1IC TMe+/−succumbed to disease. N1IC TMe−/− chimeras developed preleukemic DP T cells but did not progress to lethal disease. These preleukemic DP cells did not express detectable levels of Myc, suggesting that TMe is required for expression and upregulation of Myc and thus disease progression. The conceptual framework of our study can help to identify additional regulators of the preleukemic to leukemic transition of relevance to therapeutic approaches.
Acknowledgments
The authors would like to acknowledge the staff from the Flow Cytometry Facility at the University of Lausanne for their excellent technical support and thank the technical staff from the CPG of EPFL.
This work was in part supported by the Swiss National Science Foundation (SNF 31003A_1467198 and SNF 310030_188505), the Swiss Cancer League, and the US National Institutes of Health NIH grants P30 CA013696 and R35 CA210065.
Footnotes
Detailed bioinformatics tools used for the analysis of highthroughput data are provided in supplements. ATAC-seq, RNAseq, ChIP-seq, and Hi-C data sets of murine LSK and human ATAC-seq were deposited (GSE169121). Publicly available ATAC-seq (GSM4743251 and GSM4743252) datasets from noninfected and noncultured CD34+ human CB cells were used as Controls. Publicly available ATAC-seq (GSM5259037) dataset on DND-41 was analyzed as well.
The online version of this article contains a data supplement.
There is a Blood Commentary on this article in this issue.
The publication costs of this article were defrayed in part by page charge payment. Therefore, and solely to indicate this fact, this article is hereby marked “advertisement” in accordance with 18 USC section 1734.
New therapeutic modalities for T-cell acute lymphoblastic leukemia (T-ALL) are needed to improve outcomes. Using elegant mouse modeling and differential analysis of chromatin structure in hematopoietic progenitor cells, Antoszewski and colleagues demonstrate how the transcription factor Tcf1, encoded by the Notch target gene Tcf7, shapes chromatin architecture to facilitate initiation of Notch-induced T-ALL. Their data point to alternative targets to NOTCH1 in this disease.
Contributor Information
Ute Koch, Email: ute.koch@epfl.ch.
Freddy Radtke, Email: freddy.radtke@epfl.ch.
Authorship
Contribution: F.R. and U.K. provided conceptualization; M.A., T.S., U.K., C.E.J.P., I.J.H., and A.P.W. provided methodology; N.F., G.A.R.B., J.L., and Y.L. provided software analysis; M.A., T.S., M.N., C.D., U.K., N.F., G.A.R.B., J.L., Y.L., G.C.S., S.A.-M., and E.S. provided formal analysis and investigation; C.E.J.P., I.J.H., G.C.S., A.P.W., L.B., and G.C. provided resources; F.R., M.A., and U.K. wrote the original draft; F.R., M.A., U.K., G.A.R.B., J.L., Y.L., L.B., A.A.F., G.C., and A.P.W. reviewed and edited the manuscript; M.A., U.K., and F.R. provided visualization; F.R. and U.K. provided supervision; U.K. and M.A. provided project administration; and F.R. handled funding acquisition.
Conflict-of-interest disclosure: The authors declare no competing financial interests.
Supplementary Material
REFERENCES
- 1.Stadhouders R, Filion GJ, Graf T. Transcription factors and 3D genome conformation in cell-fate decisions. Nature. 2019;569(7756):345–354. doi: 10.1038/s41586-019-1182-7. [DOI] [PubMed] [Google Scholar]
- 2.Flavahan WA, Drier Y, Liau BB, et al. Insulator dysfunction and oncogene activation in IDH mutant gliomas. Nature. 2016;529(7584):110–114. doi: 10.1038/nature16490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Katainen R, Dave K, Pitkänen E, et al. CTCF/cohesin-binding sites are frequently mutated in cancer. Nat Genet. 2015;47(7):818–821. doi: 10.1038/ng.3335. [DOI] [PubMed] [Google Scholar]
- 4.Hnisz D, Weintraub AS, Day DS, et al. Activation of proto-oncogenes by disruption of chromosome neighborhoods. Science. 2016;351(6280):1454–1458. doi: 10.1126/science.aad9024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Jiang S. Tet2 at the interface between cancer and immunity. Commun Biol. 2020;3(1):667. doi: 10.1038/s42003-020-01391-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Heilmann AM, Schrock AB, He J, et al. Novel PDGFRB fusions in childhood B- and T-acute lymphoblastic leukemia. Leukemia. 2017;31(9):1989–1992. doi: 10.1038/leu.2017.161. [DOI] [PubMed] [Google Scholar]
- 7.Morrow MA, Mayer EW, Perez CA, Adlam M, Siu G. Overexpression of the Helix-Loop-Helix protein Id2 blocks T cell development at multiple stages. Mol Immunol. 1999;36(8):491–503. doi: 10.1016/s0161-5890(99)00071-1. [DOI] [PubMed] [Google Scholar]
- 8.Richter-Pechańska P, Kunz JB, Hof J, et al. Identification of a genetically defined ultra-high-risk group in relapsed pediatric T-lymphoblastic leukemia. Blood Cancer J. 2017;7(2):e523. doi: 10.1038/bcj.2017.3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Radtke F, Wilson A, Stark G, et al. Deficient T cell fate specification in mice with an induced inactivation of Notch1. Immunity. 1999;10(5):547–558. doi: 10.1016/s1074-7613(00)80054-0. [DOI] [PubMed] [Google Scholar]
- 10.Wilson A, MacDonald HR, Radtke F. Notch 1-deficient common lymphoid precursors adopt a B cell fate in the thymus. J Exp Med. 2001;194(7):1003–1012. doi: 10.1084/jem.194.7.1003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Weng AP, Ferrando AA, Lee W, et al. Activating mutations of NOTCH1 in human T cell acute lymphoblastic leukemia. Science. 2004;306(5694):269–271. doi: 10.1126/science.1102160. [DOI] [PubMed] [Google Scholar]
- 12.Ma X, Liu Y, Liu Y, et al. Pan-cancer genome and transcriptome analyses of 1,699 paediatric leukaemias and solid tumours. Nature. 2018;555(7696):371–376. doi: 10.1038/nature25795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Aster JC, Pear WS, Blacklow SC. Notch signaling in leukemia. Annu Rev Pathol. 2008;3(1):587–613. doi: 10.1146/annurev.pathmechdis.3.121806.154300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Koch U, Radtke F. Notch in T-ALL: new players in a complex disease. Trends Immunol. 2011;32(9):434–442. doi: 10.1016/j.it.2011.06.005. [DOI] [PubMed] [Google Scholar]
- 15.Wang H, Zang C, Taing L, et al. NOTCH1-RBPJ complexes drive target gene expression through dynamic interactions with superenhancers. Proc Natl Acad Sci USA. 2014;111(2):705–710. doi: 10.1073/pnas.1315023111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Herranz D, Ambesi-Impiombato A, Palomero T, et al. A NOTCH1-driven MYC enhancer promotes T cell development, transformation and acute lymphoblastic leukemia. Nat Med. 2014;20(10):1130–1137. doi: 10.1038/nm.3665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Yashiro-Ohtani Y, Wang H, Zang C, et al. Long-range enhancer activity determines Myc sensitivity to Notch inhibitors in T cell leukemia. Proc Natl Acad Sci USA. 2014;111(46):E4946–E4953. doi: 10.1073/pnas.1407079111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Xing S, Li F, Zeng Z, et al. Tcf1 and Lef1 transcription factors establish CD8(+) T cell identity through intrinsic HDAC activity. Nat Immunol. 2016;17(6):695–703. doi: 10.1038/ni.3456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Raghu D, Xue H-H, Mielke LA. Control of lymphocyte fate, infection, and tumor immunity by TCF-1. Trends Immunol. 2019;40(12):1149–1162. doi: 10.1016/j.it.2019.10.006. [DOI] [PubMed] [Google Scholar]
- 20.Johnson JL, Georgakilas G, Petrovic J, et al. Lineage-determining transcription factor TCF-1 initiates the epigenetic identity of T cells. Immunity. 2018;48(2):243–257.e10. doi: 10.1016/j.immuni.2018.01.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Giambra V, Jenkins CE, Lam SH, et al. Leukemia stem cells in T-ALL require active Hif1α and Wnt signaling. Blood. 2015;125(25):3917–3927. doi: 10.1182/blood-2014-10-609370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Weber BN, Chi AW-S, Chavez A, et al. A critical role for TCF-1 in T-lineage specification and differentiation. Nature. 2011;476(7358):63–68. doi: 10.1038/nature10279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Germar K, Dose M, Konstantinou T, et al. T-cell factor 1 is a gatekeeper for T-cell specification in response to Notch signaling. Proc Natl Acad Sci USA. 2011;108(50):20060–20065. doi: 10.1073/pnas.1110230108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Brault V, Moore R, Kutsch S, et al. Inactivation of the β-catenin gene by Wnt1-Cre-mediated deletion results in dramatic brain malformation and failure of craniofacial development. Development. 2001;128(8):1253–1264. doi: 10.1242/dev.128.8.1253. [DOI] [PubMed] [Google Scholar]
- 25.Kühn R, Schwenk F, Aguet M, Rajewsky K. Inducible gene targeting in mice. Science. 1995;269(5229):1427–1429. doi: 10.1126/science.7660125. [DOI] [PubMed] [Google Scholar]
- 26.Murtaugh LC, Stanger BZ, Kwan KM, Melton DA. Notch signaling controls multiple steps of pancreatic differentiation. Proc Natl Acad Sci USA. 2003;100(25):14920–14925. doi: 10.1073/pnas.2436557100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Verbeek S, Izon D, Hofhuis F, et al. An HMG-box-containing T-cell factor required for thymocyte differentiation. Nature. 1995;374(6517):70–74. doi: 10.1038/374070a0. [DOI] [PubMed] [Google Scholar]
- 28.Yu S, Zhou X, Steinke FC, et al. The TCF-1 and LEF-1 transcription factors have cooperative and opposing roles in T cell development and malignancy. Immunity. 2012;37(5):813–826. doi: 10.1016/j.immuni.2012.08.009. [published correction appears in Immunity. 2014;40(1):166]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Wang F, Qi Z, Yao Y, et al. Exploring the stage-specific roles of Tcf-1 in T cell development and malignancy at single-cell resolution. Cell Mol Immunol. 2021;18(3):644–659. doi: 10.1038/s41423-020-00527-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Gudmundsson KO, Oakley K, Han Y, Du Y. Analyzing gene function in adult long-term hematopoietic stem cells using the interferon inducible Mx1-Cre mouse system. Methods Mol Biol. 2014;1194:313–325. doi: 10.1007/978-1-4939-1215-5_17. [DOI] [PubMed] [Google Scholar]
- 31.Iwasaki H, Mizuno S, Arinobu Y, et al. The order of expression of transcription factors directs hierarchical specification of hematopoietic lineages. Genes Dev. 2006;20(21):3010–3021. doi: 10.1101/gad.1493506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Laiosa CV, Stadtfeld M, Graf T. Determinants of lymphoid-myeloid lineage diversification. Annu Rev Immunol. 2006;24(1):705–738. doi: 10.1146/annurev.immunol.24.021605.090742. [DOI] [PubMed] [Google Scholar]
- 33.Rao SSP, Huntley MH, Durand NC, et al. A 3D map of the human genome at kilobase resolution reveals principles of chromatin looping. Cell. 2014;159(7):1665–1680. doi: 10.1016/j.cell.2014.11.021. [published correction appears in Cell. 2015:162(3):687-8] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Durand NC, Shamim MS, Machol I, et al. Juicer provides a one-click system for analyzing loop-resolution Hi-C experiments. Cell Syst. 2016;3(1):95–98. doi: 10.1016/j.cels.2016.07.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Chen C, Yu W, Tober J, et al. Spatial genome re-organization between fetal and adult hematopoietic stem cells. Cell Rep. 2019;29(12):4200–4211.e7. doi: 10.1016/j.celrep.2019.11.065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Dixon JR, Jung I, Selvaraj S, et al. Chromatin architecture reorganization during stem cell differentiation. Nature. 2015;518(7539):331–336. doi: 10.1038/nature14222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Schmitt AD, Hu M, Jung I, et al. A compendium of chromatin contact maps reveals spatially active regions in the human genome. Cell Rep. 2016;17(8):2042–2059. doi: 10.1016/j.celrep.2016.10.061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Liu Y, Nanni L, Sungalee S, et al. Systematic inference and comparison of multi-scale chromatin sub-compartments connects spatial organization to cell phenotypes. Nat Commun. 2021;12(1):2439. doi: 10.1038/s41467-021-22666-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Wendorff AA, Quinn SA, Rashkovan M, et al. Phf6 loss enhances HSC self-renewal driving tumor initiation and leukemia stem cell activity in T-ALL. Cancer Discov. 2019;9(3):436–451. doi: 10.1158/2159-8290.CD-18-1005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Guo Y, Xu Q, Canzio D, et al. CRISPR inversion of CTCF sites alters genome topology and enhancer/promoter function. Cell. 2015;162(4):900–910. doi: 10.1016/j.cell.2015.07.038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.León TE, Rapoz-D'Silva T, Bertoli C, et al. EZH2-deficient T-cell acute lymphoblastic leukemia is sensitized to CHK1 inhibition through enhanced replication stress. Cancer Discov. 2020;10(7):998–1017. doi: 10.1158/2159-8290.CD-19-0789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Chiaretti S, Li X, Gentleman R, et al. Gene expression profile of adult T-cell acute lymphocytic leukemia identifies distinct subsets of patients with different response to therapy and survival. Blood. 2004;103(7):2771–2778. doi: 10.1182/blood-2003-09-3243. [DOI] [PubMed] [Google Scholar]
- 43.Dunham I, Kundaje A, Aldred SF, et al. ENCODE Project Consortium An integrated encyclopedia of DNA elements in the human genome. Nature. 2012;489(7414):57–74. doi: 10.1038/nature11247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Lee W. A model to study the function of NPM-MLF1 in myelodysplasia [abstract] Blood. 2010;116(21):1571. bstract. [Google Scholar]
- 45.Li L, Leid M, Rothenberg EV. An early T cell lineage commitment checkpoint dependent on the transcription factor Bcl11b. Science. 2010;329(5987):89–93. doi: 10.1126/science.1188989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Trinquand A, Dos Santos NR, Tran Quang C, et al. Triggering the TCR developmental checkpoint activates a therapeutically targetable tumor suppressive pathway in T-cell leukemia. Cancer Discov. 2016;6(9):972–985. doi: 10.1158/2159-8290.CD-15-0675. [DOI] [PubMed] [Google Scholar]
- 47.Belver L, Yang AY, Albero R, et al. GATA3-controlled nucleosome eviction drives MYC enhancer activity in T-cell development and leukemia. Cancer Discov. 2019;9(12):1774–1791. doi: 10.1158/2159-8290.CD-19-0471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Kusakabe M, Sun AC, Tyshchenko K, et al. Synthetic modeling reveals HOXB genes are critical for the initiation and maintenance of human leukemia. Nat Commun. 2019;10(1):2913. doi: 10.1038/s41467-019-10510-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Cobas M, Wilson A, Ernst B, et al. β-catenin is dispensable for hematopoiesis and lymphopoiesis. J Exp Med. 2004;199(2):221–229. doi: 10.1084/jem.20031615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Jeannet G, Scheller M, Scarpellino L, et al. Long-term, multilineage hematopoiesis occurs in the combined absence of β-catenin and γ-catenin. Blood. 2008;111(1):142–149. doi: 10.1182/blood-2007-07-102558. [DOI] [PubMed] [Google Scholar]
- 51.Koch U, Wilson A, Cobas M, Kemler R, Macdonald HR, Radtke F. Simultaneous loss of beta- and gamma-catenin does not perturb hematopoiesis or lymphopoiesis. Blood. 2008;111(1):160–164. doi: 10.1182/blood-2007-07-099754. [DOI] [PubMed] [Google Scholar]
- 52.Graf T, Enver T. Forcing cells to change lineages. Nature. 2009;462(7273):587–594. doi: 10.1038/nature08533. [DOI] [PubMed] [Google Scholar]
- 53.Ha VL, Luong A, Li F, et al. The T-ALL related gene BCL11B regulates the initial stages of human T-cell differentiation. Leukemia. 2017;31(11):2503–2514. doi: 10.1038/leu.2017.70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Sivakumar A, de Las Heras JI, Schirmer EC. Spatial genome organization: from development to disease. Front Cell Dev Biol. 2019;7:18. doi: 10.3389/fcell.2019.00018. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.