

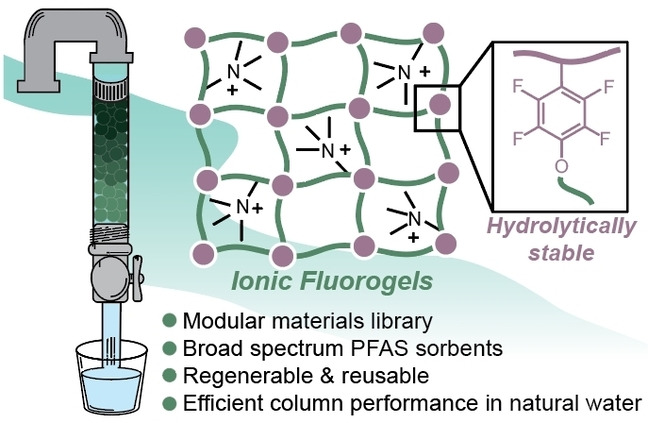
Abstract
PFAS are known bioaccumulative and persistent chemicals which pollute natural waters globally. There exists a lack of granular sorbents to efficiently remove both legacy and emerging PFAS at environmentally relevant concentrations. Herein, we report a class of polymer networks with a synergistic combination of ionic and fluorous components that serve as granular materials for the removal of anionic PFAS from water. A library of Ionic Fluorogels (IFs) with systematic variation in charge density and polymer network architecture was synthesized from hydrolytically stable fluorous building blocks. The IFs were demonstrated as effective sorbents for the removal of 21 legacy and emerging PFAS from a natural water and were regenerable over multiple cycles of reuse. Comparison of one IF to a commercial ion exchange resin in mini‐rapid small‐scale column tests demonstrated superior performance for the removal of short‐chain PFAS from natural water under operationally relevant conditions.
Keywords: Networks, PFAS, Polymers, Water Purification
The design and synthesis of a library of ion exchange resins that are selective for PFAS removal from real water enabled the evaluation of structure–property relationships of the chemically stable polymer networks with high PFAS affinity. The granular Ionic Fluorogel resin is a potent PFAS remediation sorbent and demonstrated excellent performance in translationally relevant flow‐through packed bed columns using natural water.
Introduction
Per‐ and polyfluoroalkyl substances (PFAS) are commercially relevant molecules which are widely used in consumer products and fire‐fighting foams, and as surfactants in production of fluorinated polymers.[ 1 , 2 , 3 ] PFAS are known environmental pollutants which contaminate ground and surface water worldwide.[ 4 , 5 , 6 , 7 ] Many PFAS are persistent in the environment and bioaccumulative, having been detected in human and other animal tissue across the globe,[ 8 , 9 , 10 , 11 ] and exposure to some PFAS leads to adverse health and environmental outcomes.[ 12 , 13 , 14 , 15 ]
The potential health effects of PFAS exposure, along with the expected regulation of these pollutants, has led to increased interest in PFAS remediation technologies. Recently, the U.S. Environmental Protection Agency (EPA) announced a PFAS Strategic Roadmap,[ 16 , 17 ] which includes PFAS testing and reporting requirements, toxicity assessments, and remediation efforts. Many states have regulated PFAS levels in drinking water, and six states have set enforceable PFAS Maximum Contaminant Limits (MCLs) for a range of PFAS. As an example, New Jersey set MCLs of 14 part‐per‐trillion (ng L−1 or ppt) for perfluorooctanoic acid (PFOA) and 13 ppt each for perfluoro‐1‐octanesulfonic acid (PFOS) and perfluorononanoic acid (PFNA) in drinking water. [18] Other states have set non‐enforceable Health Advisory Limits (HALs), such as North Carolina, which set an HAL of 140 ppt for hexafluoropropylene oxide‐dimer acid (GenX) in 2017. The regulatory pressure generated by these MCLs and HALs has initiated significant interest in removing a broad range of PFAS from contaminated waters.
Both state environmental agencies and the EPA have identified activated carbon, ion exchange resins (IX), and nanofiltration or reverse osmosis (RO) as potential solutions to remediate impacted water.[ 19 , 20 ] None of these technologies, however, is regarded as a platform solution to achieve high‐performance removal of PFAS in all cases. Although RO systems remove over 99 % of many PFAS from water, [21] these non‐selective separation processes produce large volumes of PFAS‐contaminated brine, suffer from fouling, and require significant energy input. Conversely, granular sorbents such as granular activated carbon (GAC) and IX are implemented in large‐scale flow‐through columns which can purify many volumes of water with little relative waste, and the required infrastructure for GAC and IX is established in many municipal water treatment plants. Therefore, granular sorbents are poised as drop‐in solutions for PFAS remediation. However, while state‐of‐the‐art GAC and IX technologies demonstrate adequate removal of long‐chain PFAS, they suffer from low selectivity for emerging short‐chain and branched PFAS.[ 22 , 23 , 24 , 25 , 26 ] Additionally, these technologies often suffer from limited regenerability, slow kinetics of sorption, and breakthrough of short‐chain PFAS into effluent water at relatively low treatment bed volumes.[ 27 , 28 , 29 , 30 , 31 , 32 , 33 ] Taken together, the state‐of‐the‐art indicates that there remains an outstanding challenge to develop new materials for remediation of short‐chain PFAS from water and understand the structure‐property‐performance relationships of the materials under complex conditions relevant to commercial use.
The presence of organic matter in natural waters at three to six orders magnitude higher concentration than PFAS makes removing PFAS from water to low part‐per‐trillion levels challenging. The lack of selectivity inherent to GAC and IX results in saturation of the resin binding sites by non‐fluorinated solutes, which leads to breakthrough of short‐chain PFAS in flow‐through columns used in home and municipal water treatment.[ 27 , 28 , 29 , 30 , 32 , 34 , 35 ] Many emerging PFAS remediation technologies have demonstrated efficient PFAS removal in batch studies conducted in pure water at low to moderate PFAS concentrations, but few have been studied in real water matrices or in flow‐through columns. In general, emerging PFAS remediation technologies leverage intermolecular interactions such as ion exchange,[ 36 , 37 , 38 , 39 , 40 ] hydrophobic or fluorous interactions,[ 39 , 40 , 41 , 42 , 43 , 44 , 45 ] or encapsulation by supramolecular receptors[ 46 , 47 , 48 , 49 , 50 , 51 , 52 ] to bind PFAS.
Recently, we reported a platform approach for granular resin design termed Ionic Fluorogels (IF), which exhibited highly efficient PFAS remediation from natural waters (Figure 1A). [53] We hypothesized that a synergistic combination of fluorous interactions[ 54 , 55 ] and ion exchange capability would result in selective sorption of anionic PFAS. Partitioning of fluorinated compounds into a perfluoropolyether (PFPE)‐based fluorous resin improved PFAS sorption selectivity relative to GAC or IX resins, and ammonium‐based electrostatic interactions resulted in stable PFAS binding, even in the presence of 20 000 times excess non‐fluorinated organic matter. However, the PFPE used in the initial proof‐of‐concept study, Fluorolink® MD700, was connected to polymerizable methacrylate end‐groups via ester and carbamate linkages, which we hypothesized would degrade over long lifetimes in flow‐through columns. Additionally, the use of only one PFPE limited systematic evaluation of the influence of network architecture on PFAS sorption.
Figure 1.
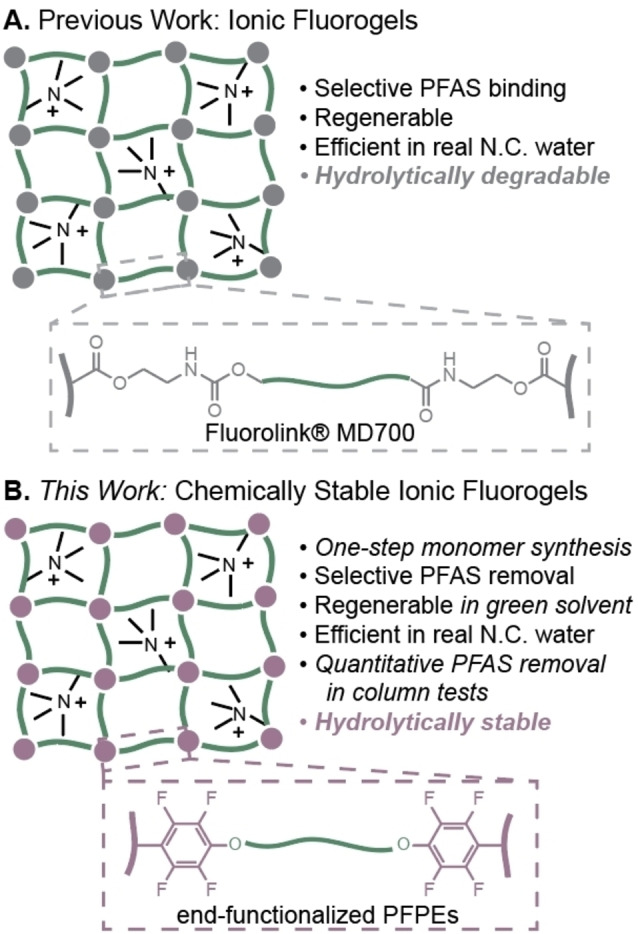
A) Previously reported Ionic Fluorogels leveraged carbamate‐terminated Fluorolink® MD700 for selective PFAS remediation from water. B) This work uses chain‐end modified PFPEs to enable an IF that demonstrates chemical stability and excellent performance under conditions relevant to commercial use.
Herein, we report structurally‐tunable and chemically‐stable Ionic Fluorogels that efficiently remove short‐chain PFAS from water in both batch experiments and flow‐through columns (Figure 1B). Chemical stability was achieved by end‐functionalization of PFPEs with polymerizable functionality connected through aryl‐ether linkages. Copolymerization of these PFPEs with an amine‐containing monomer resulted in a library of IFs with systematic variations of network architecture and cation density. Investigation of this library enabled insights into structure‐property relationships important to high affinity PFAS binding, wherein IFs with similar composition and different crosslink densities demonstrated disparate performance in PFAS sorption. In accelerated degradation studies, the hydrolytically stable IF maintained a low swelling ratio and consistent mass, while Fluorolink® MD700‐based IF degraded significantly, as measured via mass loss and nontargeted analysis using high‐resolution mass spectrometry. Finally, in a translationally relevant proof‐of‐concept experiment, mini‐Rapid Small‐Scale Column Tests (RSSCTs) of one IF significantly outperformed a state‐of‐the‐art ion exchange resin by demonstrating effective PFAS removal from natural North Carolina water over 1 year of simulated use.
Results and Discussion
In order to explore the influence of polymer network architecture, we identified two PFPE oligomers—Fluorolink® E10H (E10H) and fluorinated tetraethylene glycol (FTEG)—which have analogous chemical structures and different molecular weights (Figure 2A). Fluorolink® E10H has a similar molecular weight to the material used in our previous study (≈1600 g mol−1) [56] while FTEG is a short PFPE oligomer (410 g mol−1). Nucleophilic aromatic substitution of pentafluorostyrene (PFS) using the hydroxy end groups of the PFPEs (Figure 2A) yielded PFS‐capped oligomers of E10H and FTEG. The same nucleophilic aromatic substitution approach was used to synthesize an amine‐containing monomer 1 (Scheme S2) to ensure similar copolymerization kinetics between IF building blocks.
Figure 2.

A) Synthesis of PFPE crosslinkers (PFPEs starting materials shown in shaded inset). B) Synthesis of Ionic Fluorogel formulations (denoted as IF‐X), with varied compositions of PFS‐E10H, PFS‐FTEG, and 1. Resulting crosslinked materials after polymerization were ground and sieved to obtain resin particles 75–125 um.
Synthesis of IF was achieved through the thermally initiated radical copolymerization of PFS‐E10H, PFS‐FTEG, and 1 (Figure 2B) to form polymer networks, which were subsequently quaternized with methyl iodide, crushed into granules, and sieved in order to isolate IF grains of a desired size. The IF compositions in Figure 3A represent a library of materials that systematically vary the incorporation of fluorophilic and electrostatic components, as well as vary network architecture. The amine content of the IF was held constant at either 20 or 40 weight % amine comonomer, a range of amine incorporation that has provided efficient PFAS removal in previous studies. [53] The resulting materials were elastomers with low swelling ratios in 0.1 M NaCl. Notably, the swelling ratio was generally higher for materials with higher incorporation of ammonium ion, which is presumably due to both increased hydrophilicity and decreased crosslink density. The glass transition temperatures (T g) of the IFs did not change considerably with the subtle differences in chemical composition, and remained between 3.7 and 14 °C (Figures 3A and S2). Scanning electron microscopy images of the materials (Figure S31) demonstrated their granular nature and lack of appreciable porosity.
Figure 3.

A) Library of Ionic Fluorogels. a Calculated by Differential Scanning Calorimetry during the second heating cycle at a rate of 10 °C/min. b Ratio of swollen to dry mass of IF after swelling in 0.1 M NaCl. B) Batch equilibrium PFAS sorption by IFs. IX=Purolite PFA694AE. Water constituents: 200 mg L−1 NaCl and 20 mg L−1 humic acid; pH 6.4; Sorbent: 10 mg L−1; PFAS: (PFOA, PFHxA, GenX, 1 μg L−1 each); Equilibrium time: 21 h. Error bars: Standard deviation of 3 experiments. Some error bars are obscured by the columns.
Evaluation of structure‐property‐performance relationships of IFs was achieved through batch equilibrium PFAS sorption experiments under simulated natural water conditions (details in Supporting Information). Each IF was tested at environmentally‐relevant concentrations of three analytes that represent anionic long‐chain (PFOA), short‐chain (perfluorohexanoic acid, PFHxA), and branched (GenX) PFAS in water that contained a high concentration of organic matter (20 mg L−1 humic acid) and salt (200 mg L−1 sodium chloride). Notably, many IFs removed over 75 % each of PFOA, PFHxA, and GenX, and all IFs outperformed Purolite PFA694AE, a commercial IX used for PFAS remediation (Figure 3B). Varying network architecture had a marked effect on PFAS remediation; IF‐1 demonstrated nearly identical performance to IF‐20+, which is a structurally similar analogue from our previous work. [53] However IF‐3, which contained the same ammonium content but contained 40 wt % of the shorter PFS‐FTEG as a fluorous component, demonstrated enhanced performance for the remediation of the short‐chain PFAS substrates PFHxA and GenX. Using only the shorter PFS‐FTEG as a fluorous component (IF‐5, IF‐10), however, led to a decrease in performance compared to all the other materials. From these data, we conclude that a heterogeneous distribution of mesh size within the polymer network improves PFAS sorption. Direct comparison to other emerging PFAS remediation materials is challenging due to varied experimental conditions. However, optimal IF performance under these conditions was on par with the performance of other fluorinated PFAS remediation materials at higher sorbent loading [44] or in pure water spiked with PFAS. [50] In addition, we found that IFs synthesized with PFS incorporated as a comonomer resulted in poor PFAS sorption (Figure S3), likely due to the increased hydrophobicity limiting the swelling of these resins. Taken together, these data reveal that nuanced changes in resin microstructure and chemical composition can yield broad differences in PFAS removal.
Evaluation of the kinetics of sorption and binding capacity using IF‐1, a high performing resin, and GenX, a model anionic PFAS, provided more detailed analysis of IF performance. IF‐1 was chosen due to its structural similarity to previously reported resin IF‐20+, [53] which enabled a direct comparison between these first generation and second generation materials. IF‐1 demonstrated rapid, irreversible adsorption of GenX in pure water over 72 hours (Figure 4A). The construction of a GenX binding isotherm with IF‐1 enabled analysis of the capacity of the resin for PFAS and the mechanism of sorption. The concentration of IF‐1 was fixed at 100 mg L−1 in deionized water while the GenX concentration was varied from 0.20 to 50 mg L−1. The IF‐1 isotherm data demonstrated a better fit to the Langmuir relative to the Freundlich isotherm (Figure 4B, Table S2), indicating that surface adsorption of GenX to the IF particle is likely the dominant method of PFAS removal from water. [57] The Langmuir adsorption model indicated a binding capacity for GenX of 280 mg g−1 of resin, which is on par with the highest capacity for GenX in the literature at an environmentally relevant pH (278 mg g−1). [53]
Figure 4.
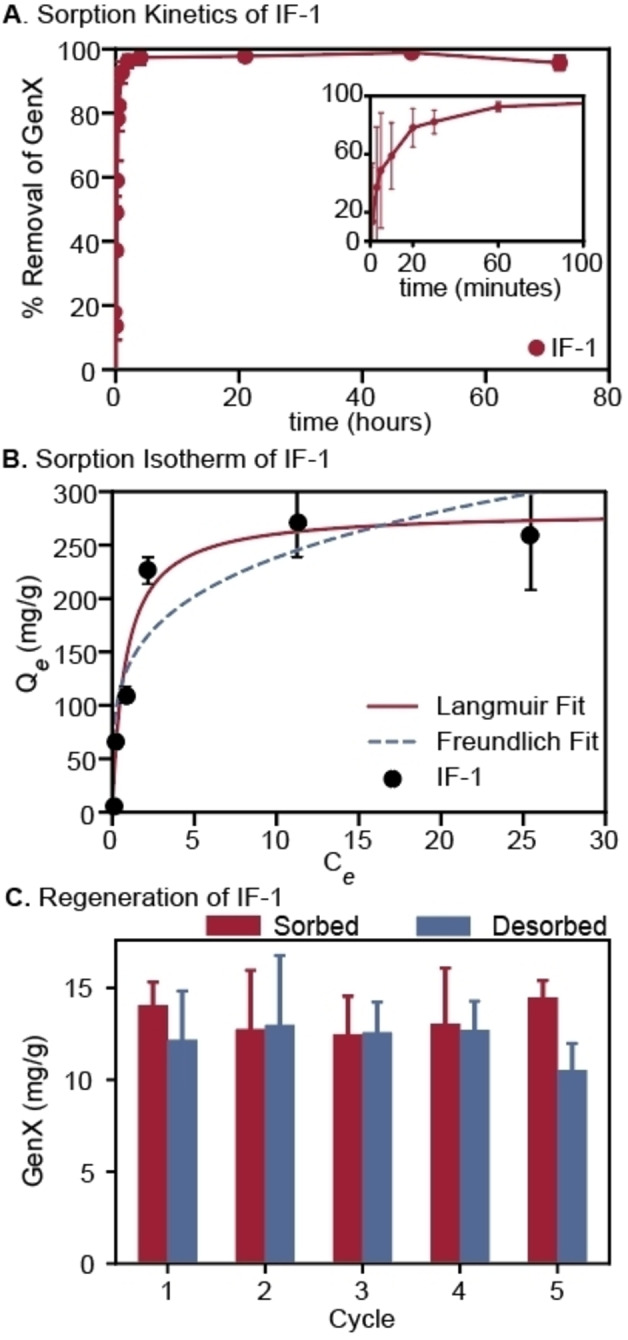
A) Time dependent GenX sorption by IF‐1 ([GenX]0=1 μg L−1; [IF‐1]=10 mg L−1). pH 9.7. B) GenX Sorption Isotherm by IF‐1 ([IF‐1]=100 mg L−1; [GenX]0=0.2–50 mg L−1). Lines show fit to Langmuir (red, solid) and Freundlich (blue, dashed) models. C) Regeneration and reuse over 5 cycles of IF‐1. IF‐1=20 mg, [GenX]=18 mg L−1 (20 mL); extraction with 400 mM ammonium acetate in 1 : 1 EtOH:H2O (20 mL). Error bars: Standard deviation of 3 experiments.
The regenerability of IF‐1 over multiple cycles of GenX sorption and desorption was investigated. Sorption experiments were performed by loading IF‐1 (20 mg) onto a PTFE syringe filter (0.45 μm, 25 mm diameter). GenX solution (18 mg L−1, 20 mL, quantified by LC‐MS) was passed through the filter over 2 minutes, achieving over 75 % removal of GenX per LC‐MS analysis of the filtrate, equivalent to 10–15 mg g−1 PFAS loading of the IF material. IF‐1 was regenerated by passing a solution of 400 mM ammonium acetate in 50 % aqueous ethanol (20 mL) through the filter. In this proof‐of‐concept experiment, GenX was adsorbed and desorbed from IF‐1 over 5 cycles (Figure 4C) with no decrease in GenX sorption efficiency, using an environmentally‐friendly solvent. [58] The ability to regenerate these IF enables their reuse and creates a concentrated solution of PFAS readily disposed of or destroyed through traditional or emerging technologies. [59]
Despite the performance similarities for PFAS remediation between IF‐1 and the previously reported resin (IF‐20+), we hypothesized that the aryl‐ether linker in these next‐generation materials would enhance their chemical stability in aqueous environments and prevent leaching of fluoropolymer from the resin. To probe the hydrolytic stability of the IF under practical conditions, we subjected the crosslinked polymer networks to accelerated degradation conditions,[ 60 , 61 ] which entailed submerging IF‐1 and IF‐20+ in basic solution (pH 8.7) at 50 °C. Resin samples were removed at specific time intervals over 56 days, washed with methanolic ammonium acetate to remove fluorinated oligomers, dried, and analyzed gravimetrically. As expected based on the chemical composition, IF‐1 remained stable in these aggressive conditions. In contrast, IF‐20+ demonstrated precipitous mass loss, as well as 38 % mass loss over 56 days (Figure 5A).
Figure 5.
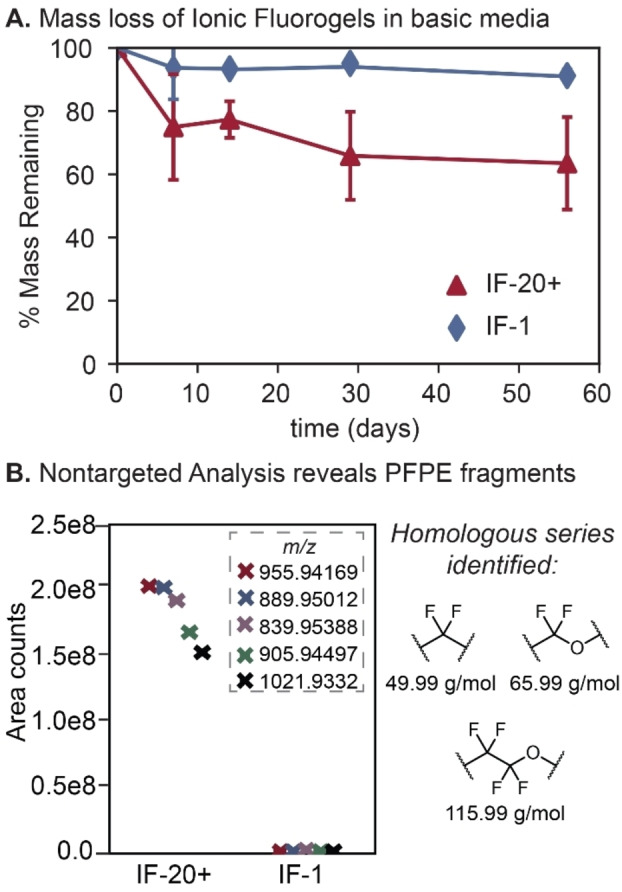
A) Mass loss of IF‐20+ (red triangles) and IF‐1 (blue diamonds) over 56 days. pH 8.7, 50 °C. Error bars: standard deviation of 3 experiments. Discs were washed with methanol before drying to determine mass loss. B) Averaged area counts (duplicate samples) of the 5 most‐intense peaks detected via nontargeted analysis of the extracts after washing the samples from day 28 of the mass loss study in methanol. The m/z values varied by combinations of 49.99, 65.99, and 115.99, indicative of PFPE repeat units.
To identify the degradation products of IF hydrolysis, nontargeted analysis via high‐resolution mass spectrometry was used to analyze the aqueous media and methanol extracts isolated from each degradation time point.[ 62 , 63 ] The nontargeted analysis of IF‐20+ revealed characteristic homologous series of m/z values for PFPEs in the IF‐20+ methanol extract (Figure 5B, Figures S12–S15). The five m/z values with the highest area counts via MS varied by approximately 49.99, 65.99 or 115.99, characteristic of the difluoromethylene (CF2), difluoromethylene glycol (CF2O), and tetrafluoroethylene glycol (CF2CF2O) (Figure 5B) repeat units present within Fluorolink® PFPEs.[ 62 , 63 ] Minimal peak areas were observed for the same m/z values in the aqueous IF‐20+ degradation media, indicating that degraded fluoropolymer remained trapped in the IF matrix until desorption with organic solvent (Figure S16). In contrast, nontargeted analysis of the aqueous media and methanol extracts of IF‐1 demonstrated over two orders of magnitude lower area counts of characteristic PFPE peaks relative to IF‐20+. These data support the hypothesis that end‐modification of Fluorolink® E10H with perfluorostyrene enables synthesis of more hydrolytically‐stable IFs.
After benchmarking this new class of IF resins with three representative anionic PFAS in simulated water (Figure 3B), IF performance in real water spiked with a chemically‐diverse mixture of PFAS at environmentally‐relevant concentrations was investigated. To that end, settled conventional (surface) water from the Orange County Water and Sewer Authority (OWASA, Chapel Hill, NC, pH 5.34, TOC≤0.50 mg L−1, conductivity=180 uS cm−1) was spiked with 21 emerging and legacy PFAS (Table S1) at 1 μg L−1 each. Batch equilibrium adsorption of each PFAS using IF‐1 (100 mg L−1) resulted in greater than 77 % removal for 18 of the 21 PFAS (Figure 6, acronyms defined in Table S1). Closer analysis of this data elucidated the relationship between PFAS chemical structure and adsorption efficiency. Notably, analytes that contain only two (PFMOAA) or three (PFBA, PMPA, and PFO2HxA) perfluorocarbons, respectively, demonstrate less selective removal efficiency, which we hypothesize is due to their limited fluorophilic interaction with the IF resin. PFAS with four perfluorocarbons (PFPeA, PFBS, PEPA, PFO3OA, NVHOS) demonstrated 87 to 97 % removal, and legacy PFAS with seven perfluorocarbons (PFOA) demonstrated over 98 % removal. Additionally, these data demonstrate clear trends wherein perfluorinated sulfonic acids are adsorbed more efficiently than their perfluorinated carboxylic acid counterparts, and linear PFAS are adsorbed more efficiently than their branched counterparts. These analyte structure‐adsorption relationships align with our previous work [53] and provide more detailed support for our proposed mechanism of PFAS adsorption.
Figure 6.

Batch equilibrium adsorption of 21 PFAS by IF‐1. [PFAS]0=1 μg L−1 each. Settled conventional water, OWASA, Chapel Hill, NC. Experiment time: 24 hours. Error bars: Standard deviation of 3 experiments.
Water purification using granular resins for household or municipal applications is most often conducted in a flow through packed bed geometry, where contaminated water is passed through a resin‐containing column for purification. [30] While batch experiments provide a fundamental understanding of the adsorption properties of granular resins, their performance in columns ultimately determines their translational relevance. Mini‐Rapid Small‐Scale Column Tests (mini‐RSSCTs) are a validated method to probe resin performance in a flow through packed bed column geometry, [64] where the figure of merit is the number of bed volumes of water that pass through the resin before 10 % of the influent solute is detected in the effluent water. [28] Comparative studies using mini‐RSSCTs were conducted using a high performing material, IF‐8, and Purolite PFA694AE IX resin. The influent water was composed of conventional settled water (OWASA, Chapel Hill, NC) spiked with environmentally relevant levels of PFOA, PFHxA, and GenX ([PFAS]0=500 ng L−1 each), and the effluent water was monitored for PFAS breakthrough over 72 hours (Figure 7). Less than 10 % breakthrough of the long‐chain PFOA was observed in either column over 150 000 bed volumes. The enhanced performance of the IF was demonstrated by its selectivity for short‐chain PFAS, PFHxA and GenX. These two PFAS demonstrated breakthrough for the IX resin at approximately 50 000 bed volumes of water. In contrast, the mini‐RSSCT that contained IF‐8 displayed no breakthrough of either PFHxA or GenX up to 180 000 bed volumes, which corresponds to 375 days of simulated continuous operation in a pilot‐scale treatment plant (Table S7). These are among the first flow‐through packed bed results reported for an emerging material for PFAS remediation from water. [65]
Figure 7.
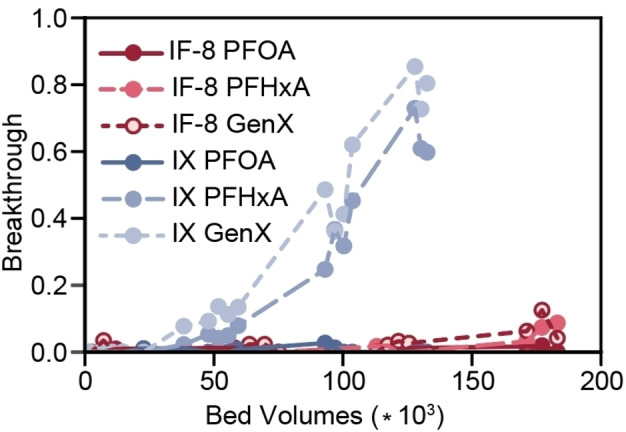
Mini‐Rapid Small‐Scale column results of IF‐8 (red traces) and IX (blue traces). IX=Purolite PFA694AE. [PFAS]0=500 ng L−1. Run time=72 hours. Breakthrough=[PFAS]effluent/[PFAS]0.
Conclusion
Hydrolytically stable IFs are poised as high‐performing materials for the remediation of both legacy and emerging anionic PFAS from water. The design and synthesis of a library of materials indicated that changes in network architecture translate to large differences in PFAS remediation in simulated natural water, while careful end group modification improves hydrolytic stability. These IFs remove a structurally diverse mixture of anionic PFAS at environmentally relevant concentrations from natural waters under batch equilibrium adsorption conditions, which enabled the identification of relationships between PFAS chemical structure and adsorption efficiency. One IF (IF‐8) demonstrated superior performance in a flow through column geometry for the removal of short‐chain PFAS from natural water compared with a commercial ion exchange resin, thus indicating the potential of this material to provide a solution for PFAS remediation that is superior and/or complementary to state‐of‐the‐art technologies.
Conflict of interest
Two provisional patent applications have been filed by the University of North Carolina at Chapel Hill on technology related to Ionic Fluorogels, including PCT/US2020/047365 and US #63/140306.
1.
Supporting information
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supporting Information
Acknowledgements
This work was supported by the Strategic Environmental Research and Development Program of the U.S. Department of Defense (ER20‐1211, F.A.L.), the NC Policy Collaboratory (F.A.L. and O.C.), the National institute of Environmental Health Sciences (P42ES031007, O.C.), and the NC Water Resources Research Institute (WRRI) and US Geological Survey (USGS) (2021‐2501‐01 and 2021‐3441‐02, O.C. and F.A.L.). I.M.M. acknowledges the Dobbins Fellowship. The authors thank Dr. Elango Kumarasamy for valuable discussions. The authors thank The Chemours Company for their donation of standards. Additionally, we thank James McCord for his R code that sorts masses into homologous series. The authors thank Orange Water and Sewer Authority (OWASA) for donation of settled conventional water. The UNC Department of Chemistry's NMR Core Laboratory provided expertise and instrumentation that enabled this study with support from National Science Foundation (CHE‐1828183 and CHE‐0922858). The views expressed in this publication are those of the authors and do not necessarily represent the views or policies of the U.S. Environmental Protection Agency. Any mention of trade names, manufacturers or products does not imply an endorsement by the United States Government or the U.S. Environmental Protection Agency. EPA and its employees do not endorse any commercial products, services, or enterprises.
I. M. Manning, N. Guan Pin Chew, H. P. Macdonald, K. E. Miller, M. J. Strynar, O. Coronell, F. A. Leibfarth, Angew. Chem. Int. Ed. 2022, 61, e202208150; Angew. Chem. 2022, 134, e202208150.
Contributor Information
Assoc. Prof. Orlando Coronell, Email: coronell@unc.edu.
Assist. Prof. Frank A. Leibfarth, Email: frankl@unc.edu.
Data Availability Statement
The data that support the findings of this study are available in the Supporting Information of this article.
References
- 1. Lindstrom A. B., Strynar M. J., Libelo E. L., Environ. Sci. Technol. 2011, 45, 7954–7961. [DOI] [PubMed] [Google Scholar]
- 2. Whitehead H. D., Venier M., Wu Y., Eastman E., Urbanik S., Diamond M. L., Shalin A., Schwartz-Narbonne H., Bruton T. A., Blum A., Wang Z., Green M., Tighe M., Wilkinson J. T., McGuinness S., Peaslee G. F., Environ. Sci. Technol. Lett. 2021, 8, 538–544. [Google Scholar]
- 3. Kotthoff M., Müller J., Jürling H., Schlummer M., Fiedler D., Environ. Sci. Pollut. Res. Int. 2015, 22, 14546–14559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Kunacheva C., Fujii S., Tanaka S., Seneviratne S. T. M. L. D., Lien N. P. H., Nozoe M., Kimura K., Shivakoti B. R., Harada H., Water Sci. Technol. 2012, 66, 2764–2771. [DOI] [PubMed] [Google Scholar]
- 5. Rostkowski P., Yamashita N., So I. M. K., Taniyasu S., Lam P. K. S., Falandysz J., Lee K. T., Kim S. K., Khim J. S., Im S. H., Newsted J. L., Jones P. D., Kannan K., Giesy J. P., Environ. Toxicol. Chem. 2006, 25, 2374–2380. [DOI] [PubMed] [Google Scholar]
- 6. Houtz E. F., Higgins C. P., Field J. A., Sedlak D. L., Environ. Sci. Technol. 2013, 47, 8187–8195. [DOI] [PubMed] [Google Scholar]
- 7. de Solla S. R., De Silva A. O., Letcher R. J., Environ. Int. 2012, 39, 19–26. [DOI] [PubMed] [Google Scholar]
- 8. Kotlarz N., McCord J., Collier D., Lea C., Strynar M., Lindstron A., Wilkie A., Islam J., Matney K., Tarte P., Polera M., Burdette K., DeWitt J., May K., Smart R., Knappe D., Hoppin J. A., Environ. Health Perspect. 2020, 128, 077005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Yao J., Pan Y., Sheng N., Su Z., Guo Y., Wang J., Dai J., Environ. Sci. Technol. 2020, 54, 13389–13398. [DOI] [PubMed] [Google Scholar]
- 10. Robuck A. R., McCord J. P., Strynar M. J., Cantwell M. G., Wiley D. N., Lohmann R., Environ. Sci. Technol. Lett. 2021, 8, 457–462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Kannan K., Corsolini S., Falandysz J., Fillmann G., Kumar K. S., Loganathan B. G., Mohd M. A., Olivero J., Van Wouwe N., Yang J. H., Aldous K. M., Environ. Sci. Technol. 2004, 38, 4489–4495. [DOI] [PubMed] [Google Scholar]
- 12. Steenland K., Fletcher T., Savitz D. A., Environ. Health Perspect. 2010, 118, 1100–1108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Barry V., Winquist A., Steenland K., Environ. Health Perspect. 2013, 121, 1313–1318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Fact Sheet: Human Health Toxicity Assessment for GenX Chemicals, 2021.
- 15. Evich M. G., Davis M. J. B., McCord J. P., Acrey B., Awkerman J. A., Knappe D. R. U., Lindstrom A. B., Speth T. F., Tebes-Stevens C., Strynar M. J., Wang Z., Weber E. J., Henderson W. M., Washington J. W., Science 2022, 375, eabg9065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. PFAS Strategic Roadmap: EPA's Commitments to Action 2021–2024, 2021.
- 17.L. Friedman, New York Times 2021.
- 18.New Jersey Division of Water Supply and Geoscience, 2020, 2–19.
- 19.L. Cummings, A. Matarazzo, N. Nelson, F. Sickels, C. Storms, New Jersey Drink. Water Qual. Inst. Treat. Subcomm. Rep. 2015.
- 20.“Treating PFAS in Drinking Water,” can be found under https://www.epa.gov/pfas/treating-pfas-drinking-water, n.d.
- 21. Tang C. Y., Fu Q. S., Robertson A. P., Criddle C. S., Leckie J. O., Environ. Sci. Technol. 2006, 40, 7343–7349. [DOI] [PubMed] [Google Scholar]
- 22. Herkert N. J., Merrill J., Peters C., Bollinger D., Zhang S., Hoffman K., Ferguson P. L., Knappe D. R. U., Stapleton H. M., Environ. Sci. Technol. Lett. 2020, 7, 178–184. [Google Scholar]
- 23. Ross I., McDonough J., Miles J., Storch P., Thelakkat Kochunarayanan P., Kalve E., Hurst J., Dasgupta S. S., Burdick J., Remediat. J. 2018, 28, 101–126. [Google Scholar]
- 24. Zaggia A., Conte L., Falletti L., Fant M., Chiorboli A., Water Res. 2016, 91, 137–146. [DOI] [PubMed] [Google Scholar]
- 25. Newell C. J., Adamson D. T., Kulkarni P. R., Nzeribe B. N., Stroo H., Remediat. J. 2020, 30, 7–26. [Google Scholar]
- 26. Vu C. T., Wu T., Environ. Sci. Water Res. Technol. 2020, 6, 2958–2972. [Google Scholar]
- 27. Appleman T. D., Dickenson E. R. V., Bellona C., Higgins C. P., J. Hazard. Mater. 2013, 260, 740–746. [DOI] [PubMed] [Google Scholar]
- 28. Park M., Wu S., Lopez I. J., Chang J. Y., Karanfil T., Snyder S. A., Water Res. 2020, 170, 115364. [DOI] [PubMed] [Google Scholar]
- 29. Zeng C., Atkinson A., Sharma N., Ashani H., Hjelmstad A., Venkatesh K., Westerhoff P., AWWA Water Sci. 2020, 2, e1172. [Google Scholar]
- 30. Liu C. J., Werner D., Bellona C., Environ. Sci. Water Res. Technol. 2019, 5, 1844–1853. [Google Scholar]
- 31. McCleaf P., Englund S., Östlund A., Lindegren K., Wiberg K., Ahrens L., Water Res. 2017, 120, 77–87. [DOI] [PubMed] [Google Scholar]
- 32. Woodard S., Berry J., Newman B., Remediat. J. 2017, 27, 19–27. [Google Scholar]
- 33. Du Z., Deng S., Chen Y., Wang B., Huang J., Wang Y., Yu G., J. Hazard. Mater. 2015, 286, 136–143. [DOI] [PubMed] [Google Scholar]
- 34. Belkouteb N., Franke V., McCleaf P., Köhler S., Ahrens L., Water Res. 2020, 182, 115913. [DOI] [PubMed] [Google Scholar]
- 35. Appleman T. D., Higgins C. P., Quiñones O., Vanderford B. J., Kolstad C., Zeigler-Holady J. C., Dickenson E. R. V., Water Res. 2014, 51, 246–255. [DOI] [PubMed] [Google Scholar]
- 36. Ji W., Xiao L., Ling Y., Ching C., Matsumoto M., Bisbey R. P., Helbling D. E., Dichtel W. R., J. Am. Chem. Soc. 2018, 140, 12677–12681. [DOI] [PubMed] [Google Scholar]
- 37. Ateia M., Attia M. F., Maroli A., Tharayil N., Alexis F., Environ. Sci. Technol. Lett. 2018, 5, 764–769. [Google Scholar]
- 38. Ateia M., Arifuzzaman M., Pellizzeri S., Attia M. F., Tharayil N., Anker J. N., Karanfil T., Water Res. 2019, 163, 114874. [DOI] [PubMed] [Google Scholar]
- 39. Chen P.-Y., Wang B., Zhuang S., Chen Y., Wei Y.-P., Colloids Surf. A 2021, 619, 126539. [Google Scholar]
- 40. Huang P. J., Hwangbo M., Chen Z., Liu Y., Kameoka J., Chu K. H., ACS Omega 2018, 3, 17447–17455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Tan X., Zhong J., Fu C., Dang H., Han Y., Král P., Guo J., Yuan Z., Peng H., Zhang C., Whittaker A. K., Macromolecules 2021, 54, 3447–3457. [Google Scholar]
- 42. Koda Y., Terashima T., Sawamoto M., J. Am. Chem. Soc. 2014, 136, 15742–15748. [DOI] [PubMed] [Google Scholar]
- 43. Koda Y., Terashima T., Takenaka M., Sawamoto M., ACS Macro Lett. 2015, 4, 377–380. [DOI] [PubMed] [Google Scholar]
- 44. Quan Q., Wen H., Han S., Wang Z., Shao Z., Chen M., ACS Appl. Mater. Interfaces 2020, 12, 24319–24327. [DOI] [PubMed] [Google Scholar]
- 45. Mantripragada S., Deng D., Zhang L., Chemosphere 2021, 283, 131235. [DOI] [PubMed] [Google Scholar]
- 46. Alsbaiee A., Smith B. J., Xiao L., Ling Y., Helbling D. E., Dichtel W. R., Nature 2016, 529, 190–194. [DOI] [PubMed] [Google Scholar]
- 47. Xiao L., Ling Y., Alsbaiee A., Li C., Helbling D. E., Dichtel W. R., J. Am. Chem. Soc. 2017, 139, 7689–7692. [DOI] [PubMed] [Google Scholar]
- 48. Ling Y., Klemes M. J., Xiao L., Alsbaiee A., Dichtel W. R., Helbling D. E., Environ. Sci. Technol. 2017, 51, 7590–7598. [DOI] [PubMed] [Google Scholar]
- 49. Xiao L., Ching C., Ling Y., Nasiri M., Klemes M. J., Reineke T. M., Helbling D. E., Dichtel W. R., Macromolecules 2019, 52, 3747–3752. [Google Scholar]
- 50. Klemes A. M. J., Ling Y., Ching C., Wu V., Helbling E., Dichtel W. R., Angew. Chem. Int. Ed. 2019, 58, 12049–12053; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2019, 131, 12177–12181. [Google Scholar]
- 51. Shetty D., Jahović I., Skorjanc T., Erkal T. S., Ali L., Raya J., Asfari Z., Olson M. A., Kirmizialtin S., Yazaydin A. O., Trabolsi A., ACS Appl. Mater. Interfaces 2020, 12, 43160–43166. [DOI] [PubMed] [Google Scholar]
- 52. Cao F., Wang L., Ren X., Sun H., J. Appl. Polym. Sci. 2016, 133, 43912. [Google Scholar]
- 53. Kumarasamy E., Manning I. M., Collins L. B., Coronell O., Leibfarth F. A., ACS Cent. Sci. 2020, 6, 487–492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Pollice R., Chen P., J. Am. Chem. Soc. 2019, 141, 3489–3506. [DOI] [PubMed] [Google Scholar]
- 55. Horváth I. T., Acc. Chem. Res. 1998, 31, 641–650. [Google Scholar]
- 56. Choi Y.-S., Kim N. K., Kang H., Jang H.-K., Noh M., Kim J., Shon D.-J., Kim B.-S., Lee J.-C., RSC Adv. 2017, 7, 38091–38099. [Google Scholar]
- 57. Swenson H., Stadie N. P., Langmuir 2019, 35, 5409–5426. [DOI] [PubMed] [Google Scholar]
- 58. Capello C., Fischer U., Hungerbühler K., Green Chem. 2007, 9, 927–934. [Google Scholar]
- 59. Cui J., Gao P., Deng Y., Environ. Sci. Technol. 2020, 54, 3752–3766. [DOI] [PubMed] [Google Scholar]
- 60. Woodard L. N., Grunlan M. A., ACS Macro Lett. 2018, 7, 976–982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Xu X.-J., Sy J. C., Shastri V. P., Biomaterials 2006, 27, 3021–3030. [DOI] [PubMed] [Google Scholar]
- 62. Washington J. W., Rosal C. G., Mccord J. P., Strynar M. J., Lindstrom A. B., Bergman E. L., Goodrow S. M., Tadesse H. K., Pilant A. N., Washington B. J., Davis M. J., Stuart B. G., Jenkins T. M., Science 2020, 368, 1103–1107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. McCord J. P., Strynar M. J., Washington J. W., Bergman E. L., Goodrow S. M., Environ. Sci. Technol. Lett. 2020, 7, 903–908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Crittenden J. C., Reddy P. S., Arora H., Trynoski J., Hand D. W., Perram D. L., Summers R. S., J. AWWA 1991, 83, 77–87. [Google Scholar]
- 65. Najm I., Gallagher B., Vishwanath N., Blute N., Gorzalski A., Feffer A., Richardson S., AWWA Water Sci. 2021, 3, e1245. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supporting Information
Data Availability Statement
The data that support the findings of this study are available in the Supporting Information of this article.