

Abstract
Reconstitution of membrane proteins into large unilamellar vesicles is an essential approach for their functional analysis under chemically defined conditions. The orientation of the protein in the liposomal membrane after reconstitution depends on many parameters, and its assessment is important prior to functional measurements. Common approaches for determining the orientation of a membrane-inserted protein are based on limited proteolytic digest, impermeable labeling reagents for specific amino acids, or membrane-impermeable quenchers for fluorescent proteins. Here, we describe a simple site-specific fluorescent assay based on self-labeling enzyme tags to determine the orientation of membrane proteins after reconstitution, exemplified on a reconstituted SNAP-tag plant H + -ATPase. This versatile method should benefit the optimization of reconstitution conditions and the analysis of many types of membrane proteins.
Graphical abstract:

Keywords: Proteoliposomes , Sidedness , SNAP-tag , Reconstitution , Membrane protein , Fluorescence
Background
Reconstitution of purified proteins into model membranes is an essential approach for investigating membrane protein function under defined conditions outside the complex cellular environment ( Rigaud and Lévy, 2003 ; Murray et al., 2014; Amati et al., 2020). Typically, proteins are reconstituted with phospholipids into large unilamellar vesicles with 100–200 nm diameters. The orientation of the protein in the liposomal membrane after reconstitution depends on many parameters, including the type of protein, the lipid composition, and the reconstitution procedure ( Tunuguntla et al., 2013 ; Amati et al., 2020 ). Knowledge of the orientation distribution of the protein is of crucial significance for interpreting its function in the proteoliposomal in vitro system and for the design of proteoliposome experiments. A common approach for determining the orientation of a membrane-inserted protein is the protease-mediated cleavage (e.g., Schuette et al., 2004; Serek et al., 2004; Islam et al., 2013; Eisinger et al., 2018). This assay is based on the principle that an externally added protease can cleave only the accessible portions of the membrane-inserted protein, which requires several critical steps such as limited proteolytic digest, stopping the proteolytic reaction, and fragment analysis (e.g., by SDS-PAGE).
Alternative approaches are based on impermeable labeling reagents for specific amino acids or membrane-impermeable quenchers for fluorescent proteins (e.g., Zhang et al., 2003; Marek et al., 2011; Deutschmann et al., 2022). The protocol described here takes advantage of the SNAP-tag technology (Keppler et al., 2004; Cole, 2013 ), whereby a protein can be labeled site-specific after expression or purification with a diversity of dyes, which are either membrane-permeable or impermeable ( Figure 1 ). This approach can be extended to other self-labeling enzyme tags including SNAP-tag, HALO-tag, or CLIP-tag and, thus, can be easily customized for the protein of interest. In contrast to other labels such as cysteine labeling, side-specific, stoichiometric 1:1 labeling without changing the amino acid sequence is ensured. Compared to protease-based approaches, labeling with dyes of different permeability allows analysis under nondestructive conditions for the vesicle and the protein. The presented strategy will be useful for the optimization of reconstitution conditions and the analysis of many types of membrane proteins. Thus, this new assay expands the toolbox of fluorescence-based methods for the estimation of membrane protein orientation in liposomes.
Figure 1. SNAP-labeling process.

The membrane protein of interest is expressed with SNAP-tag enabling site-specific labeling. Based on the human DNA repair enzyme O 6 -alkylguanine-DNA-alkyltransferase, the SNAP enzyme undergoes self-labeling via a covalent bond with O 6 -benzylguanine derivatives. Depending on the characteristics of the derivative, the labeling substrate is either membrane-permeable like SNAP-647-SIR or membrane-impermeable like SNAP-Alexa488 [chemical structures based on Lukinavičius et al. (2013) and Wilhelm et al. (2021), and drawn with https://chem-space.com ].
Things to consider before starting
-
Choice of labeling substrate
Here, we used a SNAP-tagged fused membrane protein and benzylguanine-derived substrates. Labeling of the SNAP-tag is irreversible and quantitative, and thus well suited for the detection and quantitation of labeled proteins via in-gel fluorescence scanning of SDS-PAGE gels. If alternative labeling systems based on, for example, CLIP-tag or ACP-tag, are used, the according substrate has to be chosen as basis for the labeling dye.
-
Choice of self-labeling fluorophore
The assay relies on the use of a membrane-impermeable dye in combination with a membrane-permeable dye. In addition, both dyes should be selected for low spectral overlap and based on the imaging system available.
-
Choice of lipid environment
Here, we describe labeling conditions optimized for proteins reconstituted in 1-palmitoyl-2-oleoyl- sn -glycero-3-phosphocholine (POPC) / 1-palmitoyl-2-oleoyl- sn -glycero-3-phospho-(1′-rac-glycerol) (POPG) (9:1 mol/mol) vesicles. In other lipid environments, labeling duration and label concentrations might need adjustment.
-
Additives in buffer
A reducing agent (e.g., DTT) is added to the labeling buffer to increase labeling efficiency, due to a reactive cysteine in the SNAP-tag. However, if its presence is affecting the protein, the experiment could be tested without DTT, with optimized labeling time and label concentrations. However, under oxidizing conditions, labeling might not be achieved efficiently. The presence of chelating reagents such as EDTA should be avoided as the SNAP-tag protein contains a structural Zn 2+ ion.
Biological materials
The exemplary detergent-solubilized SNAP-tagged membrane protein is a C-terminally truncated version (∆73) of the Arabidopsis thaliana auto-inhibited H + -ATPase isoform 2 (AHA2-SNAP) with additional StrepII and hexahistidine N-terminal tags. AHA2-SNAP was heterologously expressed in the Saccharomyces cerevisiae strain RS-72 ( MATa, ade1-100 his4-519 leu2-3,112 ; endogenous proton pump PMA1 gene is under control of GAL1 promoter) ( Cid et al., 1987 ) and purified via the His-tag resulting in 5–10 mg/mL protein in storage buffer (see Recipe 3), stored at -80 °C ( Lanfermeijer et al., 1998 ).
Note: The protocol can also be applied to purified membrane proteins prepared differently in combination with liposomal reconstitution procedures not based on preformed liposomes, as described here. However, these membrane proteins may have different preferences for lipids, with respect to the phospholipid headgroup and the lipid fatty acid composition.
Materials and Reagents
All catalog numbers provided below shall serve as guide; alternative sources can be used as well.
Materials
2 mL disposable syringe (Henry Schein, catalog number: 9003017)
Pipette tips 10 µL, 200 µL, and 1,000 µL (Sarstedt, catalog numbers: 70.760.002, 70.3030.020, and 70.3050.020)
Reaction tubes 1.5 mL, 2 mL, 15 mL, and 50 mL (Sarstedt, catalog numbers: 72.690.001, 72.691, 62.554.502, and 62.547.254)
Glass beads, 3 mm (Supelco, catalog number: 1040150500)
Round bottom glass tube (Roth, catalog number: NY90.1)
Screw cap, ND8 (Roth, catalog number: NL96.1)
Screw neck ND8 vial (Roth, catalog number: KE27.1)
Sterican disposable 26 gauge needle, 0.45 × 12 mm (Braun, catalog number: 4665457)
Chemicals
Chloroform, ethanol-stabilized and certified for absence of phosgene and HCl (Roth, catalog number: 7331.2)
Methanol (VWR, catalog number: 20847.307)
4-Morpholinepropanesulfonic acid, 3-(N-Morpholino)propanesulfonic acid (MOPS) (Sigma, catalog number: M3183)
Dimethylsulfoxide (DMSO) (Sigma, catalog number: 34943)
KOH (Fisher Chemical, catalog number: P/5640/60)
K 2 SO 4 (VWR, catalog number: 26997.293)
Sodium acetate (Sigma, catalog number: S2889)
2-( N -Morpholino)-ethane sulphonic acid (MES) (Roth, catalog number: 4256.2)
Glycerol (VWR, catalog number: 24388.295)
KCl (Honeywell, catalog number: 31248)
Ethylenediaminetetraacetic acid (EDTA) (Sigma, catalog number: E6758)
threo -1,4-dimercapto-2,3-butanediol (DTT) (Sigma, catalog number: 43819)
Liquid nitrogen
N-dodecyl β-maltoside (DDM) (Glycon, catalog number: D97002)
N-octyl β-D-glucopyranoside (OG) (Glycon, catalog number: D97001)
Sephadex G50, fine (Cytiva, catalog number: 17004201)
BioBeads (Bio-Rad, catalog number: 152-3920).
Bromophenol blue (Roth, catalog number: A512.3)
Tris(hydroxymethyl)aminomethan (Tris) (Sigma, catalog number: T1503)
Sucrose (Fisher Chemical, catalog number: S/8600/60)
N,N,N′,N′-tetramethyl ethylenediamine (TEMED) (Merck, catalog number: 8.08742.0250)
Ammonium peroxydisulfate (APS) (Roth, catalog number: 9592.2)
Sodium dodecyl sulfate (SDS) (Sigma, catalog number: L3771)
Aluminum sulfate hydrate (Roth, catalog number: 3731.1)
Ethanol absolute ≥99.8% (VWR, catalog number: 20821.321)
Acrylamide (Roth, catalog number: 3029.1)
Orthophosphoric acid (VWR, catalog number: 20624.295)
Coomassie brilliant blue G-250 (Serva, catalog number: 35050)
PageRuler TM Plus Prestained Protein Ladder, 10–250 kDa (Thermo Scientific, catalog number: 26619)
Reconstitution buffer (see Recipes)
Protein storage buffer (see Recipes)
1 M OG stock (see Recipes)
0.5 M DTT stock (see Recipes)
Sephadex G50 fine slurry (see Recipes)
Pre-washed BioBeads (see Recipes)
Laemmli buffer (see Recipes)
20% SDS stock (see Recipes)
SDS-PAGE running buffer (see Recipes)
SDS gel (see Recipes)
10% APS stock (see Recipes)
1 M Tris (see Recipes)
Colloidal Coomassie staining solution (see Recipes)
Colloidal Coomassie destaining solution (see Recipes)
Lipids
1-palmitoyl-2-oleoyl- sn -glycero-3-phosphocholine (POPC) (Avanti Polar Lipids, catalog number: 850457)
1-palmitoyl-2-oleoyl- sn -glycero-3-phospho-(1'-rac-glycerol) (sodium salt) (POPG) (Avanti Polar Lipids, catalog number: 840457)
SNAP dyes in DMSO
SNAP-Surface Alexa488 (New England BioLabs Inc., catalog number: S9129S)
-
SNAP-Cell 647-SiR (New England BioLabs Inc., catalog number: S9102S)
Note: The assay has also been successfully performed using SNAP-Surface 647 (New England BioLabs Inc., catalog number: S9136S) and SNAP-Cell Oregon Green (New England BioLabs Inc., catalog number: S9104S)
Equipment
Analytical balance (Sartorius Entris-i II, 220 g/0.1 mg, Buch Holm, catalog number: 4669128)
-
Avanti mini extruder set (Avanti Polar Lipids, catalog number: 610000)
Filter supporter for extruder (Polyester Drain Disc, 10 mm; Cytiva, catalog number: 230300)
Membrane 200 nm for extruder (Cytiva, catalog number: 10417004)
1,000 μL gas-tight syringe (Avanti Polar Lipids, catalog number: 610017)
Centrifuge with rotor for 15 and 50 mL polypropylene tubes (Eppendorf 5810 R; Wesseling, Germany)
Chemidoc MP imaging system (Bio-Rad) with illumination at 460–490 nm and 520–545 nm with 530/28 nm and 695/55 nm emission filters, respectively.
Eppendorf Research ® plus pipettes P2.5, P20, P200, and P1000 (Eppendorf, catalog numbers: 3123000012, 3123000039, 3123000055, and 3123000063)
Glass desiccator Boro 3.3 with socket in lid, 20 cm, including stopcock (BRAND GmbH, catalog number: 65238)
Hamilton 700 Series syringes 25 µL, 100 µL, and 1,000 µL (Hamilton Company, Nevada, USA)
Head-over-head turning device (neoLab, catalog number: 7-0045)
Mini-PROTEAN ® Tetra cell system (Bio-Rad, catalog number: 1658000)
Vacuum Pump V-100 with interface I-100 and rotary evaporator Rotavapor ® R-100, SJ29/32, V, 220–240V (Buchi, catalog numbers: 11593636, 11593655D and 11100V111, 11061895)
Vortex mixer (Scientific Industries Inc., model: Vortex Genie 2, catalog number: SI-0236)
Water bath (Julabo, CORIO C-BT5, catalog number: 9011305)
Software
ImageLab software version 5.2.1 (Bio-Rad)
Procedure
When using other liposomal reconstitution procedures not based on preformed liposomes as described here, skip sections A and B and start at section C.
-
Liposome preparation
Since lipids stick to plasticware, glassware has to be used until liposomes are formed. Furthermore, chloroform can extract components from plasticware, reinforcing the need to use glassware. Glass tubes as well as Hamilton syringes are used for the lipid film preparation. To reduce unwanted evaporation of chloroform during pipetting, lipid stocks are handled on ice.
Note: Chloroform is a hazardous solvent. Conduct all work in a fume hood while wearing proper protective clothing.
-
Lipids are received in chloroform packaged in sealed glass ampoules; store at -20 °C until use.
Note: For long-term storage, evaporate the solvent and store the lipids at -80 °C to avoid oxidation of unsaturated lipids.
To prepare lipid stocks, transfer 10 mg aliquots from the Avanti glass ampoule into glass screw neck vials. Evaporate the solvent from the glass vials in a desiccator at 250 mbar for 3 h with an additional incubation for 1 h at 30 mbar. Close the vials with screw caps and store at -80 °C until further use.
-
Remove desired lipid stocks from freezer, place on ice, and dissolve in methanol:chloroform (1:1, v/v) to a final lipid concentration of 10 mg/mL.
Note: Lipids other than POPC and POPG may have limited or very poor solubility in chloroform:methanol and require a mixture of chloroform/methanol/water.
For a ratio of 9:1, transfer 450 µL of 10 mg/mL POPC and 50 µL of 10 mg/mL POPG into a grounded round bottom glass tube for a final lipid film of 5 mg.
Dry lipid film in a rotary evaporator at 150 mbar until no residual solvent is visible.
Go down to the lowest possible pressure (here 30 mbar) and dry lipid film for an additional 1 h.
-
Rehydrate the lipid film (5 mg, greasy film in the bottom of the tube, appearance ranges from clear to whitish depending on lipid composition) with 334 μL of reconstitution buffer (see Recipe 1) to yield a final lipid concentration of 15 mg/mL (approximately 20 mM total phospholipid). Complete rehydration is achieved by addition of a glass bead and vortexing until no lipid film is visible on the tube bottom anymore, but for a minimum of 5 min.
Note: The mixing of the lipid film with aqueous solution results in spontaneous formation of liposomes .
-
Subject the lipid solution to five freeze-thawing cycles by placing the tube alternating into liquid nitrogen for 30 s for freezing and in a water bath at 50 °C for 90 s for thawing.
Note: Wear face shield and insulating gloves when handling liquid nitrogen. Use glass tubes with high thermal shock resistance.
Assemble the Avanti mini extruder according to the manual with two filter supporters on each side and two 200 nm pore sized polycarbonate membranes in between.
-
Check tightness by flushing with 1 mL of reconstitution buffer (see Recipe 1), i.e., pass back and forth between syringes three to four times. Only continue if the buffer volume stays the same for each passing.
Note: In the first passage, buffer volume can be reduced due to the void volume of the extruder.
Fill one Hamilton glass syringe with approximately 334 µL of liposome solution and place the filled syringe into one end of the mini extruder.
-
Carefully place an empty syringe into the opposite end of the mini extruder. The plunger of the
empty syringe should be moved completely into the syringe barrel.
-
Perform manual extrusion through assembled Avanti mini extruder with 21 passages.
Note: Number of passages needs to be uneven to finish in the other syringe, as residual suspension in the starting syringe might have never passed the extruder.
Final liposomes can be stored in 1.5 mL microcentrifuge tubes in the fridge for up to two weeks.
-
-
Reconstitution
Note: Reconstitution is performed here by using detergent-mediated vesicle destabilization ( Rigaud et al., 1988 ). The method was optimized for the needs of the used protein. For detailed reviews on this topic see Amati et al. (2020).
-
Dilute 66 μL liposome suspension (4–5 mM lipid concentration) with 124 μL of reconstitution buffer (see Recipe 1) in a 1.5 mL microcentrifuge tube.
Destabilize liposomes with 9 μL of detergent (45 mM final concentration, OG stock, see Recipe 2) with 5 min head-over-head rotation.
Note: Addition of the appropriate amount of detergent can be monitored by measuring light scattering of the liposome-containing solution at 600 nm using a spectrofluorometer. With increasing concentrations of detergent added to the vesicles, the absorbance starts to decrease (point of detergent saturation) until complete solubilization of the vesicles. The best stage for protein insertion for AHA2 was on the turning point between detergent saturation and total solubilization of the liposomes.
Add 25 μg of purified AHA2 (in storage buffer, see Recipe 3) to the liposome–detergent mixture and incubate with head-over-head rotation for another 5 min.
Meanwhile, prepare the G50 filtration column: cut two filter supporters from the side to the middle and place in a disposable 2 mL syringe without the plunger to create a stopper. Place syringe into a disposable 15 mL reaction tube, add 3 mL Sephadex G50 fine slurry (see Recipe 5), centrifuge at 180 × g for 5 min, and transfer column to a new 15 mL reaction tube (see Figure 2 ).
Add lipid–protein–detergent suspension on top of the column and incubate for 5 min.
-
Centrifuge at 180 × g for 8 min.
Note: This size-exclusion column is trapping free detergent, i.e., OG.
-
Transfer the flow-through to a 2 mL microcentrifuge tube.
Note: The wider bottom of the 2 mL compared to the 1.5 mL tube is important for better rotation and mixing of the liquid in the next step.
-
Add 50 mg of pre-washed BioBeads (see Recipe 6) to the sample and incubate for 60 min, with head-over-head rotation.
Note: BioBeads are hydrophobic and extract the leftover detergent from the liposomes by adsorption.
-
Punch a small hole in the bottom of the 2 mL microcentrifuge tube with a 26-gauge needle and place it in a 15 mL reaction tube.
Note: The plastic is thickest at the center of the bottom; punch next to this for easier handling.
Punch a hole in the lid of the 2 mL microcentrifuge tube for pressure equilibrium and centrifuge at 180 × g for 30 s to separate liposome sample from pre-washed BioBeads.
Move the flow-through to a fresh 1.5 mL microcentrifuge tube. Store in the fridge for up to four days, use as soon as possible.
-
-
Asymmetric protein labeling and analysis
Liposomes containing the reconstituted SNAP-tagged membrane protein are labeled separately or sequentially with membrane-impermeable and permeable SNAP dyes as outlined in Figure 3 .
Dilute SNAP dye stock 1:100 with reconstitution buffer supplemented with 1 mM DTT (added from a 0.5 M DTT stock, see Recipe 4) to yield a 10 µM diluted stock.
Pipette 10 µL of liposome suspension (approximately 62.5–125 µg protein, 5–10 pmol) in six 1.5 mL microcentrifuge tubes. This corresponds to the three different sample types (I, II, III) in duplicates.
Add the impermeable SNAP-dye in a 1:2 (mol protein:mol dye) excess (1–2 µL from 10 µM SNAP-Surface Alexa488 diluted stock) to sample I and II.
-
Incubate for 2 h at 25 °C.
Note: To achieve maximum labeling of the protein, use an excess of the dyes (at least 1:2, mol protein:mol dye) and test different incubation times until the labeling intensity does not further increase. We have experienced slower labeling with the red SNAP-Cell 647-SIR compared to the green dye SNAP-Surface Alexa488.
Add membrane-permeable dye (1–2 µL from 10 µM SNAP-Cell 647-SiR diluted stock) to sample II and III.
-
Incubate for 2 h at 25 °C.
Note: Consider testing different times to reach maximum labeling.
Stop reaction by adding 5 µL of Laemmli buffer (see Recipe 7).
-
Load samples directly on 10% 1.5 mm SDS gel (see Recipe 10).
Note: Membrane protein samples are not boiled before loading to prevent aggregation.
Use marker for the molecular weight (e.g., 3 µL of PageRuler TM Plus Prestained Protein Ladder, 10–250 kDa).
Run SDS-PAGE for approximately 2 h at 100 V, until running front is 0.25 cm from the end of the gel.
-
Take the gel out of the cassette and image with Chemidoc imager with pre-programmed option for Alexa488 (460–490 nm, 530/28 nm filter) and Alexa647 (625–675 nm, 695/50 nm filter).
Note: Adjust exposure times so that the signal is not saturated, since we want to quantify it. Due to the weak signal of some SNAP dyes exposure, times up to 10 s might be used. First, take an image of the entire gel to see the free dye; most of the actual protein–dye conjugate is not yet visible. Afterwards, cut the lower part containing the free dye. Take a second image with higher exposure time to properly visualize the protein-dye bands.
Save the images as ImageLab inbuilt data format “.scn.” Exemplary results from dual color labeling experiment are shown in Figure 5 .
-
Colloidal Coomassie staining, according to Dyballa and Metzger (2009)
Transfer the gel into a bowl with ddH 2 O and shake twice for 10 min to get rid of residual SDS.
Stain shaking in Colloidal Coomassie solution (see Recipe 13) for up to 4 h.
Destain for several hours with destaining solution (see Recipe 14).
Use pre-programmed option for Coomassie stained gels (white trans illumination) in Chemidoc.
Save the images as ImageLab inbuilt data format “.scn.”
Figure 2. Preparation of G50 column.

(1) Two filter papers are cut from the side up to the middle. (2) After wetting the papers with the reconstitution buffer, they are inserted into a plastic syringe without the plunger. (3) The pre-soaked G50 slurry is applied to the syringe, which is placed in a 15 mL tube. (4) After centrifugation (180 × g for 5 min) the G50 column is transferred to a fresh 15 mL tube and is ready to use.
Figure 3. Double labeling assay workflow.

The proteoliposome sample is split into conditions I, II, and III. Sample I is only labeled with the membrane-impermeable SNAP-Alexa488 and serves later as bleed-through correction. Sample II is labeled with the membrane-permeable SNAP-647-SIR for the 100% labeling signal. Sample III is labeled with the membrane-impermeable SNAP-Alexa488 first, to block all the outward-facing proteins, and in a second step with the cell-permeable SNAP-647-SIR to obtain a signal corresponding to the inward-facing proteins. Samples are prepared in duplicates and loaded on an SDS-PAGE. Gel is sequentially imaged for in-gel fluorescence in green and red channels and subsequently stained for total protein amount via colloidal Coomassie staining.
Figure 5. Exemplary results from dual color labeling experiment.

Proteoliposomes are separated in samples I, II, and III, and, after SNAP-labeling, in-gel fluorescence is measured in both channels. Subsequently, colloidal Coomassie staining is performed for normalization on total protein amount. All samples are prepared and loaded in duplicates. Sample I is labeled with the membrane-impermeable SNAP-Cell Alexa488. Sample II is labeled with the membrane-permeable SNAP-Cell 647-SIR and served as 100% protein signal. Sample III is blocked on the outside with membrane-impermeable SNAP-Surface Alexa488 first, followed by the labeling of inward-facing proteins only with the membrane-permeable SNAP-Cell 647-SIR. All fluorescence intensities are normalized on corresponding Coomassie signal. The normalized signal intensities in the SNAP-Cell 647-SIR channel are used to determine the percentage of inside-oriented protein by division of the signals derived from sample III (only inside labeling) by sample II (all protein labeled), showing here 25%. Bleed-through in the SNAP-Cell 647-SIR channel can be determined by division of the signals derived from sample I (no label) by sample II (all protein labeled), showing here less than 1%.
Data analysis
-
Extract signal intensities with ImageLab software
Open the gel images with the red dyes, green dyes, and the Coomassie image as “.scn” with ImageLab Version 5.2.1.
-
From each image, extract the band intensities:
Click on “Lane and bands” menu.
-
Click on the tab “Lanes” and use the lane finder (type number of lanes in the “manual” field) to detect lanes. Adjust lane positions, fitting to the actual lane position.
Note: All lanes should have an equal width, so that errors in background correction are minimized.
Click on the tab “Bands” and use “Band Finder” by clicking on “detect bands” (default settings: band sensitivity – low; lanes – all). Thereby, all prominent bands are detected. Add additional bands via “Bands” – “add.”
Check your result by using the “Lane Profiler”: the intensity of the bands over the lane is displayed as intensity function.
-
Do quality control: the entire area under the band peak has to be covered by the band borders. Adjust by moving the band boundaries (see Figure 4 ).
Note: It is important to treat all bands equally, thus the same area fraction should be chosen. To avoid mistakes by manual selection of band borders, include the entire area.
-
Adjust the background correction by going back to the “Lanes” tab and changing “Background Subtraction.” Specify the disk size, as background correction is done by rolling ball method (see Figure 4 ). Do not tick the “apply to selected lane” as all lanes should be treated equal.
Note: Too high disk size leads to non-separated peaks or too long tails increasing the area too much. Too low disk size might lead to cropping of the area under the peak as partly considered as background.
After iterating steps A2e and A2f until the areas under the peak are marked properly, export the peak data to an Excel file.
Combine the exported Excel sheets and use the extracted volume intensities.
-
Normalize the intensities of the SNAP dyes signal (volume intensities) by dividing by the Coomassie signal (volume intensities).
Note: In more current versions of the software, the background corrected data needs to be used. In the version used here, only the background corrected value is given as Volume (Int).
-
The quotient of inside-oriented protein is obtained by division of the signals from the membrane-permeable SNAP-Cell 647-SIR dye derived from sample III (only inside labeling) by sample II (all protein labeled).
Fluorescence values of the red channel are used here after normalization to the signal of a Coomassie staining. F III : fluorescence of the inward-labeled protein (average); F II : fluorescence of total labeled protein (average); F I : bleed-through, fluorescence of the lane without red dye (average).
Note: As control for bleed-through, determine also the quotient of the signals from the membrane-permeable SNAP-Cell 647-SIR dye derived from sample I (no label) by sample II (all protein labeled). For a robust assay, bleed-through of less than 1% is recommended to ensure minimal interference with image analysis.

Figure 4. Gel bands quantification in ImageLab.
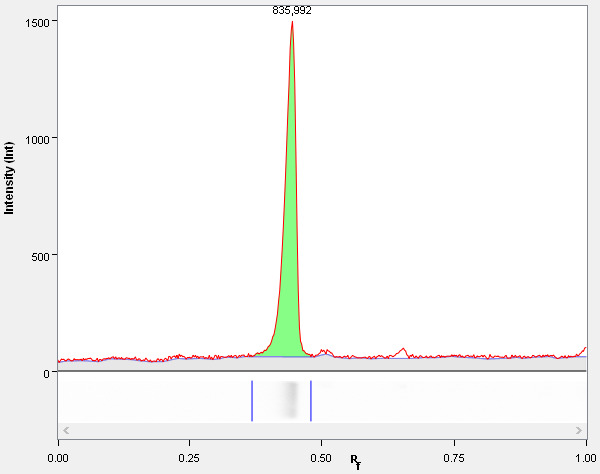
ImageLab graphical user interface showing the data analysis. The boundaries of the selected gel lane were adjusted so that the entire peak is covered between the boundaries. In addition, the background subtraction was done by a rolling ball correction (upper panel, blue line), optimizing the disk size to make it fit to the red line (actual signal).
Recipes
-
Reconstitution buffer (1 L)
20 mM MOPS-KOH (4.185 g), pH 7, 50 mM K 2 SO 4 (8.713 g)
-
1 M OG stock
Dissolve 4.386 g OG to a final volume of 15 mL in ddH 2 O. Freeze aliquots at -20 °C.
-
Protein storage buffer (1 L)
50 mM MES-KOH (9.76 g; pH 7), 50 mM KCl (3.728 g), 20% (v/v) glycerol, 1 mM EDTA (0.292 g), and 1 mM DTT (2 mL from 0.5 M stock) supplemented with 0.04% (w/v) DDM.
-
0.5 M DTT stock
Dissolve 1 g of DTT in 42.81 µL of 3 M Na-acetate (pH 5.2), and add 12.93 mL of ddH 2 O. Freeze aliquots at -20 °C.
Note: DTT is volatile; thaw and freeze directly before and after use.
-
Sephadex G50 fine slurry
Dissolve 2 g of Sephadex G50 to a final volume of 50 mL of reconstitution buffer, soak for 3 h at room temperature or overnight in the fridge.
-
Pre-washed BioBeads
Weight approximately 1 g of BioBeads in a 50 mL reaction tube. Wash twice for 10 min in methanol, twice for 10 min in ddH 2 O, and once for 10 min in reconstitution buffer at room temperature. BioBeads can be stored in reconstitution buffer for up to one week in the fridge. For usage, the buffer is removed to have moist BioBeads.
-
Laemmli buffer (100 mL)
60 mM Tris (0.727 g), 0.75% (v/v) SDS (3.75 mL from 20% stock), 10 mM DTT (2 mL from 0.5 M stock), 2.6% (w/v) sucrose (2.6 g), 0.005% (w/v) bromophenol blue (0.005 g), and 2 mM EDTA (0.058 g), pH 6.8
Note: DTT is volatile; thaw and freeze directly before and after use.
-
20% SDS stock (100 mL)
Dissolve 20 g of SDS to a total volume of 100 mL in ddH 2 O.
-
SDS-PAGE running buffer (1 L)
25 mM Tris (3.029 g), 192 mM Glycine (14.413 g), and 1% SDS (50 mL from 20% stock), pH 8.6
-
SDS gel
-
10% 1.5 mm SDS-polyacrylamide separation gel (50 mL)
10% (v/v) acrylamide (5 mL), 375 mM Tris, pH 8.8 (18.75 mL of 1 M stock), 0.1% (v/v) SDS (250 µL of 20% stock), 0.1% (v/v) APS (500 µL of 10% stock), and 0.01% (v/v) TEMED (5 µL)
-
5% stacking gel (10 mL)
5 % (v/v) acrylamide (2.5 mL), 125 mM Tris, pH 6.8 (6.25 mL of 1 M stock), 0.1 % (v/v) SDS (250 µL of 20 % stock), 0.1 % (v/v) APS (100 µL of 10 % stock), and 0.01 % (v/v) TEMED (5 µL)
-
-
10% APS stock
Dissolve 1 g to a total volume of 10 mL in ddH 2 O.
-
1 M Tris (100 mL)
Dissolve 12.114 g Tris to a total volume of 100 mL in ddH 2 O, adjust the pH to either 8.8 or 6.8, respectively.
-
Colloidal Coomassie staining solution (1 L)
0.02% (w/v) Coomassie brilliant blue G-250 (0.2 g), 0.02% (w/v) aluminum sulfate hydrate (0.2 g), 10% (v/v) ethanol (100 mL), and 2% (v/v) 85% orthophosphoric acid (20 mL)
-
Colloidal Coomassie destaining solution (1 L)
10% (v/v) ethanol (100 mL), 2% (v/v) 85% orthophosphoric acid (20 mL)
Acknowledgments
This protocol was adapted from our previous work ( Veit et al., 2022 ). The work was funded by grants from the German Research 545 Foundation (GU 1133/11-1) and DAAD (57386621) to TGP, LCP and SV acknowledge funding by the Studienstiftung des deutschen Volkes. We thank Huriye Deniz Uzun and Bo Højen Justesen for the generation of the AHA2-SNAP construct. All figures are prepared using Biorender.com.
Competing interests
The authors declare that no competing interests exist.
Citation
Readers should cite both the Bio-protocol article and the original research article where this protocol was used.
Q&A
Post your question about this protocol in Q&A and get help from the authors of the protocol and some of its users.
References
- 1. Amati A. M. , Graf S. , Deutschmann S. , Dolder N. and von Ballmoos C. ( 2020 . ). Current problems and future avenues in proteoliposome research . Biochem Soc Trans 48 ( 4 ): 1473 - 1492 . [DOI] [PubMed] [Google Scholar]
- 2. Cid A. , Perona R. and Serrano R. ( 1987 . ). Replacement of the promoter of the yeast plasma membrane ATPase gene by a galactose-dependent promoter and its physiological consequences . Curr Genet 12 ( 2 ): 105 - 110 . [DOI] [PubMed] [Google Scholar]
- 3. Cole N. B. ( 2013 . ). Site-specific protein labeling with SNAP-tags . Curr Protoc Protein Sci 73 : 30 31 31-30 31 16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Deutschmann S. , Rimle L. and von Ballmoos C. ( 2022 . ). Rapid Estimation of Membrane Protein Orientation in Liposomes . Chembiochem 23 ( 2 ): e202100543 . [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Dyballa N. and Metzger S. ( 2009 . ). Fast and sensitive colloidal coomassie G-250 staining for proteins in polyacrylamide gels . J Vis Exp ( 30 ): 1431 . [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Eisinger M. L. , Nie L. , Dorrbaum A. R. , Langer J. D. and Michel H. ( 2018 . ). The Xenobiotic Extrusion Mechanism of the MATE Transporter NorM_PS from Pseudomonas stutzeri . J Mol Biol 430 ( 9 ): 1311 - 1323 . [DOI] [PubMed] [Google Scholar]
- 7. Islam S. T. , Eckford P. D. , Jones M. L. , Nugent T. , Bear C. E. , Vogel C. and Lam J. S. ( 2013 . ). Proton-dependent gating and proton uptake by Wzx support O-antigen-subunit antiport across the bacterial inner membrane . mBio 4 ( 5 ): e00678 - 00613 . [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Keppler A. , Pick H. , Arrivoli C. , Vogel H. and Johnsson K. ( 2004 . ). Labeling of fusion proteins with synthetic fluorophores in live cells . Proc Natl Acad Sci U S A 101 ( 27 ): 9955 - 9959 . [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Lanfermeijer F. C. , Venema K. and Palmgren M. G. ( 1998 . ). Purification of a histidine-tagged plant plasma membrane H + -ATPase expressed in yeast . Protein Expr Purif 12 ( 1 ): 29 - 37 . [DOI] [PubMed] [Google Scholar]
- 10. Lukinavičius G. , Umezawa K. , Olivier N. , Honigmann A. , Yang G. , Plass T. , Mueller V. , Reymond L. , Correa I. R. Jr. Luo Z. G. , et al. .( 2013 . ). A near-infrared fluorophore for live-cell super-resolution microscopy of cellular proteins . Nat Chem 5 ( 2 ): 132 - 139 . [DOI] [PubMed] [Google Scholar]
- 11. Marek M. , Milles S. , Schreiber G. , Daleke D. L. , Dittmar G. , Herrmann A. , Muller P. and Pomorski T. G. ( 2011 . ). The yeast plasma membrane ATP binding cassette(ABC) transporter Aus1: purification, characterization, and the effect of lipids on its activity . J Biol Chem 286 ( 24 ): 21835 - 21843 . [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Murray D. T. , Griffin J. and Cross T. A. ( 2014 . ). Detergent optimized membrane protein reconstitution in liposomes for solid state NMR . Biochemistry 53 ( 15 ): 2454 - 2463 . [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Rigaud J. L. and Lévy D. ( 2003 . ). Reconstitution of membrane proteins into liposomes . Methods Enzymol 372 : 65 - 86 . [DOI] [PubMed] [Google Scholar]
- 14. Rigaud J. L. , Paternostre M. T. and Bluzat A. ( 1988 . ). Mechanisms of membrane protein insertion into liposomes during reconstitution procedures involving the use of detergents. 2. Incorporation of the light-driven proton pump bacteriorhodopsin . Biochemistry 27 ( 8 ): 2677 - 2688 . [DOI] [PubMed] [Google Scholar]
- 15. Schuette C. G. , Hatsuzawa K. , Margittai M. , Stein A. , Riedel D. , Kuster P. , Konig M. , Seidel C. and Jahn R. ( 2004 . ). Determinants of liposome fusion mediated by synaptic SNARE proteins . Proc Natl Acad Sci U S A 101 ( 9 ): 2858 - 2863 . [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Serek J. , Bauer-Manz G. , Struhalla G. , van den Berg L. , Kiefer D. , Dalbey R. and Kuhn A. ( 2004 . ). Escherichia coli YidC is a membrane insertase for Sec-independent proteins . EMBO J 23 ( 2 ): 294 - 301 . [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Tunuguntla R. , Bangar M. , Kim K. , Stroeve P. , Ajo-Franklin C. M. and Noy A. ( 2013 . ). Lipid bilayer composition can influence the orientation of proteorhodopsin in artificial membranes . Biophys J 105 ( 6 ): 1388 - 1396 . [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Veit S. , Paweletz L. C. , Bohr S. S. , Menon A. K. , Hatzakis N. S. and Pomorski T. G. ( 2022 . ). Single Vesicle Fluorescence-Bleaching Assay for Multi-Parameter Analysis of Proteoliposomes by Total Internal Reflection Fluorescence Microscopy . ACS Appl Mater Interfaces 14 ( 26 ): 29659 - 29667 . [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Wilhelm J. , Kuhn S. , Tarnawski M. , Gotthard G. , Tunnermann J. , Tanzer T. , Karpenko J. , Mertes N. , Xue L. , Uhrig U. , et al. .( 2021 . ). Kinetic and Structural Characterization of the Self-Labeling Protein Tags HaloTag7, SNAP-tag, and CLIP-tag . Biochemistry 60 ( 33 ): 2560 - 2575 . [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Zhang W. , Bogdanov M. , Pi J. , Pittard A. J. and Dowhan W. ( 2003 . ). Reversible topological organization within a polytopic membrane protein is governed by a change in membrane phospholipid composition . J Biol Chem 278 ( 50 ): 50128 - 50135 . [DOI] [PubMed] [Google Scholar]