

Abstract
Hyperdiploidy is the largest genetic entity B-cell precursor acute lymphoblastic leukemia in children. The diagnostic hallmark of its two variants that will be discussed in detail herein is a chromosome count between 52 and 67, respectively. The classical HD form consists of heterozygous di-, tri-, and tetrasomies, whereas the nonclassical one (usually viewed as “duplicated hyperhaploid”) contains only disomies and tetrasomies. Despite their apparently different clinical behavior, we show that these two sub-forms can in principle be produced by the same chromosomal maldistribution mechanism. Moreover, their respective array, gene expression, and mutation patterns also indicate that they are biologically more similar than hitherto appreciated. Even though in-depth analyses of the genomic intricacies of classical HD leukemias are indispensable for the elucidation of the disease process, the ensuing results play at present surprisingly little role in treatment stratification, a fact that can be attributed to the overall good prognoses and low relapse rates of the concerned patients and, consequently, their excellent treatment outcome. Irrespective of this underutilization, however, the detailed genetic characterization of HD leukemias may, especially in planned treatment reduction trials, eventually become important for further treatment stratification, patient management, and the clinical elucidation of outcome data. It should therefore become an integral part of all upcoming treatment studies.
Subject terms: Cancer genetics, Acute lymphocytic leukaemia, Genetics research
Introduction
Hyperdiploidy (HD) was first described by Fritz Lampert in 1967 when he measured the DNA content of leukemic cells by meticulously comparing their diameters with those of normal lymphocytes [1]. Despite the rather crude cytogenetic methods that were available at that time, he also succeeded in quite accurately defining the nature of the acquired chromosomes and realized that the affected patients lived longer than others. Eventually, it became clear that a specific variant of HD is, with a prevalence of up to 35%, also the largest genetic entity of childhood B-cell precursor acute lymphoblastic leukemia (BCP ALL). Lampert’s remarkable insights have not only continued to prove accurate, but they have also become the subject of many research and clinical studies since then. Today, continuously evolving sophisticated diagnostic and research tools produce a plethora of information that unravels genomic peculiarities in previously unimaginable detail, and thereby helps to refine prognostic stratification and treatment.
Despite all these extraordinary achievements, many fundamental questions about the various aneuploid sub-forms of BCP ALL remain unresolved. These primarily concern their origins, the causes and biological meaning of the various nonrandom and disease-inherent chromosome configurations, and, not least, their varied disease development and clinical behavior. Although only a small proportion of the main HD variant experience relapses, it still makes up 25% of all relapses that occur in childhood BCP ALL. At present, the processes that drive these mainly late-disease recurrences are only vaguely understood [2].
Since the last comprehensive review of HD ALL by Paulsson and Johansson was published in 2009 [3], we consider it worthwhile to recapitulate the remarkable progress that has been made since then, especially in decoding the genomic and biological features of this extraordinary disease as well as in its prognostic stratification and treatment.
Genomic features of HD ALL
Chromosome copy number abnormalities
Childhood ALL cases with gross ploidy changes are formally based on their DNA content and the overall number of chromosomes in their karyotypes. They comprise seven biologically related yet distinct categories whose common feature is a ploidy-related overrepresentation of chromosome 21, respectively (Fig. 1A). Although their categorization is primarily based on karyotype patterns, it takes increasingly also selected genomic features into account. Some of these new parameters are largely corroborating, whereas others uncover glitches and discrepancies in the current classification system, insights that justify a careful modification of the taxonomy as well as the terminology of these diseases that should take these novel findings into account [3–10]. Because the ploidy-related overrepresentation of chromosomes 21 serves as their essential and overarching hallmark, one can use the definitions of the “International System of Cytogenetic Nomenclature”, namely hyperhaploid (25–34 chromosomes), hypodiploid (35–45 chromosomes), hyperdiploid (47–58 chromosomes), hypotriploid (59–68 chromosomes) and hypertriploid (70–80 chromosomes) for their further subclassification without obscuring any otherwise pertinent information [11]. Just for convenience’s sake and practical reasons, the hypo- and hypertriploid may still be merged into a “near-triploid” group (59–80 chromosomes). In our review we will avoid the term “duplicated or doubled up haploids” because it insinuates a presumed but never proven mode that is supposed to generate this specific karyotype pattern. Since the more appropriate yet still not entirely correct descriptive term “hyperdiploidy due to a genome-wide loss of heterozygosity” would be rather impracticable to use, we opted to introduce the neutral terms “classical” and “nonclassical” for the two dissimilar but nevertheless closely related forms of genuine hyperdiploid forms that are the specific focus of our review. Both these types are defined by an agreed-upon chromosome number that lies on the lower side between 50 and 52 and on the upper side between 58 and 67, cutoffs that vary slightly in different studies (Fig. 1B, C). However, given that more than 80% of HD cases fall into the range of 52–58 with a modal peak of 55–56, we propose that an upper limit of 58 would be more appropriate, especially because such a cutoff eases the delineation of genuine HD forms from near-triploid ones (Fig. 1A) [4–8, 12–16].
Fig. 1. Defining criteria of classical and nonclassical HD forms of childhood ALL.

A Based on their number of chromosomes, aneuploid forms of childhood ALL can be subdivided into seven distinct categories. The defining thresholds for HD cases range from 52 to 58 or 67 chromosomes. Classical HD and near-triploid karyotypes contain di-, tri, and tetrasomies, hyperhaploid and hypodiploid ones only mono- and disomies. Nonclassical HD karyotypes contain only di- and tetrasomies. 28% of the hyperhaploid and 32% of the nonclassical HD cases are monoclonal, whereas 40% of them share both clones. Likewise, 34% of the hypodiploid and 24% of the near-triploid cases are monoclonal, whereas 42% of them are bi-clonal [9]. The projection of representative karyotypes (highlighted chromosomes) of a classical (B) and a nonclassical (C) case onto the 92 chromosomes of a diploid mitotic cell illustrates that both patterns can be produced by the same yet-undefined nondisjunction mechanism and, in principle, even in a single step. The daughter cells that obtain the highlighted set of chromosomes, which always contain a tetrasomy 21, can survive, whereas the ones which only receive the dimmed set that lacks chromosomes 21 will perish as a result. The karyotype of the classical case (B) is 57,XX,+X,+X,+4,+6,CN-LOH(9),+10,+14,+14,+17,+18,+21,+21 and that of the nonclassical case (C) is 52,XX,+X,+X,CN-LOH(1–8,10–13,15–20,22),+14,+14,+21,+21.
The typical karyotype of the most prevalent classical HD variant is always composed of di-, tri-, and tetrasomies. Trisomies always result from the duplication of either one of the parental chromosomes in an apparently random fashion (“2+1” pattern) and most commonly affect chromosomes X, 4, 6, 10, 14, 17, and 18 (Fig. 1B) [3, 6]. Tetrasomies, on the other hand, always result from the duplication of both parental homologs (“2+2” pattern). The most common, in addition to the obligatory tetrasomy 21 are those of chromosomes X, 14, and 18. Although the individual chromosomal composition of the remarkably stable karyotypes varies from case to case, particular chromosomes appear in a predetermined, statistically hierarchical order [4, 6]. The probability that one or the other is seen depends on the chromosomes that are already present and, therefore, also on the overall modal chromosome number. Tetrasomy 21 is always the first change, which can then be followed, in a decreasing likelihood, by gains of chromosomes X, 14, 6, 18, 4, 17, and 10 [3, 6]. The composition of the various HD genomes is thus governed by the functional interdependence and indispensable compatibility of the respective combinations of chromosomes.
The karyotypes of the second, much rarer nonclassical HD variant, contain only disomies and tetrasomies. This unique pattern was already recognized and described in the early days of cytogenetics (Fig. 1C) [17]. Array analyses revealed that the disomic chromosomes are always homozygous (“2+0” pattern), whereas the tetrasomic ones remain heterozygous (“2+2 pattern”) (Fig. 2). The same pattern is also seen in exceptionally rare cases with 48, 50, or 52 chromosomes. Notwithstanding their low chromosome numbers, however, we propose that they likewise belong to the nonclassical HD group. Although uniparental isodisomies may also appear in classical HD forms, they only involve a single or few chromosomes in those cases [18]. Nonclassical HD karyotypes are exact duplicates of hyperhaploid ones, and both can appear either alone or in combination. This circumstance led to the understandable but hitherto unproven view that nonclassical HD cases are merely duplicated hyperhaploid ones and that they can therefore be equated with hyperhaploidy, irrespective of whether a hyperhaploid clone is indeed identified or not [3, 7, 9, 19–25]. Thus, this uncritical synonymous use of these terms in this context is confusing, may often lead to misunderstandings, and, as we argue herein, it may probably also not be the correct label for how this hyperdiploid variant is formed.
Fig. 2. What the comparative analysis of germline and acquired chromosome copy number and/or sequence variants reveals about the origin, development, and biology of HD leukemias.

Left side, top: the minimal common denominator of HD leukemia is always a bi-parental derived tetrasomy 21, irrespective of whether it arises in a constitutional normal or trisomic individual. Left side, middle: the duplication of either the wild-type or variant allele of pharmacologically relevant heterozygous genes, such as the thiopurine S-methyltransferase (TPMT) on chromosome 6 and/or the γ-glutamyl hydrolase (GGH) on chromosome 8, will produce two distinct leukemia genotypes with opposite drug sensitivities [34]. Left side, bottom: the ARID5B rs7090445-C risk allele is preferentially duplicated in HD blast cells [40]. Right side, top: the recombination of the immunoglobulin heavy chain (IGH) gene on chromosome 14 follows discrete consecutive steps during B-cell maturation. A clone with a disomy 14 can thus harbor a maximum of two unique rearrangements, whereas a clone with trisomy 14 can have three unique or one unique and two related rearrangements. Systematic analyses of such rearrangement patterns have shown that trisomy 14 is usually already present before the initiation of IGH recombination and thus prove that the maldistribution of the chromosomes is indeed the essential transforming event [162, 163]. Right side, middle: the analysis of acquired trisomy-associated heterozygous mutations informs about the sequence of events and the latency period between the nondisjunction event and their emergence. Mutations acquired before trisomy formation may affect either 2/3 or 1/3 of the duplicated homologs, whereas those that are acquired after trisomy formation can merely be present in 1/3 of the non-duplicated homologs [16, 55]. Right side, bottom: X inactivation (Xi) is a dosage-compensation mechanism in females that silences either the maternal or paternal chromosome with an equal likelihood during early fetal development. As in other HD-related trisomies, either one of the two parental X chromosomes can therefore be duplicated [164]. Notwithstanding this fact, however, it is always the active X (Xa) that is nonrandomly gained [165]. This outcome concords with the high expression of X-encoded genes and suggests that specific X-linked factors help to jumpstart and maintain the disease process [61–63].
The chromosome number of near-triploid cases lies between 60 and 78, which means that at least those with a chromosome number below 67, overlap with those classical HD ones with a chromosome number above 60 (Fig. 1) [7, 8, 13–15]. Because of the dissimilar clinical impact of classical HD and near-triploid cases, the proper assignment of such ambiguous cases to one or the other group is important for appropriate treatment stratification, a requirement that is, however, virtually impossible to fulfill based on chromosome counts alone [13]. Helpful karyotypic parameters that may delineate most of the near-triploid cases are the presence of a tri- or tetrasomy 1 together with a relative underrepresentation of chromosomes 7 and 14 [13]. Nevertheless, by far the best discriminators are either germline or somatic TP53 mutations, which are seen in virtually all near-triploid and hypodiploid cases, but hardly ever in classical or nonclassical HD ones (Fig. 1) [7, 8, 13–15].
Although constitutional trisomy 21 is by far the most common leukemia-predisposing factor, it is odd that classical HD forms are significantly underrepresented in this condition. To the best of our knowledge, none of the other aneuploid leukemia types have ever been reported in Down syndrome patients either [10, 26–28]. Yet, once formed, the karyotype patterns of such classical HD cases resemble exactly the ones, which are seen in constitutionally normal patients. In contrast to what one might intuitively expect, they never contain six chromosomes 21, but only the usual four or, as may occasionally be the case in constitutionally normal HD patients, five copies of chromosome 21. This suggests that a preexistent trisomy 21 impedes a priori the formation of the essential tetrasomy and/or that daughter cells with six chromosomes 21 cannot survive (Fig. 2) [10].
Of further interest in this context is DYRK1A, a serine/threonine kinase that is encoded on chromosome 21 and whose copy number determined overexpression is implicated in several pathologies in Down syndrome. Notably, this overexpression increases the expression and phosphorylation of two transcription factors, FOXO1 and STAT3, that are indispensable for B-cell development and therefore also contribute to the development and maintenance of BCP leukemias [29]. These effects are obviously especially pronounced in HD cases (https://pecan.stjude.cloud/proteinpaint/DYRK1A) and render both DYRK1A and FOXO1 worthwhile therapeutic targets in this specific subset of ALL [29].
Nonrandom secondary events in the form of structural abnormalities include chromosome 1q duplications (10–15%), 6q deletions (5%) and isochromosomes 17q (2–5%) and 7q (1–2%) [3]. They occur in a mutually exclusive manner and, may also be present in monoclonal nonclassical HD cases. However, to the best of our knowledge, they have never been observed in bi- or monoclonal hyperhaploid cases [5, 9, 10].
Another notable phenomenon is the co-occurrence of classical HD and an additional class-specific abnormality within the same cell clone, such as a t(9;22)(q34;q11)/BCR::ABL1, a t(1;19)/TCF3::PBX1 or a t(4;11)/KMT2A::AFF1 [22, 30, 31]. Either one of these can act as a primary or secondary change. In children, such “double hit” events comprise 2–3% of all HD cases, but they are 10–15 times more common in adults and therefore make up half of the total 13% of classical HD cases [31]. Although there are even a few mono- or bi-clonal nonclassical and/or hyperhaploid cases that concur with a t(9;22), we are not aware of any hypodiploid and/or near-triploid ones with such a constellation [28, 32, 33].
Germline predisposing sequence alterations
Germline predisposition factors comprise genome-wide association study–ascertained allelic variants, which function as genetic modifiers, and distinct pathogenic gene defects (Table 1). An HD-inherent unique phenomenon is those heterozygous variants in pharmacologically relevant genes that are a priori disease-unrelated but nevertheless functionally important because their alternate allelic duplication will distort the concordance of germline and leukemia genotypes (Fig. 2) [34].
Table 1.
Overview of susceptibility factors as well as predisposing germline and somatically acquired pathogenic genetic lesions that relate to classical and nonclassical forms of HD in childhood ALL (adapted from [10]).
| Chromosome location | Candidate genes | Function | SNP/defect | Classical HD | Nonclassical HD and hyperhaploid | References | |
|---|---|---|---|---|---|---|---|
| Germline | Somatic | Somatic | |||||
| 1p13.2 | NRAS | RTK/RAS | PV | + | + | [7, 48, 53, 55, 56, 128, 129] | |
| 2p22.1 | SOS1 | RTK/RAS | PV | + | [48] | ||
| 3p21.31 | SETD2 | Histone methyltransferase | PV | + | [7, 55, 56] | ||
| 4p16.3 | NSD2 (WHSC1) | Histone methyltransferase | PV | + | [53, 55] | ||
| 5p14.2 | PRDM9 | Histone methyltransferase, meiotic recombination | Rare allele | + | [130, 131] | ||
| 5q31.1 | C5orf56 | lncRNA | rs886285 | + | [132] | ||
| 6p21.31 | BAK1 | Pro-apoptotic BCL2 antagonist | rs210143 | + | [132] | ||
| 6p22 | Histone cluster | DNA replication and packaging | Deletions | + | + | [7] | |
| 6q22.1 | ROS1 | RTK/RAS | PV | + | [56] | ||
| 7p12.2. | IKZF1 | Lymphoid transcription factor | Deletions | + | + | [2, 7, 52, 129, 133] | |
| 7q36.1 | EZH2 | Histone methyltransferase | PV | + | [7, 55, 56] | ||
| 8q21.13 | PAG1 | Ras signaling inhibitor | Deletions | + | + | [7] | |
| 9p21.3 | (TLE1) | Transcription co-repressor | rs76925697 | + | [132] | ||
| 9p21.3 | CDKN2A and CDKN2Ba | Cyclin-dependent kinase inhibitors | rs3731217, rs77728904 | + | [134–136] | ||
| 9p21.3 | CDKN2A and CDKN2Ba | Cyclin-dependent kinase inhibitors | Deletionsb | + | + | [52, 132, 134, 135] | |
| 10p12.2 | BMI1 | Chromatin remodeling epigenetic repressor | rs7088318 | + | [36, 37] | ||
| 10q21.2 | ARID5B | Histone demethylase, transcription coactivator | rs7090445 | + | [35, 40, 137, 138] | ||
| 10q21.2 | ARID5B | Histone demethylase, transcription coactivator | Deletions | + | [139] | ||
| 10p12.2 | PIP4K2A | PI-3-kinase regulator | rs2296624 | + | [36, 37] | ||
| 11q13.2 | KMT5B (SUV420H1) | Transcription regulator, histone methyltransferase | PV | + | [7, 55] | ||
| 11q14.1 | GAB2 | PI-3-kinase regulator | PV | + | [140] | ||
| 12p12 | KRAS | RTK/RAS | PV | + | + | [7, 48, 53, 55, 56, 128] | |
| 12p13.2 | ETV6 | ETS transcription factor, transcription repressor | Multiple SNPs | + | + | [47, 49] | |
| 12p13.2 | ETV6 | ETS transcription factor, transcription repressor | PV, deletionsb | +c | + | + | [47, 49, 50, 141–143] |
| 12q24.1 | PTPN11 | RTK/RAS | PV | +d | + | + | [48, 53, 55, 56] |
| 13q12.2 | FLT3 | RTK/RAS | PV, duplications | + | + | [7, 48, 53, 55, 56, 144, 145] | |
| 13q12.2 | PAN3 | RTK/RAS | Deletions | +e | + | [146] | |
| 13q14.2 | RB1 | Transcription factor, negative cell-cycle regulator | PV, deletions | + | [7, 129] | ||
| 14q11.2 | CEBPE | Transcription activator | rs2239630 | + | + | [35, 40, 132, 138, 147] | |
| 16p13.3 | CREBBP | Transcription coactivator | PV, deletions | +f | + | + | [52, 53, 55, 56, 129] |
| 16q22.1 | CTCF | Zinc finger transcriptional repressor | PV, deletions | + | [57] | ||
| 17q11.2 | NF1 | RTK/RAS | PV, deletions | + | + | + | [7, 55] |
| 17q21.1 | IKZF3 | Lymphoid transcription factor | Haplotype | + | + | [147] | |
| 17q21.1 | IKZF3 | Lymphoid transcription factor | Deletions | + | [7] | ||
| 19p13.3 | DOT1L | Histone methyltransferase | PV | + | [56] | ||
| 21q22.2 | ERG | ETS transcription factor, transcription regulator | rs9976326 | + | [132] | ||
Most of the listed somatic mutations were also identified in the recent study by Brady et al. in 324 hyperdiploid and 24 hyperhaploid ALL cases [16].
PV pathogenic variants, SNP single-nucleotide polymorphism, RTK/RAS component of the receptor tyrosine kinase signaling pathway.
aGeneric B and T ALL predisposing SNPs.
bCommon in virtually all types of B and T ALL.
cETV6-linked leukemia predisposition and familial thrombocytopenia syndrome.
dNoonan syndrome-type Rasopathy.
eFLT3 upregulation via PAN3 deletion-associated enhancer hijacking.
fRubinstein–Taybi syndrome.
The four most relevant susceptibility loci reside within or in close proximity to the ARID5B, CEPBE, BMI1, and PIP4K2A genes [35–41]. ARID5B plays an essential role in the epigenetic activation of gene expression, cell-cycle regulation, and 6-mercatopurine and methotrexate (MTX) metabolism [37, 40, 42, 43]. The intronic rs7090445-C risk allele of this gene is less expressed than that of the wild-type one. Functional studies and analyses of many carriers have shown that its presence will impede normal lymphocyte development, facilitate the clonal expansion of the affected BCPs, confer drug resistance, and increase the relapse risk [37, 40, 42–46]. CEBPE encodes one of six basic leucine transcription factors. The risk-defining SNP rs2239635 in its promotor disrupts the binding of the Ikaros transcriptional repressor [39]. CEBPE is on chromosome 14, which is incidentally also one of the most common tri- and tetrasomic chromosomes in HD. Its copy-number-linked overexpression may thus be one of the critical contributing leukemia-promoting factors [39]. BMI1 is a negative regulator of the cell-cycle checkpoint proteins p16 and p14ARF, encoded by CDKN2A, which is the most frequently deleted gene in ALL. The risk-defining SNP rs11591377 is in a predicted hematopoietic stem cell enhancer and reinforces the preferential binding of the MYBL2 and p300 transcription factors [41]. PIP4K2A encodes an enzyme that is part of the phosphoinositide signal transduction pathways that co-regulate cell proliferation, differentiation, and motility. The risk-defining SNP rs4748812 lies within a PIP4K2A regulatory element and is predicted to alter the binding of the RUNX1 transcription factor [41].
A not yet exactly determined proportion of HD ALL cases have predisposing pathogenic germline defects in genes that encode members of B-cell development, receptor tyrosine kinase/RAS (RTK/RAS), epigenetic regulatory, and DNA repair pathways (Table 1). The two most common conditions concern the ETV6 and the PTPN11 genes [16, 47–49]. Approximately 70% of all BCP ALL cases with germline defects in either one of these two genes are hyperdiploid [48, 49]. ETV6 germline-mutated cases frequently also acquire somatic mutations in NRAS, KRAS, and PTPN11 [49]. Although we have no specific information about secondary changes in cases with RASopathy, it is worth noting that in two of them the PTPN11 mutation became duplicated in the form of a uniparental disomy 12 [48].
Predisposing germline factors are unlikely to be directly responsible for the formation of HD precursor cells per se but rather alleviate the immediate survival of independently created cells by equipping them with essential elements that founder cells in non-predisposed individuals are forced to acquire as secondary changes [10]. The extent to which the occasional concurrence of two or even more such predisposition factors, for instance, ARID5B and ETV6, will augment the respective risk remains to be determined [50].
Somatically acquired sequence alterations
Virtually all classical and nonclassical HD cases also acquire somatic mutations, primarily in the RTK/RAS and phosphoinositide 3-kinase-signaling pathway genes KRAS, NRAS, FLT3, SOS, and PTPN11, as well as in the chromatin-modifying genes CREBBP, NSD2, SUV420H1, SETD2, and EZH2 (Table 1) [16, 51–57]. Other abnormalities that are typically enriched in nonclassical HD forms are NF1, CDKN2A/B, IKZF3, PAG1, and the 6p22 histone gene cluster [7, 22]. Most of these genes are located on chromosomes that usually remain disomic. Although the frequency of mutations is much higher in disease recurrences, the originally extraordinary inter- and intragenic heterogeneity of mutually exclusive RTK/RAS pathway mutations is then essentially lost. KRAS mutations are the only ones that are preferentially retained and then commonly coexist with the prevalent CREBBP mutations in the predominant relapse clones [52, 53, 58].
Of note in this context are the comparative analyses of the numbers of monoallelic and biallelic mutations on disomic and on trisomic chromosomes, which revealed that monoallelic mutations on trisomic and homozygous ones on disomic chromosomes are significantly more common than biallelic ones. These observations provide compelling evidence that such sequence alterations emerge only quite some time after the formation of the hyperdiploid genome (Fig. 2) [16, 55].
Mutation signatures
Mutation signatures can reveal environmental and endogenous sources of mutagenesis in affected tissues. At present, there are 49 accepted single base substitution signatures, whose causative factor has been established with a certain security [59, 60]. Of these, Signature7 is the one that is induced by exposure to ultraviolet light [59, 60]. Brady et al. found this signature especially enriched in 17% of hyperdiploid, 35% of hyperhaploid as well as 46% of iAMP21-positive leukemias exclusively in patients of non-African descent [16]. The fact that these mutations emerged only after the aneuploidization event had taken place was therefore taken as an indication that it can only be postnatally induced when the respective cells become trans-dermally exposed to skin-penetrating light. As interesting as these intriguing observations together with the interpretation of their emergence are, they certainly need to be further scrutinized and functionally evaluated [59].
Methylome, transcriptome, and proteome
The methylation, gene expression, and protein structure of HD leukemias are tightly interlocked and mainly shaped by dosage effects that are exerted by the surplus chromosomes [30, 61–68]. Compared to other types of ALL, HD leukemias are remarkably hypomethylated, a feature that remains constant regardless of the number and types of chromosomes that are present in the individual karyotypes. This observation has therefore been taken as an indication that this peculiar signature must either predate or at least concur with the acquisition of the extra set of chromosomes [68]. The six different pathways that are enriched in the expression signature of HD ALL include translation and ribosomes, innate immunity, cell adhesion, cytokines and activated signaling, protein folding and proteolysis, and the endosome pathway [63, 69]. Although the expressed genes and the associated range of proteins largely correlate with the number of chromosomes, the overall effect is nevertheless determined by those almost 70% of genes on chromosomes 21, X, and 14 [30, 61–63, 65]. Conversely, approximately 16% of the transcript and 25% of the protein levels are significantly lower than their corresponding gene copy number would indicate [63]. The top-downregulated genes and proteins are IGF2BP1, CLIC5, RAG1, and RAG2 [63]. Such a diminished IGF2BP1 and CLIC5 gene expression is one of the outstanding features of HD leukemias with CTCF alterations and histone gene cluster 1 deletions [57]. The transcriptional repressor CTCF and the cohesion complex are not only master regulators of the chromatin architecture but also of transcription [63, 70]. By binding to chromatin insulators, CTFC prevents the interaction between promoters and nearby enhancers and silencers. Its encoding gene, CTFC, is on chromosome 16, which usually remains disomic, whereas the core members of the cohesion complex are on trisomic chromosomes (RAD21 on 8q24, SMC3 on 10q25, SMC1A on Xp11, and STAG2 on Xq25). It was therefore proposed that this copy number discrepancy may likewise unbalance the essential expression equilibrium between these genes [63]. Although CTCF depletion and cohesin loss will first of all impair the proper cohesion, alignment, and segregation of chromosomes, it will also weaken the insulation at the borders of topologically associating domains [63, 69–71]. Since these chromatin structures control the timing of DNA replication, the destruction of their framework has also a severe effect on the genome-wide coordination of gene expression [63, 72]. Together with the faulty activity of Aurora B kinase and Survivin, which normally fine-tune the spindle assembly checkpoint, this genomic turmoil slows down the mitotic process, decreases the proliferative rate of HD cells, and is also responsible for the poor morphological appearance of metaphase chromosomes, which often makes them extremely difficult to analyze. Since neither cohesin nor condensin complex encoding genes are consistently mutated in HD leukemias, these effects can only be caused by the subordinate inadequate expression and resultant dysfunction of cohesin and/or condensin complex components [69, 71].
Notably, the gene expression profiles of classical and nonclassical HD forms are virtually identical. In unsupervised hierarchical clustering and principal component analyses, monoclonal nonclassical HD, bi-clonal, and monoclonal hyperhaploid entities form a single discrete cluster that clearly separates them from the hypodiploid/near-triploid entities, which, in turn, form their own cluster [7]. Moreover, classical and nonclassical HD forms even cluster together in an indistinguishable manner in a t-distributed stochastic neighbor embedding blot analysis [16, 73].
Finally, we need to mention the copy-number-related overexpression of the SLC19A1 gene on chromosome 21q22.3, which encodes the predominant folate and MTX uptake transporter [74, 75]. The larger amount of this transporter also increases the quantity of polyglutamates, the active metabolites of MTX and mercaptopurine, as well as thioguanine nucleotides, in blast cells. The presence of a tetrasomy 21 is therefore thought to explain, in part, the good outcome of patients with HD leukemias [76–80].
The roots of HD leukemias
HD leukemias evolve from a single immature BCP cell, which is transformed very early during fetal development [81–89]. Although there is general agreement that the initiating event is a flawed cell division during which the leukemic precursor cell is supposed to receive all extra chromosomes instantaneously, the actual cause of the triggering nondisjunction error is currently still unknown [3, 16, 90]. Since decades of extensive research have not uncovered any potentially responsible genetic defect, we proposed that the critical nondisjunction and segregation errors may be due to a physical disruption of the intricate spindle scaffold of a mitotic cell, which could be due to the untimely cytoplasmic influx from a second, partially fused interphase cell [10].
The current and intensely scrutinized view suggests that a nonclassical HD karyoytpe can only derive from the duplication of a previously generated hyperhaploid one [3, 7, 8, 14]. However, as we show in Fig. 1, it may indeed be more likely that the nonclassical HD clone is generated first in a similar fashion as the classical HD one. Reversing the order of appearance of nonclassical HD and hyperhaploid clones, simplifies the entire concept of how classical, nonclassical, and hyperhaploid HD cases are interrelated. If the nonclassical HD clone comes first, the hyperhaploid clone can only be its descendant, and then, may either coexist as a secondary change or even outperform the original nonclassical HD clone [10]. Another albeit more remote alternative possibility could be that the hyperhaploid clone originates from the cell that is supposed to cause the nondisjunction error [10].
Clinical and biological features
In the western world, up to 35% of childhood leukemias are hyperdiploid, but with 15–25%, they are far less prevalent in patients of Asian, African, and Native American descent [91–93]. Children with HD ALL are young (median age of about 4 years). At diagnosis, they have a white blood cell count that is typically below 109/l and no extramedullary disease. HD blast cells show a high expression of CD9, CD20, CD22 CD58 CD 66c, CD86, and CD123, and a low expression of CD45 [94]. Their most relevant immunophenotypic feature is the aberrant expression of CD123, the interleukin-3 receptor alpha chain, which is encoded in the pseudo-autosomal regions on Xp22.3 and Yp11.3 [95–97]. This marker is therefore also commonly used as an indicative surrogate flow-cytometric predictor of classical HD leukemias.
One thought-provoking discovery is the fact that heterozygous HLA-DPB1*0201 alleles are significantly enriched in patients with HD leukemias [98]. HLA-DPB1*0201 belongs to the HLA class II genes that are important in adaptive immune responses to infection. This preponderance has therefore been taken as evidence that yet-undefined intrauterine immunological processes execute selective forces and mediate a proliferative stress on preleukemic HD cells, effects that may then again be somewhat mitigated by protective life-style factors, such as breastfeeding and day-care attendance [45, 99, 100]. Supporting these HLA data are now recent epidemiological data and laboratory findings. They revealed that prenatal cytomegalovirus infections of the patients’ mothers are probably one of the most relevant etiologically factors for initiating and/or promoting especially the development of HD leukemias [101–103]. Moreover, this hazard is particularly pronounced in carriers of an ARID5B risk allele [101–103].
HD ALL blast cells are inherently difficult to maintain and propagate in culture, which makes it extremely difficult to perform not only cytogenetic analyses, but also any other type of research that depends on viable cells [104]. Underlining this problem is the fact that there are no established cell lines from patients with HD leukemias, and only one that derives from a nonclassical HD (MHH-CALL-2) and a second from a hyperhaploid case (NALM-16) [3, 25, 105–107]. The only way to maintain and propagate HD blasts is therefore to either cultivate them on autologous feeder layers or to xenograft them [104, 108].
Diagnostic assessment, disease stratification, and treatment outcome
Table 2 summarizes the pros and cons of the various technologies that are instrumental not only for the identification and delineation of HD ALL cases, but also for the in-depth analyses of their genomic structure. Several of these technologies are quite sophisticated, so it is quite surprising that those being used almost exclusively for treatment stratification today are still the rather basic options that have been already in use for many decades. The reason for this is that these diagnostic tools are simple to perform, cheap, and fast [109–112]. However, the fact that they are still considered sufficient also implies that, for clinical purposes, a more in-depth evaluation of the intricate features of such cases is deemed completely unnecessary, since these patients have in any case an overall very good outlook (Table 3). Yet, as alluded to above, this tactic does certainly neither suffice for the demarcation and clear assignment of classical and nonclassical HD cases nor for that of classical HD and near-triploid ones, respectively. Such delineation ambiguities can to a certain extent obscure the outcome results that are obtained by different treatment studies. Moreover, applying the usual DNA content of equal to or more than 1.16 (equivalent to approximately 54 chromosomes) as the lower defining HD threshold will fail to secure cases with karyotypes that contain only a smaller and/or lower number of chromosomes. As pointed out by Carroll et al., such allocation problems cause the misclassification of a considerable proportion (at least 25%) of nonclassical HD cases [9]. They showed that studies that evaluated the treatment outcome of mono- and bi-clonal hyperhaploid cases never included monoclonal nonclassical HD cases, even though they are always equated with hyperhaploid ones and automatically assigned to the high-risk group, in case they are indeed identified [5, 9, 113–115]. Carroll’s observation can only mean that monoclonal nonclassical HD cases were either recognized but purposely not included or not recognized and consequently stratified and handled the same way as classical HD cases.
Table 2.
Advantages and disadvantages of various diagnostic technologies for the diagnostic assessment of HD ALL.
| Method | Advantages | Disadvantages |
|---|---|---|
| DNA index measurement |
• Fast, cheap, and easy • Can to a certain extent identify multiple abnormal clones to a certain extent |
• Provides only a rough estimate of the DNA content but no information about chromosomal composition of the respective clones • Cannot differentiate between classical and nonclassical HD forms |
| Cytogenetics | • Provides information about types and numbers of chromosomes |
• Relies on the availability of blast cell metaphases • Information derives only from single dividing cells |
| Fluorescence in situ hybridization (FISH) |
• Allows interphase screening • Provides information about composition and size of cell clones |
• Utilized to screen for selected relevant chromosomes only • Cannot elucidate the chromosomal composition of the entire genome |
| CGH/SNP array analysis | • Provides a representative genome-wide profile of all large- and small-scale copy number alterations as well as their allelic pattern | • Cannot distinguish between monoclonal and/or bi-clonal forms of hyperhaploid and nonclassical HD forms |
| Whole-exome sequencing (WES) | • Provides exome-wide information about copy number as well as sequence alterations |
• Primarily a research tool • Mutations hardly diagnostically or therapeutically relevant in HD |
| Whole-genome sequencing (WGS) |
• Provides genome-wide information about all relevant copy number, structural, and sequence alterations simultaneously • Could replace all other methods, in principle |
• Cannot distinguish between monoclonal and/or bi-clonal forms of hyperhaploid and nonclassical HD forms • Not yet implemented for routine diagnostics |
| Gene expression (GEP) analysis |
• Reflects chromosome copy number changes • May be utilized for mutation calling • Can identify fusion genes • Uncovers the close relationship between mono- and/or bi-clonal nonclassical HD and hyperhaploid forms |
• Diagnostically not relevant • Primarily a research tool |
| Optical genome mapping |
• Provides genome-wide information about copy number and structural abnormalities • Reveals allele-specific patterns • May eventually replace karyotype, FISH, and array analyses |
• Research tool only • Not yet implemented in the diagnostic work-up • Cannot detect sequence variants |
Table 3.
Standard risk stratification and outcome of children with HD ALL in contemporary treatment protocols.
| Study protocol | Age (years) | WBC ×109/L | CNS status | Treatment response | Genetic parameters | Outcome | Ref. | ||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| Steroid responsea | Day 8 | Day 15 | End of induction | Later timepoints | |||||||
| AIEOP-BFM 2000 ALL | 1–17 | Any | Any | + | <1.000 blast/µl (PB) | n.a. | PCR MRD negative |
Day 78 PCR negative |
– |
Standard DI PII: 4-year OS 97.9% 8-year DFS 92% reduced intensity DI PIII: 4-year OS 93.4% 8-year DFS 90% |
[148, 149] |
| ALL IC-BFM | 1–6 | <20 | Any | + | <1.000 blast/µl (PB) | M1/M2 | – | – | – |
5-year EFS 81% 5-year OS 90% (all SR patients combined) |
[150] |
| CoALL 07–03 | <10 | <25 | Any | – | – | MRD <0.01% | MRD <0.01%b | – | – | Number of identified HD patients is too low | [151] |
| COG AALL 0331 LRS | 1–10 | <50 | CNS1 only | – | M1c | M1c | MRD <0.01%b | – | +4, +10, +17 |
6-year CCR 96.3% 6-year OS 99.4% |
[116, 118] |
| COG AALL 0932 LR-C | 1–10 | <50 | CNS1 only | – |
MRD <0.01% (PB) |
– | MRD <0.01%b | – | +4, +10 |
5-year EFS 98.8% 5-year OS 100% (all LR patients combined) |
[117] |
| COG AALL 0932 LR-M | 1–10 | <50 | CNS1 only | – |
MRD <0.01% (PB) |
– | MRD <0.01%b | – | +4, +10 |
5-year EFS 98.5% 5-year OS 100% (all LR patients combined) |
[117] |
| COG P9904 | 1–10 | <50 | No CNS3 | – | MRD <0.01% (PB) | MRD <0.01%b | – | +4, +10 |
5-year EFS 97% 5-year OS 98% (all LR patients combined) |
[117] | |
| RELLA05 | 1–9 | <50 | CNS1/CNS2 or traumatic LP | – | MRD <0.01%d | – | – | DNA index >1, 16 |
5-year EFS 92% 5-year OS 96% (LR group combined) |
[152] | |
| JACLS-ALL-02 | 1–9 | <10 | No CNS3 | + | <1.000 blast/µl (PB) | M1/M2 BM | M1 BM | – | – |
4-year EFS 89.4% 4-year OS 94.9% |
[153] |
| JACLS-ALL-02 SR | 1–9 | <10 | No CNS3 | + |
<1.000 blast/µl (PB) |
M1/M2 BM | M1 BM | – | – |
4-year EFS 100% (+4, +10, +17) 4-year EFS 88.2% (other HD) |
[93] |
| St. Jude Total XVI | 1–10 | <50 | Any | – | – | MRD <1% | D46 MRD <0.01% | – | DNA index >1, 16 |
5-year EFS 95.3% 5-year OS 99.4% |
[154, 155] |
| UKALL 2003 | 1–10 | <50 | Any | – | – | M1/M2 BM | MRD <0.01% | – | – |
10-year survival: all HD patients 90% EFS, 94% OS good risk HD 92% EFS 96% OS poor risk HD 81% EFS, 86% OS |
[120, 156] |
| UKALL 97/99 | 1–10 | <50 | Any | – | – | M1/M2 BM | – | – | – |
10-year survival: all HD patients 84% EFS, 93% OS good risk HD 86% EFS, 94% OS poor risk HD 71% EFS, 89% OS |
[120] |
| NOPHO ALL 1992/2000 |
2–10 (1992) 1–10 (2000) |
<10 | CNS1 | – | – | M1/M2 BM | M1 BM | – |
Karyotype (central review) |
5-year EFS 82% 10-year EFS 80% 5-year OS 91% 10-year OS 89% (both studies combined) |
[157, 158] |
| DCOG 10 | 1–18 | Any | CNS1 only | + |
<1.000 blast/µl (PB) |
– | PCR negative |
Day 80 PCR negative |
– |
5-year EFS 87.9%, 5-year OS 93.3% |
[159] |
| CCLG-ALL 2008 | 1–10 | <50 | No CNS3 | + | <1.000 blast/µl (PB) | M1/M2 BM | PCR negative or flow MRD <0.01% |
Week 12 PCR negative or flow MRD <0.01% |
– |
5-year EFS 81.6% 5-year OS 91.5% (SR group combined) |
[160] |
| TCCSG L04–16 | 1–6 | <20 | No CNS3 | + | <1.000 blast/µl (PB) | – |
Day 43 M1 |
– | – |
5-year EFS 85% 5-year OS 95.4% |
[161] |
CNS central nervous system involvement, PB peripheral blood, BM bone marrow, MRD measurable residual disease, DFS disease-free survival, ES event-free survival, OS overall survival, CCR continuous complete remission, PII/PIII protocol II or protocol III of AIEOP/BFM, respectively, DI delayed intensification.
aPatients were exposed for 1 week to prednisone monotherapy before chemotherapy on day 8 has been initiated. On day 0, one additional dose of methotrexate was given intrathecally. The presence of less or more than 1.000 blasts/μl PB on day 8 was defined as prednisone good or poor response, respectively.
bDay 29 responses.
cDay 8 or day 15 M1 bone marrow.
dDay 19 MRD <0.01%.
+ day 29 MRD detectable but <0.01% and undetectable before start of interim maintenance.
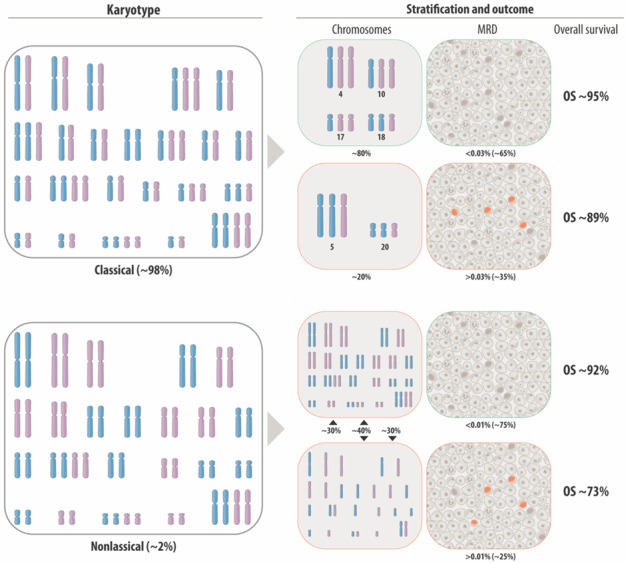
The two central challenges that clinicians are nowadays confronted with in HD leukemias are how to identify patients with a high propensity to relapse already at diagnosis and how to reduce treatment in low-risk patients without jeopardizing their good outcome. Virtually all contemporary, ongoing, and planned treatment studies rely on the assessment of the measurable residual disease (MRD), either based on the PCR-based quantification of immunoglobulin and T-cell receptor gene rearrangements or based on the flow-cytometric assessment of the immunophenotypic criteria of blast cells. Only some of these studies also require the identification of HD cases, which is then achieved by determining the DNA content, the overall chromosome number and/or the copy numbers of selected chromosomes with fluorescence in situ hybridization (FISH) that are considered appropriate for the delineation of good risk cases. Such a chromosomal risk classification has, for instance, already been used successfully in the Children’s Oncology Group study for over 25 years (Table 3) [116–118]. O’Connor et al. and Enshaei et al. have recently succeeded in significantly refining and improving the prognostic value of this genetic stratification system. First, they reported that at least in the UKALL treatment studies, the optimal MRD threshold for HD cases, which derives from a retrospective statistical analysis of data, is 0.03% rather than the 0.01% cutoff that is normally used [119, 120]. This elaborated MRD threshold of 0.03% for HD cases will now be prospectively evaluated in the newly established ALLTogether consortium, which comprises the Nordic countries, Estonia, Lithuania, the United Kingdom, the Netherlands, Belgium, Ireland, and France (https://clinicaltrials.gov/ct2/show/NCT03911128). Whether such a challenging detailed threshold definition is indeed also technically feasible to routinely achieve and therefore worthwhile to implement in future clinical studies, remains to be seen. Second, Enshaei et al. convincingly proved that the copy number assessment of four chromosomes, whose prognostic relevance was already appreciated previously is sufficient to demarcate two distinct risk groups. The low-risk group, which in their study makes up 80%, is defined by trisomies 17 and/or 18, whereas their poor risk group makes up 20% and is defined by trisomies 5 and 20 [120]. This poor risk group includes nearly half of the relapse cases. Since trisomies 5 and 20 are rarely seen in cases with less than 58 chromosomes [121], it will be important to evaluate to which extent their prognostic value depends on or is influenced by the overall chromosome number. Nevertheless, the respective UKALL profile still outperforms other trisomy-based ones and is independent of the results of MRD measurements, although it might be expected that combining both will increase the value of this risk score even further. Especially in the context of the planned treatment reduction for low-risk HD cases, we consider it appropriate to further substantiate these findings in prospective studies of large cohorts of genetically thoroughly defined HD cases.
Of further note in this context is the recent observation that nonclassical HD cases with SETD2 mutations had an inferior event-free (8/280 cases; 47% versus 95%) and overall survival than nonaffected ones [16].
Owing to the lack of more refined genetic discriminators, all mono- and bi-clonal hyperhaploid and nonclassical HD forms are hitherto stratified as high risk. However, Mullighan et al. showed that even in those cases MRD is the most important prognostic indicator since all cases with a negative MRD status (<0.01%) are highly curable with intensive chemotherapy alone [54].
The high cure rates of low-risk classical HD ALL cases imply that many of them are probably overtreated. The daunting challenge is now to reduce treatment intensity to diminish side effects and avoid ensuing long-term sequelae without endangering the hitherto achieved excellent overall treatment outcome. Some of the trials that have addressed this issue so far have been quite successful (even without utilizing any genetic risk score), whereas those trials which reduced the duration of maintenance therapy to 6 months were not, at least as regards HD ALL [55, 117, 122–125]. Although Kato et al. found that one year of maintenance therapy is probably sufficient for TCF3::PBX1- and ETV6::RUNX1-positive cases, it is definitely not adequate for all HD leukemias. As a result of relapses, their disease-free survival was only 56.6 ± 10.3% [123]. Nevertheless, viewed in reverse a remarkable proportion of patients also benefited; most relapses were salvageable, and the overall survival of the entire cohort still reached 91.7 ± 5.6% after 12 years [123].
The emergence of specific mutations in disease recurrences suggests that many of them are treatment induced. These mutations comprise, for instance, those in NR3C1/2, CREBBP, and WHSC1 for glucocorticoids; NT5C2, MSH2/6, PMS2, and PRPS1/2 for thiopurines; and FPGS for MTX [126]. Examining such mutation patterns in 103 ALL germline/diagnosis/relapse trios, Li et al. identified two novel relapse-specific signatures in 25% of children with an early or late relapse [126]. They were able to prove that one of these signatures was caused by thiopurine treatment, whereas for the other, which was significantly enriched in HD leukemias, they could not ascertain the respective cause.
Together these observations confirm that an adequate length of maintenance therapy is essential for preventing early relapses in classical HD leukemias but also, that this treatment component cannot be held accountable for inducing late ones, as seems to be the case in other genetic subtypes.
Summary
HD leukemias have puzzled and confused researchers and clinicians for more than 50 years now. Novel findings that derive from array, mutation, and gene expression analyses offer now new opportunities to re-examine deeply engrained, yet largely unproven and unquestioned views about their origin, their mode of creation, and the interrelatedness of the diverse aneuploid sub-forms. Such fresh insights will eventually help to reconsider and refine the current classification system and thereby also influence the prognostic stratification and treatment of these subgroups. One example concerns the elucidation of the biological relatedness of classical, nonclassical (“duplicated hyperhaploids”), and pure hyperhaploid cases, which will help to better understand what drives their apparent different clinical behaviors. Another one concerns hyperdiploid forms with a high chromosome number. Near-triploid cases as well as mono- and bi-clonal hypodiploid cases can nowadays be easily ascertained, because over 90% of them are TP53 mutated. However, it is less clear whether the remaining TP53 wild-type ones with a similar high chromosome count should be also stratified as near-triploid or rather as classical hyperdiploid ones. Although this concerns only a few cases, their diagnostic clarification, and most appropriate allocation requires further scrutiny. Array analyses deliver not only a detailed information about copy number changes, but also about allele distribution alterations, such as the presence of otherwise unidentifiable uniparental disomies. Bearing the ongoing trend to reduce treatment for very low-risk patients in mind, which especially also concerns HD leukemias, we advocate to implement this state-of-the-art technology together with mutation screening in treatment studies to comprehensively characterize the detailed genomic make-up of HD leukemias [127]. Only such an approach, rather than, as is current practice, merely typifying them with DNA content measurements, cytogenetics, and/or selected FISH probes, can better our understanding of the biology and the clinical behavior of the karyotypically heterogeneous subsets of classical and nonclassical HD leukemias as well as oversee in a more individual fashion the effects of various treatment interventions. The invaluable results that have already been obtained with such thorough analyses in selected yet still less well-characterized cohorts clearly prove, that the study-wide implementation of such a policy will significantly foster basic as well as clinical research in this particular group of patients and thereby provide benefits for their management that will go far beyond the simple identification and prognostic grading of HD cases, as is still done today.
Acknowledgements
This paper is dedicated to our mentor and friend Fritz Lampert, former Head of the Department of Pediatric Hematology and Oncology, Children’s University Hospital Giessen, Germany, who described the first hyperdiploid ALL cases. We thank Fikret Rifatbegovic for his invaluable help in designing and drawing the figures.
Author contributions
OAH and AB jointly contributed to conceptualization, screened, and analyzed literature, and wrote the manuscript.
Funding
Open Access funding enabled and organized by Projekt DEAL.
Competing interests
The authors declare no competing interests.
Footnotes
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Contributor Information
Oskar A. Haas, Email: oskar.haas@labdia.at
Arndt Borkhardt, Email: arndt.borkhardt@med.uni-duesseldorf.de.
References
- 1.Lampert F. Cellulärer DNS-Gehalt und Chromosomenzahl bei der akuten Leukämie im Kindesalter und ihre Bedeutung für Chemotherapie und Prognose. Klinische Wochenschr. 1967;45:763–8. doi: 10.1007/BF01747643. [DOI] [PubMed] [Google Scholar]
- 2.Irving JA, Enshaei A, Parker CA, Sutton R, Kuiper RP, Erhorn A, et al. Integration of genetic and clinical risk factors improves prognostication in relapsed childhood B-cell precursor acute lymphoblastic leukemia. Blood. 2016;128:911–22. doi: 10.1182/blood-2016-03-704973.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Paulsson K, Johansson B. High hyperdiploid childhood acute lymphoblastic leukemia. Genes Chromosomes Cancer. 2009;48:637–60. doi: 10.1002/gcc.20671.. [DOI] [PubMed] [Google Scholar]
- 4.Mertens F, Johansson B, Mitelman F. Dichotomy of hyperdiploid acute lymphoblastic leukemia on the basis of the distribution of gained chromosomes. Cancer Genet Cytogenet. 1996;92:8–10. doi: 10.1016/s0165-4608(96)00124-0.. [DOI] [PubMed] [Google Scholar]
- 5.Harrison CJ, Moorman AV, Broadfield ZJ, Cheung KL, Harris RL, Reza Jalali G, et al. Three distinct subgroups of hypodiploidy in acute lymphoblastic leukaemia. Br J Haematol. 2004;125:552–9. doi: 10.1111/j.1365-2141.2004.04948.x.. [DOI] [PubMed] [Google Scholar]
- 6.Heerema NA, Raimondi SC, Anderson JR, Biegel J, Camitta BM, Cooley LD, et al. Specific extra chromosomes occur in a modal number dependent pattern in pediatric acute lymphoblastic leukemia. Genes Chromosomes Cancer. 2007;46:684–93. doi: 10.1002/gcc.20451.. [DOI] [PubMed] [Google Scholar]
- 7.Holmfeldt L, Wei L, Diaz-Flores E, Walsh M, Zhang J, Ding L, et al. The genomic landscape of hypodiploid acute lymphoblastic leukemia. Nat Genet. 2013;45:242–52. doi: 10.1038/ng.2532.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Safavi S, Paulsson K. Near-haploid and low-hypodiploid acute lymphoblastic leukemia: two distinct subtypes with consistently poor prognosis. Blood. 2017;129:420–3. doi: 10.1182/blood-2016-10-743765.. [DOI] [PubMed] [Google Scholar]
- 9.Carroll AJ, Shago M, Mikhail FM, Raimondi SC, Hirsch BA, Loh ML, et al. Masked hypodiploidy: hypodiploid acute lymphoblastic leukemia (ALL) mimicking hyperdiploid ALL in children: a report from the Children’s Oncology Group. Cancer Genet. 2019;238:62–8. doi: 10.1016/j.cancergen.2019.07.009.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Haas OA. Somatic sex: on the origin of neoplasms with chromosome counts in uneven ploidy ranges. Front Cell Dev Biol. 2021;9:631946. doi: 10.3389/fcell.2021.631946.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.McGowan-Jordan J, Hastings RJ, Moore S. ISCN 2020: an international system for human cytogenomic nomenclature. Taipei, Taiwan: Kager; 2020. [DOI] [PubMed]
- 12.Dastugue N, Suciu S, Plat G, Speleman F, Cave H, Girard S, et al. Hyperdiploidy with 58-66 chromosomes in childhood B-acute lymphoblastic leukemia is highly curable: 58951 CLG-EORTC results. Blood. 2013;121:2415–23. doi: 10.1182/blood-2012-06-437681.. [DOI] [PubMed] [Google Scholar]
- 13.Creasey T, Enshaei A, Nebral K, Schwab C, Watts K, Cuthbert G, et al. Single nucleotide polymorphism array-based signature of low hypodiploidy in acute lymphoblastic leukemia. Genes Chromosomes Cancer. 2021;60:604–15. doi: 10.1002/gcc.22956.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Molina O, Bataller A, Thampi N, Ribera J, Granada I, Velasco P, et al. Near-haploidy and low-hypodiploidy in B-cell acute lymphoblastic leukemia: when less is too much. Cancers (Basel). 2021;14:32. 10.3390/cancers14010032. [DOI] [PMC free article] [PubMed]
- 15.Molina O, Abad MA, Sole F, Menendez P. Aneuploidy in cancer: lessons from acute lymphoblastic leukemia. Trends Cancer. 2021;7:37–47. doi: 10.1016/j.trecan.2020.08.008.. [DOI] [PubMed] [Google Scholar]
- 16.Brady SW, Roberts KG, Gu Z, Shi L, Pounds S, Pei D, et al. The genomic landscape of pediatric acute lymphoblastic leukemia. Nat Genet. 2022 doi: 10.1038/s41588-022-01159-z.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Onodera N, McCabe NR, Rubin CM. Formation of a hyperdiploid karyotype in childhood acute lymphoblastic leukemia [see comments] Blood. 1992;80:203–8. doi: 10.1182/blood.V80.1.203.203.. [DOI] [PubMed] [Google Scholar]
- 18.Lundin KB, Olsson L, Safavi S, Biloglav A, Paulsson K, Johansson B. Patterns and frequencies of acquired and constitutional uniparental isodisomies in pediatric and adult B-cell precursor acute lymphoblastic leukemia. Genes Chromosomes Cancer. 2016;55:472–9. doi: 10.1002/gcc.22349.. [DOI] [PubMed] [Google Scholar]
- 19.Charrin C, Thomas X, Ffrench M, Le QH, Andrieux J, Mozziconacci MJ, et al. A report from the LALA-94 and LALA-SA groups on hypodiploidy with 30 to 39 chromosomes and near-triploidy: 2 possible expressions of a sole entity conferring poor prognosis in adult acute lymphoblastic leukemia (ALL) Blood. 2004;104:2444–51. doi: 10.1182/blood-2003-04-1299.. [DOI] [PubMed] [Google Scholar]
- 20.Gisselsson D. Mechanisms of whole chromosome gains in tumors–many answers to a simple question. Cytogenet Genome Res. 2011;133:190–201. doi: 10.1159/000322480.. [DOI] [PubMed] [Google Scholar]
- 21.Gisselsson D. Aneuploidy in cancer: sudden or sequential? Cell Cycle. 2011;10:359–61. doi: 10.4161/cc.10.3.14518.. [DOI] [PubMed] [Google Scholar]
- 22.Safavi S, Forestier E, Golovleva I, Barbany G, Nord KH, Moorman AV, et al. Loss of chromosomes is the primary event in near-haploid and low-hypodiploid acute lymphoblastic leukemia. Leukemia. 2013;27:248–50. doi: 10.1038/leu.2012.227.. [DOI] [PubMed] [Google Scholar]
- 23.Baughn LB, Biegel JA, South ST, Smolarek TA, Volkert S, Carroll AJ, et al. Integration of cytogenomic data for furthering the characterization of pediatric B-cell acute lymphoblastic leukemia: a multi-institution, multi-platform microarray study. Cancer Genet. 2015;208:1–18. doi: 10.1016/j.cancergen.2014.11.003.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Chen C, Bartenhagen C, Gombert M, Okpanyi V, Binder V, Rottgers S, et al. Next-generation-sequencing of recurrent childhood high hyperdiploid acute lymphoblastic leukemia reveals mutations typically associated with high risk patients. Leuk Res. 2015;39:990–1001. doi: 10.1016/j.leukres.2015.06.005.. [DOI] [PubMed] [Google Scholar]
- 25.Lundin-Strom KB, Strom K, Biloglav A, Barbany G, Behrendtz M, Castor A, et al. Parental origin of monosomic chromosomes in near-haploid acute lymphoblastic leukemia. Blood Cancer J. 2020;10:51. doi: 10.1038/s41408-020-0317-2.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Maloney KW, Carroll WL, Carroll AJ, Devidas M, Borowitz MJ, Martin PL, et al. Down syndrome childhood acute lymphoblastic leukemia has a unique spectrum of sentinel cytogenetic lesions that influences treatment outcome: a report from the Children’s Oncology Group. Blood. 2010;116:1045–50. doi: 10.1182/blood-2009-07-235291.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Forestier E, Izraeli S, Beverloo B, Haas O, Pession A, Michalova K, et al. Cytogenetic features of acute lymphoblastic and myeloid leukemias in pediatric patients with Down syndrome: an iBFM-SG study. Blood. 2008;111:1575–83. doi: 10.1182/blood-2007-09-114231.. [DOI] [PubMed] [Google Scholar]
- 28.Abbasi MR, Nebral K, Haslinger S, Inthal A, Zeitlhofer P, Konig M, et al. Copy number changes and allele distribution patterns of chromosome 21 in B cell precursor acute lymphoblastic leukemia. Cancers (Basel). 2021;13:4597. 10.3390/cancers13184597. [DOI] [PMC free article] [PubMed]
- 29.Bhansali RS, Rammohan M, Lee P, Laurent AP, Wen Q, Suraneni P, et al. DYRK1A regulates B cell acute lymphoblastic leukemia through phosphorylation of FOXO1 and STAT3. J Clin Invest. 2021;131:e135937. 10.1172/JCI135937. [DOI] [PMC free article] [PubMed]
- 30.Ross ME, Zhou X, Song G, Shurtleff SA, Girtman K, Williams WK, et al. Classification of pediatric acute lymphoblastic leukemia by gene expression profiling. Blood. 2003;102:2951–9. doi: 10.1182/blood-2003-01-0338.. [DOI] [PubMed] [Google Scholar]
- 31.Chilton L, Buck G, Harrison CJ, Ketterling RP, Rowe JM, Tallman MS, et al. High hyperdiploidy among adolescents and adults with acute lymphoblastic leukaemia (ALL): cytogenetic features, clinical characteristics and outcome. Leukemia. 2014;28:1511–8. doi: 10.1038/leu.2013.379.. [DOI] [PubMed] [Google Scholar]
- 32.Sunil SK, Prakash PN, Hariharan S, Vinod G, Preethi RT, Geetha N, et al. Adult acute lymphoblastic leukemia with near haploidy, hyperdiploidy and Ph positive lines: a rare entity with poor prognosis. Leuk Lymphoma. 2006;47:561–3. doi: 10.1080/10428190500361094.. [DOI] [PubMed] [Google Scholar]
- 33.Peterson JF, Ketterling RP, Huang L, Finn LE, Shi M, Hoppman NL, et al. A near-haploid clone harboring a BCR/ABL1 gene fusion in an adult patient with newly diagnosed B-lymphoblastic leukemia. Genes Chromosomes Cancer. 2019;58:665–8. doi: 10.1002/gcc.22744.. [DOI] [PubMed] [Google Scholar]
- 34.Cheng Q, Yang W, Raimondi SC, Pui CH, Relling MV, Evans WE. Karyotypic abnormalities create discordance of germline genotype and cancer cell phenotypes. Nat Genet. 2005;37:878–82. doi: 10.1038/ng1612.. [DOI] [PubMed] [Google Scholar]
- 35.Papaemmanuil E, Hosking FJ, Vijayakrishnan J, Price A, Olver B, Sheridan E, et al. Loci on 7p12.2, 10q21.2 and 14q11.2 are associated with risk of childhood acute lymphoblastic leukemia. Nat Genet. 2009;41:1006–10. doi: 10.1038/ng.430.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Walsh KM, de Smith AJ, Chokkalingam AP, Metayer C, Dahl GV, Hsu LI, et al. Novel childhood ALL susceptibility locus BMI1-PIP4K2A is specifically associated with the hyperdiploid subtype. Blood. 2013;121:4808–9. doi: 10.1182/blood-2013-04-495390.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Xu H, Yang W, Perez-Andreu V, Devidas M, Fan Y, Cheng C, et al. Novel susceptibility variants at 10p12.31-12.2 for childhood acute lymphoblastic leukemia in ethnically diverse populations. J Natl Cancer Inst. 2013;105:733–42. doi: 10.1093/jnci/djt042.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Hsu LI, Chokkalingam AP, Briggs FB, Walsh K, Crouse V, Fu C, et al. Association of genetic variation in IKZF1, ARID5B, and CEBPE and surrogates for early-life infections with the risk of acute lymphoblastic leukemia in Hispanic children. Cancer Causes Control. 2015;26:609–19. doi: 10.1007/s10552-015-0550-3.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Wiemels JL, de Smith AJ, Xiao J, Lee ST, Muench MO, Fomin ME, et al. A functional polymorphism in the CEBPE gene promoter influences acute lymphoblastic leukemia risk through interaction with the hematopoietic transcription factor Ikaros. Leukemia. 2016;30:1194–7. doi: 10.1038/leu.2015.251.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Studd JB, Vijayakrishnan J, Yang M, Migliorini G, Paulsson K, Houlston RS. Genetic and regulatory mechanism of susceptibility to high-hyperdiploid acute lymphoblastic leukaemia at 10p21.2. Nat Commun. 2017;8:14616. doi: 10.1038/ncomms14616.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.de Smith AJ, Walsh KM, Francis SS, Zhang C, Hansen HM, Smirnov I, et al. BMI1 enhancer polymorphism underlies chromosome 10p12.31 association with childhood acute lymphoblastic leukemia. Int J Cancer. 2018;143:2647–58. doi: 10.1002/ijc.31622.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Xu H, Cheng C, Devidas M, Pei D, Fan Y, Yang W, et al. ARID5B genetic polymorphisms contribute to racial disparities in the incidence and treatment outcome of childhood acute lymphoblastic leukemia. J Clin Oncol. 2012;30:751–7. doi: 10.1200/JCO.2011.38.0345.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Xu H, Zhao X, Bhojwani D, E S, Goodings C, Zhang H, et al. ARID5B influences antimetabolite drug sensitivity and prognosis of acute lymphoblastic leukemia. Clin Cancer Res. 2020;26:256–64. doi: 10.1158/1078-0432.CCR-19-0190.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Ma SK, Chan GC, Wan TS, Lam CK, Ha SY, Lau YL, et al. Near-haploid common acute lymphoblastic leukaemia of childhood with a second hyperdiploid line: a DNA ploidy and fluorescence in-situ hybridization study. Br J Haematol. 1998;103:750–5. doi: 10.1046/j.1365-2141.1998.01044.x.. [DOI] [PubMed] [Google Scholar]
- 45.Williams LA, Yang JJ, Hirsch BA, Marcotte EL, Spector LG. Is there etiologic heterogeneity between subtypes of childhood acute lymphoblastic leukemia? A review of variation in risk by subtype. Cancer Epidemiol Biomark Prev. 2019;28:846–56. doi: 10.1158/1055-9965.EPI-18-0801.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Zhao X, Qian M, Goodings C, Zhang Y, Yang W, Wang P, et al. Molecular mechanisms of ARID5B-mediated genetic susceptibility to acute lymphoblastic leukemia. J Natl Cancer Inst. 2022;114:1287–95. 10.1093/jnci/djac101. [DOI] [PMC free article] [PubMed]
- 47.Moriyama T, Metzger ML, Wu G, Nishii R, Qian M, Devidas M, et al. Germline genetic variation in ETV6 and risk of childhood acute lymphoblastic leukaemia: a systematic genetic study. Lancet Oncol. 2015;16:1659–66. doi: 10.1016/S1470-2045(15)00369-1.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Cave H, Caye A, Strullu M, Aladjidi N, Vignal C, Ferster A, et al. Acute lymphoblastic leukemia in the context of RASopathies. Eur J Med Genet. 2016;59:173–8. doi: 10.1016/j.ejmg.2016.01.003.. [DOI] [PubMed] [Google Scholar]
- 49.Nishii R, Baskin-Doerfler R, Yang W, Oak N, Zhao X, Yang W, et al. Molecular basis of ETV6-mediated predisposition to childhood acute lymphoblastic leukemia. Blood. 2021;137:364–73. doi: 10.1182/blood.2020006164.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Karastaneva A, Nebral K, Schlagenhauf A, Baschin M, Palankar R, Juch H, et al. Novel phenotypes observed in patients with ETV6-linked leukaemia/familial thrombocytopenia syndrome and a biallelic ARID5B risk allele as leukaemogenic cofactor. J Med Genet. 2020;57:427–33. doi: 10.1136/jmedgenet-2019-106339.. [DOI] [PubMed] [Google Scholar]
- 51.Paulsson K, Horvat A, Strombeck B, Nilsson F, Heldrup J, Behrendtz M, et al. Mutations of FLT3, NRAS, KRAS, and PTPN11 are frequent and possibly mutually exclusive in high hyperdiploid childhood acute lymphoblastic leukemia. Genes Chromosomes Cancer. 2008;47:26–33. doi: 10.1002/gcc.20502.. [DOI] [PubMed] [Google Scholar]
- 52.Inthal A, Zeitlhofer P, Zeginigg M, Morak M, Grausenburger R, Fronkova E, et al. CREBBP HAT domain mutations prevail in relapse cases of high hyperdiploid childhood acute lymphoblastic leukemia. Leukemia. 2012;26:1797–803. doi: 10.1038/leu.2012.60.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Malinowska-Ozdowy K, Frech C, Schonegger A, Eckert C, Cazzaniga G, Stanulla M, et al. KRAS and CREBBP mutations: a relapse-linked malicious liaison in childhood high hyperdiploid acute lymphoblastic leukemia. Leukemia. 2015;29:1656–67. doi: 10.1038/leu.2015.107.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Mullighan CG, Jeha S, Pei D, Payne-Turner D, Coustan-Smith E, Roberts KG, et al. Outcome of children with hypodiploid ALL treated with risk-directed therapy based on MRD levels. Blood. 2015;126:2896–9. doi: 10.1182/blood-2015-09-671131.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Paulsson K, Lilljebjorn H, Biloglav A, Olsson L, Rissler M, Castor A, et al. The genomic landscape of high hyperdiploid childhood acute lymphoblastic leukemia. Nat Genet. 2015;47:672–6. doi: 10.1038/ng.3301.. [DOI] [PubMed] [Google Scholar]
- 56.de Smith AJ, Ojha J, Francis SS, Sanders E, Endicott AA, Hansen HM, et al. Clonal and microclonal mutational heterogeneity in high hyperdiploid acute lymphoblastic leukemia. Oncotarget. 2016;7:72733–45. doi: 10.18632/oncotarget.12238.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Studd JB, Cornish AJ, Hoang PH, Law P, Kinnersley B, Houlston R. Cancer drivers and clonal dynamics in acute lymphoblastic leukaemia subtypes. Blood Cancer J. 2021;11:177. doi: 10.1038/s41408-021-00570-9.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Mullighan CG, Zhang J, Kasper LH, Lerach S, Payne-Turner D, Phillips LA, et al. CREBBP mutations in relapsed acute lymphoblastic leukaemia. Nature. 2011;471:235–9. doi: 10.1038/nature09727.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Koh G, Degasperi A, Zou X, Momen S, Nik-Zainal S. Mutational signatures: emerging concepts, caveats and clinical applications. Nat Rev Cancer. 2021;21:619–37. doi: 10.1038/s41568-021-00377-7.. [DOI] [PubMed] [Google Scholar]
- 60.Alexandrov LB, Kim J, Haradhvala NJ, Huang MN, Tian Ng AW, Wu Y, et al. The repertoire of mutational signatures in human cancer. Nature. 2020;578:94–101. doi: 10.1038/s41586-020-1943-3.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Yeoh EJ, Ross ME, Shurtleff SA, Williams WK, Patel D, Mahfouz R, et al. Classification, subtype discovery, and prediction of outcome in pediatric acute lymphoblastic leukemia by gene expression profiling. Cancer Cell. 2002;1:133–43. doi: 10.1016/s1535-6108(02)00032-6.. [DOI] [PubMed] [Google Scholar]
- 62.Andersson A, Olofsson T, Lindgren D, Nilsson B, Ritz C, Eden P, et al. Molecular signatures in childhood acute leukemia and their correlations to expression patterns in normal hematopoietic subpopulations. Proc Natl Acad Sci USA. 2005;102:19069–74. doi: 10.1073/pnas.0506637102.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Yang M, Vesterlund M, Siavelis I, Moura-Castro LH, Castor A, Fioretos T, et al. Proteogenomics and Hi-C reveal transcriptional dysregulation in high hyperdiploid childhood acute lymphoblastic leukemia. Nat Commun. 2019;10:1519. doi: 10.1038/s41467-019-09469-3.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Weeks RJ, Morison IM. Detailed methylation analysis of CpG islands on human chromosome region 9p21. Genes Chromosomes Cancer. 2006;45:357–64. doi: 10.1002/gcc.20297.. [DOI] [PubMed] [Google Scholar]
- 65.Hertzberg L, Betts DR, Raimondi SC, Schafer BW, Notterman DA, Domany E, et al. Prediction of chromosomal aneuploidy from gene expression data. Genes Chromosomes Cancer. 2007;46:75–86. doi: 10.1002/gcc.20391.. [DOI] [PubMed] [Google Scholar]
- 66.Davidsson J, Lilljebjorn H, Andersson A, Veerla S, Heldrup J, Behrendtz M, et al. The DNA methylome of pediatric acute lymphoblastic leukemia. Hum Mol Genet. 2009;18:4054–65. doi: 10.1093/hmg/ddp354.. [DOI] [PubMed] [Google Scholar]
- 67.Nordlund J, Backlin CL, Zachariadis V, Cavelier L, Dahlberg J, Ofverholm I, et al. DNA methylation-based subtype prediction for pediatric acute lymphoblastic leukemia. Clin Epigenetics. 2015;7:11. doi: 10.1186/s13148-014-0039-z.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Nordlund J, Syvanen AC. Epigenetics in pediatric acute lymphoblastic leukemia. Semin Cancer Biol. 2018;51:129–38. doi: 10.1016/j.semcancer.2017.09.001.. [DOI] [PubMed] [Google Scholar]
- 69.Molina O, Vinyoles M, Granada I, Roca-Ho H, Gutierrez-Aguera F, Valledor L, et al. Impaired condensin complex and Aurora B kinase underlie mitotic and chromosomal defects in hyperdiploid B-cell ALL. Blood. 2020;136:313–27. doi: 10.1182/blood.2019002538.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Merkenschlager M, Odom DT. CTCF and cohesin: linking gene regulatory elements with their targets. Cell. 2013;152:1285–97. doi: 10.1016/j.cell.2013.02.029.. [DOI] [PubMed] [Google Scholar]
- 71.Moura-Castro LH, Pena-Martinez P, Castor A, Galeev R, Larsson J, Jaras M, et al. Sister chromatid cohesion defects are associated with chromosomal copy number heterogeneity in high hyperdiploid childhood acute lymphoblastic leukemia. Genes Chromosomes Cancer. 2021;60:410–7. doi: 10.1002/gcc.22933.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Pope BD, Ryba T, Dileep V, Yue F, Wu W, Denas O, et al. Topologically associating domains are stable units of replication-timing regulation. Nature. 2014;515:402–5. doi: 10.1038/nature13986.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Gu Z, Churchman ML, Roberts KG, Moore I, Zhou X, Nakitandwe J, et al. PAX5-driven subtypes of B-progenitor acute lymphoblastic leukemia. Nat Genet. 2019;51:296–307. doi: 10.1038/s41588-018-0315-5.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Synold TW, Relling MV, Boyett JM, Rivera GK, Sandlund JT, Mahmoud H, et al. Blast cell methotrexate-polyglutamate accumulation in vivo differs by lineage, ploidy, and methotrexate dose in acute lymphoblastic leukemia. J Clin Invest. 1994;94:1996–2001. doi: 10.1172/JCI117552.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Kager L, Cheok M, Yang W, Zaza G, Cheng Q, Panetta JC, et al. Folate pathway gene expression differs in subtypes of acute lymphoblastic leukemia and influences methotrexate pharmacodynamics. J Clin Invest. 2005;115:110–7. doi: 10.1172/JCI22477.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Whitehead VM, Vuchich MJ, Cooley LD, Lauer SJ, Mahoney DH, Shuster JJ, et al. Accumulation of methotrexate polyglutamates, ploidy and trisomies of both chromosomes 4 and 10 in lymphoblasts from children with B-progenitor cell acute lymphoblastic leukemia: a Pediatric Oncology Group Study. Leuk Lymphoma. 1998;31:507–19. doi: 10.3109/10428199809057610.. [DOI] [PubMed] [Google Scholar]
- 77.Belkov VM, Krynetski EY, Schuetz JD, Yanishevski Y, Masson E, Mathew S, et al. Reduced folate carrier expression in acute lymphoblastic leukemia: a mechanism for ploidy but not lineage differences in methotrexate accumulation. Blood. 1999;93:1643–50. doi: 10.1182/blood.V93.5.1643.. [DOI] [PubMed] [Google Scholar]
- 78.Laverdiere C, Chiasson S, Costea I, Moghrabi A, Krajinovic M. Polymorphism G80A in the reduced folate carrier gene and its relationship to methotrexate plasma levels and outcome of childhood acute lymphoblastic leukemia. Blood. 2002;100:3832–4. doi: 10.1182/blood.V100.10.3832.. [DOI] [PubMed] [Google Scholar]
- 79.Gregers J, Christensen IJ, Dalhoff K, Lausen B, Schroeder H, Rosthoej S, et al. The association of reduced folate carrier 80G>A polymorphism to outcome in childhood acute lymphoblastic leukemia interacts with chromosome 21 copy number. Blood. 2010;115:4671–7. doi: 10.1182/blood-2010-01-256958.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Wojtuszkiewicz A, Peters GJ, van Woerden NL, Dubbelman B, Escherich G, Schmiegelow K, et al. Methotrexate resistance in relation to treatment outcome in childhood acute lymphoblastic leukemia. J Hematol Oncol. 2015;8:61. doi: 10.1186/s13045-015-0158-9.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Maia AT, van der Velden VH, Harrison CJ, Szczepanski T, Williams MD, Griffiths MJ, et al. Prenatal origin of hyperdiploid acute lymphoblastic leukemia in identical twins. Leukemia. 2003;17:2202–6. doi: 10.1038/sj.leu.2403101.. [DOI] [PubMed] [Google Scholar]
- 82.Greaves MF, Maia AT, Wiemels JL, Ford AM. Leukemia in twins: lessons in natural history. Blood. 2003;102:2321–33. doi: 10.1182/blood-2002-12-3817.. [DOI] [PubMed] [Google Scholar]
- 83.Maia AT, Tussiwand R, Cazzaniga G, Rebulla P, Colman S, Biondi A, et al. Identification of preleukemic precursors of hyperdiploid acute lymphoblastic leukemia in cord blood. Genes Chromosomes Cancer. 2004;40:38–43. doi: 10.1002/gcc.20010.. [DOI] [PubMed] [Google Scholar]
- 84.Greaves M. In utero origins of childhood leukaemia. Early Hum Dev. 2005;81:123–9. doi: 10.1016/j.earlhumdev.2004.10.004.. [DOI] [PubMed] [Google Scholar]
- 85.Gruhn B, Taub JW, Ge Y, Beck JF, Zell R, Hafer R, et al. Prenatal origin of childhood acute lymphoblastic leukemia, association with birth weight and hyperdiploidy. Leukemia. 2008;22:1692–7. doi: 10.1038/leu.2008.152.. [DOI] [PubMed] [Google Scholar]
- 86.Bateman CM, Alpar D, Ford AM, Colman SM, Wren D, Morgan M, et al. Evolutionary trajectories of hyperdiploid ALL in monozygotic twins. Leukemia. 2015;29:58–65. doi: 10.1038/leu.2014.177.. [DOI] [PubMed] [Google Scholar]
- 87.Hein D, Borkhardt A, Fischer U. Insights into the prenatal origin of childhood acute lymphoblastic leukemia. Cancer Metastasis Rev. 2020;39:161–71. doi: 10.1007/s10555-019-09841-1.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Jackson TR, Ling RE, Roy A. The origin of B-cells: human fetal B cell development and implications for the pathogenesis of childhood acute lymphoblastic leukemia. Front Immunol. 2021;12:637975. doi: 10.3389/fimmu.2021.637975.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Marcotte EL, Spector LG, Mendes-de-Almeida DP, Nelson HH. The prenatal origin of childhood leukemia: potential applications for epidemiology and newborn screening. Front Pediatr. 2021;9:639479. doi: 10.3389/fped.2021.639479.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Paulsson K, Morse H, Fioretos T, Behrendtz M, Strombeck B, Johansson B. Evidence for a single-step mechanism in the origin of hyperdiploid childhood acute lymphoblastic leukemia. Genes Chromosomes Cancer. 2005;44:113–22. doi: 10.1002/gcc.20222.. [DOI] [PubMed] [Google Scholar]
- 91.Liang DC, Shih LY, Yang CP, Hung IJ, Liu HC, Jaing TH, et al. Frequencies of ETV6-RUNX1 fusion and hyperdiploidy in pediatric acute lymphoblastic leukemia are lower in far east than west. Pediatr Blood Cancer. 2010;55:430–3. doi: 10.1002/pbc.22628.. [DOI] [PubMed] [Google Scholar]
- 92.Lee SHR, Antillon-Klussmann F, Pei D, Yang W, Roberts KG, Li Z, et al. Association of genetic ancestry with the molecular subtypes and prognosis of childhood acute lymphoblastic leukemia. JAMA Oncol. 2022;8:354–63. doi: 10.1001/jamaoncol.2021.6826.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Takahashi Y, Ishida H, Imamura T, Tamefusa K, Suenobu S, Usami I, et al. JACLS ALL-02 SR protocol reduced-intensity chemotherapy produces excellent outcomes in patients with low-risk childhood acute lymphoblastic leukemia. Int J Hematol. 2022;115:890–7. 10.1007/s12185-022-03315-x. [DOI] [PubMed]
- 94.Pierzyna-Switala M, Sedek L, Kulis J, Mazur B, Muszynska-Roslan K, Koltan A, et al. Multicolor flow cytometry immunophenotyping and characterization of aneuploidy in pediatric B-cell precursor acute lymphoblastic leukemia. Cent Eur J Immunol. 2021;46:365–74. doi: 10.5114/ceji.2021.109794.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Li Z, Chu X, Gao L, Ling J, Xiao P, Lu J, et al. High expression of interleukin-3 receptor alpha chain (CD123) predicts favorable outcome in pediatric B-cell acute lymphoblastic leukemia lacking prognosis-defining genomic aberrations. Front Oncol. 2021;11:614420. doi: 10.3389/fonc.2021.614420.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Bras AE, de Haas V, van Stigt A, Jongen-Lavrencic M, Beverloo HB, Te Marvelde JG, et al. CD123 expression levels in 846 acute leukemia patients based on standardized immunophenotyping. Cytom B Clin Cytom. 2019;96:134–42. doi: 10.1002/cyto.b.21745.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Djokic M, Bjorklund E, Blennow E, Mazur J, Soderhall S, Porwit A. Overexpression of CD123 correlates with the hyperdiploid genotype in acute lymphoblastic leukemia. Haematologica. 2009;94:1016–9. doi: 10.3324/haematol.2008.000299.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Taylor GM, Dearden S, Ravetto P, Ayres M, Watson P, Hussain A, et al. Genetic susceptibility to childhood common acute lymphoblastic leukaemia is associated with polymorphic peptide-binding pocket profiles in HLA-DPB1*0201. Hum Mol Genet. 2002;11:1585–97. doi: 10.1093/hmg/11.14.1585.. [DOI] [PubMed] [Google Scholar]
- 99.Gilham C, Peto J, Simpson J, Roman E, Eden TO, Greaves MF, et al. Day care in infancy and risk of childhood acute lymphoblastic leukaemia: findings from UK case-control study. BMJ. 2005;330:1294. doi: 10.1136/bmj.38428.521042.8F.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Greenop KR, Bailey HD, Miller M, Scott RJ, Attia J, Ashton LJ, et al. Breastfeeding and nutrition to 2 years of age and risk of childhood acute lymphoblastic leukemia and brain tumors. Nutr Cancer. 2015;67:431–41. doi: 10.1080/01635581.2015.998839.. [DOI] [PubMed] [Google Scholar]
- 101.Francis SS, Wallace AD, Wendt GA, Li L, Liu F, Riley LW, et al. In utero cytomegalovirus infection and development of childhood acute lymphoblastic leukemia. Blood. 2017;129:1680–4. doi: 10.1182/blood-2016-07-723148.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Wiemels JL, Talback M, Francis S, Feychting M. Early infection with cytomegalovirus and risk of childhood hematologic malignancies. Cancer Epidemiol Biomark Prev. 2019;28:1024–7. doi: 10.1158/1055-9965.EPI-19-0044.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Gallant RE, Arroyo K, Bracci PM, Li S, Metayer C, Kogan SC, et al. Clinical characteristics of cytomegalovirus-positive pediatric acute lymphoblastic leukemia at diagnosis. Am J Hematol. 2022;97:E198–E201. doi: 10.1002/ajh.26528.. [DOI] [PubMed] [Google Scholar]
- 104.Manabe A, Coustan-Smith E, Behm FG, Raimondi SC, Campana D. Bone marrow-derived stromal cells prevent apoptotic cell death in B-lineage acute lymphoblastic leukemia. Blood. 1992;79:2370–7. doi: 10.1182/blood.V79.9.2370.2370. [DOI] [PubMed] [Google Scholar]
- 105.Kohno S, Minowada J, Sandberg AA. Chromosome evolution of near-haploid clones in an established human acute lymphoblastic leukemia cell line (NALM-16) J Natl Cancer Inst. 1980;64:485–93. [PubMed] [Google Scholar]
- 106.Tomeczkowski J, Yakisan E, Wieland B, Reiter A, Welte K, Sykora KW. Absence of G-CSF receptors and absent response to G-CSF in childhood Burkitt’s lymphoma and B-ALL cells. Br J Haematol. 1995;89:771–9. doi: 10.1111/j.1365-2141.1995.tb08414.x.. [DOI] [PubMed] [Google Scholar]
- 107.Aburawi HE, Biloglav A, Johansson B, Paulsson K. Cytogenetic and molecular genetic characterization of the ‘high hyperdiploid’ B-cell precursor acute lymphoblastic leukaemia cell line MHH-CALL-2 reveals a near-haploid origin. Br J Haematol. 2011;154:275–7. doi: 10.1111/j.1365-2141.2011.08601.x.. [DOI] [PubMed] [Google Scholar]
- 108.Townsend EC, Murakami MA, Christodoulou A, Christie AL, Koster J, DeSouza TA, et al. The public repository of xenografts enables discovery and randomized phase II-like trials in mice. Cancer Cell. 2016;29:574–86. doi: 10.1016/j.ccell.2016.03.008.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Rachieru-Sourisseau P, Baranger L, Dastugue N, Robert A, Genevieve F, Kuhlein E, et al. DNA Index in childhood acute lymphoblastic leukaemia: a karyotypic method to validate the flow cytometric measurement. Int J Lab Hematol. 2010;32:288–98. doi: 10.1111/j.1751-553X.2009.01189.x.. [DOI] [PubMed] [Google Scholar]
- 110.Gupta N, Parihar M, Banerjee S, Brahma S, Pawar R, Rath A, et al. FxCycle based ploidy correlates with cytogenetic ploidy in B-cell acute lymphoblastic leukemia and is able to detect the aneuploid minimal residual disease clone. Cytom B Clin Cytom. 2019;96:359–67. doi: 10.1002/cyto.b.21765.. [DOI] [PubMed] [Google Scholar]
- 111.Gupta T, Arun SR, Babu GA, Chakrabarty BK, Bhave SJ, Kumar J, et al. A systematic cytogenetic strategy to identify masked hypodiploidy in precursor B acute lymphoblastic leukemia in low resource settings. Indian J Hematol Blood Transfus. 2021;37:576–85. doi: 10.1007/s12288-021-01409-w.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Chaturvedi A, Shetty D, Ghogale SG, Deshpande N, Badrinath Y, Chatterjee G, et al. Detecting hypodiploidy with endoreduplication and masked hypodiploidy in B-cell acute lymphoblastic leukemia using multicolor flow cytometry. Cytom B Clin Cytom. 2022;102:199–208. doi: 10.1002/cyto.b.22063.. [DOI] [PubMed] [Google Scholar]
- 113.Raimondi SC, Zhou Y, Mathew S, Shurtleff SA, Sandlund JT, Rivera GK, et al. Reassessment of the prognostic significance of hypodiploidy in pediatric patients with acute lymphoblastic leukemia. Cancer. 2003;98:2715–22. doi: 10.1002/cncr.11841.. [DOI] [PubMed] [Google Scholar]
- 114.Nachman JB, Heerema NA, Sather H, Camitta B, Forestier E, Harrison CJ, et al. Outcome of treatment in children with hypodiploid acute lymphoblastic leukemia. Blood. 2007;110:1112–5. doi: 10.1182/blood-2006-07-038299.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Pui CH, Rebora P, Schrappe M, Attarbaschi A, Baruchel A, Basso G, et al. Outcome of children with hypodiploid acute lymphoblastic leukemia: a retrospective multinational study. J Clin Oncol. 2019;37:770–9. doi: 10.1200/JCO.18.00822.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Maloney KW, Devidas M, Wang C, Mattano LA, Friedmann AM, Buckley P, et al. Outcome in children with standard-risk B-cell acute lymphoblastic leukemia: results of Children’s Oncology Group Trial AALL0331. J Clin Oncol. 2020;38:602–12. doi: 10.1200/JCO.19.01086.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Schore RJ, Angiolillo AJ, Kairalla JA, Devidas M, Rabin KR, Zweidler-McKay PA, et al. Outcomes with reduced intensity therapy in a low-risk subset of children with National Cancer Institute (NCI) standard-risk (SR) B-lymphoblastic leukemia (B-ALL): a report from Children’s Oncology Group (COG) AALL0932. J Clin Oncol. 2020;38:10509. doi: 10.1200/JCO.2020.38.15_suppl.10509.. [DOI] [Google Scholar]
- 118.Mattano LA, Jr., Devidas M, Maloney KW, Wang C, Friedmann AM, Buckley P, et al. Favorable trisomies and ETV6-RUNX1 predict cure in low-risk B-cell acute lymphoblastic leukemia: results from Children’s Oncology Group Trial AALL0331. J Clin Oncol. 2021;39:1540–52. doi: 10.1200/JCO.20.02370.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.O’Connor D, Enshaei A, Bartram J, Hancock J, Harrison CJ, Hough R, et al. Genotype-specific minimal residual disease interpretation improves stratification in pediatric acute lymphoblastic leukemia. J Clin Oncol. 2018;36:34–43. doi: 10.1200/JCO.2017.74.0449.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Enshaei A, Vora A, Harrison CJ, Moppett J, Moorman AV. Defining low-risk high hyperdiploidy in patients with paediatric acute lymphoblastic leukaemia: a retrospective analysis of data from the UKALL97/99 and UKALL2003 clinical trials. Lancet Haematol. 2021;8:e828–e39. doi: 10.1016/S2352-3026(21)00304-5.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Heerema NA, Sather HN, Sensel MG, Zhang T, Hutchinson RJ, Nachman JB, et al. Prognostic impact of trisomies of chromosomes 10, 17, and 5 among children with acute lymphoblastic leukemia and high hyperdiploidy (> 50 chromosomes) J Clin Oncol. 2000;18:1876–87. doi: 10.1200/JCO.2000.18.9.1876.. [DOI] [PubMed] [Google Scholar]
- 122.Toyoda Y, Manabe A, Tsuchida M, Hanada R, Ikuta K, Okimoto Y, et al. Six months of maintenance chemotherapy after intensified treatment for acute lymphoblastic leukemia of childhood. J Clin Oncol. 2000;18:1508–16. doi: 10.1200/JCO.2000.18.7.1508.. [DOI] [PubMed] [Google Scholar]
- 123.Kato M, Ishimaru S, Seki M, Yoshida K, Shiraishi Y, Chiba K, et al. Long-term outcome of 6-month maintenance chemotherapy for acute lymphoblastic leukemia in children. Leukemia. 2017;31:580–4. doi: 10.1038/leu.2016.274.. [DOI] [PubMed] [Google Scholar]
- 124.Angiolillo AL, Schore RJ, Kairalla JA, Devidas M, Rabin KR, Zweidler-McKay P, et al. Excellent outcomes with reduced frequency of vincristine and dexamethasone pulses in standard-risk B-lymphoblastic leukemia: results from Children’s Oncology Group AALL0932. J Clin Oncol. 2021;39:1437–47. doi: 10.1200/JCO.20.00494.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Toksvang LN, Lee SHR, Yang JJ, Schmiegelow K. Maintenance therapy for acute lymphoblastic leukemia: basic science and clinical translations. Leukemia. 2022;36:1749–58. doi: 10.1038/s41375-022-01591-4.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Li B, Brady SW, Ma X, Shen S, Zhang Y, Li Y, et al. Therapy-induced mutations drive the genomic landscape of relapsed acute lymphoblastic leukemia. Blood. 2020;135:41–55. doi: 10.1182/blood.2019002220.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Borkhardt A, Haas OA. Chromosomal risk classification in high hyperdiploid acute lymphocytic leukaemia: the beginning of a new chapter. Lancet Haematol. 2022;9:e9. doi: 10.1016/S2352-3026(21)00348-3.. [DOI] [PubMed] [Google Scholar]
- 128.Wiemels JL, Zhang Y, Chang J, Zheng S, Metayer C, Zhang L, et al. RAS mutation is associated with hyperdiploidy and parental characteristics in pediatric acute lymphoblastic leukemia. Leukemia. 2005;19:415–9. doi: 10.1038/sj.leu.2403641.. [DOI] [PubMed] [Google Scholar]
- 129.Groeneveld-Krentz S, Schroeder MP, Reiter M, Pogodzinski MJ, Pimentel-Gutierrez HJ, Vagkopoulou R, et al. Aneuploidy in children with relapsed B-cell precursor acute lymphoblastic leukaemia: clinical importance of detecting a hypodiploid origin of relapse. Br J Haematol. 2019;185:266–83. doi: 10.1111/bjh.15770.. [DOI] [PubMed] [Google Scholar]
- 130.Hussin J, Sinnett D, Casals F, Idaghdour Y, Bruat V, Saillour V, et al. Rare allelic forms of PRDM9 associated with childhood leukemogenesis. Genome Res. 2013;23:419–30. doi: 10.1101/gr.144188.112.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Woodward EL, Olsson ML, Johansson B, Paulsson K. Allelic variants of PRDM9 associated with high hyperdiploid childhood acute lymphoblastic leukaemia. Br J Haematol. 2014;166:947–9. doi: 10.1111/bjh.12914.. [DOI] [PubMed] [Google Scholar]
- 132.Vijayakrishnan J, Qian M, Studd JB, Yang W, Kinnersley B, Law PJ, et al. Identification of four novel associations for B-cell acute lymphoblastic leukaemia risk. Nat Commun. 2019;10:5348. doi: 10.1038/s41467-019-13069-6.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Stanulla M, Dagdan E, Zaliova M, Moricke A, Palmi C, Cazzaniga G, et al. IKZF1(plus) defines a new minimal residual disease-dependent very-poor prognostic profile in pediatric B-cell precursor acute lymphoblastic leukemia. J Clin Oncol. 2018;36:1240–9. doi: 10.1200/JCO.2017.74.3617.. [DOI] [PubMed] [Google Scholar]
- 134.Xu H, Zhang H, Yang W, Yadav R, Morrison AC, Qian M, et al. Inherited coding variants at the CDKN2A locus influence susceptibility to acute lymphoblastic leukaemia in children. Nat Commun. 2015;6:7553. doi: 10.1038/ncomms8553.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Hungate EA, Vora SR, Gamazon ER, Moriyama T, Best T, Hulur I, et al. A variant at 9p21.3 functionally implicates CDKN2B in paediatric B-cell precursor acute lymphoblastic leukaemia aetiology. Nat Commun. 2016;7:10635. doi: 10.1038/ncomms10635.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Vijayakrishnan J, Henrion M, Moorman AV, Fiege B, Kumar R, da Silva Filho MI, et al. The 9p21.3 risk of childhood acute lymphoblastic leukaemia is explained by a rare high-impact variant in CDKN2A. Sci Rep. 2015;5:15065. doi: 10.1038/srep15065.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Trevino LR, Yang W, French D, Hunger SP, Carroll WL, Devidas M, et al. Germline genomic variants associated with childhood acute lymphoblastic leukemia. Nat Genet. 2009;41:1001–5. doi: 10.1038/ng.432.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Prasad RB, Hosking FJ, Vijayakrishnan J, Papaemmanuil E, Koehler R, Greaves M, et al. Verification of the susceptibility loci on 7p12.2, 10q21.2, and 14q11.2 in precursor B-cell acute lymphoblastic leukemia of childhood. Blood. 2010;115:1765–7. doi: 10.1182/blood-2009-09-241513.. [DOI] [PubMed] [Google Scholar]
- 139.Paulsson K, Forestier E, Lilljebjorn H, Heldrup J, Behrendtz M, Young BD, et al. Genetic landscape of high hyperdiploid childhood acute lymphoblastic leukemia. Proc Natl Acad Sci USA. 2010;107:21719–24. doi: 10.1073/pnas.1006981107.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.de Smith AJ, Lavoie G, Walsh KM, Aujla S, Evans E, Hansen HM, et al. Predisposing germline mutations in high hyperdiploid acute lymphoblastic leukemia in children. Genes Chromosomes Cancer. 2019;58:723–30. doi: 10.1002/gcc.22765.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Topka S, Vijai J, Walsh MF, Jacobs L, Maria A, Villano D, et al. Germline ETV6 mutations confer susceptibility to acute lymphoblastic leukemia and thrombocytopenia. PLoS Genet. 2015;11:e1005262. doi: 10.1371/journal.pgen.1005262.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Noetzli L, Lo RW, Lee-Sherick AB, Callaghan M, Noris P, Savoia A, et al. Germline mutations in ETV6 are associated with thrombocytopenia, red cell macrocytosis and predisposition to lymphoblastic leukemia. Nat Genet. 2015;47:535–8. doi: 10.1038/ng.3253.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Duployez N, Abou Chahla W, Lejeune S, Marceau-Renaut A, Letizia G, Boyer T, et al. Detection of a new heterozygous germline ETV6 mutation in a case with hyperdiploid acute lymphoblastic leukemia. Eur J Haematol. 2018;100:104–7. doi: 10.1111/ejh.12981.. [DOI] [PubMed] [Google Scholar]
- 144.Taketani T, Taki T, Sugita K, Furuichi Y, Ishii E, Hanada R, et al. FLT3 mutations in the activation loop of tyrosine kinase domain are frequently found in infant ALL with MLL rearrangements and pediatric ALL with hyperdiploidy. Blood. 2004;103:1085–8. doi: 10.1182/blood-2003-02-0418.. [DOI] [PubMed] [Google Scholar]
- 145.Stam RW, den Boer ML, Schneider P, Meier M, Beverloo HB, Pieters R. D-HPLC analysis of the entire FLT3 gene in MLL rearranged and hyperdiploid acute lymphoblastic leukemia. Haematologica. 2007;92:1565–8. doi: 10.3324/haematol.11220.. [DOI] [PubMed] [Google Scholar]
- 146.Yang M, Safavi S, Woodward EL, Duployez N, Olsson-Arvidsson L, Ungerback J, et al. 13q12.2 deletions in acute lymphoblastic leukemia lead to upregulation of FLT3 through enhancer hijacking. Blood. 2020;136:946–56. doi: 10.1182/blood.2019004684.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Wiemels JL, Walsh KM, de Smith AJ, Metayer C, Gonseth S, Hansen HM, et al. GWAS in childhood acute lymphoblastic leukemia reveals novel genetic associations at chromosomes 17q12 and 8q24.21. Nat Commun. 2018;9:286. doi: 10.1038/s41467-017-02596-9.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Moricke A, Zimmermann M, Valsecchi MG, Stanulla M, Biondi A, Mann G, et al. Dexamethasone vs prednisone in induction treatment of pediatric ALL: results of the randomized trial AIEOP-BFM ALL 2000. Blood. 2016;127:2101–12. doi: 10.1182/blood-2015-09-670729.. [DOI] [PubMed] [Google Scholar]
- 149.Schrappe M, Bleckmann K, Zimmermann M, Biondi A, Moricke A, Locatelli F, et al. Reduced-intensity delayed intensification in standard-risk pediatric acute lymphoblastic leukemia defined by undetectable minimal residual disease: results of an International Randomized Trial (AIEOP-BFM ALL 2000) J Clin Oncol. 2018;36:244–53. doi: 10.1200/JCO.2017.74.4946.. [DOI] [PubMed] [Google Scholar]
- 150.Stary J, Zimmermann M, Campbell M, Castillo L, Dibar E, Donska S, et al. Intensive chemotherapy for childhood acute lymphoblastic leukemia: results of the randomized intercontinental trial ALL IC-BFM 2002. J Clin Oncol. 2014;32:174–84. doi: 10.1200/JCO.2013.48.6522.. [DOI] [PubMed] [Google Scholar]
- 151.Schramm F, Zur Stadt U, Zimmermann M, Jorch N, Pekrun A, Borkhardt A, et al. Results of CoALL 07-03 study childhood ALL based on combined risk assessment by in vivo and in vitro pharmacosensitivity. Blood Adv. 2019;3:3688–99. doi: 10.1182/bloodadvances.2019000576.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152.Pedrosa F, Coustan-Smith E, Zhou Y, Cheng C, Pedrosa A, Lins MM, et al. Reduced-dose intensity therapy for pediatric lymphoblastic leukemia: long-term results of the Recife RELLA05 pilot study. Blood. 2020;135:1458–66. doi: 10.1182/blood.2019004215.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153.Hasegawa D, Imamura T, Yumura-Yagi K, Takahashi Y, Usami I, Suenobu SI, et al. Risk-adjusted therapy for pediatric non-T cell ALL improves outcomes for standard risk patients: results of JACLS ALL-02. Blood Cancer J. 2020;10:23. doi: 10.1038/s41408-020-0287-4.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154.Jeha S, Pei D, Choi J, Cheng C, Sandlund JT, Coustan-Smith E, et al. Improved CNS control of childhood acute lymphoblastic leukemia without cranial irradiation: St Jude Total Therapy Study 16. J Clin Oncol. 2019;37:3377–91. doi: 10.1200/JCO.19.01692.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155.Jeha S, Choi J, Roberts KG, Pei D, Coustan-Smith E, Inaba H, et al. Clinical significance of novel subtypes of acute lymphoblastic leukemia in the context of minimal residual disease-directed therapy. Blood Cancer Discov. 2021;2:326–37. doi: 10.1158/2643-3230.BCD-20-0229.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156.Vora A, Goulden N, Wade R, Mitchell C, Hancock J, Hough R, et al. Treatment reduction for children and young adults with low-risk acute lymphoblastic leukaemia defined by minimal residual disease (UKALL 2003): a randomised controlled trial. Lancet Oncol. 2013;14:199–209. doi: 10.1016/S1470-2045(12)70600-9.. [DOI] [PubMed] [Google Scholar]
- 157.Paulsson K, Forestier E, Andersen MK, Autio K, Barbany G, Borgstrom G, et al. High modal number and triple trisomies are highly correlated favorable factors in childhood B-cell precursor high hyperdiploid acute lymphoblastic leukemia treated according to the NOPHO ALL 1992/2000 protocols. Haematologica. 2013;98:1424–32. doi: 10.3324/haematol.2013.085852.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158.Schmiegelow K, Forestier E, Hellebostad M, Heyman M, Kristinsson J, Soderhall S, et al. Long-term results of NOPHO ALL-92 and ALL-2000 studies of childhood acute lymphoblastic leukemia. Leukemia. 2010;24:345–54. doi: 10.1038/leu.2009.251.. [DOI] [PubMed] [Google Scholar]
- 159.Pieters R, de Groot-Kruseman H, Van der Velden V, Fiocco M, van den Berg H, de Bont E, et al. Successful therapy reduction and intensification for childhood acute lymphoblastic leukemia based on minimal residual disease monitoring: study ALL10 From the Dutch Childhood Oncology Group. J Clin Oncol. 2016;34:2591–601. doi: 10.1200/JCO.2015.64.6364.. [DOI] [PubMed] [Google Scholar]
- 160.Cui L, Li ZG, Chai YH, Yu J, Gao J, Zhu XF, et al. Outcome of children with newly diagnosed acute lymphoblastic leukemia treated with CCLG-ALL 2008: The first nation-wide prospective multicenter study in China. Am J Hematol. 2018;93:913–20. doi: 10.1002/ajh.25124.. [DOI] [PubMed] [Google Scholar]
- 161.Takahashi H, Kajiwara R, Kato M, Hasegawa D, Tomizawa D, Noguchi Y, et al. Treatment outcome of children with acute lymphoblastic leukemia: the Tokyo Children’s Cancer Study Group (TCCSG) Study L04-16. Int J Hematol. 2018;108:98–108. doi: 10.1007/s12185-018-2440-4.. [DOI] [PubMed] [Google Scholar]
- 162.Panzer-Grumayer ER, Fasching K, Panzer S, Hettinger K, Schmitt K, Stockler-Ipsiroglu S, et al. Nondisjunction of chromosomes leading to hyperdiploid childhood B-cell precursor acute lymphoblastic leukemia is an early event during leukemogenesis. Blood. 2002;100:347–9. doi: 10.1182/blood-2002-01-0144.. [DOI] [PubMed] [Google Scholar]
- 163.Csinady E, van der Velden VH, Joas R, Fischer S, de Vries JF, Beverloo HB, et al. Chromosome 14 copy number-dependent IGH gene rearrangement patterns in high hyperdiploid childhood B-cell precursor ALL: implications for leukemia biology and minimal residual disease analysis. Leukemia. 2009;23:870–6. doi: 10.1038/leu.2008.390.. [DOI] [PubMed] [Google Scholar]
- 164.Paulsson K, Panagopoulos I, Knuutila S, Jee KJ, Garwicz S, Fioretos T, et al. Formation of trisomies and their parental origin in hyperdiploid childhood acute lymphoblastic leukemia. Blood. 2003;102:3010–5. doi: 10.1182/blood-2003-05-1444.. [DOI] [PubMed] [Google Scholar]
- 165.Weinhaeusel A, Koenig M, Pfeilstoecker M, Strehl S, Zeitlhofer P, Haas OA. A novel DNA/RNA FISH X inactivation assay reveals a nonrandom, ploidy-dependent acquisition of the active and inactive X chromosomes in childhood hyperdiploid acute lymphoblastic leukemia (ALL) and non-Hodgkin lymphoma (NHL) Blood. 2004;104:1080. doi: 10.1182/blood.V104.11.1080.1080.. [DOI] [Google Scholar]