

Abstract
Wildlife reservoirs of broad-host-range viruses have the potential to enable evolution of viral variants that can emerge to infect humans. In North America, there is phylogenomic evidence of continual transmission of severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) from humans to white-tailed deer (Odocoileus virginianus) through unknown means, but no evidence of transmission from deer to humans. We carried out an observational surveillance study in Ontario, Canada during November and December 2021 (n = 300 deer) and identified a highly divergent lineage of SARS-CoV-2 in white-tailed deer (B.1.641). This lineage is one of the most divergent SARS-CoV-2 lineages identified so far, with 76 mutations (including 37 previously associated with non-human mammalian hosts). From a set of five complete and two partial deer-derived viral genomes we applied phylogenomic, recombination, selection and mutation spectrum analyses, which provided evidence for evolution and transmission in deer and a shared ancestry with mink-derived virus. Our analysis also revealed an epidemiologically linked human infection. Taken together, our findings provide evidence for sustained evolution of SARS-CoV-2 in white-tailed deer and of deer-to-human transmission.
Subject terms: SARS-CoV-2, Evolution
White-tailed deer are a potential reservoir of SARS-CoV-2 variants.
Main
Human-pathogenic coronaviruses, such as severe acute respiratory syndrome coronavirus (SARS-CoV), SARS-CoV-2 and Middle East respiratory syndrome coronavirus, are likely to have been transmitted to humans either directly from reservoir wild animal hosts (for example, horseshoe bats) or through intermediate hosts such as civets or camels1–6. Emergence of human-pathogenic coronaviruses may result in sustained human-to-human transmission, with ongoing viral evolution, as has been observed with SARS-CoV-2 and the emergence of variants, including variants of concern (VOCs). Viral diversity may also occur from inter-species transmission and evolution in new hosts, as was observed during human–mink–human transmission of SARS-CoV-2 (ref. 7).
The evolution of divergent viral lineages can affect immunology, biology and epidemiology of any virus, as well as altering detection, vaccine efficacy, disease severity and transmission. Virus evolution can impact individual and population health. For example, the SARS-CoV-2 Omicron variant had 59 genome-wide mutations when it emerged, including 37 in the spike protein, which resulted in reduced efficacy of vaccines to the original emergent strain due to antibody evasion8. There are several competing hypotheses for the evolution of Omicron, with one hypothesis considering it may have evolved after SARS-CoV-2 transmission from humans to rodents (or another, as yet unidentified, animal reservoir9) and back into humans again (‘spillback’)10.
As of September 2022, observational and experimental reports in free-living, captive, domestic and farmed animals have shown that SARS-CoV-2 can infect at least 54 non-human mammalian species11–14. The high degree of similarity of the primary SARS-CoV-2 host cell receptor, human angiotensin converting enzyme 2, among mammals may explain the broad host-range of SARS-CoV-2 (ref. 15). Zooanthroponosis (reverse zoonotic disease transmission) has been documented in outbreaks of SARS-CoV-2 among farmed mink (Neovison vison)16,17 and pet hamsters (Mesocricetus auratus)18. The Netherlands experienced outbreaks of SARS-CoV-2 in mink farms19, and whole genome sequencing (WGS) provided evidence for the emergence of a ‘cluster 5’ variant among farmed mink with a unique combination of mutations, and identified spillback from mink to humans19. These mutations raised concerns about the potential to erode vaccine efficacy, contributing to a decision in Denmark to cull mink20. Similarly, the finding of SARS-CoV-2 in pet hamsters led Hong Kong authorities to cull thousands of animals21.
There have been suggestions (based on experimental data for VOCs) that SARS-CoV-2 host cell receptor tropism has expanded over time, increasing concerns about the potential for spillover into animals. For example, the Alpha variant can infect mice (Mus musculus), and the Omicron variant spike glycoprotein can bind to avian ACE2 receptors, unlike ancestral SARS-CoV-2 (refs. 22,23). This underscores the potential for ongoing expansion of susceptible host species as VOCs continue to emerge.
The white-tailed deer (Odocoileus virginianus) is a common North American ungulate that is susceptible to SARS-CoV-2. An experimental study reported that deer develop subclinical infection24. A subsequent study found that 40% of free-ranging deer sampled in Michigan, Illinois, New York and Pennsylvania, United States were positive for SARS-CoV-2 antibodies25. Transmission of SARS-CoV-2 among deer, and multiple spillovers from humans to deer, have also been reported26–28. So far, most of the SARS-CoV-2 isolated and reported in deer has been similar to lineages circulating among humans in the same region, which has led to proposal of multiple, recent, spillover events27,28. Also, a divergent Alpha-like virus has been reported in deer, providing some evidence of sustained transmission in deer29. Persistence of SARS-CoV-2 in wild animals might conceivably result in viral evolution through adaptation to the animal host. Although virus isolates that are circulating in animals may become less fit for humans, a risk of future emergence into the human population, with unknown consequences, remains.
In this Article, to assess the extent of infection in white-tailed deer, the potential for a deer reservoir of SARS-CoV-2 and the risk of deer-to-human transmission, we initiated a SARS-CoV-2 surveillance programme of white-tailed deer in Ontario, Canada.
Results
Divergent SARS-CoV-2 found in deer
From 1 November to 31 December 2021, 300 white-tailed deer were sampled from Southwestern (N = 249, 83%) and Eastern (N = 51, 17%) Ontario, Canada during the annual hunting season (Fig. 1). The majority of sampled white-tailed deer were adults (94%), with comparable numbers of females (N = 135, 45%) and males (N = 165, 55%). We collected 213 nasal swabs and tissue from 294 retropharyngeal lymph nodes (RPLN), which were tested for SARS-CoV-2 RNA using reverse transcription polymerase chain reaction (RT–PCR).
Fig. 1. SARS-CoV-2 in white-tailed deer sampled in Ontario, 2021.

Circle size indicates the relative number of positive white-tailed deer (n = 17/298), with crosses showing samples from which viral genomes were recovered (n = 7). Four-digit genomic sequence ID labels are shown in yellow boxes. Location of negative samples are indicated using grey as per the legend. The detailed map depicts Southwestern Ontario (red rectangle on inset map). SARS-CoV-2 RNA was not detected in samples from Eastern Ontario.
Five of 213 (2.3%) nasal swabs were positive by two independent RT–PCR analyses at separate institutes (untranslated region (UTR) and envelope (E) RT–PCR targets Ct <40; and E and N2 RT–PCR targets Ct <36). Sixteen RPLN were also confirmed by PCR. Overall, SARS-CoV-2 RNA was detected in 21 samples representing 6% (17/298) of hunter-harvested white-tailed deer; all positive animals were adult deer from Southwestern Ontario and the majority (65%) were female (Fig. 1 and Supplementary Table 1). Two deer were excluded from further analysis owing to indeterminate RPLN results with no corresponding nasal swab.
To determine viral lineage and potentially infer key epidemiological relationships, we sequenced three high-quality SARS-CoV-2 consensus genomes from the five positive nasal swabs using a standard amplicon-based approach. All samples were also independently extracted and sequenced using a capture-probe-based approach for confirmation. By combining the amplicon and capture-probe sequencing data, five high-quality genomes (white-tailed deer nasal swabs: 4581, 4645, 4649, 4658 and 4662) and two partial genomes (white-tailed deer RPLNs: 4538 and 4534) were recovered (Supplementary Table 1). The samples were negative for human RNAse P by PCR and the majority (median 79.7%; Supplementary Table 1) of non-SARS-CoV-2 reads mapped to the white-tailed deer reference genome, demonstrating that contamination from human-derived SARS-CoV-2 sequences was highly unlikely.
Maximum-likelihood (ML) and parsimony-based phylogenetic analyses were used to examine lineage origins and relatedness, and showed that these deer-derived viral genomes formed a highly divergent clade within the B.1 PANGO lineage/20C Nextstrain clade (100% ultrafast bootstrap (UFB)) that shared a most recent common ancestor (MRCA) between May and July 2021 (95% highest posterior density (HPD) node age bounds; Extended Data Fig. 1). The B.1 lineage encompasses substantial diversity and was the genetic backbone from which the Beta VOC, Epsilon and Iota variants and notable mink (Neovison) outbreaks emerged (Fig. 2). The Ontario deer lineage forms a very long branch with 76 conserved nucleotide mutations relative to ancestral SARS-CoV-2 (Wuhan Hu-1) and 49 relative to their closest common ancestor with other genomes in GISAID (as of March 2022). The closest branching genomes in GISAID are human-derived sequences from Michigan, United States, sampled approximately 1 year prior (November/December 2020), which were inferred to share an MRCA with the Ontario deer lineage between May and August 2020 (95% HPD node age bounds; Extended Data Fig. 1). These human-derived sequences in turn are closely related to a mixed clade of human and mink sequences from Michigan collected in September and October 2020. The Ontario white-tailed deer lineage has been designated as PANGO lineage B.1.641.
Extended Data Fig. 1. A BEAST inferred time tree of B.1.641.

The tree was estimated using a HKY + G4 substitution model, log-normal distributed clock rates based on a strict molecular clock of 9.5 × 10−4 (inferred from tip-to-root regression) and a constant-sized coalescent. Analysis was performed used a fixed topology derived from the global ML analysis with internal node heights (and root height) inferred by BEAST. The 95% HPD node bars are shown in blue for the tree root and selected nodes of interest.
Fig. 2. ML phylogeny of white-tailed deer viral genomes.

Included are Ontario deer-derived genomes and an associated human sample (which have been collectively designated as lineage B.1.641) and a representative sample of the global diversity of human and animal-derived SARS-CoV-2 (n = 3,645). This phylogeny represents all non-human animal-derived samples (with the exception of domestic mink from Europe, which were subsampled) in GISAID at the time of sampling along with a representative subsample of human-derived genomes. VOCs and variants previously designated as variants under investigation within the tree are annotated and nodes are coloured by host genus (as indicated in the legend). The dotted line indicates the samples selected for the local ML analysis (Fig. 3).
Given the distorting effects of long-branch attraction and incomplete sampling, there is a degree of uncertainty in the phylogenetic placement of the white-tailed deer samples. However, the geographical proximity (Michigan is adjacent to Southwestern Ontario) and the mix of human and other animal cases (for example, human and mink cases) provide compelling evidence supporting this placement. Given the degree of divergence and potential for phylogenetic biases, we conducted three analyses to examine the possibility of recombination. Using 3Seq30, bolotie31 and HyPhy’s32 genetic algorithm recombination detection method33 with datasets representative of human and animal SARS-CoV-2 diversity in GISAID (as of March 2022) there was no indication of recombination within, or having given rise, to this lineage.
Potential deer-to-human transmission
Our phylogenetic analysis also identified a human-derived sequence from Ontario (ON-PHL-21-44225) that was highly similar (80/90 shared mutations; Supplementary Table 2) and formed a well-supported monophyletic group (100% UFB) with the white-tailed deer samples (Fig. 3). The small number of samples and relative diversity within B.1.641 make it difficult to determine the exact relationship between the human sample and the white-tailed deer samples (78% UFB for a MRCA with 4,658). However, global (Fig. 2) and local (Fig. 3) ML analyses and an UShER-based34 (Supplementary Fig. 1) parsimony analysis all support this human sample belonging to B.1.641.
Fig. 3. Phylogeny of the B.1.641 Ontario white-tailed deer lineage.

Genomes are annotated with the presence/absence of amino acid mutations relative to SARS-CoV-2 Wuhan Hu-1. Genomes were selected to characterize the relationship between Ontario white-tailed deer samples, the related Ontario human sample and closest B.1 human and mink samples from Michigan, United States (dotted segment in Fig. 2). Internal nodes in the phylogeny are annotated with UFB values ≥95%, and leaves with identical amino acid profiles were collapsed as indicated. Host species for each sample is shown by the leaf label colour and first annotation column as per the legend, with geographic location in the second annotation column. Amino acid mutations are coloured by corresponding gene, with grey indicating sites that were too poorly covered to determine presence/absence (for example, sites in partial deer-derived genomes 4538 and 4534).
The human-derived viral sequence also has a plausible epidemiological link to the white-tailed deer samples since it was collected in the same geographical region (Southwestern Ontario), during the same time period (autumn 2021). The human case had known close contact with deer in the week before symptom onset and had no known contact with any individuals that had tested positive for SARS-CoV-2 before or after contact with deer. At the time of the human case detection, the Ontario COVID-19 Genomic Network aimed to sequence 100% of eligible confirmed PCR-positive SARS-CoV-2 samples collected from human cases, and no other genetically related human-derived samples were identified. It should be noted that not all requested human samples are successfully sequenced, and the Omicron surge necessitated a reduction in the human-derived SARS-CoV-2 sampled for sequencing in Ontario in late 2021 (ref. 35).
Zoonosis-associated mutations
Using the five high-quality, complete deer-derived sequences and related human-derived sequence, we analysed the prevalence of mutations across GISAID in general as well as within VOC and animal-derived samples (Supplementary Table 2) to identify and contextualize key mutations. Of the 76 mutations shared among the 6 high-quality B.1.641 sequences, 51 are in ORF1ab (with 11 and 9 each in Nsp3 and Nsp4, respectively) and 9 are in the spike (S) gene. The six non-synonymous mutations in S correspond to a six-nucleotide deletion (V143–Y145), and five substitutions (H49Y, T95I, F486L, N501T and D614G) (Fig. 4a). With the exception of H49Y, these S mutations originated before the divergence of B.1.641 from the MRCA shared with the Michigan samples. These mutations have previously been observed in animal-derived viral sequences. Not all S mutations were conserved across B.1.641; S:613H and S:L1265I were found only in the human sample, three other non-synonymous mutations were found in either 4658 (T22I and S247G) or 4662 (V705A) white-tailed deer samples, and there was a frameshift in 4662S:L959.
Fig. 4. Evolution of the B.1.641 Ontario white-tailed deer lineage.

Evolution of the B.1.641 lineage is presented relative to other animal-derived genomes, the ancestral B.1 lineage and the global SARS-CoV-2 diversity. a, Spike mutations present within the Ontario white-tailed deer (WTD) lineage. Amino acid changes present in all five Ontario WTD sequences and associated human case (orange); only in the human sample (yellow); and only in a single WTD genome (purple). Animal symbols indicate mutations in bat-, deer-, pangolin- and hamster-derived SARS-CoV-2 sequences. ‘+’ indicates presence of the mutation in additional non-human animal species, and green indicates those in Michigan mink samples. ‘*’ indicates spike mutations that were inferred to have originated subsequent to the divergence of B.1.641 from their MRCA with the Michigan-derived human samples. Spike annotations were derived from UniProt P0DTC2 (DEL, deletion; FS, frameshift; TM, transmembrane; RBD, receptor binding domain; NTD, N-terminal domain) and are not shown to scale. A complete list of mutations from across the entire genome can be found in Supplementary Table 2. b, Root-to-tip regression analysis based on the representative SARS-CoV-2 diversity in the global ML phylogeny (Fig. 2). Substitutions per site per year trends (and 95% confidence intervals) from ordinary least squares regression analyses are shown for all human samples (0.9 × 10−3 to 1.0 × 10−3), animal-derived samples (1.0 × 10−3 to 1.1 × 10−3), the B.1 lineage (0.4 × 10−3 to 0.6 × 10−3) and the Ontario WTD clade (0 to 8 × 10−3). c, Consensus substitutions (%) corresponding to a change from a reference C allele to an alternative U allele. Boxes represents the 25% quartile, median and 75% quartile, with error bars capturing the minimum and maximum values within 1.5× interquartile range. This was calculated from consensus sequences across a subsample of global human SARS-CoV-2 diversity (earliest and most recent genomes from each PANGO lineage, n = 3,127), global animal diversity (all animal genomes in GISAID at time of sampling, n = 1,522), B.1 lineage (all genomes assigned to this lineage in GISAID as of January 2022, n = 206) and B.1.641.
Many non-synonymous mutations had previously been identified in white-tailed deer, including 16 in at least 3/5 of the Ontario deer samples, S:613H and ORF8:Q27* (human sample only), and S:T22I (1/5 Ontario deer samples only, but also noted in Delta-like SARS-CoV-2 from deer in Quebec26). However, there were also five conserved non-synonymous mutations that had not been previously observed in white-tailed deer and were relatively rare in GISAID (<1,000 sequences as of 14 March 2022): ORF1a:insertion2038N/MRASD (n = 32, including 31 mink from Michigan, United States), ORF1b:V364L (G14557T, n = 442, all human sequences), S:F486L (T23020G, n = 455), ORF3a:L219V (T26047G, n = 886) and ORF10:L37F (C29666T, n = 0).
Mutational signatures of deer adaptation
We examined evolutionary parameters to garner insights into selective pressures on B.1.641. We identified a potentially elevated mutation rate (3.7 × 10−3 versus 0.9 × 10−3 substitutions per site per year; Fig. 4b) relative to other SARS-CoV-2 on the basis of a root-to-tip regression of the global phylogeny (Fig. 2). To characterize signatures of selection within B.1.641 relative to background B.1 samples, we generated codon-alignment phylogenies for S, E, M, N, ORF3a, ORF6 and ORF1ab sequences and applied selection analysis methods from HyPhy32. The adaptive branch-site random effects likelihood36 and branch-site unrestricted statistical test for episodic diversification37 branch-site methods identified no B.1.641 branches with evidence of episodic diversifying positive selection relative to background B.1 branches. Interestingly, the ORF1ab analysis identified significant relaxation of selection amongst the white-tailed deer lineage (P = 0.0032). These signatures of neutrality were further supported by the even distribution of conserved mutations in proportion to gene/product length (Extended Data Fig. 2). Together, this suggests sustained viral transmission with minimal immune pressure in a susceptible animal population. However, further investigation into the host response and disease course of SARS-CoV-2 in white-tailed deer is required to confirm these inferences.
Extended Data Fig. 2. Distribution of conserved mutations in B.1.641.

Observed vs expected mutation counts (if uniformly distributed according to gene-length) within shared B.1.641 mutations for each gene/product.
Changes in the mutational signature of SARS-CoV-2 can be used to trace and understand its spread among hosts, and provide insights into mechanistic processes (for example, positive selection, RNA-dependent RNA polymerase activity or host cell modification through RNA editing). An analysis of base substitution frequencies within B.1.641 showed an elevated proportion of mutations involving C > U changes relative to other global, B.1 lineage and animal-derived viral sequences (Fig. 4c and Extended Data Fig. 3). Further investigation using non-parametric distance-based Welch multivariate analysis of variance (dbWMANOVA) found that the mutational spectra between human, deer and mink hosts differs significantly when considering all clades (W*d = 91.04, P < 0.001) and within clade 20C, which contains the B.1 lineage and Ontario B.1.641 sequences (W*d = 160.47, P < 0.001) (Supplementary Table 3 and Extended Data Fig. 4). Principal component analysis indicated that the majority of this variation (62.2%) corresponded to C > U (PC1) and G > A (PC2) frequencies. Notably, compared with the recently collected deer-derived virus samples from Quebec, the location of B.1.641 suggests that the Ontario lineage has been evolving within deer (Extended Data Fig. 5).
Extended Data Fig. 3. Comparing mutational spectra among hosts.

Boxen plots illustrating differences in the mutational spectra among hosts when considering only (a) clade 20 C (ndeer = 42, nmink = 91, nhuman = 108) and (b) all clades (ndeer = 105, nmink = 432, nhuman = 103). These plots show differences in both the frequency and overall distribution of each mutation within each host species. Samples from each host species were isolated from individuals in North America. The measure of center for each plot is the 50th percentile. The first boxes above and below the 50th percentile represent 50% of the data and each additional box accounts for one-quarter of the remaining data. The total number of boxes and outliers (diamonds) are determined using Tukey’s method.
Extended Data Fig. 4. Principal Components Analysis of mutational spectra in Clade 20 C.

Principal Components Analysis of the mutational spectra of SARS-CoV-2 genomes isolated from different hosts within Clade 20 C. The first two components account for 62.2% of the variation between the samples. Variation in the spectra along the first principal component associated with changes in the frequency of C > U. Samples appear to be spread along the first component by host-type. Ontario WTD samples (pink) and a human sample from Ontario (blue) appear close together in the projection, suggesting that they share a very similar mutation spectrum.
Extended Data Fig. 5. Principal Component Analysis of the mutational spectra from different SARS-CoV-2 variants.

The first two principal components account for 65.5% of the variation among the samples. Variation in the spectra along the first principal component are associated with changes in the frequency of C > U mutations. Interestingly, samples along this component are also differentiated by host-type. Variation along the second principal component reflect changes in the frequency of G > U and G > A mutations. (A) The biplot showing a projection of all samples. (B) A simplified version of plot A highlighting the positions of white-tailed deer samples isolated from Ontario (Blue) and Quebec (Pink). A human isolate from Ontario appears in the same area of the plot (teal) as Ontario deer samples. Arrows are scaled to 30% of their original size to create a cleaner plot.
Analyses of genome composition and codon usage bias may provide information on virus evolution and adaptation to host. We assessed similarity of B.1.641 codon usage signatures to other SARS-CoV-2 sequences (samples isolated from Wuhan-Hu-1, Quebec white-tailed deer, and mink from Canada and the United States), cervid viruses (epizootic haemorrhagic disease virus (EHDV), cervid atadenovirus A and elk circovirus) and the Odocoileus virginianus genome. No apparent differences were observed in codon usage bias between B.1.641 and other SARS-CoV-2 sequences across the entire coding region of the viral genome. Although some similarity in codon usage bias to cervid atadenovirus A was observed, generally there were clear differences between SARS-CoV-2 and non-SARS-CoV-2 sequences (Supplementary Table 4).
Virus isolation and S antigenicity
To determine the infectivity of positive samples, virus isolation was carried out using Vero E6 cells expressing human transmembrane protease serine 2 (TMPRSS2) with cathepsin L knocked out. At 4 days post-infection, cytopathic effect of 50% or less of the cell monolayer was observed for four of the samples (4581, 4645, 4658 and 4649) and virus supernatants were harvested. Confirmatory quantitative PCR for SARS-CoV-2 was carried out using 5′ UTR and E RT–PCR targets. The Ct for the four isolated samples (1/7 dilutions), 4581, 4645, 4658 and 4649, were 14.89, 16.39, 12.80 and 13.89 and 16.17, 24.18, 13.06 and 13.91, respectively, for 5′ UTR and E amplicons, respectively. Confirmatory sequencing was carried out successfully for isolates from samples 4581, 4645 and 4658. These showed only minor frequency variations of one single-nucleotide polymorphism (SNP) change (4581: gain of ORF3a Pro42Leu) or two SNP changes (4,658, loss of n.13 T > C and gain of ORF1a p.His3125Tyr) compared with the original swab consensus sequences.
Considering that S-gene mutations may lead to immune evasion to antibody responses generated by vaccination or previous infection, we measured spike recognition and neutralizing activity of plasma from vaccinated recipients or convalescent individuals to S glycoproteins identified in this study to broach a key element of risk to human health. Cells were transfected with codon-optimized S expression constructs corresponding to the S genes of samples 4581/4645, 4658, ON-PHL-21-44225 or SARS-CoV-2 S:D614G or Omicron (BA.1) and incubated with sera to analyse antibody recognition of S (Fig. 5a). We found that all white-tailed deer S were recognized to a similar extent to the S:D614G by sera from vaccinated or convalescent individuals, while Omicron S was less recognized overall (Fig. 5a and Supplementary Table 5). In addition, lentiviral pseudotypes were incubated with serial dilutions of sera, and neutralization half-maximal inhibitory serum dilution (ID50) was determined (Fig. 5b and Supplementary Table 6). We found that sera from vaccinated recipients, after either two or three doses, and from convalescent individuals efficiently neutralized all B.1.641S proteins, unlike Omicron, which required three doses for neutralization (Fig. 5b). Importantly, we did not observe a difference between the ability of sera to neutralize SARS-CoV-2 D614G or any of the Ontario white-tailed deer SARS-CoV-2. Taken together, these results suggest that the white-tailed deer S-gene mutations do not have substantial antigenic impact on antigenicity.
Fig. 5. Neutralization of the B.1.641 Ontario white-tailed deer spike.

a, 293T cells transfected with plasmids encoding the indicated spike variants were incubated with 1:250 diluted plasma from vaccinated (two or three doses of BNT162b2), convalescent or naïve individuals (n = 10 for each group) or with the conformationally independent anti-S2 CV3-25 antibody, followed by staining with fluorescently labelled anti-human IgG and flow cytometry analysis. MFI was normalized by surface expression of spike variants on the basis of CV3-25 binding (Supplementary Table 5). Differences in MFI between Omicron and ancestral D614G samples were significant for two-dose (P = 0.0284) and three-dose (P = 0.001) sera. Similarly, Omicron versus deer 4581/4645 (P = 0.0157) and deer 4658 (P = 0.0097) were significant for three-dose sera. b, Lentiviral pseudotypes encoding luciferase and harbouring the indicated spike variants were incubated with serial dilutions of plasma for 1 h at 37 °C and then used to infect 293T-ACE2. Infection was measured by quantitating luciferase activity 72 h post-infection. Neutralization ID50 for the sera from vaccinated or convalescent individuals was determined using a normalized non-linear regression using GraphPad Prism (Supplementary Table 6). Limit of detection is indicated by a dotted line (ID50 = 50). Distributions across replicates are represented by box plots with a central median value and whiskers showing the 1.5× interquartile range. Significant group differences (from Welch’s one-way ANOVA with Tukey’s post hoc testing) are indicated using brackets and asterisks (*P < 0.05, **P < 0.01, ***P < 0.001).
Discussion
We identified a divergent lineage of SARS-CoV-2 in white-tailed deer, and report evidence of host adaptation and unsustained deer-to-human transmission. White-tailed deer present many attributes important for a sustainable virus reservoir, including social behaviour, high density, highly transient populations with multiple human–deer interfaces and sylvatic interactions with other animals. A stable reservoir in white-tailed deer means that there is potential for spillover into human and sylvatic wildlife populations over a broad geographic distance. Unlike SARS-CoV-2 in farmed and contained mink, mitigation of onward transmission from wild white-tailed deer to humans is more challenging.
Phylogenetic analysis revealed that B.1.641 shares a relatively recent common ancestor with mink- and human-derived viral sequences from nearby Michigan. This includes specific mutational similarities such as a subset of mink sequences from Michigan exhibiting a rare 12-nucleotide insertion in the ORF1a gene that was also present in B.1.641. Two S-gene mutations, F486L and N501T, have been associated with mustelid (mink or ferret) host adaptations, and N501T has been associated with enhanced ACE2 binding and entry into human (Huh7) cells7,19,38,39. Notably, the Ontario white-tailed deer SARS-CoV-2 genomes did not harbour the relatively well-described S:Y453F mutation associated with mink and increased replication and morbidity in ferrets, but reduced replication in primary human airway epithelial cells40. Many of the mutations in the B.1.641 genomes have not been described previously or are infrequent and uncharacterized. These deer-derived genomes provide new insights into viral evolution and inferred virus mobility in animal species outside of the human population.
The mutational spectra of SARS-CoV-2 genomes from white-tailed deer, mink and humans vary between hosts, as highlighted by differences within clade 20C. Importantly, this provides evidence supporting the hypothesis that mutational spectra can be used to infer viral host species9,40. Furthermore, the frequency of C > U and A > G mutations differed between hosts, which may reflect host cell activity, such as restriction factors (for example, apolipoprotein B messenger RNA editing enzyme, catalytic polypeptide-like or APOBEC family of mRNA editing proteins), RNA editing enzymes (ADAR1) and reactive oxygen species42–45. We also observed that the mutational spectrum of B.1.641 is similar to that of other deer. However, this observation does not imply that mutations found in these sequences are related to those found in other deer. Rather, it is likely that the interaction between the viral genome and various host factors will alter the mutation spectra in broad, host-specific ways9,41. When placed in the context of the broader literature, our results provide further evidence this lineage of SARS-CoV-2 probably evolved in deer over time.
The absence of detectable positive selection in B.1.641, evidence of relaxation of selection within ORF1ab and the distribution of mutations across the genome contrasts with the signatures of strong selection in the equivalently divergent Omicron VOC. While additional complete deer-derived genomes could enable a more nuanced analysis46, it is clear that the evolutionary forces acting within B.1.641 are considerably different to those in Omicron. From these results, and a phylogenetically distant MRCA from 2020, we can infer that the B.1.641 lineage likely diverged in 2020 and has been maintained in wildlife under minimal selection pressure. It is possible that the absence of pre-existing host immunity permitted genetic drift to drive accumulation of neutral mutations (in combination with accumulation of mutations associated with animal adaptation).
It is unclear whether the initial spillover occurred directly from humans to deer, or an intermediate host such as mink or other yet undefined species was involved. The long branch length and period of unsampled evolution provide a number of possible scenarios (Fig. 6). The human-derived sample in the B.1.641 lineage lies within a relatively small number of deer-derived samples, rendering the precise relationship between the human- and deer-derived viruses uncertain (78% UFB). B.1.641 could represent a spillover into deer with a human spillback or the emergence of a virus reservoir in another wildlife species infecting both human and deer. However, the epidemiological data, evidence of infectious virus from deer, and the paucity of SARS-CoV-2 surveillance in white-tailed deer relative to human cases suggest spillover in deer followed by deer-to-human transmission is the most likely scenario (Fig. 6, scenario 1).
Fig. 6. Hypothetical zoonoses and evolution of the B.1.641 lineage.

The timeline and approximate relationship between the Beta VOC (bold), Iota/Epsilon former VUIs, and viral samples in white-tailed deer, humans and mink from both Michigan (green) and Ontario (orange) are displayed. As it likely emerged during one of the indicated poorly sampled periods of viral evolution, it is unclear whether the viral ancestor of B.1.641 was from an unknown animal (for example, mink, white-tailed deer or other species) or human reservoir. From this ancestor, there was either a spillback transmission from deer to human (scenario 1) or the emergence of a virus infecting both human and deer (scenario 2).
At this time, there is no evidence of recurrent deer-to-human or sustained human-to-human transmission of B.1.641. However, there has been considerable reduction in human and deer testing and genomic surveillance since the study samples were collected, and we cannot exclude the possibility of sustained transmission within or between these host populations. Enhanced surveillance is critical given human population density and mobility in the region, coupled with white-tailed deer population dynamics. In addition, rapid characterization of this new lineage from biological and epidemiological perspectives is critical to understanding viral transmission, immune evasion and disease in both wildlife and humans. Therefore, we assessed the ability of antibodies elicited following vaccination or infection to recognize and neutralize S and found that the mutations in B.1.641 do not have significant impact on S antigenicity (acknowledging the limited number of plasma tested) (Fig. 5). More work is needed to determine the potential roles of the mutations on spike functions, and to understand the pathogenesis and transmission phenotypes of this virus.
Secondary wildlife reservoirs have the potential to fundamentally alter the ecology of SARS-CoV-2. Our work underscores the need for a broad international One Health focus to identify new intermediate or reservoir hosts capable of driving sustained transmission and divergent viral evolution. An examination of human drivers of spillover and spillback and knock-on effects on wildlife and human health is urgently needed to identify, develop and implement mitigation strategies, beginning with reducing viral activity in humans.
Methods
Deer sample collection and study area
Between 1 November and 31 December 2021, adult and yearling free-ranging white-tailed deer were sampled as part of the Ontario Ministry of Natural Resources and Forestry’s (MNRF) annual Chronic Wasting Disease (CWD) surveillance programme. Samples were collected from hunter-harvested deer in Southwestern and Eastern Ontario and included nasal swabs and RPLNs. Samples were collected by staff wearing masks and disposable gloves. Nasal swabs were stored in individual 2 ml tubes with ~1 ml of universal transport medium (Sunnybrook Research Institute (SRI)), and RPLN tissues were stored dry in 2 ml tubes. After collection, samples were immediately chilled on ice packs then transferred to a −20 °C freezer where they were held for up to 1 week. Samples were then transferred to a −80 °C freezer where they were held until analysis. Location, date of harvest, and demographic data (age/sex) were recorded for each animal when available.
PCR screening and detection
RNA extractions and PCR testing of samples collected from deer were performed at the SRI in Toronto, Ontario. RNA extractions were conducted with 140 µl of nasal swab sample spiked with Armored RNA enterovirus (Asuragen; https://www.asuragen.com) via the Nuclisens EasyMag using Generic Protocol 2.0.1 (bioMérieux Canada) according to the manufacturer’s instructions; RNA was eluted in 50 µl. Tissue samples were thawed, weighed, minced with a scalpel and homogenized in 600 µl of lysis buffer using the Next Advance Bullet Blender (Next Advance) and a 5 mm stainless steel bead at 5 m s−1 for 3 min. RNA from 30 mg tissue samples was extracted using Specific Protocol B 2.0.1 via Nuclisens EasyMag; RNA was eluted in 50 µl. RT–PCR was performed using the Luna Universal Probe One-Step RT–qPCR kit (New England BioLabs, NEB; https://www.neb.ca). A SARS-CoV-2 5′ UTR and E specific multiplex RT–PCR were used for RNA detection47. Quantstudio 3 software (Thermo Fisher Scientific; https://www.thermofisher.com) was used to determine the cycle threshold (Ct). All samples were run in duplicate, and samples with Ct <40 for both RT–PCR targets and Armored RNA enterovirus in at least one replicate were considered presumed positive. For tissue samples, the presence of inhibitors was assessed by a 1:5 dilution of one of the replicates. Samples were considered inconclusive if no Armored enterovirus was detected or if only one RT–PCR target was detected and re-extracted for additional analysis. Samples were considered indeterminate if inconclusive after re-extraction or if no original material was left. Presumed positive samples were further analysed for human RNAse P to rule out potential human contamination9. Original material from presumed positive samples detected at SRI were sent to the Canadian Food Inspection Agency (CFIA) for confirmatory PCR testing. The MagMax CORE Nucleic Acid Purification Kit (Thermo Fisher Scientific) and the automated KingFisher Duo Prime magnetic extraction system was used to extract total RNA spiked with Armored RNA enterovirus. The enteroviral armored RNA was used as an exogenous extraction control. A SARS-CoV-2 E and nucleocapsid (N) specific multiplex RT–PCR was used for confirmatory RNA detection7. Master mix for qRT–PCR was prepared using TaqMan Fast Virus 1-step Master Mix (Thermo Fisher Scientific) according to the manufacturer’s instructions. Reaction conditions were 50 °C for 5 min, 95 °C for 20 s and 40 cycles of 95 °C for 3 s then 60 °C for 30 s. Runs were performed by using a 7500 Fast Real-Time PCR System (Thermofisher, ABI). Samples with Ct <36 for both RT–PCR targets were considered positive.
WGS
WGS was performed at both SRI and CFIA using independent extractions and sequencing methods. At SRI, DNA was synthesized from extracted RNA using 4 μl LunaScript RT SuperMix 5× (NEB) and 8 μl nuclease free water, and was added to 8 μl extracted RNA. Complementary DNA synthesis was performed under the following conditions: 25 °C for 2 min, 55 °C for 20 min, 95 °C for 1 min and holding at 4 °C.
The ARTIC V4 primer pool (https://github.com/artic-network/artic-ncov2019) was used to generate amplicons from the cDNA. Specifically, two multiplex PCR tiling reactions were prepared by combining 2.5 μl cDNA with 12.5 μl Q5 High-Fidelity 2× Master Mix (NEB), 6 μl nuclease-free water and 4 μl of respective 10 μM ARTIC V4 primer pool (Integrated DNA Technologies). PCR cycling was then performed in the following manner: 98 °C for 30 s followed by 35 cycles of 98 °C for 15 s and 63 °C for 5 min.
PCR reactions were then both combined and cleaned using 1× ratio sample purification beads (Illumina) at a 1:1 bead to sample ratio. The amplicons were quantified using the Qubit 4.0 fluorometer using the 1× dsDNA High Sensitivity (HS) Assay Kit (Thermo Fisher Scientific) and sequencing libraries prepared using the Nextera DNA Flex Prep kit (Illumina) as per the manufacturer’s instructions. Paired-end (2 × 150 bp) sequencing was performed on a MiniSeq with a 300-cycle reagent kit (Illumina) with a negative-control library with no input SARS-CoV-2 RNA extract.
WGS performed at CFIA used extracted nucleic acid quantified using the Qubit RNA HS Assay Kit on a Qubit Flex Fluorometer (Thermo Fisher Scientific). Eleven microlitres or 200 ng of total RNA was subject to DNase treatment using the ezDNase enzyme (Thermo Fisher Scientific) according to the manufacturer’s instructions. DNase-treated RNA was then used for library preparation and target sequence capture according to the ONETest Coronaviruses Plus Assay protocol (Fusion Genomics48). The enriched libraries were quantified using the Qubit 1× dsDNA HS Assay Kit on a Qubit Flex Fluorometer (Thermo Fisher Scientific) and subsequently pooled in equimolar amounts before fragment analysis on 4200 TapeStation System using the D5000 ScreenTape Assay (Agilent). The final pooled library was sequenced on an Illumina MiSeq using a V3 flowcell and 600 cycle kit (Illumina).
Human specimens are received at Public Health Ontario Laboratory for routine SARS-CoV-2 diagnostic testing (RT–PCR) from multiple healthcare settings, including hospitals, clinics and coronavirus disease 2019 (COVID-19) assessment centres. The human sample (ON-PHL-21-44225) was sequenced at Public Health Ontario Laboratory using an Illumina-based ARTIC V4 protocol (10.17504/protocols.io.b5ftq3nn), similar to the deer sequencing methods. Briefly, cDNA was synthesized using LunaScript reverse transcriptase (NEB). Amplicons were generated with premixed ARTIC V4 primer pools (Integrated DNA Technologies). Amplicons from the two pools were combined, purified with AMPure XP beads (Beckman Coulter) and quantified. Genomic libraries were prepared using the Nextera XT DNA Library Preparation Kit (Illumina), and genomes were sequenced as paired-end (2 × 150 bp) reads on an Illumina MiSeq instrument.
Genomic analysis
Paired-end illumina reads from ARTIC V4 and Fusion Genomics sequencing were initially analysed separately with the nf-core/viralrecon Nextflow workflow (v2.3) (refs. 49–51) that ran: FASTQC (v0.11.9) (ref. 52) read-level quality control, fastp (v0.20.1) (ref. 53) quality filtering and adapter trimming, Bowtie2 (v2.4.2) (ref. 54) read mapping to Wuhan-Hu-1 (MN908947.3) (ref. 55) SARS-CoV-2 reference, Mosdepth (v0.3.1) (ref. 56)/Samtools (v.1.12) (ref. 57) read mapping statistics calculation, iVar (v1.3.1) (ref. 58) ARTIC V4 primer trimming, variant calling and consensus generation; SnpEff (v5.0) (ref. 59)/SnpSift (v4.3t) (ref. 60) for variant effect prediction and annotation; and Pangolin (v3.1.20) (ref. 61) with PangoLEARN (2022-01-05), Scorpio (v0.3.16) (ref. 62), and Constellations (v.0.1.1) was used for PANGO lineage63 assignment. iVar primer trimmed soft-clipped read alignments were converted to hard-clipped alignments with fgbio ClipBam (http://fulcrumgenomics.github.io/fgbio/). Reads from hard-clipped BAM files were output to FASTQ files with ‘samtools fastq’. nf-core/viralrecon was re-run in amplicon mode without iVar primer trimming on the combined Fusion Genomics and ARTIC V4 primer trimmed FASTQ reads to generate the variant calling, variant effect and consensus sequence results used in downstream analyses. Additional quality-control steps to check for negative-control validity, drop-out, sample cross-contamination and excess ambiguity were performed using ncov-tools v1.8.0 (ref. 64). The mutations identified by the Nextclade (v1.10.2) (ref. 65) with 2022-01-05 database and xlavir (v0.6.1) report were manually searched in outbreak.info’s ‘Lineage | Mutation Tracker’ (on 2 February 2022) (ref. 66) to get information on the prevalence of observed mutations globally and within Canada. Mutations were also investigated for presence in specific lineages including VOCs, Michigan mink samples and other animal samples. Finally, mutations were searched in GISAID (on 2 February 2022) to tally the number of non-human hosts each mutation had been observed in.
Some limitations in genome quality and coverage existed that may have resulted in failure to detect additional mutations. All B.1.641 samples had missing terminal domains and contained internal regions with no or low coverage when sequenced using the ARTIC v4 amplicon scheme. This is a widespread issue that may explain the rarity of the 3′ proximal ORF10:L37F in GISAID. Significantly in our samples this meant there was no or <10× coverage in all five deer-derived sequences from ~27000 to 27177 (drop-out of ARTICv4 amplicons 90-91), which includes regions of the M gene. However, by combining the ARTIC v4 sequencing with additional sequencing using probe-based enrichment we were able to compensate for this drop-out and generate high coverage and completeness (<100 positions with no coverage in all deer and <100 positions with <10× coverage in 3/5 deer genomes; Supplementary Table 7).
Phylogenetics
To evaluate possible sampling biases due to the poorly defined and diverse B.1 and B.1.311 lineages and select closely related publicly available sequences for further phylogenetic analysis, a phylogenetic placement analysis based on UShER (https://genome.ucsc.edu/cgi-bin/hgPhyloPlace)34 was performed using the 7,217,299 sample tree (derived from UShER placement of GISAID, GenBank, COG-UK and CNCB onto 13-11-20 sarscov2phylo ML tree) via the SHUShER web-portal (shusher.gi.ucsc.edu). Phylogenetic analyses were performed using CFIA-NCFAD/scovtree Nextflow workflow (v1.6.0) (https://github.com/CFIA-NCFAD/scovtree/) with the consensus sequences contextualized with closely related sequences identified by UShER and randomly sampled representative sequences from major WHO SARS-CoV-2 clades from GISAID12 (downloaded 10 February 2022). This workflow generated a multiple sequence alignment using Nextalign CLI (v1.10.1) (ref. 65) and inferred an ML phylogeny using IQ-TREE (v2.2.0_beta)67 using the general time-reversible (GTR) model for visualisation with Phylocanvas68 via shiptv (v0.4.1) (https://github.com/CFIA-NCFAD/shiptv) and ggtree69. Divergence times for the inferred global ML topology were estimated using BEAST v1.10.4 (ref. 70). This analysis used a coalescent model with constant population size, a Hasegawa–Kishono–Yano substitution model with four discrete gamma categories, and a log-normal distributed strict molecular clock rate of 9.5 × 10−4 substitutions per site per year (based on a tip-to-root regression performed using TempEst71). Internal node heights and root node height were sampled by BEAST over 50 million MCMC generations (recorded every 1,000 iterations) before collation using BEAST’s LogCombiner. The final maximum clade credibility tree was generated using BEAST’s TreeAnnotator with node heights set to median values before final visualization in FigTree and Inkscape.
A subset of 157 taxa from an ancestral clade of B.1.641 were selected from the global phylogenetic tree shown in Fig. 2 to generate the phylogenetic tree shown in Fig. 3. Multiple sequence alignment of this subset of sequences was performed with MAFFT (v7.490) (ref. 72). An ML phylogenetic tree was inferred with IQ-TREE (v2.2.0_beta) using the GTR model and 1,000 UFB replicates73. Nextclade (v1.10.2) analysis was used to determine amino acid mutations and missing or low/no coverage regions from the sample genome sequences. Amino acid mutation profiles were determined relative to the B.1.641 samples, discarding mutations that were not present in any of the Ontario samples. Taxa with duplicated amino acid mutation profiles were pruned from the tree, keeping only the first observed taxa with a duplicated profile.
Recombination analyses were performed using 3Seq (v1.7) (ref. 30) and Bolotie (e039c01) (ref. 31). Specifically, 3Seq was executed with B.1.641 sequences and the most recent example of each lineage found in Canada and closest samples in GISAID in subtree (n = 595). Bolotie was executed using the B.1.641 sequences and two datasets, the provided pre-computed 87,695 probability matrix and a subsample of the earliest and latest example of each lineage in GISAID with all animal-derived samples and closest UShER samples (n = 4,688). Additionally, HyPhy’s32 (v2.5.31) genetic algorithm recombination detection method33 was applied to local alignment and ML phylogeny (Fig. 3) for all possible sites. Phylogenies were inferred using IQTree for segments either side of the identified putative breakpoint, and the B.1.641 clade and local topology was unchanged. Sequence statistics such as C > T rate were directly calculated from nextclade results (v1.10.2 with 2022-01-05 database). Additional figures were generated and annotated using BioRender74 and Inkscape75.
A phylogenetic approach with HyPhy32 (v2.5.31) was used to investigate signatures of selection within B.1.641 relative to the wider B.1 background lineage. To ensure necessary genomic completeness for codon alignment, all B.1 sequences in GISAID (as of 8 March 2022) were filtered to those with <0.1% N with full date information. Genomes with 0% N were removed to avoid biases from consensus workflows that replace undetermined sequence with reference genome. From this, all animal-derived (49 mink and 1 cat) sequences and 100 randomly sampled human B.1 sequences were extracted (n = 150). Finally, the WH0-1 reference genome was added to this alignment along with the five complete Ontario deer-derived genomes, associated human sequence, and the two most closely related Michigan human samples (MI-MDHSS-SC23517 and M-MDHSS-SC22669). Virulign76 (v1.0.1) was then used to generate codon alignments for E, M, N, S, ORF1ab, ORF3a, ORF6, ORF7a, ORF7b, ORF8 and ORF10 genes relative to the Wuhan-Hu-1 (MN908947.3) reference. ML phylogenies were inferred for these alignments using raxml-ng (v1.0.2) (ref. 77) with the GTR model and three parsimony-based starting trees. These phylogenies were manually inspected and rooted on Hu-1, and the B.1.641 branches were labelled using phylowidget37,78. Genes for which the phylogeny did not have a resolved B.1.641 clade (ORF7a and ORF7b) or a viable codon alignment without any internal stop codons (ORF8 and ORF10) were excluded from further analyses. For each gene, signatures of positive selection were evaluated using HyPhy’s adaptive branch-site random effects likelihood (aBSREL) method36 and signatures of gene-wide episodic diversification were evaluated using the branch-site unrestricted statistical test for episodic diversification (BUSTED) method37 with ten starting points. Finally, evidence of intensification or relaxation of selection was investigated using the RELAX method79 with ten starting points and synonymous rate variation. Additional code for divergence dating, recombination and selection analyses can be found under 10.5281/zenodo.7086599.
Analysis of mutational spectrum
The mutational spectra were created using a subset of 3,645 sequences used to create the high-quality global phylogeny. Included in this dataset are the seven unique deer samples from Ontario (samples 4538, 4534, 4662, 4649, 4581, 4645 and 4658; the five high-quality genomes plus two genomes with lower coverage), three white-tailed deer samples from Quebec (samples 4055, 4022 and 4249) and one human sample from Ontario (ON-PHL-21-44225), and any remaining human, mink and deer sequences from North America. The counts for each type of nucleotide change, with respect to the reference strain, were compiled and used to create a 12-dimensional vector. This subset was then filtered to remove sequences with fewer than 15 nucleotide changes. The counts were converted into the mutation spectrum by simply dividing each count by the sum of the counts in each sample10,41. As the mutation spectrum summarizes how host factors act upon the genome of SARS-CoV-2, it can potentially be used as an independent source of evidence supporting the evolution of the virus in a different host10,41. To investigate this, we conducted experiments using a dbWMANOVA80. If a significant difference between hosts was detected, a pairwise distance-based Welch t-test was used to identify which pair of hosts differed81. This approach was used because it is more robust on unbalanced and heteroscedastic data80,81. In the first experiment, samples from all lineages were used. The second experiment used only the subset of samples belonging to Nextstrain clade 20C (which contains the B.1.641 sequences). An analysis using the entire Pangolin B.1 lineage was not appropriate since this lineage also includes Nextstrain clades 20A and 20B and the inclusion of these samples could potentially obfuscate important patterns since the results would have to be interpreted in a context wider than necessary.
Codon usage analysis
Consensus sequences of SARS-CoV-2 samples from this and previous studies and additional sequences gathered from public databases were used. The sequences include the reference SARS-CoV-2 Wuhan-Hu-1 (NCBI NC_045512), SARS-CoV-2 mink/Canada/D/2020 (GISAID EPI_ISL_717717), SARS-CoV-2 mink/USA/MI-20-028629-004/2020 (GISAID EPI_ISL_2834697), Cervid atadenovirus A 20-5608 (NCBI OM470968)82, EHDV serotype 2/strain Alberta (NCBI AM744997 - AM745006), epizootic haemorrhagic disease virus, EHDV serotype 1/New Jersey (NCBI NC_013396 - NC_013405), EHDV 6 isolate OV208 (NCBI MG886400 - MG886409) and elk circovirus Banff/2019 (NCBI MN585201) (ref. 83) were imported into Geneious (v.9.1.8) (ref. 84). Annotations for the coding sequences of SARS-CoV-2 samples were transferred from the reference sequence SARS-CoV-2 Wuhan-Hu-1 (NC_ 045512) using the Annotate from Database tool. The coding sequences were extracted using the Extract Annotations tool for all viral sequences. An annotated file of the coding sequences for the Odocoileus virginianus texanus isolate animal Pink-7 (GCF_002102435.1) genome was downloaded from NCBI (https://ftp.ncbi.nlm.nih.gov/genomes/all/annotation_releases/9880/100/GCF_002102435.1_Ovir.te_1.0/). Coding sequences were input into CodonW (http://codonw.sourceforge.net/) with settings set to concatenate genes and output to file set to codon usage. Codon usage indices were set to the effective number of codons (ENc), GC content of gene (G + C), GC of silent third codon position (GC3s), silent base composition, number of synonymous codons (L_sym), total number of amino acids (L_aa), hydrophobicity of protein (Hydro) and aromaticity of protein (Aromo).
Virus isolation
For virus isolation, T25 flasks were seeded to confluency 1 day before infection with cathepsin L knock-out Vero E6 cells overexpressing TMPRSS2. The following day, swab samples were vortexed and spun down and 200 μl of the swab samples medium was combined with 16 μg ml−1 working concentration of TPCK-treated trypsin (NEB), 2× A/A/p/s antifungal/antibiotic solution (Wisent) and a 0.1% working concentration of BSA (Thermo Fisher Scientific) and added to the cell monolayer after removal of the medium. Samples were subjected to a 45 min adsorption with rocking every 5 min, after which the inoculum was removed and discarded and the monolayer was either washed once with 2 ml of D1 to remove blood cells present in the samples (4581, 4649 and 4676) or not washed (4645, 4658 and 4662) and 5 ml of DMEM with 1% FBS and antibiotics was added to the flask and incubated at 37 °C with 5% CO2. At 4 days post-infection, samples with visible cytopathic effect (partial, 50% or less rounded or detached cells) were harvested followed by collection and centrifugation at 4,000g for 10 min at 20 °C. The harvested supernatants were aliquoted and stored at −80 °C or inactivated and removed from the CL3 laboratory and RNA was extracted with the QIAamp Viral RNA Mini Kit (Qiagen), and stored at −20 °C until downstream analyses were carried out. All infectious work was performed under biosafety level 3 conditions.
Codon-optimized spike constructs, cells, sera and antibodies
Expression constructs of S mutants corresponding to samples 4581/4645 (S:H49Y, S:T95I, S:Δ143-145InsD, S:F486L, S:N501T, S:D614G), 4658 (S:T22I, S:H49Y, S:T95I, S:Δ143-145InsD, S:S247G, S:F486L, S:N501T, S:D614G) and ON-PHL-21-44225 (S:H49Y, S:T95I, S:Δ143-145InsD, S:F486L, S:N501T, S:Q613H, S:D614G) were generated by overlapping PCR as described previously85. S:D614G and S Omicron (BA.1) constructs were described elsewhere86. All constructs were cloned in pCAGGS and verified by Sanger sequencing.
HEK293T cells (ATCC) were cultured in DMEM supplemented with 10% FBS (Sigma), 100 U ml−1 penicillin, 100 µg ml−1 streptomycin and 0.3 mg ml−1 l-glutamine (Invitrogen) and maintained at 37 °C, 5% CO2 and 100% relative humidity.
Serum samples were obtained from consenting participants in several cohort studies with sample collection and sharing for this analysis approved by the Sinai Health System Research Ethics Board (#22-0030-E). Plasma of SARS-CoV-2 naïve, naïve-vaccinated (28–40 days after two or three doses of BNT162b2) and unvaccinated SARS-CoV-2 Delta previously infected donors was collected (Supplementary Table 8), heat inactivated for 1 h at 56 °C, aliquoted and stored at −80 °C until use. The conformation-independent monoclonal anti-S2 CV3-25 from a convalescent individual was described and produced as described previously87,88. The goat anti-human IgG conjugated with Alexa Fluor-647 was purchased from Invitrogen (A21445).
Spike binding assays
Hek293T cells seeded in a 10 cm Petri dish at 70% confluency were transfected with 10 µg of SARS-CoV-2 spike protein plasmid, 1 µg of lentiviral vector bearing green fluorescent protein (GFP) (PLV-eGFP) (gift from Pantelis Tsoulfas, Addgene plasmid number 36083) (ref. 89) using Jetprime transfection reagent (Polyplus, catalogue number 101000046) according to the manufacturer’s instructions. At 16 h post-transfection, the cells were stained with sera samples (1:250 dilution) for 45 min at 37 °C. Alexa Fluor-647-conjugated goat anti-human IgG (H + L) was used to detect plasma binding of the treated cells following 1 h incubation at room temperature. Samples were washed once with PBS, fixed in 1% paraformaldehyde and acquired using BD LSR Fortessa Flow cytometer (BD Biosciences). The seropositivity threshold was defined on the basis of mean fluorescence intensity (MFI) for naïve samples plus three standard deviations. The data were normalized by surface expression on the basis of the MFI of the monoclonal antibody CV3-25 (5 μg ml−1). The data analysis was performed using FlowJo 10.8.1 (Extended Data Fig. 6). For each set of sera, binding was compared across samples using Welch’s (unequal variance) one-way ANOVA procedure and a post-hoc Tukey’s honestly significant difference test (using a family-wise error rate of 0.05) via the statsmodel library (v0.14.0) (ref. 90).
Extended Data Fig. 6. Flow cytometry gating strategy for IgG plasma binding analysis.
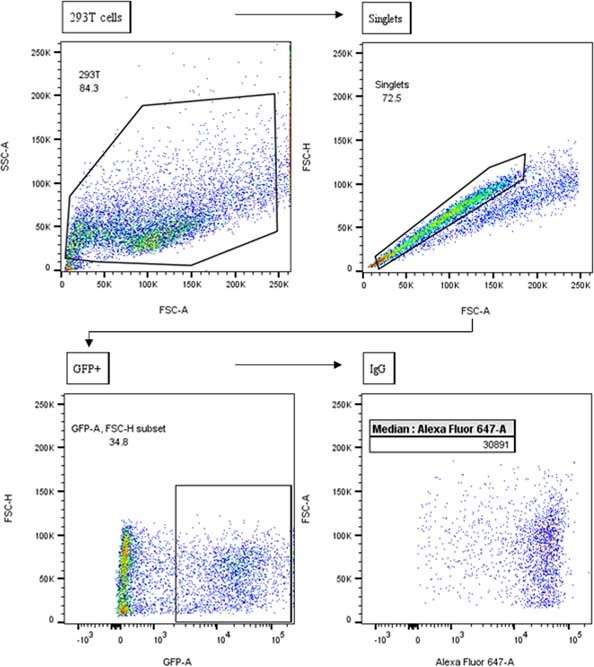
HEK 293 T cells were identified in forward scatter (FSC) and side scatter (SSC). Single cells (Singlets) were selected for analysis using FSC-A and FSC-H. Cells transfected with SARS-CoV-2 spike protein plasmid were gated (GFP+), negative and auto-fluorescent cells were excluded. Median fluorescence intensity for IgG (AlexaFluor 647-A) was analyzed in GFP + cells. Data was analyzed using FlowJo software.
Pseudotype production and neutralization assays
HEK293T seeded in 10 cm dishes were co-transfected with lentiviral packaging plasmid psPAX2 (gift from Didier Trono, Addgene number 12260), lentiviral vector pLentipuro3/TO/V5-GW/EGFP-Firefly Luciferase (gift from Ethan Abela, Addgene number 119816) and plasmid encoding the indicated S construct at a 5:5:1 ratio using jetPRIME transfection reagent according to the manufacturer’s protocol. Twenty-four hours post-transfection, media were changed, and supernatants containing lentiviral pseudotypes were harvested 48 h post-transfection, filtered with a 0.45 µM filter and stored at −80 °C until use.
HEK293T stably expressing human ACE2 (293T-ACE2, kind gift of Hyeryun Choe, Scripps Research91) were seeded in poly-d-lysine-coated 96-well plates. The next day, supernatants containing lentiviral pseudotypes were incubated with sera (serially diluted by five-fold, from 1:50 to 156,250) for 1 h at 37 °C and then added to cells in the presence of 5 µg ml−1 polybrene. Seventy-two hours later, media were removed, and cells were rinsed in phosphate-buffered saline and lysed by the addition of 40 µl passive lysis buffer (Promega) followed by one freeze–thaw cycle. A Synergy Neo2 Multi-Mode plate reader (BioTek) was used to measure the luciferase activity of each well after the addition of 50–100 µl of reconstituted luciferase assay buffer (Promega) as per the manufacturer’s protocol. Neutralization ID50 was calculated using Graphpad Prism and represents the plasma dilution that inhibits 50% of pseudotype transduction in 293T-ACE2. For each set of sera, neutralization was compared across samples using Welch’s (unequal variance) one-way ANOVA procedure and a post-hoc Tukey’s honestly significant difference test (using a family-wise error rate of 0.05) via the statsmodel library (v0.14.0) (ref. 90).
Reporting summary
Further information on research design is available in the Nature Research Reporting Summary linked to this article.
Supplementary information
Legends for Supplementary Tables 1–9.
Supplementary Fig. 1. UShER-based phylogenetic tree with Ontario white-tailed deer-derived high quality (n = 5) and partial (n = 2) genomes and the 2022-02-21 USCS UShER build (shared in Newick format).
A Microsoft Excel Workbook containing Supplementary Tables 1–9.
Acknowledgements
We acknowledge the contributions of the Virus Detection, Molecular Diagnostics, DNA Core sections and the Biocomputing Centre of Public Health Ontario, and in particular S. Teatero for leading the genome sequencing efforts. We gratefully acknowledge contributions of SARS-CoV-2 genome sequences from other laboratories through GISAID (Supplementary Table 9). We thank the licensed Ontario deer hunters who submitted samples for wildlife disease surveillance, the staff of MNRF’s CWD surveillance programme for their assistance with sample collection, and S. Hagey for GIS support. We also acknowledge the contributions of CFIA NCFAD’s Genomics Unit for their assistance with additional laboratory support and sequencing. S.M. and B.P. are members of the Canada and the Canadian Institutes for Health Research Coronavirus Variants Rapid Response Network (CoVaRR-Net). We are grateful to D. Bulir from McMaster University for providing the primer and probe sequences for the 5′ UTR RT–PCR SARS-CoV-2 PCR assay, and to M. Taipale for cathepsin L knock-out TMPRSS2-overexpressing Vero cells. We appreciate the Public Health Agency of Canada for uploading the raw sequencing data for the Public Health Ontario human sequence to the Short Read Archive. The CV3-25 antibody was produced using the pTT vector kindly provided by the Canada Research Council. We also thank D. Leclair, J. Provencher, C. Soos and A. Wilcox (Environment and Climate Change Canada) for comments on an earlier draft of the manuscript. Ethical approvals for this work were provided by Sinai Health System Research Ethics Board (#22-0030-E), Sunnybrook HSC Research Ethics Board (#SUN-2218) and University of Ottawa Research Ethics Board (#H-01-22-7842). Funding to S.M. was provided by the Public Health Agency of Canada and the Canadian Institutes for Health Research Operating grant: Emerging COVID-19 Research Gaps and Priorities #466984. J.D.K. was supported by the Association of Medical Microbiology and Infectious Diseases (AMMI) Canada 2020 AMMI Canada/Biomérieux Post Residency Fellowship in Microbial Diagnostics (unrestricted). Funding and computing resources for F.M. were provided by the Shared Hospital Laboratory, Dalhousie University and the Donald Hill Family. Funding for B.P. was provided by the Canadian Food Inspection Agency and the Canadian Safety and Security Program. Funding to J.B., T.B. and L.N. was provided by MNRF and the Public Health Agency of Canada. Funding for O.L. was from the Canadian Safety and Security Program, Laboratories Canada and CFIA. A.F. is the recipient of Canada Research Chair on Retroviral Entry no. RCHS0235 950-232424. M.C. is a Canada Research Chair in Molecular Virology and Antiviral Therapeutics (950-232840) and a recipient of an Ontario Ministry of Research, Innovation and Science Early Researcher Award (ER18-14-09). Funding to M.C. was provided by a COVID-19 Rapid Research grant from the Canadian Institutes for Health Research (CIHR, OV3 170632), CIHR stream 1 for SARS-CoV-2 Variant Research. Work from M.C. and A.F. is also supported by the Sentinelle COVID Quebec network led by the Laboratoire de Santé Publique du Québec (LSPQ) in collaboration with Fonds de Recherche du Québec-Santé (FRQS) and Genome Canada—Génome Québec, and by the Ministère de la Santé et des Services Sociaux (MSSS) and the Ministère de l’Économie et Innovation (MEI).
Extended data
Author contributions
Conceptualization: B.P., O.L., J.B., S.M. and T.B; methodology: F.M., O.L., J.D.K., B.P. and D.S.; software: F.M., P.K., O.L. and J.B.-S.; formal analysis: F.M., P.K., J.L.G., O.L. and J.B.-S.; investigation: M.C., K.N., P.A., G.L., A.A., B.V., J.B.-S., H.-Y.C, E.C., W.Y., M.G., M.S., M.P., G.S., O.V. and A.M.-A.; data curation: J.D.K., P.K., L.N. and H.M.; resources: A.M., E.A., A.F. and G.G.; writing—original draft: F.M., J.B., O.L., S.M., B.P. and J.L.G.; writing—review and editing: all authors; visualization: F.M., P.K. and J.B.-S.; supervision: J.B., S.M. and O.L.; funding: B.P., S.M., M.C., J.D.K., O.L., J.B., T.B. and L.N.
Peer review
Peer review information
Nature Microbiology thanks the anonymous reviewers for their contribution to the peer review of this work.
Data availability
All genomic sequence data are publicly available data through GISAID (https://gisaid.org/), and SRA accession numbers are provided in the supplementary material (Supplementary Table 1). Computer code and analysis scripts can be accessed at 10.5281/zenodo.7086599. All other data are available in the supplementary material (Supplementary Tables 1–9).
Code availability
All computer code and analysis scripts used in the manuscript are archived at https://github.com/fmaguire/on_deer_spillback_analyses/ and can be accessed at 10.5281/zenodo.7086599.
Competing interests
The authors declare no competing interests.
Footnotes
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
These authors contributed equally: Bradley Pickering, Oliver Lung, Finlay Maguire.
These authors jointly supervised this work: Samira Mubareka, Jeff Bowman.
Change history
12/12/2022
A Correction to this paper has been published: 10.1038/s41564-022-01298-3
Contributor Information
Bradley Pickering, Email: bradley.pickering@canada.ca.
Samira Mubareka, Email: samira.mubareka@sunnybrook.ca.
Jeff Bowman, Email: jeff.bowman@ontario.ca.
Extended data
is available for this paper at 10.1038/s41564-022-01268-9.
Supplementary information
The online version contains supplementary material available at 10.1038/s41564-022-01268-9.
References
- 1.Andersen KG, Rambaut A, Lipkin WI, Holmes EC, Garry RF. The proximal origin of SARS-CoV-2. Nat. Med. 2020;26:450–452. doi: 10.1038/s41591-020-0820-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Boni MF, et al. Evolutionary origins of the SARS-CoV-2 sarbecovirus lineage responsible for the COVID-19 pandemic. Nat. Microbiol. 2020;5:1408–1417. doi: 10.1038/s41564-020-0771-4. [DOI] [PubMed] [Google Scholar]
- 3.Cui J, Li F, Shi Z-L. Origin and evolution of pathogenic coronaviruses. Nat. Rev. Microbiol. 2019;17:181–192. doi: 10.1038/s41579-018-0118-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Haagmans BL, et al. Middle East respiratory syndrome coronavirus in dromedary camels: an outbreak investigation. Lancet Infect. Dis. 2014;14:140–145. doi: 10.1016/S1473-3099(13)70690-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Hu B, Guo H, Zhou P, Shi Z-L. Characteristics of SARS-CoV-2 and COVID-19. Nat. Rev. Microbiol. 2021;19:141–154. doi: 10.1038/s41579-020-00459-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Memish ZA, et al. Middle East respiratory syndrome coronavirus in bats, Saudi Arabia. Emerg. Infect. Dis. 2013;19:1819–1823. doi: 10.3201/eid1911.131172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Lu L, et al. Adaptation, spread and transmission of SARS-CoV-2 in farmed minks and associated humans in the Netherlands. Nat. Commun. 2021;12:6802. doi: 10.1038/s41467-021-27096-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Mannar, D. et al. SARS-CoV-2 Omicron variant: antibody evasion and cryo-EM structure of spike protein–ACE2 complex. Science10.1126/science.abn7760 (2022). [DOI] [PMC free article] [PubMed]
- 9.Hallmaier-Wacker LK, Munster VJ, Knauf S. Disease reservoirs: from conceptual frameworks to applicable criteria. Emerg. Microbes Infect. 2017;6:1–5. doi: 10.1038/emi.2017.65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Wei, C. et al. Evidence for a mouse origin of the SARS-CoV-2 Omicron variant. J. Genet. Genomics10.1016/j.jgg.2021.12.003 (2021). [DOI] [PMC free article] [PubMed]
- 11.Abdel-Moneim AS, Abdelwhab EM. Evidence for SARS-CoV-2 infection of animal hosts. Pathogens. 2020;9:E529. doi: 10.3390/pathogens9070529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Shu Y, McCauley J. GISAID: global initiative on sharing all influenza data—from vision to reality. Eurosurveillance. 2017;22:30494. doi: 10.2807/1560-7917.ES.2017.22.13.30494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Tan CCS, et al. Transmission of SARS-CoV-2 from humans to animals and potential host adaptation. Nat. Commun. 2022;13:2988. doi: 10.1038/s41467-022-30698-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.SARS-CoV-2 in animals situation update. FAOhttps://www.fao.org/animal-health/situation-updates/sars-cov-2-in-animals/en (2022).
- 15.Damas J, et al. Broad host range of SARS-CoV-2 predicted by comparative and structural analysis of ACE2 in vertebrates. Proc. Natl Acad. Sci. USA. 2020;117:22311–22322. doi: 10.1073/pnas.2010146117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Molenaar RJ, et al. Clinical and pathological findings in SARS-CoV-2 disease outbreaks in farmed mink (Neovison vison) Vet. Pathol. 2020;57:653–657. doi: 10.1177/0300985820943535. [DOI] [PubMed] [Google Scholar]
- 17.Shriner SA, et al. SARS-CoV-2 exposure in escaped mink, Utah, USA. Emerg. Infect. Dis. J. 2021;27:988–990. doi: 10.3201/eid2703.204444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Yen H-L, et al. Transmission of SARS-CoV-2 delta variant (AY.127) from pet hamsters to humans, leading to onward human-to-human transmission: a case study. Lancet. 2022;399:1070–1078. doi: 10.1016/S0140-6736(22)00326-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Oude Munnink BB, et al. Transmission of SARS-CoV-2 on mink farms between humans and mink and back to humans. Science. 2021;371:172–177. doi: 10.1126/science.abe5901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Frutos R, Devaux CA. Mass culling of minks to protect the COVID-19 vaccines: is it rational? New Microbes New Infect. 2020;38:100816. doi: 10.1016/j.nmni.2020.100816. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Pang, J. & Siu, T. Hong Kong to cull 2,000 hamsters after COVID-19 outbreak. Reuters (2022).
- 22.Peacock, T. P. et al. The altered entry pathway and antigenic distance of the SARS-CoV-2 Omicron variant map to separate domains of spike protein. 10.1101/2021.12.31.474653v2 (2022).
- 23.Shuai H, et al. Emerging SARS-CoV-2 variants expand species tropism to murines. eBioMedicine. 2021;73:103643. doi: 10.1016/j.ebiom.2021.103643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Palmer MV, et al. Susceptibility of white-tailed deer (Odocoileus virginianus) to SARS-CoV-2. J. Virol. 2021;95:e00083–21. doi: 10.1128/JVI.00083-21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Chandler JC, et al. SARS-CoV-2 exposure in wild white-tailed deer (Odocoileus virginianus) Proc. Natl Acad. Sci. USA. 2021;118:e2114828118. doi: 10.1073/pnas.2114828118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Hale VL, et al. SARS-CoV-2 infection in free-ranging white-tailed deer. Nature. 2021;602:481–486. doi: 10.1038/s41586-021-04353-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Kotwa, J. D. et al. First detection of SARS-CoV-2 infection in Canadian wildlife identified in free-ranging white-tailed deer (Odocoileus virginianus) from southern Québec, Canada. Preprint at bioRxiv10.1101/2022.01.20.476458 (2022).
- 28.Kuchipudi SV, et al. Multiple spillovers and onward transmission of SARS-CoV-2 in free-living and captive white-tailed deer. Proc. Nat. Acad. Sci. USA. 2022;119:e2121644119. doi: 10.1073/pnas.2121644119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Marques, A. D. et al. Multiple introductions of SARS-CoV-2 Alpha and Delta variants into white-tailed deer in Pennsylvania. mBio10.1128/mbio.02101-22 (2022). [DOI] [PMC free article] [PubMed]
- 30.Lam HM, Ratmann O, Boni MF. Improved algorithmic complexity for the 3SEQ recombination detection algorithm. Mol. Biol. Evol. 2018;35:247–251. doi: 10.1093/molbev/msx263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Varabyou A, Pockrandt C, Salzberg SL, Pertea M. Rapid detection of inter-clade recombination in SARS-CoV-2 with Bolotie. Genetics. 2021;218:iyab074. doi: 10.1093/genetics/iyab074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Kosakovsky Pond SL, et al. HyPhy 2.5—a customizable platform for evolutionary hypothesis testing using phylogenies. Mol. Biol. Evol. 2020;37:295–299. doi: 10.1093/molbev/msz197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Kosakovsky Pond SL, Posada D, Gravenor MB, Woelk CH, Frost SDW. Automated phylogenetic detection of recombination using a genetic algorithm. Mol. Biol. Evol. 2006;23:1891–1901. doi: 10.1093/molbev/msl051. [DOI] [PubMed] [Google Scholar]
- 34.Turakhia Y, et al. Ultrafast sample placement on existing trees (UShER) enables real-time phylogenetics for the SARS-CoV-2 pandemic. Nat. Genet. 2021;53:809–816. doi: 10.1038/s41588-021-00862-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Public Health Ontario. SARS-CoV-2 Whole Genome Sequencing in Ontario (weekly report) https://www.publichealthontario.ca/-/media/Documents/nCoV/epi/covid-19-sars-cov2-whole-genome-sequencing-epi-summary.pdf (2022).
- 36.Smith MD, et al. Less is more: an adaptive branch-site random effects model for efficient detection of episodic diversifying selection. Mol. Biol. Evol. 2015;32:1342–1353. doi: 10.1093/molbev/msv022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Murrell B, et al. Gene-wide identification of episodic selection. Mol. Biol. Evol. 2015;32:1365–1371. doi: 10.1093/molbev/msv035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Starr TN, et al. Deep mutational scanning of SARS-CoV-2 receptor binding domain reveals constraints on folding and ACE2 binding. Cell. 2020;182:1295–1310.e20. doi: 10.1016/j.cell.2020.08.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Han P, et al. Molecular insights into receptor binding of recent emerging SARS-CoV-2 variants. Nat. Commun. 2021;12:6103. doi: 10.1038/s41467-021-26401-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Zhou J, et al. Mutations that adapt SARS-CoV-2 to mink or ferret do not increase fitness in the human airway. Cell Rep. 2022;38:110344. doi: 10.1016/j.celrep.2022.110344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Shan K-J, Wei C, Wang Y, Huan Q, Qian W. Host-specific asymmetric accumulation of mutation types reveals that the origin of SARS-CoV-2 is consistent with a natural process. Innov. N. Y. N. 2021;2:100159. doi: 10.1016/j.xinn.2021.100159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.De Maio N, et al. Mutation rates and selection on synonymous mutations in SARS-CoV-2. Genome Biol. Evol. 2021;13:evab087. doi: 10.1093/gbe/evab087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Mourier T, et al. Host-directed editing of the SARS-CoV-2 genome. Biochem. Biophys. Res. Commun. 2021;538:35–39. doi: 10.1016/j.bbrc.2020.10.092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Ringlander J, et al. Impact of ADAR-induced editing of minor viral RNA populations on replication and transmission of SARS-CoV-2. Proc. Natl Acad. Sci. USA. 2022;119:e2112663119. doi: 10.1073/pnas.2112663119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Simmonds P, Ansari MA. Extensive C->U transition biases in the genomes of a wide range of mammalian RNA viruses; potential associations with transcriptional mutations, damage- or host-mediated editing of viral RNA. PLoS Pathog. 2021;17:e1009596. doi: 10.1371/journal.ppat.1009596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Pond SLK, et al. Adaptation to different human populations by HIV-1 revealed by codon-based analyses. PLoS Comput. Biol. 2006;2:e62. doi: 10.1371/journal.pcbi.0020062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.LeBlanc JJ, et al. Real-time PCR-based SARS-CoV-2 detection in Canadian laboratories. J. Clin. Virol. 2020;128:104433. doi: 10.1016/j.jcv.2020.104433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Zhan SH, et al. Target capture sequencing of SARS-CoV-2 genomes using the ONETest Coronaviruses Plus. Diagn. Microbiol. Infect. Dis. 2021;101:115508. doi: 10.1016/j.diagmicrobio.2021.115508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Di Tommaso P, et al. Nextflow enables reproducible computational workflows. Nat. Biotechnol. 2017;35:316–319. doi: 10.1038/nbt.3820. [DOI] [PubMed] [Google Scholar]
- 50.Ewels PA, et al. The nf-core framework for community-curated bioinformatics pipelines. Nat. Biotechnol. 2020;38:276–278. doi: 10.1038/s41587-020-0439-x. [DOI] [PubMed] [Google Scholar]
- 51.Patel, H. et al. nf-core/viralrecon: nf-core/viralrecon v2.3 - Copper Coatimundi. Zenodo10.5281/zenodo.5974693 (2022).
- 52.Andrews, S. FastQC: a quality control tool for high throughput sequence data. (2010).
- 53.Chen S, Zhou Y, Chen Y, Gu J. fastp: an ultra-fast all-in-one FASTQ preprocessor. Bioinformatics. 2018;34:i884–i890. doi: 10.1093/bioinformatics/bty560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Langmead B, Salzberg SL. Fast gapped-read alignment with Bowtie 2. Nat. Methods. 2012;9:357–359. doi: 10.1038/nmeth.1923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Wu F, et al. A new coronavirus associated with human respiratory disease in China. Nature. 2020;579:265–269. doi: 10.1038/s41586-020-2008-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Pedersen BS, Quinlan AR. Mosdepth: quick coverage calculation for genomes and exomes. Bioinformatics. 2018;34:867–868. doi: 10.1093/bioinformatics/btx699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Li H, et al. The sequence alignment/map format and SAMtools. Bioinformatics. 2009;25:2078–2079. doi: 10.1093/bioinformatics/btp352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Grubaugh ND, et al. An amplicon-based sequencing framework for accurately measuring intrahost virus diversity using PrimalSeq and iVar. Genome Biol. 2019;20:8. doi: 10.1186/s13059-018-1618-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Cingolani P, et al. A program for annotating and predicting the effects of single nucleotide polymorphisms, SnpEff: SNPs in the genome of Drosophila melanogaster strain w1118; iso-2; iso-3. Fly. 2012;6:80–92. doi: 10.4161/fly.19695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Cingolani P, et al. Using Drosophila melanogaster as a model for genotoxic chemical mutational studies with a new program, SnpSift. Front. Genet. 2012;3:35. doi: 10.3389/fgene.2012.00035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.O’Toole Á, et al. Assignment of epidemiological lineages in an emerging pandemic using the pangolin tool. Virus Evol. 2021;7:veab064. doi: 10.1093/ve/veab064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Colquhoun, R. & Jackson, B. Scorpio. (2021).
- 63.Rambaut A, et al. A dynamic nomenclature proposal for SARS-CoV-2 lineages to assist genomic epidemiology. Nat. Microbiol. 2020;5:1403–1407. doi: 10.1038/s41564-020-0770-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Jared S. & de Borja, R. ncov-tools. (2020).
- 65.Aksamentov I, Roemer C, Hodcroft E, Neher R. Nextclade: clade assignment, mutation calling and quality control for viral genomes. J. Open Source Softw. 2021;6:3773. doi: 10.21105/joss.03773. [DOI] [Google Scholar]
- 66.Tsueng, G. et al. Outbreak.info Research Library: a standardized, searchable platform to discover and explore COVID-19 resources and data. Preprint at bioRxiv10.1101/2022.01.20.477133 (2022). [DOI] [PMC free article] [PubMed]
- 67.Minh BQ, et al. IQ-TREE 2: new models and efficient methods for phylogenetic inference in the genomic era. Mol. Biol. Evol. 2020;37:1530–1534. doi: 10.1093/molbev/msaa015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Abudahab, K., Underwood, A., Taylor, B., Yeats, C. & Aanensen, D. M. Phylocanvas.gl: A WebGL-powered JavaScript library for large tree visualisation. Preprint at OSF Preprints10.31219/osf.io/nfv6m (2021).
- 69.Yu G, Smith DK, Zhu H, Guan Y, Lam TT-Y. ggtree: an R package for visualization and annotation of phylogenetic trees with their covariates and other associated data. Methods Ecol. Evol. 2017;8:28–36. doi: 10.1111/2041-210X.12628. [DOI] [Google Scholar]
- 70.Suchard MA, et al. Bayesian phylogenetic and phylodynamic data integration using BEAST 1.10. Virus Evol. 2018;4:vey016. doi: 10.1093/ve/vey016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Rambaut A, et al. Exploring the temporal structure of heterochronous sequences using TempEst (formerly Path-O-Gen) Virus Evol. 2016;2:vew007. doi: 10.1093/ve/vew007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Katoh K, Standley DM. MAFFT multiple sequence alignment software version 7: improvements in performance and usability. Mol. Biol. Evol. 2013;30:772–780. doi: 10.1093/molbev/mst010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Hoang DT, Chernomor O, von Haeseler A, Minh BQ, Vinh LS. UFBoot2: improving the ultrafast bootstrap approximation. Mol. Biol. Evol. 2018;35:518–522. doi: 10.1093/molbev/msx281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.BioRender (BioRender, 2022).
- 75.Inkscape Project (Inkscape, 2020).
- 76.Libin PJK, Deforche K, Abecasis AB, Theys K. VIRULIGN: fast codon-correct alignment and annotation of viral genomes. Bioinformatics. 2019;35:1763–1765. doi: 10.1093/bioinformatics/bty851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Kozlov AM, Darriba D, Flouri T, Morel B, Stamatakis A. RAxML-NG: a fast, scalable and user-friendly tool for maximum likelihood phylogenetic inference. Bioinformatics. 2019;35:4453–4455. doi: 10.1093/bioinformatics/btz305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Jordan GE, Piel WH. PhyloWidget: web-based visualizations for the tree of life. Bioinformatics. 2008;24:1641–1642. doi: 10.1093/bioinformatics/btn235. [DOI] [PubMed] [Google Scholar]
- 79.Wertheim JO, Murrell B, Smith MD, Kosakovsky Pond SL, Scheffler K. RELAX: detecting relaxed selection in a phylogenetic framework. Mol. Biol. Evol. 2015;32:820–832. doi: 10.1093/molbev/msu400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Hamidi B, Wallace K, Vasu C, Alekseyenko AV. $W_{d}^{*}$-test: robust distance-based multivariate analysis of variance. Microbiome. 2019;7:51. doi: 10.1186/s40168-019-0659-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Alekseyenko AV. Multivariate Welch t-test on distances. Bioinformatics. 2016;32:3552–3558. doi: 10.1093/bioinformatics/btw524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Lung O, et al. Whole-genome sequence of Cervid atadenovirus A from the initial cases of an adenovirus hemorrhagic disease epizootic of black-tailed deer in Canada. Microbiol. Resour Announc. 2022;11:e0066222. doi: 10.1128/mra.00662-22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Fisher M, et al. Discovery and comparative genomic analysis of elk circovirus (ElkCV), a novel circovirus species and the first reported from a cervid host. Sci. Rep. 2020;10:19548. doi: 10.1038/s41598-020-75577-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Kearse M, et al. Geneious Basic: an integrated and extendable desktop software platform for the organization and analysis of sequence data. Bioinformatics. 2012;28:1647–1649. doi: 10.1093/bioinformatics/bts199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Chatterjee D, et al. Antigenicity of the Mu (B.1.621) and A.2.5 SARS-CoV-2 spikes. Viruses. 2022;14:144. doi: 10.3390/v14010144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Chatterjee D, et al. SARS-CoV-2 Omicron spike recognition by plasma from individuals receiving BNT162b2 mRNA vaccination with a 16-week interval between doses. Cell Rep. 2022;38:110429. doi: 10.1016/j.celrep.2022.110429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Jennewein MF, et al. Isolation and characterization of cross-neutralizing coronavirus antibodies from COVID-19+ subjects. Cell Rep. 2021;36:109353. doi: 10.1016/j.celrep.2021.109353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Li W, et al. Structural basis and mode of action for two broadly neutralizing antibodies against SARS-CoV-2 emerging variants of concern. Cell Rep. 2022;38:110210. doi: 10.1016/j.celrep.2021.110210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Enomoto M, Bunge MB, Tsoulfas P. A multifunctional neurotrophin with reduced affinity to p75NTR enhances transplanted Schwann cell survival and axon growth after spinal cord injury. Exp. Neurol. 2013;248:170–182. doi: 10.1016/j.expneurol.2013.06.013. [DOI] [PubMed] [Google Scholar]
- 90.Seabold, S. & Perktold, J. Statsmodels: econometric and statistical modeling with Python. SciPy10.25080/MAJORA-92BF1922-011 (2010).
- 91.Moore MJ, et al. Retroviruses pseudotyped with the severe acute respiratory syndrome coronavirus spike protein efficiently infect cells expressing angiotensin-converting enzyme 2. J. Virol. 2004;78:10628–10635. doi: 10.1128/JVI.78.19.10628-10635.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Legends for Supplementary Tables 1–9.
Supplementary Fig. 1. UShER-based phylogenetic tree with Ontario white-tailed deer-derived high quality (n = 5) and partial (n = 2) genomes and the 2022-02-21 USCS UShER build (shared in Newick format).
A Microsoft Excel Workbook containing Supplementary Tables 1–9.
Data Availability Statement
All genomic sequence data are publicly available data through GISAID (https://gisaid.org/), and SRA accession numbers are provided in the supplementary material (Supplementary Table 1). Computer code and analysis scripts can be accessed at 10.5281/zenodo.7086599. All other data are available in the supplementary material (Supplementary Tables 1–9).
All computer code and analysis scripts used in the manuscript are archived at https://github.com/fmaguire/on_deer_spillback_analyses/ and can be accessed at 10.5281/zenodo.7086599.