

Abstract
Dopamine (DA) and glutamate neurotransmission are strongly implicated in schizophrenia pathophysiology. While most studies focus on contributions of neurons that release only DA or glutamate, neither DA nor glutamate models alone recapitulate the full spectrum of schizophrenia pathophysiology. Similarly, therapeutic strategies limited to either system cannot effectively treat all three major symptom domains of schizophrenia: positive, negative, and cognitive symptoms. Increasing evidence suggests extensive interactions between the DA and glutamate system and more effective treatments may therefore require the targeting of both DA and glutamate signaling. This offers the possibility that disrupting DA-glutamate circuitry between these two systems, particularly in the striatum and forebrain, culminate in schizophrenia pathophysiology. Yet, the mechanisms behind these interactions and their contributions to schizophrenia remain unclear. In addition to circuit- or system-level interactions between neurons that solely release either DA or glutamate, here we posit that functional alterations involving a subpopulation of neurons that co-release both DA and glutamate provide a novel point of integration between DA and glutamate systems, offering a key missing link in our understanding of schizophrenia pathophysiology. Better understanding of mechanisms underlying DA/glutamate co-release from these neurons may therefore shed new light on schizophrenia pathophysiology and lead to more effective therapeutics.
Keywords: Dopamine, glutamate, VGLUT2, co-transmission, schizophrenia
Introduction
Schizophrenia is a prevalent serious mental illness, affecting ~1% of the global population1. The combination of positive (e.g., delusions, hallucinations, thought disorganization), negative (e.g., social withdrawal, amotivation), and cognitive (e.g., attention deficits, impaired working memory) symptoms causes substantial disabilities2. Antipsychotic drugs remain the primary therapeutic option in schizophrenia. Nevertheless, these drugs remain limited in their abilities to effectively improve negative symptoms and cognition, and often produce metabolic disturbances that limit compliance1, 3–5. Furthermore, the mechanisms underlying schizophrenia pathophysiology remain unclear. Thus, an improved understanding of the biology of schizophrenia is needed to generate more effective treatments that ameliorate all three major symptom domains.
Despite increasing awareness of the complex polygenic and environmental interrelationships that contribute to schizophrenia pathogenesis, dysfunction in brain dopamine (DA) and glutamate neurotransmission have remained mainstays of our understanding of the disease. We will examine DA and glutamate systems as well as their interactions in the context of schizophrenia. We will additionally focus on a distinct subpopulation of midbrain DA neurons that co-transmit both DA and glutamate in the striatum and forebrain, regions highly relevant to schizophrenia pathogenesis and its treatment.
Dopamine and glutamate models of schizophrenia
Dysregulated modulation of striatal DA neurotransmission is the central tenet of the DA hypothesis of schizophrenia, the predominant theory of schizophrenia for the last several decades6–9. This theory suggests that altered striatal DA neurotransmission within the mesolimbic pathway leads to positive symptoms7. Early postmortem human brain studies demonstrated increased striatal DA, later confirmed by in vivo PET imaging7, 8, 10. Similarly, striatal presynaptic DA function is elevated in individuals with prodromal symptoms of schizophrenia, and conversion from the prodrome to first-episode psychosis is associated with increased striatal DA synthesis11, 12. Indeed, mouse models that raise striatal DA signaling via psychostimulants or DA D2 receptor (D2R) overexpression produce behaviors associated with psychosis (e.g., working memory deficits)13–16. Conversely, blockade of striatal DA D2-like receptors improves positive symptoms, further supporting the importance of DA signaling in schizophrenia symptomatology7, 17.
Glutamate has also been implicated in schizophrenia pathogenesis18. Antagonists of N-methyl-D-aspartate (NMDA) glutamate receptors including ketamine and phencyclidine produce positive, negative, and cognitive symptoms clinically; animal models similarly exhibit schizophrenia-related impairments (e.g., sensorimotor gating and social deficits)13, 19, 20. Animals with diminished levels of d-serine, an NDMA receptor co-agonist, exhibit decreased hippocampal volume and dendritic spine density; these pathologic changes are similar to those in brains of individuals with schizophrenia (see Box)21. Additionally, induced pluripotent stem cell (iPSC) studies employing neurons derived from individuals with schizophrenia show dysfunctional glutamate receptor signaling, diminished glutamate release, delayed maturation of glutamate neurons, and reduced synaptic connectivity22–24. Postmortem brain studies of individuals with schizophrenia also show that expression of excitatory amino transporter 2 (EAAT2), a transporter required for glutamate uptake, is reduced in temporal and frontal regions versus unaffected comparison subjects25. This is accompanied with diminished glutaminase expression in dorsolateral prefrontal cortex (DLPFC) and thalamus in schizophrenia25. Overall, these data point to the relevance of both DA and glutamate systems in schizophrenia.
Box: d-Serine.
d-Serine is an amino acid synthesized in astrocytes, microglia, and neurons from its enantiomer, l-Serine, by serine racemase26. d-Serine is concentrated in human and rodent forebrain regions including PFC, hippocampus, and striatum27–29. d-serine’s anatomic distribution is closely correlated with expression of its target, the NMDA receptor28. d-serine is a potent and selective co-agonist of NMDA receptors30–32, binding the receptor’s glycine site33. Given that 1) d-serine is the primary co-agonist of pyramidal neuron NMDA receptors in the prefrontal cortex29, 34, and 2) genes encoding serine racemase and d-amino acid oxidase (which degrades d-serine) are linked to schizophrenia risk35, 36, d-serine has become a popular novel candidate for schizophrenia therapeutics26, 37.
Limitations of DA and glutamate models of schizophrenia
Many questions remain concerning respective contributions of DA and glutamate models to schizophrenia pathophysiology. For the DA model, while antipsychotic drug blockade of D2-like receptor signaling is linked to positive symptom reduction, these medications do not effectively address either the negative or cognitive symptoms of the illness38, 39. Likewise, despite the glutamate model’s reproduction of some negative and cognitive symptoms in various animal systems, it is limited in effectively reproducing the disturbances in DA neurotransmission indicative of schizophrenia or in explaining the clinical efficacy of DAergic antipsychotic drugs13. This suggests that, individually, these animal models are unlikely to fully recapitulate the primary pathology related to schizophrenia resulting in both DAergic and glutamatergic dysfunction. Additionally, evidence for the efficacy of glutamatergic drugs in schizophrenia treatment has been mixed38. Though some clinical studies suggest that NMDA receptor modulators including d-serine and sarcosine improve positive and negative symptoms, other work shows no benefit38, 40, 41. Similarly, there were initially promising results in a small randomized, double blind, placebo-controlled clinical trial comparing efficacy of LY2140023, a metabotropic glutamate 2/3 receptor (mGluR2/3) agonist42. However, a subsequent larger multicenter, randomized, double-blind, parallel, placebo-controlled trial was inconclusive since neither LY2140023 nor the olanzapine positive control differed significantly versus placebo42, 43. These data suggest that neither DA nor glutamate models on their own sufficiently explain the full spectrum of schizophrenia pathogenesis and that our existing pharmacological and animal models provide only partial answers. Instead, we posit that pathogenic alterations that modify the interactions between DA and glutamate systems alter striatal signaling, ultimately culminating in psychosis as well as negative and cognitive symptoms.
Interactions between DA and glutamate systems in schizophrenia
There is increasing evidence of extensive bidirectional communication between DA and glutamate systems, particularly in the PFC, midbrain, and striatum of humans, nonhuman primates, and rodents44–48. Each system impacts its counterpart and disturbances in these interactions have been implicated in schizophrenia pathology9, 49. Furthermore, DA-glutamate system interactions are part of a larger framework where additional neurotransmitter systems intersect. For example, serotonin signaling in PFC and striatum modulates both DAergic and glutamatergic systems and interactions between these systems mediates therapeutic efficacy of atypical antipsychotic drugs50–52. Similarly, the interplay of glutamatergic and cholinergic systems in cortex and hippocampus is important for working memory and linked to the cognitive deficits of schizophrenia both clinically and in rodent schizophrenia models53–56.
In DA/glutamate system interactions, midbrain DA neuron activity is modulated by glutamatergic inputs from the frontal cortex57–59. In this circuit: 1) Cortical pyramidal glutamatergic projections stimulate inhibitory GABA interneurons; 2) Release of GABA from the interneurons lowers the firing rate of cortical glutamate neurons that project onto midbrain DA neurons; 3) Modulating glutamate tone from these midbrain projections provides homeostatic control over DAergic firing in the mesolimbic pathway57 (Figure 1a). Disruptions in DA-glutamate communications may drive schizophrenia pathology, leading to abnormally elevated striatal DA13, 38, 57. This forms the foundation of the ‘NMDA receptor hypofunction hypothesis’ which asserts that hypofunctional NMDA receptor signaling in cortical GABAergic interneurons decrease GABAergic inhibitory tone to increase firing by midbrain-projecting secondary glutamatergic neurons (Figure 1b). This increases midbrain DA neuron stimulation to raise striatal DA associated with positive symptoms57, 59.
Figure 1. Interplay between dopamine and glutamate systems is disrupted in schizophrenia.
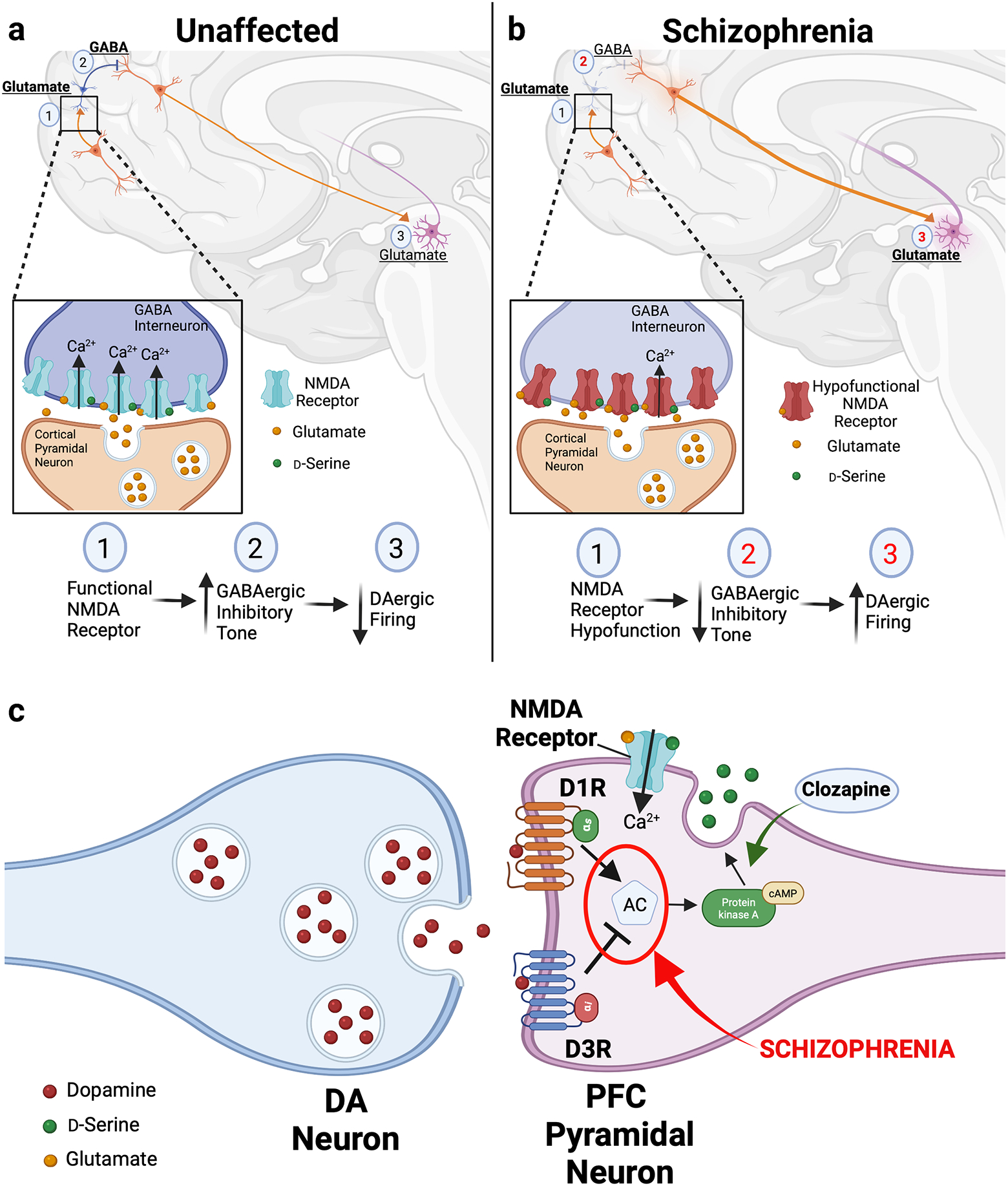
(a) In unaffected individuals, striatal dopamine (DA) levels are modulated via a polysynaptic circuit: (1) Cortical pyramidal glutamatergic projections stimulate GABAergic interneurons via stimulatory NMDA receptors. Upon activation by local increases in synaptic glutamate and co-substrate d-serine, the NMDA receptors conduct Ca2+, triggering increased GABAergic interneuron firing (inset). (2) GABA released by these interneurons lowers the firing rate of cortical glutamate neurons that project onto DAergic midbrain neurons. (3) The resulting glutamate tone from these stimulatory projections to the midbrain enables homeostatic control over DAergic firing in the mesolimbic pathway to finely modulate striatal DA levels. (b) According to the NMDA receptor hypofunction hypothesis, in schizophrenia, disruptions in DA-glutamate communications within this circuit drive abnormal increases in striatal DA to produce schizophrenia pathology. Hypofunctional NMDA receptors expressed by GABAergic interneurons diminish the amount of Ca2+ influx in response to glutamatergic stimulation (inset). This significantly dampens GABAergic inhibitory tone and increases firing by the midbrain-projecting secondary glutamatergic neurons. The resulting increases in midbrain DA neuron activity raise striatal DA levels, contributing to the positive symptoms of schizophrenia. Red numbers indicate pathway steps affected by schizophrenia pathology. (c) In pyramidal neurons of the prefrontal cortex (PFC), the combined actions of dopamine (DA) on stimulatory D1 (D1R) and inhibitory D3 (D3R) receptors modulates levels the activity of adenylate cyclase, the enzyme responsible for cAMP synthesis. Tight control over intracellular cAMP enables neurons to control release of d-serine and finely tune local glutamatergic neurotransmission. In schizophrenia, we propose that disturbances in coordination between D1R- and D3R-mediated cAMP signaling alter dopaminergic regulation of d-serine release (in red). This consequently produces pathologic changes in the downstream glutamatergic circuitry. Conversely, treatment with the antipsychotic drug clozapine raises d-serine release (in green), providing a potential therapeutic mechanism for ameliorating the disturbances in the dopaminergic modulation of d-serine release.
The DA system plays a similarly important role in modulating glutamatergic circuitry. Ascending DAergic projections to the PFC impact the glutamatergic circuitry that coordinate working memory and executive function60, 61. This DA signaling occurs via stimulatory D1 (D1R) as well as inhibitory D2R and D3 (D3R) receptors, which are commonly co-expressed in glutamatergic PFC neurons62. This combination of stimulatory and inhibitory DA receptors exerts opposing effects on extracellular levels of d-serine to fine-tune the local glutamatergic neurotransmission60. Such DA-mediated fine-tuning in the PFC has important functional implications where DA levels and cognitive performance follow an inverted U-shaped function: either too little or too much DA receptor activation impairs cognition63, 64. Therefore, we posit that changes in the coordination between DAergic signals may precipitate disturbances in the downstream glutamatergic circuitry to fuel schizophrenia pathology (Figure 1c). Such a mechanism provides a key link between DA- and d-serine-mediated NMDA receptor signaling and the cognitive symptoms in schizophrenia. Moreover, since the antipsychotic drug clozapine raises d-serine release in the frontal cortex and modifies local PFC D2-like receptor signaling, this may reestablish the balance of DAergic fine-tuning, providing targets for development of more drugs that can better target the cognitive sequelae of schizophrenia60, 65–67 (Figure 1c).
Abnormal sensorimotor gating and accompanying hippocampal hyperactivity, robust schizophrenia endophenotypes68, provide additional evidence of interactions between DA and glutamate systems. Data from rodent models shows a mechanistic relationship between hippocampal hyperactivity involving glutamatergic circuits and dysregulated DA release from midbrain DA neurons projecting to the striatum. Raising ventral hippocampus or ventral subiculum output leads to increased DA signaling, resulting disruptions in sensorimotor gating including reduced pre-pulse inhibition (PPI)68–70. Similarly, in a 22q11 deletion syndrome mouse model of schizophrenia, elevated DA D2 receptor-mediated DA signaling in thalamic neurons disrupts glutamatergic neurotransmission from thalamocortical neurons that project to the auditory cortex71. Conversely, acute treatment with NMDA receptor antagonist ketamine raises striatal and cortical DA release in rodents, consistent with the NMDA receptor hypofunction hypothesis72. In humans, magnetic resonance spectroscopy (1H-MRS) studies showed that amphetamines which raise striatal DA also increase glutamate levels in the dorsal anterior cingulate cortex (dACC) in a sex-specific manner in healthy subjects73. While amphetamine increases dACC glutamate similarly in men and women, methamphetamine differentially augments glutamate levels in women versus men73. This is consistent with increased vulnerability to methamphetamine and methamphetamine-induced euphoria in women74, 75. Overall, close integration between DA and glutamate systems may also more accurately explain why alterations primarily to DA or glutamate signaling alone are insufficient to reproduce the deficits in all three schizophrenia symptom domains.
Overview of DA/glutamate co-transmission
To date, most studies examining DA and glutamate systems have focused on neurons that primarily release either DA or glutamate. We propose that co-transmission of glutamate alongside DA from a population of the same midbrain DA neurons may represent an important but previously underappreciated aspect of schizophrenia pathophysiology. Historically, Dale’s principle maintained that a single neurotransmitter is released from all synaptic terminals of a neuron – a concept accepted as dogma for decades76. It has since been demonstrated that more than one neurotransmitter can be released from a single neuron77, 78. This paradigm is embodied by co-transmission of both DA and glutamate from a distinct subpopulation of midbrain DA neurons that also express machinery of glutamatergic neurotransmission including the vesicular glutamate transporter 2 (VGLUT2). VGLUT2-expressing DA/glutamate neurons are mainly found in the medial ventral tegmental area (VTA) and project to the nucleus accumbens (NAc) medial shell; an additional population of DA/glutamate cells localizes to the lateral substantia nigra pars compacta (SNc) and projects to the tail of the striatum79–85.
VGLUT2 expression is dynamic across DA neuron development86. In mouse mesencephalon and diencephalon development, VGLUT2 is detected by embryonic day 9.5, which precedes expression of DA neuron markers NURR1 and tyrosine hydroxylase (TH)87, suggesting that VGLUT2 is an early marker of DA neuron development. Importantly, mouse studies show that the majority of SNc DA neurons repress VGLUT2 expression by postnatal day 14, raising the possibility that DA neuron VGLUT2 expression is part of a regulated neurodevelopmental program linking DA and glutamate systems87, 88.
Compared to rodents, considerably less is known about the developmental course of VGLUT2 in human brain. Availability of human brain gene expression repositories enabled us to examine regional VGLUT2 expression across human brain development and the lifespan. Per the Brainspan Atlas of the Developing Human Brain, VGLUT2’s expression levels in striatum are high early in development in utero and prior to parturition, followed by a significant drop for the rest of life. This pattern is consistent with the Human Brain Transcriptome atlas where VGLUT2 expression is high in fetal brain across most brain regions (e.g., neocortex, hippocampus, amygdala, striatum, cerebellar cortex), followed by a rapid decrease in expression shortly following birth. A limitation of these analyses is the absence of data for VGLUT2 expression specifically in DA neurons. Future studies will focus on mapping VGLUT2 expression in human midbrain TH+/VGLUT2+ DA neurons.
Dynamic changes in DA neuron VGLUT2 expression also occur in the adult human brain as a neuroprotective response to insults. For example, in subjects with Parkinson’s disease, VGLUT2 expression is upregulated in surviving midbrain DA neurons89. Similar findings in rodent and Drosophila models show DA neurons that upregulate VGLUT are more resilient84, 85, 88, 90. Overall, these models show that temporal control over DA neuron VGLUT2 expression is highly regulated and conserved87, 88, 91–95. Such control is likely relevant to the overlapping circuitry between DA and glutamate systems since VGLUT2 expression increases DA axon arborization92. Subsets of VTA DA neurons that retain VGLUT2 expression through adulthood employ VGLUT2 to maintain the complex axonal branching necessary for synaptic communication. Conversely, disruption of VGLUT2’s regulatory program in DA neurons either in development or postnatally may lead to dysfunction in striatal circuits that rely on DA and glutamate neurotransmission. These data raise the question: what roles might co-transmission of DA and glutamate play in the context of schizophrenia?
Relevance of VGLUT2 and DA/glutamate co-transmission in schizophrenia
Important clues concerning the relevance of DA/glutamate co-release to schizophrenia have emerged in recent years based on work in Drosophila and mouse models. These studies suggest that VGLUT2-driven regional alterations in patterns of DA neuron activity and the associated DA release from striatal terminals may drive schizophrenia pathogenesis96. In Drosophila, we showed Drosophila VGLUT (dVGLUT) modulates activity-dependent vesicular DA loading and release, allowing neurons to tune presynaptic DA release to changes in cell firing97. These VGLUT-mediated effects are also evident in mouse midbrain DA neurons97, reflecting VGLUT’s importance in modulating DA release. The mechanism for this conserved phenomenon is based on VGLUT’s ability to hyperacidify DA synaptic vesicles (SVs) in response to increased activity via vesicular synergy96, 98. Since the vesicular pH gradient is the primary driving force for vesicular DA loading, VGLUT2-mediated vesicle hyperacidification elevates DA loading and increases release during neuronal depolarization97, 99, 100. Disrupting DA neuron VGLUT expression via conditional knockout (cKO) in mice or RNAi knockdown in flies significantly alters vesicular DA loading and psychostimulant-induced behaviors, emphasizing VGLUT’s relevance to DAergic neurotransmission81, 97, 101. Moreover, in Drosophila, dVGLUT functions in DA neurons as a rheostat of DAergic neurotransmission. When synaptic DA levels drop, DA neurons upregulate dVGLUT expression as a homeostatic compensatory response to maintain relatively stable DA neurotransmission85. While the Drosophila model has provided many insights, caution must be taken in directly extrapolating observations in the fly to human psychiatric disorders. The fly model lacks homologs of some vertebrate-specific pathogenic factors evident in schizophrenia such as DISC1102. It is also more difficult to model complex behaviors including those related to cognition and fly brain architecture differs from mammalian models. Finally, monoamine pharmacology differs between Drosophila and vertebrates, with flies substituting the trace amines octopamine and tyramine for norepinephrine. Together, these factors underscore potential drawbacks in modeling human disease in Drosophila and emphasize the need to validate findings from the fly with mammalian models.
Additional questions remain concerning VGLUT2’s localization in DA neurons. Initial studies in rat ventral striatum assessing SV colocalization of VGLUT2 and vesicular monoamine transporter 2 (VMAT2), which concentrates DA into SVs, demonstrated that most VMAT2+ immunoreactive vesicles possessed VGLUT2 in reciprocal immunoprecipitation experiments. These data suggested that VGLUT2 colocalizes with a discrete fraction of VMAT2+ vesicles, consistent with evidence that glutamate stimulates VMAT2-mediated uptake in rat SVs103. Subsequent studies in Drosophila similarly showed co-localization of Drosophila VMAT and dVGLUT at specific sites in adult fly brain97. However, other work in mice, rats, and hamsters found negligible co-localization between VGLUT2 and VMAT2 in midbrain DA neuron projections to striatum. Instead, these studies demonstrated that most glutamatergic and DAergic SVs segregate to separate release sites84, 104–106. Insights into reconciling these discrepancies come from new combined super-resolution imaging and immunolabeling methods that accurately resolve colocalization of vesicular transporters on single SVs107. These methods showed that VGLUT2+/VMAT2+ SVs comprise 3–4% of the total VMAT2+ SV pool107. Since these results used SV fractions from total rat brain homogenates, we posit that some of the transporter colocalization differences between studies may be related to species- and/or brain region-specificity where some species/brain regions are enriched in VGLUT2+/VMAT2+ SVs whereas other regions are sparser. Emergence of more sensitive methodologies enables future comprehensive mapping of this SV subpopulation in the context of schizophrenia, particularly in regions such as PFC where VGLUT2/VMAT2 vesicular colocalization has yet to be identified.
We propose that the co-release of DA and glutamate from the same neurons enables finely tuned integration of DAergic and glutamatergic signaling with exquisite temporal and spatial specificity. We also speculate that elevations in DA neuron VGLUT2 expression further boost striatal DA levels during periods of neuronal activity to contribute to positive symptoms (Figure 2). This agrees with work showing that VGLUT2 levels are elevated in the NAc of individuals with schizophrenia108–110. Furthermore, analyses examining single nucleotide polymorphisms (SNPs) within the VGLUT2 promoter and exons of VGLUT2 from genomic data of schizophrenia patients revealed 9 rare genetic variants in the patient group with none in controls111. This suggests that some of these SNPs may increase the genetic burden that contributes to schizophrenia pathogenesis. Additionally, in human postmortem brain studies, VGLUT2 transcript expression is elevated in the dorsal thalamus and inferior temporal gyrus in schizophrenia112, 113. These elevations in VGLUT2 expression are region-specific as the anterior cingulate cortex, dorsolateral prefrontal cortex, and hippocampus do not exhibit these expression differences versus controls114, 115. However, since these studies relied on brain homogenates to detect changes in VGLUT2 expression, the identities of the VGLUT2-expressing projections into these regions remains unclear. Future work will require in situ mapping of the pre- and postsynaptic circuitry within the intact respective brain regions.
Figure 2. Model for vesicular glutamate transporter 2 (VGLUT2)-mediated role in elevated striatal dopaminergic neurotransmission in schizophrenia.
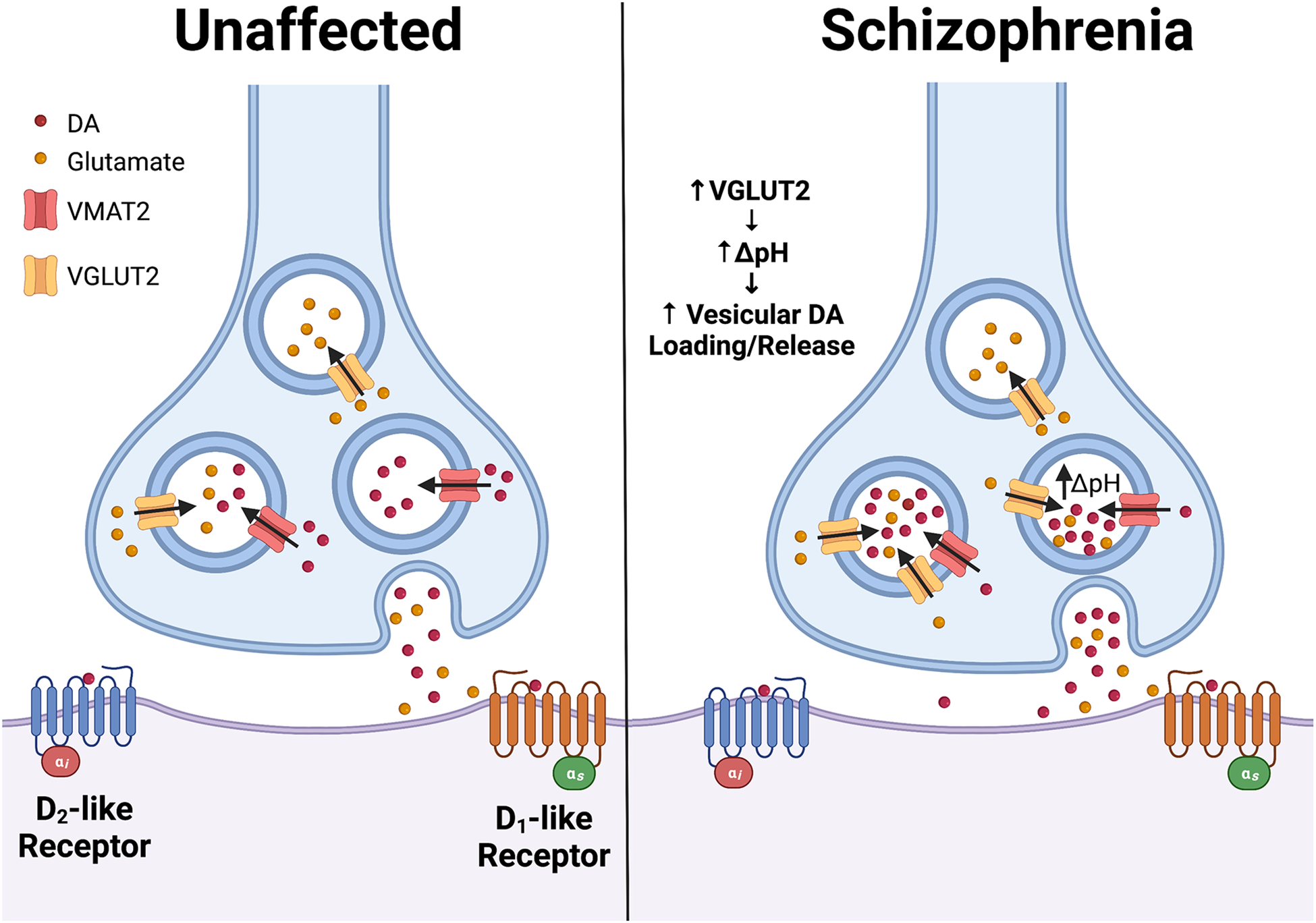
In addition to its role in loading glutamate into synaptic vesicles, VGLUT2 mediates activity-dependent vesicular hyperacidification in midbrain dopamine (DA) neurons that co-transmit glutamate. The resulting drop in DA vesicle pH increases the vesicular proton gradient (ΔpH), the main driving force for VMAT2-mediated DA vesicle loading. This mechanism confers the ability of neurons to tune vesicular DA loading in response to changes in neuronal activity. In schizophrenia, we propose that abnormal increases in VGLUT2 expression in midbrain DA neurons may raise activity-dependent hyperacidification by increasing ΔpH. The resulting elevation in vesicular DA loading and release boosts striatal DA levels, contributing to schizophrenia pathology including positive symptoms.
In addition to DA, striatal glutamate co-released by projections of VGLUT2+ VTA DA/glutamate neurons is relevant both physiologically and in the context of schizophrenia. While glutamatergic inputs to the VTA are well-studied, less is known about roles of glutamatergic inputs to the striatum, particularly in terms of their regulation of DA release and/or medium spiny neuron activation. Work in rodents and guinea pigs shows glutamate released from these inputs inhibits striatal DA release via indirect effects on nearby striatal medium spiny neurons (MSNs)116, 117. Glutamate released by the striatal glutamatergic inputs causes the following sequence of events: 1) AMPA receptor-mediated MSN activation which 2) increases mitochondrial production of membrane permeable H2O2, which 3) rapidly diffuses into adjacent DAergic projections where it inhibits DA release116–118. Glutamate released by striatal glutamatergic projections also drives burst firing of striatal cholinergic interneurons (ChI) via AMPA/kainate and NMDA receptor signaling, especially in the NAc medial shell. In mice, this glutamate-driven firing is important for behavioral plasticity that is pathologically impacted by drugs of abuse such as amphetamines119. Moreover, VGLUT2 expressed by VTA DA/glutamate neurons mediates this process since VGLUT2 cKO in DA/glutamate neurons attenuates subsequent ChI burst firing119. Similarly, glutamate released from VTA neurons that project to the NAc medial shell is important for positive reinforcement and in predicting responses to reward and reward-mediated cues120–122. Notably, even when DA co-release is blocked, the glutamate released from striatal projections of VTA DA/glutamate neurons promotes reinforcement122. This demonstrates a role for glutamate independent of concurrent DA co-release and further implicates glutamate as having its own novel roles in reward-seeking behaviors. In schizophrenia, though abnormalities in reward processing are classically associated with dysfunctional DAergic activity by VTA projections to the NAc and elevated DA levels, glutamate is increasingly shown to be a key modulator of these processes123, 124. Therefore, we speculate that alterations in glutamate release from VTA DA/glutamate neuronal projections to the NAc may contribute to mechanisms mediating reward-related abnormalities in schizophrenia. Given the distinct functional properties of DA and glutamate detailed above, this also raises the question of whether DA/glutamate neurons behave more like purely DAergic neurons versus glutamatergic neurons or have an entirely distinct identity which further work will need to disentangle.
In addition to their striatal projections, midbrain DA/glutamate neurons also project to forebrain regions, including the PFC86, 125. In tracing studies, cell bodies of DA/glutamate neurons are situated in specific VTA subdivisions, specifically the rostral VTA, the rostral linear nucleus of the raphe and the parabrachial pigmented nucleus126, 127. These cells’ projections elicit excitatory postsynaptic potentials in PFC GABAergic interneurons and cingulate cortical pyramidal neurons86, 128–130. This raises that possibility that altered DA/glutamate co-transmission in the forebrain leads to the dysfunctional interactions between DA and glutamate signaling that contributes to cognitive symptoms associated with schizophrenia.
Impairments in working memory are established cognitive symptoms in schizophrenia131, 132. Recent work in mouse models implicated DA/glutamate co-transmission in working memory deficits, showing that VGLUT2 cKO in DA neurons impairs spatial working memory and disrupts oscillatory activity in the hippocampal CA3 region133. Since loss of oscillatory synchrony between hippocampal and prefrontal regions is implicated in schizophrenia-related working memory deficits134, this offers a new pathological mechanism linking DA neuron VGLUT2-mediated deficits to altered coordination between hippocampal-prefrontal activity. Another key cognitive symptom is disruption in latent inhibition, the ability to discriminate cue saliency. Latent inhibition models attentional abnormalities in schizophrenia and in individuals at high risk for psychosis135–137. In DA/glutamate neurons, reducing expression of glutaminase, the enzyme responsible for glutamate biosynthesis, causes potentiation of latent inhibition and leads to a schizophrenia resilience phenotype in mice138. This suggests a role for DA neuron glutamate in hippocampus-mediated spatial memory, and raises the possibility that DA neuron glutamate biosynthesis may be impaired in schizophrenia139–144. Finally, while the relevance of DA/glutamate neuron projections to the PFC on PPI have yet to be directly investigated in human subjects, DA and glutamate systems in the PFC are known to play a major role in mediating deficits in PPI in rodent models of schizophrenia145–148. Altogether, reducing glutamatergic co-transmission in DA neurons may provide a novel therapeutic strategy to improve cognitive deficits in schizophrenia. Future work should therefore manipulate DA and glutamate co-transmission in this distinct neuronal subpopulation to target the cognitive symptoms in schizophrenia more effectively. However, to date, all studies of DA/glutamate co-release, VGLUT2 localization on DAergic vesicles and DA/glutamate vesicular synergy have been conducted in the VTA/NAc circuit. Thus, while DA/glutamate neurons are known to project to the PFC and cingulate cortex, the interplay between DA and glutamate transmission of DA/glutamate neurons in these regions is unknown and therefore merits future study.
Conclusions
The original DA hypothesis of schizophrenia asserts that alterations in the balance of DAergic neurotransmission in the mesolimbic and mesocortical pathways are responsible for positive and negative symptoms, respectively. This hypothesis has provided key insights into the circuitry and signaling associated with the positive symptoms of schizophrenia and their treatment with DA D2-like receptor antagonists. Yet, the DA hypothesis has been insufficient in providing a robust framework for understanding the mechanisms underlying negative or cognitive symptoms which often predominate. Instead, more recent preclinical and clinical studies have given way to a more complete picture – that the DA system extensively interacts with other brain systems including the glutamate system. Thus, disturbances in this DA/glutamate interplay may drive the development of not only positive but also negative and cognitive schizophrenia symptoms. Consistent with this, treatments primarily geared only to targets within the DA or glutamate systems have not been effective in improving all three core domains of schizophrenia. Rather, disruptions in the communications between these systems likely play significant roles in shaping the pathophysiology of schizophrenia.
Nevertheless, many questions remain concerning the roles of the DA and glutamatergic systems in schizophrenia biology. Foremost, it remains unclear whether alterations in striatal DA levels and signaling are the primary drivers of schizophrenia pathophysiology or are a downstream consequence of a dysfunctional glutamate system. Given the heterogeneity of schizophrenia, it is likely that both scenarios are correct. There may be a subset of individuals where primary defects in DA signaling drive the main symptoms of the illness. In other cases, alterations in the DA system may be downstream of other primary causes including within the glutamate system. Another outstanding question concerns identifying the precise circuits linking the DA and glutamate systems and their functional relevance. Lastly, the precise contributions of DA/glutamate co-releasing neurons to schizophrenia remain largely unknown. Therefore, a key aim of this Perspective is to raise the speculative hypothesis that DA/glutamate neurons may have a role in schizophrenia and thus motivate the field to actively pursue these important questions. Future work will offer critical new insights on these questions as well as provide new targets that lead to the development of new, more effective therapies for schizophrenia.
Acknowledgements
All figures were created with BioRender.com.
Funding
This work is supported by the National Institutes of Health F31NS118811 (SAB), R21AG068607 (ZF), R21DA052419 (ZF and RWL), R21AA028800 (ZF and RWL), and R01DK124219 (ZF).
Footnotes
Conflicts
The authors declare no competing financial interest.
References
- 1.Karam CS, Ballon JS, Bivens NM, Freyberg Z, Girgis RR, Lizardi-Ortiz JE et al. Signaling pathways in schizophrenia: emerging targets and therapeutic strategies. Trends Pharmacol Sci 2010; 31(8): 381–390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Lewis DA, Lieberman JA. Catching up on schizophrenia: natural history and neurobiology. Neuron 2000; 28(2): 325–334. [DOI] [PubMed] [Google Scholar]
- 3.MacKenzie NE, Kowalchuk C, Agarwal SM, Costa-Dookhan KA, Caravaggio F, Gerretsen P et al. Antipsychotics, Metabolic Adverse Effects, and Cognitive Function in Schizophrenia. Frontiers in psychiatry 2018; 9: 622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Newcomer JW. Metabolic considerations in the use of antipsychotic medications: a review of recent evidence. J Clin Psychiatry 2007; 68 Suppl 1: 20–27. [PubMed] [Google Scholar]
- 5.McEvoy JP, Meyer JM, Goff DC, Nasrallah HA, Davis SM, Sullivan L et al. Prevalence of the metabolic syndrome in patients with schizophrenia: baseline results from the Clinical Antipsychotic Trials of Intervention Effectiveness (CATIE) schizophrenia trial and comparison with national estimates from NHANES III. Schizophr Res 2005; 80(1): 19–32. [DOI] [PubMed] [Google Scholar]
- 6.Seeman P Antipsychotic drugs, dopamine receptors, and schizophrenia. Clinical Neuroscience Research 2001; 1(1): 53–60. [Google Scholar]
- 7.McCutcheon RA, Abi-Dargham A, Howes OD. Schizophrenia, Dopamine and the Striatum: From Biology to Symptoms. Trends Neurosci 2019; 42(3): 205–220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.McCutcheon R, Beck K, Jauhar S, Howes OD. Defining the Locus of Dopaminergic Dysfunction in Schizophrenia: A Meta-analysis and Test of the Mesolimbic Hypothesis. Schizophr Bull 2018; 44(6): 1301–1311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Brisch R, Saniotis A, Wolf R, Bielau H, Bernstein HG, Steiner J et al. The role of dopamine in schizophrenia from a neurobiological and evolutionary perspective: old fashioned, but still in vogue. Frontiers in psychiatry 2014; 5: 47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Bird ED, Spokes EG, Barnes J, MacKay AV, Iversen LL, Shepherd M. Increased brain dopamine and reduced glutamic acid decarboxylase and choline acetyl transferase activity in schizophrenia and related psychoses. Lancet 1977; 2(8049): 1157–1158. [DOI] [PubMed] [Google Scholar]
- 11.Howes OD, Montgomery AJ, Asselin MC, Murray RM, Valli I, Tabraham P et al. Elevated striatal dopamine function linked to prodromal signs of schizophrenia. Archives of general psychiatry 2009; 66(1): 13–20. [DOI] [PubMed] [Google Scholar]
- 12.Howes O, Bose S, Turkheimer F, Valli I, Egerton A, Stahl D et al. Progressive increase in striatal dopamine synthesis capacity as patients develop psychosis: a PET study. Mol Psychiatry 2011; 16(9): 885–886. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.McCutcheon RA, Krystal JH, Howes OD. Dopamine and glutamate in schizophrenia: biology, symptoms and treatment. World psychiatry : official journal of the World Psychiatric Association (WPA) 2020; 19(1): 15–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Featherstone RE, Kapur S, Fletcher PJ. The amphetamine-induced sensitized state as a model of schizophrenia. Progress in neuro-psychopharmacology & biological psychiatry 2007; 31(8): 1556–1571. [DOI] [PubMed] [Google Scholar]
- 15.Kellendonk C, Simpson EH, Polan HJ, Malleret G, Vronskaya S, Winiger V et al. Transient and selective overexpression of dopamine D2 receptors in the striatum causes persistent abnormalities in prefrontal cortex functioning. Neuron 2006; 49(4): 603–615. [DOI] [PubMed] [Google Scholar]
- 16.Schmack K, Bosc M, Ott T, Sturgill JF, Kepecs A. Striatal dopamine mediates hallucination-like perception in mice. Science 2021; 372(6537). [DOI] [PubMed] [Google Scholar]
- 17.Freyberg Z, Ferrando SJ, Javitch JA. Roles of the Akt/GSK-3 and Wnt signaling pathways in schizophrenia and antipsychotic drug action. Am J Psychiatry 2010; 167(4): 388–396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Krystal JH, Anticevic A, Yang GJ, Dragoi G, Driesen NR, Wang XJ et al. Impaired Tuning of Neural Ensembles and the Pathophysiology of Schizophrenia: A Translational and Computational Neuroscience Perspective. Biol Psychiatry 2017; 81(10): 874–885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Schobel SA, Chaudhury NH, Khan UA, Paniagua B, Styner MA, Asllani I et al. Imaging patients with psychosis and a mouse model establishes a spreading pattern of hippocampal dysfunction and implicates glutamate as a driver. Neuron 2013; 78(1): 81–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Javitt DC, Zukin SR, Heresco-Levy U, Umbricht D. Has an angel shown the way? Etiological and therapeutic implications of the PCP/NMDA model of schizophrenia. Schizophr Bull 2012; 38(5): 958–966. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Balu DT, Li Y, Puhl MD, Benneyworth MA, Basu AC, Takagi S et al. Multiple risk pathways for schizophrenia converge in serine racemase knockout mice, a mouse model of NMDA receptor hypofunction. Proc Natl Acad Sci U S A 2013; 110(26): E2400–2409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Robicsek O, Karry R, Petit I, Salman-Kesner N, Müller FJ, Klein E et al. Abnormal neuronal differentiation and mitochondrial dysfunction in hair follicle-derived induced pluripotent stem cells of schizophrenia patients. Mol Psychiatry 2013; 18(10): 1067–1076. [DOI] [PubMed] [Google Scholar]
- 23.Brennand KJ, Simone A, Jou J, Gelboin-Burkhart C, Tran N, Sangar S et al. Modelling schizophrenia using human induced pluripotent stem cells. Nature 2011; 473(7346): 221–225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Wen Z, Nguyen HN, Guo Z, Lalli MA, Wang X, Su Y et al. Synaptic dysregulation in a human iPS cell model of mental disorders. Nature 2014; 515(7527): 414–418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Hu W, MacDonald ML, Elswick DE, Sweet RA. The glutamate hypothesis of schizophrenia: evidence from human brain tissue studies. Ann N Y Acad Sci 2015; 1338(1): 38–57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Van Horn MR, Sild M, Ruthazer ES. D-serine as a gliotransmitter and its roles in brain development and disease. Front Cell Neurosci 2013; 7: 39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Hashimoto A, Oka T, Nishikawa T. Anatomical distribution and postnatal changes in endogenous free D-aspartate and D-serine in rat brain and periphery. The European journal of neuroscience 1995; 7(8): 1657–1663. [DOI] [PubMed] [Google Scholar]
- 28.Hashimoto A, Kumashiro S, Nishikawa T, Oka T, Takahashi K, Mito T et al. Embryonic development and postnatal changes in free D-aspartate and D-serine in the human prefrontal cortex. J Neurochem 1993; 61(1): 348–351. [DOI] [PubMed] [Google Scholar]
- 29.Schell MJ, Brady RO Jr., Molliver ME, Snyder SH. D-serine as a neuromodulator: regional and developmental localizations in rat brain glia resemble NMDA receptors. J Neurosci 1997; 17(5): 1604–1615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Kleckner NW, Dingledine R. Requirement for glycine in activation of NMDA-receptors expressed in Xenopus oocytes. Science 1988; 241(4867): 835–837. [DOI] [PubMed] [Google Scholar]
- 31.Mothet JP, Parent AT, Wolosker H, Brady RO Jr., Linden DJ, Ferris CD et al. D-serine is an endogenous ligand for the glycine site of the N-methyl-D-aspartate receptor. Proc Natl Acad Sci U S A 2000; 97(9): 4926–4931. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Papouin T, Ladepeche L, Ruel J, Sacchi S, Labasque M, Hanini M et al. Synaptic and extrasynaptic NMDA receptors are gated by different endogenous coagonists. Cell 2012; 150(3): 633–646. [DOI] [PubMed] [Google Scholar]
- 33.MacKay MB, Kravtsenyuk M, Thomas R, Mitchell ND, Dursun SM, Baker GB. D-Serine: Potential Therapeutic Agent and/or Biomarker in Schizophrenia and Depression? Front Psychiatry 2019; 10: 25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Fossat P, Turpin FR, Sacchi S, Dulong J, Shi T, Rivet JM et al. Glial D-serine gates NMDA receptors at excitatory synapses in prefrontal cortex. Cereb Cortex 2012; 22(3): 595–606. [DOI] [PubMed] [Google Scholar]
- 35.Morita Y, Ujike H, Tanaka Y, Otani K, Kishimoto M, Morio A et al. A genetic variant of the serine racemase gene is associated with schizophrenia. Biol Psychiatry 2007; 61(10): 1200–1203. [DOI] [PubMed] [Google Scholar]
- 36.Verrall L, Burnet PW, Betts JF, Harrison PJ. The neurobiology of D-amino acid oxidase and its involvement in schizophrenia. Mol Psychiatry 2010; 15(2): 122–137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Chouinard ML, Gaitan D, Wood PL. Presence of the N-methyl-D-aspartate-associated glycine receptor agonist, D-serine, in human temporal cortex: comparison of normal, Parkinson, and Alzheimer tissues. J Neurochem 1993; 61(4): 1561–1564. [DOI] [PubMed] [Google Scholar]
- 38.Howes O, McCutcheon R, Stone J. Glutamate and dopamine in schizophrenia: an update for the 21st century. J Psychopharmacol 2015; 29(2): 97–115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Javitt DC, Zukin SR. Recent advances in the phencyclidine model of schizophrenia. Am J Psychiatry 1991; 148(10): 1301–1308. [DOI] [PubMed] [Google Scholar]
- 40.Tsai GE, Lin PY. Strategies to enhance N-methyl-D-aspartate receptor-mediated neurotransmission in schizophrenia, a critical review and meta-analysis. Current pharmaceutical design 2010; 16(5): 522–537. [DOI] [PubMed] [Google Scholar]
- 41.Weiser M, Heresco-Levy U, Davidson M, Javitt DC, Werbeloff N, Gershon AA et al. A multicenter, add-on randomized controlled trial of low-dose d-serine for negative and cognitive symptoms of schizophrenia. J Clin Psychiatry 2012; 73(6): e728–734. [DOI] [PubMed] [Google Scholar]
- 42.Patil ST, Zhang L, Martenyi F, Lowe SL, Jackson KA, Andreev BV et al. Activation of mGlu2/3 receptors as a new approach to treat schizophrenia: a randomized Phase 2 clinical trial. Nature medicine 2007; 13(9): 1102–1107. [DOI] [PubMed] [Google Scholar]
- 43.Kinon BJ, Zhang L, Millen BA, Osuntokun OO, Williams JE, Kollack-Walker S et al. A multicenter, inpatient, phase 2, double-blind, placebo-controlled dose-ranging study of LY2140023 monohydrate in patients with DSM-IV schizophrenia. Journal of clinical psychopharmacology 2011; 31(3): 349–355. [DOI] [PubMed] [Google Scholar]
- 44.Sesack SR, Carr DB, Omelchenko N, Pinto A. Anatomical substrates for glutamate-dopamine interactions: evidence for specificity of connections and extrasynaptic actions. Ann N Y Acad Sci 2003; 1003: 36–52. [DOI] [PubMed] [Google Scholar]
- 45.Tseng KY, O’Donnell P. Dopamine-glutamate interactions controlling prefrontal cortical pyramidal cell excitability involve multiple signaling mechanisms. J Neurosci 2004; 24(22): 5131–5139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Del Arco A, Mora F. Prefrontal cortex-nucleus accumbens interaction: in vivo modulation by dopamine and glutamate in the prefrontal cortex. Pharmacol Biochem Behav 2008; 90(2): 226–235. [DOI] [PubMed] [Google Scholar]
- 47.Gleich T, Deserno L, Lorenz RC, Boehme R, Pankow A, Buchert R et al. Prefrontal and Striatal Glutamate Differently Relate to Striatal Dopamine: Potential Regulatory Mechanisms of Striatal Presynaptic Dopamine Function? J Neurosci 2015; 35(26): 9615–9621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Frankle WG, Laruelle M, Haber SN. Prefrontal cortical projections to the midbrain in primates: evidence for a sparse connection. Neuropsychopharmacology 2006; 31(8): 1627–1636. [DOI] [PubMed] [Google Scholar]
- 49.Brisch R, Bernstein HG, Krell D, Dobrowolny H, Bielau H, Steiner J et al. Dopamine-glutamate abnormalities in the frontal cortex associated with the catechol-O-methyltransferase (COMT) in schizophrenia. Brain Res 2009; 1269: 166–175. [DOI] [PubMed] [Google Scholar]
- 50.Shah UH, Gonzalez-Maeso J. Serotonin and Glutamate Interactions in Preclinical Schizophrenia Models. ACS Chem Neurosci 2019; 10(7): 3068–3077. [DOI] [PubMed] [Google Scholar]
- 51.de Bartolomeis A, Buonaguro EF, Iasevoli F. Serotonin-glutamate and serotonin-dopamine reciprocal interactions as putative molecular targets for novel antipsychotic treatments: from receptor heterodimers to postsynaptic scaffolding and effector proteins. Psychopharmacology (Berl) 2013; 225(1): 1–19. [DOI] [PubMed] [Google Scholar]
- 52.López-Gil X, Artigas F, Adell A. Unraveling monoamine receptors involved in the action of typical and atypical antipsychotics on glutamatergic and serotonergic transmission in prefrontal cortex. Current pharmaceutical design 2010; 16(5): 502–515. [DOI] [PubMed] [Google Scholar]
- 53.Field JR, Walker AG, Conn PJ. Targeting glutamate synapses in schizophrenia. Trends Mol Med 2011; 17(12): 689–698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Timofeeva OA, Levin ED. Glutamate and nicotinic receptor interactions in working memory: importance for the cognitive impairment of schizophrenia. Neuroscience 2011; 195: 21–36. [DOI] [PubMed] [Google Scholar]
- 55.Yang AC, Tsai SJ. New Targets for Schizophrenia Treatment beyond the Dopamine Hypothesis. International journal of molecular sciences 2017; 18(8). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Hamilton HK, D’Souza DC, Ford JM, Roach BJ, Kort NS, Ahn KH et al. Interactive effects of an N-methyl-d-aspartate receptor antagonist and a nicotinic acetylcholine receptor agonist on mismatch negativity: Implications for schizophrenia. Schizophr Res 2018; 191: 87–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Schwartz TL, Sachdeva S, Stahl SM. Glutamate neurocircuitry: theoretical underpinnings in schizophrenia. Frontiers in pharmacology 2012; 3: 195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Schwartz TL, Sachdeva S, Stahl SM. Genetic data supporting the NMDA glutamate receptor hypothesis for schizophrenia. Current pharmaceutical design 2012; 18(12): 1580–1592. [DOI] [PubMed] [Google Scholar]
- 59.Lisman JE, Coyle JT, Green RW, Javitt DC, Benes FM, Heckers S et al. Circuit-based framework for understanding neurotransmitter and risk gene interactions in schizophrenia. Trends Neurosci 2008; 31(5): 234–242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Dallérac G, Li X, Lecouflet P, Morisot N, Sacchi S, Asselot R et al. Dopaminergic neuromodulation of prefrontal cortex activity requires the NMDA receptor coagonist d-serine. Proc Natl Acad Sci U S A 2021; 118(23). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Robbins TW, Arnsten AF. The neuropsychopharmacology of fronto-executive function: monoaminergic modulation. Annual review of neuroscience 2009; 32: 267–287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Clarkson RL, Liptak AT, Gee SM, Sohal VS, Bender KJ. D3 Receptors Regulate Excitability in a Unique Class of Prefrontal Pyramidal Cells. J Neurosci 2017; 37(24): 5846–5860. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Seamans JK, Yang CR. The principal features and mechanisms of dopamine modulation in the prefrontal cortex. Prog Neurobiol 2004; 74(1): 1–58. [DOI] [PubMed] [Google Scholar]
- 64.Cools R, D’Esposito M. Inverted-U-shaped dopamine actions on human working memory and cognitive control. Biol Psychiatry 2011; 69(12): e113–125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Yamamori H, Hashimoto R, Fujita Y, Numata S, Yasuda Y, Fujimoto M et al. Changes in plasma D-serine, L-serine, and glycine levels in treatment-resistant schizophrenia before and after clozapine treatment. Neurosci Lett 2014; 582: 93–98. [DOI] [PubMed] [Google Scholar]
- 66.Takeuchi S, Hida H, Uchida M, Naruse R, Yoshimi A, Kitagaki S et al. Blonanserin ameliorates social deficit through dopamine-D3 receptor antagonism in mice administered phencyclidine as an animal model of schizophrenia. Neurochem Int 2019; 128: 127–134. [DOI] [PubMed] [Google Scholar]
- 67.Calabrese F, Tarazi FI, Racagni G, Riva MA. The role of dopamine D3 receptors in the mechanism of action of cariprazine. CNS Spectr 2020; 25(3): 343–351. [DOI] [PubMed] [Google Scholar]
- 68.Kätzel D, Wolff AR, Bygrave AM, Bannerman DM. Hippocampal Hyperactivity as a Druggable Circuit-Level Origin of Aberrant Salience in Schizophrenia. Frontiers in pharmacology 2020; 11: 486811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Wolff AR, Bygrave AM, Sanderson DJ, Boyden ES, Bannerman DM, Kullmann DM et al. Optogenetic induction of the schizophrenia-related endophenotype of ventral hippocampal hyperactivity causes rodent correlates of positive and cognitive symptoms. Sci Rep 2018; 8(1): 12871. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Nguyen R, Morrissey MD, Mahadevan V, Cajanding JD, Woodin MA, Yeomans JS et al. Parvalbumin and GAD65 interneuron inhibition in the ventral hippocampus induces distinct behavioral deficits relevant to schizophrenia. J Neurosci 2014; 34(45): 14948–14960. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Chun S, Westmoreland JJ, Bayazitov IT, Eddins D, Pani AK, Smeyne RJ et al. Specific disruption of thalamic inputs to the auditory cortex in schizophrenia models. Science 2014; 344(6188): 1178–1182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Kokkinou M, Ashok AH, Howes OD. The effects of ketamine on dopaminergic function: meta-analysis and review of the implications for neuropsychiatric disorders. Mol Psychiatry 2018; 23(1): 59–69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.White TL, Monnig MA, Walsh EG, Nitenson AZ, Harris AD, Cohen RA et al. Psychostimulant drug effects on glutamate, Glx, and creatine in the anterior cingulate cortex and subjective response in healthy humans. Neuropsychopharmacology 2018; 43(7): 1498–1509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Dluzen DE, Liu B. Gender differences in methamphetamine use and responses: a review. Gend Med 2008; 5(1): 24–35. [DOI] [PubMed] [Google Scholar]
- 75.Cohen JB, Greenberg R, Uri J, Halpin M, Zweben JE. Women with methamphetamine dependence: research on etiology and treatment. J Psychoactive Drugs 2007; Suppl 4: 347–351. [DOI] [PubMed] [Google Scholar]
- 76.Sulzer D, Rayport S. Dale’s principle and glutamate corelease from ventral midbrain dopamine neurons. Amino Acids 2000; 19(1): 45–52. [DOI] [PubMed] [Google Scholar]
- 77.Hnasko TS, Edwards RH. Neurotransmitter corelease: mechanism and physiological role. Annu Rev Physiol 2012; 74: 225–243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Trudeau LE, Hnasko TS, Wallen-Mackenzie A, Morales M, Rayport S, Sulzer D. The multilingual nature of dopamine neurons. Prog Brain Res 2014; 211: 141–164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Kouwenhoven WM, Fortin G, Penttinen AM, Florence C, Delignat-Lavaud B, Bourque MJ et al. VGluT2 Expression in Dopamine Neurons Contributes to Postlesional Striatal Reinnervation. J Neurosci 2020; 40(43): 8262–8275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Dal Bo G, Berube-Carriere N, Mendez JA, Leo D, Riad M, Descarries L et al. Enhanced glutamatergic phenotype of mesencephalic dopamine neurons after neonatal 6-hydroxydopamine lesion. Neuroscience 2008; 156(1): 59–70. [DOI] [PubMed] [Google Scholar]
- 81.Mingote S, Amsellem A, Kempf A, Rayport S, Chuhma N. Dopamine-glutamate neuron projections to the nucleus accumbens medial shell and behavioral switching. Neurochem Int 2019; 129: 104482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Chuhma N, Choi WY, Mingote S, Rayport S. Dopamine neuron glutamate cotransmission: frequency-dependent modulation in the mesoventromedial projection. Neuroscience 2009; 164(3): 1068–1083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Root DH, Wang HL, Liu B, Barker DJ, Mod L, Szocsics P et al. Glutamate neurons are intermixed with midbrain dopamine neurons in nonhuman primates and humans. Scientific reports 2016; 6: 30615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Buck SA, Miranda BR, Logan RW, Fish KN, Greenamyre JT, Freyberg Z. VGLUT2 is a determinant of dopamine neuron resilience in a rotenone model of dopamine neurodegeneration. J Neurosci 2021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Buck SA, Steinkellner T, Aslanoglou D, Villeneuve M, Bhatte SH, Childers VC et al. Vesicular glutamate transporter modulates sex differences in dopamine neuron vulnerability to age-related neurodegeneration. Aging Cell 2021: e13365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Eskenazi D, Malave L, Mingote S, Yetnikoff L, Ztaou S, Velicu V et al. Dopamine Neurons That Cotransmit Glutamate, From Synapses to Circuits to Behavior. Frontiers in neural circuits 2021; 15: 665386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Dumas S, Wallen-Mackenzie A. Developmental Co-expression of Vglut2 and Nurr1 in a Mes-Di-Encephalic Continuum Preceeds Dopamine and Glutamate Neuron Specification. Front Cell Dev Biol 2019; 7: 307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Steinkellner T, Zell V, Farino ZJ, Sonders MS, Villeneuve M, Freyberg RJ et al. Role for VGLUT2 in selective vulnerability of midbrain dopamine neurons. J Clin Invest 2018; 128(2): 774–788. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Steinkellner T, Conrad WS, Kovacs I, Rissman RA, Lee EB, Trojanowski JQ et al. Dopamine neurons exhibit emergent glutamatergic identity in Parkinson’s disease. Brain 2021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Shen H, Marino RAM, McDevitt RA, Bi GH, Chen K, Madeo G et al. Genetic deletion of vesicular glutamate transporter in dopamine neurons increases vulnerability to MPTP-induced neurotoxicity in mice. Proc Natl Acad Sci U S A 2018; 115(49): E11532–E11541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Papathanou M, Creed M, Dorst MC, Bimpisidis Z, Dumas S, Pettersson H et al. Targeting VGLUT2 in Mature Dopamine Neurons Decreases Mesoaccumbal Glutamatergic Transmission and Identifies a Role for Glutamate Co-release in Synaptic Plasticity by Increasing Baseline AMPA/NMDA Ratio. Frontiers in neural circuits 2018; 12: 64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Kouwenhoven WM, Fortin G, Penttinen AM, Florence C, Delignat-Lavaud B, Bourque MJ et al. VGluT2 Expression in Dopamine Neurons Contributes to Postlesional Striatal Reinnervation. J Neurosci 2020; 40(43): 8262–8275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Berube-Carriere N, Riad M, Dal Bo G, Levesque D, Trudeau LE, Descarries L. The dual dopamine-glutamate phenotype of growing mesencephalic neurons regresses in mature rat brain. The Journal of comparative neurology 2009; 517(6): 873–891. [DOI] [PubMed] [Google Scholar]
- 94.Fougere M, van der Zouwen CI, Boutin J, Ryczko D. Heterogeneous expression of dopaminergic markers and Vglut2 in mouse mesodiencephalic dopaminergic nuclei A8–A13. The Journal of comparative neurology 2021; 529(7): 1273–1292. [DOI] [PubMed] [Google Scholar]
- 95.Mendez JA, Bourque MJ, Dal Bo G, Bourdeau ML, Danik M, Williams S et al. Developmental and target-dependent regulation of vesicular glutamate transporter expression by dopamine neurons. J Neurosci 2008; 28(25): 6309–6318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Chuhma N, Mingote S, Kalmbach A, Yetnikoff L, Rayport S. Heterogeneity in Dopamine Neuron Synaptic Actions Across the Striatum and Its Relevance for Schizophrenia. Biol Psychiatry 2017; 81(1): 43–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Aguilar JI, Dunn M, Mingote S, Karam CS, Farino ZJ, Sonders MS et al. Neuronal Depolarization Drives Increased Dopamine Synaptic Vesicle Loading via VGLUT. Neuron 2017; 95(5): 1074–1088 e1077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.El Mestikawy S, Wallen-Mackenzie A, Fortin GM, Descarries L, Trudeau LE. From glutamate co-release to vesicular synergy: vesicular glutamate transporters. Nat Rev Neurosci 2011; 12(4): 204–216. [DOI] [PubMed] [Google Scholar]
- 99.Freyberg Z, Sonders MS, Aguilar JI, Hiranita T, Karam CS, Flores J et al. Mechanisms of amphetamine action illuminated through optical monitoring of dopamine synaptic vesicles in Drosophila brain. Nat Commun 2016; 7: 10652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Johnson RG Jr. Accumulation of biological amines into chromaffin granules: a model for hormone and neurotransmitter transport. Physiol Rev 1988; 68(1): 232–307. [DOI] [PubMed] [Google Scholar]
- 101.Bimpisidis Z, Wallen-Mackenzie A. Neurocircuitry of Reward and Addiction: Potential Impact of Dopamine-Glutamate Co-release as Future Target in Substance Use Disorder. J Clin Med 2019; 8(11). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Shao L, Lu B, Wen Z, Teng S, Wang L, Zhao Y et al. Disrupted-in-Schizophrenia-1 (DISC1) protein disturbs neural function in multiple disease-risk pathways. Hum Mol Genet 2017; 26(14): 2634–2648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Hnasko TS, Chuhma N, Zhang H, Goh GY, Sulzer D, Palmiter RD et al. Vesicular glutamate transport promotes dopamine storage and glutamate corelease in vivo. Neuron 2010; 65(5): 643–656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Silm K, Yang J, Marcott PF, Asensio CS, Eriksen J, Guthrie DA et al. Synaptic Vesicle Recycling Pathway Determines Neurotransmitter Content and Release Properties. Neuron 2019; 102(4): 786–800 e785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Zhang S, Qi J, Li X, Wang HL, Britt JP, Hoffman AF et al. Dopaminergic and glutamatergic microdomains in a subset of rodent mesoaccumbens axons. Nat Neurosci 2015; 18(3): 386–392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Been LE, Staffend NA, Tucker A, Meisel RL. Vesicular glutamate transporter 2 and tyrosine hydroxylase are not co-localized in Syrian hamster nucleus accumbens afferents. Neurosci Lett 2013; 550: 41–45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Upmanyu N, Jin J, Emde HV, Ganzella M, Bosche L, Malviya VN et al. Colocalization of different neurotransmitter transporters on synaptic vesicles is sparse except for VGLUT1 and ZnT3. Neuron 2022. [DOI] [PubMed] [Google Scholar]
- 108.Schoonover KE, McCollum LA, Roberts RC. Protein Markers of Neurotransmitter Synthesis and Release in Postmortem Schizophrenia Substantia Nigra. Neuropsychopharmacology 2017; 42(2): 540–550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Roberts RC, McCollum LA, Schoonover KE, Mabry SJ, Roche JK, Lahti AC. Ultrastructural evidence for glutamatergic dysregulation in schizophrenia. Schizophr Res 2020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.McCollum LA, Roberts RC. Uncovering the role of the nucleus accumbens in schizophrenia: A postmortem analysis of tyrosine hydroxylase and vesicular glutamate transporters. Schizophr Res 2015; 169(1–3): 369–373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Shen YC, Liao DL, Lu CL, Chen JY, Liou YJ, Chen TT et al. Resequencing of the vesicular glutamate transporter 2 gene (VGLUT2) reveals some rare genetic variants that may increase the genetic burden in schizophrenia. Schizophr Res 2010; 121(1–3): 179–186. [DOI] [PubMed] [Google Scholar]
- 112.Smith RE, Haroutunian V, Davis KL, Meador-Woodruff JH. Vesicular glutamate transporter transcript expression in the thalamus in schizophrenia. Neuroreport 2001; 12(13): 2885–2887. [DOI] [PubMed] [Google Scholar]
- 113.Uezato A, Meador-Woodruff JH, McCullumsmith RE. Vesicular glutamate transporter mRNA expression in the medial temporal lobe in major depressive disorder, bipolar disorder, and schizophrenia. Bipolar Disord 2009; 11(7): 711–725. [DOI] [PubMed] [Google Scholar]
- 114.Oni-Orisan A, Kristiansen LV, Haroutunian V, Meador-Woodruff JH, McCullumsmith RE. Altered vesicular glutamate transporter expression in the anterior cingulate cortex in schizophrenia. Biol Psychiatry 2008; 63(8): 766–775. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.De Rosa A, Fontana A, Nuzzo T, Garofalo M, Di Maio AD, Punzo D et al. Machine Learning algorithm unveils glutamatergic alterations in the post-mortem schizophrenia brain. Schizophr 2022; 8(8). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Sulzer D, Cragg SJ, Rice ME. Striatal dopamine neurotransmission: regulation of release and uptake. Basal ganglia 2016; 6(3): 123–148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Lee CR, Patel JC, O’Neill B, Rice ME. Inhibitory and excitatory neuromodulation by hydrogen peroxide: translating energetics to information. J Physiol 2015; 593(16): 3431–3446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Avshalumov MV, Patel JC, Rice ME. AMPA receptor-dependent H2O2 generation in striatal medium spiny neurons but not dopamine axons: one source of a retrograde signal that can inhibit dopamine release. Journal of neurophysiology 2008; 100(3): 1590–1601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Chuhma N, Mingote S, Moore H, Rayport S. Dopamine neurons control striatal cholinergic neurons via regionally heterogeneous dopamine and glutamate signaling. Neuron 2014; 81(4): 901–912. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Root DH, Estrin DJ, Morales M. Aversion or Salience Signaling by Ventral Tegmental Area Glutamate Neurons. iScience 2018; 2: 51–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Root DH, Barker DJ, Estrin DJ, Miranda-Barrientos JA, Liu B, Zhang S et al. Distinct Signaling by Ventral Tegmental Area Glutamate, GABA, and Combinatorial Glutamate-GABA Neurons in Motivated Behavior. Cell Rep 2020; 32(9): 108094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Zell V, Steinkellner T, Hollon NG, Warlow SM, Souter E, Faget L et al. VTA Glutamate Neuron Activity Drives Positive Reinforcement Absent Dopamine Co-release. Neuron 2020; 107(5): 864–873 e864. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Bossong MG, Wilson R, Appiah-Kusi E, McGuire P, Bhattacharyya S. Human Striatal Response to Reward Anticipation Linked to Hippocampal Glutamate Levels. Int J Neuropsychopharmacol 2018; 21(7): 623–630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Schultz W Neuronal Reward and Decision Signals: From Theories to Data. Physiol Rev 2015; 95(3): 853–951. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Poulin JF, Caronia G, Hofer C, Cui Q, Helm B, Ramakrishnan C et al. Mapping projections of molecularly defined dopamine neuron subtypes using intersectional genetic approaches. Nat Neurosci 2018; 21(9): 1260–1271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Gorelova N, Mulholland PJ, Chandler LJ, Seamans JK. The glutamatergic component of the mesocortical pathway emanating from different subregions of the ventral midbrain. Cerebral cortex (New York, NY : 1991) 2012; 22(2): 327–336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Li X, Qi J, Yamaguchi T, Wang HL, Morales M. Heterogeneous composition of dopamine neurons of the rat A10 region: molecular evidence for diverse signaling properties. Brain Struct Funct 2013; 218(5): 1159–1176. [DOI] [PubMed] [Google Scholar]
- 128.Mingote S, Chuhma N, Kusnoor SV, Field B, Deutch AY, Rayport S. Functional Connectome Analysis of Dopamine Neuron Glutamatergic Connections in Forebrain Regions. J Neurosci 2015; 35(49): 16259–16271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Kabanova A, Pabst M, Lorkowski M, Braganza O, Boehlen A, Nikbakht N et al. Function and developmental origin of a mesocortical inhibitory circuit. Nat Neurosci 2015; 18(6): 872–882. [DOI] [PubMed] [Google Scholar]
- 130.Perez-Lopez JL, Contreras-Lopez R, Ramirez-Jarquin JO, Tecuapetla F. Direct Glutamatergic Signaling From Midbrain Dopaminergic Neurons Onto Pyramidal Prefrontal Cortex Neurons. Frontiers in neural circuits 2018; 12: 70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Kahn RS, Keefe RS. Schizophrenia is a cognitive illness: time for a change in focus. JAMA psychiatry 2013; 70(10): 1107–1112. [DOI] [PubMed] [Google Scholar]
- 132.Guo JY, Ragland JD, Carter CS. Memory and cognition in schizophrenia. Mol Psychiatry 2019; 24(5): 633–642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Nordenankar K, Smith-Anttila CJ, Schweizer N, Viereckel T, Birgner C, Mejia-Toiber J et al. Increased hippocampal excitability and impaired spatial memory function in mice lacking VGLUT2 selectively in neurons defined by tyrosine hydroxylase promoter activity. Brain Struct Funct 2015; 220(4): 2171–2190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Speers LJ, Bilkey DK. Disorganization of Oscillatory Activity in Animal Models of Schizophrenia. Front Neural Circuits 2021; 15: 741767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Weiner I, Arad M. Using the pharmacology of latent inhibition to model domains of pathology in schizophrenia and their treatment. Behav Brain Res 2009; 204(2): 369–386. [DOI] [PubMed] [Google Scholar]
- 136.Lodge DJ, Behrens MM, Grace AA. A loss of parvalbumin-containing interneurons is associated with diminished oscillatory activity in an animal model of schizophrenia. J Neurosci 2009; 29(8): 2344–2354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Kraus M, Rapisarda A, Lam M, Thong JYJ, Lee J, Subramaniam M et al. Disrupted latent inhibition in individuals at ultra high-risk for developing psychosis. Schizophr Res Cogn 2016; 6: 1–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Mingote S, Chuhma N, Kalmbach A, Thomsen GM, Wang Y, Mihali A et al. Dopamine neuron dependent behaviors mediated by glutamate cotransmission. Elife 2017; 6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Arnold SJ, Ivleva EI, Gopal TA, Reddy AP, Jeon-Slaughter H, Sacco CB et al. Hippocampal volume is reduced in schizophrenia and schizoaffective disorder but not in psychotic bipolar I disorder demonstrated by both manual tracing and automated parcellation (FreeSurfer). Schizophr Bull 2015; 41(1): 233–249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Bobilev AM, Perez JM, Tamminga CA. Molecular alterations in the medial temporal lobe in schizophrenia. Schizophr Res 2019. [DOI] [PubMed] [Google Scholar]
- 141.Heckers S, Rauch SL, Goff D, Savage CR, Schacter DL, Fischman AJ et al. Impaired recruitment of the hippocampus during conscious recollection in schizophrenia. Nat Neurosci 1998; 1(4): 318–323. [DOI] [PubMed] [Google Scholar]
- 142.Li W, Ghose S, Gleason K, Begovic A, Perez J, Bartko J et al. Synaptic proteins in the hippocampus indicative of increased neuronal activity in CA3 in schizophrenia. Am J Psychiatry 2015; 172(4): 373–382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Steen RG, Mull C, McClure R, Hamer RM, Lieberman JA. Brain volume in first-episode schizophrenia: systematic review and meta-analysis of magnetic resonance imaging studies. Br J Psychiatry 2006; 188: 510–518. [DOI] [PubMed] [Google Scholar]
- 144.Tamminga CA, Stan AD, Wagner AD. The hippocampal formation in schizophrenia. Am J Psychiatry 2010; 167(10): 1178–1193. [DOI] [PubMed] [Google Scholar]
- 145.Brown RW, Varnum CG, Wills LJ, Peeters LD, Gass JT. Modulation of mGlu5 improves sensorimotor gating deficits in rats neonatally treated with quinpirole through changes in dopamine D2 signaling. Pharmacol Biochem Behav 2021; 211: 173292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Yan L, Shamir A, Skirzewski M, Leiva-Salcedo E, Kwon OB, Karavanova I et al. Neuregulin-2 ablation results in dopamine dysregulation and severe behavioral phenotypes relevant to psychiatric disorders. Mol Psychiatry 2018; 23(5): 1233–1243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Didriksen M, Fejgin K, Nilsson SR, Birknow MR, Grayton HM, Larsen PH et al. Persistent gating deficit and increased sensitivity to NMDA receptor antagonism after puberty in a new mouse model of the human 22q11.2 microdeletion syndrome: a study in male mice. J Psychiatry Neurosci 2017; 42(1): 48–58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Heidbreder CA, Foxton R, Cilia J, Hughes ZA, Shah AJ, Atkins A et al. Increased responsiveness of dopamine to atypical, but not typical antipsychotics in the medial prefrontal cortex of rats reared in isolation. Psychopharmacology (Berl) 2001; 156(2–3): 338–351. [DOI] [PubMed] [Google Scholar]