

Abstract
Ischemia-reperfusion injury is an inevitable event during organ transplantation and represents a primary risk factor for the development of early graft dysfunction in lung, heart, liver, and kidney transplant recipients. Recent studies have implicated recipient neutrophils as key mediators of this process and have also found that early innate immune responses after transplantation can ultimately augment adaptive alloimmunity and impact late graft outcomes. Here, we discuss signaling pathways involved in neutrophil recruitment and activation after ischemia-mediated graft injury in solid organ transplantation with an emphasis on lung allografts, which have been the focus of recent studies. These findings suggest novel therapeutic interventions that target ischemia-reperfusion injury-mediated graft dysfunction in transplant recipients.
INTRODUCTION
The interruption of blood flow is inherent to organ transplantation and often followed by prolonged cold ischemic storage during transport to the recipient. Once blood flow is restored, allograft tissues become enriched in oxygen and substrates that exaggerate the ischemic injury, a phenomenon known as ischemia-reperfusion injury (IRI). Early graft dysfunction after transplantation is the clinical presentation of molecular and cellular events largely induced by IRI and encompasses a wide variety of terms, including primary graft dysfunction (PGD), early graft failure, primary dysfunction, primary nonfunction, initial poor function, reimplantation response and delayed graft function. Throughout this review, we will refer to the syndrome of early graft dysfunction after lung, heart, liver, or kidney transplantation as PGD.
When IRI is assessed by histologic markers (e.g. neutrophilic infiltration) in human biopsies of transplanted organs, approximately 40–50% of lung and kidney allografts and 87% of liver allografts show evidence of IRI within the first 3 hours after reperfusion1–3. This early inflammatory response significantly impacts graft viability and is evidenced clinically by early graft dysfunction in 20–30% of all transplanted lungs, hearts, livers, and kidneys2–12. PGD can adversely affect short-term post-transplant outcomes and remains the leading cause for early mortality following lung and heart transplantation6,13,14. Mortality following the development of PGD after kidney or liver transplantation is often circumvented by re-initiation of dialysis or immediate hepatic re-transplantation, with an estimated 2–6% liver transplant recipients requiring early re-transplantation15–19. Furthermore, PGD is associated with reduced long-term allograft function and recipient survival, and is a risk factor for rejection after lung, heart, liver, and kidney transplantation6,9,20–28.
While much remains to be discovered regarding the cellular and molecular pathways underlying IRI, recipient neutrophils have been shown to play a central role in this process29,30. Multiple studies in both human and animal models have demonstrated that IRI-mediated injury is associated with neutrophilic infiltration into allografts and inhibition of this process may protect against post-transplant IRI30–34. In this review, we synthesize an expanding body of experimental findings on neutrophil recruitment, extravasation and activation following solid organ transplantation with a focus on pulmonary transplantation. These studies pave the way for future investigations and novel strategies to improve allograft function following solid organ transplantation.
Neutrophil Recruitment
In the setting of IRI, recipient neutrophils are recruited to newly reperfused tissues through numerous cellular and molecular pathways that differ between organ systems. Novel techniques of in vivo multiphoton microscopy have allowed visualization of neutrophil infiltration in various animal models of IRI, including mouse lung, heart, and kidney transplantation, as well as mouse models of warm ischemia induced by transient interruption of arterial flow to the lung, kidney, and liver31,34–44. Following syngeneic or allogeneic lung transplantation in mice, recipient neutrophils are rapidly recruited to the vessels within the graft, many of which extravasate during the first 2 to 3 hours after reperfusion38,39. Similarly, rapid recruitment of neutrophils has also been observed by intravital two-photon microscopy after syngeneic heart transplantation in mice, where neutrophils slow down, adhere to the walls of large coronary veins, and subsequently enter the myocardial tissue where they form clusters35,36. In mouse models of lung IRI induced by 60 minutes of hilar clamping, robust neutrophil influx occurs within the first few hours40,41. However, in mice that undergo transient occlusion of the renal hilum for 45 minutes, neutrophils immediately infiltrate glomeruli from the afferent arterioles early after reperfusion and can be observed infiltrating inside and outside of the peritubular capillaries and filling the tubules44. In a mouse model of liver IRI induced by 45 minutes of hepatic arterial clamping, Honda reported that the number of neutrophils that accumulate in the vasculature of the affected lobules steadily increases until 4 hours after reperfusion42. Similarly, Jaeschke reported an 80-fold increase in neutrophilic accumulation at 24 hours in a rat model of warm hepatic IRI34.
Unlike most other organs, resting human lungs contain a neutrophil reservoir that equates to approximately three times as many neutrophils as the peripheral circulation45,46. Multiple in vivo microscopic studies in animal models have illustrated this marginated pool of neutrophils within the lungs of ventilated mice and dogs at steady state as well31,47. While the role of this marginated pool of neutrophils remains unknown, recent work has suggested that this pulmonary reservoir of neutrophils may contribute to the defense against intravascular pathogens48. Other studies have found that this neutrophil pool regulates diurnal oscillations of transcription patterns in the lung49. Additionally, these marginated neutrophils may serve as an alternate reservoir for circulating neutrophils. To this end, Devi found that administration of plerixafor, a CXCR4 antagonist, triggered neutrophil mobilization from the pulmonary vasculature50. Marginated pools of neutrophils may also be encountered in other organs (i.e. liver), albeit to a lesser extent. Although most solid organs are flushed with a cold solution at the time of organ recovery, this is unlikely to eliminate many sequestered neutrophils adherent to vascular endothelium. Thus, the role of marginated neutrophil pools within donor organs, especially lungs, warrants further explorations.
The stimulus for neutrophil recruitment following IRI-mediated injury is multifactorial and largely driven by a sterile inflammatory process that is triggered after reperfusion. Inflammatory cascades after reperfusion can be initiated by several pathways of cell death, both apoptotic and non-apoptotic. The predominant pathway of cell death following transplantation appears to vary among organs and may be impacted by the severity of injury. We have previously reported that administration of ferrostatin-1, a specific ferroptosis inhibitor, inhibits neutrophil recruitment following myocardial IRI, including heart transplantation in mice35. Neutrophil trafficking was not impacted when cardiac grafts were deficient in receptor-interacting protein kinase 3 (RIPK3), a kinase essential in mediating necroptotic cell death. It is worth noting that the predominant pathway of cell death may change as time progresses following transplantation. To this end, Pavlosky has shown that RIPK3-deficient cardiac grafts develop lower levels of tissue necrosis, high-mobility group box 1 (HMGB1) production, and cell infiltration 12 days after transplantation (vs. earlier post-operative time points in our study35)51.
We have recently shown that necroptosis triggers the recruitment of neutrophils to lung allografts during severe injury induced by cold and warm ischemia and leads to impaired graft function52. Here, neutrophil recruitment is mediated by CXCL1 produced by donor-derived non-classical monocytes (NCMs). Donor lungs deficient in RIPK3 had significantly improved function and decreased neutrophilic extravasation52. Moreover, in a rat model of lung transplantation with prolonged cold ischemia, inhibition of necroptosis through administration of necrostatin-1 to both the donor and recipient resulted in significantly reduced tissue injury and improved graft function53.
Necroptosis has also been shown to play an important role after kidney IRI. Following renal hilar clamping in mice, RIPK3 expression is markedly upregulated in proximal and distal tubules as early as 4 hours after IRI and remains elevated at 48 hours post-injury54. Similar to Pavlosky’s findings in hearts, RIPK3-deficient kidney allografts demonstrated markedly decreased injury following transplant-mediated IRI54,55. However, other cell death pathways may contribute to renal IRI including cyclophilin-D-mediated necrosis and ferroptosis54,55,56,57. Thus, modulation of multiple cell death pathways may provide maximal protection from renal transplant-mediated IRI.
Following IRI, dying cells release damage-associated molecular patterns (DAMPs) which stimulate the release of pro-inflammatory cytokines and chemokines. For example, elevated levels of extracellular ATP are present in human samples of bronchoalveolar lavage (BAL) fluid following lung transplantation, and higher levels of ATP are found in BAL fluid from patients who develop early PGD58. The binding of DAMPs to toll-like receptors (TLRs) results in activation of pro-inflammatory signaling pathways that promote neutrophil recruitment. TLR4 is one such receptor that has been recognized as an important mediator of neutrophil trafficking following IRI in lung, heart, liver, and kidney transplantation35,59–62. Prakish showed that TLR4 expression is significantly upregulated following transient pulmonary artery ligation in mice and correlates with neutrophil infiltration in the affected lung59. In our previously mentioned study using a mouse orthotopic lung transplant model, we showed that neutrophil-mediated vascular leakage is dependent on TLR4 expression on vascular endothelium52 (Figure 1). Additionally, we showed that the downstream expression of NADPH oxidase 4 (NOX4) leads to production of reactive oxygen species (ROS). TLR4-dependent signaling promotes the release of IL-1β, CXCL1, and CXCL2, as well as enhancing neutrophil recruitment to the lung graft52,59.
Figure 1:
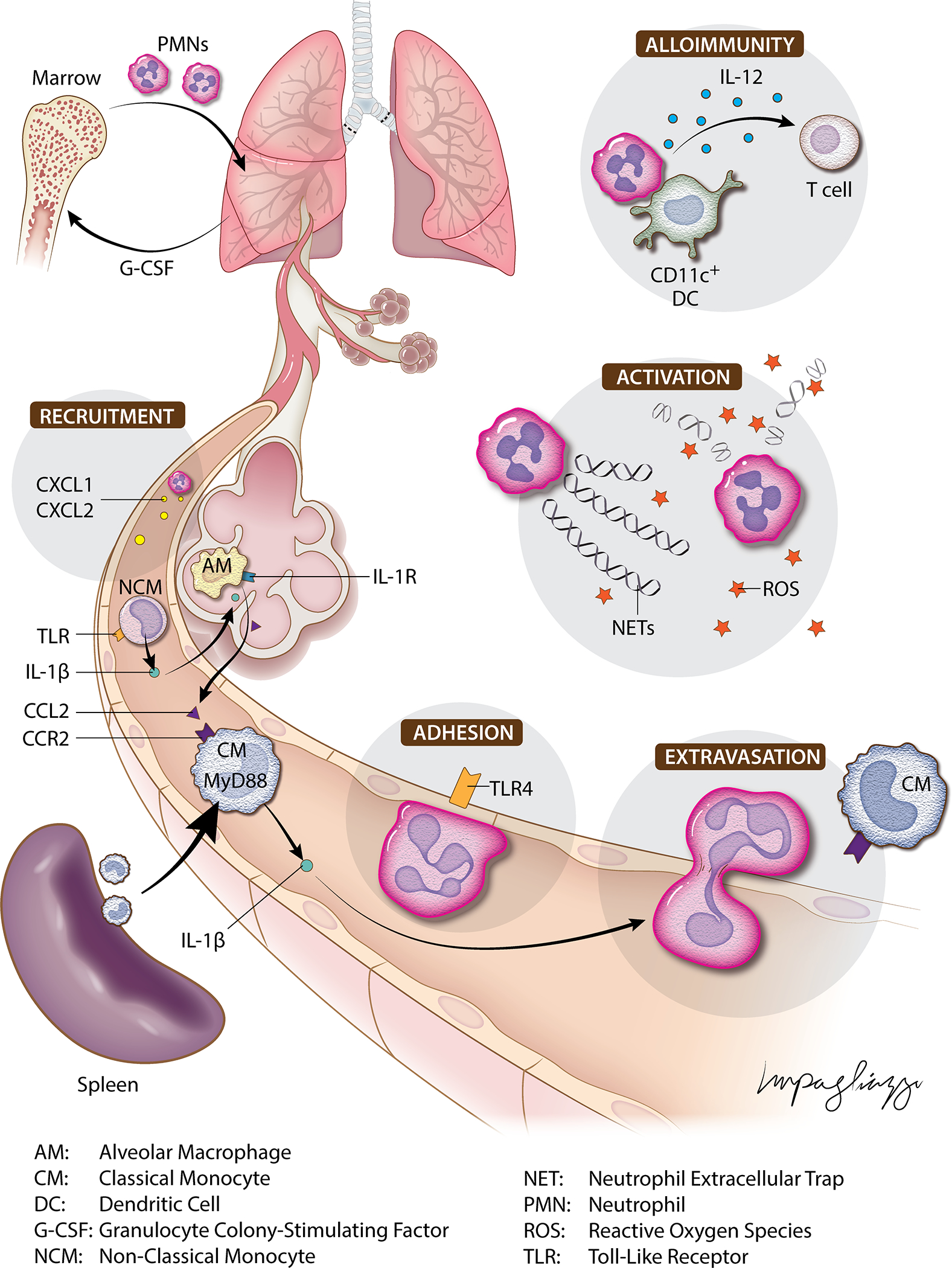
Schematic illustrating neutrophil-mediated immune responses within the lung following ischemia-reperfusion injury.
The importance of TLR4 in neutrophil recruitment after IRI has been demonstrated in other organs as well. We have previously shown that neutrophil adhesion to the walls of coronary veins after murine heart transplantation is mediated by TLR4/Trif-dependent signaling in graft endothelial cells35. TLR4 is also highly upregulated on renal tubular epithelial cells after ischemic injury, triggering multiple downstream effects that exaggerate allograft injury61. Activation of TLR4 in renal IRI promotes the recruitment of neutrophils, illustrated by markedly reduced neutrophil infiltration in TLR4-deficient mice subjected to renal IRI63. Following renal IRI, TLR4-dependent signaling stimulates the synthesis of neutrophil chemoattractants in response to inflammatory cytokines such as IL-1β, IL-6 and tumor necrosis factor alpha (TNF-α). TLR4 signaling is also an important mediator of neutrophil recruitment in hepatic IRI. Upregulation of TLR4 following hepatic IRI is associated with increased levels of IL-6 and TNF-α62. Tsung employed TLR4 chimeric mice to demonstrate that TLR4 engagement on phagocytic nonparenchymal cells is required for warm IRI-mediated injury in the liver. They found that administration of HMGB1, a TLR4 ligand, resulted in increased IRI-induced hepatic injury in a TLR4-dependent fashion. Similarly, TLR4-deficient mice or treatment with an HMGB1-neutralizing antibody attenuated injury after hepatic warm IRI64. In a mouse model of renal hilar clamping, HMGB1 release after reperfusion was localized to areas with increased susceptibility to ischemic injury including renal tubules and peritubular capillaries65. Importantly, administration of a HMGB1-blocking antibody 30 minutes prior to renal ischemia markedly reduced inflammation and preserved renal function. These findings show that TLR4 signaling is an important mediator in early responses to IRI after transplantation and suggest that targeting HMGB1 or other endogenous TLR4 ligands may minimize IRI-mediated graft dysfunction. In addition to TLR4, other TLRs have also been shown to play a role in neutrophil recruitment after transplantation. For example, we have shown that TLR9 expression by cardiac macrophages is crucial for promoting neutrophil trafficking into the graft following ischemic injury in a mouse cardiac transplantation model36. Using this model, we found that neutrophil influx was regulated by TLR9/MyD88-mediated production of CXCL2 and CXCL5. Thus, targeting TLR signaling pathways may provide a valuable therapeutic opportunity to minimize IRI-mediated graft injury.
Several CXC chemoattractants, namely CXCL1 and CXCL2, have been identified as key mediators of neutrophil recruitment to allografts following reperfusion. In a rat model of orthotopic lung transplantation, Belperio found that allografts and isografts exposed to 6 hours of cold ischemia had increased levels of CXCL1 and CXCL2, which peaked 8 to 16 hours after transplantation and correlated with graft neutrophilia30. Notably, treatment with an anti-CXCR2 antibody resulted in decreased neutrophil infiltration and attenuated lung graft injury. Expression of CXCL1 and CXCL2 also increases in mouse livers after IRI66. Of note, CXCL2 expression is induced within 3 hours after reperfusion, before any detectable increase in neutrophil accumulation; this is in contrast to CXCL1 expression, which increases 9 hours after reperfusion, suggesting that CXCL2 mediates the initial recruitment of neutrophils following reperfusion. In a study of canine heart transplantation, Birdsall found that potent neutrophil chemotactic factors such as C5a appear in cardiac lymphatic fluid shortly following reperfusion and correlate with rapid and robust neutrophil infiltration into the myocardium67. Other chemoattractants, such as IL-8, appear to aid in neutrophil recruitment to allografts as well. Oz analyzed human blood samples from the following reperfusion after elective cardiac surgery and cardiac transplantation and found significantly higher levels of IL-8 among transplant patients. Furthermore, human endothelial cells subjected to ischemia in vivo triggered a time-dependent release of IL-8, demonstrating a direct role of tissue ischemia to the local release of a neutrophil chemoattractant in humans68.
Prior work from our laboratory has identified molecular targets that regulate acute granulopoietic responses induced by pulmonary transplant-associated IRI. Following transplantation of syngeneic or allogeneic murine lung grafts, we have discovered that a surge of granulocyte colony-stimulating factor (G-CSF) stimulates expansion of bone-marrow-resident neutrophil progenitors and leads to accumulation of neutrophils in the peripheral blood and graft tissues32,69. Additionally, transplantation of mouse lung allografts following prolonged cold ischemic storage is associated with significantly higher serum concentrations of G-CSF, increased neutrophils in the graft and periphery, and more severe acute graft injury69. Importantly, G-CSF blockade or neutrophil-depleting antibodies in the setting of extended cold ischemic storage resulted in diminished neutrophilic graft infiltration and improved allograft function. In renal transplantation, similar findings of transient granulopoiesis have been documented. For example, in human kidney transplant recipients, ischemic injury induces a transient rise in G-CSF expression in renal tubular epithelium which correlates with increased neutrophil expression of the surface glycoprotein CD17770. Following kidney IRI in mice, renal G-CSF expression significantly increases, triggering systemic neutrophilia. Thus, regulation of the G-CSF-mediated granulopoietic response following IRI holds potential to minimize allograft injury after transplantation.
Many graft-resident cell populations play an important role in mediating neutrophil recruitment to newly reperfused tissues, the majority of which involve monocyte or macrophage equivalents. In lung allografts, donor-derived Ly6G−, NK1.1+, CD11b+, Ly6Clow, CD64− NCMs are retained within the vasculature of human and murine lungs procured for transplantation and have been identified as key facilitators of neutrophil recruitment after IRI39. In a mouse model of lung transplantation, lung-resident NCMs are necessary for early neutrophil recruitment through the production of CXCL2 in a MyD88/TRIF-dependent fashion39. Here, neutralization of CXCL2 attenuates neutrophil infiltration of lung grafts, and depletion of NCMs in donor lungs, either pharmacologically or genetically, results in significantly impaired neutrophil influx and attenuated lung graft injury. Some studies suggest that alveolar macrophages (AMs) play a role in neutrophil recruitment to lungs as well. Using a mouse model of warm IRI, Prakash showed that depletion of CD11c+ AMs via treatment with liposomal clodronate or diphtheria toxin in CD11c-DTR donors resulted in markedly diminished pulmonary neutrophil recruitment59. Kupffer cells, the analogous resident macrophages of the liver, appear to facilitate neutrophilic infiltration and subsequent injury following liver transplantation. In a study by Mosher using a mouse model of warm hepatic IRI, Kupffer cells were found to be major contributors to the production pro-inflammatory cytokines, which correlated with increased neutrophil infiltration into the injured liver71. When Kupffer cell activity was inhibited by gadolinium chloride administration 24 to 48 hours prior to the onset of ischemia, there was a marked reduction in CXC chemokine production and IRI-mediated liver injury. In a mouse model of renal IRI induced by 20 minutes of hilar clamping, Ferenbach similarly demonstrated that macrophages promote neutrophil recruitment through production of various cytokines72. Interestingly, they found contrasting effects between two different strategies of macrophage depletion, with significant protection induced by clodronate administration but no protective effect seen with DT-mediated depletion in CD11b-DTR mice, presumably due to preservation of a protective CD11c+ cell population with clodronate treatment (CD206+ CD11c+ M2 macrophages and/or CD11c+ resident monocytes). While this CD206+ CD11c+ cell population has not yet been identified as playing a protective role in the IRI of other organs, other CD11c+ cell types have been shown to be protective after transplant-mediated ischemic injury. In a mouse model of renal transplantation, Aiello found that intragraft donor-derived CD11c+ F4/80+ renal macrophages transition into a reparative phenotype that orchestrates tissue repair in an IL-1 receptor 8-dependent manner73. Studies have suggested a dichotomous role for tissue-resident and recruited CD11c+ dendritic cells (DCs) following liver IRI. Utilizing hepatic portal clamping, Zhang found that DC-deficient mice experienced significantly less tissue injury and reduced neutrophil infiltration, and adoptive transfer of wild-type DCs increased hepatic injury74. Interestingly, when CD11c+ DC-deficient livers were transplanted into wild-type mice, hepatic necrosis and neutrophil infiltration were significantly increased compared to controls. Thus, tissue-resident and circulating DCs appear to play important and distinct roles in neutrophil recruitment and tissue injury following IRI.
Following cardiac IRI, IL-6 is generated from hypoxic myocytes and associated with increased neutrophil influx into ischemic myocardium75. Furthermore, IL-6 production is regulated by TNF-α secretion by mast cells in a canine model of cardiac IRI following transient coronary occlusion76. While the precise mechanistic pathway underlying this enhancement of neutrophil migration remains unclear, these findings highlight the multifaceted stimuli for neutrophil recruitment from both hematopoietic and non-hematopoietic tissues following IRI.
Neutrophil Extravasation
Neutrophils are rapidly recruited to the vasculature of reperfused tissues, where they subsequently extravasate out of the vessel lumen into the surrounding interstitial tissues. Neutrophils interact with the vascular endothelium through tethering, rolling, adherence, and crawling before extravasating through vessel walls in either a paracellular or transcellular fashion. IRI can induce altered expression of endothelial proteins that enhance neutrophil adhesion and subsequent extravasation. In ischemic heart grafts, endothelial cells exhibit increased expression of intercellular adhesion molecule-1 (ICAM-1), largely due to the release of proinflammatory cytokines such as TNF-α77,78. Using intravital two-photon microscopy, we have demonstrated that blockade of ICAM-1 receptors LFA-1 (CD11a/CD18) and Mac-1 (CD11b/CD18) prevented neutrophil adherence to and crawling on the endothelium, respectively, markedly inhibiting neutrophilic extravasation37. Furthermore, consistent with the notion that CXCL2 enhances integrin affinity, administration of anti-CXCL2 antibodies prevents adhesion of neutrophils to coronary veins. In a mouse model of myocardial IRI induced by transient coronary artery occlusion for 30 minutes, Bowden demonstrated that vascular cell adhesion molecule-1 (VCAM-1) is significantly upregulated on cardiac endothelium and associated with transendothelial migration of neutrophils79. In rat renal grafts, the expression of ICAM-1, VCAM-1, and P-selectin increases rapidly following transplantation in a TLR4-dependent manner and correlates with neutrophil influx and impaired renal function80,81. During hepatic IRI, ICAM-1 is transcriptionally upregulated on the surfaces of hepatocytes and sinusoidal endothelial cells82–84. However, ICAM-1 expression was only partially required for neutrophil migration due to the extensive vascular injury present. To this end, murine models of hepatic IRI only demonstrate partially attenuated injury after treatment with anti-ICAM-1 antibodies85–87. Dipeptidase-1 has also been identified as a major adhesion receptor on lung and liver endothelium for neutrophil trafficking in response to endotoxemia88. However, we have shown that immune responses can differ in sterile and pathogen-driven inflammation40,89. Thus, this receptor should be explored in transplant models to investigate its role as a therapeutic target. These findings illustrate the situational variability and tissue-specificity which exists to regulate neutrophil trafficking to injured tissues.
In both human lung allografts and murine models, we have found that IRI is associated with the mobilization of recipient classical monocytes (CMs) into injured lungs40,90. In mice, CMs are hallmarked by the expression of CCR2 and high levels of Ly6C. We have reported that recipient CCR2 expression mediates lung allograft dysfunction91. Using intravital two-photon microscopy, we have observed neutrophils tracking behind CMs (CD115+, CD11b+, Ly6Chi cells) during transendothelial migration in reperfused lung grafts31 (Figure 1). Furthermore, spleen-derived recipient CMs serve a crucial role for neutrophil extravasation through MyD88-dependent production of IL-1β, resulting in increased endothelial permeability through downregulation of ZO-2, a cytoplasmic protein that interacts with transmembrane tight junction proteins in pulmonary vascular endothelial cells40. We found that depletion of recipient CMs abrogated neutrophil extravasation and ameliorated IRI. Interestingly, in lung grafts devoid of donor NCMs, which was achieved by administration of airway liposomal clodronate prior to lung procurement or by using donors deficient in NR4A1, a transcription factor required for NCM formation, CM recruitment was significantly reduced. Accordingly, reconstitution of NR4A1-deficient grafts with NCMs resulted in CM recruitment and neutrophil extravasation into the transplanted lung. Recently, we have shown that donor NCMs following lung transplantation release IL-1β, which activates AMs and results in the release of CCL2, in turn recruiting recipient CCR2+ CMs to the lung graft92. Thus, in addition to the aforementioned role of NCMs in mediating neutrophil recruitment, donor NCMs serve an essential role in recruiting recipient CMs from the spleen, which then mediate neutrophil extravasation (Figure 1).
Donor-derived tissue resident macrophages also play an important role in neutrophil extravasation in lung allografts following IRI. We have shown that tissue-resident macrophages in both mouse and human lungs subjected to cold ischemic storage express DNAX-activation protein 12 (DAP12), a cell-membrane associated protein that regulates the production of various neutrophil chemokines including CXCL1, CXCL2, IL-6, and TNF-α39. While transplantation of DAP12-deficient murine lungs resulted in similar recruitment of neutrophils in the pulmonary vessels, neutrophil extravasation was markedly impaired. Furthermore, we found that administration of wild-type macrophages into the donor bronchus of DAP12-deficient lung grafts immediately prior to reperfusion reconstituted neutrophil extravasation to levels observed in wild-type grafts. These findings highlight the necessity of DAP12 expression by pulmonary resident cells for neutrophil extravasation following IRI and that different steps of neutrophil extravasation are orchestrated by cells of both donor and recipient origin.
Tissue-resident macrophages also promote neutrophil extravasation into ischemic myocardium following cardiac transplantation. Using intravital two-photon imaging in a mouse model of IRI after cardiac transplantation, we have demonstrated that donor tissue-resident CCR2+ monocyte-derived macrophages orchestrate neutrophil extravasation through TLR9/MyD88-mediated production of CXCL5. Here, MyD88 deficiency in heart-resident macrophages alone was sufficient to impair the extravasation of neutrophils36. Interestingly, sub-types of tissue-resident macrophages can serve a protective role against neutrophil infiltration during ischemic kidney injury93. In this study, the authors found that depletion of kidney-resident CD169+ macrophages exacerbated neutrophil migration into injured tissue due to elevated expression of endothelial ICAM-1, as well as increased CXCL1 and CXCL2, resulting in more severe IRI-mediated tissue injury and functional impairment. Additional in vitro co-culture experiments confirmed that direct interactions between tissue-resident macrophages and endothelial cells suppress the adhesive and transmigratory activity of neutrophils through ICAM-1 suppression. As such, various tissue-resident macrophages appear to play different roles among solid organ systems, but additional investigations are warranted before these studies can be translated clinically.
Neutrophil Activation
Following neutrophil extravasation in the setting of IRI, molecular cues guide neutrophil migration through extravascular tissues and stimulate their activation. Neutrophil activation can trigger various effector pathways, including the oxidative response that consists of ROS production, such as superoxide anion and hydrogen peroxide following NADPH-oxidase complex activation, and production of halogenated oxidants, such as hypochlorous acid and chloramines via myeloperoxidase (MPO). Neutrophil granules provide a reservoir of inflammatory mediators, including azurophilic granules (MPO, neutrophil elastase) and specific granules (metalloproteinase-8 (MMP-8), MMP-9). In vitro experiments have demonstrated that following neutrophil activation some of these stored enzymes, such as MMPs, must receive additional activation signals to trigger their release, though these mechanisms remain incompletely understood94–96. In support of these findings, a mouse model of warm lung IRI demonstrated that recruited neutrophils remain dormant unless activated by additional signals within the lung tissue97. These findings were corroborated by results in a mouse model of ventilated warm lung IRI, which showed that despite the early surge of inflammatory cytokines and rapid recruitment of neutrophils to injured lungs, there was quick resolution of this process within 24 hours without any evidence of tissue damage59. Furthermore, in studies of IRI in rat lungs subjected to 90 minutes of hilar clamping or transplanted after cold ischemic storage, levels of MPO activity in lung tissues increased during the first 16 hours following reperfusion and fell thereafter30,98. These findings suggest that IRI-mediated injury is associated with rapid recruitment of neutrophils that undergo extravasation and activation within ischemic tissues, followed by a phase of inflammation resolution mediated by specialized pro-resolving lipid mediators, such as resolvins, that can be released from a variety of cells including macrophages and neutrophils99,100.
In canine heart and rat liver IRI models, NADPH oxidase-mediated ROS production by neutrophils has been identified as an important effector arm of tissue damage101,102. Neutrophil elastase release by activated neutrophils can also mediate tissue damage in animal models of lung and hepatic IRI. In a mouse model of warm liver IRI, neutrophil transmigration from the vascular lumen into liver parenchyma triggers neutrophil elastase release, which degrades the extracellular matrix of the endothelium. The resultant tissue injury leads to the release of pro-inflammatory cytokines and chemokines and upregulation of TLR4, resulting in further propagation of neutrophil-mediated graft injury via the mechanisms discussed above103. The damaging effects of neutrophil elastase have also been demonstrated in lung IRI. In a study by Ishikawa using a rabbit model of lung IRI induced by 2 hours of hilar clamping, intravenous administration of a neutrophil elastase inhibitor resulted in reduced tissue injury and neutrophil infiltration104. Similarly, in a canine model of single lung transplantation, continuous infusion of a neutrophil elastase inhibitor during reperfusion resulted in significantly reduced MPO activity and successfully ameliorated lung injury, as evidenced by pulmonary function and histology105. Moreover, in a rat model of lung transplantation, addition of a neutrophil elastase inhibitor, sivelestat, to the organ flushing solution prior to cold ischemic storage resulted in significantly improved graft function and reduced lung tissue MPO activity106. These results were corroborated in a porcine lung transplant model, where a neutrophil elastase inhibitor given during ex vivo lung perfusion resulted in improved lung function both during and after transplantation107.
Increased levels of MMP-9 is present in human hepatic allograft tissues following reperfusion108. Animal studies have illustrated that increased levels of MMP-9 in acutely injured liver tissues stimulates tissue destruction through the degradation of extracellular matrix proteins109. In a mouse model of warm liver IRI, both neutrophils and Mac-1-positive leukocytes were found to be the major sources of MMP-9 in damaged hepatic tissues, and the expression of MMP-9 was specifically localized to leukocytes that had already initiated the process of extravasation110. In a rat liver model of syngeneic liver transplantation following 24 hours of ex vivo cold ischemia, vascular expression of fibronectin was significantly increased in sinusoidal endothelial cells shortly following reperfusion, and fibronectin binding to the integrin receptor α4β1 regulated the expression of MMP-9 by infiltrating neutrophils110,111. Moreover, blockade of fibronectin interactions disrupted the extravasation of neutrophils after IRI and down-regulated MMP-9 expression. To further evaluate the role of MMP-9 of infiltrating leukocytes, MMP-9-deficient mice and wild-type mice treated with an anti-MMP-9 neutralizing antibody were subjected to warm liver IRI. MMP-9 inhibition prevented neutrophil migration across fibronectin and reduced MPO activation in vivo112. Additionally, mice treated with a broad inhibitor for both MMP-9 and MMP-2 showed inferior protection against IRI-mediated hepatic damage compared to MMP-9 specific inhibition. These findings suggest that MMP-2 and MMP-9 likely serve distinct roles in liver IRI, with MMP-2 potentially promoting anti-inflammatory pathways to reduce tissue destruction.
The effects of MMPs may be organ-specific. In a rat model of orthotopic lung transplantation, MMP-2 and MMP-9 were found to be elevated following reperfusion and were associated with increased alveolar capillary permeability and neutrophil infiltration113. Notably, when a nonselective MMP inhibitor was administered to both donors and recipients, there was significantly reduced capillary leakage and neutrophil infiltration. In another study by Yano utilizing a rat lung transplantation model, elevated enzymatic activity of both MMP-2 and MMP-9 were seen following prolonged cold storage which correlated with markers of pulmonary graft injury114. In another rat model of lung transplantation utilizing warm IRI, increased levels of both MMP-2 and MMP-9 were observed, but MMP-9 expression was transient while MMP-2 expression was sustained115. Thus, MMP inhibition may serve as a potential target to reduce lung injury following IRI and future investigations should further define the role and appropriate timing for specific MMP inhibitors in this context.
The role of MMPs in cardiac transplantation also appears unique compared to other solid organ transplants. In a study of isolated, perfused rat hearts subjected to 20 minutes of warm ischemia, MMP-2 was found to be elevated in the coronary effluent during reperfusion and correlated with the duration of ischemia and the degree of myocardial dysfunction116. Furthermore, inhibition of MMP-2 improved myocardial function, whereas MMP-2 administration worsened myocardial recovery during reperfusion. In vitro studies have revealed that human troponin I and troponin T are susceptible to proteolytic degradation by MMP-2117. In rat hearts subjected to 20 minutes of ischemia followed by reperfusion via the Langendorff method, MMP-2 co-localizes with troponin I in cardiomyocytes, and MMP-2 inhibition prevented the IRI-induced degradation of troponin I and improved recovery of myocardial function117. In canine hearts that underwent coronary occlusion for 60 minutes, levels of MMP-9 were increased in cardiac lymph and myocardium, and dual-labeling immunofluorescence determined that neutrophils were the main source of MMP-9118. Using a mouse model of warm cardiac IRI, Carbone found elevated myocardial and serum levels of receptor activator of nuclear factor-κB ligand (RANKL), and administration of a neutralizing anti-RANKL antibody resulted in decreased neutrophil infiltration and degranulation and significantly reduced MMP-9119. The authors also demonstrated in vitro that RANKL abrogated neutrophil chemotaxis and release of MMP-9.
Clinical studies of lung transplant recipients have demonstrated that patients with severe PGD following lung transplantation have higher levels of circulating mitochondrial DNA (mtDNA) than patients without PGD120. Notably, in a mouse model of PGD following orthotopic lung transplantation, graft injury was inhibited by pharmacologic inhibition or genetic deletion of formyl peptide receptor 1 (FPR1), a chemotactic receptor that drives neutrophil migration towards free peptides released by bacteria and mitochondria. Further analysis revealed that FPR1-mediated neutrophil trafficking is associated with the engulfment of damaged mitochondria and the activation of ROS-induced tissue injury. A novel method of in vivo single-photon emission computed tomography (SPECT) imaging was recently developed which allows visualization of activated neutrophils through administration of a synthetic FPR1 ligand (polyethylene glycolated cinnamoyl-F-(D)L-F-(D)L-F-K)41. Using this imaging technique in a mouse model of pulmonary hilar clamping, there is increased signal intensity from activated neutrophils, which peaks 2 hours after reperfusion and resolves by 24 hours.
Neutrophils can also promote graft injury by undergoing a unique form of cell death that involves extrusion of neutrophil extracellular traps (NETs) comprised of extracellular chromatin decorated with histones, antibacterial peptides, and serine proteases. NETs appear to serve as a marker of severity of inflammation, and increased levels of NETs have been reported in the BAL fluid of human lung transplant recipients with PGD as well as renal transplant recipients with acute antibody-mediated rejection121,122. Multiple experimental models of lung and liver transplantation have similarly demonstrated that NETs play a role in transplant-mediated IRI121–124. These studies showed that inhibition of NET formation through administration of DNAse, Cl-Amidine, or recombinant human thrombomodulin improved organ function and attenuated inflammation. In a mouse model of orthotopic lung transplantation, Mallavia found that prolonged cold ischemia triggered mtDNA release into BAL fluid125. Accordingly, intrabronchial DNase treatment resulted in degradation of mtDNA in BAL fluid, and in vitro studies of mtDNA-stimulated neutrophils incubated with DNase failed to produce NETs. Additionally, adoptive transfer of mtDNA in a minimal ischemia model during transplantation induced NET formation and lung injury, which was prevented in mice with TLR9 deficiency in either the lung donor or recipient.
MicroRNAs (miRNAs), particularly miR-223, have been identified as important regulators of neutrophil activation and function. However, the role of miR-223 appears to vary among different experimental settings of acute injury. In both humans and mice with acute lung injury, pulmonary expression of miR-223 is significantly increased126,127. In mouse models of acute lung injury induced by infection or barotrauma, Neudecker found that miR-223 deficiency is associated with severe inflammation, while pulmonary miR-223 overexpression results in lung protection128. Furthermore, activated neutrophils were found to be a major source of miR-223, and in vitro co-culture of human neutrophils with epithelial cells resulted in transfer of miR-223 from neutrophils to the epithelial cells. These results collectively suggest that miR-223 may serve as a negative feedback molecule to limit acute inflammation. However, this protective effect is less clear in the setting of IRI. In a mouse model of lung IRI induced by transient pulmonary hilar occlusion, miR-223 was similarly found to be expressed at high levels in injured tissue, but here it was shown to inhibit expression of hypoxia-inducible factor-2α and β-catenin, which promoted autophagy and aggravated IRI-induced injury127. In a mouse model of liver IRI induced by portal clamping, Yu found that miR-223 expression levels were upregulated following reperfusion and significantly correlated with serum transaminases129. As such, the mechanisms whereby miRNA expression alters IRI-induced injury warrants further investigation to determine the potential of this pathway as a therapeutic target.
Purine signaling has also been implicated in guiding neutrophil responses during acute inflammation. Eltzschig showed that activated human neutrophils can promote vascular integrity by releasing adenine nucleotides which subsequently undergo metabolism and activate endothelial adenosine receptors, particularly the adenosine A2B receptor130. A study by Li reported increased levels of circulating netrin-1, a protective neuronal guidance protein, in patients after myocardial infarction and mice following myocardial IRI131. Neutrophils were found to be the main source of serum netrin-1 levels following IRI, and netrin-1-mediated cardioprotection was dependent on purinergic signaling through the myeloid adenosine A2B receptor. Similarly, a protective role for adenosine A2B receptor has been described after renal and hepatic IRI. Utilizing a mouse model of renal IRI with ischemic preconditioning, Grenz demonstrated that mice deficient in the adenosine A2B receptor had increased kidney injury, while treatment of wild-type mice with an adenosine A2B receptor agonist attenuated kidney injury132. However, contrasting results reported by other studies suggest that adenosine A2B receptor may play a pro-inflammatory role in other instances after IRI. For example, Anvari found that adenosine A2B receptor-deficient mice had significantly reduced lung injury following IRI induced by hilar clamping133. Further experiments using bone marrow chimeras revealed that these effects were due to A2B receptor activation on pulmonary resident cells. Thus, the pro- and anti-inflammatory effects associated with adenosine A2B receptor signaling is likely dependent on the specific organ and inflammatory model studied.
Other innate immune cells, such as NK cells, contribute to tissue injury following IRI as well. In a mouse model of hepatic IRI induced by portal vein clamping, NK cell-deficient mice develop reduced liver injury134. Similarly, tissue-resident NK cells influence immune responses following renal IRI. For example, Victorino demonstrated that depletion of conventional and resident NK cells protected against renal IRI, while selective depletion of only conventional NK cells failed to confer protection135. These studies highlight that cellular mediators other than neutrophils also contribute to IRI-induced injury, and while outside the scope of this article, these alternate pathways should also be considered in future investigations.
Facilitation of Alloimmunity
Cellular depletion experiments have illustrated that neutrophils provide an important link between innate immune activation and the development of alloimmunity. However, the pathways that facilitate alloimmunity as a result of IRI are less understood than the innate immune processes discussed above and likely differ between organs. For example, we have previously demonstrated that alloimmune responses following lung transplantation are initiated within the allograft itself, independent of secondary lymphoid tissues and setting it apart from other solid organs. Following lung transplantation in mice, T cell clusters are observed around donor-derived graft-resident CD11c+ DCs as early as 30 hours after transplantation136. Using intravital two-photon microscopy, we have shown that graft-infiltrating neutrophils stimulate donor CD11c+ DCs in lung allografts in a contact-dependent fashion shortly after transplantation69. We have demonstrated that neutrophil depletion or G-CSF inhibition in the setting of severe IRI results in decreased IL-12 production by DCs, reduced interferon-gamma (IFN-γ+) T cells, and attenuated rejection. As described above, ischemic injury stimulates the release of inflammatory mediators which trigger neutrophil activation. To this end, higher levels of mtDNA have been shown to be associated with PGD in lung transplant recipients, thereby increasing their risk for chronic rejection23,120. Several other pathways have been suggested that may further clarify the mechanisms linking early neutrophil responses and lung transplant rejection, but much remains to be discovered before such pathways can be targeted clinically137.
Little mechanistic evidence currently exists regarding how neutrophils may regulate humoral responses in transplanted organs. As mentioned previously, prior studies have elucidated that increased levels of NETs are associated with the development of acute antibody-mediated rejection in renal transplant recipients121. In studies of acute antibody-mediated rejection in CCR5-deficient mice, there is an initial wave of neutrophil infiltration into kidney allografts immediately after transplantation followed by a later surge of neutrophil infiltration that occurs in parallel to rising titers of donor-specific antibody titers138,139. Depletion of neutrophils prior to this second wave of infiltration reduces natural killer cell and monocyte proliferation within the graft and abrogates natural killer cell activation, a key mechanism underlying acute antibody-mediated rejection of kidney grafts. These findings suggest that depletion of neutrophils may inhibit myeloid cell activation and indirectly suppress alloimmune responses. In a study of cardiac allograft rejection in mice, recipients with genetic or pharmacologic inhibition of the neutrophil chemokine receptor CXCR2 exhibited significantly attenuated neutrophilic graft infiltration and inhibited of T cell infiltration into the transplanted hearts140. Additionally, when costimulatory blockade was combined with either peri-transplant neutrophil depletion or anti-CXCL1/2 antibodies, cardiac allograft survival was significantly prolonged. Neutrophils may also augment alloimmunity by transporting donor antigens from injured tissues to peripheral lymph nodes in a process termed ‘reverse neutrophil migration’141,142. Furthermore, neutrophils may acquire antigen-presenting capabilities or enhance the secretion of immunomodulatory molecules that influence lymphocyte activity143,144.
Conclusion
Neutrophils play a pivotal role in IRI following heart, lung, kidney and liver transplantation. The recruitment, extravasation and activation of recipient-derived neutrophils is orchestrated through a variety of graft-resident and recipient-derived cell populations. Recent technological advances, including novel intravital imaging techniques, have enabled a more accurate characterization of neutrophilic responses during IRI. These newly recognized immune pathways warrant further investigation and translation to clinical trials in hopes of ameliorating IRI-mediated graft dysfunction.
Funding:
DK is supported by National Institutes of Health grants 1P01AI116501, R01HL094601, R01HL151078, Veterans Administration Merit Review grant 1I01BX002730, The Cystic Fibrosis Foundation and The Foundation for Barnes-Jewish Hospital.
Abbreviations
- AM
Alveolar macrophage
- BAL
Bronchoalveolar lavage
- CM
Classical monocyte
- DAMP
Damage-associated molecular pattern
- DAP12
DNAX-activation protein 12
- DC
Dendritic cell
- FPR1
Formyl peptide receptor 1
- G-CSF
Granulocyte colony-stimulating factor
- HMGB1
High-mobility group box 1
- ICAM-1
Intercellular adhesion molecule-1
- IFN-γ+
Interferon-gamma
- IRI
Ischemia-reperfusion injury
- miRNA
microRNA
- MMP
Metalloproteinase
- MPO
Myeloperoxidase
- MPT
Mitochondrial permeability transition
- mtDNA
Mitochondrial DNA
- NCM
Non-classical monocyte
- NET
Neutrophil extracellular trap
- NK
natural killer
- NOX4
NADPH oxidase 4
- PGD
Primary graft dysfunction
- RANKL
Receptor activator of nuclear factor-κB ligand
- RIPK1
Receptor-interacting protein kinase 1
- RIPK3
Receptor-interacting protein kinase 3
- ROS
Reactive oxygen species
- SPECT
Single-photon emission computed tomography
- TLR
Toll-like receptor
- TNF-α
Tumor necrosis factor alpha
- VCAM-1
Vascular cell adhesion molecule-1
Footnotes
Disclosure: DK has a pending patent entitled “Compositions and methods for detecting CCR2 receptors” (application number 15/611,577).
references:
- 1.Colombat M, Castier Y, Leseche G, et al. Early expression of adhesion molecules after lung transplantation: evidence for a role of aggregated P-selectin-positive platelets in human primary graft failure. J Heart Lung Transplant. 2004;23(9): 1087–1092. [DOI] [PubMed] [Google Scholar]
- 2.Koo DD, Welsh KI, Roake JA, et al. Ischemia/reperfusion injury in human kidney transplantation: an immunohistochemical analysis of changes after reperfusion. Am J Pathol. 1998;153(2): 557–566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Ito T, Naini BV, Markovic D, et al. Ischemia-reperfusion injury and its relationship with early allograft dysfunction in liver transplant patients. Am J Transplant. 2021;21(2): 614–625. [DOI] [PubMed] [Google Scholar]
- 4.King RC, Binns OA, Rodriguez F, et al. Reperfusion injury significantly impacts clinical outcome after pulmonary transplantation. Ann Thorac Surg. 2000;69(6): 1681–1685. [DOI] [PubMed] [Google Scholar]
- 5.Fiser SM, Tribble CG, Long SM, et al. Ischemia-reperfusion injury after lung transplantation increases risk of late bronchiolitis obliterans syndrome. Ann Thorac Surg. 2002;73(4): 1041–1047; discussion 1047–1048. [DOI] [PubMed] [Google Scholar]
- 6.Balsara KR, Krupnick AS, Bell JM, et al. A single-center experience of 1500 lung transplant patients. J Thorac Cardiovasc Surg. 2018;156(2): 894–905 e893. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.D’Alessandro C, Aubert S, Golmard JL, et al. Extra-corporeal membrane oxygenation temporary support for early graft failure after cardiac transplantation. Eur J Cardiothorac Surg. 2010;37(2): 343–349. [DOI] [PubMed] [Google Scholar]
- 8.Lima B, Rajagopal K, Petersen RP, et al. Marginal cardiac allografts do not have increased primary graft dysfunction in alternate list transplantation. Circulation. 2006;114(1 Suppl): I27–32. [DOI] [PubMed] [Google Scholar]
- 9.Olthoff KM, Kulik L, Samstein B, et al. Validation of a current definition of early allograft dysfunction in liver transplant recipients and analysis of risk factors. Liver Transpl. 2010;16(8): 943–949. [DOI] [PubMed] [Google Scholar]
- 10.Hoyer DP, Paul A, Gallinat A, et al. Donor information based prediction of early allograft dysfunction and outcome in liver transplantation. Liver Int. 2015;35(1): 156–163. [DOI] [PubMed] [Google Scholar]
- 11.Bastos-Neves D, Salvalaggio PRO, Almeida MD. Risk factors, surgical complications and graft survival in liver transplant recipients with early allograft dysfunction. Hepatobiliary Pancreat Dis Int. 2019;18(5): 423–429. [DOI] [PubMed] [Google Scholar]
- 12.Pfaff WW, Howard RJ, Patton PR, Adams VR, Rosen CB, Reed AI. Delayed graft function after renal transplantation. Transplantation. 1998;65(2): 219–223. [DOI] [PubMed] [Google Scholar]
- 13.Chambers DC, Cherikh WS, Goldfarb SB, et al. The International Thoracic Organ Transplant Registry of the International Society for Heart and Lung Transplantation: Thirty-fifth adult lung and heart-lung transplant report-2018; Focus theme: Multiorgan Transplantation. J Heart Lung Transplant. 2018;37(10): 1169–1183. [DOI] [PubMed] [Google Scholar]
- 14.Kobashigawa J, Zuckermann A, Macdonald P, et al. Report from a consensus conference on primary graft dysfunction after cardiac transplantation. J Heart Lung Transplant. 2014;33(4): 327–340. [DOI] [PubMed] [Google Scholar]
- 15.Briggs JD. Causes of death after renal transplantation. Nephrol Dial Transplant. 2001;16(8): 1545–1549. [DOI] [PubMed] [Google Scholar]
- 16.Baganate F, Beal EW, Tumin D, et al. Early mortality after liver transplantation: Defining the course and the cause. Surgery. 2018;164(4): 694–704. [DOI] [PubMed] [Google Scholar]
- 17.D’Alessandro AM, Ploeg RJ, Knechtle SJ, et al. Retransplantation of the liver--a seven-year experience. Transplantation. 1993;55(5): 1083–1087. [DOI] [PubMed] [Google Scholar]
- 18.Markmann JF, Markowitz JS, Yersiz H, et al. Long-term survival after retransplantation of the liver. Ann Surg. 1997;226(4): 408–418; discussion 418–420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Uemura T, Randall HB, Sanchez EQ, et al. Liver retransplantation for primary nonfunction: analysis of a 20-year single-center experience. Liver Transpl. 2007;13(2): 227–233. [DOI] [PubMed] [Google Scholar]
- 20.Girgis RE, Tu I, Berry GJ, et al. Risk factors for the development of obliterative bronchiolitis after lung transplantation. J Heart Lung Transplant. 1996;15(12): 1200–1208. [PubMed] [Google Scholar]
- 21.Daud SA, Yusen RD, Meyers BF, et al. Impact of immediate primary lung allograft dysfunction on bronchiolitis obliterans syndrome. Am J Respir Crit Care Med. 2007;175(5): 507–513. [DOI] [PubMed] [Google Scholar]
- 22.Whitson BA, Prekker ME, Herrington CS, et al. Primary graft dysfunction and long-term pulmonary function after lung transplantation. J Heart Lung Transplant. 2007;26(10): 1004–1011. [DOI] [PubMed] [Google Scholar]
- 23.Huang HJ, Yusen RD, Meyers BF, et al. Late primary graft dysfunction after lung transplantation and bronchiolitis obliterans syndrome. Am J Transplant. 2008;8(11): 2454–2462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Yamani MH, Haji SA, Starling RC, et al. Myocardial ischemic-fibrotic injury after human heart transplantation is associated with increased progression of vasculopathy, decreased cellular rejection and poor long-term outcome. J Am Coll Cardiol. 2002;39(6): 970–977. [DOI] [PubMed] [Google Scholar]
- 25.Hariharan S, McBride MA, Cherikh WS, et al. Post-transplant renal function in the first year predicts long-term kidney transplant survival. Kidney Int. 2002;62(1): 311–318. [DOI] [PubMed] [Google Scholar]
- 26.Yarlagadda SG, Coca SG, Formica RN Jr.,et al. Association between delayed graft function and allograft and patient survival: a systematic review and meta-analysis. Nephrol Dial Transplant. 2009;24(3): 1039–1047. [DOI] [PubMed] [Google Scholar]
- 27.Kouwenhoven EA, de Bruin RW, Heemann UW, et al. Late graft dysfunction after prolonged cold ischemia of the donor kidney: inhibition by cyclosporine. Transplantation. 1999;68(7): 1004–1010. [DOI] [PubMed] [Google Scholar]
- 28.Salvalaggio PR, Felga GE, Afonso RC, et al. Early allograft dysfunction and liver transplant outcomes: a single center retrospective study. Transplant Proc. 2012;44(8): 2449–2451. [DOI] [PubMed] [Google Scholar]
- 29.Schofield ZV, Woodruff TM, Halai R, et al. Neutrophils--a key component of ischemia-reperfusion injury. Shock. 2013;40(6): 463–470. [DOI] [PubMed] [Google Scholar]
- 30.Belperio JA, Keane MP, Burdick MD, et al. CXCR2/CXCR2 ligand biology during lung transplant ischemia-reperfusion injury. J Immunol. 2005;175(10): 6931–6939. [DOI] [PubMed] [Google Scholar]
- 31.Kreisel D, Nava RG, Li W, et al. In vivo two-photon imaging reveals monocyte-dependent neutrophil extravasation during pulmonary inflammation. Proc Natl Acad Sci U S A. 2010;107(42): 18073–18078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Kreisel D, Sugimoto S, Tietjens J, et al. Bcl3 prevents acute inflammatory lung injury in mice by restraining emergency granulopoiesis. J Clin Invest. 2011;121(1): 265–276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Tomizawa N, Ohwada S, Ohya T, et al. The effects of a neutrophil elastase inhibitor (ONO-5046.Na) and neutrophil depletion using a granulotrap (G-1) column on lung reperfusion injury in dogs. J Heart Lung Transplant. 1999;18(7): 637–645. [DOI] [PubMed] [Google Scholar]
- 34.Jaeschke H, Farhood A, Smith CW. Neutrophils contribute to ischemia/reperfusion injury in rat liver in vivo. FASEB J. 1990;4(15): 3355–3359. [PubMed] [Google Scholar]
- 35.Li W, Feng G, Gauthier JM, et al. Ferroptotic cell death and TLR4/Trif signaling initiate neutrophil recruitment after heart transplantation. J Clin Invest. 2019;129(6): 2293–2304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Li W, Hsiao HM, Higashikubo R, et al. Heart-resident CCR2(+) macrophages promote neutrophil extravasation through TLR9/MyD88/CXCL5 signaling. JCI Insight. 2016;1(12). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Li W, Nava RG, Bribriesco AC, et al. Intravital 2-photon imaging of leukocyte trafficking in beating heart. J Clin Invest. 2012;122(7): 2499–2508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Zheng Z, Chiu S, Akbarpour M, et al. Donor pulmonary intravascular nonclassical monocytes recruit recipient neutrophils and mediate primary lung allograft dysfunction. Sci Transl Med. 2017;9(394). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Spahn JH, Li W, Bribriesco AC, et al. DAP12 expression in lung macrophages mediates ischemia/reperfusion injury by promoting neutrophil extravasation. J Immunol. 2015;194(8): 4039–4048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Hsiao HM, Fernandez R, Tanaka S, et al. Spleen-derived classical monocytes mediate lung ischemia-reperfusion injury through IL-1beta. J Clin Invest. 2018;128(7): 2833–2847. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Charles EJ, Chordia MD, Zhao Y, et al. SPECT imaging of lung ischemia-reperfusion injury using [(99m)Tc]cFLFLF for molecular targeting of formyl peptide receptor 1. Am J Physiol Lung Cell Mol Physiol. 2020;318(2): L304–L313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Honda M, Takeichi T, Asonuma K, et al. Intravital imaging of neutrophil recruitment in hepatic ischemia-reperfusion injury in mice. Transplantation. 2013;95(4): 551–558. [DOI] [PubMed] [Google Scholar]
- 43.Camirand G, Li Q, Demetris AJ, et al. Multiphoton intravital microscopy of the transplanted mouse kidney. Am J Transplant. 2011;11(10): 2067–2074. [DOI] [PubMed] [Google Scholar]
- 44.Maruyama Y, Araki M, Kidokoro K, et al. Evaluation of Neutrophil Dynamics Change by Protective Effect of Tadalafil After Renal Ischemia/Reperfusion Using In Vivo Real-time Imaging. Transplantation. 2022;106(2): 280–288. [DOI] [PubMed] [Google Scholar]
- 45.Staub NC, Schultz EL, Albertine KH. Leucocytes and pulmonary microvascular injury. Ann N Y Acad Sci. 1982;384: 332–343. [DOI] [PubMed] [Google Scholar]
- 46.Peters AM, Saverymuttu SH, Bell RN, et al. Quantification of the distribution of the marginating granulocyte pool in man. Scand J Haematol. 1985;34(2): 111–120. [DOI] [PubMed] [Google Scholar]
- 47.Lien DC, Wagner WW Jr., Capen RL, et al. Physiological neutrophil sequestration in the lung: visual evidence for localization in capillaries. J Appl Physiol (1985). 1987;62(3): 1236–1243. [DOI] [PubMed] [Google Scholar]
- 48.Yipp BG, Kim JH, Lima R, et al. The Lung is a Host Defense Niche for Immediate Neutrophil-Mediated Vascular Protection. Sci Immunol. 2017;2(10). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Casanova-Acebes M, Nicolas-Avila JA, Li JL, et al. Neutrophils instruct homeostatic and pathological states in naive tissues. J Exp Med. 2018;215(11): 2778–2795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Devi S, Wang Y, Chew WK, et al. Neutrophil mobilization via plerixafor-mediated CXCR4 inhibition arises from lung demargination and blockade of neutrophil homing to the bone marrow. J Exp Med. 2013;210(11): 2321–2336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Pavlosky A, Lau A, Su Y, et al. RIPK3-Mediated Necroptosis Regulates Cardiac Allograft Rejection. Am J Transplant. 2014;14(8): 1778–1790. [DOI] [PubMed] [Google Scholar]
- 52.Li W, Terada Y, Tyurina YY, et al. Necroptosis triggers spatially restricted neutrophil-mediated vascular damage during lung ischemia reperfusion injury. Proc Natl Acad Sci U S A. 2022;119(10): e2111537119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Kanou T, Ohsumi A, Kim H, et al. Inhibition of regulated necrosis attenuates receptor-interacting protein kinase 1-mediated ischemia-reperfusion injury after lung transplantation. J Heart Lung Transplant. 2018;37(10): 1261–1270. [DOI] [PubMed] [Google Scholar]
- 54.Lau A, Wang S, Jiang J, et al. RIPK3-mediated necroptosis promotes donor kidney inflammatory injury and reduces allograft survival. Am J Transplant. 2013;13(11): 2805–2818. [DOI] [PubMed] [Google Scholar]
- 55.Linkermann A, Brasen JH, Darding M, et al. Two independent pathways of regulated necrosis mediate ischemia-reperfusion injury. Proc Natl Acad Sci U S A. 2013;110(29): 12024–12029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Tonnus W, Meyer C, Steinebach C, et al. Dysfunction of the key ferroptosis-surveilling systems hypersensitizes mice to tubular necrosis during acute kidney injury. Nat Commun. 2021;12(1): 4402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Linkermann A, Skouta R, Himmerkus N, et al. Synchronized renal tubular cell death involves ferroptosis. Proc Natl Acad Sci U S A. 2014;111(47): 16836–16841. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Ibrahim M, Wang X, Puyo CA, et al. Human recombinant apyrase therapy protects against canine pulmonary ischemia-reperfusion injury. J Heart Lung Transplant. 2015;34(2): 247–253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Prakash A, Mesa KR, Wilhelmsen K, et al. Alveolar macrophages and Toll-like receptor 4 mediate ventilated lung ischemia reperfusion injury in mice. Anesthesiology. 2012;117(4): 822–835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Kaczorowski DJ, Nakao A, McCurry KR, et al. Toll-like receptors and myocardial ischemia/reperfusion, inflammation, and injury. Curr Cardiol Rev. 2009;5(3): 196–202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Kruger B, Krick S, Dhillon N, et al. Donor Toll-like receptor 4 contributes to ischemia and reperfusion injury following human kidney transplantation. Proc Natl Acad Sci U S A. 2009;106(9): 3390–3395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Tsung A, Hoffman RA, Izuishi K, et al. Hepatic ischemia/reperfusion injury involves functional TLR4 signaling in nonparenchymal cells. J Immunol. 2005;175(11): 7661–7668. [DOI] [PubMed] [Google Scholar]
- 63.Wu H, Chen G, Wyburn KR, et al. TLR4 activation mediates kidney ischemia/reperfusion injury. J Clin Invest. 2007;117(10): 2847–2859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Tsung A, Sahai R, Tanaka H, et al. The nuclear factor HMGB1 mediates hepatic injury after murine liver ischemia-reperfusion. J Exp Med. 2005;201(7): 1135–1143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Li J, Gong Q, Zhong S, et al. Neutralization of the extracellular HMGB1 released by ischaemic damaged renal cells protects against renal ischaemia-reperfusion injury. Nephrol Dial Transplant. 2011;26(2): 469–478. [DOI] [PubMed] [Google Scholar]
- 66.Lentsch AB, Yoshidome H, Cheadle WG, et al. Chemokine involvement in hepatic ischemia/reperfusion injury in mice: roles for macrophage inflammatory protein-2 and KC. Hepatology. 1998;27(4): 1172–1177. [DOI] [PubMed] [Google Scholar]
- 67.Birdsall HH, Green DM, Trial J, et al. Complement C5a, TGF-beta 1, and MCP-1, in sequence, induce migration of monocytes into ischemic canine myocardium within the first one to five hours after reperfusion. Circulation. 1997;95(3): 684–692. [DOI] [PubMed] [Google Scholar]
- 68.Oz MC, Liao H, Naka Y, et al. Ischemia-induced interleukin-8 release after human heart transplantation. A potential role for endothelial cells. Circulation. 1995;92(9 Suppl): II428–432. [DOI] [PubMed] [Google Scholar]
- 69.Kreisel D, Sugimoto S, Zhu J, et al. Emergency granulopoiesis promotes neutrophil-dendritic cell encounters that prevent mouse lung allograft acceptance. Blood. 2011;118(23): 6172–6182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Volkmann J, Schmitz J, Nordlohne J, et al. Kidney injury enhances renal G-CSF expression and modulates granulopoiesis and human neutrophil CD177 in vivo. Clin Exp Immunol. 2020;199(1): 97–108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Mosher B, Dean R, Harkema J, et al. Inhibition of Kupffer cells reduced CXC chemokine production and liver injury. J Surg Res. 2001;99(2): 201–210. [DOI] [PubMed] [Google Scholar]
- 72.Ferenbach DA, Sheldrake TA, Dhaliwal K, et al. Macrophage/monocyte depletion by clodronate, but not diphtheria toxin, improves renal ischemia/reperfusion injury in mice. Kidney Int. 2012;82(8): 928–933. [DOI] [PubMed] [Google Scholar]
- 73.Aiello S, Podesta MA, Rodriguez-Ordonez PY, et al. Transplantation-Induced Ischemia-Reperfusion Injury Modulates Antigen Presentation by Donor Renal CD11c(+)F4/80(+) Macrophages through IL-1R8 Regulation. J Am Soc Nephrol. 2020;31(3): 517–531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Zhang M, Ueki S, Kimura S, et al. Roles of dendritic cells in murine hepatic warm and liver transplantation-induced cold ischemia/reperfusion injury. Hepatology. 2013;57(4): 1585–1596. [DOI] [PubMed] [Google Scholar]
- 75.Sawa Y, Ichikawa H, Kagisaki K, et al. Interleukin-6 derived from hypoxic myocytes promotes neutrophil-mediated reperfusion injury in myocardium. J Thorac Cardiovasc Surg. 1998;116(3): 511–517. [DOI] [PubMed] [Google Scholar]
- 76.Frangogiannis NG, Lindsey ML, Michael LH, et al. Resident cardiac mast cells degranulate and release preformed TNF-alpha, initiating the cytokine cascade in experimental canine myocardial ischemia/reperfusion. Circulation. 1998;98(7): 699–710. [DOI] [PubMed] [Google Scholar]
- 77.Kukielka GL, Hawkins HK, Michael L, et al. Regulation of intercellular adhesion molecule-1 (ICAM-1) in ischemic and reperfused canine myocardium. J Clin Invest. 1993;92(3): 1504–1516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Yamazaki T, Seko Y, Tamatani T, et al. Expression of intercellular adhesion molecule-1 in rat heart with ischemia/reperfusion and limitation of infarct size by treatment with antibodies against cell adhesion molecules. Am J Pathol. 1993;143(2): 410–418. [PMC free article] [PubMed] [Google Scholar]
- 79.Bowden RA, Ding ZM, Donnachie EM, et al. Role of alpha4 integrin and VCAM-1 in CD18-independent neutrophil migration across mouse cardiac endothelium. Circ Res. 2002;90(5): 562–569. [DOI] [PubMed] [Google Scholar]
- 80.Dragun D, Hoff U, Park JK, et al. Ischemia-reperfusion injury in renal transplantation is independent of the immunologic background. Kidney Int. 2000;58(5): 2166–2177. [DOI] [PubMed] [Google Scholar]
- 81.Chen J, John R, Richardson JA, et al. Toll-like receptor 4 regulates early endothelial activation during ischemic acute kidney injury. Kidney Int. 2011;79(3): 288–299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Nagendra AR, Mickelson JK, Smith CW. CD18 integrin and CD54-dependent neutrophil adhesion to cytokine-stimulated human hepatocytes. Am J Physiol. 1997;272(3 Pt 1): G408–416. [DOI] [PubMed] [Google Scholar]
- 83.Farhood A, McGuire GM, Manning AM, et al. Intercellular adhesion molecule 1 (ICAM-1) expression and its role in neutrophil-induced ischemia-reperfusion injury in rat liver. J Leukoc Biol. 1995;57(3): 368–374. [PubMed] [Google Scholar]
- 84.McKeown CM, Edwards V, Phillips MJ, et al. Sinusoidal lining cell damage: the critical injury in cold preservation of liver allografts in the rat. Transplantation. 1988;46(2): 178–191. [PubMed] [Google Scholar]
- 85.Nishimura Y, Takei Y, Kawano S, et al. The F(ab’)2 fragment of an anti-ICAM-1 monoclonal antibody attenuates liver injury after orthotopic liver transplantation. Transplantation. 1996;61(1): 99–104. [DOI] [PubMed] [Google Scholar]
- 86.Rentsch M, Post S, Palma P, et al. Anti-ICAM-1 blockade reduces postsinusoidal WBC adherence following cold ischemia and reperfusion, but does not improve early graft function in rat liver transplantation. J Hepatol. 2000;32(5): 821–828. [DOI] [PubMed] [Google Scholar]
- 87.Vollmar B, Glasz J, Menger MD, et al. Leukocytes contribute to hepatic ischemia/reperfusion injury via intercellular adhesion molecule-1-mediated venular adherence. Surgery. 1995;117(2): 195–200. [DOI] [PubMed] [Google Scholar]
- 88.Choudhury SR, Babes L, Rahn JJ, et al. Dipeptidase-1 Is an Adhesion Receptor for Neutrophil Recruitment in Lungs and Liver. Cell. 2019;178(5): 1205–1221 e1217. [DOI] [PubMed] [Google Scholar]
- 89.Akbarpour M, Lecuona E, Chiu SF, et al. Residual endotoxin induces primary graft dysfunction through ischemia/reperfusion-primed alveolar macrophages. J Clin Invest. 2020;130(8): 4456–4469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Gelman AE, Okazaki M, Sugimoto S, et al. CCR2 regulates monocyte recruitment as well as CD4 T1 allorecognition after lung transplantation. Am J Transplant. 2010;10(5): 1189–1199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Liu Y, Li W, Luehmann HP, et al. Noninvasive Imaging of CCR2(+) Cells in Ischemia-Reperfusion Injury After Lung Transplantation. Am J Transplant. 2016;16(10): 3016–3023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Kurihara C, Lecuona E, Wu Q, et al. Crosstalk between nonclassical monocytes and alveolar macrophages mediates transplant ischemia-reperfusion injury through classical monocyte recruitment. JCI Insight. 2021;6(6). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Karasawa K, Asano K, Moriyama S, et al. Vascular-resident CD169-positive monocytes and macrophages control neutrophil accumulation in the kidney with ischemia-reperfusion injury. J Am Soc Nephrol. 2015;26(4): 896–906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Knauper V, Murphy G, Tschesche H. Activation of human neutrophil procollagenase by stromelysin 2. Eur J Biochem. 1996;235(1–2): 187–191. [DOI] [PubMed] [Google Scholar]
- 95.Murphy G, Ward R, Gavrilovic J, et al. Physiological mechanisms for metalloproteinase activation. Matrix Suppl. 1992;1: 224–230. [PubMed] [Google Scholar]
- 96.Peppin GJ, Weiss SJ. Activation of the endogenous metalloproteinase, gelatinase, by triggered human neutrophils. Proc Natl Acad Sci U S A. 1986;83(12): 4322–4326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Tian X, Sun H, Casbon AJ, et al. NLRP3 Inflammasome Mediates Dormant Neutrophil Recruitment following Sterile Lung Injury and Protects against Subsequent Bacterial Pneumonia in Mice. Front Immunol. 2017;8: 1337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Eppinger MJ, Jones ML, Deeb GM, et al. Pattern of injury and the role of neutrophils in reperfusion injury of rat lung. J Surg Res. 1995;58(6): 713–718. [DOI] [PubMed] [Google Scholar]
- 99.Ganesan R, Henkels KM, Shah K, et al. D-series Resolvins activate Phospholipase D in phagocytes during inflammation and resolution. FASEB J. 2020;34(12): 15888–15906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Dalli J, Serhan CN. Specific lipid mediator signatures of human phagocytes: microparticles stimulate macrophage efferocytosis and pro-resolving mediators. Blood. 2012;120(15): e60–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Duilio C, Ambrosio G, Kuppusamy P, et al. Neutrophils are primary source of O2 radicals during reperfusion after prolonged myocardial ischemia. Am J Physiol Heart Circ Physiol. 2001;280(6): H2649–2657. [DOI] [PubMed] [Google Scholar]
- 102.Kimura K, Shirabe K, Yoshizumi T, et al. Ischemia-Reperfusion Injury in Fatty Liver Is Mediated by Activated NADPH Oxidase 2 in Rats. Transplantation. 2016;100(4): 791–800. [DOI] [PubMed] [Google Scholar]
- 103.Uchida Y, Freitas MC, Zhao D, Bet al. The inhibition of neutrophil elastase ameliorates mouse liver damage due to ischemia and reperfusion. Liver Transpl. 2009;15(8): 939–947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Ishikawa N, Oda M, Kawaguchi M, et al. The effects of a specific neutrophil elastase inhibitor (ONO-5046) in pulmonary ischemia-reperfusion injury. Transpl Int. 2003;16(5): 341–346. [DOI] [PubMed] [Google Scholar]
- 105.Aoki T, Tsuchida M, Takekubo M, et al. Neutrophil elastase inhibitor ameliorates reperfusion injury in a canine model of lung transplantation. Eur Surg Res. 2005;37(5): 274–280. [DOI] [PubMed] [Google Scholar]
- 106.Mori H, Nagahiro I, Osaragi T, et al. Addition of a neutrophil elastase inhibitor to the organ flushing solution decreases lung reperfusion injury in rat lung transplantation. Eur J Cardiothorac Surg. 2007;32(5): 791–795. [DOI] [PubMed] [Google Scholar]
- 107.Harada M, Oto T, Otani S, et al. A neutrophil elastase inhibitor improves lung function during ex vivo lung perfusion. Gen Thorac Cardiovasc Surg. 2015;63(12): 645–651. [DOI] [PubMed] [Google Scholar]
- 108.Kuyvenhoven JP, Molenaar IQ, Verspaget HW, et al. Plasma MMP-2 and MMP-9 and their inhibitors TIMP-1 and TIMP-2 during human orthotopic liver transplantation. The effect of aprotinin and the relation to ischemia/reperfusion injury. Thromb Haemost. 2004;91(3): 506–513. [DOI] [PubMed] [Google Scholar]
- 109.Uchida Y, Freitas MC, Zhao D, et al. The protective function of neutrophil elastase inhibitor in liver ischemia/reperfusion injury. Transplantation. 2010;89(9): 1050–1056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Moore C, Shen XD, Gao F, et al. Fibronectin-alpha4beta1 integrin interactions regulate metalloproteinase-9 expression in steatotic liver ischemia and reperfusion injury. Am J Pathol. 2007;170(2): 567–577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Amersi F, Shen XD, Moore C, et al. Fibronectin-alpha 4 beta 1 integrin-mediated blockade protects genetically fat Zucker rat livers from ischemia/reperfusion injury. Am J Pathol. 2003;162(4): 1229–1239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Hamada T, Fondevila C, Busuttil RW, et al. Metalloproteinase-9 deficiency protects against hepatic ischemia/reperfusion injury. Hepatology. 2008;47(1): 186–198. [DOI] [PubMed] [Google Scholar]
- 113.Soccal PM, Gasche Y, Miniati DN, et al. Matrix metalloproteinase inhibition decreases ischemia-reperfusion injury after lung transplantation. Am J Transplant. 2004;4(1): 41–50. [DOI] [PubMed] [Google Scholar]
- 114.Yano M, Omoto Y, Yamakawa Y, et al. Increased matrix metalloproteinase 9 activity and mRNA expression in lung ischemia-reperfusion injury. J Heart Lung Transplant. 2001;20(6): 679–686. [DOI] [PubMed] [Google Scholar]
- 115.Iwata T, Chiyo M, Yoshida S, et al. Lung transplant ischemia reperfusion injury: metalloprotease inhibition down-regulates exposure of type V collagen, growth-related oncogene-induced neutrophil chemotaxis, and tumor necrosis factor-alpha expression. Transplantation. 2008;85(3): 417–426. [DOI] [PubMed] [Google Scholar]
- 116.Cheung PY, Sawicki G, Wozniak M, et al. Matrix metalloproteinase-2 contributes to ischemia-reperfusion injury in the heart. Circulation. 2000;101(15): 1833–1839. [DOI] [PubMed] [Google Scholar]
- 117.Wang W, Schulze CJ, Suarez-Pinzon WL, et al. Intracellular action of matrix metalloproteinase-2 accounts for acute myocardial ischemia and reperfusion injury. Circulation. 2002;106(12): 1543–1549. [DOI] [PubMed] [Google Scholar]
- 118.Lindsey M, Wedin K, Brown MD, et al. Matrix-dependent mechanism of neutrophil-mediated release and activation of matrix metalloproteinase 9 in myocardial ischemia/reperfusion. Circulation. 2001;103(17): 2181–2187. [DOI] [PubMed] [Google Scholar]
- 119.Carbone F, Crowe LA, Roth A, et al. Treatment with anti-RANKL antibody reduces infarct size and attenuates dysfunction impacting on neutrophil-mediated injury. J Mol Cell Cardiol. 2016;94: 82–94. [DOI] [PubMed] [Google Scholar]
- 120.Scozzi D, Ibrahim M, Liao F, et al. Mitochondrial damage-associated molecular patterns released by lung transplants are associated with primary graft dysfunction. Am J Transplant. 2019;19(5): 1464–1477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Torres-Ruiz J, Villca-Gonzales R, Gomez-Martin D, et al. A potential role of neutrophil extracellular traps (NETs) in kidney acute antibody mediated rejection. Transpl Immunol. 2020;60: 101286. [DOI] [PubMed] [Google Scholar]
- 122.Sayah DM, Mallavia B, Liu F, et al. Neutrophil extracellular traps are pathogenic in primary graft dysfunction after lung transplantation. Am J Respir Crit Care Med. 2015;191(4): 455–463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Liu Y, Qin X, Lei Z, et al. Tetramethylpyrazine inhibits neutrophil extracellular traps formation and alleviates hepatic ischemia/reperfusion injury in rat liver transplantation. Exp Cell Res. 2021;406(1): 112719. [DOI] [PubMed] [Google Scholar]
- 124.Liu Y, Lei Z, Chai H, et al. Thrombomodulin-mediated Inhibition of Neutrophil Extracellular Trap Formation Alleviates Hepatic Ischemia-reperfusion Injury by Blocking TLR4 in Rats Subjected to Liver Transplantation. Transplantation. 2022;106(2): e126–e140. [DOI] [PubMed] [Google Scholar]
- 125.Mallavia B, Liu F, Lefrancais E, et al. Mitochondrial DNA Stimulates TLR9-Dependent Neutrophil Extracellular Trap Formation in Primary Graft Dysfunction. Am J Respir Cell Mol Biol. 2020;62(3): 364–372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Feng Z, Qi S, Zhang Y, et al. Ly6G+ neutrophil-derived miR-223 inhibits the NLRP3 inflammasome in mitochondrial DAMP-induced acute lung injury. Cell Death Dis. 2017;8(11): e3170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Ye C, Qi W, Dai S, et al. microRNA-223 promotes autophagy to aggravate lung ischemia-reperfusion injury by inhibiting the expression of transcription factor HIF2alpha. Am J Physiol Lung Cell Mol Physiol. 2020. [DOI] [PubMed] [Google Scholar]
- 128.Neudecker V, Brodsky KS, Clambey ET, et al. Neutrophil transfer of miR-223 to lung epithelial cells dampens acute lung injury in mice. Sci Transl Med. 2017;9(408). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Yu CH, Xu CF, Li YM. Association of MicroRNA-223 expression with hepatic ischemia/reperfusion injury in mice. Dig Dis Sci. 2009;54(11): 2362–2366. [DOI] [PubMed] [Google Scholar]
- 130.Eltzschig HK, Ibla JC, Furuta GT, et al. Coordinated adenine nucleotide phosphohydrolysis and nucleoside signaling in posthypoxic endothelium: role of ectonucleotidases and adenosine A2B receptors. J Exp Med. 2003;198(5): 783–796. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Li J, Conrad C, Mills TW, et al. PMN-derived netrin-1 attenuates cardiac ischemia-reperfusion injury via myeloid ADORA2B signaling. J Exp Med. 2021;218(6). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Grenz A, Osswald H, Eckle T, et al. The reno-vascular A2B adenosine receptor protects the kidney from ischemia. PLoS Med. 2008;5(6): e137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Anvari F, Sharma AK, Fernandez LG, et al. Tissue-derived proinflammatory effect of adenosine A2B receptor in lung ischemia-reperfusion injury. J Thorac Cardiovasc Surg. 2010;140(4): 871–877. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Shimamura K, Kawamura H, Nagura T, et al. Association of NKT cells and granulocytes with liver injury after reperfusion of the portal vein. Cell Immunol. 2005;234(1): 31–38. [DOI] [PubMed] [Google Scholar]
- 135.Victorino F, Sojka DK, Brodsky KS, et al. Tissue-Resident NK Cells Mediate Ischemic Kidney Injury and Are Not Depleted by Anti-Asialo-GM1 Antibody. J Immunol. 2015;195(10): 4973–4985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Gelman AE, Li W, Richardson SB, et al. Cutting edge: Acute lung allograft rejection is independent of secondary lymphoid organs. J Immunol. 2009;182(7): 3969–3973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Scozzi D, Wang X, Liao F, et al. Neutrophil extracellular trap fragments stimulate innate immune responses that prevent lung transplant tolerance. Am J Transplant. 2019;19(4): 1011–1023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Miyairi S, Ueda D, Yagisawa T, et al. Recipient myeloperoxidase-producing cells regulate antibody-mediated acute versus chronic kidney allograft rejection. JCI Insight. 2021;6(13). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Kohei N, Tanaka T, Tanabe K, et al. Natural killer cells play a critical role in mediating inflammation and graft failure during antibody-mediated rejection of kidney allografts. Kidney Int. 2016;89(6): 1293–1306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.El-Sawy T, Belperio JA, Strieter RM, Remick DG, Fairchild RL. Inhibition of polymorphonuclear leukocyte-mediated graft damage synergizes with short-term costimulatory blockade to prevent cardiac allograft rejection. Circulation. 2005;112(3): 320–331. [DOI] [PubMed] [Google Scholar]
- 141.Duffy D, Perrin H, Abadie V, et al. Neutrophils transport antigen from the dermis to the bone marrow, initiating a source of memory CD8+ T cells. Immunity. 2012;37(5): 917–929. [DOI] [PubMed] [Google Scholar]
- 142.Maletto BA, Ropolo AS, Alignani DO, et al. Presence of neutrophil-bearing antigen in lymphoid organs of immune mice. Blood. 2006;108(9): 3094–3102. [DOI] [PubMed] [Google Scholar]
- 143.Matsushima H, Geng S, Lu R, et al. Neutrophil differentiation into a unique hybrid population exhibiting dual phenotype and functionality of neutrophils and dendritic cells. Blood. 2013;121(10): 1677–1689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Kish DD, Min S, Dvorina N, Baldwin WM, 3rd, Stohlman SA, Fairchild RL. Neutrophil Cathepsin G Regulates Dendritic Cell Production of IL-12 during Development of CD4 T Cell Responses to Antigens in the Skin. J Immunol. 2019;202(4): 1045–1056. [DOI] [PMC free article] [PubMed] [Google Scholar]