

Abstract
The high prevalence of neurodegenerative diseases has become a major public health challenge and is associated with a tremendous burden on individuals, society and federal governments worldwide. Protein misfolding and aggregation are the major pathological hallmarks of several neurodegenerative disorders. The cells have evolved several regulatory mechanisms to deal with aberrant protein folding, namely the classical ubiquitin pathway, where ubiquitination of protein aggregates marks their degradation via lysosome and the novel autophagy or mitophagy pathways. Autophagy is a catabolic process in eukaryotic cells that allows the lysosome to recycle the cell’s own contents, such as organelles and proteins, known as autophagic cargo. Their most significant role is to keep cells alive in distressed situations. Mitophagy is also crucial for reducing abnormal protein aggregation and increasing organelle clearance and partly accounts for maintaining cellular homeostasis. Furthermore, substantial data indicate that any disruption in these homeostatic mechanisms leads to the emergence of several age-associated metabolic and neurodegenerative diseases. So, targeting autophagy and mitophagy might be a potential therapeutic strategy for a variety of health conditions.
Keywords: Autophagy, Mitophagy, Neurodegenerative disorders, Misfolding, Aggregation, Autophagic cargo, Homeostasis, Functional foods
Graphical Abstract
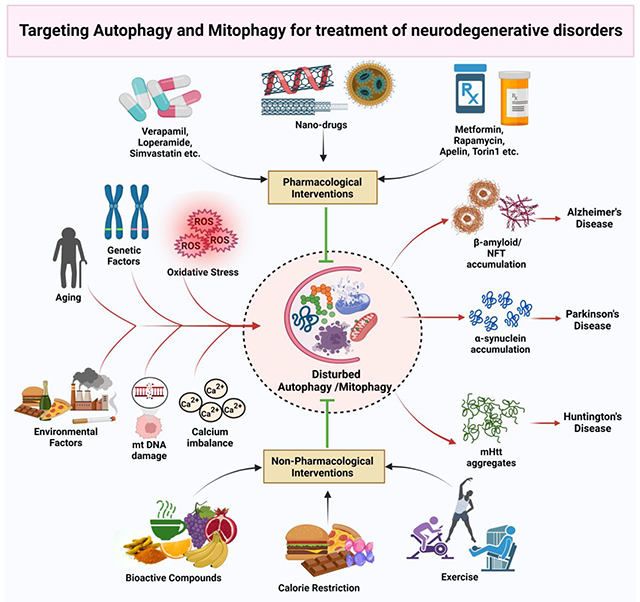
Introduction
Population ageing is a global phenomenon that must be recognized to combat the risk of neurodegeneration in society and promote healthier and longer lifespans. Multiple comorbidities are included in the definition of neurodegenerative disorders as heterogeneous diseases. Escalating neurodegeneration is a crippling ailment defined as the progressive loss of nerve connections and neuronal cells required for normal functioning. Dysfunction of cognitive ability increases the vulnerability of the appearance of other metabolic functions. The increasing burden of neurodegenerative diseases like Alzheimer’s disease (AD), Parkinson’s disease (PD), Huntington’s disease (HD), and Amyotrophic lateral sclerosis (ALS) primarily affect the elderly population by hampering their socioeconomic status. Hence, the gleaming phase of research is now focused on understanding the disease pathophysiology, diagnosis and therapeutics for numerous debilitating neuro disorders. Many neurodegenerative diseases manifest symptoms in their late stages, so the global burden of some fatal and frequently occurring neurodegenerative diseases is still an enigma for researchers. Currently, more than 55 million people have dementia, among which 6.5 million are living with Alzheimer’s dementia, and 10 million new cases are reported yearly. This figure is expected to rise to 78 million by 2030 and 139 million by 2050 (Gauthier et al., 2021). A study on the US population suggests that about 1 million people are living with PD, projected to rise to 1.2 million by 2030 (Bloem et al., 2021). Similarly, HD, a monogenetic neurodegenerative disease, appears in 1-15 people per million (Martí-Martínez and Valor, 2022).
Both genetic and environmental risk factors are known to play a critical role in the pathophysiology of these neurological. Genetic mutations are a significant risk factor as they lead to the production of unfolded and toxic proteins involved in disease progression (Basavarajappa et al., 2017). However, oxidative stress, endocrine dysfunction, infections and inflammatory responses, depression, trauma, vascular conditions and type of nutrition are remarkable risk factors for neurodegenerative diseases (Brown et al., 2005). Amateurish biological molecule synthesis results in the dysregulation of various cellular functions, including oxidative phosphorylation, ATP generation, and other metabolic pathways. Unchecked phospholipid metabolism and enzymatic pathways resulted in excessive generation of free radicals, which then alter the antioxidant system, and organelle dysfunction. Exponential increase in the reactive oxygen species (ROS) compromise calcium homeostasis, which affects the regulation of ETC, lysosomal processes, neuronal integrity, and mitochondrial function. Furthermore, it causes DNA damage and exhibits detrimental effects on cellular senescence (Singh et al., 2019). These dysregulated cellular changes obstruct essential cellular functions including autophagy and mitophagy. Failure to keep cells in a state of homeostasis and produce vital biological molecules results in the synthesis of abnormal proteins, resulted in dysfunctional autophagy/mitophagy in neurons. Additionally, sedentary life style, junk foods or dietary components low in antioxidants play a significant role in the free radical formation, which interact with the autophagic pathways by blocking or activating several autophagic regulators (Hannigan and Gorski, 2009). Figure 1 demonstrates an overall view of the risk determinants contributing to neuron degeneration.
Figure 1: An overview of risk factors contributing to neuron degeneration in pathogenic conditions.
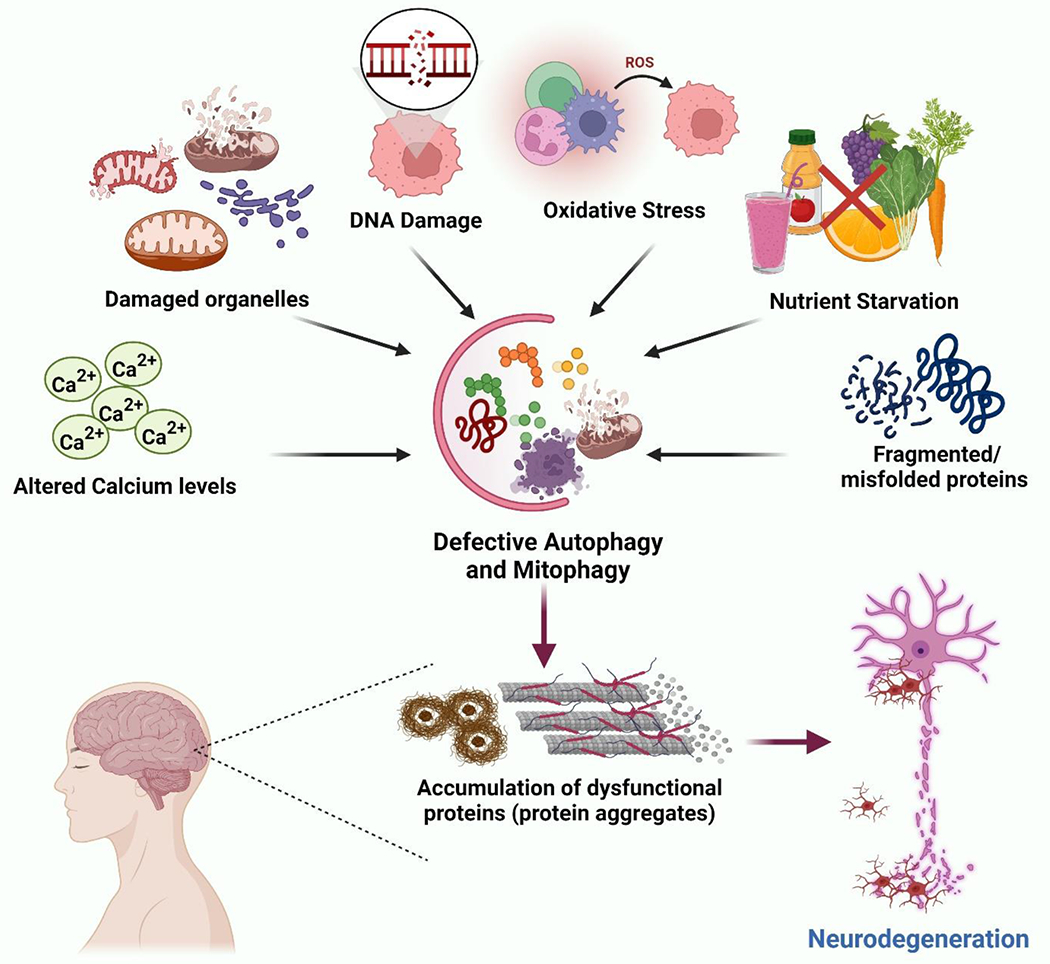
Multiple risk factors include increased ROS production, nutrient deprivation, oxidative stress, DNA damage, accumulation of misfolded proteins, damaged organelles etc., representing the complex nature of neurodegenerative diseases. Likewise, the interaction between these factors further targets basic homeostatic mechanisms such as autophagy and mitophagy, thereby promoting the pathogenesis of various neurodegenerative disorders.
Mitochondrial function is critical for optimal health and brain functions. Its dysfunction can result in neurotoxic chemicals, which can actually deteriorate brain health. Excessive ROS generation and oxidative stress can damage mitochondrial and nuclear DNA and alter the cell cycle, apoptosis, and senescence, negatively impacting the cell viability. Further, environmental and internal stimuli also significantly affect calcium homeostasis, electron transport chain, and energy homeostasis, leading to defective autophagy and mitophagy, which causes neurodegeneration (Facecchia et al., 2011). A substantial corpus of research has focused on the pathophysiology of neurodegenerative disorders and their risk determinants. Thus, molecular signalling pathways can now be targeted. Autophagy and mitophagy are essential mechanisms implicated in homeostasis regulation and modulating many signalling pathways linked to various disorders, including neurodegenerative diseases. These distinct processes are critical for individuals suffering from neurodegenerative diseases. Damaged cells show impaired cellular functions and undergo clearance via autophagic pathways (Yim and Mizushima, 2020). Most neurological disorders share a common molecular mechanism known as proteinopathies. Proteotoxic stress and its accompanying aberrations in the ubiquitin–proteasomal and autophagosomal/lysosomal systems, apoptosis, oxidative stress, and inflammation in the nervous system are all linked with the dysfunction and death of neuronal cells (Dugger and Dickson, 2017). Acquisition of aggregated-prone proteins, misfolded proteins, and distorted or toxic proteins leads to the progression of many neurodegenerative diseases (Taylor et al., 2002).
The ubiquitin-mediated proteasome system eliminates altered variants of normal proteins prone to aggregation and misfolded proteins and lysosomal degradation, accomplished through autophagy (Limanaqi et al., 2020). After the degradation of these macromolecules, the resulting products, like free amino acids, are also recycled and reused by the cell when the energy requirement is high. Autophagy is upregulated to protect cells against stress responses, starvation, and high energy requirements. Energy is obtained from the breakdown of protein aggregations or accumulations, organellar structures, heterophagic removal of apoptotic cells, and other cytosolic components. Several pharmacological and non-pharmacological approaches have been used to treat or delay the progression of these age-associated neurodegenerative diseases. This article mainly focuses on molecular mechanisms, modulating the autophagy and mitophagy processes in various neurological disorders via physical exercises, lifestyle modifications, and dietary supplementation of bioactive compounds.
Autophagy
Autophagy is a physiological biodegradation process that entails the transfer of cytoplasmic cargo to the lysosome for lysosomal disruption. The autophagy process is known to remove the toxic protein aggregates, contributes to the operation of recycling of residual proteins, helps in cell renewal, and generatingbuilding blocks for the regeneration of cells. Therefore, it can be considered an essential process in neurodegenerative disorders (Doherty and Baehrecke, 2018). Autophagy is regulated by evolutionarily conserved autophagy-related (ATG) genes and is necessary for cellular, tissue, and organismal integrity (Levine and Kroemer, 2008, Levine and Kroemer, 2019). The protein elements that comprise the fundamental autophagy machinery are mainly conserved from yeast to mammals. The initiation process, elongation of the phagophore, maturation of the autophagosome, autophagosome union with the lysosome, and proteolytic breakdown of the contents are the stages that occur in autophagy (Feng et al., 2014). There are three fundamental kinds of autophagy: (a) macroautophagy, (b) chaperone-mediated autophagy (CMA), and (c) microautophagy seen in mammalian cells, as depicted in Fig. 2, all of which are determined by how the protein is carried to the lysosome. The amount of the removed substrates and the extent of their degradation are used to classify various forms of autophagy. They all share the trait of lysosomal disintegration of degraded proteins, but their processes for transporting the substrate to the lysosome differ (Bednarczyk et al., 2018). Selective autophagy is the macroautophagy of a single cellular component, such as mitophagy for mitochondria, lipophagy for lipids, and pexophagy for peroxisomes.
Figure 2: An overview of various autophagic pathways.
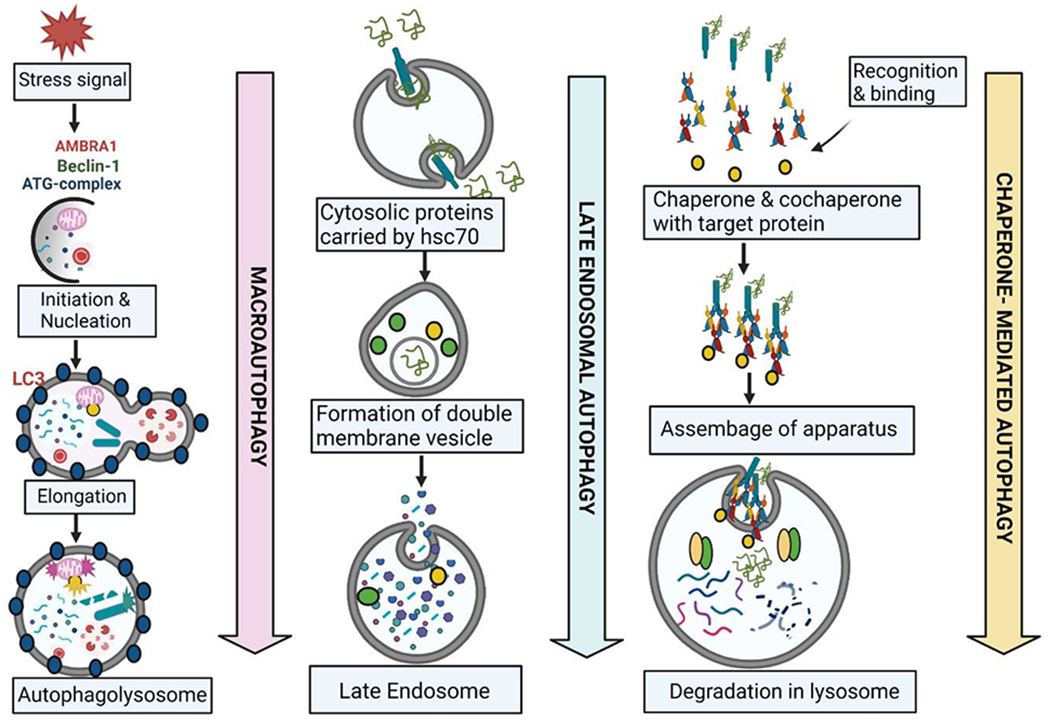
Autophagy is a protective mechanism exhibited by the cell in three ways to deteriorate the damaged organelles and proteins. (A) Macro-autophagy: substrates are sequestered in the autophagosome which are double-membrane cytosolic vesicles. ATG proteins are the core molecule of autophagosome formation (B) Late Endosomal Micro-autophagy: In response to amino acid starvation, multivesicular bodies are form known as endosomes which can be capable of engulfing the cytosolic materials (C) Chaperone-mediated autophagy: Proteolytic pathway is promoted by molecular chaperones which leads to degradation of a large body of cytosolic proteins. AMBRA1: Autophagy and Beclin-1 regulator 1; ATG: Autophagy related 1; LC3: Microtubule-associated protein 1A/1B-light chain 3; hsc70: heat shock protein 70 or DnaK
Macroautophagy is an autophagic process mediated by the autophagosome, a unique organelle that contains cytosolic double-membrane vesicles that sequester substrates. Autophagosome development relies on the coordinated activity of evolutionarily conserved ATG proteins that create different protein complexes. The combinatorial role of a series of ATG proteins regulates autophagosome assembly. For cargo breakdown to be complete, autophagosomes must combine with endosomes and lysosomes. The anchoring complex HOPS, the small GTPases Rab2 and Rab7, in addition to particular SNARE proteins such as autophagosomal Syntaxin 17 as well as its interacting members SNAP29 and Vamp8, are the key mediators of fusion (Vamp7 in Drosophila) (Furuta and Amano, 2012, Itakura et al., 2012). Extraneous and damaged organelles, cytosolic proteins, and invading microorganisms are all substrates for macroautophagy. The 26S proteasome destroys both misfolded and ubiquitinated cytoplasmic proteins and appropriately folded proteins that exhibit specific destruction signals. However, when ubiquitinated proteins continue to accumulate, they tend to form aggregates that are destroyed by macroautophagy when they attach to autophagy receptors (Galluzzi et al., 2017). Furthermore, mounting evidence shows specific autophagy subtypes such as Mitophagy, ER-phagy, Lipophagy, Ribophagy, Pexophagy, Lysophagy, Nucleophagy, Proteophagy, Aggrephagy, Ferritinophagy, Granulophagy, Myelinophagy, Xenophagy, and Virophagy and serve a variety of physiological functions (Galluzzi et al., 2017, Reggiori et al., 2012).
Chaperone-mediated autophagy describes the movement of soluble cytosolic proteins to lysosomes and disintegration straightforwardly through the lysosome membrane. This proteolytic pathway is aided by molecular chaperones found in the cytosol and lysosomal lumen. The cytosol’s molecular chaperones unfold substrate proteins before they pass the lysosomal membrane, whereas the lysosomal lumen’s chaperone necessarily drags the substrate protein throughout the membrane (Dice, 2007). Heat shock-cognate chaperone 70 KDa (HSC70) and lysosomal associated-membrane protein 2A (LAMP-2A) proteins are involved in CMA. All CMA substrate proteins are characterized by a unique KFERQ motif recognized by cytosolic HSC70. In autophagic pathways, CMA influences the metabolism of various biomolecules in neurodegenerative diseases, including proteins, lipids and glucose (Alfaro et al., 2019).
Microautophagy was previously described as the direct enwrapping and transport of cytosolic components into the lytic organelle’s lumen by lysosomal membrane dynamics (Oku and Sakai, 2018). Microautophagy is a non-selective lysosomal biodegradation process that involves autophagic tubes directly engulfing cytoplasmic cargo at a border membrane, allowing invagination and fractionization of vesicle and conformational changes into the lumen. Nitrogen deprivation or rapamycin can trigger microautophagy in soluble substrates via regulatory signalling complex pathways because of its constitutive properties (Li et al., 2012). Proteins that enter the endo-lysosomal complex via infolding are swallowed by the late endosome and lysosomal membrane in this mechanism. The continuous formation of SNARE proteins is essential for releasing neurotransmitters, and any alterations in this process might result in the development of neurodegenerative disorders (Malik et al., 2019).
Selective autophagy
Aforetime the term “autophagy” was coined, preliminary ultrastructural studies had demonstrated that cells could selectively destroy particular cargo. Despite this, at the commencement of the molecular age of autophagy study after the finding of Atg genes in the 1990s, autophagy was still mainly considered to as a bulk degradation mechanism with a minimal basis for specificity, with rare exceptions in yeast (Chu, 2019, Harding et al., 1995, Tsukada and Ohsumi, 1993). However, identification of ubiquitinated cargos through ubiquitin-binding sites is a fundamental motif among the 26S proteasome and selective autophagy, and it is the principal strategy in selective autophagy that underlying substrate recruitment in eukaryotic organisms. Findings in yeast, meanwhile, show that all known cargo adaptors are devoid of ubiquitin-binding domains (Schreiber and Peter, 2014). The ability to tie a cargo to a developing autophagosome by concurrently interacting the cargo and ATG8-family proteins on the separation membrane is a function of cargo receptor proteins, which confer selective autophagy. With the help of selective autophagy receptors, cytoplasmic components such as various intracellular organelles, protein aggregates, and microbes are recognized and marked during the process before getting sequestered into an autophagosome via several cellular pathways(Zaffagnini and Martens, 2016) and accordingly they are known as mitophagy, ribophagy, aggrephagy, xenophagy, etc. During selective autophagy, cargo adapters (LIR-motif proteins) can connect autophagic cargos to LC3. Because they attach to and/or destroy the cargo, they are now more commonly known as autophagy receptors. To specifically activate the autophagy machinery and enable the creation of autophagosomes, cargo adaptors link to both the cargo and lipidation of Atg8 or Atg family members together (Chu, 2019). Furthermore, substrate ubiquitination functions as a widespread cargo recognition signal in selective autophagy, close to the ubiquitin-proteasome process, and has been connected to a suitable degradation signal to target mitochondria, protein aggregates, intracellular pathogens, and macromolecular assemblies like the mid-body and inflammasome (Schreiber and Peter, 2014). Earlier studies demonstrated that Ubiquitin could identify protein aggregates and improperly folded proteins that 26S proteasomes cannot correctly process due to their size, increasing complexity, and other factors (Fredrickson and Gardner, 2012). Cells drive large ubiquitinated aggregates to the autolysosome using both bulk and selective autophagy to avert the build-up of hazardous structures, preventing cell damage or cell death and the process is also known as aggrephagy (Gatica et al., 2018). In addition to selective autophagy in mammals’ cellular machinery, a potentially active autophagic regulation known as p62/SQSTM1 (sequestosome-1), a neighbour of BRCA1 gene has also emerged. When p62/SQSTM1 was discovered to be a genuine autophagy receptor in mammals, it helped us to understand that macroautophagy is a selective process. First, certain payloads are directly interacted with by p62. There are instances where p62 binds to cargo that is not ubiquitinated and interacts with it using its C-terminal UBA domain. In addition, it engages in ATG8s attached to the phagophore’s inner membrane via its lipid tail. Thirdly, p62 homopolymerization. Its co-aggregation with the cargo is made easier by its PB1 domain, which mediates. The tight connection between the p62-coated cargo and lipidated ATG8s at the phagophore is also made possible by polymerization (Lamark et al., 2017). Moreover, in several neurodegenerative diseases, aggrephagy may be used to break down aggresomes, including misfolded peripheral myelin protein 22 in hereditary neuropathies commonly known as Charcot-Marie Tooth disease or aggregate-prone proteins emphasising huntingtin exon 1 gene or exon 1 of the huntingtin protein in HD, α-synuclein linked to PD and many other prion related diseases (Chu, 2019). In AD, neuronal degeneration results from a defect in the microtubule-dependent rapid axonal transport of unfolded protein aggregates and autophagy vacuoles from the neuronal distal end to the lysosome-rich microtubule-organizing centre. Typically, HDAC6 can identify the misfolded tau and Aβ proteins in AD, their aggregates, and autophagy vacuoles (Malampati et al., 2020). Therefore, neurodegeneration is greatly influenced by dysfunctional selective autophagy/aggrephagy machinery in neuronal cells. To alleviate the adverse effects of AD pathology, protective strategies may be implemented by focusing on a number of different areas, such as aggregosome clearance, lysosomal function restoration, and aggrephagy enhancers. (Lyu et al., 2022, Malampati et al., 2020).
Autophagy pathways
Autophagy is best understood in the context of hunger when it sustains survival by feeding nutrients and amino acids from digested cytosolic material. Still, it can also be induced by a range of stress, such as hypoxia, DNA damage, ER stress, and pathogen infection (King et al., 2011). It is an evolutionary conserved self-eating mechanism executed through a series of cellular events described below.
Induction of autophagy: The initiation of autophagy involves a variety of proteins and other macromolecules that forms multi-subunit complexes by undergoing phosphorylation and dephosphorylation. Macromolecules that help in the formation of initiation complexes include Unc-51 like kinase (ULK), a mammalian homolog of ATG1. The ULKs are involved in the phosphorylation of ATG13 and FIP200 (FAK-family interacting protein 200 kDa) inside a structure commonly known as phagophore. Finally, upon binding of the ATG101 to the previous complex, it becomes more stable and is sufficient to induce autophagy. Under the appropriate circumstances, mammalian rapamycin target protein complex 1 (mTOR1) can phosphorylate ULK1 and Atg13, preventing ULK1 from binding to Atg13, FIP200, and Atg101, and therefore suppressing autophagy. During starvation, mTORC1 activity on the lysosome surface is blocked, and ULK1 and Atg13 are dephosphorylated, resulting in ULK1 kinase activation, and the ULK complex localises to phagosomes to form the ULK1-Atg13-FIP200-Atg101 complex that initiates autophagy (Chen et al., 2021, King et al., 2011).
Sequestration and expansion: The Beclin1 complex regulates the production of preautophagosomes and the commencement of autophagy, as well as recruiting-related autophagy proteins (such as Atg12-Atg5, Atg6, and LC3) that facilitate phagosome extension (Chen et al., 2021). The LC3 ubiqutin-like lipidation complex converts cytosolic LC3-I to membrane-associated LC3-II by conjugating it to a phosphoethanolamine. SQSTM1 and other autophagy cargo receptors carry cargo into the phagophore as it forms and joins LC3 through LC3 interaction region (LIR) domain (Velazquez and Jackson, 2018).
Maturation is the next process where the autophagosome becomes an amphisome after fusing with an acidic late endosome. The autolysosome is formed when an autophagosome or amphisome fuses with a lysosome, resulting in the final breakdown of cargo. Furthermore, LAMP1 (lysosome-associated membrane protein 1) and LAMP2 (lysosome-associated membrane protein 2) are required for autophagic lysosome fusion (Chen et al., 2021, Galluzzi et al., 2017). The autolysosome either degrades the damaged molecules resulting in cell death or it may recycle them, promoting cell survival. The autophagy signalling pathways are briefly illustrated in Fig. 3.
Figure 3: Signaling pathways involved in the autophagy process.
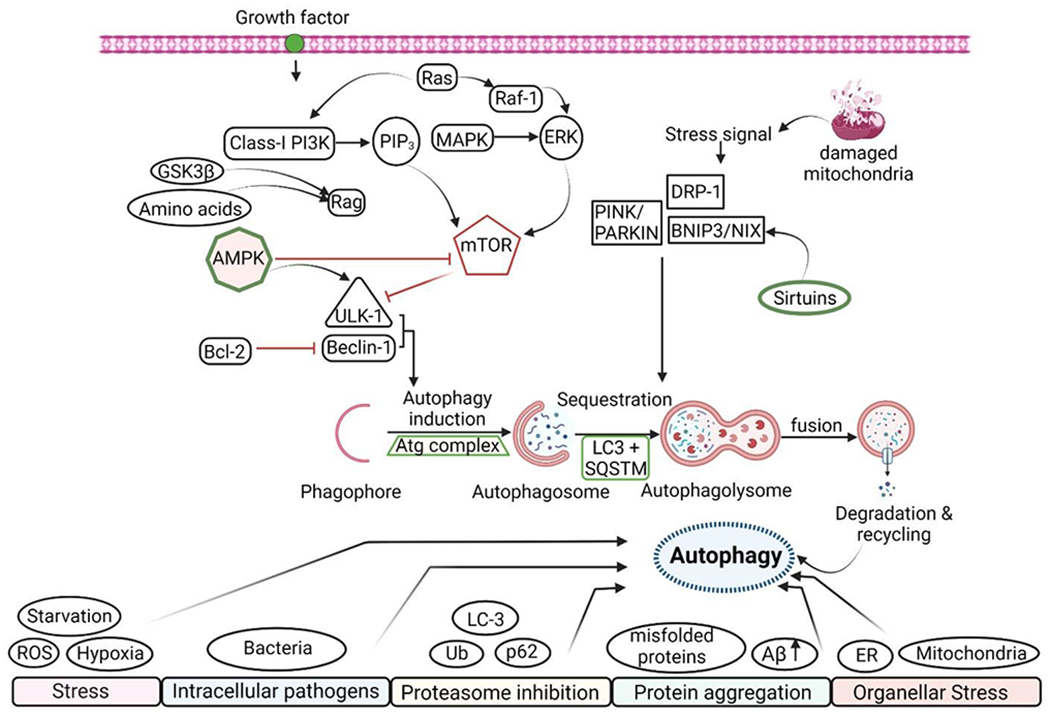
Autophagy is a highly selective process triggered by extracellular and intracellular stress and signals such as growth factor shortage, nutrient scarcity, organellar stress, and pathogen infection. Growth factors can usually start the signalling cascade by activating Class-l PI3K, activating the ERK signalling pathway. These can act as a positive regulator of mTOR, a negative regulator of the autophagy process. AMPK is a central regulator of autophagy induction, suppressing mTOR activation and thus ULK-1 and Beclin-1 expression. The phagophore then binds to the Atg-complex, causing the autophagosome to be sequestered, resulting in the breakdown and removal of the damaged protein. Damaged mitochondria produce a stress signal that is controlled by Sirtuins and causes mitophagy to occur.
Autophagy and neurodegenerative disorders
Autophagy is an intracellular protein degradation pathway sustained by lysosomes to perform homeostatic functions in response to cellular stress. Aggresomes play a pivotal part in the appearance of neurodegenerative disorders resulting from autophagy failure in neurons. There are several possibilities involved in proteinopathies that are raised from various sources, like any deformations in protein synthesis, misfolding and aggregation of proteins, damages after synthesis of polypeptide chains, and improper attachment of cofactors. The Ubiquitin-proteasome system (UPS) or the autophagy-lysosome system, which has been reconfigured as a quality control mechanism for cellular homeostasis, degrades such hazardous substrates (Lee et al., 2013). Autophagy plays a predominant role in ageing and age-associated neurological disorders such as HD, ALS, AD, and PD (De Gaetano et al., 2021). Fig. 4 shows the critical aspects of impaired autophagy in neurodegenerative illnesses. In the cellular stress and damage, the autophagy process is triggered, allowing cells to revitalise through the catabolism of the cellular components and eliminating all disintegrating parts, which facilitates the restoration of cellular homeostasis. Multiple positive and negative regulators, acting powerfully as metabolic sensors, mediate the entire process (Piffoux et al., 2021). Various housekeeping tasks completed by autophagy processesincluded removing harmful proteins and organelles, preventing the accumulation of abnormal protein aggregates, and eliminating intracellular pathogens (Levine and Kroemer, 2008). However, excess of these anomalies leads to autophagy defects, which leads tovarious disease conditions, as mentioned in Table 1.
Figure 4: Dysregulated Autophagy in Neurodegenerative diseases.
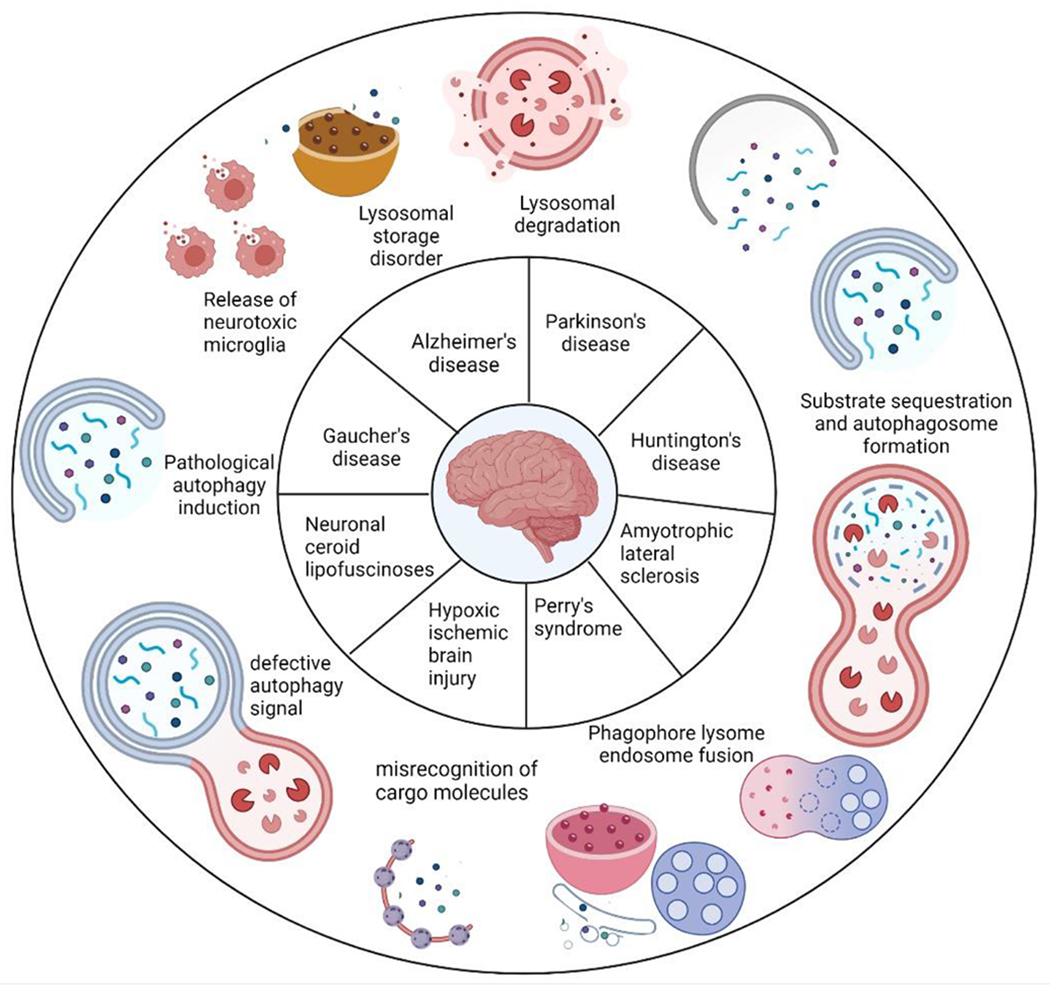
Autophagy is critical for brain homeostasis, and its dysfunction has been linked to many neurodegenerative disorders. Autophagy failure is sometimes seen as the primary pathophysiology of illness. This figure demonstrated the possible mechanisms that lead to the development of neurodegenerative diseases. Numerous variables impact autophagic machinery in some circumstances, resulting in autophagy dysfunction and neurodegeneration. Lysosomal degradation, pathological induction of autophagy, substrate sequestration and formation of autophagosome, inefficient phagophore fusion, lysosome and endosome alteration in signal transduction, and failure in cargo molecule recognition and vesicle trafficking are all disruptive pathways involved in disease pathophysiology.
Table 1:
Dysregulated Autophagy genes in neurodegenerative diseases.
| Disease | Alterations of proteins | Effect on Autophagy process | References |
|---|---|---|---|
| AD | ATG 5 and ATG 7 ↓ | preventing constitutive autophagy and ubiquitinated protein aggregates | (Funderburk et al., 2010) |
| Beclin-1 ↓ | exhibited enhanced synaptic loss, enhanced Aβ-peptide deposition, and ensuing neuronal death | (Pickford et al., 2008, Ułamek-Kozioł et al., 2013) | |
| ATG- related proteins dysregulation | Mitophagy, ER-phagy Lipophagy and other autophagy processes are hampered and accumulation of pathogenic Aβ and tau proteins promote neurodegeneration | (Mizushima et al., 2008) | |
| PICALM/CALM (phosphatidylinositol binding clathrin assembly protein) ↑ | Exacerbate tau toxicity | (Moreau et al., 2014) | |
| Toll-interacting protein (Tollip) ↓ | Endo-lysosome formation is consistently disrupted in macrophages and fails to sustain neuronal health | (Chen et al., 2017) | |
| NRBF2 (nuclear receptor binding factor 2) ↓ | PI3K-III dysregulation and couldn’t protect from ER-stress | (Lu et al., 2014) | |
| TREM2 (Triggering receptor expressed on myeloid cells 2) ↓ | Couldn’t maintain cellular energy and metabolism | (Zhou et al., 2018) | |
| ErbB2 (Erb-B2 Receptor Tyrosine Kinase 2) ↑ | Suppress the autophagic flux | (Wang et al., 2017) | |
| Transient receptor potential Mucolipin-1 (TRPML1) ↓ | Autophagic lysosome formation is halted and hampered the mTOR signaling pathway | (Zhang et al., 2017a) | |
| PD | ATG 7 ↑ | Accumulation of α-syn | (Crews et al., 2010) |
| LRRK2 (Leucine-rich repeat kinase 2) ↓ | (Singh et al., 2021) | ||
| GBA (glucocerebrosidase) ↓ | Inhibits autophagic function and glucocerebrocidase (GCase) activity which results in aggregation of glucosylceramide and α-syn | (Pang et al., 2022) | |
| PINK1 ↓ | Mitochondrial Complex-I activity is impaired | (Morais et al., 2009) | |
| Parkin ↓ | Loss of ubiquitinylation of molecules involves in several cellular functions | (Arkinson and Walden, 2018) | |
| VPS35 (Vacuolar protein sorting ortholog 35) ↓ | Lamp2a Retrieval from Endosome to Golgi couldn’t achieve which prevent chaperone mediated α-syn degradation, Decrease in MFN2 leads impairment in mitochondrial fusion | (Tang et al., 2015a, Tang et al., 2015b) | |
| SYT11 (Synaptotagmin 11) ↓ | Translational inhibition of mTORC1 activity and alters lysosomal function | (Bento et al., 2016) | |
| ATP13A2 ↓ | |||
| HD | mTOR ↓ | Induction of autophagy and prevents accumulation of Huntingtin | (Ravikumar et al., 2004) |
| P62/SQSTM1 ↑ | Sequestration process couldn’t initiate results in disruption of autophagy which increases the susceptibility of neurodegeneration | (Bjørkøy et al., 2005) | |
| HTT (Huntingtin) ↓ | (Croce and Yamamoto, 2019a, Franco-Iborra et al., 2021) | ||
| ATG7 ↓ | Accumulation of polyuiquitinated proteins leads to dysfunctional autophagy | (Collier et al., 2021, Komatsu et al., 2006) |
Alzheimer’s disease
The extracellular aggravation of amyloid-beta (Aβ) plaques and intracellular tau (τ) protein tangles characterize AD, a chronic neurodegenerative disease. ROS contributes to hallmarks of oxidative stress (lipid oxidation, protein carbonylation, and mtDNA oxidation) that rise with age and appear especially prominent in AD. After Aβ is carried into the mitochondria, it communicates with proteins present in mitochondria, resulting in a rise in ROS generation, a surplus build-up of mitochondrial Ca2+ level, and mitochondrial alteration, also limiting the number of functionally active mitochondria, and, eventually, neuronal dysfunction. Autophagy is involved in the diminishment of Aβ, and an excess of Aβ inhibits the fusion of autophagosomes with lysosomes. Aβ synthesis that escapes the cell and forms plaques in the extracellular space is aided by autophagy (De Gaetano et al., 2021, Yun et al., 2020). The autophagic build-up of mitochondria in susceptible AD neurons has been used to demonstrate anomalous mitophagy in the AD patient brain. Reduced mitophagic induction can cause a spike in malonaldehyde, which promotes Aβ deposition. As a result, the activity of Beclin1 is limited, resulting in autophagy dysfunction and, consequently, progression in AD pathology (Shefa et al., 2019).
Parkinson’s disease
The second most prevalent degenerative condition is PD, which primarily targets dopaminergic neurons in the substantia nigra pars compacta and results in dopamine depletion (Vidyadhara et al., 2019). A substantial body of evidence suggests that proteins associated with autosomal dominant PD, including a-synuclein and LRRK2, are required in the autophagy pathway. Furthermore, proteins related to recessive PD, including PINK1 and PARKIN, play a predominant role in mitophagy. However, both macroautophagy and CMA are considered to be included in the aetiology of PD. Whereas the ubiquitin-proteasome pathway and macroautophagy have both been connected to alpha-synuclein clearance, CMA has been revealed to be required for the breakdown of wild-type alpha-synuclein. Further, inhibiting CMA causes the production of high molecular weight and detergent-insoluble alpha-synuclein species, indicating that alpha-synuclein clearance by CMA is critical for limiting alpha-synuclein oligomerization in healthy neurons (Cheung and Ip, 2009). Two autosomal dominant variants in the -synuclein gene, A53T and A30P, have been discovered in familial cases of PD. In addition to point mutations, ubiquitination, phosphorylation, oxidation, nitration, and the production of dopamine-dependent adducts are the post-translational changes that result in the formation of harmful proteins. A 15-amino-acid region of the protein, a-synuclein comprises incomplete but overlapping versions of the KFERQ CMA recognizing motif. HSC70 is a chaperone protein attaches to a-synuclein and recognizes the pentapeptide sequence motif. A-synuclein interacts with the lysosomal-associated membrane protein type 2A (LAMP-2A) at the lysosomal membrane. This CMA receptor delivers a-synuclein to the lysosome, which is destroyed by proteases with the help of a lysosomal luminal HSC70. In cultured cells and autopsy tissues, mutant variants of the protein act as receptor inhibitors that can impede its breakdown through the CMA route, culminating in hazardous cytoplasmic aggregation in neurons. Specific genes linked to PD have also been revealed to engage in the clearance of disintegrated mitochondria through autophagy, implying a link between autophagy activation by abnormal a-synuclein expression and mitochondrial dysfunction. The protein PARKIN and mitochondrial disintegration are required for increased mitochondria elimination reported in the cell lines (Cheung and Ip, 2009, Lynch-Day et al., 2012). PARKIN and PINK1 have been demonstrated to interact genetically and physiologically, particularly in the areas of mitophagy and mitochondrial function, which protect the neurons.
Huntington’s disease
Huntington’s disease is a lethal neurodegenerative disorder caused by an abnormal extension of the CAG repeat of the huntingtin gene, in which mutant Huntingtin (mHtt) gene disrupts many cellular activities, resulting in neuronal malfunction and cell death. Mitochondrial dysfunction seems to play a critical function in HD pathogenesis among these changes. When malfunctioned mitochondria swamp cells, selective autophagy removes the damaged organelles. PINK1 is a molecular sensor that concentrates in the outer membrane of injured mitochondria, employing Parkin, an E3 ubiquitin ligase, and starts the mitophagy process (Khalil et al., 2015). Although fasting and subsequent suppression of the serine/threonine mTOR was the first method for activating macroautophagy, it is now accepted that macroautophagy can occur even when mTOR is activated by Beclin1 modification. Furthermore, it is unknown how much autophagy induction is produced in neurons by inhibiting mTOR. Overall, it’s unclear how macroautophagy is most efficiently engaged in the adult brain (Croce and Yamamoto, 2019b). Autophagy has been linked to the breakdown of cytosolic mutant Htt in both soluble and accumulated forms. The acetylated mutant Htt at K444 is localised explicitly to autophagosomes, making it easier for the autophagic-lysosomal route to remove it. As a result, mutant Htt has arisen as a viable paradigm for screening drugs or compounds that may have autophagy-stimulating effects and their potential therapeutic relevance (Cheung and Ip, 2011, Nixon, 2013).
Mitophagy
Mitophagy is also an essential process that is critical for maintaining cellular homeostasis. Recent studies have proposed that mitophagy is involved in various mitochondrial quality control processes, promoting degradation of the defective mitochondrion and regulating multiple functions such as mitochondrial biogenesis and dynamics (Zhang et al., 2019). During the development of the myoblast, the process of mitophagy contributes to the regulation of oxidative stress, apoptosis, and “mitochondrial network signalling.” On the other hand, the mitophagy process also helps in the degradation of the “ocular lens organelle”, and autophagic maturation of erythroid cells (Costello et al., 2013). Additionally, mitochondrial malfunction might result in cell death and oxidative damage to various micro- and macromolecules in the body. There is increasing evidence that mitophagy aids in attaining a healthier body and brain by eliminating the damage caused by defective organelles (Chen et al., 2020a). Also, defective mitophagy can be detrimental to human health and might promote the pathogenesis of various diseases, including neurodegeneration.
Mitophagy pathways
Mitophagy is triggered, and selected mitochondria are connected to autophagic vesicles when certain proteins rely on the outer mitochondrial membrane can detect and engage LC3. There are several types of mitophagy processes such as (a) “Parkin and PINK1” regulated, (b) “FUNDC1” mediated, (c) “NIX and BNIP3” dependent, and (d) “ATG32” controlled mitophagy (Marinković et al., 2021).
Parkin and PINK1 regulated mitophagy
PINK1 is a ubiquitously produced mitochondrial serine/threonine kinase protein having MTS (mitochondrial targeting sequences) at its N-terminus. The kinase domain of PINK1 could be positioned on the outer mitochondrial membrane, with the kinase domain fronting the cytoplasm, which would explain the direct interrelation with the cytosolic E3 ubiquitin ligase Parkin. Parkin’s E3 ubiquitin ligase activity promotes autophagy degradation in mitochondria. The activation of PINK1 kinase is necessary to recruit Parkin at the site of mitochondrial malfunction. Parkin becomes ubiquitinated when K6 ubiquitin chains connect to it, which may play a role in its degradation (Jin and Youle, 2012). Endogenous PINK1 is constitutively degraded at the mitochondria. Still, its localization is mainly related to a decrease in membrane potential. Under steady-state conditions, Parkin’s E3 ligase activity is repressed in the cytoplasm but is liberated by PINK 1-dependent mitochondrial localization (Matsuda et al., 2010). It is evident from earlier studies that Parkin monitors the mitochondrial quality and transfers from the cytosol to dysfunctional mitochondria, thereby promoting mitophagy (Narendra et al., 2008). Mitochondrial dysfunction has been connected to several neurodegenerative diseases (Bhatti et al., 2017). Recent studies have revealed that Parkin or PINK1 deficiency reduces mitophagy and leads to PD due to anomalies in various proteins like synuclein. It leads to the build-up of faulty mitochondria that would usually be destroyed, resulting in oxidative stress and, eventually, neurodegeneration of prone cells. PINK1 levels are reported to be higher in the brain of AD and multiple sclerosis (MS) patients. Amyotrophic Lateral Sclerosis (ALS) and HD are two more neurodegenerative diseases, linked with PINK1-Parkin pathway (Barodia et al., 2017).
FUNDC1 mediated mitophagy
The outer mitochondrial membrane contains the tertiary transmembrane protein FUNDC1 with an N-terminal LC3 interaction region motif that aids hypoxia-induced mitophagy. The outer mitochondrial membrane-anchored MARCH5/MITOL, an E3 ubiquitin ligase known to ubiquitylate numerous proteins involved in mitochondrial dynamics, regulates FUNDC1 protein levels in part (Onishi et al., 2021). FUNDC1 can activate mitophagy in mammalian cells by recruiting LC3 via its LIR motif; it also has the potential to communicate with DNM1L/DRP1 and OPA1, facilitating mitophagy and influencing mitochondrial fission or fusion. The Lys70 (K70) residue in OPA1 interacts with FUNDC1, and changing K70 to Ala (A) enhances mitochondrial fission and mitophagy. FUNDC1 is enhanced when it is dephosphorylated, separated from OPA1, and interacts with DNM1L under stressful circumstances (Chen et al., 2016). Phosphorylation of FUNDC1 can limit mitophagy in normoxic situations, whereas dephosphorylation can efficiently increase mitophagy in hypoxic conditions. FUNDC1 can thereby coordinate mitochondrial dynamics and quality control (Wang et al., 2019b).
NIX and BNIP3-dependent mitophagy
The BH3-only family of proteins that cause cell death and autophagy includes BNIP3 and NIX. Because NIX is considered necessary for mitophagy, especially in erythroid cells and BNIP3 monitors mitophagy in hypoxic conditions, BNIP3 and NIX are equally involved in autophagy induction (Zhang and Ney, 2009). The phosphorylation of BNIP3 may also contribute to the association between BNIP3 and LC3. Mitophagy function can be augmented by increased expression of NIX or BNIP3. Inducing autophagy using BNIP3 or NIX can protect cells in some situations, but it can also lead to autophagic cell death in others. Whether too much autophagy causes death or BNIP3 or NIX have their death-inducing role (Wang et al., 2019b).
ATG32 controlled mitophagy
Atg32, a 529-amino-acid protein with a single transmembrane domain, was discovered through a genome-wide search for a mutant yeast that has impaired mitophagy. The N- and C-termini of Atg32 are directed straight to the cytosol and mitochondrial intermembrane space, respectively. It can be found in the mitochondrial outer membrane. Although the significance of these interactions is unknown, Atg32 communicates with Atg8, a ubiquitin-like protein found only in the autophagosome, and Atg11, a scaffold protein involved in autophagy-related activities. (Furukawa et al., 2019). A recent study shows that Casein Kinase 2 (CK2) phosphorylates Atg32, implying phosphorylation is a significant signalling phase for Atg32 function and mitophagy. The mitophagy receptor Atg32 is vital in the quality control mechanism because it allows for the particular labelling of damaged or redundant mitochondria, and mitophagy eliminates the mitochondria labelled with Atg32 (Furukawa et al., 2019, Levchenko et al., 2016). Protein aggregation has been connected to the cellular pathology of many neurodegenerative disorders, and protein turnover has been associated with neurodegenerative diseases. These mechanisms are suitable for eliminating intracellular aggregates and misfolded proteins, as well as organelle turnover, particularly mitochondria, in the homeostasis of brain cells. Therefore, the two processes are impactful on Neurodegenerative disorders.
Mitochondrial dysfunction is induced by direct exposure to specific ambient conditions or genetic defects in both mitochondrial and nuclear DNA. Mitochondria also regulate calcium homeostasis, membrane excitability control, signal transmission, and remodelling in neurons. As a result, they are especially susceptible to mitochondrial malfunction and might damage cell viability. Among all sorts of abnormalities, one involved in maintaining the membrane permeability and regulation of ions in normal and pathological conditions is increased Ca2+ build-up in the matrix that arises in reaction to cellular damage. This is the primary trigger for the opening of the valve which further disrupt the proton electrochemical gradient, and alter membrane potential and pH gradient (Norat et al., 2020). Cytochrome c is a regulatory molecule in normal mitochondrial metabolism and the leakage leads to apoptosis activation can also be caused by the mitochondrial malfunction. The development of various neurodegenerative illnesses is linked to an increase in ROS generation and OXPHOS disruptions, which can disrupt several cellular processes and the internal environment. This dynamically changing organelle undergoes multiplication by fission and fusion, which is entirely dependent on the type of metabolic supply it receives, and it ultimately regulates the mitochondrial pool. Alterations in fission and fusion proteins result in abnormalities in mitochondrial biogenesis resulting in the development of several diseases. Fig. 5 summarizes various stimuli inducing the mitochondrial dysfunction that leads to different pathological conditions (Hroudová et al., 2014).
Figure 5: Factors promoting Mitochondrial dysfunction in neurodegenerative diseases.
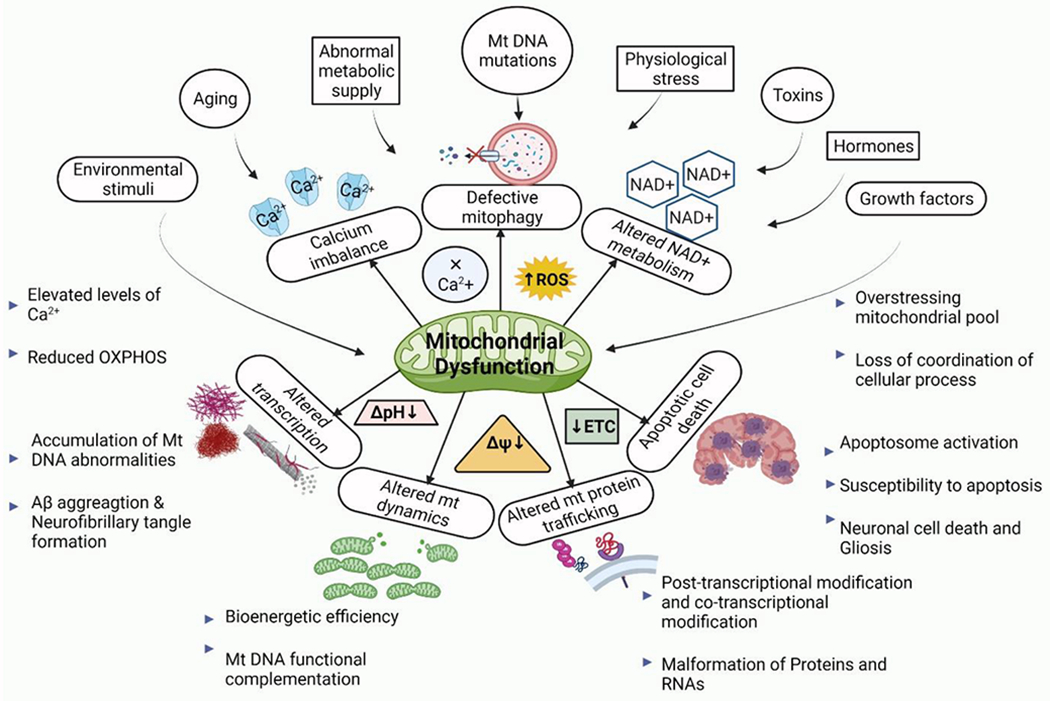
Various stimuli are directly linked to maintaining the proper morphology and physiology of mitochondria by modulating the internal environment. Aging, aberrant metabolic supply (oversupply/undersupply), mutation in mitochondrial DNA, physiological stress, exposure to some toxins or accumulated toxins, hormones, and growth factors are all stress factors that can affect mitochondrial function. Disturbances in the internal environment do not restore the pH value or the membrane potential imbalance. As a result, ETC (Electron transport chain) is severely impeded, and a lack of ATP impacts not just mitochondrial biogenesis but also overall mitochondrial dynamics. Finally, protein transport and transcription and translocation are disrupted, triggering the cell survival and death signalling (apoptosis) pathway, resulting in neuronal cell death and the progression of numerous neurodegenerative diseases.
Mitophagy has sparked great interest due to the role of damaged mitochondria in neuropathologies. Identifying specific drugs capable of regulating the cellular processes involved in removing protein clumps and defective mitochondria remains a most incredible therapeutic strategy in clinical studies (Shin et al., 2019). Mitophagy is a key mitochondrial qualitycontrol (QC) system that helps neurons stay healthy and function correctly, and it may be compromised in several neurodegenerative diseases (Chakravorty et al., 2019). The mitophagy unfolded protein response (mtUPR), a mitochondrial stress response, is set off when the amount of misfolded and damaged proteins exceeds a particular level. Nuclear genes encipher mitochondrial chaperones, and proteases are overexpressed when the mtUPR is activated, reducing the number of protein aggregates. The 26S proteasome system in the cytoplasm can also aid in degrading abnormal mitochondrial proteins. Autophagy adaptor proteins like optineurin (OPTN), nuclear dot protein 52 (NDP52), Tax1-binding protein 1 (TAX1BP1), sequestosome-1 (SQSTM1)/p62, and BRCA1 gene 1 (NBR1) are attracted to defective mitochondria when PARKIN ubiquitinates K63. These adaptor proteins communicate with the GABARAP (gamma-aminobutyric acid receptor-associated protein) or autophagosomal proteins LC3 (microtubule-associated protein 1A/1B-light chain 3) to recruit autophagy machinery to defective mitochondria, forming a mitophagosome that fuses with lysosomes, where the defective mitochondria deteriorate (Nguyen et al., 2016). FUNDC1 (FUN14 domain containing 1), NIX (Nip3-like protein X), FKBP8/FKBP38 (FK506 binding protein 8) BNIP3 (BCL2 interacting protein 3), Bcl2L13 (Bcl2-like protein 13), and the IMM phospholipid cardiolipin stimulate mitophagy in a PINK1-Parkin acts independently in regards to numerous cellular stimuli (Chakravorty et al., 2019). Pharmaceutical therapies that increase mitochondrial activity and quality control have been tried in several disease models because mitochondrial function and quality control are crucial in the aetiology of neurodegenerative disorders.
Various lifestyle interventions that have been shown to stimulate mitochondrial biosynthesis, turn down oxidative stress, and promote autophagy, such as intermittent fasting, calorie restriction, and intense exercise, could also improve mitochondrial health in neurodegenerative disorders (Halagappa et al., 2007). Several mitophagy promoters, such as NAD+ precursors, urolithin A (UA), spermidine, and the antibiotic actinonin (AC), have been investigated and found to have tremendous benefits to enhancing mitophagy, extending health span, increasing mitochondrial resistance to oxidative stress, and protect neurons in animal models and/or human cells. Enhanced activity of PI3K-Akt and MAPK/ERK1/2, as well as increased expression of the transcription factor CREB, are expected to have beneficial effects. Additional pharmaceutical strategies to encourage mitophagy, such as influencing minor bioenergetic stress or inhibiting mTOR activity, have been established to be helpful in preventing or treating neurological illnesses like AD (Cai and Jeong, 2020). Some vitamins significantly impact mitochondrial dysfunction such as vitamin B, C, E, zinc, Selenium and Coenzyme.
Auto-regulation and neuroprotection via autophagy and mitophagy
Mitochondrial quality control (MQC) is an operational system that regulates numerous activities such as mitophagy, biogenesis, proteostasis, and dynamics to maintain cellular homeostasis. With advancing age, the loss of control and coordination that results in dysfunctions and deregulation in many cellular systems is a significant step in the development of neurodegenerative diseases. Because neurons are metabolically active cells, their survival and maintenance rely on mitochondrial activity. Their primary purpose of mitochondria is to produce ATP, which is necessary for neuronal function and integrity. Mitochondria also take up and release Ca2+, which helps to maintain a stable intracellular Ca2+ level. Mitochondria that are not functioning properly, generate insufficient ATP, buffer Ca2+ ineffectively, and produce more ROS. The build-up of impaired mitochondria in axons and synapses during the lifespan of neurons is known to play a critical role in the aetiology of PD and AD (Franco-Iborra et al., 2018).
SIRT3 regulation of mitochondrial function
The sirtuins in mitochondria, such as SIRT3, SIRT4, and SIRT5 are among the seven mammalian sirtuins. SIRT3 is a deacetylase that regulates mitochondrial activity and is one of the most significant deacetylases in mitochondria. SIRT3 deacetylates the PDC (pyruvate dehydrogenase complex) in glycolysis, allowing pyruvate to take part in the Krebs cycle and increasing glucose absorption by triggering protein kinase B (Meng et al., 2019, Wang et al., 2019a). SIRT3 also protects against or delays the effects of oxidative stress by activating several antioxidants, including FOXO3, IDH2, and SOD (Meng et al., 2019). A recent study shows SIRT3 expression has been connected to neurodegenerative illnesses such as PD and AD. SIRT3 overexpression in AD not only limits ROS damage to the mitochondrial structure by deacetylating and activating SOD2, but it also regulates mitochondrial quality (Kincaid and Bossy-Wetzel, 2013, Meng et al., 2019). SIRT3 activation may thereby reduce or even prevent mitochondrial dysfunction associated with ageing and dementia. SIRT3’s antioxidant and metabolic actions indicate that it may play a neuroprotective role by improving mitochondrial function, which leads to greater neuronal survival and reduced ageing effects(Kincaid and Bossy-Wetzel, 2013).
Some disease-associated proteins are susceptible to misfolding, accumulation, and aggregation and are hypothesised to produce gain-of-function proteotoxicity via a variety of pathways. In a nutshell, protein quality control (PQC) refers to a group of pathways that are responsible for monitoring protein integrity and may play a role in neurodegenerative disease. Because neurons have a distinct cellular structure with lengthy extensions, PQC in neurons is more difficult than other cell types. PQC failure is frequently linked to neurodegenerative illnesses such as AD, HD, PD, and prion disease (Ciechanover and Kwon, 2017). The UPS, CMA, and macroautophagy are among the proteolytic systems used by mammalian cells to eliminate misfolded proteins. A brief description of the quality control system regulating different pathways involving protein molecules and cellular homeostasis is illustrated in Fig. 6.
Figure 6: Different types of Autoregulation through Mitochondrial Quality Control mechanisms.
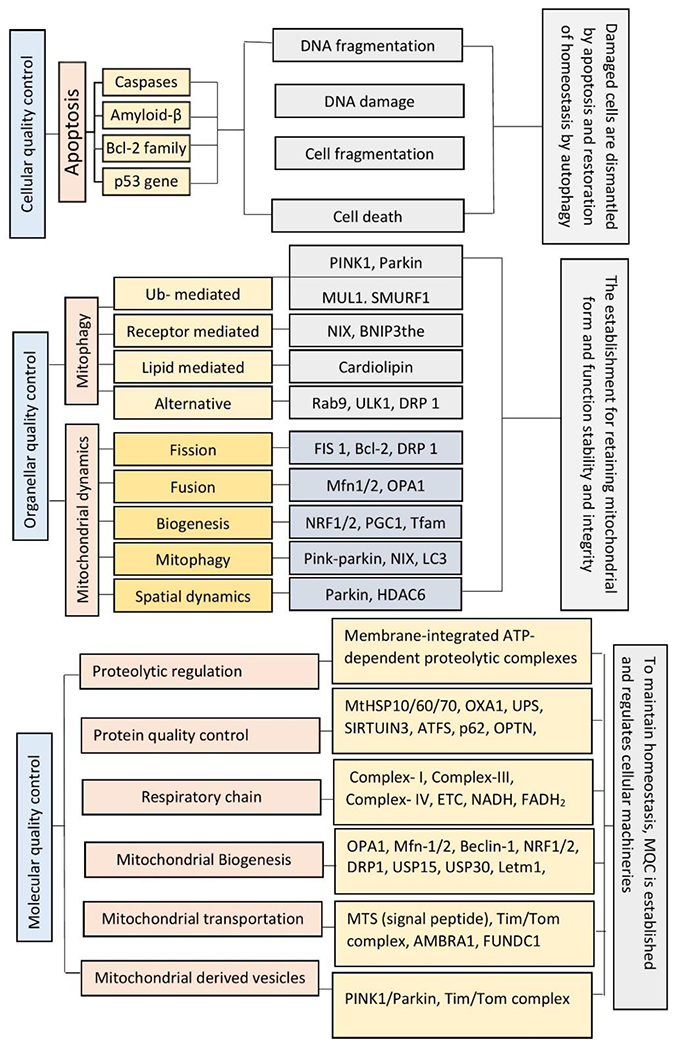
The Mitochondrial Quality Control mechanism can be seen in three different types such as: (A) Cellular Quality Control that is basically worked by apoptosis through which damaged or fragmented DNA can be eliminated and cell cycle is regulated. (B) Organellar Quality control that is worked mainly in mitochondria by regulating two major processes like mitophagy and mitochondrial dynamics to stabilize the morphology and physiology of mitochondria. (C) Molecular Quality Control is accomplished at molecular level by covering all types of mechanisms involved in quality control including proteolytic pathways, mitochondrial biogenesis and all.
Exosome secretion and autophagy pathway crosstalk
Eukaryotic cells require the disintegration and recycling programs of cellular components to maintain homeostasis. All cell types employ autophagy to resist hunger, nutrient recycling and eliminate undesired or damaged intracellular elements, such as proteins and organelles, among other things. In some cases, cells eliminate undesired or damaged material by releasing exosomes, tiny vesicles secreted by practically every cell type into the extracellular space. As exosomes can be transported from one cell to another, the release of undesirable material into the extracellular space may affect surrounding cells. Exosomes are considered an unusual secretory pathway that carries particular repertoires of biological macromolecules in the form of mRNAs and snRNAs, including miRNAs, and occurs in a reified manner despite cellular stress or stimulation signals modulating their secretion (Raposo and Stoorvogel, 2013). Furthermore, exosomes spread lethal forms of aggregated proteins, including β-amyloid, α-synuclein, and PrPc prion proteins, which areconnected to several neurological diseases (Bellingham et al., 2012). Thus exosomes might be used to advance drug administration, as it can avoid several concerns, such as immune response, target cell acceptance, and cargo destruction.
Factors affecting Autophagy and mitophagy in ageing and neurodegeneration
Oxidative stress
Exogenously or endogenously produced highly reactive oxidising molecules are always present in living cells, posing a threat. ROS are small, unstable molecules that quickly react with other molecules in a cell. They are primarily obtained as by-products of mitochondrial ETC and can damage proteins, RNA, DNA and cause cell death. Electron leaks from ETC at mitochondrial complexes (primarily complexes I and III) and results in the partial reduction of oxygen to O2−, which quickly disproportionate into H2O2 either spontaneously or via superoxide dismutase (SOD)-mediated catalysis (De Gaetano et al., 2021, Filomeni et al., 2015, Yun et al., 2020). ROS levels can act as signalling molecules when they are low enough in the body. However, several factors can alter these levels, causing oxidative stress by raising the amount of ROS inside the cell (Moreno et al., 2018). To degrade H2O2, mammalian cells have various antioxidant enzymes, including SOD in the matrix, catalase in the cytosol, and glutathione peroxidases (GSH-Px or GPx) in the mitochondria (Yun et al., 2020). ROS have been linked to autophagy induction when required nutrients such as amino acids, glucose, or serum are depleted, as well as ischemia/reperfusion (IR) and hypoxia, which can cause oxidative damage to mitochondria. Because mitochondrial dysfunction is inextricably associated with oxidative stress, mitochondria play as both producers and targets for reactive oxygen species. When the consistent production of ROS exceed their catabolism or detoxification, oxidative stress tends to occur, which can affect mitophagy on different stages (De Gaetano et al., 2021, Lee et al., 2011, Yun et al., 2020). Because the mitochondrial genome lacks histones, it is less protected than the nuclear genome and has a much higher mutation rate. Mutations in mitochondrial DNA, as well as disequilibrium in the mitochondrial respiratory chain, calcium concentration and homeostasis, and excitotoxicity, as well as damages to permeability of membranes and mitochondrial defence systems, all are common reasons of neuronal injury and deterioration caused by oxidative stress (Moreno et al., 2018). Inadequate mitochondrial activity may aggravate the oxidative stress condition, resulting in a biochemical and metabolic crisis. Due to this, the build-up of toxic proteins and a decline in mitochondrial function occur, which further leads to increased oxidative stress. Therefore, mitophagy is an essential pathway for mitochondria turnover rate, promoting its survival and function (Lee et al., 2011, Moreno et al., 2018). When mitochondria are deprived of nutrients, they produce superoxide (O) and H2O2 (Filomeni et al., 2015). They influence autophagy through three main mechanisms: (a) S-glutathionylation: specific cysteine residues in desired proteins can be specifically oxidised by ROS, and the S-glutathionylation through the reversible process might alleviate or intercede the destruction. Such a post-translational modification brings a tripeptide and a net negative charge to the protein of interest, which can cause alternations (Xiong et al., 2011). (b) Oxidation of Atg4’s Cys81 (SH → Sox): this results in the inactivation of ‘delipidating’ activity on LC3 and the accumulation of pro-autophagic LC3-II isoform. (c) Production of reduced glutathione (GSH) subsequently to the extracellular environment via the multidrug resistance protein 1 facilitates a broad change in thiol redox state (e.g., the ratio of GSH/GSSG reduction and marked raise of oxidised thiols, Sox) (Filomeni et al., 2015). The cellular redox balance modifications also can alter mitochondrial dynamics by influencing mitophagy. While reduced overall glutathione (GSH) is oxidised to glutathione (GSSG), Mfn develops oligomers and improves membrane fusion by forming disulfide bonds (De Gaetano et al., 2021). Autophagy has also been correlated with the development and aggravation of neurodegenerative diseases. Autophagy impairment caused by increased oxidative stress has also been connected to the progression and worsening of neurodegenerative disorders (Yun et al., 2020).
Neuroinflammation
Several neurodegenerative diseases share a similar thread known as neuroinflammation, which have a significant role in neuroinflammatory and non-neuroinflammatory diseases. Inflammatory responses to infection, trauma, stress and damage are reported; these responses are mediated by specific cytokines, chemokines, secondary messengers, and ROS. The generation of these molecules for the inflammatory response is primarily influenced by endothelial cells, microglia, and astrocytes. Stringent inflammatory responses are often believed to be beneficial by inducing tissue remodelling and repair-like protective mechanisms. Elevated cytokine release, including that of IL-1 and tumour necrosis factor (TNF), ROS, and other inflammatory mediators like inducible nitric oxide synthase (iNOS), is a hallmark of chronic, uncontrolled inflammation (DiSabato et al., 2016). The neuroinflammation-induced alterations in these cellular signaling pathways indirectly exacerbate neurodegenerative disorders. Because neuroinflammation is highly effective in inducing the alteration of microglia and astrocytes, it further activates the pro-inflammatory pathways along with toll-like receptors (TLRs). It promotes ROS accumulation and phagocytosis (Li et al., 2021). Interestingly, nitric oxide and ROS further mediate neuroinflammation by activating several other inflammatory pathways in the brain. Whereas increasing Ca2+ influx via ROS can stimulate the activation of caspases and calpains, and calpain activation decreases neuronal autophagy. Resorption of glutamate is also responsible for the surge of extracellular glutamate levels and, consequently, exorbitant influx of Ca2+ resulting in Ca2+ excitotoxicity by the N-methyl-D-aspartic acid (NMDA) receptor activation (Alirezaei et al., 2011). TNF-α is a significant modulator of PD-related neuroinflammation and is extremely damaging to dopaminergic neurons. It can potentially distort the autophagic flux in the glial cells (Jin et al., 2018). Furthermore, a high level of inflammatory cytokines then leads to vascular dysfunction and is linked with a wide range of metabolic diseases.
Role of microRNAs in Autophagy/Mitophagy regulation
The mammalian genome produces several categories of noncoding RNAs, which are further subdivided (based on their size) into microRNAs (miRNA), piwi interacting RNAs (piRNA) and long noncoding RNAs (lncRNA). miRNAs are the endogenous transcripts which play a significant role in disease pathogenesis (Kiss et al., 2019). miRNAs are the custodian of the cell as they are responsible for maintaining cellular systems and typical cellular functions (Mehta et al., 2020). miRNAs, are usually 21–23 nucleotides long non-coding RNAs known to regulate gene expression at post-transcriptional levels. They are known to play a role in a variety of pathological conditions. In humans, till now, more than 2000 miRNAs have been discovered, with a significant proportion of these miRNAs being intrinsically related to disease progression, acting as potential biomarkers of various diseases (Shah et al., 2018b). A recent study demonstrated that miRNA-27a and miRNA-27b play a critical role in autophagy regulation for clearing damaged mitochondria via the PTEN-induced putative kinase 1 (PINK1) gene (Kim et al., 2016). miRNA-30a directly regulate the essential autophagy-promoting protein Beclin 1 (BECN1), demonstrating that miRNAs play a role in autophagy regulation. HDAC4, a protein linked to autophagy, was likewise influenced by miRNA-206 (Zhu et al., 2009). Several miRNAs, such as miRNA-193b-3p, miRNA-29a/b-1, miRNA-16, miRNA-181a, miR-106a, miRNA-124, and miR-224, have been shown to modulate autophagy and display neuroprotective effects (Shah et al., 2018a). Researchers are currently working on identifying potential biomarkers derived from miRNAs in biological fluids such as blood, serum, cerebrospinal fluid, and plasma (Gilad et al., 2008, Shah et al., 2018a). Identifying such autophagy-modulating miRNA biomarkers could pave the way for therapeutic intervention where these can be directly targeted to either upregulate or downregulate the autophagy process (Shah et al., 2018a).
Modulations of autophagy and mitophagy in neurodegenerative disorders
Autophagy is a ubiquitous lysosomal degradation mechanism that regulates cytoplasmic integrity by removing protein aggregates and damaged organelles. Mitophagy is a cytoprotective mechanism that breaks down damaged mitochondria, reduces ROS production and releasing damaging intramitochondrial proteins. Dysregulated autophagy is observed invarious pathological conditions, including metabolic disorders, neurological disorders, viral infections, and cancer. Increased expression of autophagy may be beneficial in treating some neurological illnesses, whereas autophagy suppression is also being explored as a cancer treatment strategy. Protein inclusions inside neurons or aggregates are typically intracytoplasmic in PD, AD (which frequently involves extracellular aggregates), and HD. So, therapeutic strategies targeting the removal of these toxic protein aggregates might reduce the burden of these neurodegenerative disorders (Rubinsztein et al., 2012). Recent reports have highlighted the importance of autophagy and mitophagy in various neurodegenerative disorders (Boland et al., 2018).
Therapeutic strategies
Pharmacological regulation of Autophagy/Mitophagy
Autophagy is a key process in maintaining homeostasis and protects the cells and cellular machinery from stress-induced damage. Many drugs targeting autophagy defects are available in the market that can be used as a therapeutic strategy in neurodegenerative diseases (Table 2).
Table 2:
Regulation of autophagy through pharmacological and natural compounds
| Sl No. | Drug | Regulation | Mechanism | Reference |
|---|---|---|---|---|
| 1 | Apelin | Apoptosis and autophagy | i) PI3K/Akt/mTOR signaling pathway is influenced by Apelin/APJ(G protein-coupled receptor angiotensin-like-receptor 1) system and protect the cells from excessive autophagic flux ii) Bcl2-Bax ratio is enhanced to attenuate neuronal apoptosis |
(Shao et al., 2021) (Shao et al., 2021, Zhang et al., 2014a) |
| 2 | BH3 (Bcl-2 Homology 3) mimetic drugs | Apoptosis | Mimic BH3 only proteins and prevent their binding with Bcl2 family proteins | (Merino et al., 2018) |
| 3 | Rapamycin | Inhibit apoptosis and promote autophagy | Key regulator of cell proliferation and a downstream effector of the PI3K signalling pathway (allosteric inhibitor of mTOR) | (Takeuchi et al., 2005) |
| 4 | Torin 1 | Inducer of autophagy | Increased autophagy flux by targeting mTORC1 (inhibitor of mTORC1) | (Andersson et al., 2016, Thoreen et al., 2009) |
| 5 | Single walled carbon nanotube | Reduce lysosomal/autophagic dysfunction | alleviate lysosomal dysfunction and re-establish neuronal autophagy | (Xue et al., 2014) |
| 7 | Metformin | Stimulate autophagy | It can initiate autophagy by elevating AMPK and then limiting mTOR | (De Santi et al., 2019) |
| 8 | GTM-1 | trigger autophagy | It is reliant on the mTOR pathway | (Zhang et al., 2017b) |
| 9 | Carbamazepine (CBZ) | Induces autophagy | increases myo-inositol concentrations and activates AMPK to promote autophagy | (Vasconcelos-Ferreira et al., 2022) |
| 10 | CCI-779 | mTOR inhibitor | Triggering autophagy and regulating cell cycle by limiting p21 | (Yazbeck et al., 2006) |
| 11 | Regorafenib | Induces autophagy | Initiate PRKAA (protein kinase, AMP-activated, α catalytic subunit)-dependent autophagy and prevent RAB11A (Ras-related protein Rab-11A)-mediated autophagosome-lysosome fusion, also can effectively stabilises PSAT1 (phosphohydroxythreonine aminotransferase) | (Jiang et al., 2020) |
| 12 | PP242 (2-[4-amino-1-(1-methylethyl)-1H-pyrazolo[3,4-d]pyrimidin-3-yl]-1H-indol-5-ol) (Torkinib) | mTOR inhibitor | through reducing mTORC1 and mTORC2, inhibits the AKT/mTOR pathway | (Feng et al., 2018) |
| 13 | Xestospongin B | IP3R (Inositol trisphosphate receptor) inhibitor | Promotes cell death and by disabling the IP3R-Beclin-1 complex, Xestospongin B’s IP3R inhibition promotes autophagy | (Cruz et al., 2021) |
| 14 | Simvastatin | mTOR inhibitor | inhibits the Rac1-mTOR pathway, and thereby increases autophagy | (Wei et al., 2013) |
| 15 | Nitrendipine | Blocks Ca2+ channels | It can prevent the formation of protein inclusions produced by sustained calcium influx and reduce autophagy defects induced by the adverse role of excitotoxicity | (Park et al., 2014) |
| 16 | Loperamide | Induce autophagy | Facilitated the synthesis of autophagosomes and autophagolysosomes as well as the stimulation of autophagy inducing ATG5- and ATG7-dependent cell death in glioblastoma cells, which is preceded by a considerable induction of autophagy, is mediated by dephosphorylation of mTORC1 | (Zielke et al., 2018) |
| 17 | L-NAME | Induce autophagy | A NOS inhibitor that promotes autolysosomes formation and increases autophagic flux | (Sarkar et al., 2011) |
| 18 | Verapamil | Induce autophagy | Upregulates ATG-5 and ATG-7 and enhances autophagic flux | (Kania et al., 2017) |
Lifestyle and Dietary interventions in modulating autophagy
Promising role of Natural and synthetic compounds
Defective autophagy and dysfunctional miRNAs have recently been linked to ageing and ageing-related neurodegenerative disorders. Thus, targeting such dysfunctional miRNAs could be a potential therapeutic strategy for the treatment of AD. The role of autophagy has also been discovered in the neurons; and it is known to promote synaptic plasticity, oligodendrocyte formation, the myelination process and anti-inflammatory activity in glial cells. However, autophagy exhibits an inverse relation with age and reduces autophagic activity with ageing. Furthermore, declined autophagic action can result in the production of toxic protein aggregates and the build-up of damaged mitochondria, resulting in an increase in ROS, cell death, and neurodegeneration (Kou and Chen, 2017). The ubiquitin-proteasome system is well-known for degrading proteins having a short life span. In contrast, the autophagy-lysosome system destroys long-lived and aberrant protein complexes and organelles. Autophagy deficit or malfunction in neurons due to diminished lysosomal activity plays a role in the aetiology of neurodegenerative disorders (Cheung and Ip, 2011, Shintani and Klionsky, 2004). Dietary therapies appear to be intriguing, given that autophagy induction helps delay or prevent neurodegenerative disorders. Intake of a polyphenol-rich diet has been linked to a reduced incidence of chronic diseases (Xie et al., 2019). A wide variety of identified or unidentified bioactive compounds in natural products such as fruits and vegetables have health-promoting benefits. These bioactive compounds exhibit pleiotropic effects, such as antioxidant, anti-inflammatory, neuroprotective, hypolipidemic, glycaemic-regulating, immune-enhancing, and cytoprotective capabilities (Barnard et al., 2014, Xie et al., 2019). Due to the impaired autophagic flux, autophagic machineries are unable to exterminate the surplus organelles and aggregates. Synaptic impairment, loss of neuronal communication, damage of cellular organelle, and neuronal cell death are all related to the aggregation and accumulation of toxic proteins as well as intracytoplasmic autophagosomes resulting in the development of neurodegenerative diseases (Cheung and Ip, 2011, Menzies et al., 2017). Some of the commonly explored autophagy-associated target compounds and bioactive components in functional meals are discussed here.
Trehalose is a disaccharide widely used to kick-start autophagy by inhibiting GLUT protein and activating AMPK. Metformin and Berherine are well-known mitochondrial inhibitors that have shown beneficial effects on animal models of neurodegeneration by inducing autophagy through AMPK activation. Other reported AMPK activators include methylene blue, an anti-aggregant and nilotinib, which induces autophagy to eliminate the toxic proteins and may play key role in the treatment of neurodegenerative diseases. It has been shown that modulating cyclic AMP and Inositol 3 –phosphate (cAMP/IP3) positively regulates the autophagy process (Chen et al., 2021, Menzies et al., 2017). Several other molecules possessing autophagy-modulating capacity have been identified.
Resveratrol (RSV) is a polyphenol found in grapes, nuts, berries, and other plants that has pleiotropic effects such as antioxidant activity, neuroprotective properties, anti-inflammatory activity, and cytoprotective effect. RSV could induce autophagy via enhancing the levels of LC3-II puncta, up-regulating Bcelin1, and increasing LC3-II/LC3-I ratio. Resveratrol may influence cellular functions by triggering critical metabolic sensors/effectors such as AMPK, SIRT1, and Peroxisome proliferator-activated receptor γ co-activator-1α (PGC-1α). RSV protects against AD by promoting autophagy, thus eliminating Aβ and lipopolysaccharide (LPS) and suppressing inflammatory cytokines like as interleukin 1 beta (IL-1), tumour necrosis factor alpha (TNF-α), and nuclear factor kappa-β (NF-κβ) (Kou and Chen, 2017, Xie et al., 2019).
Curcumin, turmeric’s major medicinal element, has long been recognised for its antioxidant and anti-inflammatory properties and exerts hypolipidemic, neuroprotective, and cardiovascular protective effects. It has the ability to reduce the expression of NF-κβ and ROS significantly while also defending against mitochondrial malfunction. Curcumin can increase the expression of LC3-II and Beclin1, facilitate autophagosome formation, and speed up the deterioration of p62 to improve cell viability during oxidative stress. It promotes autophagy and cell survival by indicating the expression of Bcl2 and Bax while suppressing the expression of Beclin1 and Sirt1, respectively. Curcumin may rescue cells from oxidative damage by triggering autophagy via the Akt/mTOR signal pathway and decreasing α-synuclein formation in A53 T cells and Aβ production in AD mice by down-regulating mTOR signalling (Xie et al., 2019). Because of its antioxidant properties and ability to regulate insulin sensitivity, lipid metabolism, and inflammatory response, curcumin exerts neuroprotective effects.
Epigallocatechin-3-gallate (EGCG), a plant-derived polyphenol compound found in green tea, has been shown to slow the progression of metabolic disorders by activating autophagy and lowering lipid build-up in vascular endothelial cells. EGCG has also been demonstrated to stimulate autophagy, as evidenced by the overexpression of autophagic biomarkers such as LC3-II, Beclin1, and Atg7, restricting p62, and decreasing Bax and cytochrome C (Cyc) throughout the process of neurodegeneration and protect neurons by promoting autophagy (Lee et al., 2015, Singh et al., 2015, Xie et al., 2019). Some other natural compounds are enhancing autophagic pathways can be considered as possible therapeutics in the treatment of neurodegenerative diseases are represented in Table 3.
Table 3:
Plant derived bioactive compounds regulating autophagy in neurodegenerative diseases.
| Sl No. | Bioactive compounds | Regulation | Mechanism | Reference |
|---|---|---|---|---|
| 1 | Cannabidiol | Autophagy stimulation | It can upregulate SIRT1 expression and protect mitochondrial proteins hence limiting mitochondrial dysfunction | (Kang et al., 2021) |
| 2 | Trehalose | Accelerating the elimination of mutant huntingtin and α-syn is a unique mTOR-independent autophagy enhancer | (Sarkar et al., 2007) | |
| 3 | Resveratrol | Through the Ca2+/AMPK-mTOR signaling cascade and autophagy, it may promote autophagocytic death and cause cell death. | (Tian et al., 2019) | |
| 4 | EGCG | Induction of apoptosis | It enhances autophagy through Nrf2 nuclear translocation, increased mRNA expression of LC3 and Caspase 9. Activated autophagy might serve as a sensor for ROS that later triggers cell death. |
(Ferrari et al., 2022) |
| 5 | Delphinidin | Promotes apoptosis and induces autophagy | It blocks mTOR signaling pathway and activates AMPK pathway which activates autophagy | (Chen et al., 2018) |
| 6 | Berberine | Induction of autophagy | By stimulating SIRT1 through the NAD+ production pathway, it activated peritoneal macrophages’ autophagy while also boosting TFEB nuclear translocation and deacetylation. | (Zheng et al., 2021) |
| 7 | Spermidine | Induction of autophagy, reduce inflammation and senescence | Spermidine and spermine modulate autophagy proteins through phosphorylation of AMPK, and activate AMPK, LC3, Beclin 1 and p62. Additionally, the control of MFN1, MFN2, DRP1, COX IV, and ATP, it sustain equilibrium in the mitochondria and provide energy for the neurons. | (Xu et al., 2020) |
| 8 | Curcumin | Induction of autophagy and alleviating oxidative stress | It raised the levels of LC3, Bcl-2, beclin1, and Nrf2 while downregulating the expression of p62, PI3K/p-AKT/p-mTOR Bax and Caspase-3 proteins. | (Di Tu et al., 2020) |
| 9 | Genistein | Activating autophagy | It elevates PPAR-γ and LC3-II levels and reduces the expression of p62 | (Liu et al., 2019) |
| 10 | Piperine | Activating Autophagy | It stimulates protein phosphotase 2A (PP2A), which in turn inhibited the mTORC1 and also it increases LC3II protein level that enhances autophagy | (Liu et al., 2016) |
Calorie restriction or deprivation may have a role to play
Autophagy is a type of cellular autolysis initiated by various stimuli, one of which is cytoplasmic deprivation, which allows cells to eliminate cytoplasmic components. Autophagy’s understanding as a mechanism of resistance to metabolic stress-inducing therapies or as a means of cell death is fast developing, indicating that a new therapeutic starvation paradigm is emerging (DiPaola et al., 2008). Calorie restriction (CR) is frequently praised for several ageing intervention techniques. CR represent a reduction in average daily calorie intake below what is typical or habitual without causing malnutrition or nutrient deficiency. Organelle damage like in mitochondria and ER, hypoxia, and inflammation are biologically relevant stimuli that trigger autophagy, but nutrition and energy stress seem to be the most potent autophagy regulators. The upstream regulatory elements, such as mTOR can induce autophagy in response to shifting cellular energy levels (Dunlop and Tee, 2014, Kim and Guan, 2015). There are also a variety of other known nutrient-sensing signalling proteins governing autophagy induction in nutrient-deficient circumstances. The AMPK is another significant participant in sensing nutrition shortage. Autophagy is triggered by AMPK via establishing the link between AMPK and ULK1 that can be prevented by mTORC1-mediated ULK1 phosphorylation, which is essential for ULK1 activation and autophagy onset under nutritional depletion environments (Kim et al., 2011). CR appears to work in several ways to slow the ageing process: CR improves cell potential to restore DNA damage, stimulates anti-stress proteins, enhances glucose metabolism efficiency, and slows age-related immune system decline. Moreover, substantial oxidative stress reduction and neuroendocrine system modulation are other positive impacts of dietary restriction (Guo and Richardson, 1999). Utilizing a Sirtuin-1-dependent autophagy initiation pathway, CR has been shown to prolong longevity or defend against hypoxia. Also, extending life through methionine limitation necessitated the stimulation of autophagy. Further studies have demonstrated that CR substantially activates autophagy under diverse pathophysiologic situations and therefore has a beneficial effect in maintaining normal processes in the organism, especially lifespan (Chung and Chung, 2019).
A favourable influence of lifestyle and exercise
Autophagy is a suitable target mechanism from a therapeutic standpoint. Under various circumstances, it aids in the clearance of the underlying toxic entities associated with disease pathology, thereby targeting the root cause of the problem. It has been demonstrated in earlier studies that exercise triggers autophagy in various metabolically essential organs, including the brain, liver, adipose tissue, pancreas, and muscle. In contrast, a subdued lifestyle combined with a sedentary lifestyle may increase the risk of stroke, PD, and AD. Long-term intermittent physiological demands, such as cognitive pursuits, energy restriction, and exercise, may protect the brain from ILOD (idiopathic late-onset dementia) and enhance neuroplasticity and cellular stress tolerance. Strenuous physical activity may improve mitochondrial function in the cerebellum and cerebral cortex, lowering apoptosis-related indicators and oxidative stress. Physical activity may positively influence lysosomal disintegration and mitochondrial quality control, reducing age-related cognitive decline (Xing et al., 2019). Exercise is a well-known physiological therapy that can preserve tissue integrity, reduce inflammation, and activate the direct signalling route for the cellular response. Additionally, exercise has been shown to increase autophagy flux and initiate transcription of key autophagy genes, potentially increasing autophagy activity. It has also been shown to significantly enhance diverse mitochondrial gene transcripts and autophagy-associated genes, promote mitophagy in mitochondria and increase autophagy and mitophagy flux (chul Jang et al., 2018).
Autophagy is always present at a low level in tissues and cells, facilitating cellular level of clearance and cellular metabolism. Cellular disturbances such as energy imbalance and oxidative damage can activate this system as a survival mechanism. A pulsatile autophagy response is activated during exercise, resulting in an immediate activation and a protracted potentiation of the regulatory process. AMP, Calcium, NAD+, and ROS concentrations rise during muscular contraction. AMPK, CaMK/calcineurin, SIRT1, and p38 are the molecular messengers activating their downstream effectors. As a result, both acute autophagy and a relatively long transcriptional programme are initiated (Vainshtein and Hood, 2016, Zhang et al., 2014b). Daily exercise can promote several neurogenic activities essential for brain function; it stimulates neurogenesis and angiogenesis by up-regulating growth factors and brain-derived neurotrophic factors (Chen et al., 2020b). Proteinopathies are the underlying mechanism of increasing oxidative stress and developing various neurodegenerative diseases such as AD, HD, ALS, and PD. The proteasome, also known as the 26S, is a multi-catalytic unit with a 20S proteolytic core accompanied by regulatory units that detect ubiquitinated substrates, malformed and unfolded proteins, or unhealthy proteins which are vulnerable and direct them to breakdown (Andreotti et al., 2020). Exercise can promote synaptic remodelling and improve cell survival by upregulating BDNF in hippocampal and cortical neurons. It also lowers the release of the pro-inflammatory cytokine TNF-α, which reduces apoptotic cell death through the extrinsic route when present in small concentrations.
Exercise has long been a driving force in human evolution, allowing us to forage for food, adjust to our surroundings, and, as a result, change the physiology and continued development of our brains, demonstrating the co-evolution of neuronal plasticity signal transduction pathways (Mattson, 2012). A large body of research supports that obesity, diabetes, and cardiovascular diseases are comorbidities of neurodegenerative disorders, and they are becoming a big concern in today’s society, with a significant impact on socioeconomic status. Physical exercise, and moderate to vigorous intensity interventions as a metabolic stimulus can activate various adaptive stress responses in brain cells, including the expression of neurotrophic factors, mitochondrial biogenesis and breakdown, and protein chaperones, all of which are strongly linked to autophagy. Physical activity has been shown to have long-term advantages in both animals and humans, including enhanced cognitive ability, improved cerebral blood flow, lowered oxidative stress response, expanded neurotransmitter thresholds and synaptic plasticity, and significantly improved attention skills and process information in investigations on both animals and humans. Physical activity can also help people with cognitive disorders like anxiety and depression relieve stress and alleviate negative psychology (Chen et al., 2020b). Sleep deprivation is a common problem among the elderly and people suffering from mental illnesses like depression. Different types of aerobic exercise, yoga, and physical activity training are beneficial to the young generation suffering from insomnia and the elderly in getting a good night’s sleep. This is undoubtedly one of the most helpful lifestyle interventions for persons with mental illness. It can assist in minimizing the risk of disease progression by improving metabolism and limiting the accumulation of toxins within the body.
Conclusions
Compelling evidence can be drawn from earlier studies that dysregulated autophagy and mitophagy strongly influence the progression of neurodegenerative diseases. Modulating the signalling pathways halting autophagy and mitophagy in the brain can restore normal function. Dysregulated autophagy process can be targeted through several pharmacological and non-pharmacological interventions. Natural bioactive compounds are the budding approaches for regulating physiological processes. Over and above, intake of necessary dietary supplements, following physical exercise, and maintaining a healthy lifestyle will significantly impact physiological function and delay ageing and senescence. Ultimately preventive measures can be taken against age-related metabolic and neurodegenerative diseases. Further studies can be proposed at the level of modification and regulation of transcriptional factors and epigenetic regulations. Adoption of computational approaches also beneficial in developing novel drug discovery and pave the path in addressing multiple targets for treating neurodegenerative diseases.
Acknowledgement
This research has been supported by DST-SERB (Grant No. EEQ/2020/000188) (to JSB); DST-INSPIRE Fellowship (to JM); NIH grants AG042178, AG047812, NS205473, AG060767, AG069333, AG066347 and AG079264 (to PHR); Alzheimer’s Association through a SAGA grant, Garrison Family Foundation Grant and NIH grants AG063162 and AG071560 (to APR). BioRender software was used to create the figures appended in this manuscript.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Ethical Approval and Consent to participate: Not applicable
Consent for publication: Not applicable
Competing Interest: The authors declare that they have no competing interests
Availability of Data and Material:
All data analyzed during this study are included in this article.
References
- Alfaro IE, Albornoz A, Molina A, Moreno J, Cordero K, Criollo A, et al. Chaperone Mediated Autophagy in the Crosstalk of Neurodegenerative Diseases and Metabolic Disorders. Frontiers in Endocrinology. 2019;9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alirezaei M, Kemball CC, Whitton JL. Autophagy, inflammation and neurodegenerative disease. Eur J Neurosci. 2011;33:197–204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Andersson AM, Andersson B, Lorell C, Raffetseder J, Larsson M, Blomgran R. Autophagy induction targeting mTORC1 enhances Mycobacterium tuberculosis replication in HIV co-infected human macrophages. Sci Rep. 2016;6:28171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Andreotti DZ, Silva JdN, Matumoto AM, Orellana AM, de Mello PS, Kawamoto EM. Effects of Physical Exercise on Autophagy and Apoptosis in Aged Brain: Human and Animal Studies. Front Nutr. 2020;7:94-. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arkinson C, Walden H. Parkin function in Parkinson’s disease. Science. 2018;360:267–8. [DOI] [PubMed] [Google Scholar]
- Barnard ND, Bush Al, Ceccarelli A, Cooper J, de Jager CA, Erickson Kl, et al. Dietary and lifestyle guidelines for the prevention of Alzheimer’s disease. Neurobiology of aging. 2014;35:S74–S8. [DOI] [PubMed] [Google Scholar]
- Barodia SK, Creed RB, Goldberg MS. Parkin and PINK1 functions in oxidative stress and neurodegeneration. Brain Res Bull. 2017;133:51–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Basavarajappa BS, Shivakumar M, Joshi V, Subbanna S. Endocannabinoid system in neurodegenerative disorders. J Neurochem. 2017;142:624–48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bednarczyk M, Zmarzły N, Grabarek B, Mazurek U, Muc-Wierzgoń M. Genes involved in the regulation of different types of autophagy and their participation in cancer pathogenesis. Oncotarget. 2018;9:34413–28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bellingham S, Guo B, Coleman B, Hill A. Exosomes: Vehicles for the Transfer of Toxic Proteins Associated with Neurodegenerative Diseases? Frontiers in Physiology. 2012;3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bento CF, Ashkenazi A, Jimenez-Sanchez M, Rubinsztein DC. The Parkinson’s disease-associated genes ATP13A2 and SYT11 regulate autophagy via a common pathway. Nature Communications. 2016;7:11803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bhatti J, Kumar S, Vijayan M, Bhatti G, Reddy P. Therapeutic strategies for mitochondrial dysfunction and oxidative stress in age-related metabolic disorders. Progress in molecular biology and translational science. 2017;146:13–46. [DOI] [PubMed] [Google Scholar]
- Bjørkøy G, Lamark T, Brech A, Outzen H, Perander M, Overvatn A, et al. p62/SQSTM1 forms protein aggregates degraded by autophagy and has a protective effect on huntingtin-induced cell death. J Cell Biol. 2005;171:603–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bloem BR, Okun MS, Klein C. Parkinson’s disease. Lancet. 2021;397:2284–303. [DOI] [PubMed] [Google Scholar]
- Boland B, Yu WH, Corti O, Mollereau B, Henriques A, Bezard E, et al. Promoting the clearance of neurotoxic proteins in neurodegenerative disorders of ageing. Nature Reviews Drug Discovery. 2018;17:660–88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brown RC, Lockwood AH, Sonawane BR. Neurodegenerative diseases: an overview of environmental risk factors. Environ Health Perspect. 2005;113:1250–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cai Q, Jeong YY. Mitophagy in Alzheimer’s Disease and Other Age-Related Neurodegenerative Diseases. Cells. 2020;9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chakravorty A, Jetto CT, Manjithaya R. Dysfunctional Mitochondria and Mitophagy as Drivers of Alzheimer’s Disease Pathogenesis. Frontiers in Aging Neuroscience. 2019;11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen C, Gao H, Su X. Autophagy-related signaling pathways are involved in cancer (Review). Exp Ther Med. 2021;22:710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen G, Kroemer G, Kepp O. Mitophagy: An Emerging Role in Aging and Age-Associated Diseases. Front Cell Dev Biol. 2020a;8:200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen J, Zhu Y, Zhang W, Peng X, Zhou J, Li F, et al. Delphinidin induced protective autophagy via mTOR pathway suppression and AMPK pathway activation in HER-2 positive breast cancer cells. BMC Cancer. 2018;18:342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen K, Yuan R, Geng S, Zhang Y, Ran T, Kowalski E, et al. Toll-interacting protein deficiency promotes neurodegeneration via impeding autophagy completion in high-fat diet-fed ApoE(−/−) mouse model. Brain Behav Immun. 2017;59:200–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen M, Chen Z, Wang Y, Tan Z, Zhu C, Li Y, et al. Mitophagy receptor FUNDC1 regulates mitochondrial dynamics and mitophagy. Autophagy. 2016;12:689–702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen Z, Lan W, Yang G, Li Y, Ji X, Chen L, et al. Exercise Intervention in Treatment of Neuropsychological Diseases: A Review. Frontiers in Psychology. 2020b;11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheung ZH, Ip NY. The emerging role of autophagy in Parkinson’s disease. Molecular Brain. 2009;2:29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheung ZH, Ip NY. Autophagy deregulation in neurodegenerative diseases – recent advances and future perspectives. Journal of Neurochemistry. 2011;118:317–25. [DOI] [PubMed] [Google Scholar]
- Chu CT. Mechanisms of selective autophagy and mitophagy: Implications for neurodegenerative diseases. Neurobiol Dis. 2019;122:23–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- chul Jang Y, Hwang DJ, Koo JH, Um HS, Lee NH, Yeom DC, et al. Association of exercise-induced autophagy upregulation and apoptosis suppression with neuroprotection against pharmacologically induced Parkinson’s disease. Journal of Exercise Nutrition & Biochemistry. 2018;22:1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chung KW, Chung HY. The Effects of Calorie Restriction on Autophagy: Role on Aging Intervention. Nutrients. 2019;11:2923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ciechanover A, Kwon YT. Protein Quality Control by Molecular Chaperones in Neurodegeneration. Frontiers in Neuroscience. 2017;11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Collier JJ, Suomi F, Oláhová M, McWilliams TG, Taylor RW. Emerging roles of ATG7 in human health and disease. EMBO Molecular Medicine. 2021;13:e14824. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Costello MJ, Brennan LA, Basu S, Chauss D, Mohamed A, Gilliland KO, et al. Autophagy and mitophagy participate in ocular lens organelle degradation. Exp Eye Res. 2013;116:141–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crews L, Spencer B, Desplats P, Patrick C, Paulino A, Rockenstein E, et al. Selective molecular alterations in the autophagy pathway in patients with Lewy body disease and in models of alpha-synucleinopathy. PLoS One. 2010;5:e9313. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- Croce KR, Yamamoto A. A role for autophagy in Huntington’s disease. Neurobiol Dis. 2019a;122:16–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Croce KR, Yamamoto A. A role for autophagy in Huntington’s disease. Neurobiology of Disease. 2019b;122:16–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cruz P, Ahumada-Castro U, Bustos G, Molgó J, Sauma D, Lovy A, et al. Inhibition of InsP3R with Xestospongin B Reduces Mitochondrial Respiration and Induces Selective Cell Death in T Cell Acute Lymphoblastic Leukemia Cells. Int J Mol Sci. 2021;22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Gaetano A, Gibellini L, Zanini G, Nasi M, Cossarizza A, Pinti M. Mitophagy and Oxidative Stress: The Role of Aging. Antioxidants (Basel). 2021;10:794. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Santi M, Baldelli G, Diotallevi A, Galluzzi L, Schiavano GF, Brandi G. Metformin prevents cell tumorigenesis through autophagy-related cell death. Scientific Reports. 2019;9:66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Di Tu Q, Jin J, Hu X, Ren Y, Zhao L, He Q. Curcumin Improves the Renal Autophagy in Rat Experimental Membranous Nephropathy via Regulating the PI3K/AKT/mTOR and Nrf2/HO-1 Signaling Pathways. Biomed Res Int. 2020;2020:7069052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dice JF. Chaperone-mediated autophagy. Autophagy. 2007;3:295–9. [DOI] [PubMed] [Google Scholar]
- DiPaola RS, Dvorzhinski D, Thalasila A, Garikapaty V, Doram D, May M, et al. Therapeutic starvation and autophagy in prostate cancer: a new paradigm for targeting metabolism in cancer therapy. The Prostate. 2008;68:1743–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DiSabato DJ, Quan N, Godbout JP. Neuroinflammation: the devil is in the details. Journal of Neurochemistry. 2016;139:136–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Doherty J, Baehrecke EH. Life, death and autophagy. Nature Cell Biology. 2018;20:1110–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dugger BN, Dickson DW. Pathology of Neurodegenerative Diseases. Cold Spring Harb Perspect Biol. 2017;9:a028035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dunlop EA, Tee AR. mTOR and autophagy: a dynamic relationship governed by nutrients and energy. Semin Cell Dev Biol. 2014;36:121–9. [DOI] [PubMed] [Google Scholar]
- Facecchia K, Fochesato L-A, Ray SD, Stohs SJ, Pandey S. Oxidative Toxicity in Neurodegenerative Diseases: Role of Mitochondrial Dysfunction and Therapeutic Strategies. Journal of Toxicology. 2011;2011:683728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feng H, Yang Z, Bai X, Yang M, Fang Y, Zhang X, et al. Therapeutic potential of a dual mTORC1/2 inhibitor for the prevention of posterior capsule opacification: An in vitro study. Int J Mol Med. 2018;41:2099–107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feng Y, He D, Yao Z, Klionsky DJ. The machinery of macroautophagy. Cell Research. 2014;24:24–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferrari E, Bettuzzi S, Naponelli V. The Potential of Epigallocatechin Gallate (EGCG) in Targeting Autophagy for Cancer Treatment: A Narrative Review. Int J Mol Sci. 2022;23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Filomeni G, De Zio D, Cecconi F. Oxidative stress and autophagy: the clash between damage and metabolic needs. Cell Death & Differentiation. 2015;22:377–88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Franco-Iborra S, Plaza-Zabala A, Montpeyo M, Sebastian D, Vila M, Martinez-Vicente M. Mutant HTT (huntingtin) impairs mitophagy in a cellular model of Huntington disease. Autophagy. 2021;17:672–89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Franco-Iborra S, Vila M, Perier C. Mitochondrial Quality Control in Neurodegenerative Diseases: Focus on Parkinson’s Disease and Huntington’s Disease. Frontiers in Neuroscience. 2018;12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fredrickson EK, Gardner RG. Selective destruction of abnormal proteins by ubiquitin-mediated protein quality control degradation. Semin Cell Dev Biol. 2012;23:530–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Funderburk SF, Marcellino BK, Yue Z. Cell “self-eating” (autophagy) mechanism in Alzheimer’s disease. Mt Sinai J Med. 2010;77:59–68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Furukawa K, Innokentev A, Kanki T. Regulatory Mechanisms of Mitochondrial Autophagy: Lessons From Yeast. Frontiers in Plant Science. 2019;10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Furuta N, Amano A. SNARE mediates autophagosome–lysosome fusion. Journal of Oral Biosciences. 2012;54:83–5. [Google Scholar]
- Galluzzi L, Baehrecke EH, Ballabio A, Boya P, Bravo-San Pedro JM, Cecconi F, et al. Molecular definitions of autophagy and related processes. The EMBO Journal. 2017;36:1811–36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gatica D, Lahiri V, Klionsky DJ. Cargo recognition and degradation by selective autophagy. Nat Cell Biol. 2018;20:233–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gauthier S, Rosa-Neto P, Morais JA, Webster C. World Alzheimer Report 2021: Journey through the diagnosis of dementia. London, England: 2021. [Google Scholar]
- Gilad S, Meiri E, Yogev Y, Benjamin S, Lebanony D, Yerushalmi N, et al. Serum microRNAs are promising novel biomarkers. PLoS One. 2008;3:e3148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo Z, Richardson A. Effects of aging and dietary restriction on DNA repair. ALFRED BENZON SYMPOSIUM: MUNKSGAARD BOGHANDEL; 1999. p. 362–72. [Google Scholar]
- Halagappa VKM, Guo Z, Pearson M, Matsuoka Y, Cutler RG, LaFerla FM, et al. Intermittent fasting and caloric restriction ameliorate age-related behavioral deficits in the triple-transgenic mouse model of Alzheimer’s disease. Neurobiology of Disease. 2007;26:212–20. [DOI] [PubMed] [Google Scholar]
- Hannigan AM, Gorski SM. Macroautophagy: the key ingredient to a healthy diet? Autophagy. 2009;5:140–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harding TM, Morano KA, Scott SV, Klionsky DJ. Isolation and characterization of yeast mutants in the cytoplasm to vacuole protein targeting pathway. J Cell Biol. 1995;131:591–602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hroudová J, Singh N, Fišar Z. Mitochondrial Dysfunctions in Neurodegenerative Diseases: Relevance to Alzheimer’s Disease. BioMed Research International. 2014;2014:175062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Itakura E, Kishi-Itakura C, Mizushima N. The Hairpin-type Tail-Anchored SNARE Syntaxin 17 Targets to Autophagosomes for Fusion with Endosomes/Lysosomes. Cell. 2012;151:1256–69. [DOI] [PubMed] [Google Scholar]
- Jiang J, Zhang L, Chen H, Lei Y, Zhang T, Wang Y, et al. Regorafenib induces lethal autophagy arrest by stabilizing PSAT1 in glioblastoma. Autophagy. 2020;16:106–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jin M-m, Wang F, Qi D, Liu W-w, Gu C, Mao C-J, et al. A Critical Role of Autophagy in Regulating Microglia Polarization in Neurodegeneration. Frontiers in Aging Neuroscience. 2018;10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jin SM, Youle RJ. PINK1- and Parkin-mediated mitophagy at a glance. J Cell Sci. 2012;125:795–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kang S, Li J, Yao Z, Liu J. Cannabidiol Induces Autophagy to Protects Neural Cells From Mitochondrial Dysfunction by Upregulating SIRT1 to Inhibits NF-κB and NOTCH Pathways. Front Cell Neurosci. 2021;15:654340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kania E, Pająk B, O’Prey J, Sierra Gonzalez P, Litwiniuk A, Urbańska K, et al. Verapamil treatment induces cytoprotective autophagy by modulating cellular metabolism. Febs j. 2017;284:1370–87. [DOI] [PubMed] [Google Scholar]
- Khalil B, El Fissi N, Aouane A, Cabirol-Pol MJ, Rival T, Liévens JC. PINK1-induced mitophagy promotes neuroprotection in Huntington’s disease. Cell Death & Disease. 2015;6:e1617–e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim J, Fiesel FC, Belmonte KC, Hudec R, Wang WX, Kim C, et al. miR-27a and miR-27b regulate autophagic clearance of damaged mitochondria by targeting PTEN-induced putative kinase 1 (PINK1). Mol Neurodegener. 2016;11:55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim J, Kundu M, Viollet B, Guan KL. AMPK and mTOR regulate autophagy through direct phosphorylation of Ulk1. Nat Cell Biol. 2011;13:132–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim YC, Guan KL. mTOR: a pharmacologic target for autophagy regulation. J Clin Invest. 2015;125:25–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kincaid B, Bossy-Wetzel E. Forever young: SIRT3 a shield against mitochondrial meltdown, aging, and neurodegeneration. Front Aging Neurosci. 2013;5:48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- King JS, Veltman DM, Insall RH. The induction of autophagy by mechanical stress. Autophagy. 2011;7:1490–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kiss T, Giles CB, Tarantini S, Yabluchanskiy A, Balasubramanian P, Gautam T, et al. Nicotinamide mononucleotide (NMN) supplementation promotes anti-aging miRNA expression profile in the aorta of aged mice, predicting epigenetic rejuvenation and anti-atherogenic effects. Geroscience. 2019;41:419–39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Komatsu M, Waguri S, Chiba T, Murata S, Iwata J, Tanida I, et al. Loss of autophagy in the central nervous system causes neurodegeneration in mice. Nature. 2006;441:880–4. [DOI] [PubMed] [Google Scholar]
- Kou X, Chen N. Resveratrol as a Natural Autophagy Regulator for Prevention and Treatment of Alzheimer’s Disease. Nutrients. 2017;9:927. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lamark T, Svenning S, Johansen T. Regulation of selective autophagy: the p62/SQSTM1 paradigm. Essays in Biochemistry. 2017;61:609–24. [DOI] [PubMed] [Google Scholar]
- Lee J-H, Moon J-H, Kim S-W, Jeong J-K, Nazim UM, Lee Y-J, et al. EGCG-mediated autophagy flux has a neuroprotection effect via a class III histone deacetylase in primary neuron cells. Oncotarget. 2015;6:9701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee J, Giordano S, Zhang J. Autophagy, mitochondria and oxidative stress: cross-talk and redox signalling. Biochemical Journal. 2011;441:523–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee MJ, Lee JH, Rubinsztein DC. Tau degradation: the ubiquitin-proteasome system versus the autophagy-lysosome system. Prog Neurobiol. 2013;105:49–59. [DOI] [PubMed] [Google Scholar]
- Levchenko M, Lorenzi I, Dudek J. The degradation pathway of the mitophagy receptor Atg32 is rerouted by a posttranslational modification. PLoS One. 2016;11:e0168518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Levine B, Kroemer G. Autophagy in the pathogenesis of disease. Cell. 2008;132:27–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Levine B, Kroemer G. Biological Functions of Autophagy Genes: A Disease Perspective. Cell. 2019;176:11–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li L, Acioglu C, Heary RF, Elkabes S. Role of astroglial toll-like receptors (TLRs) in central nervous system infections, injury and neurodegenerative diseases. Brain Behav Immun. 2021;91:740–55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li WW, Li J, Bao JK. Microautophagy: lesser-known self-eating. Cell Mol Life Sci. 2012;69:1125–36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Limanaqi F, Biagioni F, Gambardella S, Familiari P, Frati A, Fornai F. Promiscuous Roles of Autophagy and Proteasome in Neurodegenerative Proteinopathies. Int J Mol Sci. 2020;21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu J, Chen M, Wang X, Wang Y, Duan C, Gao G, et al. Piperine induces autophagy by enhancing protein phosphotase 2A activity in a rotenone-induced Parkinson’s disease model. Oncotarget. 2016;7:60823–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu X, Zhang M, Zhang H, Zhao A, Sun J, Tang W. [Role of PPAR-γ-regulated autophagy in genistein-induced inhibition of hepatic stellate cell activation]. Nan Fang Yi Ke Da Xue Xue Bao. 2019;39:561–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu J, He L, Behrends C, Araki M, Araki K, Jun Wang Q, et al. NRBF2 regulates autophagy and prevents liver injury by modulating Atg14L-linked phosphatidylinositol-3 kinase III activity. Nature Communications. 2014;5:3920. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lynch-Day MA, Mao K, Wang K, Zhao M, Klionsky DJ. The role of autophagy in Parkinson’s disease. Cold Spring Harb Perspect Med. 2012;2:a009357–a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lyu L, Chen Z, McCarty N. TRIM44 links the UPS to SQSTM1/p62-dependent aggrephagy and removing misfolded proteins. Autophagy. 2022;18:783–98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Malampati S, Song JX, Chun-Kit Tong B, Nalluri A, Yang CB, Wang Z, et al. Targeting Aggrephagy for the Treatment of Alzheimer’s Disease. Cells. 2020;9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Malik BR, Maddison DC, Smith GA, Peters OM. Autophagic and endo-lysosomal dysfunction in neurodegenerative disease. Molecular Brain. 2019;12:100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marinković M, šprung M, Novak I. Dimerization of mitophagy receptor BNIP3L/NIX is essential for recruitment of autophagic machinery. Autophagy 2021;17:1232–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martí-Martínez S, Valor LM. A Glimpse of Molecular Biomarkers in Huntington’s Disease. International Journal of Molecular Sciences. 2022;23:5411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matsuda N, Sato S, Shiba K, Okatsu K, Saisho K, Gautier CA, et al. PINK1 stabilized by mitochondrial depolarization recruits Parkin to damaged mitochondria and activates latent Parkin for mitophagy. Journal of Cell Biology. 2010;189:211–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mattson MP. Evolutionary aspects of human exercise--born to run purposefully. Ageing Res Rev. 2012;11:347–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mehta SL, Dempsey RJ, Vemuganti R. Role of circular RNAs in brain development and CNS diseases. Prog Neurobiol. 2020;186:101746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meng H, Yan W-Y, Lei Y-H, Wan Z, Hou Y-Y, Sun L-K, et al. SIRT3 Regulation of Mitochondrial Quality Control in Neurodegenerative Diseases. Frontiers in aging neuroscience. 2019;11:313-. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Menzies FM, Fleming A, Caricasole A, Bento CF, Andrews SP, Ashkenazi A, et al. Autophagy and Neurodegeneration: Pathogenic Mechanisms and Therapeutic Opportunities. Neuron. 2017;93:1015–34. [DOI] [PubMed] [Google Scholar]
- Merino D, Kelly GL, Lessene G, Wei AH, Roberts AW, Strasser A. BH3-Mimetic Drugs: Blazing the Trail for New Cancer Medicines. Cancer Cell. 2018;34:879–91. [DOI] [PubMed] [Google Scholar]
- Mizushima N, Levine B, Cuervo AM, Klionsky DJ. Autophagy fights disease through cellular self-digestion. Nature. 2008;451:1069–75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morais VA, Verstreken P, Roethig A, Smet J, Snellinx A, Vanbrabant M, et al. Parkinson’s disease mutations in PINK1 result in decreased Complex I activity and deficient synaptic function. EMBO Mol Med. 2009;1:99–111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moreau K, Fleming A, Imarisio S, Lopez Ramirez A, Mercer JL, Jimenez-Sanchez M, et al. PICALM modulates autophagy activity and tau accumulation. Nat Commun. 2014;5:4998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moreno M-L, Mérida S, Bosch-Morell F, Miranda M, Villar VM. Autophagy Dysfunction and Oxidative Stress, Two Related Mechanisms Implicated in Retinitis Pigmentosa. Frontiers in Physiology. 2018;9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Narendra D, Tanaka A, Suen D-F, Youle RJ. Parkin is recruited selectively to impaired mitochondria and promotes their autophagy. Journal of cell biology. 2008;183:795–803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nguyen TN, Padman BS, Lazarou M. Deciphering the Molecular Signals of PINK1/Parkin Mitophagy. Trends in Cell Biology. 2016;26:733–44. [DOI] [PubMed] [Google Scholar]
- Nixon RA. The role of autophagy in neurodegenerative disease. Nature Medicine. 2013;19:983–97. [DOI] [PubMed] [Google Scholar]
- Norat P, Soldozy S, Sokolowski JD, Gorick CM, Kumar JS, Chae Y, et al. Mitochondrial dysfunction in neurological disorders: Exploring mitochondrial transplantation. npj Regenerative Medicine. 2020;5:22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oku M, Sakai Y. Three Distinct Types of Microautophagy Based on Membrane Dynamics and Molecular Machineries. BioEssays. 2018;40:1800008. [DOI] [PubMed] [Google Scholar]
- Onishi M, Yamano K, Sato M, Matsuda N, Okamoto K. Molecular mechanisms and physiological functions of mitophagy. The EMBO Journal. 2021;40:e104705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pang SY, Lo RCN, Ho PW, Liu HF, Chang EES, Leung CT, et al. LRRK2, GBA and their interaction in the regulation of autophagy: implications on therapeutics in Parkinson’s disease. Transl Neurodegener. 2022;11:5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park H-W, Park H, Semple IA, Jang I, Ro S-H, Kim M, et al. Pharmacological correction of obesity-induced autophagy arrest using calcium channel blockers. Nature Communications. 2014;5:4834. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pickford F, Masliah E, Britschgi M, Lucin K, Narasimhan R, Jaeger PA, et al. The autophagy-related protein beclin 1 shows reduced expression in early Alzheimer disease and regulates amyloid beta accumulation in mice. J Clin Invest. 2008;118:2190–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Piffoux M, Eriau E, Cassier PA. Autophagy as a therapeutic target in pancreatic cancer. Br J Cancer. 2021;124:333–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raposo G, Stoorvogel W. Extracellular vesicles: Exosomes, microvesicles, and friends. Journal of Cell Biology. 2013;200:373–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ravikumar B, Vacher C, Berger Z, Davies JE, Luo S, Oroz LG, et al. Inhibition of mTOR induces autophagy and reduces toxicity of polyglutamine expansions in fly and mouse models of Huntington disease. Nat Genet. 2004;36:585–95. [DOI] [PubMed] [Google Scholar]
- Reggiori F, Komatsu M, Finley K, Simonsen A. Selective Types of Autophagy. International Journal of Cell Biology. 2012;2012:156272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rubinsztein DC, Codogno P, Levine B. Autophagy modulation as a potential therapeutic target for diverse diseases. Nature Reviews Drug Discovery. 2012;11:709–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sarkar S, Davies JE, Huang Z, Tunnacliffe A, Rubinsztein DC. Trehalose, a novel mTOR-independent autophagy enhancer, accelerates the clearance of mutant huntingtin and alpha-synuclein. J Biol Chem. 2007;282:5641–52. [DOI] [PubMed] [Google Scholar]
- Sarkar S, Korolchuk VI, Renna M, Imarisio S, Fleming A, Williams A, et al. Complex inhibitory effects of nitric oxide on autophagy. Mol Cell. 2011;43:19–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schreiber A, Peter M. Substrate recognition in selective autophagy and the ubiquitin-proteasome system. Biochimica et Biophysica Acta (BBA) - Molecular Cell Research. 2014;1843:163–81. [DOI] [PubMed] [Google Scholar]
- Shah SZA, Zhao D, Hussain T, Sabir N, Yang L. Regulation of MicroRNAs-Mediated Autophagic Flux: A New Regulatory Avenue for Neurodegenerative Diseases With Focus on Prion Diseases. Frontiers in Aging Neuroscience. 2018a;10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shah SZA, Zhao D, Hussain T, Sabir N, Yang L. Regulation of MicroRNAs-Mediated Autophagic Flux: A New Regulatory Avenue for Neurodegenerative Diseases With Focus on Prion Diseases. Frontiers in aging neuroscience. 2018b;10:139–. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shao ZQ, Dou SS, Zhu JG, Wang HQ, Wang CM, Cheng BH, et al. Apelin-13 inhibits apoptosis and excessive autophagy in cerebral ischemia/reperfusion injury. Neural Regen Res. 2021;16:1044–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shefa U, Jeong NY, Song IO, Chung H-J, Kim D, Jung J, et al. Mitophagy links oxidative stress conditions and neurodegenerative diseases. Neural Regen Res. 2019;14:749–56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shin YS, Ryall JG, Britto JM, Lau CL, Devenish RJ, Nagley P, et al. Inhibition of bioenergetics provides novel insights into recruitment of PINK1-dependent neuronal mitophagy. Journal of Neurochemistry. 2019;149:269–83. [DOI] [PubMed] [Google Scholar]
- Shintani T, Klionsky DJ. Autophagy in Health and Disease: A Double-Edged Sword. Science. 2004;306:990–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Singh A, Kukreti R, Saso L, Kukreti S. Oxidative Stress: A Key Modulator in Neurodegenerative Diseases. Molecules. 2019;24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Singh F, Prescott AR, Rosewell P, Ball G, Reith AD, Ganley IG. Pharmacological rescue of impaired mitophagy in Parkinson’s disease-related LRRK2 G2019S knock-in mice. Elife. 2021;10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Singh NA, Mandal AKA, Khan ZA. Potential neuroprotective properties of epigallocatechin-3-gallate (EGCG). Nutrition journal. 2015;15:1–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takeuchi H, Kondo Y, Fujiwara K, Kanzawa T, Aoki H, Mills GB, et al. Synergistic augmentation of rapamycin-induced autophagy in malignant glioma cells by phosphatidylinositol 3-kinase/protein kinase B inhibitors. Cancer Res. 2005;65:3336–46. [DOI] [PubMed] [Google Scholar]
- Tang FL, Erion JR, Tian Y, Liu W, Yin DM, Ye J, et al. VPS35 in Dopamine Neurons Is Required for Endosome-to-Golgi Retrieval of Lamp2a, a Receptor of Chaperone-Mediated Autophagy That Is Critical for α-Synuclein Degradation and Prevention of Pathogenesis of Parkinson’s Disease. J Neurosci. 2015a;35:10613–28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tang FL, Liu W, Hu JX, Erion JR, Ye J, Mei L, et al. VPS35 Deficiency or Mutation Causes Dopaminergic Neuronal Loss by Impairing Mitochondrial Fusion and Function. Cell Rep. 2015b;12:1631–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taylor JP, Hardy J, Fischbeck KH. Toxic Proteins in Neurodegenerative Disease. Science. 2002;296:1991–5. [DOI] [PubMed] [Google Scholar]
- Thoreen CC, Kang SA, Chang JW, Liu Q, Zhang J, Gao Y, et al. An ATP-competitive Mammalian Target of Rapamycin Inhibitor Reveals Rapamycin-resistant Functions of mTORC1*. Journal of Biological Chemistry. 2009;284:8023–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tian Y, Song W, Li D, Cai L, Zhao Y. Resveratrol As A Natural Regulator Of Autophagy For Prevention And Treatment Of Cancer. Onco Targets Ther. 2019;12:8601–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsukada M, Ohsumi Y. Isolation and characterization of autophagy-defective mutants of Saccharomyces cerevisiae. FEBS Lett. 1993;333:169–74. [DOI] [PubMed] [Google Scholar]
- Ułamek-Kozioł M, Furmaga-Jabłońska W, Januszewski S, Brzozowska J, Sciślewska M, Jabłoński M, et al. Neuronal autophagy: self-eating or self-cannibalism in Alzheimer’s disease. Neurochem Res. 2013;38:1769–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vainshtein A, Hood DA. The regulation of autophagy during exercise in skeletal muscle. Journal of Applied Physiology. 2016;120:664–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vasconcelos-Ferreira A, Carmo-Silva S, Codêsso JM, Silva P, Martinez ARM, França MC Jr, et al. The autophagy-enhancing drug carbamazepine improves neuropathology and motor impairment in mouse models of Machado–Joseph disease. Neuropathology and Applied Neurobiology. 2022;48:e12763. [DOI] [PubMed] [Google Scholar]
- Velazquez AFC, Jackson WT. So Many Roads: the Multifaceted Regulation of Autophagy Induction. Molecular and Cellular Biology. 2018;38:e00303–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vidyadhara DJ, Lee JE, Chandra SS. Role of the endolysosomal system in Parkinson’s disease. J Neurochem. 2019;150:487–506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang BJ, Her GM, Hu MK, Chen YW, Tung YT, Wu PY, et al. ErbB2 regulates autophagic flux to modulate the proteostasis of APP-CTFs in Alzheimer’s disease. Proc Natl Acad Sci U S A. 2017;114:E3129–e38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang G, Fu XL, Wang JJ, Guan R, Sun Y, Tony To SS. Inhibition of glycolytic metabolism in glioblastoma cells by Pt3glc combinated with PI3K inhibitor via SIRT3-mediated mitochondrial and PI3K/Akt-MAPK pathway. J Cell Physiol. 2019a;234:5888–903. [DOI] [PubMed] [Google Scholar]
- Wang Y, Liu N, Lu B. Mechanisms and roles of mitophagy in neurodegenerative diseases. CNS Neurosci Ther. 2019b;25:859–75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wei YM, Li X, Xu M, Abais JM, Chen Y, Riebling CR, et al. Enhancement of autophagy by simvastatin through inhibition of Rac1-mTOR signaling pathway in coronary arterial myocytes. Cell Physiol Biochem. 2013;31:925–37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xie J, Liang J, Chen N. Autophagy-associated signal pathways of functional foods for chronic diseases. Food Science and Human Wellness. 2019;8:25–33. [Google Scholar]
- Xing Y, Yang S-D, Wang M-M, Feng Y-S, Dong F, Zhang F. The beneficial roles of exercise training via autophagy in neurological diseases and possible mechanisms. Life Sciences. 2019;221:130–4. [DOI] [PubMed] [Google Scholar]
- Xiong Y, Uys JD, Tew KD, Townsend DM. S-glutathionylation: from molecular mechanisms to health outcomes. Antioxid Redox Signal. 2011;15:233–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu TT, Li H, Dai Z, Lau GK, Li BY, Zhu WL, et al. Spermidine and spermine delay brain aging by inducing autophagy in SAMP8 mice. Aging (Albany NY). 2020;12:6401–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xue X, Wang L-R, Sato Y, Jiang Y, Berg M, Yang D-S, et al. Single-Walled Carbon Nanotubes Alleviate Autophagic/Lysosomal Defects in Primary Glia from a Mouse Model of Alzheimer’s Disease. Nano Letters. 2014;14:5110–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yazbeck VY, Georgakis GV, Li Y, Younes A. The mTOR inhibitor CCI-779 (temsirolimus) downregulates p21 and induces cell cycle arrest and autophagy in mantle cell lymphoma (MCL). Journal of Clinical Oncology. 2006;24:7573–. [Google Scholar]
- Yim WW-Y, Mizushima N. Lysosome biology in autophagy. Cell discovery. 2020;6:1–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yun HR, Jo YH, Kim J, Shin Y, Kim SS, Choi TG. Roles of Autophagy in Oxidative Stress. Int J Mol Sci. 2020;21:3289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zaffagnini G, Martens S. Mechanisms of Selective Autophagy. J Mol Biol. 2016;428:1714–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang H, Gong Y, Wang Z, Jiang L, Chen R, Fan X, et al. Apelin inhibits the proliferation and migration of rat PASMCs via the activation of PI3K/Akt/mTOR signal and the inhibition of autophagy under hypoxia. J Cell Mol Med. 2014a;18:542–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang J, Ney PA. Role of BNIP3 and NIX in cell death, autophagy, and mitophagy. Cell Death & Differentiation. 2009;16:939–46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang L, Fang Y, Cheng X, Lian Y, Xu H, Zeng Z, et al. TRPML1 Participates in the Progression of Alzheimer’s Disease by Regulating the PPARγ/AMPK/Mtor Signalling Pathway. Cell Physiol Biochem. 2017a;43:2446–56. [DOI] [PubMed] [Google Scholar]
- Zhang L, Niu W, He Z, Zhang Q, Wu Y, Jiang C, et al. Autophagy suppression by exercise pretreatment and p38 inhibition is neuroprotective in cerebral ischemia. Brain Research. 2014b;1587:127–32. [DOI] [PubMed] [Google Scholar]
- Zhang L, Wang L, Wang R, Gao Y, Che H, Pan Y, et al. Evaluating the Effectiveness of GTM-1, Rapamycin, and Carbamazepine on Autophagy and Alzheimer Disease. Med Sci Monit. 2017b;23:801–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Y, Yao Y, Qiu X, Wang G, Hu Z, Chen S, et al. Listeria hijacks host mitophagy through a novel mitophagy receptor to evade killing. Nature Immunology. 2019;20:433–46. [DOI] [PubMed] [Google Scholar]
- Zheng Y, Kou J, Wang P, Ye T, Wang Z, Gao Z, et al. Berberine-induced TFEB deacetylation by SIRT1 promotes autophagy in peritoneal macrophages. Aging (Albany NY). 2021;13:7096–119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou Y, Ulland TK, Colonna M. TREM2-Dependent Effects on Microglia in Alzheimer’s Disease. Front Aging Neurosci. 2018;10:202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu H, Wu H, Liu X, Li B, Chen Y, Ren X, et al. Regulation of autophagy by a beclin 1-targeted microRNA, miR-30a, in cancer cells. Autophagy. 2009;5:816–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zielke S, Meyer N, Mari M, Abou-El-Ardat K, Reggiori F, van Wijk SJL, et al. Loperamide, pimozide, and STF-62247 trigger autophagy-dependent cell death in glioblastoma cells. Cell Death Dis. 2018;9:994. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
All data analyzed during this study are included in this article.