

Abstract
Iodide-bound ruthenium-JOSIPHOS complexes catalyze the redox-neutral C-C coupling of primary alcohols with methylallene (1,2-butadiene) or 1,3-butadiene to form products of anti-crotylation with good to excellent levels of diastereo- and enantioselectivity. Distinct from other methods, direct crotylation of primary alcohols in the presence of unprotected secondary alcohols is possible, enabling generation of spirastrellolide B (C9-C15) and leucascandrolide A (C9-C15) substructures in significantly fewer steps than previously possible.
Keywords: Alcohols, Dienes, Crotylation, Enantioselectivity, Ruthenium Catalysis
Graphical Abstract
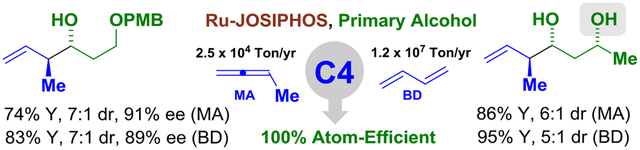
Ruthenium-JOSIPHOS-catalysts affect anti-crotylation of primary alcohols mediated by methylallene or 1,3-butadiene to provide polyketide propionate motifs. These C-C bond formations are byproduct-free and can be conducted in the presence of unprotected secondary alcohols.
Introduction
Polyketides and their derivatives are used more frequently in human medicine than any other class of natural products, and are estimated to comprise roughly 20% of top-selling small-molecule drugs.[1] Despite their importance, nearly all polyketide drugs are prepared via fermentation or semi-synthesis, as current methods for their de novo synthesis have not availed sufficiently concise or scalable manufacturing routes. As illustrated in the structure of erythromycin A, polyketides often comprise so-called “propionate subunits,” which, classically, are assembled via organometal-mediated carbonyl additions, such as the aldol reaction, or as shown, the “Brown crotylation” (Figure 1).[2,3] The use of π-unsaturated petrochemical feedstocks as pronucleophiles in metal-catalyzed carbonyl addition offers an alternative to the use of discrete organometallic reagents, which are often hazardous and functional group intolerant.[4] Inspired by the extensive use of hydrogenation in reactions used to prepare small-molecule clinical candidates,[5] we have developed a broad family of metal-catalyzed carbonyl reductive couplings mediated by hydrogen or 2-propanol, and related hydrogen auto-transfer processes in which alcohol reactants function dually as carbonyl proelectrophile and reductant.[6,7] In this context, the use of allene and diene feedstocks as allylmetal pronucleophiles was explored (Figure 1),[8,9,10] resulting in the first use of commercially available allene (propadiene), methylallene (1,2-butadiene) and dimethylallene (3-methyl-1,2-butadiene) as allylmetal pronucleophiles in metal-catalyzed carbonyl addition (2007).[10] Despite progress toward the development of enantioselective variants,[11] catalytic asymmetric crotylations using methylallene have not been reported,[8] even though it could potentially serve as an atom-efficient reagent for carbonyl crotylation. Additionally, first-generation conditions for butadiene-mediated carbonyl crotylation[12] suffer from significant limitations: (a) 2-trialkylsilyl groups[12b] or non-commercial chiral counterions[12d] are required to enforce syn-diastereoselectivity[13] and, most importantly, (b) asymmetric reactions that display anti-diastereoselectivity are restricted to benzylic alcohols.[12c] These processes and related enantioselective ruthenium-catalyzed alcohol C-C couplings relevant to polyketide construction[14] are summarized in Figure 2.
Figure 1.
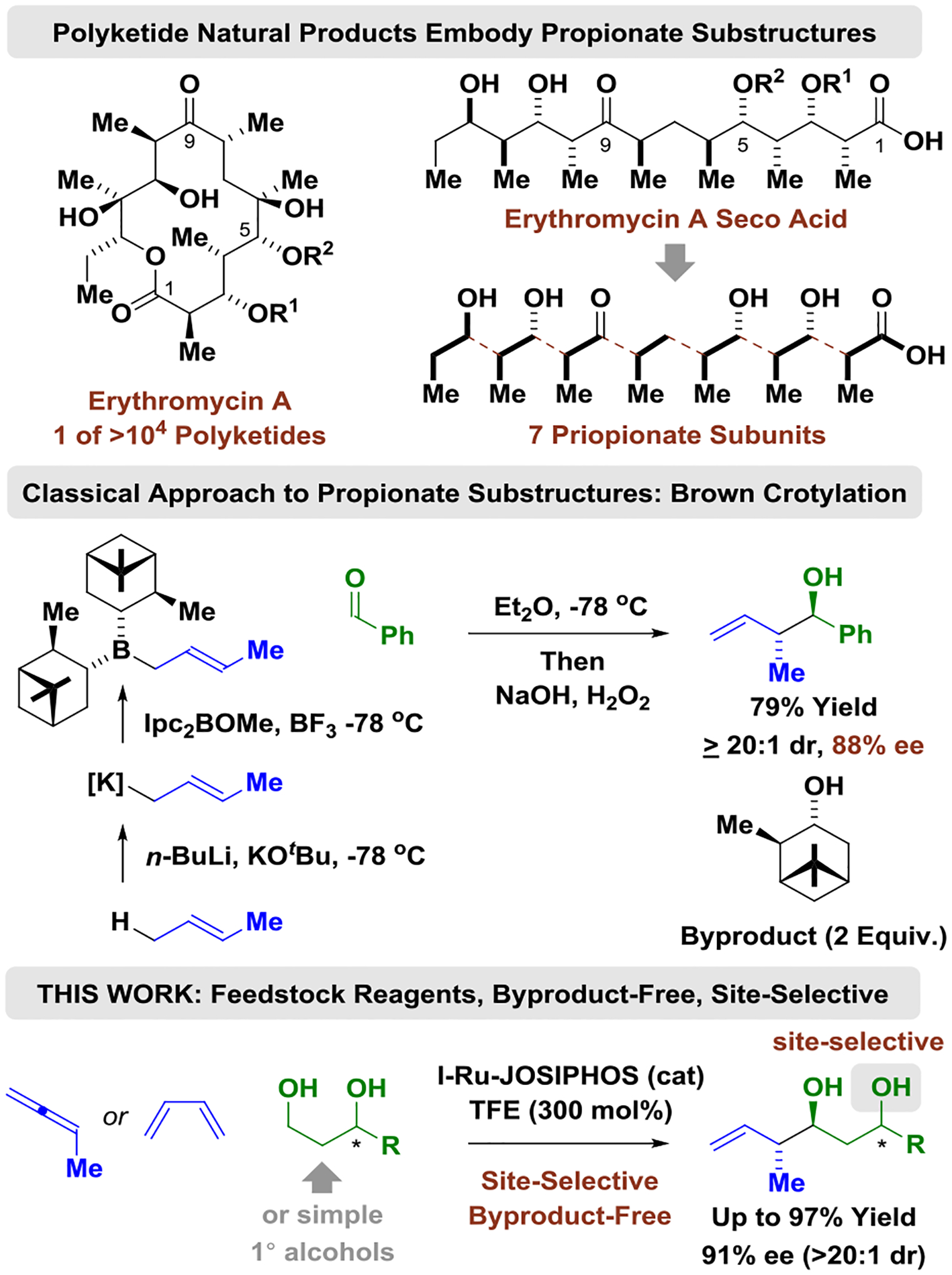
Asymmetric carbonyl crotylation: A gateway to polyketide structural motifs.
Figure 2.
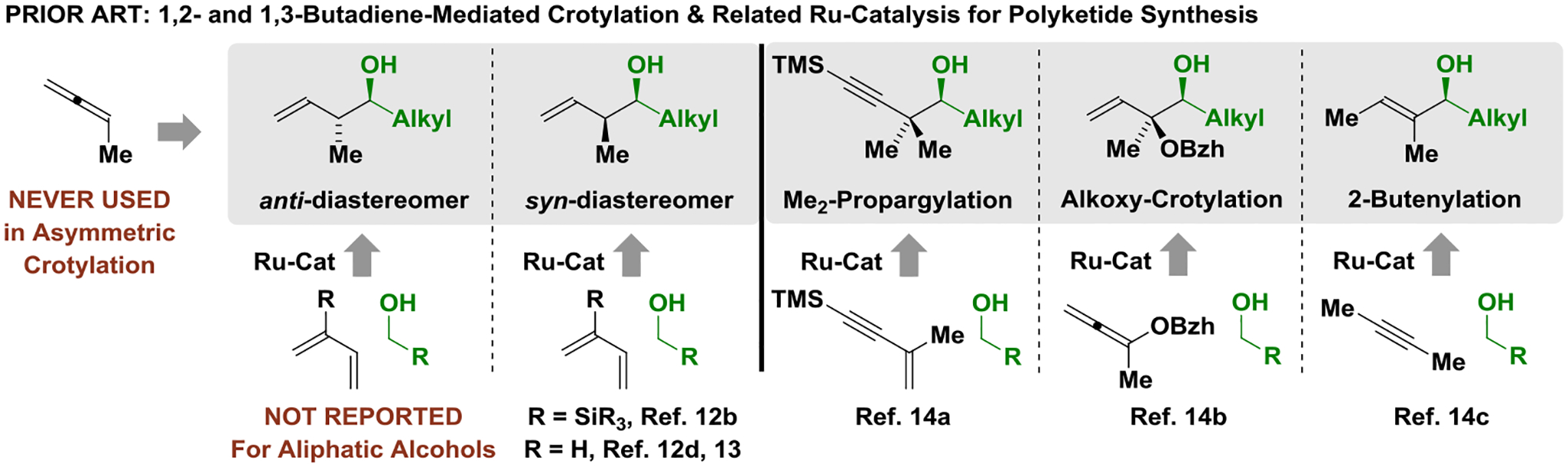
Enantioselective ruthenium-catalyzed C-C coupling of alcohols.
Here, we report that iodide-bound ruthenium-JOSIPHOS catalysts recently developed in our laboratory[15,16] promote methylallene- and butadiene-mediated carbonyl crotylations of primary alcohol reactants via hydrogen auto-transfer with good to excellent levels of anti-diastereo- and enantioselectivity. Furthermore, distinct from conventional chiral crotylmetal reagents,[2,3] the crotylation of primary alcohols can be achieved in the presence of unprotected secondary alcohols via site-selective dehydrogenation, enabling the streamlined synthesis of type-I polyketide substructures in the absence of secondary hydroxyl protecting groups or generation of stoichiometric byproducts, as demonstrated by the construction of C9-C15 of spirastrellolide B and C9-C15 of leucascandrolide A.[17,18,19]
Results and Discussion
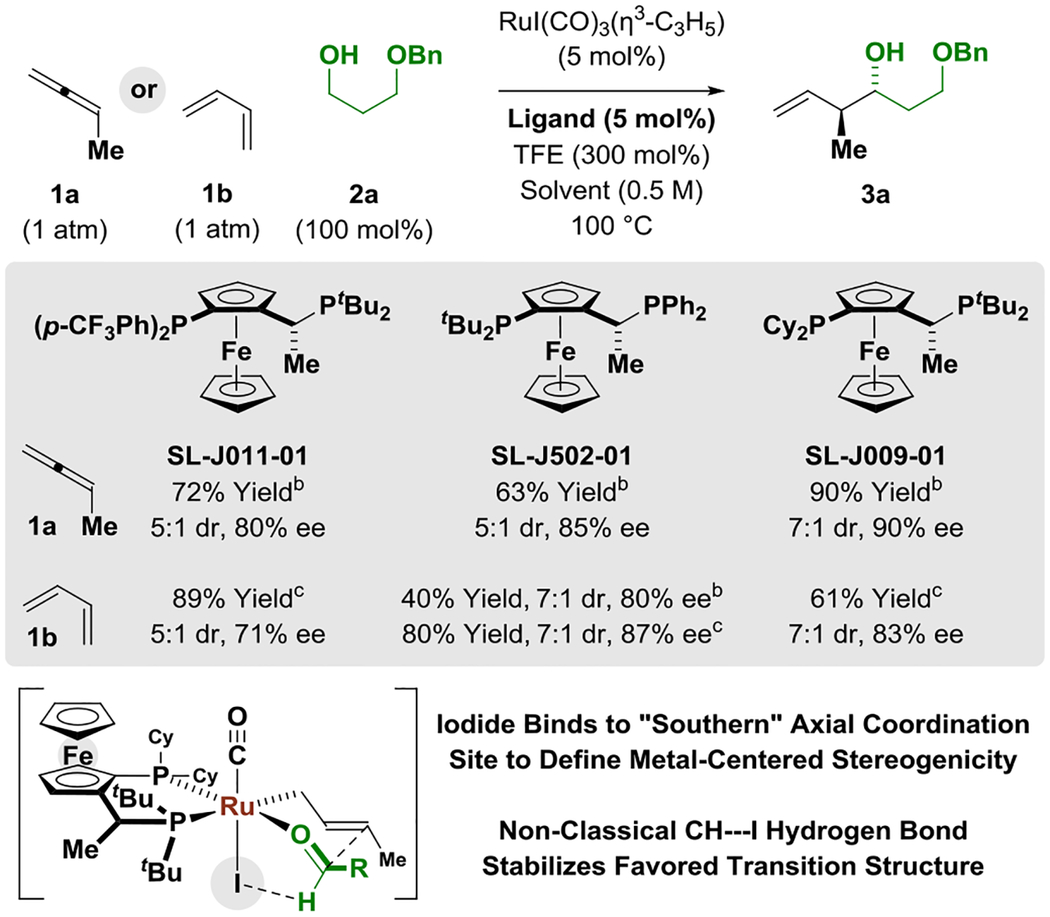
In connection with studies on the ruthenium-catalyzed C-C coupling of primary alcohols with 1-aryl-1-propynes to form products of carbonyl anti-(α-aryl)allylation, the ruthenium complexes, RuX(CO)(η3-C3H5)(JOSIPHOS), where X = Cl, Br, I, were characterized by single crystal X-ray diffraction, revealing a halide-dependent diastereomeric preference that defines stereogenicity at the ruthenium center in the solid state.[15,20] As supported by computational studies,[15] iodide counterions were unique in their ability to (a) enforce control of stereochemistry at ruthenium, which enables high levels of enantioselectivity and, (b) engage the aldehyde in formyl CH···I hydrogen-bonding, which stabilizes the favored transition state for carbonyl addition.[21] From these studies, an improved ruthenium catalyst system was developed in which the active metal complex is assembled in situ from the iodide-containing precursor RuI(CO)3(η3-C3H5) and a commercially available JOSIPHOS ligand. Eager to assess the capabilities of this improved iodide-bound ruthenium-JOSIPHOS catalyst, the redox-neutral C-C coupling of methylallene 1a with propanediol benzyl ether 2a was attempted (Table 1). To this end, a pressure tube charged with a THF solution (0.5 M) of 2a (100 mol%), trifluoroethanol (300 mol%), RuI(CO)3(η3-C3H5) (5 mol%) and the JOSIPHOS ligand SL-J011–01 (5 mol%) was backfilled with methylallene 1a and the reaction vessel was sealed and placed in a 100 °C temperature bath. To our delight, the desired product of carbonyl crotylation 3a was formed in 72% isolated yield with a 5:1 preference for the anti-diastereomer and 80% ee. Under otherwise identical conditions using the JOSIPHOS ligand SL-J502–01, the level of enantiomeric enrichment increased to 85% ee. Finally, using the JOSIPHOS ligand SL-J009–01, adduct 3a was generated in 90% isolated yield with a 7:1 preference for the anti-diastereomer and 90% ee. Deviation from these conditions did not avail any further improvement. Upon omission of trifluoroethanol (TFE), conversion to adduct 3a was not observed. These data and prior computational studies suggest TFE catalyzes exchange of the homoallylic alkoxide with the primary alcohol reactant to release product (vide infra).[15] In an analogous series of experiments, butadiene 1b and propanediol benzyl ether 2a were exposed to identical conditions. Here, optimal results were obtained using the JOSIPHOS ligand SL-J502–01, revealing methylallene 1a and butadiene 1b display distinct reactivities, although identical crotylruthenium intermediates are anticipated (vide infra).
Table 1.
Ruthenium-JOSIPHOS-catalyzed C-C coupling of methylallene 1a or butadiene 1b with alcohol 2a to form adduct 3a and stereochemical model for carbonyl addition.a
|
Yields of material isolated by silica gel chromatography. Enantioselectivities were determined by HPLC analysis. TFE = trifluoroethanol.
THF,
Isoctane.
See Supporting Information for further details.
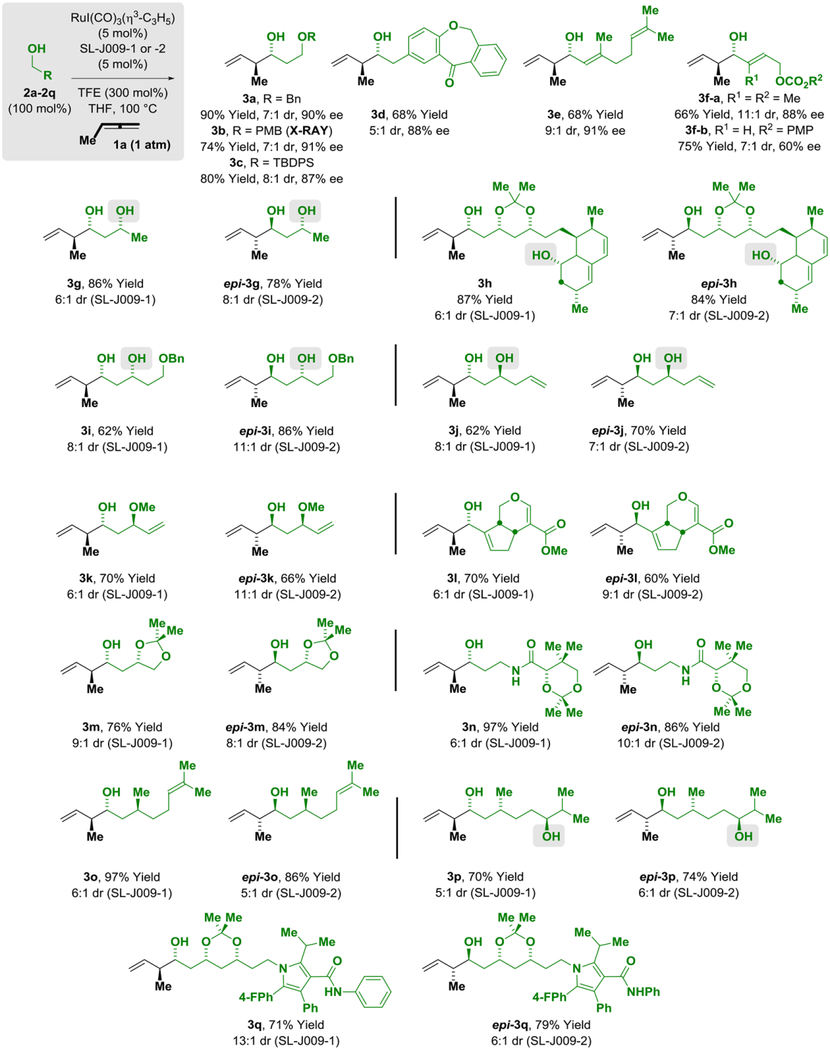
Using the iodide bound ruthenium catalyst assembled from RuI(CO)3(η3-C3H5) and the JOSIPHOS ligand SL-J009–01, a range of primary alcohols 2a-2q were surveyed as participants for methylallene-mediated crotylation via hydrogen auto-transfer (Table 2). Good levels of anti-diastereo- and enantioselectivity were observed in reactions of achiral primary alcohols 2a-2f. Additionally, methylallene-mediated crotylation of diverse chiral primary alcohols 2g-2q were explored using the enantiomeric ruthenium catalysts modified by the JOSIPHOS ligands SL-J009–01 and SL-J009–02. Adducts 3g-3q were formed with good levels of catalyst-directed diastereoselectivity in both the matched and mismatched cases. Remarkably, these data encompass reactions of chiral β-stereogenic 1°,2°-1,3-diols 2g-2j, 2p, which were found to undergo C-C coupling to form 3g-3j, 3p without competing dehydrogenation of the secondary hydroxyl groups. One limitation of the present catalytic system resides in couplings of benzylic alcohols, which occur in good yield, but with modest levels of enantiocontrol (ca. 70% ee). Additionally, 1,2-diols are inefficient reaction partners as the vicinal oxygen atom destabilizes the transition state for dehydrogenation due to its negative inductive effect. Finally, as illustrated by the conversion of dehydro-2c, dehydro-2e, and dehydro-2m to adducts 3c, 3e, 3m and epi-3m, respectively, methylallene-mediated crotylation can be conducted from the aldehyde oxidation level using 2-propanol (200 mol%) as reductant under otherwise identical conditions with roughly equivalent levels of diastereo- and enantioselectivity (Scheme 1, eq. 1–4). The stereochemical assignments of adducts 3a-3q were made in analogy to that determined for the dinitrobenzoate of 3b, which was established via single crystal X-ray diffraction.
Table 2.
Stereo- and site-selective methylallene-mediated carbonyl crotylation via ruthenium-JOSIPHOS-catalyzed hydrogen auto-transfer of primary alcohols 2a-2q hydrogen-auto transfer.a
|
Yields of material isolated by silica gel chromatography. Enantioselectivities determined by HPLC analysis. Diastereoselectivities determined by 1H NMR analysis of crude reaction mixtures and refer to the major isomer vs all other isomers. See Supporting Information for further details.
Scheme 1.
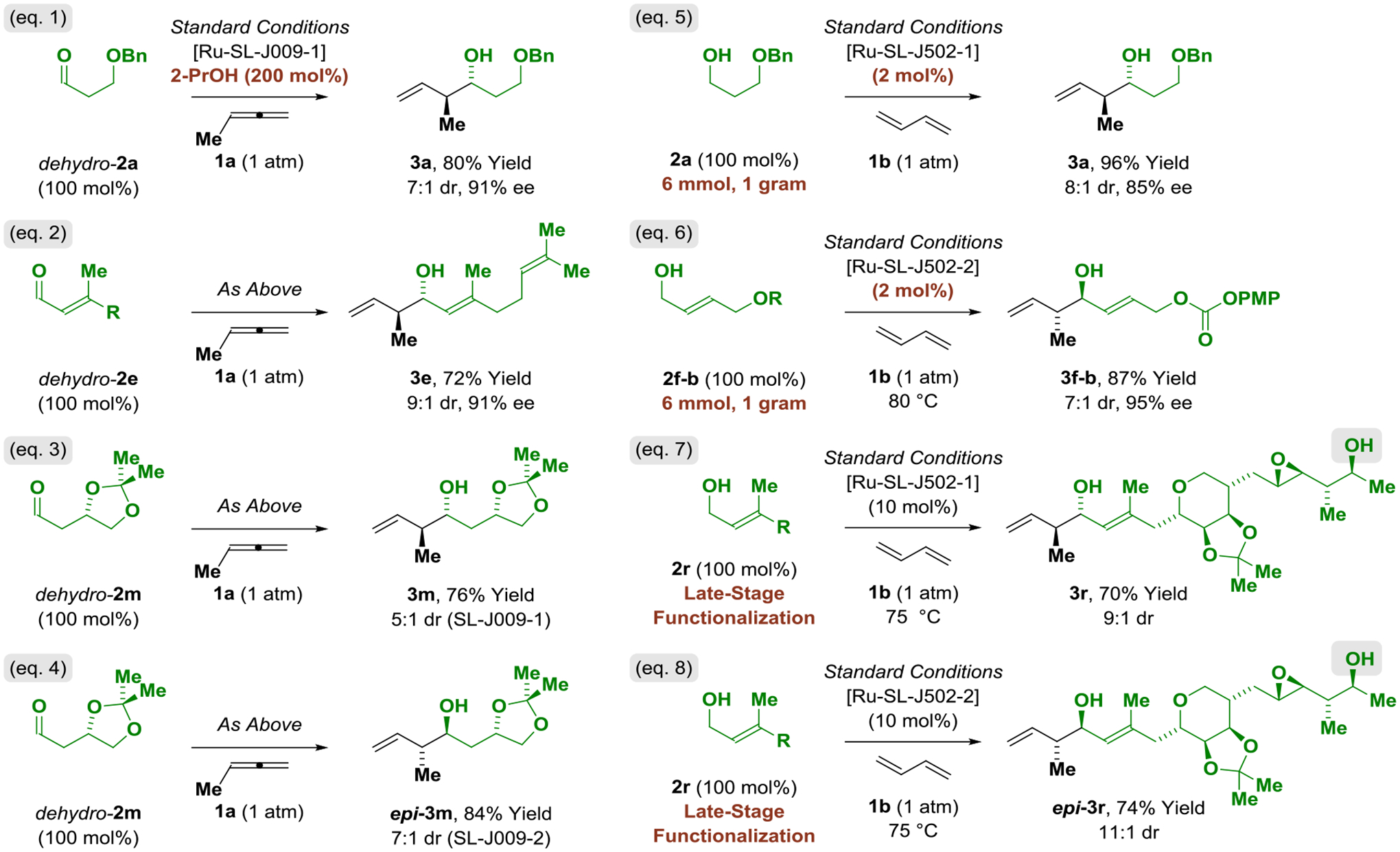
Stereo- and site-selective methylallene-mediated carbonyl crotylation from the aldehyde oxidation level, gram-scale butadiene-mediated crotylations and late-stage functionalizations.a
aYields of material isolated by silica gel chromatography. Enantioselectivities determined by HPLC analysis. Diastereoselectivities determined by 1H NMR analysis of crude reaction mixtures and refer to the major isomer vs all other isomers.
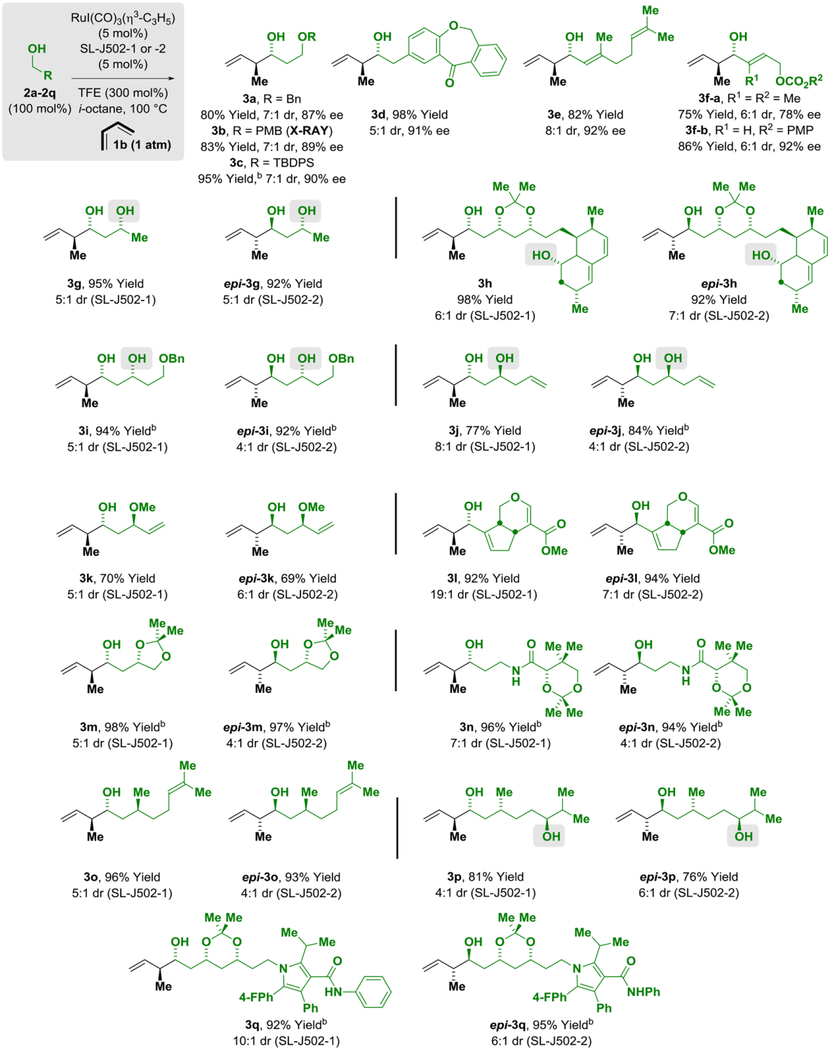
To assess the scope of related butadiene-mediated crotylations, a duplicate set of adducts 3a-3q were prepared using the ruthenium catalyst modified by JOSIPHOS ligand SL-J502–01 under otherwise identical conditions, notwithstanding use of iso-octane as solvent (Table 3). Due to issues of solubility, the solvent MTBE was substituted iso-octane in certain cases. In nearly all cases, roughly equivalent diastereoselectivities and enantioselectivities were observed. Thus, chiral primary alcohols 2g-2q were converted to adducts 3g-3q using the ruthenium catalysts modified by the enantiomeric ligands SL-J502–01 and SL-J502–02 with good levels of catalyst-directed diastereoselectivity, and chiral β-stereogenic 1°,2°-1,3-diols 2g-2j, 2p were converted to adducts 3g-3j, 3p in the absence of secondary hydroxyl protecting groups. However, as illustrated by the divergent selectivities observed in reactions of 3f-a and 3f-b, certain alcohol proelectrophiles display distinctly different selectivities in reactions of methylallene 1a vs butadiene 1b. Additionally, perhaps due to its greater stability, reactions of butadiene 1b are more efficient, often not requiring chromatographic purification. Indeed, diastereo- and enantioselective butadiene-mediated crotylation can be conducted in excellent yield on gram-scale at lower catalyst loadings (2 mol%), underscoring the practicality of this method (Scheme 1, eq. 5 and 6). As illustrated in the asymmetric crotylation of alcohols 2h and 2q, which are derived from Lovastatin (Mevacor®) and atorvastatin (Lipitor®), respectively, structurally complex reactants that incorporate diverse functional groups are tolerated, suggesting applicability of this method to even more challenging “late-stage functionalizations.”[22] To probe the limits of functional group compatibility, the enantiomeric ruthenium catalysts modified by SL-J502–01 and SL-J502–02 were reacted with an alcohol derived from the type I polyketide antibiotic mupirocin, specifically, pseudomonic acid A (Bactroban®). Despite the presence of sensitive epoxide and secondary hydroxyl functional groups, the diastereomeric adducts were obtained in good isolated yield with high levels of catalyst-directed diastereocontrol (Scheme 1, eq. 7 and 8). Finally, as demonstrated by the reaction of the p-bromophenyl-substituted allene 1c, the conditions optimized for catalytic enantioselective allene-mediated crotylation are transferable to other allene pronucleophiles (eq. 9).
Table 3.
Stereo- and site-selective butadiene-mediated carbonyl crotylation via ruthenium-JOSIPHOS-catalyzed hydrogen auto-transfer of primary alcohols 2a-2q hydrogen-auto transfer.a
|
Yields of material isolated by silica gel chromatography. Enantioselectivities determined by HPLC analysis. Diastereoselectivities determined by 1H NMR analysis of crude reaction mixtures and refer to the major isomer vs all other isomers.
MTBE (0.5 M). See Supporting Information for further details.
 |
A unique feature of the present method resides in the ability to affect the crotylation of primary alcohols in the presence of unprotected secondary hydroxyl groups. To demonstrate how this capability streamlines type I polyketide construction, two different C9-C15 spirastrellolide B substructures were prepared via stereo- and site-selective crotylation of the chiral β-stereogenic 1°,2°-1,3-diol ent-2i and a comparison of step-count (Longest Linear Sequence, LLS) to previously reported C9-C15 spirastrellolide B substructures was made (Scheme 2, Top). The C9-C15 spirastrellolide B substructure ent-epi-3i, which was previously made in 10 steps (LLS) by Hanson,[23a] is accessible in 3 steps (LLS) via butadiene-mediated crotylation. Ozonolysis-reduction of ent-epi-3i delivers compound 4a in 4 steps (LLS), whereas the related compound 4b was prepared by Chandrasekhar in 8 steps (LLS).[23b] The ability to directly deploy butadiene, an abundant feedstock chemical, in catalytic enantioselective crotylation offers another advantage. Many methods for crotylation rely on the use of chiral crotylmetal reagents, which are prepared through multi-step sequences, which impact the number of total steps (TS). To illustrate, the C9- C15 leucascandrolide A substructure prepared by Cossy requires 6 steps (LLS), but 8 total steps (TS),[24] as it uses the chiral crotyltitanium reagent developed by Hafner and Duthaler, which requires a 3 step synthesis.[25] The identical compound is prepared via butadiene-mediated crotylation in 4 steps (LLS = TS) (Scheme 2, Bottom).
Scheme 2.
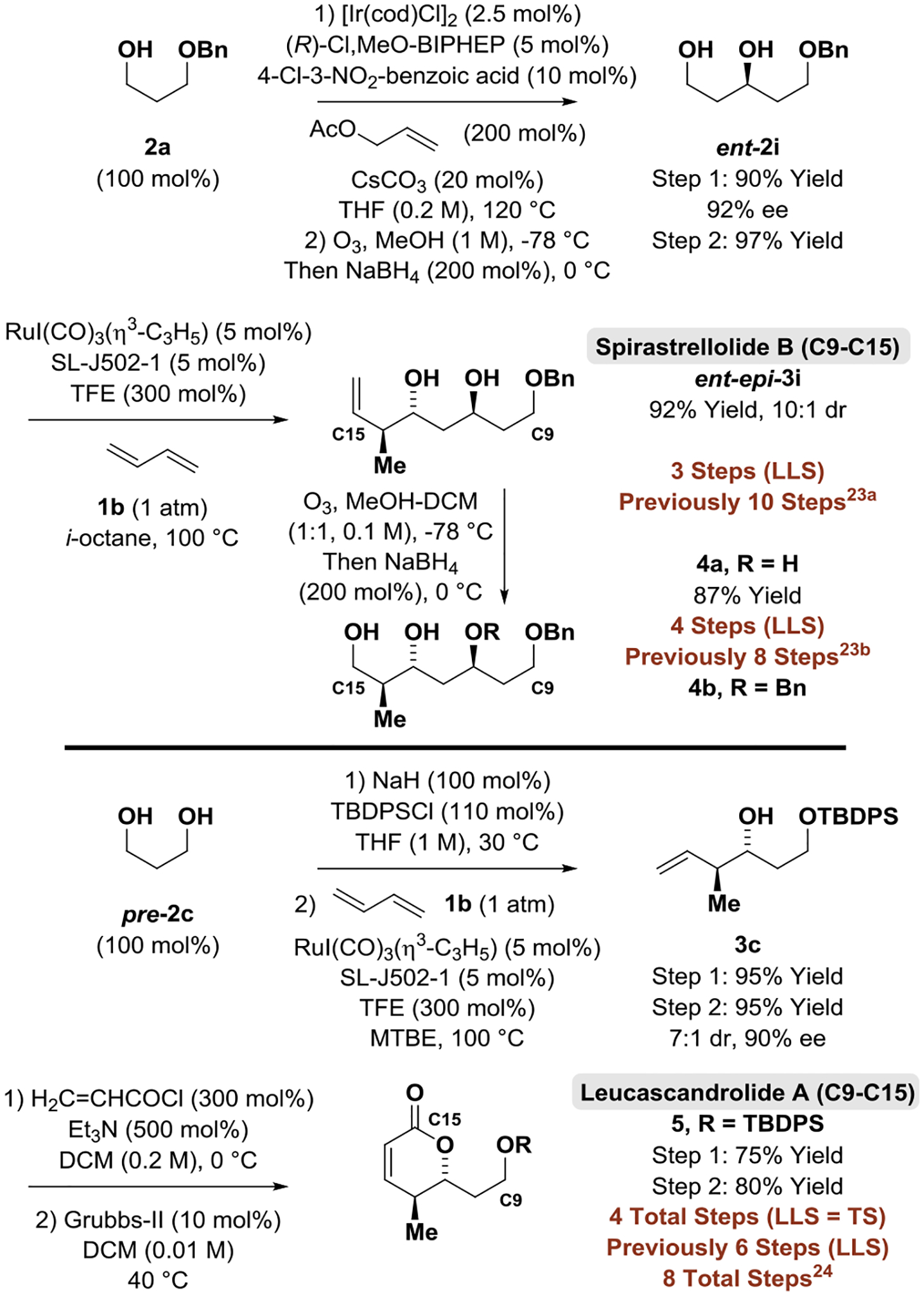
Generation of spirastrellolide B (C9-C15) and leucascandrolide A (C9-C15) substructures.
aYields of material isolated by silica gel chromatography. Enantioselectivities determined by HPLC analysis. Diastereoselectivities determined by 1H NMR analysis of crude reaction mixtures and refer to the major isomer vs all other isomers. LLS = Longest linear sequence.
A general catalytic mechanism for enantioselective methylallene-mediated carbonyl crotylation via ruthenium-JOSIPHOS-catalyzed hydrogen auto-transfer has been proposed (Scheme 3). Entry into the catalytic cycle occurs via protonolysis of the π-allylruthenium precatalyst by TFE followed by substitution with a reactant alcohol to form the primary ruthenium alkoxide I.[15] β-Hydride elimination of ruthenium alkoxide I releases aldehyde and forms the ruthenium hydride II. Hydroruthenation of methylallene provides an equilibrating mixture of π- and σ-crotylruthenium species III.[26] Carbonyl addition from the (E)-σ-crotylruthenium isomer by way of a closed Zimmerman-Traxler type transition structure[27] provides the homoallylic ruthenium alkoxide IV. The stereospecific nature of the carbonyl addition event is inferred by the anticipated influence of steric effects on the geometry of σ-allyl intermediates and, therefrom, diastereoselectivity in reactions of substituted allenes28 and dienes.[14b] TFE-assisted exchange of the homoallylic ruthenium alkoxide IV with reactant alcohol releases the product of crotylation and regenerates the primary ruthenium alkoxide I to close the catalytic cycle.[15] This mechanism is consistent with the outcome of the indicated deuterium labelling experiment, in which deuterio-2a is exposed to methylallene 1a (800 mol%) under otherwise standard conditions. The relatively low level of deuterium transfer to the internal vinylic position (11% 2H) is due to exchange of the transient ruthenium deuteride akin to II with the hydroxyl hydrogen of TFE and deuterio-2a.[29] An analogous mechanism is postulated for corresponding reactions of butadiene 1b. Slight differences in reactivity and selectivity displayed by methylallene 1a and butadiene 1b are attributed to differences in kinetic stereoselectivity in the hydrometalation event to generate the initially formed chiral-at-metal crotylruthenium intermediate. Butadiene cannot be detected in reactions of methylallene, indicating that allene-to-diene isomerization does not occur in methylallene-mediated crotylation. Attempted reactions of 1,4-pentadiene under the present conditions provides a 25% yield of C-C coupling product with low levels of stereocontrol (not shown).
Scheme 3.

Proposed mechanism for enantioselective methylallene-mediated carbonyl crotylation via ruthenium-JOSIPHOS-catalyzed hydrogen auto-transfer of primary alcohols and deuterium labelling experiment.a
aThe pattern of deuterium incorporation in deuterio-3a was assigned by 1H NMR, 2H NMR and HRMS. See Supporting Information for further details.
Conclusion
The development of practical methods for the de novo construction of polyketide natural products remains an important challenge in chemical synthesis. Here, using iodide-bound ruthenium-JOSIPHOS complexes recently developed in our laboratory, we report the reaction of primary alcohols with methylallene (1,2-butadiene) or 1,3-butadiene to form products of carbonyl crotylation with good to excellent levels of anti-diastereo- and enantioselectivity, providing byproduct-free entry to propionate substructures that are the ubiquitous among polyketides. A remarkable feature of these processes resides in the kinetic preference for primary alcohol dehydrogenation, which enables asymmetric crotylation of primary alcohols in the presence of unprotected secondary alcohols. As illustrated in concise syntheses of spirastrellolide B (C9-C15) and leucascandrolide A (C9-C15) substructures, this capability streamlines type I polyketide construction. Future studies will focus on related hydrogen auto-transfer process for byproduct-free enantioselective carbonyl addition that exploit feedstock pronucleophiles.
Supplementary Material
Acknowledgments
The Welch Foundation (F-0038) and the NIH (RO1-GM093905) are acknowledged for partial support of this research.
References
- [1].a) Rohr J, Angew. Chem. Int. Ed 2000, 39, 2847–2849; [DOI] [PubMed] [Google Scholar]; Angew. Chem 2000, 112, 2967–2969; [Google Scholar]; b) Weissman KJ, Leadlay F, Nat. Rev. Microbiol 2005, 3, 925–936; [DOI] [PubMed] [Google Scholar]; c) Rimando AM, Baerson SR, eds., Polyketides: Biosynthesis, Biological Activity, and Genetic Engineering (ACS Symp. Ser.,Vol. 955), American Chemical Society, Washington DC, 2007. [Google Scholar]; d) Li JW-H, Vederas JC, Science 2009, 325, 161–165. [DOI] [PubMed] [Google Scholar]
- [2].a) Brown HC, Bhat KS, J. Am. Chem. Soc 1986, 108, 293–294; [DOI] [PubMed] [Google Scholar]; b) Brown HC, Bhat KS, J. Am. Chem. Soc 1986, 108, 5919–5923. [DOI] [PubMed] [Google Scholar]
- [3].a) For selected reviews on catalytic enantioselective carbonyl allylation and crotylation, see: Denmark SE, Fu J, Chem. Rev 2003, 103, 2763–2794; [DOI] [PubMed] [Google Scholar]; b) Hall DG, Synlett 2007, 1644–1655; [Google Scholar]; c) Yus M, González-Gómez JC, Foubelo F, Chem. Rev 2011, 111, 7774–7854; [DOI] [PubMed] [Google Scholar]; d) Huo H-X, Duvall JR, Huang M-Y, Hong R, Org. Chem. Front 2014, 1, 303–320; [Google Scholar]; e) Spielmann K, Niel G, de Figueiredo RM, Campagne J-M, Chem. Soc. Rev 2018, 47, 1159–1173. [DOI] [PubMed] [Google Scholar]
- [4].For a recent review, see: Doerksen RS, Meyer CC, Krische MJ, Angew. Chem. Int. Ed 2019, 58, 14055–14064; [DOI] [PMC free article] [PubMed] [Google Scholar]; Angew. Chem 2019, 131, 14193–14202. [Google Scholar]
- [5].Carey JS, Laffan D, Thomson C, Williams MT, Org. Biomol. Chem 2006, 4, 2337–2347. [DOI] [PubMed] [Google Scholar]
- [6].For a recent review, see: Santana CG, Krische MJ, ACS Catal 2021, 11, 5572–5585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].a) The hydrogen auto-transfer processes we report result in carbonyl addition and, hence, are distinct from those that result in alcohol substitution (so-called borrowing hydrogen reactions). For selected reviews on alcohol substitution via hydrogen auto-transfer, see: Hamid MHSA, Slatford PA, Williams JMJ, Adv. Synth. Catal 2007, 349, 1555–1575; [Google Scholar]; b) Guillena G, Ramón DJ, Yus M, Angew. Chem. Int. Ed 2007, 46, 2358–2364; [DOI] [PubMed] [Google Scholar]; c) Dobereiner GE, Crabtree RH, Chem. Rev 2010, 110, 681–703; [DOI] [PubMed] [Google Scholar]; d) Bähn S, Imm S, Neubert L, Zhang M, Neumann H, Beller M, ChemCatChem 2011, 3, 1853–1864; [Google Scholar]; e) Yang Q, Wang Q, Yu Z, Chem. Soc. Rev 2015, 44, 2305–2329. [DOI] [PubMed] [Google Scholar]; f) Aitchison H, Wingad RL, Wass DF, ACS Catal 2016, 6, 7125–7132; [Google Scholar]; g) Quintard A, Rodriguez J, Chem. Commun 2016, 52, 10456–10473; [DOI] [PubMed] [Google Scholar]; h) Reed-Berendt BG, Polidano K, Morrill LC, Org. Biomol. Chem 2019, 17, 1595–1607; [DOI] [PubMed] [Google Scholar]; i) Kwok T, Hoff O, Armstrong RJ, Donohoe TJ, Chem. Eur. J 2020, 26, 12912–12926. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].a) For selected reviews on the use of allenes and dienes in enantioselective metal-catalyzed reductive C=X (X = O, NR) allylation, see: Holmes M, Schwartz LA, Krische MJ, Chem. Rev 2018, 118, 6026–6052; [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Xiang M, Pfaffinger DE, Krische MJ, Chem. Eur. J 2021, 27, 13107–13116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].a) For selected reviews on the use of allenes and dienes in enantioselective metal-catalyzed borylative C=X (X = O, NR) allylation, see: Semba K, Fujihara T, Terao J, Tsuji Y, Tetrahedron 2015, 71, 2183–2197; [Google Scholar]; b) Pulis AP, Yeung K, Procter DJ, Chem. Sci 2017, 8, 5240–5247; [DOI] [PMC free article] [PubMed] [Google Scholar]; c) Fuji-hara T, Tsuji Y, Synthesis 2018, 50, 1737–1749; [Google Scholar]; d) Talbot FJT, Dherbassy Q, Manna S, Shi C, Zhang S, Howell GH, Perry GJP, Procter DJ, Angew. Chem. Int. Ed 2020, 59, 20278–20289; [DOI] [PMC free article] [PubMed] [Google Scholar]; Angew. Chem 2020, 132, 202454–20465; [Google Scholar]; e) Whyte A, Torelli A, Mirabi B, Zhang A, Lautens M, ACS Catal 2020, 10, 11578–11622; [Google Scholar]; f) Das KK, Manna S, Panda S, Chem. Commun 2021, 57, 441–459. [DOI] [PubMed] [Google Scholar]
- [10].a) Skucas E, Bower JF, Krische MJ, J. Am. Chem. Soc 2007, 129, 12678–12679; [DOI] [PubMed] [Google Scholar]; b) Bower JF, Skucas E, Patman RL, Krische MJ, J. Am. Chem. Soc 2007, 129, 15134–15135. [DOI] [PubMed] [Google Scholar]
- [11].a) Han SB, Kim IS, Han H, Krische MJ, J. Am. Chem. Soc 2009, 131, 6916–6917; [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Liu RY, Zhou Y, Yang Y, Buchwald SL, J. Am. Chem. Soc 2019, 141, 2251–2256; [DOI] [PMC free article] [PubMed] [Google Scholar]; c) Kim SW, Meyer CC, Mai BK, Liu P, Krische MJ, ACS Catalysis 2019, 9, 9158–9163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].a) Shibahara F, Bower JF, Krische MJ, J. Am. Chem. Soc 2008, 130, 6338–6339; [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Zbieg JR, Moran J, Krische MJ, J. Am. Chem. Soc 2011, 133, 10582–10586; [DOI] [PMC free article] [PubMed] [Google Scholar]; c) Zbieg JR, Yamaguchi E, McInturff EL, Krische MJ, Science 2012, 336, 324–327; [DOI] [PMC free article] [PubMed] [Google Scholar]; d) McInturff EL, Yamaguchi E, Krische MJ, J. Am. Chem. Soc 2012, 134, 20628–20631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].a) For selected applications of ruthenium-catalyzed syn-diastereoselective butadiene-mediated crotylation in polyketide total synthesis, see: Gao X, Woo SK, Krische MJ, J. Am. Chem. Soc 2013, 135, 4223–4226; [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Yoo M, Krische MJ, Angew. Chem. Int. Ed 2021, 60, 13923–13928; [DOI] [PMC free article] [PubMed] [Google Scholar]; Angew. Chem 2021, 133, 14042–14047. [Google Scholar]
- [14].a) Nguyen KD, Herkommer D, Krische MJ, J. Am. Chem. Soc 2016, 138, 5238–5241; [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Xiang M, Pfaffinger D, Ortiz E, Brito GA, Krische MJ, J. Am. Chem. Soc 2021, 143, 8849–8854. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Ortiz E, Shezaf JZ, Chang Y-H, Gonçalves TP, Huang K-W, Krische MJ, J. Am. Chem. Soc 2021, 143, 16709–16717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Blaser H-U, Brieden W, Pugin B, Spindler F, Studer M, Togni A, Top. Catal 2002, 19, 3–16. [Google Scholar]
- [17].a) For selected reviews highlighting methods for type I polyketide construction, see: Koskinen AMP, Karisalmi K, Chem. Soc. Rev 2005, 34, 677–690; [DOI] [PubMed] [Google Scholar]; b) Dechert-Schmitt A-MR, Schmitt DC, Gao X, Itoh T, Krische MJ, Nat. Prod. Rep 2014, 31, 504–513; [DOI] [PMC free article] [PubMed] [Google Scholar]; c) Feng J, Kasun ZA, Krische MJ, J. Am. Chem. Soc 2016, 138, 5467–5478; [DOI] [PMC free article] [PubMed] [Google Scholar]; d) Liu H, Lin S, Jacobsen KM, Poulsen TB, Angew. Chem. Int. Ed 2019, 58, 13630–13642; [DOI] [PubMed] [Google Scholar]; Angew. Chem 2019, 131, 13764–13777; [Google Scholar]; e) Friedrich RM, Friestad GK, Nat. Prod. Rep 2020, 37, 1229–1261. [DOI] [PubMed] [Google Scholar]
- [18].a) For reviews of the catalytic site-selective modification of diols and higher polyols, see: Shin I, Krische MJ, Top. Curr. Chem 2016, 372, 85–101; [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Giuliano MW, Miller SJ, Top. Curr. Chem 2016, 372, 157–202; [DOI] [PubMed] [Google Scholar]; c) Shugrue CR, Miller SJ, Chem. Rev 2017, 117, 11894–11951; [DOI] [PMC free article] [PubMed] [Google Scholar]; d) Schwartz LA, Krische MJ, Isr. J. Chem 2018, 58, 45–51; [Google Scholar]; e) Dimakos V, Taylor MS, Chem. Rev 2018, 118, 11457–11517. [DOI] [PubMed] [Google Scholar]
- [19].a) For site-selective allylation of 1,3-diols catalyzed by iridium using allyl acetate as a pronucleophile, see: Dechert-Schmitt A-MR, Schmitt DC, Krische MJ, Angew. Chem. Int. Ed 2013, 52, 3195–3198; [DOI] [PMC free article] [PubMed] [Google Scholar]; Angew. Chem 2013, 125, 3277–3280; [Google Scholar]; b) Shin I, Wang G, Krische MJ, Chem. Eur. J 2014, 20, 13382–13389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].a) For reviews on halide effects in transition metal-catalyzed reactions, see: Maitlis PM, Haynes A, James BR, Catellani M, Chiusoli GP, Dalton Trans 2004, 3409–3419; [DOI] [PubMed] [Google Scholar]; b) Fagnou K, Lautens M, Angew. Chem. Int. Ed 2002, 41, 26–47; [DOI] [PubMed] [Google Scholar]; Angew. Chem 2002, 114, 26–49. [Google Scholar]
- [21].a) For selected studies of formyl CH hydrogen bonds, see: Corey EJ, Lee TW, Chem. Commun 2001, 1321–1329; [Google Scholar]; b) Thakur TS, Kirchner MT, Bläser D, Boese R, Desiraju GR, Phys. Chem. Chem. Phys 2011, 13, 14076–14091; [DOI] [PubMed] [Google Scholar]; c) Grayson MN, Krische MJ, Houk KN, J. Am. Chem. Soc 2015, 137, 8838–8850. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].a) For selected reviews on late-stage functionalization of complex small molecules, see: Cernak T, Dykstra KD, Tyagarajan S, Vachal P, Krska SW, Chem. Soc. Rev 2016, 45, 546–576; [DOI] [PubMed] [Google Scholar]; b) Shugrue CR, Miller SJ, Chem. Rev 2017, 117, 11894–11951; [DOI] [PMC free article] [PubMed] [Google Scholar]; c) Blakemore DC, Castro L, Churcher I, Rees DC, Thomas AW, Wilson DM, Wood A, Nat. Chem 2018, 10, 383–394; [DOI] [PubMed] [Google Scholar]; d) Guillemard L, Kaplaneris N, Ackermann L, Johansson MJ, Nat. Rev. Chem 2021, 5, 522–545. [DOI] [PubMed] [Google Scholar]
- [23].a) Maitra S, Bodugam M, Hanson PR, Org. Lett 2016, 18, 3094–3097; [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Chandrasekhar S, Rambabu C, Reddy AS, Org. Lett 2008, 10, 4355–4357. [DOI] [PubMed] [Google Scholar]
- [24].Ferrié L, Boulard L, Pradaux F, Bouzbouz S, Reymond S, Capdevielle P, Cossy J, J. Org. Chem 2008, 73, 1864–1880. [DOI] [PubMed] [Google Scholar]
- [25].Hafner A, Duthaler RO, Marti R, Rihs G, Rothe-Streit P, Schwarzenbach F, J. Am. Chem. Soc 1992, 114, 2321–2336. [Google Scholar]
- [26].a) For the stoichiometric reaction of HXRu(CO)(PR3)3 (X = Cl, Br) with allenes and dienes to form isolable π-allylruthenium species, see: Hiraki K, Ochi N, Sasada Y, Hayashida H, Fuchita Y, Yamanaka SJ, Chem. Soc., Dalton Trans 1985, 873–877; [Google Scholar]; b) Hill AF, Ho CT, Wilton-Ely JDET, Chem. Commun 1997, 2207–2208; [Google Scholar]; c) Xue P, Bi S, Sung HHY, Williams ID, Lin Z, Jia G, Organometallics 2004, 23, 4735–4743. [Google Scholar]
- [27].Zimmerman HE, Traxler MDI, J. Am. Chem. Soc 1957, 79, 1920–1923. [Google Scholar]
- [28].Skucas E, Zbieg JR, Krische MJ, J. Am. Chem. Soc 2009, 131, 5054–5055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].a) Tse SKS, Xue P, Lin Z, Jia G, Adv. Synth. Catal 2010, 352, 1512–1522; [Google Scholar]; b) Isbrandt ES, Vandavasi JK, Zhang W, Jamshidi MP, Newman SG, Synlett 2017, 28, 2851–2854. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.