

Abstract
Chronic, excessive neuroinflammation is a key feature of neurodegenerative diseases such as Alzheimer’s disease (AD) and Parkinson’s disease (PD). However, neuroinflammatory pathways have yet to be effectively targeted in clinical treatments for such diseases. Interestingly, increased inflammation and neurodegenerative disease risk have been associated with type 2 diabetes mellitus (T2DM) and insulin resistance (IR), suggesting that treatments that mitigate T2DM pathology may be successful in treating neuroinflammatory and neurodegenerative pathology as well. Glucagon-like peptide-1 (GLP-1) is an incretin hormone that promotes healthy insulin signaling, regulates blood sugar levels, and suppresses appetite. Consequently, numerous GLP-1 receptor (GLP-1R) stimulating drugs have been developed and approved by the US Food and Drug Administration (FDA) and related global regulatory authorities for the treatment of T2DM. Furthermore, GLP-1R stimulating drugs have been associated with anti-inflammatory, neurotrophic, and neuroprotective properties in neurodegenerative disorder preclinical models, and hence hold promise for repurposing as a treatment for neurodegenerative diseases. In this review, we discuss incretin signaling, neuroinflammatory pathways, and the intersections between neuroinflammation, brain IR, and neurodegenerative diseases, with a focus on AD and PD. We additionally overview current FDA-approved incretin receptor stimulating drugs and agents in development, including unimolecular single, dual, and triple receptor agonists, and highlight those in clinical trials for neurodegenerative disease treatment. We propose that repurposing already-approved GLP-1R agonists for the treatment of neurodegenerative diseases may be a safe, efficacious, and cost-effective strategy for ameliorating AD and PD pathology by quelling neuroinflammation.
Keywords: Glucagon-like peptide-1 (GLP-1), neurodegeneration, Alzheimer’s disease, Parkinson’s disease, neuroinflammation, incretin mimetic, glucose-dependent insulinotropic peptide (GIP), insulin resistance
Graphical Abstract
1. Incretin Overview
Across all neurodegenerative disorders and injuries, neuroinflammation plays a key role in disease progression and recovery. There is currently a void in viable US Food and Drug Administration (FDA)-approved medications to treat let alone mitigate disease progression of chronic brain disorders, such as Parkinson’s disease (PD) and Alzheimer’s disease (AD), and more acute brain injuries such as traumatic brain injury (TBI). A recent genome-wide association study (GWAS) provides new genetic insights into AD and related dementias indicating elevation of inflammation-related pathways as a driver of risk for these chronic conditions (1). The repurposing of FDA-approved drugs towards treating inflammation-related central nervous system (CNS) diseases and injuries would be ideal, as their safety profile in humans has already been established. Several FDA-approved incretin mimetics, originally developed for the treatment of type 2 diabetes mellitus (T2DM) (Table 1), are a promising class of drugs currently progressing towards repurposing as a new treatment strategy for a wide variety of brain/neurological disorders. A wealth of preclinical animal studies in models of stroke (2–4), PD (4–6), AD (6–8), glaucoma (9–11), and TBI (12) provide evidence for using incretin mimetics as a treatment option. In addition, several human clinical trials related to CNS diseases are progressing or have been completed (Table 2), with initial phases of trials in PD showing efficacy (13). Incretin mimetics exhibit pleiotropic signaling effects in a variety of different cell types (14), but their potent anti-inflammatory properties, together with their neurotrophic and neuroprotective features, make them ideal for treating neurodegenerative disease. Within this review article we provide a brief overview of incretin mimetic drugs, both FDA-approved and in clinical and preclinical trials (Tables 1 and 2), and underscore their potent anti-inflammatory capacity that makes them ideal candidates for repurposing and investigation in clinical trials for neurodegenerative conditions.
Table 1.
Comprehensive list of incretin receptor-targeting drugs for metabolic disease treatment. Numerous incretin receptor-targeting treatments have been approved (red if in the United States, green if abroad) or are currently in clinical trials or being developed (blue) for metabolic diseases, namely T2DM and obesity, and/or promoting weight loss. Highlighted drugs are in clinical trials for the treatment of AD or PD. While DPP-IV inhibitors do not directly stimulate incretin receptors, they increase levels of endogenous incretins and, thereby, promote incretin receptor signaling, and thus are included in the table. Only Victoza®, Bydureon®, and Byetta® have completed clinical trials for incretin analog treatment for neurodegenerative diseases; Ozempic®, Rybelsus®, Adlyxin®/Lyxumia®, PT320, and NLY01 are currently in clinical trials for neurodegenerative disease treatment.
| Mechanism of Action | Company | Drug Name | Administration | Uses | Other Notes | |
|---|---|---|---|---|---|---|
| GLP-1R Agonist | AstraZeneca | Exenatide | Bydureon ® BCise ™ | Once weekly SC | T2DM | Latest formulation of Bydureon®. |
| Bydureon ® | Once weekly SC | T2DM | 2 mg injection once each week, extended release formulation. Completed clinical trial in severely obese adolescents in 2020 (#NCT02496611). Phase 2 clinical trial (#NCT01971242) for PD patients completed in 2016 (see (a.) Athauda et al. 2017). Additional clinical trials for PD patients ongoing. | |||
| Byetta ® | Twice daily SC | T2DM | Used any time within 1 hour before morning and evening meals (or before the two main meals of the day, approximately 6 hours or more apart). In clinical trial (#NCT02442791), neuroprotective effects of Byetta® administration following cardiac arrest showed no significant outcomes (see (b.) Wiberg et al. 2016). Phase 2 clinical trial (#NCT02838589) completed investigating effects on cerebral blood flow. Clinical trials in AD (#NCT01255163; see (c.) Mullins et al. 2019) and PD (*EUCTR2009-018137-37-GB; see (d.) Aviles-Olmos et al. 2013) completed in 2016 and 2013, respectively. | |||
| Sanofi-Aventis | Lixisenatide + insulin glargine | Soliqua ® | Once daily SC | T2DM | Approved for adults with T2DM to lower A1C. 15–60 units of Soliqua® 100/33 per injection. Each unit of Solique 100/33 contains 0.33 mcg lixisenatide and 1 unit of Lantus. | |
| Lixisenatide | Adlyxin®/Lyxumia® | Once daily SC | T2DM | 10 mcg once daily injection for 14 days. On Day 15, dosage increased to 20 mcg once daily. Administered within one hour before the first meal of the day. Currently in phase 2 clinical trial (#NCT03439943) for PD treatment. | ||
| Sanofi-Aventis/Hanmi Pharmaceuticals | Efpeglenatide | HM 11260C | Once weekly SC | T2DM | Completed Phase 3 clinical trial for T2DM treatment (#NCT03353350). | |
| Novo Nordisk | Liraglutide | Victoza ® | Once daily SC | T2DM | Used at any time of day, independent of meals. Use 0.6 mg per day for one week then increase to 1.2 mg, which can be increased to 1.8 mg for additional glycemic control. Clinical trial in AD patients (#NCT01469351) completed in 2013 (see (e.) Gejl et al. 2016). Additional clinical trials for AD and PD patients ongoing. | |
| Saxenda ® | Once daily SC | Weight Loss | 3 mg daily SC injection. | |||
| Liraglutide + insulin degludec | Xultophy ® | Once daily SC | T2DM | 10 units Xultophy® 100/3.6 (10 units insulin degludec, 0.36 mg liraglutide) once daily SC. In patients converting from basal insulin or GLP 1R agonist: 16 units Xultophy® 100/3.6 (16 units insulin degludec, 0.58 mg liraglutide). | ||
| Semaglutide | Ozempic ® | Once weekly SC | T2DM | 0.25 mg (with or without meals). After 4 weeks, dose increased to 0.5 mg once weekly. Can be increased to 1 mg once weekly if needed. Phase 2 clinical trial (#NCT03659682) not yet recruiting in PD patients. | ||
| Rybelsus ® | Oral once daily | T2DM | 7 mg or 14 mg tablets. Approved to lower A1C, may help with weight loss. Currently recruiting for phase 3 clinical trials (#NCT04777409, #NCT04777396) in early AD patients. | |||
| Wegovy ® | Once weekly SC | Obesity | 2.4 mg weekly SC injection. | |||
| IcoSema | Once weekly SC | T2DM | Semaglutide + insulin icodec. Phase 3 clinical trial (#NCT05259033) currently recruiting. | |||
| NASH | Daily SC | NASH/NAFLD | Phase 2 clinical trial (#NCT02970942) completed. NASH measurements are primary outcome with NAFLD as secondary outcome measure. | |||
| ----------------- | NN9926, NN9927 | Oral | T2DM | Long acting, new molecular entities currently in development. | ||
| Eli Lilly | Dulaglutide | Trulicity ® | Once weekly SC | T2DM | 0.75 or 1.5 mg SC injection once weekly from single dose pen. | |
| Beijing Dongfang Biotech Co., Ltd. | Exendin-4 | JY09 | Once dailly SC | T2DM | Phase 1 clinical trial (#NCT04354090) completed. Exendin-4 + human Immunoglobulin G2 (IgG2) Fc fragment. | |
| Peptron | Exenatide SR | PT320 | Biweekly SC | T2DM/Obesity/PD | Formerly PT302. Proprietary formulation allows for injection with smaller needle to reduce injection pain. Phase 1 clinical trial (#NCT00964262) completed for treatment of T2DM. Phase 2 clinical trials completed in South Korea for T2DM. Phase 2 clinical tral in PD patients in progress (#NCT04269642). | |
| Intarcia | Exenatide | ITCA 650 | Long term implant | T2DM | Implant with potential to offer 6 month exenatide delivery with 1 year delivery options in development. Phase 3 clinical trials have been completed with New Drug Application (NDA) submitted to the Food and Drug Administration (FDA) (pending approval). | |
| vTv Therapeutics | ----------------- | TTP273 | Oral once or twice daily | T2DM | Non-peptide GLP-1R agonist. Completed phase 2 clinical trial (#NCT02653599) for T2DM treatment. | |
| CSPC ZhongQi Pharmaceutical Technology Co., Ltd. | Recombinant Exenatide | rExenatide-4 | SC twice daily | T2DM | Phase 3 clinical trial (#NCT03239119) in Chinese T2DM patients initiated, but not yet recruiting. | |
| Oramed | Exenatide Oral | ORMD-0901 | Oral | T2DM | US FDA cleared Investigational New Drug (IND) application for trials in humans as of September 2018. | |
| Shanghai Biolaxy | Exenatide | Nodexen | Oral | T2DM | Nanoparticle oral delivery. Clinical trials ongoing in China. | |
| Jiangsu Hengrui Medicine Co. Ltd. | Loxenatide | PEX168 | Once weekly SC | T2DM | Phase 3 clinical trial in progress (#NCT02477969). This is a PEGylated formulation. | |
| Shanghai Benemae Pharmaceutical | ----------------- | Beinaglutide | 3x daily SC | T2DM/Obesity | Phase 4 clinical trial recruiting (#NCT05005741). Approved for use in China in 2016. | |
| Zydus Lifesciences Ltd. | ----------------- | ZYD1 | Oral and SC | T2DM | Phase 1 clinical trial completed (#NCT01972893). | |
| PegBio Co., Ltd. | ----------------- | PB-119 | Once weekly SC | T2DM | Phase 3 clinical trial (#NCT04504396) recruiting. This is a PEGylated exenatide formulation. | |
| Regor Pharmaceuticals Inc. | ----------------- | RGT001-075 | Oral once daily | T2DM | Phase 2 clinical trial (#NCT05297045) in progress. | |
| CSPC Baike (Shandong) Biopharmaceutical Co., Ltd. | ----------------- | TG103 | Once weekly SC | Obesity | Phase 2 clinical trial (#NCT05299697) recruiting. | |
| Hansoh Pharmaceuticals | Polyethylene Glycol Loxenatide | Fu Laimei ® | Once weekly SC | T2DM | Approved in China for adults with poor blood glucose control. Recommended dose 0.1–0.2 mg/week. | |
| Neuraly Inc. | Exenatide | NLY01 | Once weekly SC | PD/AD | This is a PEGylated formulation. Phase 1 clinical trial completed in 2019 (#NCT03672604; see (f.) Yun et al. 2018). Phase 2 clinical trial ongoing in patients with PD (#NCT04154072). | |
| GIPR Agonist | ----------------- | GIP Peptide | ----------------- | ----------------- | T1DM | GIP peptide completed clinical trial (#NCT03556098) investigating its efficacy as a safeguard against hypoglycemia in patients with Type-1 diabetes mellitus (T1DM). |
| Zealand Pharmaceuticals | ----------------- | ZP4165 | Intra Venous (I.V.) or SC | T2DM | DPP-IV resistance and potentiates GLP-1 mediated weight loss and improved glycemic control in rats (see (g.) Nørregaard et al. 2018). | |
| GLP-1R/GcgR Dual Agonist | Hamni Pharmaceuticals/Janssen Research & Development, LLC | ----------------- | HM12525A/JNJ-64565111 | Once weekly SC | T2DM/Obesity | Phase 2 clinical trial completed (#NCT03586830). |
| Zealand Pharmaceuticals/Boehringer Ingelheim | ----------------- | BI 456906 | Once weekly SC | Obesity | Long lasting analogue of amylin and partially builds on the effects of oxyntomodulin. Completed phase 1 clinical tral (#NCT03175211) to assess safety, tolerability, and pharmacokinetics/pharmacodynamics. Phase 1 trials in Germany for obesity initiated. | |
| Astra Zeneca/MedImmune | ----------------- | MEDI0382 | Daily SC | T2DM | Completed phase 2a (see (h.) Ambery et al. 2018) and phase 2b clinical trials (#NCT03235050). | |
| Sanofi-Aventis | ----------------- | SAR425899 | Daily SC | T2DM/Obesity | Completed phase 1 clinical trial (#NCT03414736; see (i.) Goebel et al. 2018). | |
| Janssen Pharmaceuticals | ----------------- | JNJ-54728518 | ----------------- | T2DM/Obesity | Phase 1 clinical trials initiated for obesity and T2DM in 2016. Pre-clinical trials for obesity also initiated in 2016. | |
| Novo Nordisk | ----------------- | NNC9204-1177 | Once weekly SC | Obesity | Completed phase 1 clinical trial (#NCT03308721). | |
| Prolor/OPKO Biological | ----------------- | OPK-88003 | Once weekly SC | T2DM | Completed phase 2 clinical trial (#NCT03406377). | |
| Spitfire Pharma | ----------------- | SP-1373 | Once daily SC | NASH/T2DM/Obesity | Clinical trial for T2DM and obesity planned for 2019. | |
| Boehringer Ingelheim | ----------------- | BI 456906 | ----------------- | Obesity | Completed phase 1 clinical trial (#NCT03591718) in patients with obesity. | |
| GLP-1R/GLP-2R Dual Agonist | Zealand Pharmaceuticals | ----------------- | Dapiglutide | ----------------- | T2DM | Early phase 1 clinical trials completed (#NCT03994549, #NCT04612517). |
| GLP-1R/GIPR Dual Agonist | Eli Lilly | Tirzepatide | Mounjaro ™ | Once weekly SC | T2DM | Approved for adults with T2DM. Available in 2.5 mg, 5 mg, 7.5 mg, 10 mg, 12.5 mg, and 15 mg doses through auto-injector pen. |
| Novo Nordisk | ----------------- | RG 7697/NNC0090-2746/MAR709 | Once daily SC | T2DM | Completed phase 2 clinical trial (#NCT02205528; see (j.) Frias et al. 2017). | |
| Kariya Pharmaceuticals | ----------------- | KP405 | Info not available | PD/AD | Ready for phase 1 clinical trials. Toxicology studies completed. | |
| Sanofi | ----------------- | SAR438335 | Info not available | T2DM | Currently in phase 1 clinical trals in France. | |
| GLP-1R/GIPR/GcgR Triagonist | Novo Nordisk | ----------------- | NNC9204-1706 | Once daily SC | Obesity | Completed phase 1 clinical trial (#NCT03661879). |
| Hanmi Pharmaceutical Co. Lmtd. | ----------------- | HM15211 | SC | Obesity | Long acting formulation. Current phase 1 clinical trial (#NCT03374241) recruiting (see (k.) Kim et al. 2018 for latest research on compound). | |
| Sanofi | ----------------- | SAR441255 | SC | T2DM/Obesity/NASH | Preclinical. | |
| DPP-IV Inhibition | Astra Zeneca | Saxagliptin | Onglyza ™ | Once daily oral | T2DM | 2.5 or 5 mg regardless of meals. |
| Saxagliptin + metformin HCl | Kombiglyze ® XR | Once daily oral | T2DM | Take once daily with evening meal. Available in 5 mg saxagliptin/500 mg metformin HCl, 5 mg saxagliptin/1000 mg metformin HCl, or 2.5 mg saxagliptin/1000 mg metformin HCl doses. | ||
| Merck | Sitagliptin | Januvia ® | Once daily oral | T2DM | 25, 50, or 100 mg with or without food. | |
| Sitagliptin phosphate + metformin HCl | Janumet ® / Janumet ® XR | Oral twice daily/Once daily (XR formula) | T2DM | Combination drug therapy with metformin. XR formulation is an extended release version. Max dosage 100 mg Sitagliptin and 2000 mg metformin HCl. | ||
| Omarigliptin | Marizev ® | Oral once weekly | T2DM | Available in Japan. See (l.) Goldenberg et al. 2017 for phase 3 clinical trial information (#NCT01703221). Possible repositioning of drug for intran asal delivery for PD (see (m.) Ayoub et al. 2018). | ||
| Takeda | Trelagliptin | Zafatek ® | Oral once weekly | T2DM | Approved for use in Japan. Phase 2 clinical trials abandoned in the USA because of costs. | |
| Alogliptin | Nesina ® | Oral once daily | T2DM | 25 mg with or without food. | ||
| Alogliptin + metformin HCl | Kazano ® | Oral twice daily | T2DM | Take oral twce daily with food. Available in 12.5 mg alogliptin/500 mg metformin HCl or 12.5 mg alogliptin/1000 mg metformin HCl doses. Max dosage 25 mg alogliptin/2000 mg metformin HCl per day. | ||
| Alogliptin + pioglitazone | Oseni ® | Oral once daily. | T2DM | Take with or without food. Available in 12.5 or 25 mg alogliptin doses. | ||
| Boehringer Ingelheim | Linagliptin | Tradjenta ® | Oral once daily | T2DM | 5 mg once daily. | |
| Linagliptin + empagliflozin | Glyxambi ® | Oral once daily | T2DM | Take once daily in the morning with or without food. Available in 10 mg empagliflozin/5 mg linagliptin and 25 mg empagliflozin/5 mg linagliptin doses. | ||
| Linagliptin + metformin HCl | Jentadueto ® / Jentadueto ® XR | Oral twice daily/Once daily (XR formula) | T2DM | Combination drug therapy with metformin. XR formulation is an extended release version. Max dosage 2.5 mg linagliptin and 1000 mg metformin HCl. | ||
| Dong-A ST | Evogliptin | Suganon ® | Oral once daily | T2DM | 5 mg once daily. Approved for use in South Korea in 2015. Sold with extended release metformin. | |
| SatRx LLC | Gosogliptin | SatRx ® | Oral | T2DM | Approved for use in Russia. Completed phase 3 clinical trial of safety alone or with metformin compared to Vildagliptin alone or with metformin (#NCT03088670). | |
| LG Life Sciences/Sanofi | Gemigliptin | Zemiglo ™ | Oral once daily | T2DM | Long acting DPP-IV inhibitor. See (n.) Kim et al. 2016 for characterization. | |
| Zealand Pharmaceuticals | Vildagliptin | Galvus ® | Oral once daily | T2DM | 50 mg in combination with metformin (see (o.) Mathieu & Degrande 2008). | |
| Mitsubishi Tanabe Pharma and Daiichi Sankyo | Tenegliptin | Tenelia ® | Oral twice daily | T2DM | 20 mg twice daily. See (p.) Kishimoto 2013. | |
| Zealand Pharmaceuticals | Anagliptin | Suiny ® | Oral twice daily | T2DM | Approved for use in Japan, 200 mg daily. See (q.) Nishio et al. 2015 | |
Visit https://clinicaltrials.gov to locate clinical trials registered in the United States.
Visit https://trialsearch.who.int to locate clinical trials registered internationally.
NASH = nonalcoholic steatohepatitis; NAFLD = nonalcoholic fatty liver disease; SC = subcutaneous injection; T1DM = Type 1 Diabetes Mellitus. Sources: (a) ref(13); (b) ref(256); (c) ref(257); (d) ref(258); (e) ref(259); (f) ref(103); (g) ref(260); (h) ref(261); (i) ref(262); (j) ref(263); (k) ref(264); (l) ref(265); (m) ref(266); (n) ref(267); (o) ref(268); (p) ref(269); (q) ref(270). Table updated from Glotfelty et al., 2019 (12).
Table 2.
GLP-1R agonists in clinical trials for repurposing to treat neurodegenerative diseases. Drugs in red (approved in the United States) or green (approved abroad) have been approved for metabolic disease treatment, and those in blue are in clinical trials for metabolic disease treatment. Only Bydureon®, Byetta®, and Victoza® have completed interventional clinical studies and demonstrated efficacy in treating a neurodegenerative disease.
| Company | Drug Name | Use | Clinical Trial | Phase | Status | Outcomes | |
|---|---|---|---|---|---|---|---|
| AstraZeneca | Exenatide | Bydureon ® | PD | # NCT03456687 | 1 | Ongoing | N/A |
| # NCT01971242 | 2 | Completed (2020) | Treatment improved off-medication motor scores. See (a.) Athauda et al., 2017. | ||||
| # NCT04305002 | 2 | Recruiting | N/A | ||||
| * EUCTR2019-000732-26-SE | 2 | Ongoing | N/A | ||||
| # NCT04232969 | 3 | Ongoing | N/A | ||||
| * ISRCTN14552789 | 3 | Ongoing | N/A | ||||
| * ACTRN12620000627954 | 4 | Recruiting | N/A | ||||
| Byetta ® | AD | # NCT01255163 | 2 | Completed (2016) | Treatment reduced amyloid-β-42 concentration in extracellular vesicles. See (b.) Mullins et al., 2019. |
||
| PD | # NCT01174810 | 2 | Completed (2013) | Treatment improved motor abilities and cognition. See (c.) Aviles-Olmos et al., 2013. | |||
| FRDA | * EUCTR2014-003598-41-BE | Pilot | Completed (2019) | Treatment improved frataxin levels in FRDA patients. See (d.) Igoillo-Esteve et al., 2020. | |||
| Novo Nordisk | Liraglutide | Victoza ® | AD | # NCT01469351 | 2 | Completed (2013) | Treatment maintained healthy brain glucose metabolism. See (e.) Gejl et al., 2016. |
| # NCT01843075 | 2 | Ongoing | N/A | ||||
| PD | # NCT02953665 | 2 | Completed (2022) | Treatment improved non-motor symptoms, quality of life, and mobility. See (f.) Hogg et al., 2022. | |||
| Semaglutide | Ozempic ® | PD | # NCT03659682 | 2 | Not yet recruiting | N/A | |
| Rybelsus ® | AD | * ISRCTN71283871 | 2 | Recruiting | N/A | ||
| # NCT04777409 | 3 | Recruiting | N/A | ||||
| # NCT04777396 | 3 | Recruiting | N/A | ||||
| Neuraly Inc. | Exenatide | NLY01 | PD | # NCT03672604 | 1 | Ongoing | N/A |
| # NCT04154072 | 2 | Ongoing | N/A | ||||
| Peptron | Exenatide | PT320 | PD | # NCT04269642 | 2 | Ongoing | N/A |
| Sanofi-Aventis | Lixisenatide | Adlyxin®/Lyxumia® | PD | # NCT03439943 | 2 | Ongoing | N/A |
| Various | Any GLP-1R agonist approved for metabolic disease treatment | Various | Glaucoma | N/A | N/A | Completed (2021) | GLP-1R agonist treatment cohort had reduced risk of developing glaucoma. See (g.) Sterling et al., 2021. |
Visit https://clinicaltrials.gov to locate clinical trials registered in the United States.
Visit https://trialsearch.who.int to locate clinical trials registered internationally.
1.1. Incretins – focus on the endocrine system
The incretin signaling system, constituting the gut-derived metabolic peptides glucagon-like peptide-1 (GLP-1) and glucose-dependent insulinotropic peptide (GIP), is responsible for blood glucose regulation following food ingestion (15). These peptides act by stimulating the release of insulin. The incretins GLP-1 and GIP are secreted from the small intestinal enteroendocrine L and K cells, respectively, and act on their target receptors present on the β-cells of the pancreas. The GLP-1 receptor (GLP-1R) and GIP receptor (GIPR) are class B G protein-coupled receptors, each with a seven-helix transmembrane domain and an extracellular ligand binding domain. The intracellular face interacts primarily with the Gαs subunit of G proteins and with β-arrestins, as well as various other signaling molecules. GLP-1R and GIPR signaling have several known functions in the endocrine system: GLP-1R activation initiates signaling cascades that promote insulin secretion and β-cell survival; GIPR activation promotes insulin secretion after feeding, glucagon (Gcg) secretion during fasting, triacylglycerol uptake by adipose tissue (16), and reduced bone resorption (17). Both members of the glucagon superfamily, GLP-1 and GIP activity both supplement as well as oppose select actions of Gcg, a structurally related secretin (Figure 1A) synthesized in islet α-cells of the pancreas and within the small intestine, that acts by raising blood glucose levels in periods of fasting. Following their release, GLP-1, GIP, and Gcg are rapidly degraded to end their pharmacological/physiological regulatory action by the enzyme dipeptidyl peptidase-4 (DPP-IV), which is responsible for their short plasma half-lives of 1–2 minutes (18), 2–5 minutes (19), and 6–7 minutes (20), respectively. DPP-IV acts as a relatively unpromiscuous amino-peptidase and releases a dipeptide from the N-terminal end of its substrates. With a preference for an alanine or proline in the penultimate position, it additionally cleaves peptides with other amino acid residues (serine, glycine, valine) albeit more slowly. Discovery of reduced incretin actions in T2DM patients (21) led to robust research into GLP-1 and GIP as potential disease-modifying drug therapies. Although both peptides are highly insulinotropic in healthy individuals (22, 23), only GLP-1 was demonstrated to have preserved insulinotropic effects in patients with T2DM (24, 25). This, combined with the ability of truncated GLP-1 to inhibit Gcg secretion (26), prompted drug developers to target GLP-1R agonists as potential treatments for T2DM.
Figure 1.
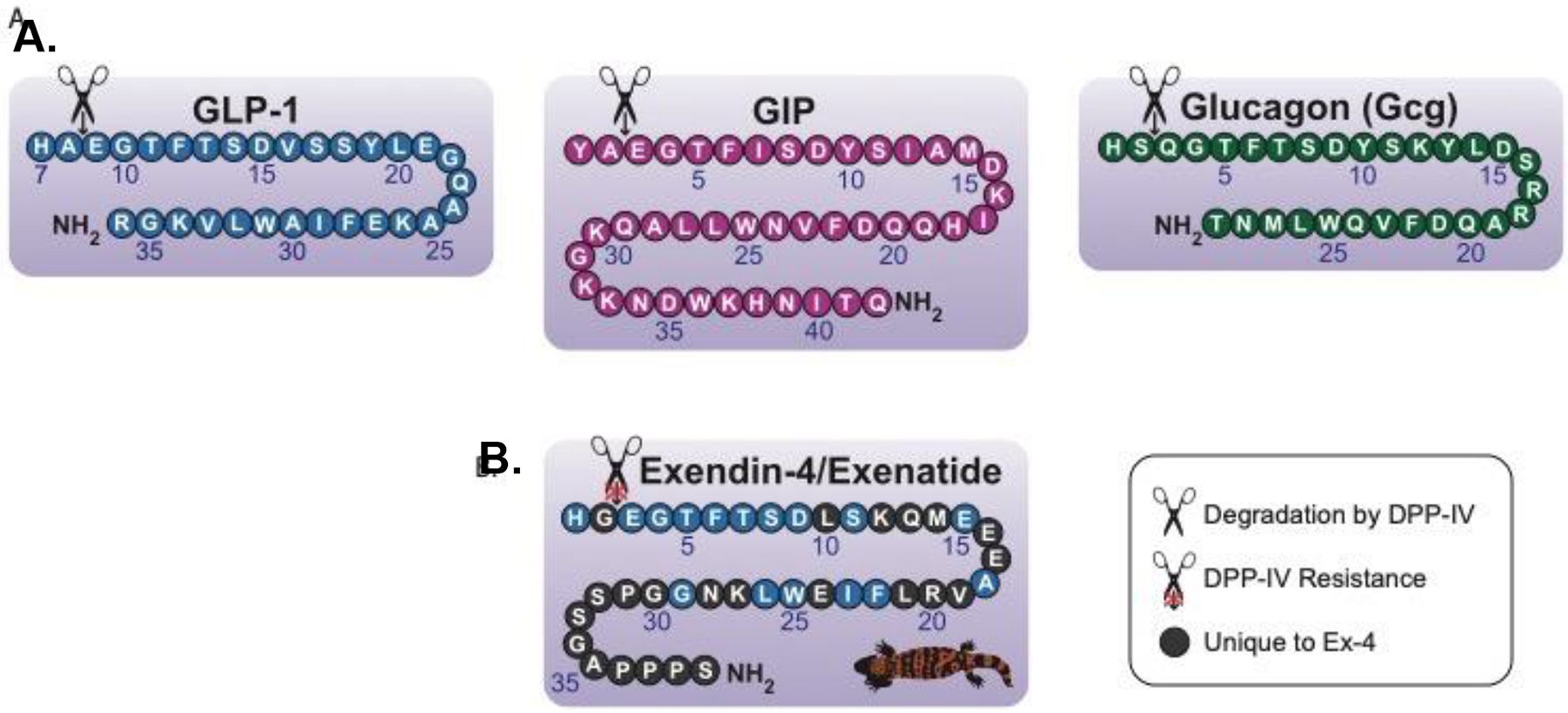
(A) The endogenous secretins have similar amino acid sequences and structures, most notably their favorable cleavage sites for DPP-IV. This results in very short half-lives for these peptides. (B) Exendin-4 is a GLP-1 analog naturally occurring in Gila monster lizard venom. The unique glycine residue at the DPP-IV cleavage site renders the protein unrecognizable by DPP-IV and prolongs the half-life of the peptide. Thus, the exendin-4 backbone has been utilized in the creation of longer-acting synthetic GLP-1R agonists. Figure adapted from Glotfelty et al. 2020 (5).
1.2. FDA approval of incretin-based therapies
Various GLP-1 analogs have been developed to lengthen the half-life of the peptide and prolong the beneficial effects of GLP-1R signaling. In 1992, during the early research of incretin effects in diabetes, an endogenous GLP-1 analog, exendin-4, was isolated from the venom of the Gila monster lizard (Heloderma suspectum) (27) (Figure 1B). Exendin-4 evades DPP-IV cleavage due to an alanine for glycine substitution at the enzyme cleavage site (Figure 1B) (28) and thus has a prolonged half-life relative to GLP-1 (2.4 hours in plasma) (29). In 2005, exendin-4, under the name “Exenatide”, became the first FDA-approved GLP-1R agonist for the treatment of T2DM. Since then, many other GLP-1 analogs have been approved by the FDA, including liraglutide, semaglutide, lixisenatide, dulaglutide, and formerly albiglutide (discontinued in July of 2017) (12) (Table 1). In addition to GLP-1R agonist adoption for T2DM treatment, DPP-IV inhibitors, which elevate the levels of endogenously produced incretins throughout the body, have proven efficacious and been FDA-approved to treat the disease (Table 1).
1.3. Multi-agonism
More recent research has investigated the use of drugs that target a combination of secretin receptors (GLP-1R, GIPR, and GcgR) to treat T2DM. In 2009, a dual GLP-1R/GcgR agonist was pioneered for treating metabolic disorders in rodents (30). Later, in 2013, a dual GLP-1R/GIPR agonist, termed “twincretin”, demonstrated efficacy in animal models and humans with T2DM in reducing glycosylated hemoglobin A1c (HbA1c), an average measure of blood sugar levels over the duration of several months (31). Further research led to the development of Tirzepatide, a GLP-1R/GIPR dual agonist, which was found to enhance insulin signaling, reduce blood glucose, and promote weight loss in preclinical and phase 1 and 2 clinical trials (32). Moreover, the beneficial effects of Tirzepatide were significantly greater than the effects of a single GLP-1R agonist or a single GIPR agonist alone (32). A meta-analysis of randomized controlled trials found that Tirzepatide treatment in patients with T2DM significantly reduced HbA1c, fasting blood glucose, postprandial blood glucose, body mass index (BMI), waist circumference, and weight, as compared to single GLP-1R agonist treatment or insulin analog treatment (33). Tirzepatide (Mounjara™) (Table 1) is the only currently FDA-approved incretin multi-agonist (approved in 2022).
Additionally, unimolecular triple incretin-based agonists, or triagonists, have been developed that target the GLP-1, GIP, and Gcg receptors (GcgR). GLP-1R/GIPR/GcgR triagonist treatment in a high-fat diet mouse model system reduced plasma glucose levels, increased plasma insulin levels, and regulated blood glucose levels following food intake (34). Another rodent study demonstrated the heightened potency of a GLP-1R/GIPR/GcgR triagonist and its superior performance to a GLP-1R/GIPR dual agonist in reducing body weight, improving blood glucose regulation, and ameliorating fatty liver disease (35). A novel GLP-1R/GIPR/GcgR triagonist, SAR441255, has been shown to promote weight loss and maintain healthy blood glucose levels in a rodent model, and outperformed a GLP-1R/GIPR dual agonist (36). In a mouse model of obesity, triagonist treatment enhanced weight loss and outperformed single and dual incretin-based receptor agonists (37). Furthermore, preclinical studies of SAR441255 revealed incretin receptor activation and maintenance of normal blood sugar levels in healthy monkeys in response to the drug, as well as the safety and tolerability of the drug in a phase 1 human clinical trial (36) (NCT04521738, 48 participants, aged 18–55 years, lean to overweight). SAR4411255 demonstrated a terminal half-life of 3.5–6.1 hours, which is a relatively short exposure for a proposed once daily subcutaneously administered drug, and doses up to 150 ug were well tolerated, with the most frequent treatment-emergent adverse events being gastrointestinal, in accord with other GLP-1R agonists. Maintaining an appropriately balanced agonism among the three receptor subtypes (GLP-1, GIP, and Gcg) is key for single molecule triagonists to optimize efficacy across measures of glucose and body weight-lowering action, as well as to maintain tolerability across organ systems. Consequent to business decisions within Sanofi, it is possible that SAR4411255 may not proceed forward in clinical development (38). The first human clinical trial (phase 1 - NCT03374241) of a novel GLP-1R/GIPR/GcgR triagonist, HM15211, was initiated in 2018 in patients with obesity (39) (Table 1). This HM15211 (NCT03374241) trial was a first-in-human study to evaluate the safety, tolerability, pharmacokinetics, and pharmacodynamics after single ascending dose in healthy obese subjects (40 participants, aged 18–65 years). A further phase 1 trial to evaluate the safety, tolerability, pharmacokinetics, and pharmacodynamics of multiple doses of HM15211 in obese subjects with nonalcoholic fatty liver disease (NAFLD) has been reported as NCT03744182 (66 participants, aged 18–65 years). Finally, an ongoing phase 2 study to evaluate efficacy, safety and tolerability of HM15211 treatment for 12 months in subjects with biopsy confirmed non-alcoholic steatohepatitis (NASH) has been reported as NCT04505436 (217 participants, aged 18–70 years, across multiple US sites with a proposed completion date of November 2025). Currently, no peer reviewed publications of results of any HM15211 clinical study appear available.
Although the dual GLP-1R/GIPR agonist Tirzepatide is the only FDA-approved incretin multi-agonist, unimolecular incretin receptor multi-agonists will likely dominate as treatment options in the future due to growing evidence of their increased efficacy over single GLP-1R agonists (15, 35, 40, 41). Several multi-agonist incretin analogues are in the preclinical or clinical trial phases (Table 1) and are likely to be approved over the coming decade to treat both obesity and diabetes. Interestingly, recent research is unraveling the nuances in intracellular biased agonism these drugs may have, including preferential cAMP and other intracellular pathway induction (42, 43). FDA approval of Tirzepatide and ongoing clinical trials indicate that the multi-agonist approach is safe in humans, which has important implications for treating a range of conditions, including nervous system-related chronic diseases and acute injuries. Preclinical animal models of chronic neurodegenerative disorders such as PD, AD, and glaucoma (9, 44) and acute injury models such as stroke and TBI (12, 45) have robustly demonstrated incretin therapy utility (both single and multi-agonist approaches) as a potential treatment option for these conditions.
1.4. Insulin resistance and introducing incretins in the CNS
In healthy individuals, there is an appropriate balance between the activity of GLP-1, GIP, and Gcg, which is continuously adjusted and maintained by homeostasis. When this fails, a disrupted balance in GLP-1, GIP, and Gcg signaling can lead to chronically elevated blood glucose and subsequent development of insulin resistance (IR) (defined broadly as reduced cellular responsiveness to insulin), a feature of T2DM and obesity (46). Chronically high blood glucose can cause excessive inhibitory phosphorylation of key serine residues of insulin receptor substrates (IRS)-1 and 2 (for example, IRS-1 S312 and S616), resulting in reduced insulin receptor binding sensitivity, and triggers the translocation of IRS-1 and 2 from the cell membrane into the cytoplasm with reduced potential activation of downstream Ak strain transforming (Akt) and extracellular signal-regulated kinase 1/2 (ERK1/2) signaling kinases (47, 48). Insulin can hence play a fundamental role in neurodegeneration through binding to its receptor, which is particularly abundant within the striatum, cerebral cortex, and hippocampus (49, 50). Postmortem studies have reported that PD patients exhibit a reduced expression of insulin receptors (49, 51) and additionally have revealed a raised IRS-1 pS312 expression within nigral dopaminergic neurons as compared to aged-matched controls (52, 53), symbolic of dysfunctional insulin signaling. Since insulin stimulates glucose uptake into cells, a reduction in insulin sensitivity causes a reduction in glucose uptake; consequently, the elevation in blood glucose levels persists, and IR becomes more severe (54).
IR may be present in the brain and is a common feature of neurodegenerative diseases (55–58). Extensive research indicates that T2DM is a risk factor for AD (8, 59–62) and PD (58, 63–65). However, brain IR may occur in AD in the absence of a T2DM diagnosis, as indicated by reduced insulin and insulin-like growth factor-1 (IGF-1) responses in patients with AD and without T2DM (66). These findings suggest that, while peripheral IR as observed in T2DM can contribute to neurodegenerative disease pathology, IR in the CNS may occur via its own independent mechanisms as a separate entity. Yet, it is notable that increased weight circumference, mid-life adiposity, and metabolic syndrome (all allied to peripheral IR and systemic inflammation) are associated with an elevated risk of PD (67–69). In relation to acute neurodegenerative disorders, T2DM in patients challenged with TBI is a significant predictor of mortality—in a study of 51,585 TBI patients, those with T2DM exhibited a significantly higher mortality rate (14.4%) than those without T2DM (8.2%) (70). Furthermore, brain glucose metabolism is dramatically altered following a TBI, with an acute increase in cerebral glucose metabolism typically followed by an extended decline in glucose metabolism (71). Aligned with this, there are multiple reports of hyperglycemia following TBI, with uncontrolled blood glucose levels resulting in poorer outcomes for recovery and an increased mortality risk (72–74). IR is a marker of increased risk for ischemic stroke (75) and, additionally, is associated with poorer functional outcomes (76).
Incretin receptors are prominent in the brain and are intricately associated with peripheral insulin regulation through increased control of feeding behaviors (77, 78). GLP-1R signaling in the hippocampus and hypothalamus improves memory function (79, 80) and is additionally linked to reduced appetite and regulation of food intake (77). GIPR is expressed in neurons and glial cells in the paraventricular nucleus (PVN), dorsomedial nucleus (DMN), and arcuate nucleus (ARC) of the hypothalamus, and reduces food intake in cooperation with GLP-1R signaling (78). GIPR signaling in cortical regions likely has a role in progenitor cell proliferation (81). The expression of incretin receptors in the CNS as well as the link between brain IR and neurodegeneration have fueled scientific interest in the use of GLP-1R and GIPR agonist treatments to promote neural health.
GLP-1R stimulation has been associated with neuroprotective and neurotrophic properties in several foundational studies conducted in the early 2000s (82–84). Rat hippocampal neurons treated with GLP-1 (82, 84) or exendin-4 (84) were more resistant to glutamate excitotoxicity, and both treatments enhanced neural growth and differentiation in a rat neuroendocrine cell model (83)—findings that have been replicated numerous times across cell types of neuronal origin and multiple animal models. In contrast, GLP-1R deficient mice have a learning deficit that can be restored by hippocampal Glp1r gene transfer (85), and have impairments in synaptic plasticity, long-term potentiation (LTP), and memory formation (86). GLP-1R overexpression results in a great resilience to a host of physiological and pathological challenges—compared with parent SH-SY5Y cells, in a human neuroblastoma cell line (SH-SY5Y) overexpressing GLP-1R (by as little as 2-fold) and challenged with hydrogen peroxide and 6-hydroxydopamine (6-OHDA), GLP-1R stimulation significantly enhanced cell viability and proliferation, clearly illustrating the neurotrophic and anti-apoptotic properties of GLP-1R signaling (87). The observed neuroprotective properties of GLP-1R-targeting drugs suggested the potential of incretin receptor agonists in treating diseases of the brain and, since the foundational studies, numerous studies with single, dual, and triple agonists have been undertaken across neurodegenerative and neuropsychiatric conditions preclinically and are now extending into clinical research trials.
An important consideration in GLP-1R/GIPR/GcgR agonist drug development and, indeed, in all neurological therapeutic development (88), is blood-brain barrier (BBB) penetration. GLP-1, GIP, and Gcg are large peptide molecules and thus have limited BBB penetration. Nevertheless, early studies by Kastin and colleagues (89, 90) as well as by Banks and colleagues (91) demonstrated that GLP-1, as well as many alike peptides (92), can access the brain in small but pharmacologically relevant amounts. GLP-1 influx was rapid, associated with simple diffusion and not a saturable transport system (89). A recent study confirmed the rapid brain uptake and pharmacological action of systemically administered GLP-1 and exendin-4, and suggested the presence of a saturable BBB transporter that potentially involves the GLP-1R, as antagonists reduced brain uptake (93). Notably, this same study confirmed the abundance of the GLP-1R on brain vascular endothelial cells (93, 94) that has been reported to potentially mediate arteriolar dilation and, thereby, regulate tissue perfusion (95). The same study additionally demonstrated that resection of the vagus nerve (i.e., complete vagotomy) had no effect on brain GLP-1 uptake and pharmacological action (as evaluated by elevated protein kinase A (PKA) in rat brain), thus suggesting that such pharmacological action required the presence of drug in the brain and was not mediated via the peripheral (vagal) nervous system (93)—as vagal afferent neuron GLP-1Rs have been shown to have a role in facilitating the actions of GLP-1 on food intake and glycemic control (96).
Certain synthetic GLP-1R agonists described below, likewise, have varying ability to cross the BBB. In a study by Banks and colleagues (97) comparing the brain uptake pharmacokinetics of GLP-1R agonists, the acylated GLP-1R ligands liraglutide and semaglutide did not measurably cross the BBB, whereas the non-acylated and non-PEGylated GLP-1R ligands exendin-4 and lixisenatide did measurably cross the BBB (97). However, despite the lack of evidence of semaglutide crossing the BBB, semaglutide has been reported to access the brain through interactions with certain ventricular sites and circumventricular organs, and can directly access the brainstem, hypothalamus, and septal nucleus (98). In separate studies involving the administration of exendin-4 (in the form of sustained-release exenatide as Bydureon®) to humans with PD (13) and as a twice daily immediate release form (BID Byetta®) in AD (99), cerebrospinal fluid (CSF) concentrations of exendin-4 were approximately 2.1% and 1.4% of concomitant plasma levels, respectively. In this regard, the maintenance of long-term steady-state plasma levels of exendin-4, as achieved by sustained-release PT320/PT302 in preclinical studies or by pump where peptide plasma and CSF levels were quantified, results in greater levels in CSF and a higher CSF/plasma ratio than the immediate release exendin-4 form (100, 101).
Several dual GLP-1R/GIPR agonists were also evaluated in the study by Banks and colleagues (97), and the non-acylated and non-PEGylated dual agonists Peptide 17 and Hölscher peptides DA3-CH and DA-JC4 were found to cross the BBB, with exendin-4 and DA-JC4 exhibiting the best BBB penetration of the peptides assessed (97). This is not to say that PEGylated GLP-1R ligands cannot cross the BBB—novel GLP-1R agonist NLY01 is a PEGylated formulation reported to be effective in treating a mouse model of multiple sclerosis (MS) (102) and of PD (103) and is currently in a phase 2 clinical trial for PD treatment (104) (Table 1), highlighting the drug’s promise for potentially treating neurological disorders. In contrast, recent studies by Hölscher and colleagues (105) indicate that the brain delivery of NLY01 is very low, far less than that of exendin-4 and dual GLP-1R/GIPR dual agonists. This is likely consequent to NLY01’s attachment to a 40 kDa PEGylation to extend its systemic half-life but simultaneously making it the size of a small protein (with a consequentially lower BBB permeability than far smaller peptides such as GLP-1 (molecular mass 3,298.7 Da) and Exenatide (4,186.6 Da)). With the many variations of GLP-1R-based drugs on the market and in development, BBB penetration is a paramount consideration for treatment of neurodegenerative diseases and injuries (5).
2. Neuroinflammation Overview
2.1. Neuroinflammation
Although inflammation is part of the body’s healthy response to pathogens and injury, excessive inflammation can have detrimental health effects. In the brain, neuroinflammation is the product of cytokine, chemokine, reactive oxygen species (ROS), and second messenger signaling, involving interactions between neurons, microglia, and astrocytes (Figure 2) (106, 107). Generally, in response to a physiological or environmental trigger (for example, pathogens/pathogen-associated molecular patterns (PAMPs), infection, neurotoxins, neuronal damage, or injury), resting microglia (i.e., in their physiological state) are activated (106) (Figure 2A, B). Microglia exist in context-dependent activation states. In a healthy brain, microglia are homeostatic or quiescent when not activated by a stimulus and patrol for aberrant signaling and other cues of damage; when an insult is detected, microglia are activated into a pro-inflammatory state (12) and express a spectrum of associated phenotypes (Figure 2B). In the literature, pro-inflammatory microglia are often termed “M1” as opposed to “M2” microglia, which comprise a subtype of activated microglia (commonly referred to as “alternatively activated” or “anti-inflammatory”) that secrete anti-inflammatory cytokines and exhibit other reparative functions; however, it is important to note that the “M1” and “M2” terms are falling out of favor due to growing evidence of a spectrum of microglial phenotypes (108). M1 microglia release inflammatory molecules, ROS, chemokines (including C-X-C motif chemokine ligand 1 (CXCL1), C-C motif chemokine ligand 1 (CCL1), and CCL5), prostaglandin E2 (PGE2), and cytokines (including interleukin (IL)-1 beta (IL-1β) and tumor necrosis factor (TNF)-alpha (TNF-α)); which, when excessive, can damage neurons by activating intracellular inflammatory pathways involving nuclear factor kappa-light-chain-enhancer of activated B cells (NF-κB), cyclooxygenases (COX) 1 and 2, and ROS, among other signaling molecules (109, 110) (Figure 2D). In response, the damaged and dying neurons release damage-associated molecular patterns (DAMPs), adenosine triphosphate (ATP), cell debris, neuroinflammatory cytokines, and microglial activators, further activating microglia and continuing the inflammatory cycle (80, 109) (Figure 2H). DAMPs include myelin sheath fragments, tau, amyloid-β, α-synuclein, advanced glycation end products (AGEs), and neuron-specific enolase (80). Astrocytes may exacerbate or ameliorate neuroinflammation depending on the signals present: interferon gamma (IFNγ), transforming growth factor β (TGF-β), signal transducer and activator of transcription 3 (STAT3), glycoprotein gp130, Fas ligand (FasL), and brain-derived neurotrophic factor (BDNF) stimulate protective pathways, but sphingolipids, IL-17, NF-κB, tropomyosin receptor kinase B (TrkB), suppressor of cytokine signaling 3 (SOCS3), vascular endothelial growth factor (VEGF), and chemokines can trigger harmful pathways that worsen inflammation (111). CNS disease or injury can induce astrocytes to convert to their reactive form (112) through a process termed reactive astrogliosis (113) (Figure 2F). More specifically, M1 microglia induce astrocytes to convert to their reactive state through the release of cytokines, particularly IL-1α, TNF-α, and complement component 1, subcomponent q (C1q) (112, 114) (Figure 2E). Reactive astrocytes are neurotoxic and can potentially destroy neurons and oligodendrocytes through release of saturated lipids in apolipoprotein (APO) E and APOJ lipoparticles (115) (Figure 2G). This subtype of astrocytes, previously termed “A1” reactive astrocytes, have been identified in human Huntington’s disease, amyotrophic lateral sclerosis (ALS), AD, PD, and MS, with common gene markers of these cells becoming significantly upregulated in several brain regions (112). This “A1” state is typified by inflammatory transcriptional responses, a downregulation of phagocytic function, and neurotoxic activity that likely involve activation of toll-like receptor (TLR) 4 on the cell membrane and the downstream triggering of the inhibitor of κB kinase (IKK)-NF-κB and mitogen-activated protein kinase (MAPK) signaling pathways. Even in the absence of disease states, normal aging induces A1 astrocyte formation (116). Although neuroinflammation involves an incredibly complex interplay between stimuli (context dependent cascades), cell types, and organism health, we have briefly overviewed several key neuroinflammatory mechanisms in the following sections.
Figure 2.
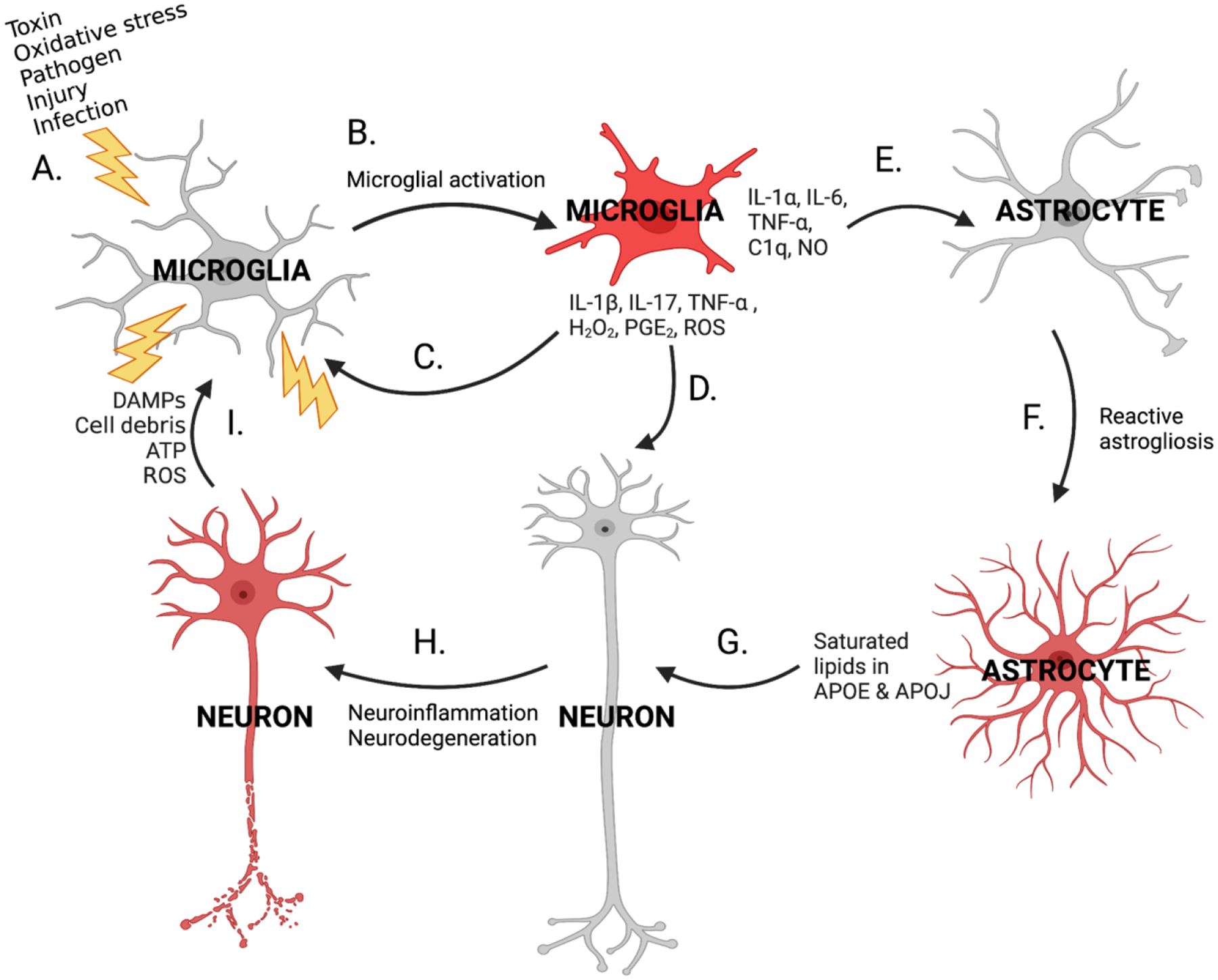
Cycle of neuroinflammation. Various cellular stressors (A) can activate microglia (B), which release cytokines, chemokines, and other pro-inflammatory molecules onto neurons (D) and astrocytes (E). These pro-inflammatory molecules can also bind to microglia in an autocrine fashion, triggering activation of additional microglia and further inflammatory signaling (C). Intracellular neuroinflammatory pathways in astrocytes and neurons are stimulated in response. As a result, a subpopulation of astrocytes undergoes reactive astrogliosis and convert to their neurotoxic reactive form (F) and release APOE and APOJ particles containing harmful saturated lipids onto neurons (G), producing further neuroinflammation and neurodegeneration (H). Damaged neurons release DAMPs, cell debris, ATP, and ROS and activate additional microglia (I), recommencing and amplifying the cycle of neuroinflammation.
2.2. NF-κB signaling
Microglia initiate the first stages of neuroinflammation through the activation of NF-κB, a Rel family transcription factor. There are 5 members of the NF-κB family; 2 subunits, alike or different, may dimerize, translocate to the nucleus, and act as a transcription factor for a wide variety of inflammation-related genes (109). In the cytoplasm, NF-κB is inhibited by its interaction with inhibitor of κB (IκB)—only free NF-κB may translocate into the nucleus (117). The canonical NF-κB signaling pathway is initiated by a pro-inflammatory molecule such as IL-1, lipopolysaccharide (LPS), or TNF-α forming rimmers and binding to its respective receptor on the cell. This induces a signaling cascade that culminates in the inhibitory phosphorylation of IκB by the IKK complex (117, 118). Phosphorylated IκB is ubiquitinated and subsequently degraded, thereby freeing NF-κB for nuclear translocation (117, 118). In this manner, NF-κB is activated by cellular stresses (such as oxidative stress, cytokines, toxins, and carcinogens). Activation of receptors belonging to the TNF superfamily induces the noncanonical pathway of NF-κB signaling, in addition to the aforementioned canonical pathway (117, 118). Following stimulation of the noncanonical pathway, NF-κB inducing kinase (NIK) is freed from its inhibitory interactions with TNF receptor-associated factors (TRAF) 2 and 3 and is thus stabilized; NIK accumulates and induces a signaling cascade through IKKα homodimers that allows a different NF-κB dimer to freely translocate into the nucleus (118). Once in the nucleus, dimerized NF-κB binds to κB elements on gene promoters, which augments the expression of downstream genes for various enzymes (such as COX-1 and COX-2, implicated in neuroinflammation and neurodegeneration), adhesion molecules, chemokines, and cytokines involved in the inflammatory response (119).
2.3. Pro-inflammatory cytokines
Pro-inflammatory cytokines, such as TNF-α and IL-1β, have important roles in neuroinflammation and apoptosis. Other cytokines are anti-inflammatory and counter these inflammatory responses to provide homeostatic regulatory control. A large imbalance between pro- and anti-inflammatory cytokines is problematic. Pro-inflammatory cytokine TNF-α activates various receptors that induce apoptosis, including TNF receptors (TNFR) 1 and 2 and cluster of differentiation (CD) 95, whereas IL-1β activates IL-1 receptor (IL-1R) which induces signaling pathways leading to inflammation, excitotoxicity, and neurodegeneration (109, 120). Various TNF-α inhibitors have been examined in rodent models of CNS diseases and were found to improve neuronal survival, minimize neuroinflammation, maintain synapses, reduce apoptosis, and enhance cognitive function (121–125), implying detrimental effects of excessive TNF-α activity on CNS health and neuronal survival. IL-1β injections in the striatum of rats increased adhesion molecule expression, broke down the BBB, and promoted diffusion of toxic nitric oxide (NO) (126). Additionally, chronic neuroinflammation, activation of microglia, impaired cognitive function, and reduced neurogenesis were observed in mice engineered to overexpress IL-1β (127). In humans, elevated levels of these pro-inflammatory cytokines have been linked with various neurological disorders, including AD, PD (128, 129), stroke (130), TBI (12), and a host of psychiatric disorders that include major depressive disorder, generalized anxiety disorder, post-traumatic stress disorder, bipolar disorder, and schizophrenia (131–133). Furthermore, pro-inflammatory cytokines increase production of inducible NO synthase (iNOS), which in turn elevates levels of NO. NO exhibits multiple mechanisms of neurotoxicity, including formation of peroxynitrite and resulting DNA damage, glutamate excitotoxicity, and activation of apoptosis signaling cascades (134). In addition to the cytotoxic effects of high concentrations of NO, greater abundance of iNOS and NO allows for enhanced nitrotyrosination of neural proteins, a common feature of neurodegenerative diseases (135, 136).
2.4. ROS
Additional consequences of neuroinflammation include DNA damage, greater oxidative stress, and increases in ROS. Conversely, neuroinflammation can be a consequence of ROS. ROS are produced by mitochondrial dysfunction (137), and frequently observed in neurodegenerative disease (138–141). Oxidative stress and ROS co-occur with neuroinflammation, AD, and T2DM (142). Ferroptosis, an iron-dependent form of cell death resulting from excess accumulation of intracellular lipids peroxidation, is linked to increased neuroinflammation, and is associated with several neurological disorders (143). ROS activate NF-κB and thus exacerbate neuroinflammatory gene and subsequent protein expression (144). Antioxidant treatments, such as N-acetyl cysteine (NAC), have demonstrated anti-ROS and anti-inflammatory capacity across a wide range of animal models of acute brain injury (TBI and stroke) and resulted in improved symptoms of military personnel who suffered a TBI in the Iraq war theater (145, 146).
2.5. Pericyte loss
In addition to neurons, astrocytes, and microglia, pericytes are a fourth cell type with evidence of GLP-1R expression and demonstrated roles in regulating neuroinflammation and neurodegeneration (147, 148). Pericytes are contractile cells found on capillaries that confer several important functions, including blood flow control through constriction and dilation of capillaries, BBB integrity through regulation of BBB protein expression, regulation of leukocyte entry into the brain, angiogenesis, and CNS injury responses (149–151). Loss or dysfunction of pericytes has been repeatedly associated with BBB damage, consequent neurodegeneration, and several CNS diseases (149, 152–156). For instance, capillary abnormalities and reduced cerebral blood flow are known characteristics of AD and are linked with pericyte dysfunction (149, 157, 158). Pericyte activity is a significant component in regulating neuroinflammation and neurodegenerative disease pathology, yet few studies examine the therapeutic potential of GLP-1R signaling in pericytes, highlighting a need for future research in this realm.
2.6. Neuroinflammation in CNS disease
Neuroinflammation can exacerbate neurodegenerative disorders, and neurodegeneration routinely feeds further neuroinflammation (159) (Figure 2). Although healthy aging can naturally stimulate neuroinflammatory signaling in the brain (160, 161), AD patients exhibit notably higher levels of neuroinflammation (128, 162). Chronic neuroinflammation can impair neuroplasticity and cognition, damage neurons, and culminate in neurodegenerative diseases epitomized by AD (106) (Figure 2H). AD is the most common neurodegenerative disorder and is identified by the accumulation of amyloid-β plaques and neurofibrillary tau tangles, which form when tau protein becomes hyperphosphorylated (163). Microglial exposure to these plaques elevates pro-inflammatory cytokine production (Figure 2A, B, C, D, E), further aggravating the disease and contributing to hippocampal dysfunction and associated memory problems in patients (164). Neurotoxic amyloid-β is produced when amyloid precursor protein (APP) is disproportionally cleaved by β-site APP cleaving enzyme 1 (BACE1) (163). Amyloid-β deposits in the AD brain activate microglia through binding to TLR4 and 6 and CD36 receptors (Figure 2A, B), initiating release of pro-inflammatory cytokines and chemokines that can then damage neurons (Figure 2D); the neuronal debris itself can also activate microglia (Figure 2C), perpetuating a cycle of chronic neuroinflammation and neurodegeneration (165) (Figure 2H). Furthermore, mass cytokine release also may disrupt microglial clearance of extracellular amyloid-β deposits by reducing the number of amyloid-β receptors expressed on the microglial cell surface (166). Transgenic mouse AD models exhibit increased markers of neuroinflammation relative to healthy controls, including elevated brain levels of IFNγ and IL-1β (166–168). Recent research proposes that treatments targeting neuroinflammation, particularly through reduction of TNF-α activity and resulting reduction in activated microglial activity and astrocyte reactivity, may outperform treatments targeting amyloid-β pathology in AD clinical trials (114). Although the amyloid-β hypothesis has persisted as a main contributor to AD pathology and morbidity, anti-amyloid therapies have not proven effective in preventing disrupted cognition (169). A recent GWAS examining common genetic features of AD has identified neuroinflammatory pathways (mainly associated with TNF) to be a key feature of AD (1), offering perhaps a more viable target for treating the disease.
PD is the second most common neurodegenerative disorder and features characteristic neuroinflammation and dopaminergic degeneration in the substantia nigra region of the midbrain—neuroinflammation exacerbates PD progression through interactions with PD-linked gene products, mitochondrial dysfunction, and oxidative stress (170). Drugs targeting inflammatory pathways have been proposed as treatments to slow PD progression (171). In the PD brain, increased pro-inflammatory cytokine levels, microglial activation, ROS, and elevated α-synuclein in neurons are indicative of a neuroinflammatory environment (5). This neuroinflammation aggravates pre-existing noxious genetic factors and contributes to degeneration of dopaminergic neurons in PD (5, 171).
3. GLP-1R Signaling and Neuroinflammation
3.1. Brain IR and neuroinflammation
In the brain, IR exacerbates neuroinflammation (Figure 3). IR produces chronically elevated blood sugar levels and an unhealthy imbalance of lipids in the blood (Figure 3A), conditions which can damage and increase the permeability of the BBB (48) (Figure 3B). In response to BBB damage, free fatty acids, and high blood glucose (Figure 3C), microglia are activated into a pro-inflammatory state (Figure 3D), which can lead to cytokine release (Figure 3E) and downstream neuroinflammatory cascades (Figure 3F) and reactive astrogliosis (Figure 3G) (48). In turn, neuroinflammation further damages the BBB, making it increasingly permeable (172)—this enhanced permeability elevates the already-high blood glucose concentrations in the brain and contributes to a vicious cycle of neuroinflammation as a result of hyperglycemia (173) (Figure 3H). Other neuroinflammatory consequences of brain IR include vascular dysfunction (which has been linked to dementia accompanying neurodegenerative disease (174)), greater NO production, oxidative stress, and consequent neuroinflammation (99). Additionally, chronic hyperglycemia and IR worsen neuroinflammation by stimulating excessive mitochondrial respiration in the brain, which leads to increased production of ROS and downstream activation of NF-κB, cytokine production, and corresponding neuroinflammatory signaling cascades (172).
Figure 3.
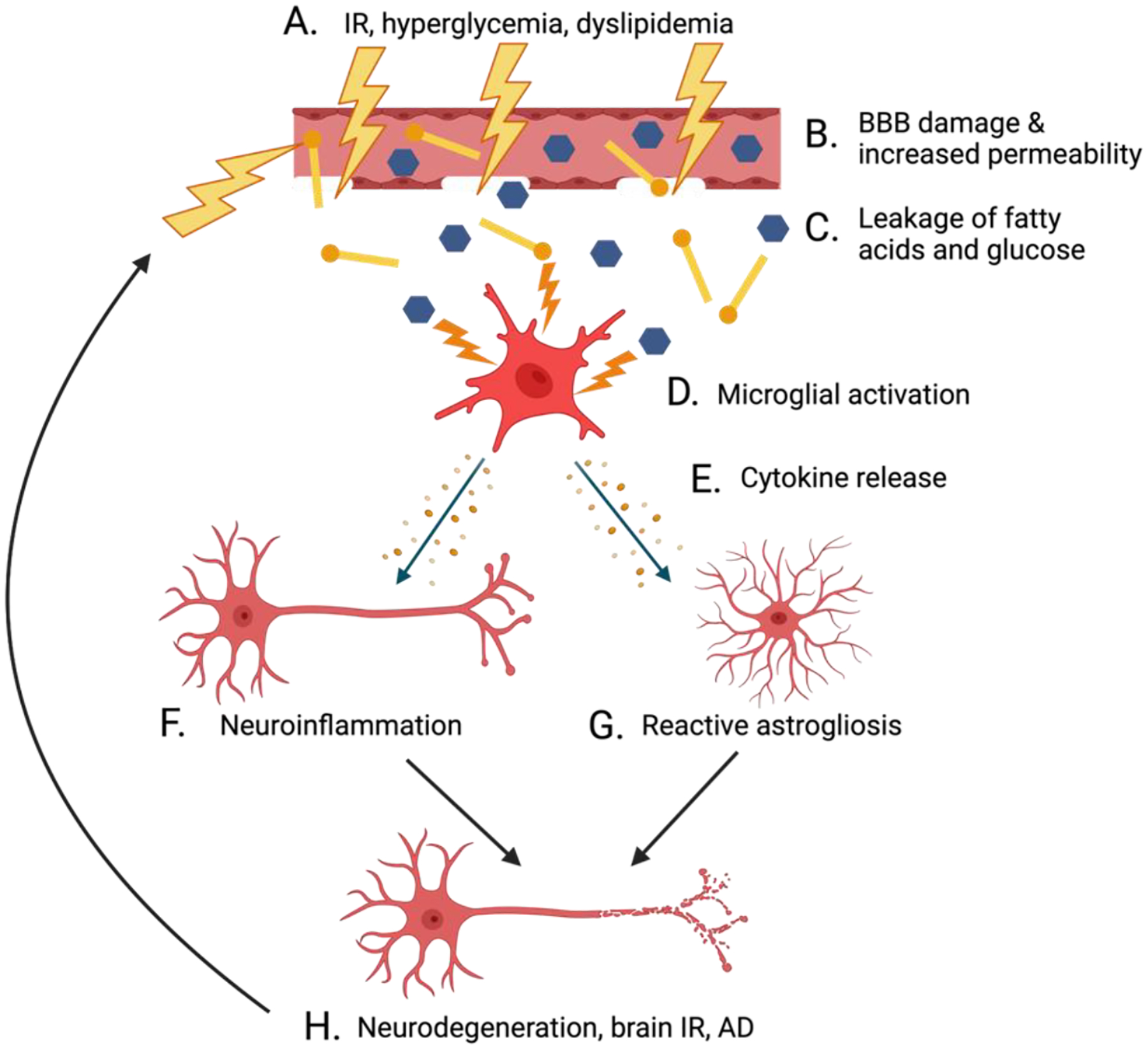
Interactions between IR, neuroinflammation, and neurodegenerative disease. Elevated blood sugar levels and dyslipidemia in the blood are consequences of IR (A) that can damage and increase permeability of the BBB (B). BBB damage results in the infiltration of the brain with free fatty acids and excessive glucose (C), thus activating microglia into a pro-inflammatory state (D). These activated microglia release cytokines (E) that stimulate neuroinflammation (F) and reactive astrogliosis (G). Chronic neuroinflammation fuels the development of brain IR, neurodegeneration, and the progression of AD (H).
3.2. Brain IR, neuroinflammation, and AD
Brain IR is a risk factor for neurodegenerative diseases such as AD as well as motor disorders such as multiple system atrophy (formerly known as Shy-Drager syndrome), a sporadic and progressive neurodegenerative condition commonly identified by motor abnormalities (ataxia and parkinsonism) and autonomic dysfunction (175), and PD (176–179). Several studies using animal models provide strong evidence that IR and neuroinflammation feed AD neuropathology (167, 180–182). For example, in a double transgenic mouse model of AD (APP/PS1), brain levels of pro-inflammatory cytokines (IFNγ, IL-1β, and IL-4), reactive astrocytes, and IRS-1 phosphorylated at serine 616 (a marker of IR (99, 178)) were raised, suggesting that neuroinflammation and IR interact in the AD brain (167). Other studies in transgenic mouse models of AD (5xFAD; APN−/− and APP23) have found that disruptions in insulin signaling increase microglial activation, neuroinflammatory signaling, cognitive deficits, and AD neuropathology (181, 182). Brain IR also promotes the hyperphosphorylation of tau, leading to the formation of tau neurofibrillary tangles characteristic of AD (99). Conversely, certain neurobiological characteristics of AD can aggravate IR and neuroinflammation. Amyloid-β oligomers stimulate serine phosphorylation of IRS-1, thus worsening brain IR, and a feed-forward cycle of IR and amyloid-β plaque formation ensues (99). Moreover, solubilized amyloid-β can trigger pro-inflammatory signaling cascades that amplify vascular inflammation and vasoconstriction (183). The unhealthy interactions between IR, inflammatory signaling cascades, amyloid-β deposition, endothelial dysfunction, and tau yperphosphorylation largely contribute to the progression of neuroinflammation and neurodegeneration in AD in both animal models and humans (66, 184).
3.3. GLP-1R agonists as treatment for neurodegeneration
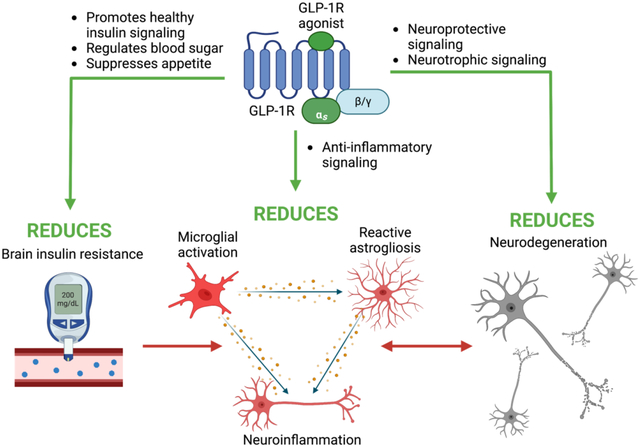
Drugs that target brain IR by promoting healthy incretin and insulin signaling are a promising research direction for the treatment of neurodegenerative disease (185–188). GLP-1R has evidenced expression in neurons, microglia, and astrocytes across key brain regions (10) (Figure 4: right panel) and GLP-1R agonists have demonstrated neuroprotective and anti-inflammatory properties (189–191). GLP-1R and other secretin receptors GIPR and GcgR signal via multiple anti-inflammatory pathways (192, 193) that could be stimulated to reduce neuroinflammation and ameliorate conditions of neurodegenerative diseases (Figure 4: left panel). Ligand binding-induced activation of the Gαs subunit of a secretin receptor’s associated G protein stimulates adenylyl cyclase and consequently elevates cyclic adenosine monophosphate (cAMP) levels. cAMP activates exchange protein activated by cAMP (Epac) which has downstream anti-inflammatory and antiapoptotic effects (194, 195). cAMP also stimulates protein kinase A (PKA), which stimulates MAPK/ERK anti-inflammatory signaling and phosphorylates and activates the transcription factor cAMP response element (CRE)-binding protein (CREB) (196, 197). Another signaling pathway, the phosphatidylinositol-3 kinase (PI3K)/Akt pathway, likewise amplifies CREB signaling (198). CREB binds to CRE promoters and enhances expression of downstream genes, several of which are anti-inflammatory and neuroprotective, such as B-cell lymphoma 2 (Bcl-2) and B-cell lymphoma-extra large (Bcl-xL) (199). Bcl-2 and Bcl-xL are antiapoptotic molecules that inhibit the activity of pro-apoptotic proteins including Bcl-2 associated agonist of cell death (BAD), p53, and caspases (12, 199, 200). CREB also activates apurinic/apyrimidinic endonuclease 1 (APE1), with downstream effects of ameliorating oxidative stress-associated DNA damage (201). Notable in Figure 4 is that key signaling proteins, particularly Akt, act as a control center and thereby are master regulators of downstream biochemical cascades that can impact cell survival, metabolism, protein homeostasis, and inflammation—these proteins are a central hub of both insulin and GLP-1 signaling.
Figure 4.
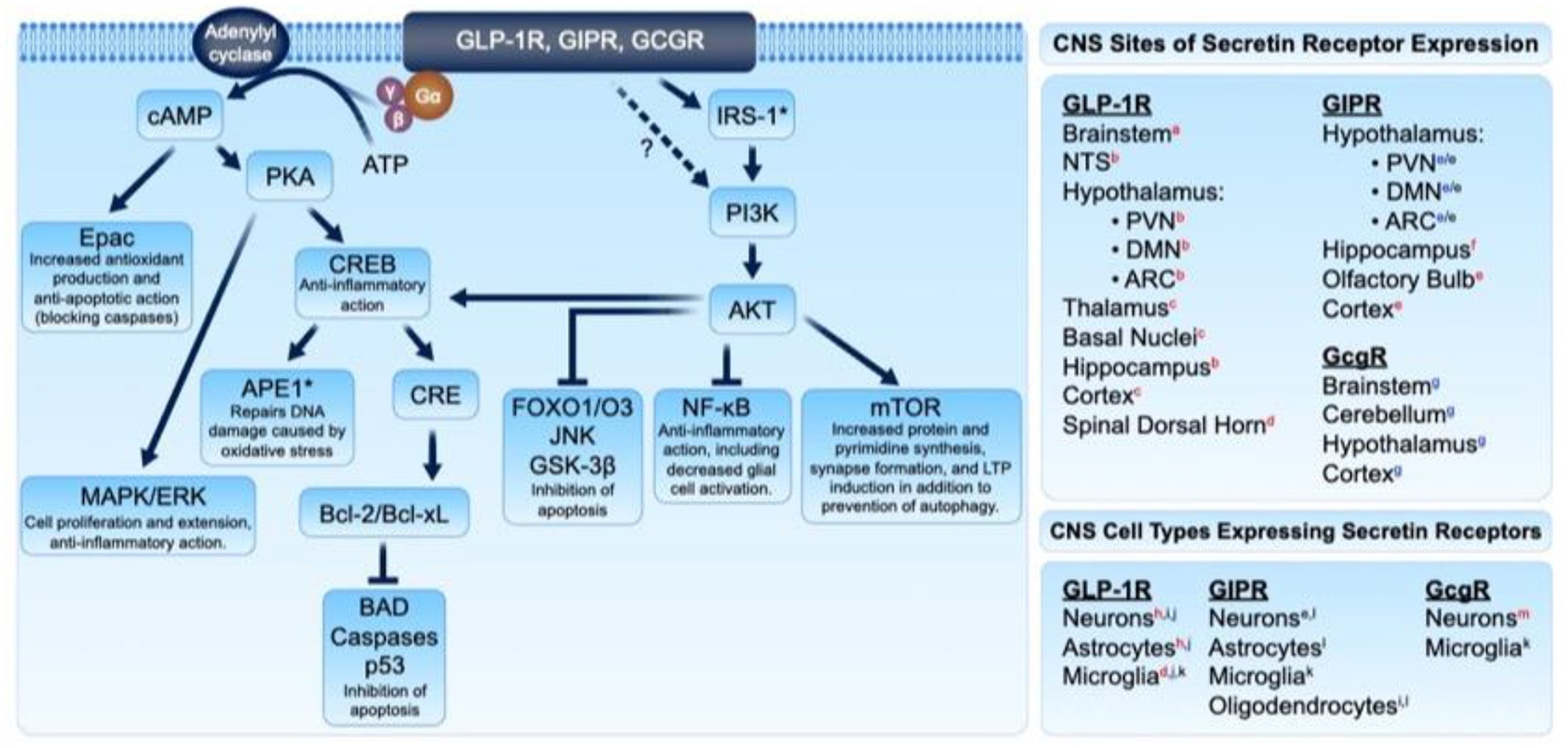
Anti-inflammatory mechanisms of GLP-1R/GIPR/GcgR signaling, and sites of receptor expression as evidenced in rats (red superscript), mice (blue superscript), and humans (black superscript). Signaling pathways downstream of secretin receptor activation minimize neuroinflammation, oxidative stress, and apoptosis and provide cytoprotective effects. Sources: (a.) ref(208); (b.) ref(77); (c.) ref(209); (d.) ref(210); (e.) ref(78); (f.) ref(81); (g.) ref(211); (h.) ref(212); (i.) ref(213); (j.) ref(214); (k.) ref(215); (l.) ref(216); (m.) ref(217). Figure adapted from Glotfelty et al. 2019 (12).
The relevance of Akt in PD development is backed by reports of selective loss of dopaminergic neurons associated with a decreased expression of Akt pS473 in the PD brain (202, 203). Additionally, PD risk factor genes, such as PINK1, appear to converge and interact with Akt; while inhibition of this cascade promotes neurodegeneration (204, 205). The site of action of incretin receptor activation on PI3K/Akt signaling has not been definitively elucidated but could be downstream of IRS-1 activation by incretin signaling (12). In addition to CREB regulation, the PI3K/Akt pathway also inhibits NF-κB inflammatory signaling; inhibits Forkhead box protein O1/O3 (FOXO1/O3)-, Jun N-terminal kinase (JNK)-, and glycogen synthase kinase-3β (GSK-3β)-mediated apoptotic signaling; and stimulates mammalian target of rapamycin (mTOR) neuroprotective signaling (12, 206, 207). As in Figure 4 and above, GLP-1R stimulation as well as insulin also trigger the MAPK pathway. This is comprised of three primary arms, the JNKs, the ERKs, and p38 kinases that, together, can either augment survival and proliferation or engender stress and apoptosis, depending on the cell type and stimulus. With considerable cross talk and feedback between these pathways, these potentially protective signaling cascades downstream of GLP-1R, GIPR, and GcgR indicate the great potential in targeting these receptors for anti-inflammatory and anti-neurodegenerative drug treatments.
Numerous GLP-1R, GIPR, and/or GcgR agonists have been developed for the treatment of T2DM and obesity and have the potential to be repurposed for the treatment of neurodegenerative diseases (Table 1). GLP-1R agonists have been tested in preclinical and clinical studies to investigate whether restoring brain insulin signaling is a viable treatment option for neuroinflammation and corresponding neurodegeneration.
3.4. GLP-1R agonism reduces neuroinflammation in preclinical studies
The link between IR and neuroinflammation is highlighted by a multitude of cell culture and animal studies that utilize various incretin receptor-stimulating treatments to maintain healthy insulin signaling and reduce markers of neuroinflammation and neurodegeneration. Such preclinical studies are briefly overviewed in the following sections.
GLP-1
In the brain, GLP-1R stimulation by GLP-1 is associated with neuroprotective benefits to the CNS. Research in SH-SY5Y human neuroblastoma cells found that GLP-1 treatment promoted neuronal viability through anti-apoptotic, anti-oxidative, and neurotrophic mechanisms, involving elevated PKA and PI3K pathway activity, decreased expression of apoptotic factors, and increased expression of anti-apoptotic factors (87, 191). Furthermore, GLP-1 activity has been demonstrated to have anti-neuroinflammatory and anti-neurodegenerative effects in preclinical model systems. Cultured rat primary hippocampal neurons treated with GLP-1 were protected against amyloid-β-induced neurotoxicity (218). In mice pretreated with LPS to simulate AD-like neuroinflammation, GLP-1-increasing treatments reversed inflammation-induced synaptic impairments in the hippocampus (219); reduced amyloid-β deposition, inflammatory glial activation, and expression of inflammatory molecules COX-2, TNF-α, IL-1β, and TLR4 (220); and inhibited activity along the inflammatory NF-κB/TLR4 and Akt/GSK-3β signaling pathways (221). Moreover, increasing GLP-1 levels in mice pretreated with LPS reduced memory impairment (221) and improved cognitive performance (219); in mice pretreated with 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine (MPTP) to simulate motor dysfunction characteristic of PD, GLP-1 augmentation improved motor abilities (221). The potential for GLP-1 to improve AD and PD pathology was reinforced in experiments using primary mouse mixed neuronal cultures challenged with amyloid-β (modeling AD) or α-synuclein (modeling PD), in which GLP-1 metabolite (9–36) enhanced cell survival and decreased M1-like microglia numbers (191). Additionally, the metabolite reduced IL-6 and TNF-α levels secreted by LPS challenged human HMC3 and mouse IMG microglial cells lines (191). Hence, multiple studies have independently demonstrated that GLP-1R stimulation by GLP-1 and, more recently, by its primary metabolite GLP-1(9–36) (191) are anti-inflammatory and neuroprotective.
Exendin-4/Exenatide
Neuroprotective and neurotrophic effects of exendin-4 treatment have been observed in mouse models of various neural diseases, including middle cerebral artery occlusion-induced stroke (4), ALS in SOD1 G93A mutant mice (222), and T2DM-related neuropathies induced by pyroxidine (223). Furthermore, a study in cultured SH-SY5Y human neuroblastoma cells demonstrated greater neuronal viability and proliferation with exendin-4 treatment (87). Exendin-4 treatment has been found to exhibit significant anti-inflammatory effects as well, especially in the context of neurodegenerative disease—exendin-4 treatment in cultured primary rat astrocytes (224) and neurons (225) reduced amyloid-β-induced oxidative stress, cytotoxicity, and neuroinflammation. In transgenic mouse models of AD (5xFAD and 3xTg-AD) as well as in human cortical neurons obtained post-mortem, GLP-1R stimulation using an engineered exendin-4 treatment reduced amyloid-β-induced microglial activation, thus limiting neuroinflammation; inhibited reactive astrogliosis through the reduction of inducers TNF-α, C1q, and IL-1α; and improved neuronal viability (226). Similar effects, using the same GLP-1 mimetic, were demonstrated in a mouse PD model (103). Furthermore, GLP-1R activation through exendin-4 treatment was found to improve recognition memory impairment by dampening signaling along the AMPK/NF-κB pathway, reducing levels of neuroinflammatory cytokines IL-1β, IL-1β p17, and TNF-α, and increasing synaptic protein levels in mice with spared nerve injury modeling neuropathic pain (227). Markers of AD (amyloid-β deposition, abnormal glycoprotein glycan expression, and cognitive and memory impairments) and of brain IR (IRS-1 serine phosphorylation and elevated blood glucose levels) were reduced with exendin-4 treatment in transgenic mouse models of AD (225, 228, 229). With relevance to PD, MPTP and 6-OHDA mouse models of PD treated with exendin-4 exhibited enhanced motor function and dopamine signaling and minimized neurodegeneration (4, 101).
Exenatide, a synthetic version of exendin-4, exhibits similar anti-neuroinflammatory and anti-neurodegenerative benefits in preclinical models. In a 3xTg-AD mouse model, exenatide treatment reversed high fat diet-induced impairments of BDNF signaling and reduced levels of NF-κB and peroxisome proliferator-activated receptor proteins (PPARs) α and γ, indicating reduced inflammation; however, there was no significant effect on systemic metabolism nor cognitive performance in response to exenatide treatment in this experiment (230). Exenatide has also been found to combat neuroinflammation and AD as indicated by reduced ‘nucleotide-binding oligomerization domain, leucine-rich repeat, and pyrin domain containing 2’ (NLRP2; a component of the inflammasome in astrocytes) levels in the piriform cortex, reduced amyloid-β deposition, and improved cognition in 5xFAD transgenic mice modeling AD (224). PD mice respond to exenatide treatment as well—6-OHDA and MitoPark PD mouse models treated with a sustained release exenatide formulation (PT320) experienced neuroprotective effects of the drug, including improved motor function (231) and enhanced dopamine signaling activity in midbrain networks (232). Additionally notable in these two PD preclinical studies was an exenatide-mediated reduction in L-3,4-dihydroxyphenylalanine (L-DOPA)-induced abnormal involuntary movements (AIMs) in the rat 6-OHDA model and a slowing of disease progression, dopamine loss, and motor deficits in the MitoPark mouse when administered a clinically translatable drug dose (231).
Liraglutide
Liraglutide is structurally similar to GLP-1; however, its acylation (supporting binding to serum albumin) increases the peptide’s half-life by slowing absorption and evading recognition by DPP-IV (233). Although FDA-approved for the treatment of T2DM, research indicates that liraglutide may provide neuroprotective effects. For example, in cultured SH-SY5Y human neuroblastoma cells, liraglutide pretreatment decreased oxidative stress and promoted neuroprotection and neurotrophy, likely through a cAMP-dependent PKA/CREB signaling pathway (234). Results of preclinical studies of liraglutide have important implications for treating AD and neuroinflammation. Interestingly, a 2016 study reported no significant effect of liraglutide treatment in reducing amyloid-β plaque load in transgenic APP/PS1 mouse models (235). This could potentially be due to relatively poor BBB penetration of liraglutide and highlights the need for the research into the development of GLP-1R-stimulating drugs that can measurably reach the brain from the periphery. However, other research has found that liraglutide appears capable of crossing the BBB in pharmacologically relevant amounts and acting on GLP-1R in the CNS (236). More recently, in a study using a streptozotocin (STZ)-induced insulin resistant and a 5xFAD mouse model, liraglutide treatment reversed markers of neuroinflammation (particularly astrocyte reactivity and microglial activity in the cortex and hippocampus) and amyloid-β plaque deposition (237). Furthermore, liraglutide pretreatment in human SH-SY5Y neuroblastoma cell culture protected against the apoptotic effects of okadaic acid and limited tau activation and BACE1 expression, and in a rat model of okadaic acid-induced AD, liraglutide improved rat memory function and cognition and reduced neuronal apoptosis, tau phosphorylation, and BACE1 levels (163). These more recent studies suggest that liraglutide may be efficacious in improving certain markers of neuroinflammation and AD when administered in a sufficiently high dose. However, since relatively few studies evaluate plasma drug levels, how drug doses selected for evaluation in preclinical studies relate to those safely tolerated in and translatable to humans remains unknown.
Lixisenatide
Fewer studies have investigated neuroprotective effects of lixisenatide, a long-lasting GLP-1R agonist confirmed to be capable of measurably crossing the BBB and exerting neuroprotective effects (236). These neuroprotective effects of lixisenatide treatment include reductions in amyloid-β plaques, tau neurofibrillary tangles, and neuroinflammation (measured by microglial activation in the hippocampus), as demonstrated by a study utilizing an APP/PS1/tau mouse model of AD (238). These effects were a result of augmented PKA/CREB pathway and inhibited p38/MAPK pathway signaling due to GLP-1R stimulation (238). Lixisenatide has also been found to enhance LTP in the hippocampus, improve working memory, maintain synapses, reduce amyloid-β plaque levels, and combat neuroinflammation in AD rodent models (APPswe/PS1ΔE9 mice and rats) (239–241). Furthermore, in a MPTP PD mouse model, lixisenatide treatment was associated with neuroprotective benefits and reduced PD-related motor impairment (242).
DPP-IV Inhibitors
Although not GLP-1R agonists, DPP-IV inhibitors block the action of DPP-IV and therefore reduce the rate of breakdown of GLP-1 and GIP, consequently raising levels of endogenous incretins and promoting healthy incretin hormone signaling. Numerous DPP-IV inhibitors are FDA-approved for the treatment of T2DM, and DPP-IV inhibition has demonstrated efficacy in reducing neuroinflammation and AD markers—DPP-IV inhibition in an STZ-induced rat model of AD elevated GLP-1 levels and decreased levels of amyloid-β, total tau, phosphorylated tau, and pro-inflammatory cytokines TNF-α and IL-1β in a dose-dependent manner (243). Furthermore, treatment of Zucker diabetic fatty (ZDF) rats with the DPP-IV inhibitor alogliptin raised the expression of CREB target genes and decreased blood glucose in the hippocampus, thus dampening the neuroinflammatory response (244). Anti-neuroinflammatory effects were also observed with DPP-IV inhibitor sitagliptin, which significantly quelled the neuroinflammatory response in various rodent models by reducing levels of the pro-inflammatory molecules TNF-α, IL-6, IL-17, and CD163 and by raising levels of the anti-inflammatory molecules TGF-β and IL-10 (207). With regard to neurodegenerative disease treatment, sitagliptin has also been found to reduce amyloid-β deposition, have antiapoptotic and antioxidative properties, and improve scores on mini-mental state exam (MMSE) tests for dementia of elderly people with and without AD (207). Linagliptin, another DPP-IV inhibitor, was found to increase levels of GLP-1 and GIP in the brain; reduce amyloid-β, tau phosphorylation, and neuroinflammation (indicated by reduced glial fibrillary acidic protein (GFAP) immunoreactivity); and improve cognitive deficits of AD in a 3xTg-AD mouse model (245). Similarly, DPP-IV inhibitor Gramcyclin A treatment in a recent APP/PS1/tau triple transgenic mouse model of AD stimulated GLP-1R signaling, promoted glucose uptake in the brain, decreased levels of activated microglia and neuroinflammation in the hippocampus, and reduced amyloid-β plaques, soluble amyloid-β–40, and soluble amyloid-β–42 levels (246). As a cautionary note, drug levels were not evaluated in majority of the above studies, and how the selected doses in these preclinical studies relate to those translatable to humans remains unknown.
Dual Agonists
Dual incretin agonists co-stimulate both GLP-1R and GIPR and, in general and when well-designed, have been found to outperform single GLP-1R agonists in ameliorating markers of neuroinflammation and neurodegeneration (247–249). Several GLP-1R/GIPR dual agonists generated by Hölscher and colleagues have been found to have anti-neuroinflammatory and anti-neurodegenerative benefits in preclinical studies (247–250). Treatment of APP/PS1 and APP/PS1/tau mouse models of AD with the Hölscher dual agonist DA-JC4 reduced pro-inflammatory cytokine levels, phosphorylated tau levels, and amyloid-β plaque load; improved memory impairments and mitochondrial volume and numbers; and strengthened synapses and LTP in the hippocampus (249, 250). DA-CH5, another of the Hölscher dual agonists, outperformed liraglutide in reducing neuroinflammation (as evaluated by reduced levels of activated microglia and astrocytes), apoptosis, and oxidative stress in a MPTP mouse model of PD (247). Similarly, DA-JC1 outperformed liraglutide in reducing oxidative stress and ROS, enhancing neuronal viability and neurogenesis, and reducing reactive astrocytes and neuroinflammation, both in an APP/PS1 mouse model of AD and in cultured SH-SY5Y human neuroblastoma cells challenged with hydrogen peroxide (248). Anti-neuroinflammatory benefits have also been observed using the original GLP-1R/GIPR agonist of DiMarchi and colleagues (termed “twincretin”). In cultured SH-SY5Y human neuroblastoma cells, twincretin has been demonstrated to enhance neurotrophic signaling through stimulating the CREB pathway and promote neuronal survival in conditions modeling neuroinflammation (251). Additionally, twincretin’s actions on elevating levels of cAMP (the initial event in the cascade triggered by receptor engagement, binding, and activation) and inducing neurotrophic activity were significantly greater than equimolar concentrations of single GLP-1R or GIPR agonists (251). Dual agonists also offer numerous other neuroprotective benefits including reduced oxidative stress and improved memory function, synaptic health, and neurogenesis in mice (252), as well as neuroprotective and antioxidant properties in rodent models of TBI (12, 100, 251). Interestingly, the human dose of liraglutide (single GLP-1R agonist) and twincretin (dual GLP-1R/GIPR agonist) in T2DM is the same: 1.8 mg subcutaneously daily. In the preclinical evaluation of twincretin in concussive head injury by Bader and colleagues (100), a direct comparison was made to liragultide treatment—a 247.6 μg/kg dose of liraglutide in mouse, which translates to a 1.8 mg dose in an 88.8 kg human, provided similar efficacy to a 5-fold lower dose of twincretin (50 μg/kg) to mitigate TBI-induced neuroinflammation, neuronal loss and behavioral impairments.
Triple Agonists
“Triagonists” are unimolecular peptide drugs that simultaneously target GLP-1R, GIPR, and GcgR. While primarily in development for treating metabolic disease (37), these triple agonists have also been linked with reduced neuroinflammation and neurodegeneration. A novel triple agonist stimulated CREB pathway signaling in hippocampal neurons in a 3xTg-AD mouse model of AD and produced improvements in cognition, working memory, and long-term spatial memory (253). The triple agonist was also found to reduce amyloid-β plaques, neuroinflammation, and oxidative stress and increase neurogenesis, number of synapses, and BDNF expression in APP/PS1 mice (254), indicating a potential for treating AD. In SH-SY5Y human neuroblastoma cells, a GLP-1R/GIPR/GcgR triagonist reduced oxidative stress, enhanced neuronal health and viability, and minimized glutamate excitotoxicity, highlighting the neurotrophic/protective effects of secretin receptor stimulation (255). The ability of the triagonist to elevate cAMP levels was dramatically greater than that of a single GLP-1R agonist on an equimolar basis (255), and selective antagonists for each of the three secretin receptors were required to inhibit the triagonist mediated neurotrophic/protective actions, thereby suggesting balanced pharmacological action across these receptors (255). Moreover, the triagonist significantly reduced TNF-α secretion in primary mouse microglia challenged with LPS (255). Additionally, in a 30-gram weight drop close head injury mouse model of mild concussive TBI, the triagonist improved memory function following injury (255). These findings support the successful translation of neuroprotective and anti-neuroinflammatory properties from cellular to animal studies for this promising drug.
3.5. GLP-1R agonists in human trials
Use of incretin receptor-targeting drugs for the treatment of CNS diseases is further supported by meta-analyses revealing that T2DM patients’ prescribed GLP-1R stimulating medications may promote CNS health. Cohort studies of T2DM patients using GLP-1R stimulating drugs, either through GLP-1R agonism or DPP-IV inhibition, exhibited decreased risk of developing PD (271, 272). Another cohort of T2DM patients taking GLP-1R agonists had a reduced risk of stroke relative to patients who were taking GLP-1R-stimulating drugs (3, 273). These analyses suggest CNS disease modifying capability of drugs that promote GLP-1R signaling. Thus, clinical studies in humans have been proposed and completed to investigate the potential for GLP-1R agonists in treating neurodegenerative diseases (Table 2) and will be overviewed in the following sections.
GLP-1R agonists liraglutide (Victoza®), semaglutide (Ozempic®, Rybelsus®), exenatide (Bydureon®, Byetta®, PT320, NLY01), and lixisenatide (Adlyxin®/Lyxumia®) have been FDA-approved or are in trials for the treatment of T2DM and are currently being investigated to determine their potential efficacy in treating neurodegenerative diseases. Only trials of liraglutide and exenatide have reached completion. In a phase 2 clinical trial investigating liraglutide treatment in patients with AD, 6 months of liraglutide treatment significantly increased glucose transport at the BBB, elevating the cerebral metabolic rate for glucose and reversing the abnormalities in brain glucose transport commonly associated with AD pathology (274). In addition, a human study found that 12 weeks of liraglutide treatment improved brain connectivity but had no measurable effect on cognition (275). Furthermore, this research revealed a negative association between fasting plasma glucose level and bilateral hippocampal and anterior medial frontal connectivity (275), indicating a potential relationship between incretin signaling (regulates plasma glucose) and neural abnormalities. Most recently, a 52-week phase 2 clinical trial of liraglutide in PD patients found that liraglutide treatment was safe and well-tolerated and improved non-motor symptoms, mobility, and quality of life (276). Currently awaited is the peer-reviewed analysis of the Evaluating Liraglutide in Alzheimer’s Disease (ELAD) study, a 12-month, multi-center, randomized, double-blind, placebo-controlled, phase 2b clinical trial of liraglutide in mild to moderate Alzheimer’s dementia (204 patients; NCT1843075) where the primary outcome is to measure changes in cortical glucose metabolic rates during the 12-month liraglutide treatment versus the placebo group (277). In a preliminary correspondence, there was no significant change in this primary marker; however, liraglutide was found to be well-tolerated, and positive numerical outcomes were evident amongst secondary outcome measures that comprise various markers of AD and neuroinflammation, including changes in several cognitive measures, brain volume and connectivity, microglial activation, tau phosphorylation, and amyloid-β levels (278). Another trial that is currently in progress investigates the safety and efficacy of liraglutide in treating patients with PD (279).
At present, exenatide is the most prevalent GLP-1R agonist in clinical trials for AD and PD treatment. In an 18-month phase 2 clinical trial investigating the safety and efficacy of exenatide (administered as BID Byetta®) in treating early AD, extracellular vesicles isolated from the patients exhibited reduced amyloid-β–42 concentrations with exenatide treatment (257). However, in the same study, there were no significant differences between exenatide treatment and placebo in patients’ cognition, cortical thickness and volume, or biomarkers of AD in CSF or plasma (257). This was a small double-blind, randomized, placebo-controlled clinical trial whose primary outcome was to assess safety and tolerability. A total of 18 patients completed the study, and partial outcomes were available on 21 patients prior to the premature termination of the study when the sponsor withdrew provision of drug and matched placebo pens. Exenatide proved to be well-tolerated but the study was underpowered to truly evaluate markers of efficacy and drug action (257). Hence, further research is required to elucidate the effects of exenatide and related drugs in AD patients.
Exenatide may also be efficacious in treating PD—a proof of concept, single-blinded phase 2 clinical trial assessed 45 moderate PD patients randomly delegated to receive subcutaneous exenatide (BID Byetta®) or to act as controls for 12 months. The trial evaluated overnight off-medication motor scores at baseline, time-dependently over the study, and following a 2-month washout period and found a significant exenatide-mediated improvement in the Movement Disorders Society Unified Parkinson’s Disease Rating Scale (MDS-UPDRS) part 3 that persisted through the washout period (258). A follow up of available patients indicated that the exenatide actions endured over 12 months following drug cessation (280). This open label clinical trial spurred a larger double-blind, randomized, placebo-controlled trial by the same group that evaluated once weekly extended-release exenatide (Bydureon®) in moderate PD patients and reported significantly improved off-medication motor scores (13) assessed by MDS-UPDRS part 3, the primary outcome measure. In accompanying studies evaluating the contents of blood sampled, neuronal-derived exosomes demonstrated that the efficacious exenatide interactions were mediated through the brain insulin, Akt, and mTOR signaling pathways in PD patients (281). Moreover, in a post-hoc analysis, 12 weeks of exenatide treatment in patients with moderate stage PD was found to improve mood and reduce indicators of depression, and these beneficial effects persisted up to 48 weeks following exenatide treatment (282). Following the success of these single-center exenatide PD clinical trials, Foltynie and colleagues have initiated a multi-center, phase 3 clinical trial to evaluate once weekly extended-release exenatide (Bydureon®) over a 96-week period to define whether exenatide’s advantageous actions are symptomatic or disease-modifying to potentially impact disease progression, or combined elements of both (Exenatide-PD3; NCT04232969). A sustained-release exenatide phase 2, multi-center clinical trial involving a once weekly and once every other week administration (PT320; Peptron; NCT04269642) has recently been completed in South Korea that largely follows the clinical protocol of Foltynie and colleagues (13), and results are awaited. This trial, likewise, evaluates the contents of brain-enriched exosomes to define biomarkers of drug response. Additional clinical trials for exenatide and alike drugs in neurodegenerative disease treatment are currently underway (Table 2).
Although no clinical trials have yet been completed, semaglutide is also being investigated for treatment potential for AD and PD. Two phase 3 clinical trials are currently recruiting patients with early AD to test the effects of oral semaglutide treatment on dementia ratings and measures of various cognitive and behavioral symptoms of AD (283, 284). In the United Kingdom, a phase 2 clinical trial is recruiting to evaluate effects of oral semaglutide treatment on tau regulation in aged individuals (285). As concerns PD, a phase 2 trial has been proposed to evaluate the effects of subcutaneous semaglutide treatment in improving motor function deficits in patients with PD (286).
The utilization of GLP-1R agonists to treat neurodegenerative diseases other than AD and PD, such as glaucoma and Friedreich ataxia (FRDA), has been investigated in very few clinical trials and requires further research. Glaucoma is a neurodegenerative disorder involving the deterioration and death of retinal ganglion cells (RGCs) and is, in large part, driven by neuroinflammation (287–289); thus, glaucoma has potential to be treated with incretin receptor agonists. In an observational retrospective study, the incidence of new glaucoma diagnoses in a cohort of approved GLP-1R agonist users was compared to that in a cohort of healthy control participants (11). The GLP-1R agonist user cohort exhibited a significantly lower incidence of new glaucoma diagnoses relative to the control cohort, suggesting a potential neuroprotective effect of GLP-1R stimulation in patients with glaucoma (11). FRDA is a heritable neurodegenerative movement disorder fueled and exacerbated by neuroinflammation (290–292), which could make it a candidate for GLP-1R agonist treatment. Exenatide treatment was found to re-elevate reduced levels of frataxin, a critical mitochondrial protein, in FRDA patients, which can enhance mitochondrial health and function and could, by extension, reduce neuroinflammation and neurodegeneration characteristic of the disease (293)—again, providing a potentially fruitful area of future basic and clinical research. Similarly, studies by Meissner and colleagues (178) demonstrated the presence of impaired insulin/IGF-1 and IR in vulnerable brain regions of multiple system atrophy patients and a related transgenic mouse model, and their mitigation in the latter by exendin-4, likewise suggesting a promising area of future research.
Finally, albeit it not a classical neurodegenerative disorder, studies by Sinclair and colleagues (294) demonstrated the efficacy of exenatide in reducing hypertension in a rat model of hydrocephalus, which has relevance to the human disorder of idiopathic intracranial hypertension for which effective pharmacological treatments are currently lacking (295). The choroid plexus expresses an abundance of GLP-1Rs and GLP-1R agonists reduce the activity of Na+/K+ ATPase, a marker of CSF secretion and, thereby, intracranial pressure (294). Whereas the primary pathogenesis of idiopathic intracranial hypertension remains to be identified, various mechanisms have been suggested to underpin the elevated CSF pressure that include heightened CSF generation, diminished CSF re-absorption, cerebral edema, and a raised cerebral venous pressure (296), and there is a proposed neuroinflammatory element (297). Early neuronal cell loss, later vision loss, and a host of sequela, such as severe chronic headache, ensue (298). A recent randomized, placebo-controlled, double-blind clinical trial of exenatide in women with idiopathic intracranial hypertension significantly decreased intracranial pressure, reduced headache frequency, and improved visual acuity in the treatment arm over the 12-week duration of the study (299). Following up on this, a 24-week randomized double-blind clinical trial of Presendin (which contains 2 mg of exenatide) versus a matching placebo group (once weekly) has just been initiated with a focus to evaluate 240 idiopathic intracranial hypertension patients (NCT05347147).
GLP-1R agonists may additionally have potential in treating neurological disorders that are not classified as neurodegenerative diseases, given the wealth of evidence for neurotrophic, neuroprotective, and anti-inflammatory properties of GLP-1R signaling. Extensive preclinical studies using cellular and animal models of TBI have revealed that GLP-1R stimulation provides neurotrophic, anti-neuroinflammatory, anti-neurodegenerative, and anti-oxidative effects and not only mitigates cognitive and memory impairments but blocks TBI-initiated gene pathways leading to longer-term neurodegenerative diseases (100, 234, 251, 255, 300–305). In addition, GLP-1R agonists may protect against ischemic stroke (4) and ameliorate biological consequences of stroke, as indicated by aforementioned meta-analyses (3, 273). Furthermore, studies in animal and cellular models of ischemic stroke demonstrated that GLP-1R stimulation reduced neuroinflammation (2, 306), endothelial leakage (2), neural apoptosis (307), and ROS (308); protected the BBB (306, 308); and enhanced cerebral blood flow (309). MS, a disease in which the immune system attacks the CNS, demyelinates axons, and disrupts nervous system signaling, may also be treatable using GLP-1R agonism. Preclinical trials in rodent models of MS report that GLP-1R agonists were neuroprotective, improved myelination, and reduced neuroinflammation, oxidative stress, and reactive astrogliosis (102, 310–312). Clinical trials in these conditions should be considered in order to investigate the therapeutic potential of GLP-1R agonists across a broader range of neurological diseases.
4. Conclusion
An abundance of research in cellular and animal model systems highlights the potential for GLP-1R agonists in reducing neuroinflammation and treating neurodegenerative diseases. Many GLP-1R agonists have already been approved by the FDA for the treatment of diabetes and obesity and could be repurposed for the treatment of neuroinflammation and neurodegeneration. This repurposing will require additional research in human clinical trials to confirm the safety, tolerability, and efficacy of each drug in reducing neuroinflammation in the human brain, as well as reveal the implications for neurodegenerative disease treatment. Several clinical trials have been completed or are currently underway to investigate the utility of GLP-1R agonists in treating neurodegenerative diseases, and the use of exosomes to evaluate biomarkers of drug target engagement and biological cascades involved in disease progression would provide valuable insight into drug action. In future research on the development of effective incretin receptor-stimulating drugs for reducing neuroinflammation as well as mitigating disease progression via multiple potential neurotrophic/protective actions, important considerations should include BBB penetration and the demonstrated enhanced efficacy of multi-agonism relative to single agonism agents. Across preclinical studies, the introduction of pharmacokinetics can provide a valuable measure as to whether chosen drug doses have any translatable relevance to the human condition.
Acknowledgements
The generation of this article was supported by the Intramural Research Program, National Institute on Aging, NIA. The authors thank Lauren Brick, Visual Media core, National Institute on Drug Abuse, National Institutes of Health, USA, in relation to artwork in Figure 1 and Figure 4. All other Figures were created using BioRender.com.
Funding
Intramural Research Program, National Institute on Aging, NIH: AG000333
Declaration of interests:
NHG is an inventor on patents related to incretin mimetics and has assigned them in entirety to the National Institute on Aging, NIH, US Government, and hence has no personal rights to these patents. The National Institute on Aging, NIH, has a Cooperative Research and Development Agreement with Peptron Inc. (S. Korea) to support the evaluation of GLP-1R agonists in neurodegenerative disorders. All other authors declare no conflict of interest.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Credit Author Statement
KOK: literature search, literature evaluation, initial draft, editing.
EJG: literature search, literature evaluation, editing
YL: literature search, literature evaluation, editing
NHG: literature search, literature evaluation, editing
References
- 1.Bellenguez C, Kucukali F, Jansen IE, Kleineidam L, Moreno-Grau S, Amin N, Naj AC, Campos-Martin R, Grenier-Boley B, Andrade V, Holmans PA, Boland A, Damotte V, van der Lee SJ, Costa MR, Kuulasmaa T, Yang Q, De Rojas I, Bis JC, Yaqub A, Prokic I, Chapuis J, Ahmad S, Giedraitis V, Aarsland D, Garcia-Gonzalez P, Abdelnour C, Alarcon-Martin E, Alcolea D, Alegret M, Alvarez I, Alvarez V, Armstrong NJ, Tsolaki A, Antunez C, Appollonio I, Arcaro M, Archetti S, Pastor AA, Arosio B, Athanasiu L, Bailly H, Banaj N, Baquero M, Barral S, Beiser A, Pastor AB, Below JE, Benchek P, Benussi L, Berr C, Besse C, Bessi V, Binetti G, Bizarro A, Blesa R, Boada M, Boerwinkle E, Borroni B, Boschi S, Bossu P, Brathen G, Bressler J, Bresner C, Brodaty H, Brookes KJ, Brusco LI, Buiza-Rueda D, Burger K, Burholt V, Bush WS, Calero M, Cantwell LB, Chene G, Chung J, Cuccaro ML, Cecchetti R, Cervera-Carles L, Charbonnier C, Chen HH, Chillotti C, Ciccone S, Claassen JAHR, Clark C, Conti E, Corma-Gomez A, Costantini E, Custodero C, Daian D, Dalmasso MC, Daniele A, Dardiotis E, Dartigues JF, de Deyn PP, Lopes KD, De Witte LD, Debette S, Deckert J, del Ser T, Denning N, Destefano A, Dichgans M, Diehl-Schmid J, Diez-Fairen M, Rossi PD, Djurovic S, Duron E, Duzel E, Dufouil C, Eiriksdottir G, Engelborghs S, Escott-Price V, Espinosa A, Ewers M, Faber KM, Fabrizio T, Nielsen SF, Fardo DW, Farotti L, Fenoglio C, Fernandez-Fuertes M, Ferrari R, Ferreira CB, Ferri E, Fin B, Fischer P, Fladby T, Fliessbach K, Fongang B, Fornage M, Fortea J, Foroud TM, Fostinelli S, Fox NC, Franco-Macias E, Bullido MJ, Frank-Garcia A, Froelich L, Fulton-Howard B, Galimberti D, Garcia-Alberca JM, Garcia-Madrona S, Garcia-Ribas G, Ghidoni R, Giegling I, Giorgio G, Goate AM, Goldhardt O, Gomez-Fonseca D, Gonzalez-Perez A, Graff C, Grande G, Green E, Grimmer T, Grunblatt E, Grunin M, Gudnason V, Guetta-Baranes T, Haapasalo A, Hadjigeorgiou G, Haines JL, Hamilton-Nelson KL, Hampel H, Hanon O, Hardy J, Hartmann AM, Hausner L, Harwood J, Heilmann-Heimbach S, Helisalmi S, Heneka MT, Hernandez I, Herrmann MJ, Hoffmann P, Holmes C, Holstege H, Vilas RH, Hulsman M, Humphrey J, Biessels GJ, Jian XQ, Johansson C, Jun GR, Kastumata Y, Kauwe J, Kehoe PG, Kilander L, Stahlbom AK, Kivipelto M, Koivisto A, Kornhuber J, Kosmidis MH, Kukull WA, Kuksa PP, Kunkle BW, Kuzma AB, Lage C, Laukka EJ, Launer L, Lauria A, Lee CY, Lehtisalo J, Lerch O, Lleo A, Longstreth W, Lopez O, de Munain AL, Love S, Lowemark M, Luckcuck L, Lunetta KL, Ma YY, Macias J, Macleod CA, Maier W, Mangialasche F, Spallazzi M, Marquie M, Marshall R, Martin ER, Montes AM, Rodriguez CM, Masullo C, Mayeux R, Mead S, Mecocci P, Medina M, Meggy A, Mehrabian S, Mendoza S, Menendez-Gonzalez M, Mir P, Moebus S, Mol M, Molina-Porcel L, Montrreal L, Morelli L, Moreno F, Morgan K, Mosley T, Nothen MM, Muchnik C, Mukherjee S, Nacmias B, Ngandu T, Nicolas G, Nordestgaard BG, Olaso R, Orellana A, Orsini M, Ortega G, Padovani A, Paolo C, Papenberg G, Parnetti L, Pasquier F, Pastor P, Peloso G, Perez-Cordon A, Perez-Tur J, Pericard P, Peters O, Pijnenburg YAL, Pineda JA, Pinol-Ripoll G, Pisanu L, Polak T, Popp J, Posthuma D, Priller J, Puerta R, Quenez O, Quintela I, Thomassen JQ, Rabano A, Rainero I, Rajabli F, Ramakers I, Real LM, Reinders MJT, Reitz C, Reyes-Dumeyer D, Ridge P, Riedel-Heller S, Riederer P, Roberto N, Rodriguez-Rodriguez E, Rongve A, Allende IR, Rosende-Roca M, Royo JL, Rubino E, Rujescu D, Saez ME, Sakka P, Saltvedt I, Sanchez-Arjona MB, Sanchez-Garcia F, Juan PS, Sanchez-Valle R, Sando SB, Sarnowski C, Satizabal CL, Scamosci M, Scarmeas N, Scarpini E, Scheltens P, Scherbaum N, Scherer M, Schmid M, Schneider A, Schott JM, Selbaek G, Seripa D, Serrano M, Sha J, Shadrin AA, Skrobot O, Slifer S, Snijders GJL, Soininen H, Solfrizzi V, Solomon A, Song Y, Sorbi S, Sotolongo-Grau O, Spalletta G, Spottke A, Squassina A, Stordal E, Tartan JP, Tarraga L, Tesi N, Thalamuthu A, Thomas T, Tosto G, Traykov L, Tremolizzo L, Tybjaeg-Hansen A, Uitterlinden A, Ullgren A, Ulstein I, Valero S, Valladares O, Van Broeckhoven C, Vance J, Vardarajan BN, van der Lugt A, Van Dongen J, van Rooij J, van Swieten J, Vandenberghe R, Verhey F, Vidal JS, Vogelgsang J, Vyhnalek M, Wagner M, Wallon D, Wang LS, Wang RQ, Weinhold L, Wiltfang J, Windle G, Woods B, Yannakoulia M, Zare H, Zhao Y, Zhang XL, Zhu CC, Zulaica M, Farrer LA, Psaty BM, Ghanbari M, Raj T, Sachdev P, Mather K, Jessen F, Ikram MA, de Mendonca A, Hort J, Tsolaki M, Pericak-Vance MA, Amouyel P, Williams J, Frikke-Schmidt R, Clarimon J, Deleuze JF, Rossi G, Seshadri S, Andreassen OA, Ingelsson M, Hiltunen M, Sleegers K, Schellenberg GD, van Duijn CM, Sims R, van der Flier WM, Ruiz A, Ramirez A, Lambert JC, EADB, ACE G, DEGESCO, EADI, GERAD, Demgene, FinnGen, ADGC, CHARGE. New insights into the genetic etiology of Alzheimer’s disease and related dementias. Nat Genet. 2022;54(4):412–+. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Marlet IR, Olmestig JNE, Vilsboll T, Rungby J, Kruuse C. Neuroprotective Mechanisms of Glucagon-like Peptide-1-based Therapies in Ischaemic Stroke: A Systematic Review based on Pre-Clinical Studies. Basic Clin Pharmacol Toxicol. 2018;122(6):559–569. [DOI] [PubMed] [Google Scholar]
- 3.Malhotra K, Katsanos AH, Lambadiari V, Goyal N, Palaiodimou L, Kosmidou M, Krogias C, Alexandrov AV, Tsivgoulis G. GLP-1 receptor agonists in diabetes for stroke prevention: a systematic review and meta-analysis. J Neurol. 2020;267(7):2117–2122. [DOI] [PubMed] [Google Scholar]
- 4.Li Y, Perry T, Kindy MS, Harvey BK, Tweedie D, Holloway HW, Powers K, Shen H, Egan JM, Sambamurti K, Brossi A, Lahiri DK, Mattson MP, Hoffer BJ, Wang Y, Greig NH. GLP-1 receptor stimulation preserves primary cortical and dopaminergic neurons in cellular and rodent models of stroke and Parkinsonism. Proc Natl Acad Sci U S A. 2009;106(4):1285–1290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Glotfelty EJ, Olson L, Karlsson TE, Li Y, Greig NH. Glucagon-like peptide-1 (GLP-1)-based receptor agonists as a treatment for Parkinson’s disease. Expert Opin Investig Drugs. 2020;29(6):595–602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Holscher C. Novel dual GLP-1/GIP receptor agonists show neuroprotective effects in Alzheimer’s and Parkinson’s disease models. Neuropharmacology. 2018;136:251–259. [DOI] [PubMed] [Google Scholar]
- 7.Yildirim Simsir I, Soyaltin UE, Cetinkalp S. Glucagon like peptide-1 (GLP-1) likes Alzheimer’s disease. Diabetes Metab Syndr. 2018;12(3):469–475. [DOI] [PubMed] [Google Scholar]
- 8.Boccardi V, Murasecco I, Mecocci P. Diabetes drugs in the fight against Alzheimer’s disease. Ageing Res Rev. 2019;54:100936. [DOI] [PubMed] [Google Scholar]
- 9.Mouhammad ZA, Vohra R, Horwitz A, Thein AS, Rovelt J, Cvenkel B, Williams PA, Azuara-Blanco A, Kolko M. Glucagon-Like Peptide 1 Receptor Agonists - Potential Game Changers in the Treatment of Glaucoma? Front Neurosci. 2022;16:824054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Cui QN, Stein LM, Fortin SM, Hayes MR. The role of glia in the physiology and pharmacology of glucagon-like peptide-1: implications for obesity, diabetes, neurodegeneration and glaucoma. Br J Pharmacol. 2022;179(4):715–726. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Sterling J, Hua PY, Dunaief JL, Cui QN, VanderBeek BL. Glucagon-like peptide 1 receptor agonist use is associated with reduced risk for glaucoma. Brit J Ophthalmol. 2021;Online ahead of print:1–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Glotfelty EJ, Delgado T, Tovar YRLB, Luo Y, Hoffer B, Olson L, Karlsson T, Mattson MP, Harvey B, Tweedie D, Li Y, Greig NH. Incretin Mimetics as Rational Candidates for the Treatment of Traumatic Brain Injury. ACS Pharmacol Transl Sci. 2019;2(2):66–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Athauda D, Maclagan K, Skene SS, Bajwa-Joseph M, Letchford D, Chowdhury K, Hibbert S, Budnik N, Zampedri L, Dickson J, Li Y, Aviles-Olmos I, Warner TT, Limousin P, Lees AJ, Greig NH, Tebbs S, Foltynie T. Exenatide once weekly versus placebo in Parkinson’s disease: a randomised, double-blind, placebo-controlled trial. Lancet. 2017;390(10103):1664–1675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Rowlands J, Heng J, Newsholme P, Carlessi R. Pleiotropic Effects of GLP-1 and Analogs on Cell Signaling, Metabolism, and Function. Front Endocrinol (Lausanne). 2018;9:672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Capozzi ME, DiMarchi RD, Tschop MH, Finan B, Campbell JE. Targeting the Incretin/Glucagon System With Triagonists to Treat Diabetes. Endocr Rev. 2018;39(5):719–738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Asmar M, Simonsen L, Madsbad S, Stallknecht B, Holst JJ, Bulow J. Glucose-dependent insulinotropic polypeptide may enhance fatty acid re-esterification in subcutaneous abdominal adipose tissue in lean humans. Diabetes. 2010;59(9):2160–2163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Nissen A, Christensen M, Knop FK, Vilsboll T, Holst JJ, Hartmann B. Glucose-dependent insulinotropic polypeptide inhibits bone resorption in humans. J Clin Endocrinol Metab. 2014;99(11):E2325–2329. [DOI] [PubMed] [Google Scholar]
- 18.Hui H, Farilla L, Merkel P, Perfetti R. The short half-life of glucagon-like peptide-1 in plasma does not reflect its long-lasting beneficial effects. Eur J Endocrinol. 2002;146(6):863–869. [DOI] [PubMed] [Google Scholar]
- 19.Tatarkiewicz K, Hargrove DM, Jodka CM, Gedulin BR, Smith PA, Hoyt JA, Lwin A, Collins L, Mamedova L, Levy OE, D’Souza L, Janssen S, Srivastava V, Ghosh SS, Parkes DG. A novel long-acting glucose-dependent insulinotropic peptide analogue: enhanced efficacy in normal and diabetic rodents. Diabetes Obes Metab. 2014;16(1):75–85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Pontiroli AE, Calderara A, Perfetti MG, Bareggi SR. Pharmacokinetics of intranasal, intramuscular and intravenous glucagon in healthy subjects and diabetic patients. Eur J Clin Pharmacol. 1993;45(6):555–558. [DOI] [PubMed] [Google Scholar]
- 21.Nauck M, Stockmann F, Ebert R, Creutzfeldt W. Reduced incretin effect in type 2 (non-insulin-dependent) diabetes. Diabetologia. 1986;29(1):46–52. [DOI] [PubMed] [Google Scholar]
- 22.Dupre J, Ross SA, Watson D, Brown JC. Stimulation of insulin secretion by gastric inhibitory polypeptide in man. J Clin Endocrinol Metab. 1973;37(5):826–828. [DOI] [PubMed] [Google Scholar]
- 23.Kreymann B, Williams G, Ghatei MA, Bloom SR. Glucagon-like peptide-1 7–36: a physiological incretin in man. Lancet. 1987;2(8571):1300–1304. [DOI] [PubMed] [Google Scholar]
- 24.Nauck MA, Heimesaat MM, Orskov C, Holst JJ, Ebert R, Creutzfeldt W. Preserved Incretin Activity of Glucagon-Like Peptide-1 [7–36 Amide] but Not of Synthetic Human Gastric-Inhibitory Polypeptide in Patients with Type-2 Diabetes-Mellitus. Journal of Clinical Investigation. 1993;91(1):301–307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Krarup T, Saurbrey N, Moody AJ, Kuhl C, Madsbad S. Effect of porcine gastric inhibitory polypeptide on beta-cell function in type I and type II diabetes mellitus. Metabolism. 1987;36(7):677–682. [DOI] [PubMed] [Google Scholar]
- 26.Orskov C, Holst JJ, Nielsen OV. Effect of truncated glucagon-like peptide-1 [proglucagon-(78–107) amide] on endocrine secretion from pig pancreas, antrum, and nonantral stomach. Endocrinology. 1988;123(4):2009–2013. [DOI] [PubMed] [Google Scholar]
- 27.Eng J, Kleinman WA, Singh L, Singh G, Raufman JP. Isolation and characterization of exendin-4, an exendin-3 analogue, from Heloderma suspectum venom. Further evidence for an exendin receptor on dispersed acini from guinea pig pancreas. J Biol Chem. 1992;267(11):7402–7405. [PubMed] [Google Scholar]
- 28.Hupesodmann K, Mcgregor GP, Bridenbaugh R, Rudiger G, Burkhard G, Thole H, Zimmermann B, Voigt K. Characterization of the Processing by Human Neutral Endopeptidase-24.11 of Glp-1(7–36) Amide and Comparison of the Substrate-Specificity of the Enzyme for Other Glucagon-Like Peptides. Regul Peptides. 1995;58(3):149–156. [DOI] [PubMed] [Google Scholar]
- 29.Cai Y, Wei L, Ma L, Huang X, Tao A, Liu Z, Yuan W. Long-acting preparations of exenatide. Drug Des Devel Ther. 2013;7:963–970. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Day JW, Ottaway N, Patterson JT, Gelfanov V, Smiley D, Gidda J, Findeisen H, Bruemmer D, Drucker DJ, Chaudhary N, Holland J, Hembree J, Abplanalp W, Grant E, Ruehl J, Wilson H, Kirchner H, Lockie SH, Hofmann S, Woods SC, Nogueiras R, Pfluger PT, Perez-Tilve D, DiMarchi R, Tschop MH. A new glucagon and GLP-1 co-agonist eliminates obesity in rodents. Nat Chem Biol. 2009;5(10):749–757. [DOI] [PubMed] [Google Scholar]
- 31.Finan B, Ma T, Ottaway N, Muller TD, Habegger KM, Heppner KM, Kirchner H, Holland J, Hembree J, Raver C, Lockie SH, Smiley DL, Gelfanov V, Yang B, Hofmann S, Bruemmer D, Drucker DJ, Pfluger PT, Perez-Tilve D, Gidda J, Vignati L, Zhang L, Hauptman JB, Lau M, Brecheisen M, Uhles S, Riboulet W, Hainaut E, Sebokova E, Conde-Knape K, Konkar A, DiMarchi RD, Tschop MH. Unimolecular dual incretins maximize metabolic benefits in rodents, monkeys, and humans. Sci Transl Med. 2013;5(209):209ra151. [DOI] [PubMed] [Google Scholar]
- 32.Min T, Bain SC. The Role of Tirzepatide, Dual GIP and GLP-1 Receptor Agonist, in the Management of Type 2 Diabetes: The SURPASS Clinical Trials. Diabetes Ther. 2021;12(1):143–157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Dutta D, Surana V, Singla R, Aggarwal S, Sharma M. Efficacy and safety of novel twincretin tirzepatide a dual GIP and GLP-1 receptor agonist in the management of type-2 diabetes: A Cochrane meta-analysis. Indian J Endocrinol Metab. 2021;25(6):475–489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Bhat VK, Kerr BD, Vasu S, Flatt PR, Gault VA. A DPP-IV-resistant triple-acting agonist of GIP, GLP-1 and glucagon receptors with potent glucose-lowering and insulinotropic actions in high-fat-fed mice. Diabetologia. 2013;56(6):1417–1424. [DOI] [PubMed] [Google Scholar]
- 35.Finan B, Yang B, Ottaway N, Smiley DL, Ma T, Clemmensen C, Chabenne J, Zhang L, Habegger KM, Fischer K, Campbell JE, Sandoval D, Seeley RJ, Bleicher K, Uhles S, Riboulet W, Funk J, Hertel C, Belli S, Sebokova E, Conde-Knape K, Konkar A, Drucker DJ, Gelfanov V, Pfluger PT, Muller TD, Perez-Tilve D, DiMarchi RD, Tschop MH. A rationally designed monomeric peptide triagonist corrects obesity and diabetes in rodents. Nat Med. 2015;21(1):27–36. [DOI] [PubMed] [Google Scholar]
- 36.Bossart M, Wagner M, Elvert R, Evers A, Hubschle T, Kloeckener T, Lorenz K, Moessinger C, Eriksson O, Velikyan I, Pierrou S, Johansson L, Dietert G, Dietz-Baum Y, Kissner T, Nowotny I, Einig C, Jan C, Rharbaoui F, Gassenhuber J, Prochnow HP, Agueusop I, Porksen N, Smith WB, Nitsche A, Konkar A. Effects on weight loss and glycemic control with SAR441255, a potent unimolecular peptide GLP-1/GIP/GCG receptor triagonist. Cell Metab. 2022;34(1):59–74 e10. [DOI] [PubMed] [Google Scholar]
- 37.Knerr PJ, Mowery SA, Douros JD, Premdjee B, Hjollund KR, He Y, Kruse Hansen AM, Olsen AK, Perez-Tilve D, DiMarchi RD, Finan B. Next generation GLP-1/GIP/glucagon triple agonists normalize body weight in obese mice. Mol Metab. 2022:101533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Finan B, Douros JD. GLP-1/GIP/glucagon receptor triagonism gets its try in humans. Cell Metab. 2022;34(1):3–4. [DOI] [PubMed] [Google Scholar]
- 39.Tsilingiris D, Liatis S, Dalamaga M, Kokkinos A. The Fight Against Obesity Escalates: New Drugs on the Horizon and Metabolic Implications. Curr Obes Rep. 2020;9(2):136–149. [DOI] [PubMed] [Google Scholar]
- 40.Khajavi N, Finan B, Kluth O, Muller TD, Mergler S, Schulz A, Kleinau G, Scheerer P, Schurmann A, Gudermann T, Tschop MH, Krude H, DiMarchi RD, Biebermann H. An incretin-based tri-agonist promotes superior insulin secretion from murine pancreatic islets via PLC activation. Cell Signal. 2018;51:13–22. [DOI] [PubMed] [Google Scholar]
- 41.Tschop MH, Finan B, Clemmensen C, Gelfanov V, Perez-Tilve D, Muller TD, DiMarchi RD. Unimolecular Polypharmacy for Treatment of Diabetes and Obesity. Cell Metab. 2016;24(1):51–62. [DOI] [PubMed] [Google Scholar]
- 42.Yuliantie E, Darbalaei S, Dai AT, Zhao PS, Yang DH, Sexton PM, Wang MW, Wootten D. Pharmacological characterization of mono-, dual- and tri-peptidic agonists at GIP and GLP-1 receptors. Biochemical Pharmacology. 2020;177:114001. [DOI] [PubMed] [Google Scholar]
- 43.Darbalaei S, Yuliantie E, Dai A, Chang R, Zhao P, Yang D, Wang MW, Sexton PM, Wootten D. Evaluation of biased agonism mediated by dual agonists of the GLP-1 and glucagon receptors. Biochem Pharmacol. 2020;180:114150. [DOI] [PubMed] [Google Scholar]
- 44.Holscher C Brain insulin resistance: role in neurodegenerative disease and potential for targeting. Expert Opin Investig Drugs. 2020;29(4):333–348. [DOI] [PubMed] [Google Scholar]
- 45.Greig NH, Tweedie D, Rachmany L, Li YZ, Rubovitch V, Schreiber S, Chiang YH, Hoffer BJ, Miller J, Lahiri DK, Sambamurti K, Becker RE, Pick CG. Incretin mimetics as pharmacologic tools to elucidate and as a new drug strategy to treat traumatic brain injury. Alzheimers & Dementia. 2014;10:S62–S75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Seino Y, Kuwata H, Yabe D. Incretin-based drugs for type 2 diabetes: Focus on East Asian perspectives. J Diabetes Investig. 2016;7 Suppl 1:102–109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Ryu J, Galan AK, Xin X, Dong F, Abdul-Ghani MA, Zhou L, Wang C, Li C, Holmes BM, Sloane LB, Austad SN, Guo S, Musi N, DeFronzo RA, Deng C, White MF, Liu F, Dong LQ. APPL1 potentiates insulin sensitivity by facilitating the binding of IRS1/2 to the insulin receptor. Cell Rep. 2014;7(4):1227–1238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Ferreira LSS, Fernandes CS, Vieira MNN, De Felice FG. Insulin Resistance in Alzheimer’s Disease. Front Neurosci. 2018;12:830. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Takahashi M, Yamada T, Tooyama I, Moroo I, Kimura H, Yamamoto T, Okada H. Insulin receptor mRNA in the substantia nigra in Parkinson’s disease. Neurosci Lett. 1996;204(3):201–204. [DOI] [PubMed] [Google Scholar]
- 50.Devaskar SU, Giddings SJ, Rajakumar PA, Carnaghi LR, Menon RK, Zahm DS. Insulin gene expression and insulin synthesis in mammalian neuronal cells. J Biol Chem. 1994;269(11):8445–8454. [PubMed] [Google Scholar]
- 51.Morris JK, Vidoni ED, Perea RD, Rada R, Johnson DK, Lyons K, Pahwa R, Burns JM, Honea RA. Insulin resistance and gray matter volume in neurodegenerative disease. Neuroscience. 2014;270:139–147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Bassil F, Delamarre A, Canron MH, Dutheil N, Vital A, Negrier-Leibreich ML, Bezard E, Fernagut PO, Meissner WG. Impaired brain insulin signalling in Parkinson’s disease. Neuropathol Appl Neurobiol. 2022;48(1):e12760. [DOI] [PubMed] [Google Scholar]
- 53.Yarchoan M, Toledo JB, Lee EB, Arvanitakis Z, Kazi H, Han LY, Louneva N, Lee VMY, Kim SF, Trojanowski JQ, Arnold SE. Abnormal serine phosphorylation of insulin receptor substrate 1 is associated with tau pathology in Alzheimer’s disease and tauopathies. Acta Neuropathologica. 2014;128(5):679–689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Yaribeygi H, Farrokhi FR, Butler AE, Sahebkar A. Insulin resistance: Review of the underlying molecular mechanisms. J Cell Physiol. 2019;234(6):8152–8161. [DOI] [PubMed] [Google Scholar]
- 55.Chen S, Zhou M, Sun J, Guo A, Fernando RL, Chen Y, Peng P, Zhao G, Deng Y. DPP-4 inhibitor improves learning and memory deficits and AD-like neurodegeneration by modulating the GLP-1 signaling. Neuropharmacology. 2019;157:107668. [DOI] [PubMed] [Google Scholar]
- 56.Verma MK, Goel R, Krishnadas N, Nemmani KVS. Targeting glucose-dependent insulinotropic polypeptide receptor for neurodegenerative disorders. Expert Opin Ther Targets. 2018;22(7):615–628. [DOI] [PubMed] [Google Scholar]
- 57.Chen Y, Zhang J, Zhang B, Gong CX. Targeting Insulin Signaling for the Treatment of Alzheimer’s Disease. Curr Top Med Chem. 2016;16(5):485–492. [DOI] [PubMed] [Google Scholar]
- 58.Uyar M, Lezius S, Buhmann C, Potter-Nerger M, Schulz R, Meier S, Gerloff C, Kuhle J, Choe CU. Diabetes, Glycated Hemoglobin (HbA1c), and Neuroaxonal Damage in Parkinson’s Disease (MARK-PD Study). Mov Disord. 2022;37(6):1299–1304. [DOI] [PubMed] [Google Scholar]
- 59.Bak AM, Egefjord L, Gejl M, Steffensen C, Stecher CW, Smidt K, Brock B, Rungby J. Targeting amyloid-beta by glucagon-like peptide −1 (GLP-1) in Alzheimer’s disease and diabetes. Expert Opin Ther Targets. 2011;15(10):1153–1162. [DOI] [PubMed] [Google Scholar]
- 60.Cummings J, Ortiz A, Castellino J, Kinney J. Diabetes: Risk factor and translational therapeutic implications for Alzheimer’s disease. Eur J Neurosci. 2022;Online ahead of print. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Holscher C. Diabetes as a risk factor for Alzheimer’s disease: insulin signalling impairment in the brain as an alternative model of Alzheimer’s disease. Biochem Soc T. 2011;39:891–897. [DOI] [PubMed] [Google Scholar]
- 62.Vagelatos NT, Eslick GD. Type 2 Diabetes as a Risk Factor for Alzheimer’s Disease: The Confounders, Interactions, and Neuropathology Associated With This Relationship. Epidemiol Rev. 2013;35:152–160. [DOI] [PubMed] [Google Scholar]
- 63.Liu W, Tang JF. Association between diabetes mellitus and risk of Parkinson’s disease: A prisma-compliant meta-analysis. Brain Behav. 2021;11(8):e02082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Biosa A, Outeiro TF, Bubacco L, Bisaglia M. Diabetes Mellitus as a Risk Factor for Parkinson’s Disease: a Molecular Point of View. Molecular Neurobiology. 2018;55(11):8754–8763. [DOI] [PubMed] [Google Scholar]
- 65.Brauer R, Wei L, Ma TT, Athauda D, Girges C, Vijiaratnam N, Auld G, Whittlesea C, Wong I, Foltynie T. Diabetes medications and risk of Parkinson’s disease: a cohort study of patients with diabetes. Brain. 2020;143:3067–3076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Talbot K, Wang HY, Kazi H, Han LY, Bakshi KP, Stucky A, Fuino RL, Kawaguchi KR, Samoyedny AJ, Wilson RS, Arvanitakis Z, Schneider JA, Wolf BA, Bennett DA, Trojanowski JQ, Arnold SE. Demonstrated brain insulin resistance in Alzheimer’s disease patients is associated with IGF-1 resistance, IRS-1 dysregulation, and cognitive decline. Journal of Clinical Investigation. 2012;122(4):1316–1338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Park KY, Nam GE, Han K, Park HK, Hwang HS. Waist circumference and risk of Parkinson’s disease. NPJ Parkinsons Dis. 2022;8(1):89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Nam GE, Kim SM, Han K, Kim NH, Chung HS, Kim JW, Han B, Cho SJ, Yu JH, Park YG, Choi KM. Metabolic syndrome and risk of Parkinson disease: A nationwide cohort study. PLoS Med. 2018;15(8):e1002640. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Abbott RD, Ross GW, White LR, Nelson JS, Masaki KH, Tanner CM, Curb JD, Blanchette PL, Popper JS, Petrovitch H. Midlife adiposity and the future risk of Parkinson’s disease. Neurology. 2002;59(7):1051–1057. [DOI] [PubMed] [Google Scholar]
- 70.Ley EJ, Srour MK, Clond MA, Barnajian M, Tillou A, Mirocha J, Salim A. Diabetic patients with traumatic brain injury: insulin deficiency is associated with increased mortality. J Trauma. 2011;70(5):1141–1144. [DOI] [PubMed] [Google Scholar]
- 71.Yoshino A, Hovda DA, Kawamata T, Katayama Y, Becker DP. Dynamic changes in local cerebral glucose utilization following cerebral conclusion in rats: evidence of a hyper- and subsequent hypometabolic state. Brain Res. 1991;561(1):106–119. [DOI] [PubMed] [Google Scholar]
- 72.Cerecedo-Lopez CD, Kim-Lee JH, Hernandez D, Acosta SA, Borlongan CV. Insulin-associated neuroinflammatory pathways as therapeutic targets for traumatic brain injury. Med Hypotheses. 2014;82(2):171–174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Shi J, Dong B, Mao Y, Guan W, Cao J, Zhu R, Wang S. Review: Traumatic brain injury and hyperglycemia, a potentially modifiable risk factor. Oncotarget. 2016;7(43):71052–71061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Korkmaz N, Kesikburun S, Atar MO, Sabuncu T. Insulin resistance and related factors in patients with moderate and severe traumatic brain injury. Ir J Med Sci. 2022;Online ahead of print. [DOI] [PubMed] [Google Scholar]
- 75.Chang Y, Kim CK, Kim MK, Seo WK, Oh K. Insulin resistance is associated with poor functional outcome after acute ischemic stroke in non-diabetic patients. Sci Rep-Uk. 2021;11(1):1229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Ago T, Matsuo R, Hata J, Wakisaka Y, Kuroda J, Kitazono T, Kamouchi M, Registry FS. Insulin resistance and clinical outcomes after acute ischemic stroke. Neurology. 2018;90(17):E1470–E1477. [DOI] [PubMed] [Google Scholar]
- 77.Baggio LL, Drucker DJ. Glucagon-like peptide-1 receptors in the brain: controlling food intake and body weight. J Clin Invest. 2014;124(10):4223–4226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Adriaenssens AE, Biggs EK, Darwish T, Tadross J, Sukthankar T, Girish M, Polex-Wolf J, Lam BY, Zvetkova I, Pan W, Chiarugi D, Yeo GSH, Blouet C, Gribble FM, Reimann F. Glucose-Dependent Insulinotropic Polypeptide Receptor-Expressing Cells in the Hypothalamus Regulate Food Intake. Cell Metab. 2019;30(5):987–996 e986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Hsu TM, Hahn JD, Konanur VR, Lam A, Kanoski SE. Hippocampal GLP-1 receptors influence food intake, meal size, and effort-based responding for food through volume transmission. Neuropsychopharmacology. 2015;40(2):327–337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Reich N, Holscher C. The neuroprotective effects of glucagon-like peptide 1 in Alzheimer’s and Parkinson’s disease: An in-depth review. Front Neurosci. 2022;16:970925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Nyberg J, Anderson MF, Meister B, Alborn AM, Strom AK, Brederlau A, Illerskog AC, Nilsson O, Kieffer TJ, Hietala MA, Ricksten A, Eriksson PS. Glucose-dependent insulinotropic polypeptide is expressed in adult hippocampus and induces progenitor cell proliferation. Journal of Neuroscience. 2005;25(7):1816–1825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Gilman CP, Perry T, Furukawa K, Grieg NH, Egan JM, Mattson MP. Glucagon-like peptide 1 modulates calcium responses to glutamate and membrane depolarization in hippocampal neurons. J Neurochem. 2003;87(5):1137–1144. [DOI] [PubMed] [Google Scholar]
- 83.Perry T, Lahiri DK, Chen D, Zhou J, Shaw KT, Egan JM, Greig NH. A novel neurotrophic property of glucagon-like peptide 1: a promoter of nerve growth factor-mediated differentiation in PC12 cells. J Pharmacol Exp Ther. 2002;300(3):958–966. [DOI] [PubMed] [Google Scholar]
- 84.Perry T, Haughey NJ, Mattson MP, Egan JM, Greig NH. Protection and reversal of excitotoxic neuronal damage by glucagon-like peptide-1 and exendin-4. J Pharmacol Exp Ther. 2002;302(3):881–888. [DOI] [PubMed] [Google Scholar]
- 85.During MJ, Cao L, Zuzga DS, Francis JS, Fitzsimons HL, Jiao XY, Bland RJ, Klugmann M, Banks WA, Drucker DJ, Haile CN. Glucagon-like peptide-1 receptor is involved in learning and neuroprotection. Nature Medicine. 2003;9(9):1173–1179. [DOI] [PubMed] [Google Scholar]
- 86.Abbas T, Faivre E, Holscher C. Impairment of synaptic plasticity and memory formation in GLP-1 receptor KO mice: Interaction between type 2 diabetes and Alzheimer’s disease. Behavioural Brain Research. 2009;205(1):265–271. [DOI] [PubMed] [Google Scholar]
- 87.Li Y, Tweedie D, Mattson MP, Holloway HW, Greig NH. Enhancing the GLP-1 receptor signaling pathway leads to proliferation and neuroprotection in human neuroblastoma cells. J Neurochem. 2010;113(6):1621–1631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Banks WA, Greig NH. Small molecules as central nervous system therapeutics: old challenges, new directions, and a philosophic divide. Future Med Chem. 2019;11(6):489–493. [DOI] [PubMed] [Google Scholar]
- 89.Kastin AJ, Akerstrom V, Pan WH. Interactions of glucagon-like peptide-1 (GLP-1) with the blood-brain barrier. J Mol Neurosci. 2002;18(1–2):7–14. [DOI] [PubMed] [Google Scholar]
- 90.Kastin AJ, Akerstrom V. Entry of exendin-4 into brain is rapid but may be limited at high doses. Int J Obesity. 2003;27(3):313–318. [DOI] [PubMed] [Google Scholar]
- 91.Banks WA, During MJ, Niehoff ML. Brain uptake of the glucagon-like peptide-1 antagonist exendin(9–39) after intranasal administration. Journal of Pharmacology and Experimental Therapeutics. 2004;309(2):469–475. [DOI] [PubMed] [Google Scholar]
- 92.Banks WA. Peptides and the blood-brain barrier. Peptides. 2015;72:16–19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Fu Z, Gong LY, Liu J, Wu J, Barrett EJ, Aylor KW, Liu ZQ. Brain Endothelial Cells Regulate Glucagon-Like Peptide 1 Entry Into the Brain via a Receptor-Mediated Process. Front Physiol. 2020;11:555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Nystrom T, Gutniak MK, Zhang QM, Zhang F, Holst JJ, Ahren B, Sjoholm A. Effects of glucagon-like peptide-1 on endothelial function in type 2 diabetes patients with stable coronary artery disease. Am J Physiol-Endoc M. 2004;287(6):E1209–E1215. [DOI] [PubMed] [Google Scholar]
- 95.Drucker DJ. The Cardiovascular Biology of Glucagon-like Peptide-1. Cell Metabolism. 2016;24(1):15–30. [DOI] [PubMed] [Google Scholar]
- 96.Krieger JP, Arnold M, Pettersen KG, Lossel P, Langhans W, Lee SJ. Knockdown of GLP-1 receptors in vagal afferents affects normal food intake and glycemia. Diabetes. 2016;65(1):34–43. [DOI] [PubMed] [Google Scholar]
- 97.Salameh TS, Rhea EM, Talbot K, Banks WA. Brain uptake pharmacokinetics of incretin receptor agonists showing promise as Alzheimer’s and Parkinson’s disease therapeutics. Biochem Pharmacol. 2020;180:114187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Gabery S, Salinas CG, Paulsen SJ, Ahnfelt-Ronne J, Alanentalo T, Baquero AF, Buckley ST, Farkas E, Fekete C, Frederiksen KS, Helms HCC, Jeppesen JF, John LM, Pyke C, Nohr J, Lu TT, Polex-Wolf J, Prevot V, Raun K, Simonsen L, Sun G, Szilvasy-Szabo A, Willenbrock H, Secher A, Knudsen LB, Hogendorf WFJ. Semaglutide lowers body weight in rodents via distributed neural pathways. JCI Insight. 2020;5(6). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Mullins RJ, Diehl TC, Chia CW, Kapogiannis D. Insulin resistance as a link between amyloid-beta and Tau pathologies in Alzheimer’s disease. Front Aging Neurosci. 2017;9:118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Bader M, Li Y, Tweedie D, Shlobin NA, Bernstein A, Rubovitch V, Tovar YRLB, DiMarchi RD, Hoffer BJ, Greig NH, Pick CG. Neuroprotective Effects and Treatment Potential of Incretin Mimetics in a Murine Model of Mild Traumatic Brain Injury. Front Cell Dev Biol. 2019;7:356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Chen S, Yu SJ, Li Y, Lecca D, Glotfelty E, Kim HK, Choi HI, Hoffer BJ, Greig NH, Kim DS, Wang Y. Post-treatment with PT302, a long-acting Exendin-4 sustained release formulation, reduces dopaminergic neurodegeneration in a 6-Hydroxydopamine rat model of Parkinson’s disease. Sci Rep. 2018;8(1):10722. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Gharagozloo M, Smith MD, Sotirchos ES, Jin J, Meyers K, Taylor M, Garton T, Bannon R, Lord HN, Dawson TM, Dawson VL, Lee S, Calabresi PA. Therapeutic Potential of a Novel Glucagon-like Peptide-1 Receptor Agonist, NLY01, in Experimental Autoimmune Encephalomyelitis. Neurotherapeutics. 2021;18(3):1834–1848. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Yun SP, Kam TI, Panicker N, Kim S, Oh Y, Park JS, Kwon SH, Park YJ, Karuppagounder SS, Park H, Kim S, Oh N, Kim NA, Lee S, Brahmachari S, Mao X, Lee JH, Kumar M, An D, Kang SU, Lee Y, Lee KC, Na DH, Kim D, Lee SH, Roschke VV, Liddelow SA, Mari Z, Barres BA, Dawson VL, Lee S, Dawson TM, Ko HS. Block of A1 astrocyte conversion by microglia is neuroprotective in models of Parkinson’s disease. Nat Med. 2018;24(7):931–938. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Neuraly I. A Clinical Study of NLY01 in Patient’s With Early Parkinson’s Disease. In. https://ClinicalTrials.gov/show/NCT04154072; 2023.
- 105.Lv MJ, Xue GF, Cheng HF, Meng PF, Lian X, Holscher C, Li DF. The GLP-1/GIP dualreceptor agonist DA5-CH inhibits the NF-kappa B inflammatory pathway in the MPTP mouse model of Parkinson’s disease more effectively than the GLP-1 single-receptor agonist NLY01. Brain Behav. 2021;11(8):e2231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.DiSabato DJ, Quan N, Godbout JP. Neuroinflammation: the devil is in the details. J Neurochem. 2016;139 Suppl 2:136–153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Heneka MT, O’Banion MK, Terwel D, Kummer MP. Neuroinflammatory processes in Alzheimer’s disease. J Neural Transm (Vienna). 2010;117(8):919–947. [DOI] [PubMed] [Google Scholar]
- 108.Hammond TR, Dufort C, Dissing-Olesen L, Giera S, Young A, Wysoker A, Walker AJ, Gergits F, Segel M, Nemesh J, Marsh SE, Saunders A, Macosko E, Ginhoux F, Chen J, Franklin RJM, Piao X, McCarroll SA, Stevens B. Single-Cell RNA Sequencing of Microglia throughout the Mouse Lifespan and in the Injured Brain Reveals Complex Cell-State Changes. Immunity. 2019;50(1):253–271 e256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Shabab T, Khanabdali R, Moghadamtousi SZ, Kadir HA, Mohan G. Neuroinflammation pathways: a general review. Int J Neurosci. 2017;127(7):624–633. [DOI] [PubMed] [Google Scholar]
- 110.Leng F, Edison P. Neuroinflammation and microglial activation in Alzheimer disease: where do we go from here? Nat Rev Neurol. 2021;17(3):157–172. [DOI] [PubMed] [Google Scholar]
- 111.Colombo E, Farina C. Astrocytes: Key regulators of neuroinflammation. Trends Immunol. 2016;37(9):608–620. [DOI] [PubMed] [Google Scholar]
- 112.Liddelow SA, Guttenplan KA, Clarke LE, Bennett FC, Bohlen CJ, Schirmer L, Bennett ML, Munch AE, Chung WS, Peterson TC, Wilton DK, Frouin A, Napier BA, Panicker N, Kumar M, Buckwalter MS, Rowitch DH, Dawson VL, Dawson TM, Stevens B, Barres BA. Neurotoxic reactive astrocytes are induced by activated microglia. Nature. 2017;541(7638):481–487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Escartin C, Galea E, Lakatos A, O’Callaghan JP, Petzold GC, Serrano-Pozo A, Steinhauser C, Volterra A, Carmignoto G, Agarwal A, Allen NJ, Araque A, Barbeito L, Barzilai A, Bergles DE, Bonvento G, Butt AM, Chen WT, Cohen-Salmon M, Cunningham C, Deneen B, De Strooper B, Diaz-Castro B, Farina C, Freeman M, Gallo V, Goldman JE, Goldman SA, Gotz M, Gutierrez A, Haydon PG, Heiland DH, Hol EM, Holt MG, Iino M, Kastanenka KV, Kettenmann H, Khakh BS, Koizumi S, Lee CJ, Liddelow SA, MacVicar BA, Magistretti P, Messing A, Mishra A, Molofsky AV, Murai KK, Norris CM, Okada S, Oliet SHR, Oliveira JF, Panatier A, Parpura V, Pekna M, Pekny M, Pellerin L, Perea G, Perez-Nievas BG, Pfrieger FW, Poskanzer KE, Quintana FJ, Ransohoff RM, Riquelme-Perez M, Robel S, Rose CR, Rothstein JD, Rouach N, Rowitch DH, Semyanov A, Sirko S, Sontheimer H, Swanson RA, Vitorica J, Wanner IB, Wood LB, Wu J, Zheng B, Zimmer ER, Zorec R, Sofroniew MV, Verkhratsky A. Reactive astrocyte nomenclature, definitions, and future directions. Nat Neurosci. 2021;24(3):312–325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Lecca D, Jung YJ, Scerba MT, Hwang I, Kim YK, Kim S, Modrow S, Tweedie D, Hsueh SC, Liu D, Luo W, Glotfelty E, Li Y, Wang JY, Luo Y, Hoffer BJ, Kim DS, McDevitt RA, Greig NH. Role of chronic neuroinflammation in neuroplasticity and cognitive function: A hypothesis. Alzheimers Dement. 2022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Guttenplan KA, Weigel MK, Prakash P, Wijewardhane PR, Hasel P, Rufen-Blanchette U, Munch AE, Blum JA, Fine J, Neal MC, Bruce KD, Gitler AD, Chopra G, Liddelow SA, Barres BA. Neurotoxic reactive astrocytes induce cell death via saturated lipids. Nature. 2021;599(7883):102–107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Clarke LE, Liddelow SA, Chakraborty C, Munch AE, Heiman M, Barres BA. Normal aging induces A1-like astrocyte reactivity. Proc Natl Acad Sci U S A. 2018;115(8):E1896–E1905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Lawrence T The nuclear factor NF-kappaB pathway in inflammation. Cold Spring Harb Perspect Biol. 2009;1(6):a001651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Shih RH, Wang CY, Yang CM. NF-kappaB Signaling Pathways in Neurological Inflammation: A Mini Review. Front Mol Neurosci. 2015;8:77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Ahn KS, Aggarwal BB. Transcription factor NF-kappaB: a sensor for smoke and stress signals. Ann N Y Acad Sci. 2005;1056:218–233. [DOI] [PubMed] [Google Scholar]
- 120.Vezzani A, Maroso M, Balosso S, Sanchez MA, Bartfai T. IL-1 receptor/Toll-like receptor signaling in infection, inflammation, stress and neurodegeneration couples hyperexcitability and seizures. Brain Behav Immun. 2011;25(7):1281–1289. [DOI] [PubMed] [Google Scholar]
- 121.Chen B, Deng X, Wang B, Liu H. Etanercept, an inhibitor of TNF-a, prevents propofol-induced neurotoxicity in the developing brain. Int J Dev Neurosci. 2016;55:91–100. [DOI] [PubMed] [Google Scholar]
- 122.Hsueh SC, Luo W, Tweedie D, Kim DS, Kim YK, Hwang I, Gil JE, Han BS, Chiang YH, Selman W, Hoffer BJ, Greig NH. N-Adamantyl Phthalimidine: A New Thalidomide-like Drug That Lacks Cereblon Binding and Mitigates Neuronal and Synaptic Loss, Neuroinflammation, and Behavioral Deficits in Traumatic Brain Injury and LPS Challenge. Acs Pharmacol Transl. 2021;4(2):980–1000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Tsai YR, Tweedie D, Navas-Enamorado I, Scerba MT, Chang CF, Lai JH, Wu JC, Chen YH, Kang SJ, Hoffer BJ, de Cabo R, Greig NH, Chiang YH, Chen KY. Pomalidomide Reduces Ischemic Brain Injury in Rodents. Cell Transplant. 2019;28(4):439–450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Huang PS, Tsai PY, Yang LY, Lecca D, Luo W, Kim DS, Hoffer BJ, Chiang YH, Greig NH, Wang JY. 3,6’-Dithiopomalidomide ameliorates hippocampal neurodegeneration, microgliosis and astrogliosis and improves cognitive behaviors in rats with a moderate traumatic brain injury. Int J Mol Sci. 2021;22(15):8276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Lin CT, Lecca D, Yang LY, Luo W, Scerba MT, Tweedie D, Huang PS, Jung YJ, Kim DS, Yang CH, Hoffer BJ, Wang JY, Greig NH. 3,6’-dithiopomalidomide reduces neural loss, inflammation, behavioral deficits in brain injury and microglial activation. Elife. 2020;9:e54726. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Blamire AM, Anthony DC, Rajagopalan B, Sibson NR, Perry VH, Styles P. Interleukin-1beta -induced changes in blood-brain barrier permeability, apparent diffusion coefficient, and cerebral blood volume in the rat brain: a magnetic resonance study. J Neurosci. 2000;20(21):8153–8159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Wu MD, Hein AM, Moravan MJ, Shaftel SS, Olschowka JA, O’Banion MK. Adult murine hippocampal neurogenesis is inhibited by sustained IL-1 beta and not rescued by voluntary running. Brain Behavior and Immunity. 2012;26(2):292–300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Alam Q, Alam MZ, Mushtaq G, Damanhouri GA, Rasool M, Kamal MA, Haque A. Inflammatory Process in Alzheimer’s and Parkinson’s Diseases: Central Role of Cytokines. Curr Pharm Des. 2016;22(5):541–548. [DOI] [PubMed] [Google Scholar]
- 129.Pickering M, O’Connor JJ. Pro-inflammatory cytokines and their effects in the dentate gyrus. Dentate Gyrus: A Comphrehensive Guide to Structure, Function, and Clinical Implications. 2007;163:339–354. [DOI] [PubMed] [Google Scholar]
- 130.Tuttolomondo A, Di Raimondo D, di Sciacca R, Pinto A, Licata G. Inflammatory cytokines in acute ischemic stroke. Curr Pharm Des. 2008;14(33):3574–3589. [DOI] [PubMed] [Google Scholar]
- 131.Dowlati Y, Herrmann N, Swardfager W, Liu H, Sham L, Reim EK, Lanctot KL. A meta-analysis of cytokines in major depression. Biol Psychiatry. 2010;67(5):446–457. [DOI] [PubMed] [Google Scholar]
- 132.Jung YJ, Tweedie D, Scerba MT, Kim DS, Palmas MF, Pisanu A, Carta AR, Greig NH. Repurposing Immunomodulatory Imide Drugs (IMiDs) in Neuropsychiatric and Neurodegenerative Disorders. Front Neurosci-Switz. 2021;15:656921. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Reale M, Costantini E, Greig NH. Cytokine imbalance in schizophrenia. From research to clinic: Potential implications for treatment. Front Psychiatry. 2021;12:536257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Dawson TM, Dawson VL. Nitric oxide signaling in neurodegeneration and cell death. Adv Pharmacol. 2018;82:57–83. [DOI] [PubMed] [Google Scholar]
- 135.Picon-Pages P, Garcia-Buendia J, Munoz FJ. Functions and dysfunctions of nitric oxide in brain. Biochim Biophys Acta Mol Basis Dis. 2019;1865(8):1949–1967. [DOI] [PubMed] [Google Scholar]
- 136.Guivernau B, Bonet J, Valls-Comamala V, Bosch-Morato M, Godoy JA, Inestrosa NC, Peralvarez-Marin A, Fernandez-Busquets X, Andreu D, Oliva B, Munoz FJ. Amyloid-beta peptide nitrotyrosination stabilizes oligomers and enhances NMDAR-mediated toxicity. J Neurosci. 2016;36(46):11693–11703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Paradies G, Paradies V, Ruggiero FM, Petrosillo G. Oxidative stress, cardiolipin and mitochondrial dysfunction in nonalcoholic fatty liver disease. World J Gastroenterol. 2014;20(39):14205–14218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Shoshan-Barmatz V, Nahon-Crystal E, Shteinfer-Kuzmine A, Gupta R. VDAC1, mitochondrial dysfunction, and Alzheimer’s disease. Pharmacol Res. 2018;131:87–101. [DOI] [PubMed] [Google Scholar]
- 139.Wang X, Wang W, Li L, Perry G, Lee HG, Zhu X. Oxidative stress and mitochondrial dysfunction in Alzheimer’s disease. Biochim Biophys Acta. 2014;1842(8):1240–1247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Rocha EM, De Miranda B, Sanders LH. Alpha-synuclein: Pathology, mitochondrial dysfunction and neuroinflammation in Parkinson’s disease. Neurobiol Dis. 2018;109(Pt B):249–257. [DOI] [PubMed] [Google Scholar]
- 141.Malpartida AB, Williamson M, Narendra DP, Wade-Martins R, Ryan BJ. Mitochondrial dysfunction and mitophagy in Parkinson’s disease: From mechanism to therapy. Trends Biochem Sci. 2021;46(4):329–343. [DOI] [PubMed] [Google Scholar]
- 142.Hauck AK, Huang Y, Hertzel AV, Bernlohr DA. Adipose oxidative stress and protein carbonylation. J Biol Chem. 2019;294(4):1083–1088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Cheng Y, Song Y, Chen H, Li Q, Gao Y, Lu G, Luo C. Ferroptosis mediated by lipid reactive oxygen species: A possible causal link of neuroinflammation to neurological disorders. Oxid Med Cell Longev. 2021;2021:5005136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Kaur U, Banerjee P, Bir A, Sinha M, Biswas A, Chakrabarti S. Reactive oxygen species, redox signaling and neuroinflammation in Alzheimer’s disease: the NF-kappaB connection. Curr Top Med Chem. 2015;15(5):446–457. [DOI] [PubMed] [Google Scholar]
- 145.Hoffer ME, Balaban C, Slade MD, Tsao JW, Hoffer B. Amelioration of acute sequelae of blast induced mild traumatic brain injury by N-acetyl cysteine: a double-blind, placebo controlled study. Plos One. 2013;8(1):e54163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Hoffer BJ, Pick CG, Hoffer ME, Becker RE, Chiang YH, Greig NH. Repositioning drugs for traumatic brain injury - N-acetyl cysteine and Phenserine. J Biomed Sci. 2017;24(1):71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Bailey J, Coucha M, Bolduc DR, Burnett FN, Barrett AC, Ghaly M, Abdelsaid M. GLP-1 receptor nitration contributes to loss of brain pericyte function in a mouse model of diabetes. Diabetologia. 2022;65(9):1541–1554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Lin WJ, Ma XF, Hao M, Zhou HR, Yu XY, Shao N, Gao XY, Kuang HY. Liraglutide attenuates the migration of retinal pericytes induced by advanced glycation end products. Peptides. 2018;105:7–13. [DOI] [PubMed] [Google Scholar]
- 149.Cheng J, Korte N, Nortley R, Sethi H, Tang Y, Attwell D. Targeting pericytes for therapeutic approaches to neurological disorders. Acta Neuropathol. 2018;136(4):507–523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.Attwell D, Mishra A, Hall CN, O’Farrell FM, Dalkara T. What is a pericyte? J Cerebr Blood F Met. 2016;36(2):451–455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151.Alarcon-Martinez L, Yemisci M, Dalkara T. Pericyte morphology and function. Histol Histopathol. 2021;36(6):633–643. [DOI] [PubMed] [Google Scholar]
- 152.Sagare AP, Bell RD, Zhao Z, Ma Q, Winkler EA, Ramanathan A, Zlokovic BV. Pericyte loss influences Alzheimer-like neurodegeneration in mice. Nat Commun. 2013;4:2932. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 153.Nikolakopoulou AM, Montagne A, Kisler K, Dai Z, Wang Y, Huuskonen MT, Sagare AP, Lazic D, Sweeney MD, Kong P, Wang M, Owens NC, Lawson EJ, Xie X, Zhao Z, Zlokovic BV. Pericyte loss leads to circulatory failure and pleiotrophin depletion causing neuron loss. Nat Neurosci. 2019;22(7):1089–1098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154.Shi H, Koronyo Y, Rentsendorj A, Regis GC, Sheyn J, Fuchs DT, Kramerov AA, Ljubimov AV, Dumitrascu OM, Rodriguez AR, Barron E, Hinton DR, Black KL, Miller CA, Mirzaei N, Koronyo-Hamaoui M. Identification of early pericyte loss and vascular amyloidosis in Alzheimer’s disease retina. Acta Neuropathol. 2020;139(5):813–836. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155.Bell RD, Winkler EA, Sagare AP, Singh I, LaRue B, Deane R, Zlokovic BV. Pericytes control key neurovascular functions and neuronal phenotype in the adult brain and during brain aging. Neuron. 2010;68(3):409–427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156.Mae MA, He L, Nordling S, Vazquez-Liebanas E, Nahar K, Jung B, Li X, Tan BC, Chin Foo J, Cazenave-Gassiot A, Wenk MR, Zarb Y, Lavina B, Quaggin SE, Jeansson M, Gu C, Silver DL, Vanlandewijck M, Butcher EC, Keller A, Betsholtz C. Single-cell analysis of blood-brain barrier response to pericyte loss. Circ Res. 2021;128(4):e46–e62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157.Nortley R, Korte N, Izquierdo P, Hirunpattarasilp C, Mishra A, Jaunmuktane Z, Kyrargyri V, Pfeiffer T, Khennouf L, Madry C, Gong H, Richard-Loendt A, Huang W, Saito T, Saido TC, Brandner S, Sethi H, Attwell D. Amyloid beta oligomers constrict human capillaries in Alzheimer’s disease via signaling to pericytes. Science. 2019;365(6450). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158.Kisler K, Nelson AR, Montagne A, Zlokovic BV. Cerebral blood flow regulation and neurovascular dysfunction in Alzheimer disease. Nat Rev Neurosci. 2017;18(7):419–434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159.Lyman M, Lloyd DG, Ji X, Vizcaychipi MP, Ma D. Neuroinflammation: the role and consequences. Neurosci Res. 2014;79:1–12. [DOI] [PubMed] [Google Scholar]
- 160.Ownby RL. Neuroinflammation and cognitive aging. Curr Psychiatry Rep. 2010;12(1):39–45. [DOI] [PubMed] [Google Scholar]
- 161.Guerrero A, De Strooper B, Arancibia-Carcamo L. Cellular senescence at the crossroads of inflammation and Alzheimer’s disease. Trends Neurosci. 2021;44(9):714–727. [DOI] [PubMed] [Google Scholar]
- 162.Bradburn S, Murgatroyd C, Ray N. Neuroinflammation in mild cognitive impairment and Alzheimer’s disease: A meta-analysis. Ageing Res Rev. 2019;50:1–8. [DOI] [PubMed] [Google Scholar]
- 163.Yu CJ, Ma D, Song LL, Zhai ZN, Tao Y, Zhang Y, Cai LY, Hou YH, Chen HY, Wang L. The role of GLP-1/GIP receptor agonists in Alzheimer’s disease. Adv Clin Exp Med. 2020;29(6):661–668. [DOI] [PubMed] [Google Scholar]
- 164.Dionisio-Santos DA, Olschowka JA, O’Banion MK. Exploiting microglial and peripheral immune cell crosstalk to treat Alzheimer’s disease. J Neuroinflammation. 2019;16(1):74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 165.Heneka MT, Carson MJ, El Khoury J, Landreth GE, Brosseron F, Feinstein DL, Jacobs AH, Wyss-Coray T, Vitorica J, Ransohoff RM, Herrup K, Frautschy SA, Finsen B, Brown GC, Verkhratsky A, Yamanaka K, Koistinaho J, Latz E, Halle A, Petzold GC, Town T, Morgan D, Shinohara ML, Perry VH, Holmes C, Bazan NG, Brooks DJ, Hunot S, Joseph B, Deigendesch N, Garaschuk O, Boddeke E, Dinarello CA, Breitner JC, Cole GM, Golenbock DT, Kummer MP. Neuroinflammation in Alzheimer’s disease. Lancet Neurol. 2015;14(4):388–405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 166.Hickman SE, Allison EK, El Khoury J. Microglial dysfunction and defective beta-amyloid clearance pathways in aging Alzheimer’s disease mice. J Neurosci. 2008;28(33):8354–8360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 167.Denver P, English A, McClean PL. Inflammation, insulin signaling and cognitive function in aged APP/PS1 mice. Brain Behav Immun. 2018;70:423–434. [DOI] [PubMed] [Google Scholar]
- 168.Saito T, Saido TC. Neuroinflammation in mouse models of Alzheimer’s disease. Clin Exp Neuroimmunol. 2018;9(4):211–218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 169.Haass C, Selkoe D. If amyloid drives Alzheimer disease, why have anti-amyloid therapies not yet slowed cognitive decline? PLoS Biol. 2022;20(7):e3001694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 170.Troncoso-Escudero P, Parra A, Nassif M, Vidal RL. Outside in: Unraveling the Role of Neuroinflammation in the Progression of Parkinson’s Disease. Front Neurol. 2018;9:860. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 171.Hirsch EC, Hunot S. Neuroinflammation in Parkinson’s disease: a target for neuroprotection? Lancet Neurol. 2009;8(4):382–397. [DOI] [PubMed] [Google Scholar]
- 172.Van Dyken P, Lacoste B. Impact of Metabolic Syndrome on Neuroinflammation and the Blood-Brain Barrier. Front Neurosci. 2018;12:930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 173.Shah K, Desilva S, Abbruscato T. The role of glucose transporters in brain disease: diabetes and Alzheimer’s Disease. Int J Mol Sci. 2012;13(10):12629–12655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 174.Govindpani K, McNamara LG, Smith NR, Vinnakota C, Waldvogel HJ, Faull RL, Kwakowsky A. Vascular Dysfunction in Alzheimer’s Disease: A Prelude to the Pathological Process or a Consequence of It? J Clin Med. 2019;8(5):651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 175.Fanciulli A, Stankovic I, Krismer F, Seppi K, Levin J, Wenning GK. Multiple system atrophy. Int Rev Neurobiol. 2019;149:137–192. [DOI] [PubMed] [Google Scholar]
- 176.Robbins J, Busquets O, Tong M, de la Monte SM. Dysregulation of Insulin-Linked Metabolic Pathways in Alzheimer’s Disease: Co-Factor Role of Apolipoprotein E varepsilon4. J Alzheimers Dis Rep. 2020;4(1):479–493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 177.Kim DS, Choi HI, Wang Y, Luo Y, Hoffer BJ, Greig NH. A New Treatment Strategy for Parkinson’s Disease through the Gut-Brain Axis: The Glucagon-Like Peptide-1 Receptor Pathway. Cell Transplant. 2017;26(9):1560–1571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 178.Bassil F, Canron MH, Vital A, Bezard E, Li Y, Greig NH, Gulyani S, Kapogiannis D, Fernagut PO, Meissner WG. Insulin resistance and exendin-4 treatment for multiple system atrophy. Brain. 2017;140(5):1420–1436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 179.Najem D, Bamji-Mirza M, Chang NN, Liu QY, Zhang WD. Insulin resistance, neuroinflammation, and Alzheimer’s disease. Rev Neuroscience. 2014;25(4):509–525. [DOI] [PubMed] [Google Scholar]
- 180.Kacirova M, Zelezna B, Blazkova M, Holubova M, Popelova A, Kunes J, Sediva B, Maletinska L. Aging and high-fat diet feeding lead to peripheral insulin resistance and sex-dependent changes in brain of mouse model of tau pathology THY-Tau22. J Neuroinflamm. 2021;18(1):141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 181.Ng RC, Jian M, Ma OK, Bunting M, Kwan JS, Zhou GJ, Senthilkumar K, Iyaswamy A, Chan PK, Li M, Leung KM, Kumar Durairajan SS, Lam KS, Chu LW, Festenstein R, Chung SK, Chan KH. Chronic oral administration of adipoRon reverses cognitive impairments and ameliorates neuropathology in an Alzheimer’s disease mouse model. Mol Psychiatry. 2021;26(10):5669–5689. [DOI] [PubMed] [Google Scholar]
- 182.Takeda S, Sato N, Uchio-Yamada K, Sawada K, Kunieda T, Takeuchi D, Kurinami H, Shinohara M, Rakugi H, Morishita R. Diabetes-accelerated memory dysfunction via cerebrovascular inflammation and Abeta deposition in an Alzheimer mouse model with diabetes. Proc Natl Acad Sci U S A. 2010;107(15):7036–7041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 183.Paris D, Town T, Mori T, Parker TA, Humphrey J, Mullan M. Soluble beta-amyloid peptides mediate vasoactivity via activation of a pro-inflammatory pathway. Neurobiol Aging. 2000;21(2):183–197. [DOI] [PubMed] [Google Scholar]
- 184.Yassine HN, Solomon V, Thakral A, Sheikh-Bahaei N, Chui HC, Braskie MN, Schneider LS, Talbot K. Brain energy failure in dementia syndromes: Opportunities and challenges for glucagon-like peptide-1 receptor agonists. Alzheimers Dement. 2022;18(3):478–497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 185.Perry T, Greig NH. Enhancing central nervous system endogenous GLP-1 receptor pathways for intervention in Alzheimer’s disease. Curr Alzheimer Res. 2005;2(3):377–385. [DOI] [PubMed] [Google Scholar]
- 186.Perry TA, Greig NH. A new Alzheimer’s disease interventive strategy: GLP-1. Curr Drug Targets. 2004;5(6):565–571. [DOI] [PubMed] [Google Scholar]
- 187.Perry T, Greig NH. The glucagon-like peptides: a new genre in therapeutic targets for intervention in Alzheimer’s disease. J Alzheimers Dis. 2002;4(6):487–496. [DOI] [PubMed] [Google Scholar]
- 188.Greig NH, Mattson MP, Perry T, Chan SL, Giordano T, Sambamurti K, Rogers JT, Ovadia H, Lahiri DK. New therapeutic strategies and drug candidates for neurodegenerative diseases: p53 and TNF-alpha inhibitors, and GLP-1 receptor agonists. Ann N Y Acad Sci. 2004;1035:290–315. [DOI] [PubMed] [Google Scholar]
- 189.Li QX, Gao H, Guo YX, Wang BY, Hua RX, Gao L, Shang HW, Lu X, Xu JD. GLP-1 and Underlying Beneficial Actions in Alzheimer’s Disease, Hypertension, and NASH. Front Endocrinol (Lausanne). 2021;12:721198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 190.Candeias EM, Sebastiao IC, Cardoso SM, Correia SC, Carvalho CI, Placido AI, Santos MS, Oliveira CR, Moreira PI, Duarte AI. Gut-brain connection: The neuroprotective effects of the anti-diabetic drug liraglutide. World J Diabetes. 2015;6(6):807–827. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 191.Li Y, Glotfelty EJ, Karlsson T, Fortuno LV, Harvey BK, Greig NH. The metabolite GLP-1 (9–36) is neuroprotective and anti-inflammatory in cellular models of neurodegeneration. J Neurochem. 2021;159(5):867–886. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 192.Athauda D, Foltynie T. Protective effects of the GLP-1 mimetic exendin-4 in Parkinson’s disease. Neuropharmacology. 2018;136:260–270. [DOI] [PubMed] [Google Scholar]
- 193.Foltynie T, Athauda D. Repurposing anti-diabetic drugs for the treatment of Parkinson’s disease: Rationale and clinical experience. Recent Advances in Parkinson’s Disease. 2020;252:493–523. [DOI] [PubMed] [Google Scholar]
- 194.Robichaux WG 3rd, Cheng X. Intracellular cAMP Sensor EPAC: Physiology, Pathophysiology, and Therapeutics Development. Physiol Rev. 2018;98(2):919–1053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 195.Holz GG. Epac: A new cAMP-binding protein in support of glucagon-like peptide-1 receptor-mediated signal transduction in the pancreatic beta-cell. Diabetes. 2004;53(1):5–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 196.Delghandi MP, Johannessen M, Moens U. The cAMP signalling pathway activates CREB through PKA, p38 and MSK1 in NIH 3T3 cells. Cell Signal. 2005;17(11):1343–1351. [DOI] [PubMed] [Google Scholar]
- 197.Jolivalt CG, Fineman M, Deacon CF, Carr RD, Calcutt NA. GLP-1 signals via ERK in peripheral nerve and prevents nerve dysfunction in diabetic mice. Diabetes Obes Metab. 2011;13(11):990–1000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 198.Zhang S, Xue R, Geng Y, Wang H, Li W. Fisetin Prevents HT22 Cells From High Glucose-Induced Neurotoxicity via PI3K/Akt/CREB Signaling Pathway. Front Neurosci. 2020;14:241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 199.Wicinski M, Socha M, Malinowski B, Wodkiewicz E, Walczak M, Gorski K, Slupski M, Pawlak-Osinska K. Liraglutide and its Neuroprotective Properties-Focus on Possible Biochemical Mechanisms in Alzheimer’s Disease and Cerebral Ischemic Events. Int J Mol Sci. 2019;20(5):1050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 200.Chen LH, Hsu CY, Weng CF. Involvement of P53 and Bax/Bad triggering apoptosis in thioacetamide-induced hepatic epithelial cells. World J Gastroenterol. 2006;12(32):5175–5181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 201.Yang JL, Chen WY, Chen YP, Kuo CY, Chen SD. Activation of GLP-1 Receptor Enhances Neuronal Base Excision Repair via PI3K-AKT-Induced Expression of Apurinic/Apyrimidinic Endonuclease 1. Theranostics. 2016;6(12):2015–2027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 202.Malagelada C, Jin ZH, Greene LA. RTP801 Is Induced in Parkinson’s Disease and Mediates Neuron Death by Inhibiting Akt Phosphorylation/Activation. Journal of Neuroscience. 2008;28(53):14363–14371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 203.Timmons S, Coakley MF, Moloney AM, C ON. Akt signal transduction dysfunction in Parkinson’s disease. Neurosci Lett. 2009;467(1):30–35. [DOI] [PubMed] [Google Scholar]
- 204.Furlong RM, Lindsay A, Anderson KE, Hawkins PT, Sullivan AM, O’Neill C. The Parkinson’s disease gene PINK1 activates Akt via PINK1 kinase-dependent regulation of the phospholipid PI(3,4,5)P-3. J Cell Sci. 2019;132(20). [DOI] [PubMed] [Google Scholar]
- 205.Soutar MPM, Kempthorne L, Miyakawa S, Annuario E, Melandri D, Harley J, O’Sullivan GA, Wray S, Hancock DC, Cookson MR, Downward J, Carlton M, Plun-Favreau H. AKT signalling selectively regulates PINK1 mitophagy in SHSY5Y cells and human iPSC-derived neurons. Sci Rep-Uk. 2018;8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 206.Hemmings BA, Restuccia DF. The PI3K-PKB/Akt pathway. Cold Spring Harb Perspect Biol. 2015;7(4):a011189. [Google Scholar]
- 207.Wicinski M, Wodkiewicz E, Slupski M, Walczak M, Socha M, Malinowski B, Pawlak-Osinska K. Neuroprotective Activity of Sitagliptin via Reduction of Neuroinflammation beyond the Incretin Effect: Focus on Alzheimer’s Disease. Biomed Res Int. 2018;2018:6091014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 208.Katsurada K, Nakata M, Saito T, Zhang B, Maejima Y, Nandi SS, Sharma NM, Patel KP, Kario K, Yada T. Central Glucagon-like Peptide-1 Receptor Signaling via Brainstem Catecholamine Neurons Counteracts Hypertension in Spontaneously Hypertensive Rats. Sci Rep. 2019;9(1):12986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 209.Alvarez E, Martinez MD, Roncero I, Chowen JA, Garcia-Cuartero B, Gispert JD, Sanz C, Vazquez P, Maldonado A, de Caceres J, Desco M, Pozo MA, Blazquez E. The expression of GLP-1 receptor mRNA and protein allows the effect of GLP-1 on glucose metabolism in the human hypothalamus and brainstem. J Neurochem. 2005;92(4):798–806. [DOI] [PubMed] [Google Scholar]
- 210.Ma L, Ju P, Wang W, Wei J, Wang W, Zhao M, Ahmad KA, Wang Y, Chen J. Microglial Activation of GLP-1R Signaling in Neuropathic Pain Promotes Gene Expression Adaption Involved in Inflammatory Responses. Neural Plast. 2021;2021:9923537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 211.Campos RV, Lee YC, Drucker DJ. Divergent tissue-specific and developmental expression of receptors for glucagon and glucagon-like peptide-1 in the mouse. Endocrinology. 1994;134(5):2156–2164. [DOI] [PubMed] [Google Scholar]
- 212.Reiner DJ, Mietlicki-Baase EG, McGrath LE, Zimmer DJ, Bence KK, Sousa GL, Konanur VR, Krawczyk J, Burk DH, Kanoski SE, Hermann GE, Rogers RC, Hayes MR. Astrocytes Regulate GLP-1 Receptor-Mediated Effects on Energy Balance. J Neurosci. 2016;36(12):3531–3540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 213.Zhang Y, Sloan SA, Clarke LE, Caneda C, Plaza CA, Blumenthal PD, Vogel H, Steinberg GK, Edwards MS, Li G, Duncan JA 3rd, Cheshier SH, Shuer LM, Chang EF, Grant GA, Gephart MG, Barres BA. Purification and Characterization of Progenitor and Mature Human Astrocytes Reveals Transcriptional and Functional Differences with Mouse. Neuron. 2016;89(1):37–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 214.Zhang Y, Chen K, Sloan SA, Bennett ML, Scholze AR, O’Keeffe S, Phatnani HP, Guarnieri P, Caneda C, Ruderisch N, Deng S, Liddelow SA, Zhang C, Daneman R, Maniatis T, Barres BA, Wu JQ. An RNA-sequencing transcriptome and splicing database of glia, neurons, and vascular cells of the cerebral cortex. J Neurosci. 2014;34(36):11929–11947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 215.Olah M, Patrick E, Villani AC, Xu J, White CC, Ryan KJ, Piehowski P, Kapasi A, Nejad P, Cimpean M, Connor S, Yung CJ, Frangieh M, McHenry A, Elyaman W, Petyuk V, Schneider JA, Bennett DA, De Jager PL, Bradshaw EM. A transcriptomic atlas of aged human microglia. Nat Commun. 2018;9(1):539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 216.Darmanis S, Sloan SA, Croote D, Mignardi M, Chernikova S, Samghababi P, Zhang Y, Neff N, Kowarsky M, Caneda C, Li G, Chang SD, Connolly ID, Li YM, Barres BA, Gephart MH, Quake SR. Single-Cell RNA-Seq Analysis of Infiltrating Neoplastic Cells at the Migrating Front of Human Glioblastoma. Cell Reports. 2017;21(5):1399–1410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 217.Abraham MA, Lam TKT. Glucagon action in the brain. Diabetologia. 2016;59(7):1367–1371. [DOI] [PubMed] [Google Scholar]
- 218.Perry T, Lahiri DK, Sambamurti K, Chen D, Mattson MP, Egan JM, Greig NH. Glucagon-like peptide-1 decreases endogenous amyloid-beta peptide (Abeta) levels and protects hippocampal neurons from death induced by Abeta and iron. J Neurosci Res. 2003;72(5):603–612. [DOI] [PubMed] [Google Scholar]
- 219.Iwai T, Sawabe T, Tanimitsu K, Suzuki M, Sasaki-Hamada S, Oka J. Glucagon-like peptide-1 protects synaptic and learning functions from neuroinflammation in rodents. J Neurosci Res. 2014;92(4):446–454. [DOI] [PubMed] [Google Scholar]
- 220.Chen T, Tian P, Huang Z, Zhao X, Wang H, Xia C, Wang L, Wei H. Engineered commensal bacteria prevent systemic inflammation-induced memory impairment and amyloidogenesis via producing GLP-1. Appl Microbiol Biotechnol. 2018;102(17):7565–7575. [DOI] [PubMed] [Google Scholar]
- 221.Fang X, Zhou X, Miao Y, Han Y, Wei J, Chen T. Therapeutic effect of GLP-1 engineered strain on mice model of Alzheimer’s disease and Parkinson’s disease. AMB Express. 2020;10(1):80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 222.Li Y, Chigurupati S, Holloway HW, Mughal M, Tweedie D, Bruestle DA, Mattson MP, Wang Y, Harvey BK, Ray B, Lahiri DK, Greig NH. Exendin-4 ameliorates motor neuron degeneration in cellular and animal models of amyotrophic lateral sclerosis. Plos One. 2012;7(2):e32008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 223.Perry T, Holloway HW, Weerasuriya A, Mouton PR, Duffy K, Mattison JA, Greig NH. Evidence of GLP-1-mediated neuroprotection in an animal model of pyridoxine-induced peripheral sensory neuropathy. Exp Neurol. 2007;203(2):293–301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 224.Zhang M, Wu Y, Gao R, Chen X, Chen R, Chen Z. Glucagon-like peptide-1 analogs mitigate neuroinflammation in Alzheimer’s disease by suppressing NLRP2 activation in astrocytes. Mol Cell Endocrinol. 2022;542:111529. [DOI] [PubMed] [Google Scholar]
- 225.Li Y, Duffy KB, Ottinger MA, Ray B, Bailey JA, Holloway HW, Tweedie D, Perry T, Mattson MP, Kapogiannis D, Sambamurti K, Lahiri DK, Greig NH. GLP-1 receptor stimulation reduces amyloid-beta peptide accumulation and cytotoxicity in cellular and animal models of Alzheimer’s disease. J Alzheimers Dis. 2010;19(4):1205–1219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 226.Park JS, Kam TI, Lee S, Park H, Oh Y, Kwon SH, Song JJ, Kim D, Kim H, Jhaldiyal A, Na DH, Lee KC, Park EJ, Pomper MG, Pletnikova O, Troncoso JC, Ko HS, Dawson VL, Dawson TM, Lee S. Blocking microglial activation of reactive astrocytes is neuroprotective in models of Alzheimer’s disease. Acta Neuropathol Commun. 2021;9(1):78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 227.Zhang LQ, Zhang W, Li T, Yang T, Yuan X, Zhou Y, Zou Q, Yang H, Gao F, Tian Y, Mei W, Tian XB. GLP-1R activation ameliorated novel-object recognition memory dysfunction via regulating hippocampal AMPK/NF-kappaB pathway in neuropathic pain mice. Neurobiol Learn Mem. 2021;182:107463. [DOI] [PubMed] [Google Scholar]
- 228.Bomfim TR, Forny-Germano L, Sathler LB, Brito-Moreira J, Houzel JC, Decker H, Silverman MA, Kazi H, Melo HM, McClean PL, Holscher C, Arnold SE, Talbot K, Klein WL, Munoz DP, Ferreira ST, De Felice FG. An anti-diabetes agent protects the mouse brain from defective insulin signaling caused by Alzheimer’s disease- associated Abeta oligomers. J Clin Invest. 2012;122(4):1339–1353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 229.Wang Y, Chen S, Xu Z, Chen S, Yao W, Gao X. GLP-1 receptor agonists downregulate aberrant GnT-III expression in Alzheimer’s disease models through the Akt/GSK-3beta/beta-catenin signaling. Neuropharmacology. 2018;131:190–199. [DOI] [PubMed] [Google Scholar]
- 230.Bomba M, Granzotto A, Castelli V, Onofrj M, Lattanzio R, Cimini A, Sensi SL. Exenatide Reverts the High-Fat-Diet-Induced Impairment of BDNF Signaling and Inflammatory Response in an Animal Model of Alzheimer’s Disease. J Alzheimers Dis. 2019;70(3):793–810. [DOI] [PubMed] [Google Scholar]
- 231.Yu SJ, Chen S, Yang YY, Glotfelty EJ, Jung J, Kim HK, Choi HI, Choi DS, Hoffer BJ, Greig NH, Wang Y. PT320, Sustained-Release Exendin-4, Mitigates L-DOPA-Induced Dyskinesia in a Rat 6-Hydroxydopamine Model of Parkinson’s Disease. Front Neurosci. 2020;14:785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 232.Wang V, Kuo TT, Huang EY, Ma KH, Chou YC, Fu ZY, Lai LW, Jung J, Choi HI, Choi DS, Li Y, Olson L, Greig NH, Hoffer BJ, Chen YH. Sustained Release GLP-1 Agonist PT320 Delays Disease Progression in a Mouse Model of Parkinson’s Disease. ACS Pharmacol Transl Sci. 2021;4(2):858–869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 233.Meier JJ. GLP-1 receptor agonists for individualized treatment of type 2 diabetes mellitus. Nat Rev Endocrinol. 2012;8(12):728–742. [DOI] [PubMed] [Google Scholar]
- 234.Li YZ, Bader M, Tamargo I, Rubovitch V, Tweedie D, Pick CG, Greig NH. Liraglutide is neurotrophic and neuroprotective in neuronal cultures and mitigates mild traumatic brain injury in mice. Journal of Neurochemistry. 2015;135(6):1203–1217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 235.Hansen HH, Fabricius K, Barkholt P, Kongsbak-Wismann P, Schlumberger C, Jelsing J, Terwel D, Termont A, Pyke C, Knudsen LB, Vrang N. Long-Term Treatment with Liraglutide, a Glucagon-Like Peptide-1 (GLP-1) Receptor Agonist, Has No Effect on beta-Amyloid Plaque Load in Two Transgenic APP/PS1 Mouse Models of Alzheimer’s Disease. Plos One. 2016;11(7):e0158205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 236.Hunter K, Holscher C. Drugs developed to treat diabetes, liraglutide and lixisenatide, cross the blood brain barrier and enhance neurogenesis. BMC Neurosci. 2012;13:33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 237.Paladugu L, Gharaibeh A, Kolli N, Learman C, Hall TC, Li L, Rossignol J, Maiti P, Dunbar GL. Liraglutide Has Anti-Inflammatory and Anti-Amyloid Properties in Streptozotocin-Induced and 5xFAD Mouse Models of Alzheimer’s Disease. Int J Mol Sci. 2021;22(2):860. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 238.Cai HY, Yang JT, Wang ZJ, Zhang J, Yang W, Wu MN, Qi JS. Lixisenatide reduces amyloid plaques, neurofibrillary tangles and neuroinflammation in an APP/PS1/tau mouse model of Alzheimer’s disease. Biochem Biophys Res Commun. 2018;495(1):1034–1040. [DOI] [PubMed] [Google Scholar]
- 239.McClean PL, Holscher C. Lixisenatide, a drug developed to treat type 2 diabetes, shows neuroprotective effects in a mouse model of Alzheimer’s disease. Neuropharmacology. 2014;86:241–258. [DOI] [PubMed] [Google Scholar]
- 240.Cai HY, Holscher C, Yue XH, Zhang SX, Wang XH, Qiao F, Yang W, Qi JS. Lixisenatide rescues spatial memory and synaptic plasticity from amyloid beta protein-induced impairments in rats. Neuroscience. 2014;277:6–13. [DOI] [PubMed] [Google Scholar]
- 241.Cai HY, Wang ZJ, Holscher C, Yuan L, Zhang J, Sun P, Li J, Yang W, Wu MN, Qi JS. Lixisenatide attenuates the detrimental effects of amyloid beta protein on spatial working memory and hippocampal neurons in rats. Behav Brain Res. 2017;318:28–35. [DOI] [PubMed] [Google Scholar]
- 242.Liu W, Jalewa J, Sharma M, Li G, Li L, Holscher C. Neuroprotective effects of lixisenatide and liraglutide in the 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine mouse model of Parkinson’s disease. Neuroscience. 2015;303:42–50. [DOI] [PubMed] [Google Scholar]
- 243.Kosaraju J, Madhunapantula SV, Chinni S, Khatwal RB, Dubala A, Muthureddy Nataraj SK, Basavan D. Dipeptidyl peptidase-4 inhibition by Pterocarpus marsupium and Eugenia jambolana ameliorates streptozotocin induced Alzheimer’s disease. Behav Brain Res. 2014;267:55–65. [DOI] [PubMed] [Google Scholar]
- 244.Qin L, Chong T, Rodriguez R, Pugazhenthi S. Glucagon-Like Peptide-1-Mediated Modulation of Inflammatory Pathways in the Diabetic Brain: Relevance to Alzheimer’s Disease. Curr Alzheimer Res. 2016;13(12):1346–1355. [DOI] [PubMed] [Google Scholar]
- 245.Kosaraju J, Holsinger RMD, Guo L, Tam KY. Linagliptin, a Dipeptidyl Peptidase-4 Inhibitor, Mitigates Cognitive Deficits and Pathology in the 3xTg-AD Mouse Model of Alzheimer’s Disease. Mol Neurobiol. 2017;54(8):6074–6084. [DOI] [PubMed] [Google Scholar]
- 246.Li Z, Zhang Y, Meng X, Li M, Cao W, Yang J, Xu X, Liu W, Li W, Cai Q, Wang S, Ma G, Liu Z, Huang G. A novel DPP-4 inhibitor Gramcyclin A attenuates cognitive deficits in APP/PS1/tau triple transgenic mice via enhancing brain GLP-1-dependent glucose uptake. Phytother Res. 2022;36(3):1297–1309. [DOI] [PubMed] [Google Scholar]
- 247.Zhang LY, Zhang LP, Li YW, Li L, Melchiorsen JU, Rosenkilde M, Holscher C. The Novel Dual GLP-1/GIP Receptor Agonist DA-CH5 Is Superior to Single GLP-1 Receptor Agonists in the MPTP Model of Parkinson?s Disease. J Parkinson Dis. 2020;10(2):523–542. [DOI] [PubMed] [Google Scholar]
- 248.Salles GN, Calio ML, Holscher C, Pacheco-Soares C, Porcionatto M, Lobo AO. Neuroprotective and restorative properties of the GLP-1/GIP dual agonist DA-JC1 compared with a GLP-1 single agonist in Alzheimer’s disease. Neuropharmacology. 2020;162:107813. [DOI] [PubMed] [Google Scholar]
- 249.Maskery M, Goulding EM, Gengler S, Melchiorsen JU, Rosenkilde MM, Holscher C. The Dual GLP-1/GIP Receptor Agonist DA4-JC Shows Superior Protective Properties Compared to the GLP-1 Analogue Liraglutide in the APP/PS1 Mouse Model of Alzheimer’s Disease. Am J Alzheimers Dis Other Demen. 2020;35:1533317520953041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 250.Cai HY, Yang D, Qiao J, Yang JT, Wang ZJ, Wu MN, Qi JS, Holscher C. A GLP-1/GIP Dual Receptor Agonist DA4-JC Effectively Attenuates Cognitive Impairment and Pathology in the APP/PS1/Tau Model of Alzheimer’s Disease. J Alzheimers Dis. 2021;83(2):799–818. [DOI] [PubMed] [Google Scholar]
- 251.Tamargo IA, Bader M, Li Y, Yu SJ, Wang Y, Talbot K, DiMarchi RD, Pick CG, Greig NH. Novel GLP-1R/GIPR co-agonist “twincretin” is neuroprotective in cell and rodent models of mild traumatic brain injury. Exp Neurol. 2017;288:176–186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 252.Pathak NM, Pathak V, Gault VA, McClean S, Irwin N, Flatt PR. Novel dual incretin agonist peptide with antidiabetic and neuroprotective potential. Biochem Pharmacol. 2018;155:264–274. [DOI] [PubMed] [Google Scholar]
- 253.Jiao JJ, Holscher C, Li T, Dong XF, Qu XS, Cao Y, Wu MN, Wang ZJ, Qi JS. [GLP-1/GIP/Gcg receptor Triagonist improves the cognitive behaviors in triple-transgenic mice of Alzheimer’s disease]. Sheng Li Xue Bao. 2017;69(2):135–145. [PubMed] [Google Scholar]
- 254.Tai J, Liu W, Li Y, Li L, Holscher C. Neuroprotective effects of a triple GLP-1/GIP/glucagon receptor agonist in the APP/PS1 transgenic mouse model of Alzheimer’s disease. Brain Res. 2018;1678:64–74. [DOI] [PubMed] [Google Scholar]
- 255.Li Y, Glotfelty EJ, Namdar I, Tweedie D, Olson L, Hoffer BJ, DiMarchi RD, Pick CG, Greig NH. Neurotrophic and neuroprotective effects of a monomeric GLP-1/GIP/Gcg receptor triagonist in cellular and rodent models of mild traumatic brain injury. Exp Neurol. 2020;324:113113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 256.Wiberg S, Hassager C, Schmidt H, Thomsen JH, Frydland M, Lindholm MG, Hofsten DE, Engstrom T, Kober L, Moller JE, Kjaergaard J. Neuroprotective Effects of the Glucagon-Like Peptide-1 Analog Exenatide After Out-of-Hospital Cardiac Arrest: A Randomized Controlled Trial. Circulation. 2016;134(25):2115–2124. [DOI] [PubMed] [Google Scholar]
- 257.Mullins RJ, Mustapic M, Chia CW, Carlson O, Gulyani S, Tran J, Li Y, Mattson MP, Resnick S, Egan JM, Greig NH, Kapogiannis D. A Pilot Study of Exenatide Actions in Alzheimer’s Disease. Curr Alzheimer Res. 2019;16(8):741–752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 258.Aviles-Olmos I, Dickson J, Kefalopoulou Z, Djamshidian A, Ell P, Soderlund T, Whitton P, Wyse R, Isaacs T, Lees A, Limousin P, Foltynie T. Exenatide and the treatment of patients with Parkinson’s disease. J Clin Invest. 2013;123(6):2730–2736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 259.Gejl M, Gjedde A, Egefjord L, Moller A, Hansen SB, Vang K, Rodell A, Braendgaard H, Gottrup H, Schacht A, Moller N, Brock B, Rungby J. In Alzheimer’s Disease, 6-Month Treatment with GLP-1 Analog Prevents Decline of Brain Glucose Metabolism: Randomized, Placebo-Controlled, Double-Blind Clinical Trial. Front Aging Neurosci. 2016;8:108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 260.Norregaard PK, Deryabina MA, Tofteng Shelton P, Fog JU, Daugaard JR, Eriksson PO, Larsen LF, Jessen L. A novel GIP analogue, ZP4165, enhances glucagon-like peptide-1-induced body weight loss and improves glycaemic control in rodents. Diabetes Obes Metab. 2018;20(1):60–68. [DOI] [PubMed] [Google Scholar]
- 261.Ambery P, Parker VE, Stumvoll M, Posch MG, Heise T, Plum-Moerschel L, Tsai LF, Robertson D, Jain M, Petrone M, Rondinone C, Hirshberg B, Jermutus L. MEDI0382, a GLP-1 and glucagon receptor dual agonist, in obese or overweight patients with type 2 diabetes: a randomised, controlled, double-blind, ascending dose and phase 2a study. Lancet. 2018;391(10140):2607–2618. [DOI] [PubMed] [Google Scholar]
- 262.Goebel B, Schiavon M, Visentin R, Riz M, Man CD, Cobelli C, Klabunde T. Effects of the Novel Dual GLP-1R/GCGR Agonist SAR425899 on Postprandial Glucose Metabolism in Overweight/Obese Subjects with Type 2 Diabetes. Diabetes. 2018;67:72-OR. [Google Scholar]
- 263.Frias JP, Bastyr EJ 3rd, Vignati L, Tschop MH, Schmitt C, Owen K, Christensen RH, DiMarchi RD. The Sustained Effects of a Dual GIP/GLP-1 Receptor Agonist, NNC0090–2746, in Patients with Type 2 Diabetes. Cell Metab. 2017;26(2):343–352 e342. [DOI] [PubMed] [Google Scholar]
- 264.Kim J, Lee S, Lee SH, Jung S, Kim Y, Choi I, Kim S. Neuroprotective effects of HM15211, a novel long-acting GLP-1/GIP/glucagon triple agonist in the neurodegenerative disease models. Diabetologia. 2018;61:S84–S85. [Google Scholar]
- 265.Goldenberg R, Gantz I, Andryuk PJ, O’Neill EA, Kaufman KD, Lai E, Wang YN, Suryawanshi S, Engel SS. Randomized clinical trial comparing the efficacy and safety of treatment with the once-weekly dipeptidyl peptidase-4 (DPP-4) inhibitor omarigliptin or the once-daily DPP-4 inhibitor sitagliptin in patients with type 2 diabetes inadequately controlled on metformin monotherapy. Diabetes Obes Metab. 2017;19(3):394–400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 266.Ayoub BM, Mowaka S, Safar MM, Ashoush N, Arafa MG, Michel HE, Tadros MM, Elmazar MM, Mousa SA. Repositioning of Omarigliptin as a once-weekly intranasal Anti-parkinsonian Agent. Sci Rep-Uk. 2018;8:8959. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 267.Kim SH, Yoo JH, Lee WJ, Park CY. Gemigliptin: An Update of Its Clinical Use in the Management of Type 2 Diabetes Mellitus. Diabetes Metab J. 2016;40(5):339–353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 268.Mathieu C, Degrande E. Vildagliptin: a new oral treatment for type 2 diabetes mellitus. Vasc Health Risk Man. 2008;4(6):1349–1360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 269.Kishimoto M Teneligliptin: a DPP-4 inhibitor for the treatment of type 2 diabetes. Diabetes Metab Syndr. 2013;6:187–195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 270.Nishio S, Abe M, Ito H. Anagliptin in the treatment of type 2 diabetes: safety, efficacy, and patient acceptability. Diabetes Metab Syndr. 2015;8:163–171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 271.Svenningsson P, Wirdefeldt K, Yin L, Fang F, Markaki I, Efendic S, Ludvigsson JF. Reduced incidence of Parkinson’s disease after dipeptidyl peptidase-4 inhibitors-A nationwide case-control study. Mov Disord. 2016;31(9):1422–1423. [DOI] [PubMed] [Google Scholar]
- 272.Brauer R, Wei L, Ma T, Athauda D, Girges C, Vijiaratnam N, Auld G, Whittlesea C, Wong I, Foltynie T. Diabetes medications and risk of Parkinson’s disease: a cohort study of patients with diabetes. Brain. 2020;143(10):3067–3076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 273.Barkas F, Elisaf M, Milionis H. Protection against stroke with glucagon-like peptide 1 receptor agonists: a systematic review and meta-analysis. Eur J Neurol. 2019;26(4):559–565. [DOI] [PubMed] [Google Scholar]
- 274.Gejl M, Brock B, Egefjord L, Vang K, Rungby J, Gjedde A. Blood-Brain Glucose Transfer in Alzheimer’s disease: Effect of GLP-1 Analog Treatment. Sci Rep. 2017;7(1):17490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 275.Watson KT, Wroolie TE, Tong G, Foland-Ross LC, Frangou S, Singh M, McIntyre RS, Roat-Shumway S, Myoraku A, Reiss AL, Rasgon NL. Neural correlates of liraglutide effects in persons at risk for Alzheimer’s disease. Behav Brain Res. 2019;356:271–278. [DOI] [PubMed] [Google Scholar]
- 276.Hogg E, Wu T, Bresee C, Wertheimer J, Malatt C, Tan E, Pomeroy H, Nuno M, Wyse R, Taglioti M. A Phase II, Randomized, Double-Blinded, Placebo-Controlled Trial of Liraglutide in Parkinson’s Disease. In. The Lancet; 2022. [Google Scholar]
- 277.Femminella GD, Frangou E, Love SB, Busza G, Holmes C, Ritchie C, Lawrence R, McFarlane B, Tadros G, Ridha BH, Bannister C, Walker Z, Archer H, Coulthard E, Underwood BR, Prasanna A, Koranteng P, Karim S, Junaid K, McGuinness B, Nilforooshan R, Macharouthu A, Donaldson A, Thacker S, Russell G, Malik N, Mate V, Knight L, Kshemendran S, Harrison J, Holscher C, Brooks DJ, Passmore AP, Ballard C, Edison P. Correction to: Evaluating the effects of the novel GLP-1 analogue liraglutide in Alzheimer’s disease: study protocol for a randomised controlled trial (ELAD study). Trials. 2020;21(1):660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 278.Edison P, Femminella GD, Ritchie CW, Holmes C, Walker Z, Ridha BH, Raza S, Livingston NR, Nowell J, Busza G, Frangou E, Love S, Williams G, Lawrence RM, McFarlane B, Archer H, Coulthard E, Underwood B, Koranteng P, Karim S, Bannister C, Perneczky R, Prasanna A, Junaid K, McGuinness B, Nilforooshan R, Macharouthu A, Donaldson A, Thacker S, Russell G, Malik N, Mate V, Knight L, Kshemendran S, Holscher C, Mansouri A, Chester-Jones M, Holmes J, Williams SCR, Brooks DJ, Harrison JE, Tadros G, Passmore AP, Ballard C. Evaluation of liraglutide in the treatment of Alzheimer’s disease. Alzheimers Dement. 2021;17:e057848. [Google Scholar]
- 279.Center C-SM, Trust TCPs, S. NNA. Safety and Efficacy of Liraglutide in Parkinson’s Disease. In. https://ClinicalTrials.gov/show/NCT02953665; 2022.
- 280.Aviles-Olmos I, Dickson J, Kefalopoulou Z, Djamshidian A, Kahan J, Ell P, Whitton P, Wyse R, Isaacs T, Lees A, Limousin P, Foltynie T. Motor and Cognitive Advantages Persist 12 Months After Exenatide Exposure in Parkinson’s Disease. J Parkinson Dis. 2014;4(3):337–344. [DOI] [PubMed] [Google Scholar]
- 281.Athauda D, Gulyani S, Karnati HK, Li Y, Tweedie D, Mustapic M, Chawla S, Chowdhury K, Skene SS, Greig NH, Kapogiannis D, Foltynie T. Utility of Neuronal-Derived Exosomes to Examine Molecular Mechanisms That Affect Motor Function in Patients With Parkinson Disease: A Secondary Analysis of the Exenatide-PD Trial. JAMA Neurol. 2019;76(4):420–429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 282.Athauda D, Maclagan K, Budnik N, Zampedri L, Hibbert S, Skene SS, Chowdhury K, Aviles-Olmos I, Limousin P, Foltynie T. What Effects Might Exenatide have on Non-Motor Symptoms in Parkinson’s Disease: A Post Hoc Analysis. J Parkinsons Dis. 2018;8(2):247–258. [DOI] [PubMed] [Google Scholar]
- 283.Novo Nordisk AS. A Research Study Investigating Semaglutide in People With Early Alzheimer’s Disease (EVOKE Plus). In. https://ClinicalTrials.gov/show/NCT04777409; 2024.
- 284.Novo Nordisk AS. A Research Study Investigating Semaglutide in People With Early Alzheimer’s Disease (EVOKE). In. https://ClinicalTrials.gov/show/NCT04777396; 2024.
- 285.Oxford Uo. Does semaglutide change the build up of Alzheimer’s disease proteins in people at risk? In. ISRCTN registry; 2022. [Google Scholar]
- 286.Oslo University H. GLP1R in Parkinson’s Disease. In. https://ClinicalTrials.gov/show/NCT03659682; 2024.
- 287.Quaranta L, Bruttini C, Micheletti E, Konstas AGP, Michelessi M, Oddone F, Katsanos A, Sbardella D, De Angelis G, Riva I. Glaucoma and neuroinflammation: An overview. Surv Ophthalmol. 2021;66(5):693–713. [DOI] [PubMed] [Google Scholar]
- 288.Wei X, Cho KS, Thee EF, Jager MJ, Chen DF. Neuroinflammation and microglia in glaucoma: time for a paradigm shift. J Neurosci Res. 2019;97(1):70–76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 289.Pinazo-Duran MD, Munoz-Negrete FJ, Sanz-Gonzalez SM, Benitez-Del-Castillo J, Gimenez-Gomez R, Valero-Vello M, Zanon-Moreno V, Garcia-Medina JJ. The role of neuroinflammation in the pathogenesis of glaucoma neurodegeneration. Prog Brain Res. 2020;256(1):99–124. [DOI] [PubMed] [Google Scholar]
- 290.Apolloni S, Milani M, D’Ambrosi N. Neuroinflammation in Friedreich’s Ataxia. Int J Mol Sci. 2022;23(11):6297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 291.Khan W, Corben LA, Bilal H, Vivash L, Delatycki MB, Egan GF, Harding IH. Neuroinflammation in the Cerebellum and Brainstem in Friedreich Ataxia: An [18F]-FEMPA PET Study. Mov Disord. 2022;37(1):218–224. [DOI] [PubMed] [Google Scholar]
- 292.Imbault V, Dionisi C, Naeije G, Communi D, Pandolfo M. Cerebrospinal Fluid Proteomics in Friedreich Ataxia Reveals Markers of Neurodegeneration and Neuroinflammation. Front Neurosci. 2022;16:885313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 293.Igoillo-Esteve M, Oliveira AF, Cosentino C, Fantuzzi F, Demarez C, Toivonen S, Hu A, Chintawar S, Lopes M, Pachera N, Cai Y, Abdulkarim B, Rai M, Marselli L, Marchetti P, Tariq M, Jonas JC, Boscolo M, Pandolfo M, Eizirik DL, Cnop M. Exenatide induces frataxin expression and improves mitochondrial function in Friedreich ataxia. JCI Insight. 2020;5(2):e134221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 294.Botfield HF, Uldall MS, Westgate CSJ, Mitchell JL, Hagen SM, Gonzalez AM, Hodson DJ, Jensen RH, Sinclair AJ. A glucagon-like peptide-1 receptor agonist reduces intracranial pressure in a rat model of hydrocephalus. Science Translational Medicine. 2017;9(404):eaan0972. [DOI] [PubMed] [Google Scholar]
- 295.Grech O, Mollan SP, Wakerley BR, Alimajstorovic Z, Lavery GG, Sinclair AJ. Emerging themes in idiopathic intracranial hypertension. J Neurol. 2020;267(12):3776–3784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 296.Sudhakar P Commentary: The role of inflammation in idiopathic intracranial hypertension. Indian J Ophthalmol. 2021;69(6):1506–1507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 297.Sinclair AJ, Ball AK, Burdon MA, Clarke CE, Stewart PM, Cumow SJ, Rauz S. Exploring the pathogenesis of IIH: An inflammatory perspective. J Neuroimmunol. 2008;201:212–220. [DOI] [PubMed] [Google Scholar]
- 298.Mollan SP, Grech O, Sinclair AJ. Headache attributed to idiopathic intracranial hypertension and persistent post-idiopathic intracranial hypertension headache: A narrative review. Headache. 2021;61(6):808–816. [DOI] [PubMed] [Google Scholar]
- 299.Mitchell JL, Lyons H, Walker J, Yiangou A, Grech O, Alimajstorovic Z, Greig NH, Li Y, Tsermoulas G, Brock K, Mollan SP, Sinclair AJ. The effect of exenatide on intracranial pressure in idiopathic intracranial hypertension, IIH pressure: A randomised clinical trial. In. MedRxiv; 2022. [Google Scholar]
- 300.Bader M, Li Y, Lecca D, Rubovitch V, Tweedie D, Glotfelty E, Rachmany L, Kim HK, Choi HI, Hoffer BJ, Pick CG, Greig NH, Kim DS. Pharmacokinetics and efficacy of PT302, a sustained-release Exenatide formulation, in a murine model of mild traumatic brain injury. Neurobiol Dis. 2019;124:439–453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 301.Tweedie D, Rachmany L, Rubovitch V, Lehrmann E, Zhang YQ, Becker KG, Perez E, Miller J, Hoffer BJ, Greig NH, Pick CG. Exendin-4, a glucagon-like peptide-1 receptor agonist prevents mTBI-induced changes in hippocampus gene expression and memory deficits in mice. Experimental Neurology. 2013;239:170–182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 302.Tweedie D, Rachrnany L, Rubovitch V, Li YZ, Holloway HW, Lehrmann E, Zhang YQ, Becker KG, Perez E, Hoffer BJ, Pick CG, Greig NH. Blast traumatic brain injury-induced cognitive deficits are attenuated by preinjury or postinjury treatment with the glucagon-like peptide-1 receptor agonist, exendin-4. Alzheimers & Dementia. 2016;12(1):34–48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 303.Eakin K, Li YZ, Chiang YH, Hoffer BJ, Rosenheim H, Greig NH, Miller JP. Exendin-4 Ameliorates Traumatic Brain Injury-Induced Cognitive Impairment in Rats. Plos One. 2013;8(12):e82016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 304.Rachmany L, Tweedie D, Li YZ, Rubovitch V, Holloway HW, Miller J, Hoffer BJ, Greig NH, Pick CG. Exendin-4 induced glucagon-like peptide-1 receptor activation reverses behavioral impairments of mild traumatic brain injury in mice. Age. 2013;35(5):1621–1636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 305.Rachmany L, Tweedie D, Rubovitch V, Li YZ, Holloway HW, Kim DS, Ratliff WA, Saykally JN, Citron BA, Hoffer BJ, Greig NH, Pick CG. Exendin-4 attenuates blast traumatic brain injury induced cognitive impairments, losses of synaptophysin and in vitro TBI-induced hippocampal cellular degeneration. Sci Rep-Uk. 2017;7:3735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 306.Shan Y, Tan S, Lin Y, Liao S, Zhang B, Chen X, Wang J, Deng Z, Zeng Q, Zhang L, Wang Y, Hu X, Qiu W, Peng L, Lu Z. The glucagon-like peptide-1 receptor agonist reduces inflammation and blood-brain barrier breakdown in an astrocyte-dependent manner in experimental stroke. J Neuroinflammation. 2019;16(1):242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 307.Xie Z, Enkhjargal B, Wu L, Zhou K, Sun C, Hu X, Gospodarev V, Tang J, You C, Zhang JH. Exendin-4 attenuates neuronal death via GLP-1R/PI3K/Akt pathway in early brain injury after subarachnoid hemorrhage in rats. Neuropharmacology. 2018;128:142–151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 308.Liu C, Sun S, Xie J, Li H, Li T, Wu Q, Zhang Y, Bai X, Wang J, Wang X, Li Z, Wang W. GLP-1R Agonist Exendin-4 Protects Against Hemorrhagic Transformation Induced by rtPA After Ischemic Stroke via the Wnt/beta-Catenin Signaling Pathway. Mol Neurobiol. 2022;59(6):3649–3664. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 309.Nizari S, Basalay M, Chapman P, Korte N, Korsak A, Christie IN, Theparambil SM, Davidson SM, Reimann F, Trapp S, Yellon DM, Gourine AV. Glucagon-like peptide-1 (GLP-1) receptor activation dilates cerebral arterioles, increases cerebral blood flow, and mediates remote (pre)conditioning neuroprotection against ischaemic stroke. Basic Res Cardiol. 2021;116(1):32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 310.Song S, Guo RY, Mehmood A, Zhang L, Yin BW, Yuan CC, Zhang HI, Guo L, Li B. Liraglutide attenuate central nervous inflammation and demyelination through AMPK and pyroptosis-related NLRP3 pathway. Cns Neurosci Ther. 2022;28(3):422–434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 311.Ammar RA, Mohamed AF, Kamal MM, Safar MM, Abdelkader NF. Neuroprotective effect of liraglutide in an experimental mouse model of multiple sclerosis: role of AMPK/SIRT1 signaling and NLRP3 inflammasome. Inflammopharmacology. 2022;30(3):919–934. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 312.DellaValle B, Brix GS, Brock B, Gejl M, Landau AM, Moller A, Rungby J, Larsen A. Glucagon-Like Peptide-1 Analog, Liraglutide, Delays Onset of Experimental Autoimmune Encephalitis in Lewis Rats. Front Pharmacol. 2016;7:433. [DOI] [PMC free article] [PubMed] [Google Scholar]