

Abstract
Purpose of Review
Synthesize the clinical, epidemiological, and preclinical evidence for antenatal programming of hypertension and critically appraise paradigms and paradoxes to improve translation.
Recent Findings
Clinical and epidemiological studies persistently demonstrate that antenatal factors contribute to programmed hypertension under the developmental origins of health and disease framework, including lower birth weight, preterm birth, and fetal growth restriction. Preclinical mechanisms include preeclampsia, maternal diabetes, maternal undernutrition, and antenatal corticosteroid exposure. However, clinical and epidemiological studies to date have largely failed to adequately identify, discuss, and mitigate many sources and types of bias in part due to heterogeneous study designs and incomplete adherence to scientific rigor. These limitations have led to incomplete and biased paradigms as well as persistent paradoxes that have significantly limited translation into clinical and population health interventions.
Summary
Improved understanding of these paradigms and paradoxes will allow us to substantially move the field forward.
Keywords: cardiovascular disease, causal inference, developmental origins of health and disease, growth restriction, life course, low birth weight, nephron number, preterm birth
INTRODUCTION
Hypertension remains the leading modifiable risk factor for cardiovascular disease, contributing to over 7.6 million premature deaths worldwide each year [1]. High blood pressure in childhood and adolescence is associated with an increased risk of adult hypertension and worse cardiovascular outcomes later in life [2, 3]. Globally, prevalence of youth-onset hypertension before 20 years of age has increased over the past two decades from 1.3% in the 1990s to 6% from 2010–2014 [4]. An additional 10% of children and adolescents are estimated to have elevated blood pressure (i.e., high blood pressure that does not meet criteria for hypertension) and are thus at greater risk for progression to hypertension [5]. This increase in blood pressure early in life has been associated with a 43% increase in hypertension-attributable deaths and an 86% increase in the years of life lived with disability among young adults from 1990–2019 [6].
Despite its importance, and the depth and breadth of hypertension research over the past 100 years, key mechanisms for hypertension development remain undefined. In particular, the mechanisms related to key early-life risk factors are incompletely understood. While the developmental origins of health and disease theory has been crucial to our understanding of hypertension development, many of these early hypotheses have proved overly simplistic and have created new paradoxes [7, 8]. Succinctly put, programming of hypertension can occur when antenatal—the period from conception to birth—adaptive alterations occur in response to a deleterious fetal environment to improve short-term survival (termed developmental plasticity), but that in the long term lead to maladaptive physiological changes that program an increased risk of hypertension (Table 1) [9, 10]. While certain factors such as growth restriction or maternal preeclampsia may increase the risk of subsequent hypertension in affected offspring, the precise clinical relevance of such early-life factors remains poorly defined [11]. There is a subsequent disconnect between key preclinical data and epidemiological evidence [12]. Consequently, despite the rapid advancements in the field, there has been relatively little translation of these important findings to clinical practice and health outcomes.
Table 1.
Common Terms Relevant to Programmed Hypertension
| Term | Definition |
|---|---|
| Antenatal | The time period from conception to birth |
| Perinatal | Generally defined as the time period consisting of the four weeks prior to and the four weeks after birth |
| Developmental plasticity | Antenatal fetal physiological, structural, and metabolic adaptive alterations in response to a deleterious fetal environment to improve short-term survival [9] |
| Developmental origins of health and disease | Antenatal and early-life exposures that induce developmental plasticity of sufficient severity, number, timing, or duration to lead to persistent, maladaptive physiological, structural, and metabolic changes that contribute to disease development later in life [10] |
| Small for gestational age | Statistical classification of lower than expected growth given a newborn infant’s genetic potential. Typically defined as birth weight less than the 10th percentile for gestational age and sex [29] |
| Fetal growth restriction (also known as intrauterine growth restriction) | Pathologic reduced growth potential given a fetus’s genetic potential. Defined as both low standardized growth parameters combined with abnormal blood flow in the uterine artery [29]. Subdivided into symmetric (global impairment of cellular hyperplasia) versus asymmetric (greater decrease in abdominal size relative to head circumference) or early versus late (±32 weeks’ gestation) |
This review will summarize the clinical, epidemiological, and preclinical evidence for antenatal programming of hypertension and emphasize gaps in the field in the context of inadequate scientific rigor within current paradigms. In particular, we will highlight persistent paradoxes, such as the interplay among preterm birth, lower birth weight, and growth restriction, and make suggestions on how we can move the field forward with the goal to resolve paradoxes and define new paradigms. This will assist us with improving translational research by better aligning preclinical models with clinical conditions, encouraging novel and rigorous epidemiological and statistical methods, and supporting clinical trials focused on primordial and primary prevention strategies, including at the population level [13, 14]. We will then be in a much better position to prevent, mitigate, and treat hypertension and improve cardiovascular health outcomes across the life course.
CLINICAL AND EPIDEMIOLOGICAL EVIDENCE
Key Studies of Hypertension Programming over the Life Course
A multitude of studies have demonstrated that exposures across the life course contribute to age-related increases in blood pressure and ultimately the onset of hypertension. Lower birth weight (<2500 g) and preterm birth (gestational age <37 weeks at birth) are among the most well-studied early-life exposures related to hypertension in the developmental origins of health and disease field. Across populations, birth weight has consistently had an inverse relationship with blood pressure and this relationship strengthens over the life course [15]. Systematic reviews have generally estimated that systolic blood pressure is approximately 2 mmHg higher for every 1-kg lower birth weight [16]. However, there remains robust debate about the functional form of this relationship such that some studies have observed an inverse linear association while others have observed a J (or U)-shaped association [17, 18]. Temporal trends in maternal nutrition and birth weights may in part explain these differences across studies. In addition, the impact of birth weight on subsequent blood pressure appears to be influenced in part by early-life growth trajectories such that children who experience too rapid “catch-up” growth may have greater increases in systolic blood pressure compared to those who maintain relatively smaller growth trajectories throughout their lifetime [16]. However, we have not been able to precisely define “adverse” growth trajectories, particularly trajectories that are clinically meaningful and evidence based.
In addition to absolute blood pressure values, blood pressure variability (itself an independent risk factor for cardiovascular disease in adults) has been associated with birth weight [19]. More recent studies have examined other potential pathways related to birth weight by which early-life exposures influence blood pressure. In particular, maternal weight and adiposity are associated with offspring blood pressure, which may be independent of birth weight [20], thereby supporting the concept that multiple pathways exist linking early-life exposures with hypertension programming.
Intimately related to birth weight, preterm birth has been studied extensively as a predictor of childhood blood pressure and adult hypertension. Most studies have shown that individuals born preterm have, on average, an approximately 3–4 mmHg higher systolic blood pressure compared to peers born at term [21, 22], with some studies suggesting potential dose-dependent effects in individuals born very or extremely preterm. Importantly, this increased risk of hypertension is observed in females as well as males [23]. In addition to having higher blood pressure in adolescence, individuals born extremely preterm (<28 weeks’ gestation) or extremely low birth weight (<1000 g) had a greater increase in asleep ambulatory blood pressure from age 18 to 25 years compared to controls with normal birth weight [24]. This is of particular interest because isolated nocturnal hypertension is an independent risk factor for cardiovascular disease in adults. When preterm birth and lower birth weight have been investigated simultaneously as risk factors, including among those who had fetal growth restriction/were small for gestational age (i.e., birth weight relative to gestational age), these relationships become more complex but often suggest an additive or multiplicative effect on hypertension risk. The Young Finns study observed that individuals who were born preterm and were small for gestational age had over 7-mmHg higher systolic blood pressure in middle age than those born preterm and appropriate for gestational age [25]. Individuals born very preterm or extremely preterm also appear to have greater adverse left ventricular remodeling associated with higher systolic blood pressure [26], which could also contribute to more rapid cardiovascular disease progression. However, the exact risk conferred by birth weight and gestational age are unclear, and the paradox remains of whether it is birth weight or gestational age alone or rather the antecedent causes of preterm birth, low birth weight, and/or growth restriction that are the true causal risk factors. Finally, it is important to remember that clinical definitions of lower birth weight and preterm birth are somewhat arbitrary, and that studies should always analyze birth weight, gestational age, and weight-for-gestational age z-scores on the continuous scale whenever possible.
Existing Clinical and Epidemiological Paradoxes
Despite all of these advancements in the field, the evidence remains controversial for a variety of methodological reasons, and it remains to be determined how these major factors interact to ultimately program hypertension. Indeed, the vast majority of our studies investigating this important topic have had significant limitations to scientific rigor (Table 2). Clinically, lower birth weight, preterm birth, and growth restriction have been studied extensively as implicitly causative exposures for hypertension programming. In reality, they are likely better conceptualized as intermediate factors on the causal pathway and as such, it has been quite difficult to parse out the relative contributions among various maternal, fetal, and environmental factors to hypertension programming without inducing bias along the way [27] (Figure 1). This has particularly been the case when we try to apply preclinical data to clinical and epidemiological research (see Overview of Preclinical Evidence). Take, for example, preeclampsia: is the simple fact that a woman who is pregnant develops preeclampsia enough to program hypertension? If so, to what extent is this causally necessary and sufficient? What about the effects of preeclampsia on the placenta and fetus—does the offspring have to be born preterm to cause hypertension in the future? Is fetal growth restriction a prerequisite? What about low birth weight in isolation? Simply put, we do not know whether and to what extent birth weight, gestational age, or growth restriction are necessary and sufficient to program hypertension independent of their antecedent causes [28]. These and other paradoxes have not been addressed adequately in the field, let alone are they even considered.
Table 2.
Examples of Bias to Consider in Clinical and Epidemiological Studies of Antenatal Programming of Hypertension
| Bias | Definition |
|---|---|
| Exchangeability | The assumption that individuals with the exposure of interest are exchangeable with unexposed individuals on all relevant factors except for their exposure status [27] |
| Collapsibility | The assumption that the marginal measure of association is equal to a weighted average of the stratum-specific measures of association [63] |
| Confounding bias | In causal language, an open, non-causal, biasing pathway due to a factor (covariate) that causes the exposure and the outcome |
| Selection bias | Conditioning the study sample (selecting or restricting) based on a common ancestor factor (cause) of the exposure and outcome (i.e., a confounding factor), which violates exchangeability |
| Collider bias | Conditioning the study sample based on, or adjusting for or stratifying on, a common descendent factor of the exposure and outcome. It can induce a spurious association or distort the magnitude or direction of an association between the exposure and outcome [32] |
| Time-dependent bias | Failure to appropriately account for the start of study time (i.e., birth) or time during follow up, which erroneously favors one exposure group over another [27] |
| Immeasurable time bias | Exposure groups are assigned erroneously the same exposure time due to lack of detail about how exposure status changes over time. For example, when preeclampsia develops [27] |
| Table 2 fallacy | Presenting multiple effect (parameter) estimates for the exposures and covariates from the same multivariable model that confounds direct-effect and total-effect estimates and encourages misinterpretation of results [32] |
Figure 1.
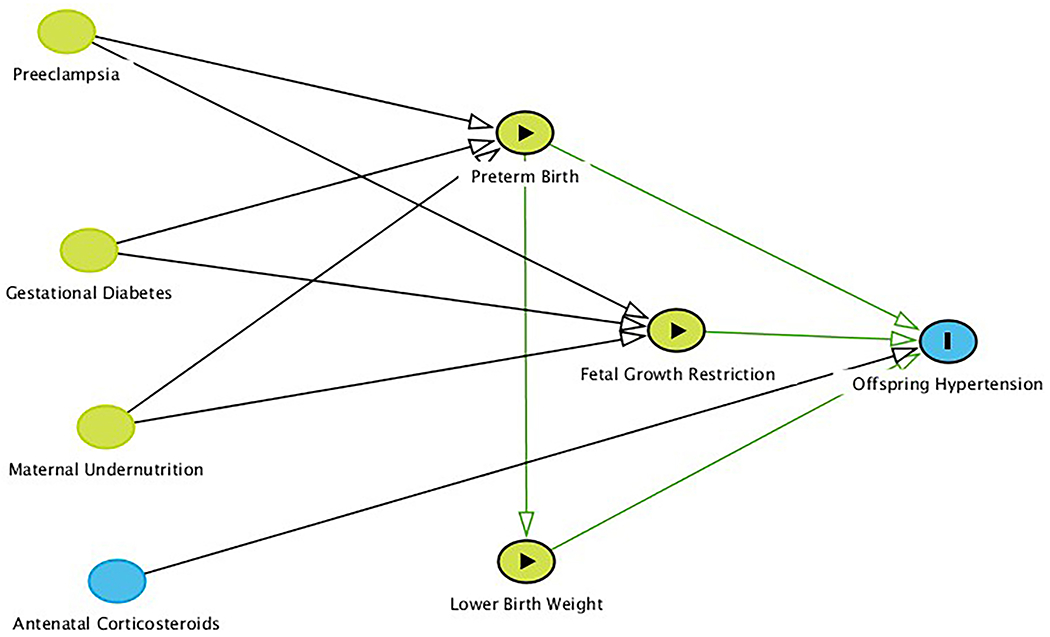
Graphical causal model (directed acyclic graph) representing the concept that traditionally defined exposures such as lower birth weight, preterm birth, and fetal growth restriction may actually be intermediate factors on the causal pathway. Research question-defined exposures are denoted by green nodes with black arrowheads; the outcome is denoted as a blue node with a black bar. Green nodes denote ancestors of the exposures while the blue node denotes an ancestor of the outcome. Causal pathways are denoted with green arrows, while undefined pathways are denoted with black arrows. Arrows represent true causal relationships. Additional arrows are omitted for clarity, such as that preeclampsia and preterm birth likely would cause antenatal corticosteroid exposure.
Small for gestational age (a statistical classification defined as birth weight less than the 10th percentile for gestational age and sex) is not synonymous with fetal growth restriction (also known as intrauterine growth restriction), which implies pathologic growth and is defined as both low standardized growth parameters in addition to abnormal Doppler flow in the umbilical artery[29]. For example, small for gestational age does not differentiate the infant that was constitutionally small as a fetus but who achieves their normal growth potential and thus is likely not at risk of hypertension programming [30, 31]. In addition, it is possible for an infant to have had fetal growth restriction but also a birth weight greater than or equal to the 10th percentile for gestational age and sex. Many studies have not differentiated further among symmetric (global impairment of cellular hyperplasia) versus asymmetric (greater decrease in abdominal size relative to head circumference) or early versus late (±32 weeks’ gestation) fetal growth restriction. While there is likely much overlap among these classifications, it is probable that asymmetric, late fetal growth restriction is most characteristic of what is often assumed to be developmental plasticity-associated growth restriction leading to programmed hypertension, wherein the fetal trade-off of increased short-term survival at the expense of an increased risk of disease later on is enhanced.
Many of our traditional analytic approaches that have attempted to disentangle the respective contributions of birth weight and gestational age may actually induce bias by conditioning on a collider or mediator, or the authors and readers may commit the Table 2 fallacy by making inappropriate inferences from covariate parameter estimates from the multivariable models [27, 32, 33]. In a large retrospective population-based cohort study in Sweden, the investigators used separate models to estimate the risk of ischemic heart disease due to birth weight or gestational age [34]. The unadjusted models for both exposures demonstrated no association with ischemic heart disease. When adjusting the birth weight model for gestational age, the authors observed that lower birth weight was associated with an increased risk of ischemic heart disease. However, when adjusting the gestational age model for birth weight, they observed a “protective” association wherein lower gestational age was associated with a lower risk of ischemic heart disease. When birth weight and gestational age were set as exposures with the outcomes systolic or diastolic blood pressure at age 6 years, both exposures were associated with higher blood pressure when modeled separately in adjusted analyses, but when modeled together, the authors observed that “only birth weight remained significantly” associated with systolic blood pressure [35].
In a similar vein, how we can best define adiposity as a covariate in the association between early-life exposures and future blood pressure has been a controversial topic, and many recommended approaches have significant flaws [36, 37]. We must first consider what this covariate may represent when we intend to account for high adiposity—weight, body mass index, or more specific (and accurate) adiposity estimates. While, in theory, incorporating multiple measures of adiposity within one model may be beneficial, without a causal framework this may induce more bias [38]. Next we must consider how best to define this adiposity measure for the given research question and subsequently the study design and analytic plan—is it a confounder, a mediator, or an effect modifier? This arises not only when “adjusting for” this covariate in a multivariable model, but also whether we decide to stratify the models as well as whether we restrict or match the study population conditional on adiposity [39]. Yun et al attempted to estimate the direct and indirect effects of offspring current body mass index on the association between birth weight and blood pressure using mediation analysis in structural equation models using data from the Bogalusa Heart Study, restricted to participants born at term [40, 41]. Using a series of cross-sectional observations, they observed that body mass index might have a suppression effect rather than a mediation effect on the association of birth weight with both systolic and diastolic blood pressure in preadolescents, adolescents, young adults, and mid-adults, and that older age attenuated these effects. While a few studies have attempted to address these important considerations when investigating hypertension programming, many studies have been limited by a lack of an adequate causal inference framework—and specifically graphical causal modeling—and have instead relied on more traditional statistical covariate selection criteria [42, 43].
Numerous statistical and epidemiological paradoxes have been described within the field of the developmental origins of health and disease, but they remain largely underappreciated [44, 45]. For a given outcome measure assessed over time (e.g., weight, blood pressure), incorporation of that measure’s baseline values as a covariate in a model, or defining the outcome as the change score from baseline, can induce bias [44, 46, 47]. Additionally, adjusting for variables inappropriately (i.e., for those factors that may not actually be confounders that lie on biasing, non-causal paths), publication bias, and random error are important, but oft overlooked, limitations in the field as well, though to date studies have been limited in their ability to parse out these types and sources of bias in the first place [48, 49].
It is important for us to be knowledgeable about how normative blood pressure values, and the definitions of high blood pressure classifications such as hypertension, have changed over time in both adults and youth, which has implications for studies analyzing blood pressure measured longitudinally or, especially, when comparing different studies from different eras in meta-analyses [50–53]. For youth in particular, to date normative blood pressure values have not been based on risk of cardiovascular outcomes, as they are in adults, but rather on epidemiologically determined normative distributions from large cross-sectional studies that are based on age, sex, and height. These normative values were obtained in approximately 40,000–50,000 healthy children using the auscultatory (manual) technique in the right upper extremity [54]. Further, in youth <18 years of age, in order to confirm a hypertension diagnosis, we must measure at least three manual blood pressures in the right upper extremity on different days, while adults require at least two independent measurements. This is due in part to high intra-individual blood pressure variability in youth and adults [55, 56]. For example, a study which measures blood pressure at only one study visit on one day cannot estimate hypertension disorder prevalence.
Finally, it is important to appraise critically the methods we use to measure blood pressure in the first place, as this has significant ramifications for measurement error and thus information bias. Many larger population-based cohort studies have measured blood pressure automatically with oscillometric devices in part due to feasibility considerations. Compared to manual measurement, oscillometric devices measure mean arterial pressure directly and subsequently calculate systolic and diastolic blood pressure using proprietary algorithms. However, automated blood pressure measurement devices are at risk for poor precision and reproducibility; the vast majority of devices are not well validated, if at all, to measure blood pressure in adults, let alone in youth [57, 58]. In addition, many devices differ in their accuracy and validity between youth and adults [59]. Manual measurement with a sphygmomanometer is the preferred method, but it confers similar measurement error risk dependent upon study staff training and appropriate measurement methods [60]. We must assess whether a given study closely adhered to established guidelines for blood pressure measurement, as technique deviations are an incredibly common source of measurement error in youth and adults, most notably due to blood pressure cuff size mismatch and incorrect positioning [61]. Ambulatory blood pressure monitoring is considered the gold-standard non-invasive blood pressure measurement method in youth and adults, but it is not without methodological limitations, including minimal device validation and lack of outcomes-based diagnostic definitions in youth [50, 53, 62]. Studies should report the specific methods and interpretation criteria used in sufficient detail to meet scientific rigor standards.
MOVING FORWARD TO ADDRESS PARADOXES AND EVOLVE PARADIGMS
It All Starts with a Great Research Question
So how do we advance the field forward in a meaningful and impactful way to more rapidly, and appropriately, translate these important discoveries to our patients? It all begins with asking the right research question—we must be intentional and thoughtful about the question we want to ask and the question we actually ask. The question should truly adhere to scientific rigor—it should be significant (actually addresses existing knowledge gaps in meaningful ways) and innovative (applies novel methods and approaches in new ways to new populations) in order to advance the field in a way that our other studies have not been able to do. Our aims and hypotheses should similarly be well thought out and explicitly stated. The appropriate study design and analytic plan for each specific hypothesis is crucial to mitigate these concerns of observational studies that we have discussed, particularly in regards to confounding bias and its relation to exchangeability (i.e., the assumption that individuals with the exposure of interest are exchangeable with unexposed individuals on all relevant factors except for their exposure status) and collapsibility (i.e., the assumption that the marginal measure of association is equal to a weighted average of the stratum-specific measures of association) [27, 63].
Moving forward, and depending on the research question, it is crucial that those of us who study early-life hypertension programming rely heavily on causal reasoning and background biological and epidemiological knowledge a priori [45]. We should make the goal of the research question clear: are we interested in the total or direct effects [39]? Methods rooted in causal inference such as causal mediation analysis and sensitivity analyses are examples that may allow us to better estimate the true causal relationships at play. In an elegant analysis, Chiolero et al applied a causal inference framework to analyze data from a large, school-based study of sixth-graders in Switzerland to estimate the potential biasing effect of an unmeasured common cause of current weight and systolic blood pressure on the controlled direct effect of birth weight on systolic blood pressure and concluded that collider bias due to conditioning on current weight was unlikely to be the sole explanation for their main findings [64]. Figure 2 is a graphical causal model that conceptualizes this research question. The authors first used causal mediation analysis to estimate the controlled direct effect of birth weight on systolic blood pressure not mediated through current weight. In other words, if all rigorous model assumptions are met, this can be interpreted as the difference in systolic blood pressure for a 1-kg difference in birth weight if one holds constant the values for current weight and all other confounding factors. The authors included directed acyclic graph-informed minimally sufficient adjustment sets in the multivariable models to close biasing pathways that confounded the exposure-outcome and mediator-outcome associations, based on background knowledge [65]. These included breastfeeding, child diet, sedentary behaviors, and physical activity. They then conducted sensitivity analyses to estimate the confounding effect of an unmeasured common cause of the mediator and the outcome, U, which is a core assumption of causal mediation analysis and specifically four-way decomposition [66]. They observed initially that (1) adjustment for potentially confounding factors revealed a statistically non-significant association between lower birth weight and higher systolic blood pressure; (2) further adjustment for current weight substantially magnified the association; and (3) additional adjustment for measured common causes of current weight and the outcome did not change the association substantively. In their sensitivity analysis, they observed that an unmeasured confounding factor U would only move the association of birth weight and systolic blood pressure—conditional on current weight—close to null if U was strongly associated with systolic blood pressure (i.e., at least ±2.5 mmHg) and if children with low birth weight had a strong difference in the prevalence of U compared to children with normal birth weight (i.e., at least ±20%).
Figure 2.
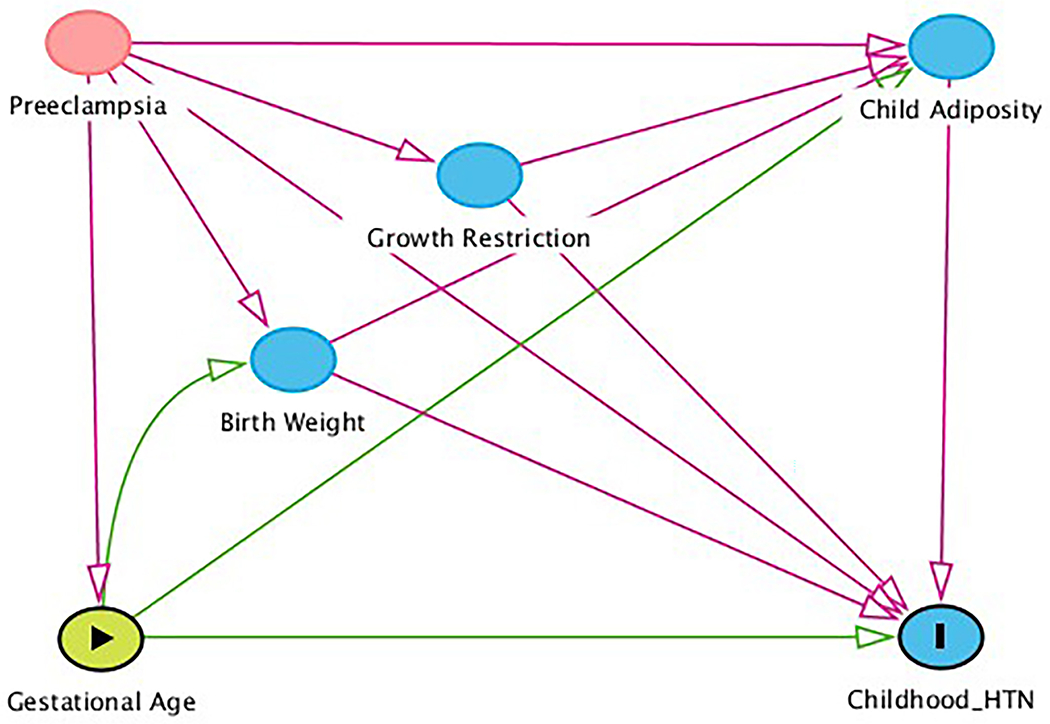
Directed acyclic graph of the association of gestational age with youth-onset hypertension with childhood adiposity as a mediator. Gestational age has a direct effect of hypertension and an indirect effect mediated through adiposity. The exposure gestational age is denoted with a green node with black arrowhead; the outcome hypertension is denoted as a blue node with a black bar. Blue nodes denote ancestors of the outcome, while the red node denotes a confounding factor and red arrows denote non-causal, biasing pathways. Causal pathways are denoted with green arrows.
While for the purposes of this paper we have limited our discussion to antenatal exposures generally, antenatal exposures can occur throughout gestation—from conception through birth—and can come from a variety of sources (paternal as well as maternal, fetal as well as environmental) [67, 68]. Preconception and postnatal exposures are likely to have equally important programming effects that may alter further the deleterious effects of antenatal exposures [69–74]. Moving forward, it is crucial that studies incorporate postnatal exposures appropriately in models of hypertension programming, especially when attempting to inform interventions. Revising our adiposity in later life paradox, clinical evidence suggests that the risk of programmed hypertension may be enhanced in people who develop obesity in later childhood (supporting the second-hit hypothesis); future studies must better define whether interventions to target obesity may fundamentally work differently in individuals with antenatal exposures such as low birth weight [75]. Finally, we must always remember that nothing is simple—the interplay amongst antenatal and other early-life exposures likely leads to cumulative effects to program hypertension on the additive and multiplicative scales [76].
Clinical Trials—Primordial and Primary Prevention
Clinical trials aimed at primordial and primary prevention of high blood pressure in the general pediatric population are relatively few and have largely been behavioral/lifestyle interventions targeted to high-risk youth—mostly those with obesity [77]. These trials have shown modest success in the short term with the magnitude of benefit often tied to the intensity of the intervention and tailoring to the population under study. The most well-known population-wide approach to primary prevention of hypertension in children is the Special Turku Coronary Risk Factor Intervention Project (STRIP) study in which 540 infants were randomized to at least biannual individualized, low-saturated fat, low-cholesterol dietary and lifestyle counseling and 522 infants randomized to a control diet at seven month of age [78]. The infants underwent biannual or annual assessments, including oscillometric blood pressure measurements at each visit, through 15 years of age. By age 15 years, participants in the low-saturated fat, low-cholesterol dietary group had 1-mmHg lower systolic blood pressure and diastolic blood pressure compared to the control participants as well as a 17% lower risk of higher blood pressure between ages 15–20 years [79]. These findings have laid the groundwork emphasizing the importance of large population-based interventions to prevent youth-onset hypertension, which likely will ultimately translate to meaningful improvements in cardiovascular disease risk later in life.
Primordial prevention of high blood pressure in childhood has focused predominantly on preventing childhood obesity and stopping accelerated weight gain, a major risk factor for subsequent increased blood pressure. Additional risk factors such as poor diet, limited physical activity, excessive screen time and poor sleep patterns may also be important targets for future intervention trials [14]. Studies that have focused on preventing antenatal risk factors (i.e., low birth weight and preterm birth) have been much more limited, especially those specifically examining the intervention’s effectiveness on preventing childhood high blood pressure. Given the epidemiological findings that the impact of low birth weight and preterm birth are moderated and/or mediated by subsequent growth patterns throughout childhood and adolescence, it is likely that focusing primordial prevention efforts on affecting change in these postnatal factors will be the most impactful and feasible, although preventing poor birth outcomes is an important goal which will have long-term benefits to the health and wellbeing of both mother and child.
OVERVIEW OF PRECLINICAL EVIDENCE
In order to better appreciate how far our field has come, and to better understand why these biased paradigms and paradoxes persist, we need to review the wealth of data from preclinical studies. Experimental data over the past 40 years has contributed substantially to our understanding of how antenatal exposures may alter fetal structural, physiological, and metabolic development to ultimately program adverse cardiovascular health, including hypertension [80]. It is believed that these adaptive alterations occur in response to a deleterious fetal environment to improve short-term survival, so-called developmental plasticity, but in the long term, they lead to maladaptive physiological changes that program an increased risk of hypertension [9]. To date, programming models have induced placental insufficiency, maternal stress, and inflammation, among other mechanisms. These models attempt to replicate such clinical conditions as preeclampsia, maternal diabetes mellitus, and maternal undernutrition as well as exogenous corticosteroid administration, either through or independent of induced fetal growth restriction. We will briefly review several of these preclinical models that investigate programming mechanisms via numerous alterations to macro and micro organ structure and function, including blood supply, cellular expression, and substrate, receptor, and enzyme function of several key hormonal pathways in a variety of cardiovascular organs, most notably the kidneys, vasculature, heart, and brain.
Preeclampsia
Preeclampsia has been one of the most well studied conditions in preclinical models as well as in clinical and epidemiological research. Rat offspring exposed to L-NAME-induced preeclampsia developed hypertension by adulthood that was associated with kidney damage consisting of proteinuria and reduced creatinine clearance [81]. In a well-studied rat model of preeclampsia, reduced uteroplacental perfusion pressure, exposed offspring developed growth restriction, programmed hypertension, and vascular hyperresponsiveness that persisted in future generations [82]. However, many studies induced fetal growth restriction by limiting placental blood flow generally rather than inducing overt preeclampsia. Fetal growth restriction induced by bilateral uterine artery ligation in pregnant rats was associated with adult-onset hypertension in offspring; this can occur in the presence or absence of associated alterations to kidney structure and function in affected offspring [83, 84]. In these exposed rats, bilateral renal denervation prevented hypertension development and reduced blood pressure significantly in those rats who had developed hypertension [85]. Rat offspring exposed to this uteroplacental insufficiency demonstrated left ventricular hypertrophy at age 35 days that preceded hypertension development at age 6 months [86]. In addition, blood pressure programming can occur in the absence of fetal growth restriction: baboon offspring exposed to preeclampsia, but without growth restriction, developed salt-sensitive blood pressure associated with alterations to aldosterone [87]. It remains unclear whether and to what extent preeclampsia per se or its antecedent causes, subsequent decreased nutrient delivery to the fetus, or growth restriction are necessary and sufficient to program hypertension.
Gestational Diabetes
Gestational diabetes, as well as preexisting maternal diabetes mellitus, has long been associated with adverse growth and metabolic health in the offspring. There has been increasing preclinical evidence that exposure to a hyperglycemic environment in utero can program hypertension through a variety of mechanisms. Streptozotocin-induced gestational diabetes was associated with hypertension as well as reduced nephron number and kidney function in rat adult offspring [88]. Rat offspring exposed to maternal hyperglycemia induced via glucose infusion during pregnancy developed adult-onset hypertension with associated reduced nephron number [89]. These studies also demonstrated that exposed rat offspring developed salt sensitivity of blood pressure at age 3 months that preceded overt hypertension at 6 months of age, due in part to altered distal nephron sodium handling [88]. However, discrepant data exist in regards to the type of induced diabetes and its gestational timing. As with other preclinical evidence, specific mechanisms and timing of hypertension development depend upon the animal studied and specific exposure models.
Maternal Undernutrition
Models of maternal undernutrition have provided a wealth of data regarding hypertension programming through a variety of mechanisms in several animal models. Rat offspring exposed to maternal protein restriction developed hypertension by 9 weeks of age and, in those exposed to more severe protein restriction, hypertension persisted throughout adulthood [90]. Maternal protein restriction has been shown to be associated with exaggerated renal sympathetic nerve responses to physical stress in rats and altered brain capillary rarefaction [91, 92]. Rat offspring exposed to maternal protein restriction also demonstrated reduced cardiomyocyte number and heart size as well as increased cardiac fibrosis [93, 94]. Maternal general undernutrition (50% reduction) during pregnancy in rats was associated with higher blood pressure in exposed offspring as well as reduced kidney weight and nephron number by 16 weeks of age [95]. However, maternal undernutrition models have been quite prone to heterogeneity depending upon the animal studied, the timing and severity of the nutritional exposure, and the nutritional composition of what is provided to the animals in lieu of a normal diet.
Antenatal Corticosteroids
Corticosteroids administered in the antenatal period have been an important area of investigation. Rat offspring that were exposed to dexamethasone throughout pregnancy developed growth restriction, reduced glomeruli number, and hypertension at 60 postnatal days [96]. Bilateral renal denervation normalized blood pressure at 8 weeks of age in exposed rats in part by normalized tubular sodium transporter expression [97]. Initially given to induce programming, many models then attempted to mimic the corticosteroid dose administered to pregnant women at risk of premature delivery to promote fetal lung maturation. Clinically relevant antenatal betamethasone exposure in sheep at the time of peak nephrogenesis (80 days’ gestation) was associated with subsequent hypertension in affected offspring as lambs and as adults in the absence of growth restriction [98]. Interestingly, these studies demonstrated this hypertension programming in the absence of kidney dysfunction and no correlation with severity of reduced glomeruli number [99]. In this sheep model, exposed adult female offspring developed altered cerebral vascular tone and reactivity [100]. Further, adult exposed sheep demonstrated abnormal brachial artery endothelial function and an enhanced vascular resistance-mediated blood pressure response [101, 102]. While studies that are more recent have attempted to mimic clinical antenatal corticosteroid use, heterogeneity continues to persist that limits clinical translation of these findings.
Paradoxes in the Preclinical Evidence
Despite these advances, several paradoxes remain unresolved. Numerous preclinical models have demonstrated clearly that exposed offspring can develop programmed hypertension independent of changes to kidney structure or function, most notably reduced nephron number, as well as offspring who develop kidney disease but who have normal blood pressure [83, 103, 104]. Much variation in preclinical studies is due to differences in study design, especially blood pressure measurement technique, and the specific species and genetic background of the animals [105–107]. The precise causes, timing, and severity of antenatal exposures remain nuanced and underappreciated aspects of hypertension programming [108, 109]. Preclinical studies have not yet been able to parse out fully the relative contributions of specific exposures, their direct effects on placental and fetal structure and function, or the surrogate markers of such exposures such as low birth weight, preterm birth, and growth restriction. These issues mirror many of the same limitations observed in clinical and epidemiological data to date.
CONCLUSIONS
To date, research on the antenatal origins of hypertension have revealed multiple complex mechanistic pathways that all rely on the interplay between antenatal and postnatal exposures [11]. While the epidemiological literature has consistently demonstrated the association of lower birth weight and preterm birth with blood pressure in later life, methodological issues have limited our ability to clearly map the pathways by which these early-life exposures alter physiology and impact blood pressure, as well as delineate how they are interrelated. Preclinical evidence has elucidated several pathways—preeclampsia, gestational diabetes, maternal undernutrition, and antenatal corticosteroids, to name a few—all of which have demonstrated physiological effects on fetal growth, development, and function that ultimately affect blood pressure. Despite the fact that these pathways are more clearly understood, the epidemiological literature suggests that postnatal factors such as childhood adiposity and growth patterns modify or mediate the association between antenatal factors and blood pressure across the life course. Due in part to the complexities of this issue, primordial prevention trials have had very limited and varying success.
So how do we move the field forward? We must be acutely aware of the strengths and limitations of the studies to date and systematically identify the precise knowledge gaps as we develop our future research questions. Our future research on the antenatal programming of hypertension should strongly emphasize scientific rigor, especially in regards to applying preclinical best practices and newer epidemiological and statistical methods as they relate to identifying, preventing, and mitigating bias. We should more precisely identify the relative importance of each pathway and how they simultaneously relate to each other, and better define the critical periods in which preventative interventions can be targeted to those individuals at greatest risk.
FUNDING:
AMS reports funding from NIH-NHLBI: K23HL148394, L40HL148910, and R01HL146818.
Footnotes
HUMAN AND ANIMAL RIGHTS:
All reported studies/experiments with human or animal subjects performed by the authors have been previously published and complied with all applicable ethical standards (including the Helsinki declaration and its amendments, institutional/national research committee standards, and international/national/institutional guidelines).
CONFLICTS OF INTEREST: No relevant conflicts of interest to disclose.
REFERENCES
- 1.Arima H, Barzi F, Chalmers J. Mortality patterns in hypertension. J Hypertens 2011;29:S3–7. doi: 10.1097/01.hjh.0000410246.59221.b1. [DOI] [PubMed] [Google Scholar]
- 2.Yang L, Magnussen CG, Yang L, Bovet P, Xi B. Elevated blood pressure in childhood or adolescence and cardiovascular outcomes in adulthood: a systematic review. Hypertension 2020;75(4):948–55. doi: 10.1161/hypertensionaha.119.14168. [DOI] [PubMed] [Google Scholar]
- 3.Yang L, Sun J, Zhao M, Liang Y, Bovet P, Xi B. Elevated blood pressure in childhood and hypertension risk in adulthood: a systematic review and meta-analysis. J Hypertens 2020;38(12). [DOI] [PubMed] [Google Scholar]; Important study that provided strong evidence linking high blood pressure in childhood with adult hypertension and demonstrating the importance of early intervention.
- 4.Song P, Zhang Y, Yu J, Zha M, Zhu Y, Rahimi K, et al. Global prevalence of hypertension in children: a systematic review and meta-analysis. JAMA Pediatr 2019;173(12):1–10. doi: 10.1001/jamapediatrics.2019.3310. [DOI] [PMC free article] [PubMed] [Google Scholar]; Crucial paper highlighting the global burden of hypertension in youth.
- 5.Falkner B, Lurbe E. Primary hypertension beginning in childhood and risk for future cardiovascular disease. J Pediatr 2021. doi: 10.1016/j.jpeds.2021.08.008. [DOI] [PubMed] [Google Scholar]
- 6.Liu J, Bu X, Wei L, Wang X, Lai L, Dong C, et al. Global burden of cardiovascular diseases attributable to hypertension in young adults from 1990 to 2019. J Hypertens 2021;39(12):2488–96. doi: 10.1097/hjh.0000000000002958. [DOI] [PubMed] [Google Scholar]
- 7.Barker DJ, Osmond C, Golding J, Kuh D, Wadsworth ME. Growth in utero, blood pressure in childhood and adult life, and mortality from cardiovascular disease. Br Med J 1989;298(6673):564–7. doi: 10.1136/bmj.298.6673.564. [DOI] [PMC free article] [PubMed] [Google Scholar]; Seminal epidemiological study that was one of the first to demonstrate that lower birth weight was associated with higher systolic blood pressure at age 10 years and 36 years.
- 8.Langford HG, Watson RL. Prepregnant blood pressure, hypertension during pregnancy, and later blood pressure of mothers and offspring. Hypertension 1980;2(4 Pt 2):130–3. [PubMed] [Google Scholar]
- 9.Nathanielsz PW. Animal models that elucidate basic principles of the developmental origins of adult diseases. ILAR Journal 2006;47(1):73–82. doi: 10.1093/ilar.47.1.73. [DOI] [PubMed] [Google Scholar]
- 10.Barker DJ. The origins of the developmental origins theory. J Intern Med 2007;261(5):412–7. doi: 10.1111/j.1365-2796.2007.01809.x. Barker, D J P Journal Article Review England J Intern Med. 2007 May;261(5):412-7. doi: 10.1111/j.1365-2796.2007.01809.x. [DOI] [PubMed] [Google Scholar]
- 11.Nuyt AM, Alexander BT. Developmental programming and hypertension. Curr Opin Nephrol Hypertens 2009;18(2). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.South AM. Antenatal Programming of Blood Pressure. In: Flynn JT, Ingelfinger JR, Brady TM, editors. Pediatric Hypertension. 5th ed.: Springer, Cham; 2022. [Google Scholar]
- 13.Goff DC, Buxton DB, Pearson GD, Wei GS, Gosselin TE, Addou EA, et al. Implementing the National Heart, Lung, and Blood Institute’s Strategic Vision in the Division of Cardiovascular Sciences. Circ Res 2019;124(4):491–7. doi: 10.1161/CIRCRESAHA.118.314338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Falkner B, Lurbe E. Primordial prevention of high blood pressure in childhood: an opportunity not to be missed. Hypertension 2020;75(5):1142–50. doi: 10.1161/HYPERTENSIONAHA.119.14059. [DOI] [PubMed] [Google Scholar]; Well written overview of the risk factors for hypertension in childhood and the importance of primordial prevention.
- 15.Chen W, Srinivasan SR, Berenson GS. Amplification of the association between birthweight and blood pressure with age: the Bogalusa Heart Study. J Hypertens 2010;28(10):2046–52. doi: 10.1097/HJH.0b013e32833cd31f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Huxley RR, Shiell AW, Law CM. The role of size at birth and postnatal catch-up growth in determining systolic blood pressure: a systematic review of the literature. J Hypertens 2000;18(7):815–31. doi: 10.1097/00004872-200018070-00002. [DOI] [PubMed] [Google Scholar]
- 17.Nilsson PM, Östergren P-O, Nyberg P, Söderström M, Allebeck P. Low birth weight is associated with elevated systolic blood pressure in adolescence: a prospective study of a birth cohort of 149 378 Swedish boys. J Hypertens 1997;15(12). [DOI] [PubMed] [Google Scholar]
- 18.Lai C, Hu Y, He D, Liang L, Xiong F, Liu G, et al. U-shaped relationship between birth weight and childhood blood pressure in China. BMC Pediatr 2019;19(1):264. doi: 10.1186/s12887-019-1638-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Chen W, Srinivasan SR, Yao L, Li S, Dasmahapatra P, Fernandez C, et al. Low birth weight is associated with higher blood pressure variability from childhood to young adulthood: the Bogalusa Heart Study. Am J Epidemiol 2012;176 Suppl 7(Suppl 7):S99–105. doi: 10.1093/aje/kws298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Dior UP, Karavani G, Bursztyn M, Paltiel O, Calderon-Margalit R, Friedlander Y, et al. Birth weight and maternal body size as determinants of blood pressure at age 17: results from the Jerusalem Perinatal Study Cohort. Maternal and Child Health Journal 2021;25(1):162–71. doi: 10.1007/s10995-020-03096-x. [DOI] [PubMed] [Google Scholar]
- 21.de Jong F, Monuteaux MC, van Elburg RM, Gillman MW, Belfort MB. Systematic review and meta-analysis of preterm birth and later systolic blood pressure. Hypertension 2012;59(2):226–34. doi: 10.1161/hypertensionaha.111.181784. [DOI] [PMC free article] [PubMed] [Google Scholar]; Important systematic review and meta-analysis synthesizing the available data on the association between preterm birth and systolic blood pressure.
- 22.South AM, Nixon PA, Chappell MC, Diz DI, Russell GB, Jensen ET, et al. Renal function and blood pressure are altered in adolescents born preterm. Pediatr Nephrol 2019;34(1):137–44. doi: 10.1007/s00467-018-4050-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Skudder-Hill L, Ahlsson F, Lundgren M, Cutfield WS, Derraik JGB. Preterm birth is associated with increased blood pressure in young adult women. J Am Heart Assoc 2019;8(12):e012274. doi: 10.1161/JAHA.119.012274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Haikerwal A, Doyle LW, Cheung MM, Wark JD, Opie G, Roberts G, et al. High blood pressure in young adult survivors born extremely preterm or extremely low birthweight in the post surfactant era. Hypertension 2020;75(1):211–7. doi: 10.1161/HYPERTENSIONAHA.119.13780. [DOI] [PubMed] [Google Scholar]
- 25.Juonala M, Cheung MMH, Sabin MA, Burgner D, Skilton MR, Kähönen M, et al. Effect of birth weight on life-course blood pressure levels among children born premature: the Cardiovascular Risk in Young Finns Study. J Hypertens 2015;33(8):1542–8. doi: 10.1097/hjh.0000000000000612. [DOI] [PubMed] [Google Scholar]; Important epidemiological study in a well-defined longitudinal cohort demonstrating the importance of fetal growth restriction in hypertension programming.
- 26.Mohamed A, Marciniak M, Williamson W, Huckstep OJ, Lapidaire W, McCance A, et al. Association of systolic blood pressure elevation with disproportionate left ventricular remodeling in very preterm-born young adults: the Preterm Heart and Elevated Blood Pressure. JAMA Cardiol 2021. doi: 10.1001/jamacardio.2021.0961. [DOI] [PMC free article] [PubMed] [Google Scholar]; Important study demonstrating an additional mechanism, higher burden of left ventricular remodeling, by which preterm birth may influence future cardiovascular disease risk through blood pressure programming.
- 27.Cohen JB, D’Agostino McGowan L, Jensen ET, Rigdon J, South AM. Evaluating sources of bias in observational studies of angiotensin-converting enzyme inhibitor/angiotensin II receptor blocker use during COVID-19: beyond confounding. J Hypertens 2021;39(4):795–805. doi: 10.1097/HJH.0000000000002706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Howards PP. Invited commentary: identifying the improbable, the value of incremental insights. Am J Epidemiol 2014;179(1):12–4. doi: 10.1093/aje/kwt231. [DOI] [PubMed] [Google Scholar]
- 29.Gordijn SJ, Beune IM, Thilaganathan B, Papageorghiou A, Baschat AA, Baker PN, et al. Consensus definition of fetal growth restriction: a Delphi procedure. Ultrasound Obstet Gynecol 2016;48(3):333–9. doi: 10.1002/uog.15884. [DOI] [PubMed] [Google Scholar]
- 30.Beune IM, Bloomfield FH, Ganzevoort W, Embleton ND, Rozance PJ, van Wassenaer-Leemhuis AG, et al. Consensus based definition of growth restriction in the newborn. J Pediatr 2018;196:71–6.e1. doi: 10.1016/j.jpeds.2017.12.059. [DOI] [PubMed] [Google Scholar]
- 31.Anderson MS, Hay WW. Intrauterine growth restriction and the small-for-gestational age infant. In: Avery GB, Fletcher MA, MacDonald MG, editors. Neonatology Pathophysiology and Management of the Newborn. 5th ed. Philadelphia: Lippincott Williams and Wilkins; 1999. p. 411. [Google Scholar]
- 32.Cole SR, Platt RW, Schisterman EF, Chu H, Westreich D, Richardson D, et al. Illustrating bias due to conditioning on a collider. Int J Epidemiol 2010;39(2):417–20. doi: 10.1093/ije/dyp334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Westreich D, Greenland S. The Table 2 Fallacy: presenting and interpreting confounder and modifier coefficients. Am J Epidemiol 2013;177(4):292–8. doi: 10.1093/aje/kws412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Kaijser M, Bonamy Anna-Karin E, Akre O, Cnattingius S, Granath F, Norman M, et al. Perinatal risk factors for ischemic heart disease: disentangling the roles of birth weight and preterm birth. Circulation 2008;117(3):405–10. doi: 10.1161/CIRCULATIONAHA.107.710715. [DOI] [PubMed] [Google Scholar]
- 35.Liew G, Wang JJ, Mitchell P. Which is the better marker for susceptibility to disease later in life--low birthweight or prematurity? Arch Dis Child 2008;93(5):450. doi: 10.1136/adc.2008.138263. [DOI] [PubMed] [Google Scholar]
- 36.Lucas A, Fewtrell MS, Cole TJ. Fetal origins of adult disease-the hypothesis revisited. Br Med J 1999;319(7204):245–9. doi: 10.1136/bmj.319.7204.245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Lawlor DA, Leon DA, Rasmussen F. Growth trajectory matters: interpreting the associations among birth weight, concurrent body size, and systolic blood pressure in a cohort study of 378,707 Swedish men. Am J Epidemiol 2007;165(12):1405–12. doi: 10.1093/aje/kwm028. [DOI] [PubMed] [Google Scholar]
- 38.Tu YK, Gilthorpe MS, Ellison GT. What is the effect of adjusting for more than one measure of current body size on the relation between birthweight and blood pressure? J Hum Hypertens 2006;20(9):646–57. doi: 10.1038/sj.jhh.1002044. [DOI] [PubMed] [Google Scholar]
- 39.Chiolero A, Kaufman JS, Paradis G. Why adjustment for current weight can bias the estimate of the effect of birth weight on blood pressure: shedding light using causal graphs. J Hypertens 2012;30(5):1042–5. doi: 10.1097/HJH.0b013e3283526663. [DOI] [PubMed] [Google Scholar]; Important epidemiological and statistical methods paper emphasizing sources of bias in the field when inappropriately adjusting for current weight.
- 40.Yun M, Wang X, Fan L, Yan Y, Bazzano L, He J, et al. Age-related suppression effect of current body weight on the association between birthweight and blood pressure: The Bogalusa Heart Study. Pediatr Obes 2021;16(3):e12716. doi: 10.1111/ijpo.12716. [DOI] [PubMed] [Google Scholar]
- 41.Sobel ME. Asymptotic confidence intervals for indirect effects in structural equation models. Sociol Methodol 1982;13:290–312. doi: 10.2307/270723. [DOI] [Google Scholar]
- 42.Mann KD, Pearce MS, Sayers SM, Singh GR. Pathways between birth weight and later body size in predicting blood pressure: Australian Aboriginal Cohort Study 1987-2007. J Hypertens 2015;33(5):933–9. doi: 10.1097/hjh.0000000000000514. [DOI] [PubMed] [Google Scholar]
- 43.South AM, Nixon PA, Chappell MC, Diz DI, Russell GB, Snively BM, et al. Antenatal corticosteroids and the renin-angiotensin-aldosterone system in adolescents born preterm. Pediatr Res 2017;81(1-1):88–93. doi: 10.1038/pr.2016.179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Tu YK, Gunnell D, Gilthorpe MS. Simpson’s Paradox, Lord’s Paradox, and Suppression Effects are the same phenomenon--the reversal paradox. Emerging Themes in Epidemiology 2008;5:2. doi: 10.1186/1742-7622-5-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Arah OA. The role of causal reasoning in understanding Simpson’s paradox, Lord’s paradox, and the suppression effect: covariate selection in the analysis of observational studies. Emerging Themes in Epidemiology 2008;5:5. doi: 10.1186/1742-7622-5-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Tennant PWG, Arnold KF, Ellison GTH, Gilthorpe MS. Analyses of ‘change scores’ do not estimate causal effects in observational data. Int J Epidemiol 2021:dyab050. doi: 10.1093/ije/dyab050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Tully N, South AM. Preterm birth and its association with altered renal sodium handling in response to mental stress in young adults [abstract]. J Am Soc Nephrol 2021;32:PO1760. [Google Scholar]
- 48.Huxley R, Neil A, Collins R. Unravelling the fetal origins hypothesis: is there really an inverse association between birthweight and subsequent blood pressure? Lancet 2002;360(9334):659–65. doi: 10.1016/s0140-6736(02)09834-3. [DOI] [PubMed] [Google Scholar]
- 49.Hardy R, Sovio U, King VJ, Skidmore PM, Helmsdal G, Olsen SF, et al. Birthweight and blood pressure in five European birth cohort studies: an investigation of confounding factors. Eur J Public Health 2006;16(1):21–30. doi: 10.1093/eurpub/cki171. [DOI] [PubMed] [Google Scholar]
- 50.Flynn JT, Kaelber DC, Baker-Smith CM, Blowey D, Carroll AE, Daniels SR, et al. Clinical Practice Guideline for Screening and Management of High Blood Pressure in Children and Adolescents. Pediatrics 2017;140(3):e20171904. doi: 10.1542/peds.2017-1904. [DOI] [PubMed] [Google Scholar]
- 51.National High Blood Pressure Education Program Working Group on High Blood Pressure in Children and Adolescents. The Fourth Report on the Diagnosis, Evaluation, and Treatment of High Blood Pressure in Children and Adolescents. Pediatrics 2004;114(2 Suppl 4th Report):555–76. [PubMed] [Google Scholar]
- 52.Chobanian AV, Bakris GL, Black HR, Cushman WC, Green LA, Izzo JL Jr, et al. The Seventh Report of the Joint National Committee on Prevention, Detection, Evaluation, and Treatment of High Blood Pressure: the JNC 7 Report. J Am Med Assoc 2003;289(19):2560–71. doi: 10.1001/jama.289.19.2560. [DOI] [PubMed] [Google Scholar]
- 53.Whelton PK, Carey RM, Aronow WS, Casey DE, Collins KJ, Dennison Himmelfarb C, et al. 2017 ACC/AHA/AAPA/ABC/ACPM/AGS/APhA/ASH/ASPC/NMA/PCNA Guideline for the Prevention, Detection, Evaluation, and Management of High Blood Pressure in Adults: a Report of the American College of Cardiology/American Heart Association Task Force on Clinical Practice Guidelines. Hypertension 2018;71(6):e13–e115. doi: 10.1161/hyp.0000000000000065. [DOI] [PubMed] [Google Scholar]
- 54.National High Blood Pressure Education Program Working Group on Hypertension Control in Children and Adolescents. Update on the 1987 Task Force Report on High Blood Pressure in Children and Adolescents: A Working Group Report from the National High Blood Pressure Education Program. Pediatrics 1996;98(4):649–58. [PubMed] [Google Scholar]
- 55.Koebnick C, Mohan Y, Li X, Porter AH, Daley MF, Luo G, et al. Failure to confirm high blood pressures in pediatric care—quantifying the risks of misclassification. J Clin Hypertens 2018;20(1):174–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Rhodehouse BC, Fan J, Chen W, McNeal MJ, Durham CG, Erwin III JP. Effect of repeat manual blood pressure measurement on blood pressure and stage of hypertension. Baylor University Medical Center Proceedings: Taylor & Francis; 2019. p. 498–501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Picone DS, Padwal R, Campbell NRC, Boutouyrie P, Brady TM, Olsen MH, et al. How to check whether a blood pressure monitor has been properly validated for accuracy. J Clin Hypertens 2020;22(12):2167–74. doi: 10.1111/jch.14065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Stergiou GS, Alpert B, Mieke S, Asmar R, Atkins N, Eckert S, et al. A universal standard for the validation of blood pressure measuring devices: Association for the Advancement of Medical Instrumentation/European Society of Hypertension/International Organization for Standardization (AAMI/ESH/ISO) Collaboration Statement. J Hypertens 2018;36(3). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Chiolero A, Paradis G, Lambert M. Accuracy of oscillometric devices in children and adults. Blood Press 2010;19(4):254–9. [DOI] [PubMed] [Google Scholar]
- 60.Macumber I, South AM. Hypertension: Epidemiology, Evaluation, and Blood Pressure Monitoring. In: Greenbaum L, Schaefer F, editors. Pediatric Kidney Disease. 3rd ed.: Springer; 2022. [Google Scholar]
- 61.Muntner P, Shimbo D, Carey RM, Charleston JB, Gaillard T, Misra S, et al. Measurement of Blood Pressure in Humans: A Scientific Statement from the American Heart Association. Hypertension 2019;73:e35–e66. doi: 10.1161/HYP.0000000000000087. [DOI] [PMC free article] [PubMed] [Google Scholar]; Crucial statement on the rigor of blood pressure measurement in humans.
- 62.Stergiou GS, Alamara CV, Salgami EV, Vaindirlis IN, Dacou-Voutetakis C, Mountokalakis TD. Reproducibility of home and ambulatory blood pressure in children and adolescents. Blood Press Monit 2005;10(3):143–7. [DOI] [PubMed] [Google Scholar]
- 63.Sander G, Judea P, James MR. Confounding and collapsibility in causal inference. Statistical Science 1999;14(1):29–46. doi: 10.1214/ss/1009211805. [DOI] [Google Scholar]
- 64.Chiolero A, Paradis G, Kaufman JS. Assessing the possible direct effect of birth weight on childhood blood pressure: a sensitivity analysis. Am J Epidemiol 2014;179(1):4–11. doi: 10.1093/aje/kwt228. [DOI] [PubMed] [Google Scholar]; Important epidemiological paper that used causal inference-based methods to estimate direct effects of birth weight on blood pressure during childhood.
- 65.Shrier I, Platt RW. Reducing bias through directed acyclic graphs. BMC Med Res Methodol 2008;8:70. doi: 10.1186/1471-2288-8-70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.VanderWeele TJ. Bias formulas for sensitivity analysis for direct and indirect effects. Epidemiology 2010;21(4):540–51. doi: 10.1097/EDE.0b013e3181df191c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Langley-Evans SC, Welham SJ, Sherman RC, Jackson AA. Weanling rats exposed to maternal low-protein diets during discrete periods of gestation exhibit differing severity of hypertension. Clin Sci 1996;91(5):607–15. doi: 10.1042/cs0910607. [DOI] [PubMed] [Google Scholar]
- 68.Kwong WY, Wild AE, Roberts P, Willis AC, Fleming TP. Maternal undernutrition during the preimplantation period of rat development causes blastocyst abnormalities and programming of postnatal hypertension. Development 2000;127(19):4195–202. [DOI] [PubMed] [Google Scholar]
- 69.Singhal A, Cole TJ, Fewtrell M, Kennedy K, Stephenson T, Elias-Jones A, et al. Promotion of faster weight gain in infants born small for gestational age: is there an adverse effect on later blood pressure? Circulation 2007;115(2):213–20. doi: 10.1161/circulationaha.106.617811. [DOI] [PubMed] [Google Scholar]
- 70.Singhal A, Cole TJ, Lucas A. Early nutrition in preterm infants and later blood pressure: two cohorts after randomised trials. Lancet 2001;357(9254):413–9. doi: 10.1016/s0140-6736(00)04004-6. [DOI] [PubMed] [Google Scholar]
- 71.Yzydorczyk C, Comte B, Cambonie G, Lavoie JC, Germain N, Ting Shun Y, et al. Neonatal oxygen exposure in rats leads to cardiovascular and renal alterations in adulthood. Hypertension 2008;52(5):889–95. doi: 10.1161/hypertensionaha.108.116251. [DOI] [PubMed] [Google Scholar]
- 72.Boubred F, Buffat C, Feuerstein J-M, Daniel L, Tsimaratos M, Oliver C, et al. Effects of early postnatal hypernutrition on nephron number and long-term renal function and structure in rats. Am J Physiol Renal Physiol 2007;293(6):F1944–F9. doi: 10.1152/ajprenal.00141.2007. [DOI] [PubMed] [Google Scholar]
- 73.Siddique K, Guzman GL, Gattineni J, Baum M. Effect of postnatal maternal protein intake on prenatal programming of hypertension. Reprod Sci 2014;21(12):1499–507. doi: 10.1177/1933719114530186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Lozano G, Elmaghrabi A, Salley J, Siddique K, Gattineni J, Baum M. Effect of prenatal programming and postnatal rearing on glomerular filtration rate in adult rats. Am J Physiol Renal Physiol 2014;308(5):F411–F9. doi: 10.1152/ajprenal.00593.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.South AM, Nixon PA, Chappell MC, Diz DI, Russell GB, Jensen ET, et al. Association between preterm birth and the renin−angiotensin system in adolescence: influence of sex and obesity. J Hypertens 2018;36(10):2092–101. doi: 10.1097/hjh.0000000000001801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Mackenzie HS, Brenner BM. Fewer nephrons at birth: a missing link in the etiology of essential hypertension? Am J Kidney Dis 1995;26(1):91–8. doi: 10.1016/0272-6386(95)90161-2. [DOI] [PubMed] [Google Scholar]
- 77.Cai L, Wu Y, Wilson RF, Segal JB, Kim MT, Wang Y. Effect of childhood obesity prevention programs on blood pressure. Circulation 2014;129(18):1832–9. doi: 10.1161/CIRCULATIONAHA.113.005666. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Niinikoski H, Jula A, Viikari J, Rönnemaa T, Heino P, Lagström H, et al. Blood pressure is lower in children and adolescents with a low-saturated-fat diet since infancy: the Special Turku Coronary Risk Factor Intervention Project. Hypertension 2009;53(6):918–24. doi: 10.1161/hypertensionaha.109.130146. [DOI] [PubMed] [Google Scholar]
- 79.Nupponen M, Pahkala K, Juonala M, Magnussen CG, Niinikoski H, Rönnemaa T, et al. Metabolic syndrome from adolescence to early adulthood: effect of infancy-onset dietary counseling of low saturated fat: the Special Turku Coronary Risk Factor Intervention Project (STRIP). Circulation 2015;131(7):605–13. doi: 10.1161/circulationaha.114.010532. [DOI] [PubMed] [Google Scholar]
- 80.Brenner BM, Garcia DL, Anderson S. Glomeruli and blood pressure. Less of one, more the other? Am J Hypertens 1988;1(4 Pt 1):335–47. doi: 10.1093/ajh/1.4.335. [DOI] [PubMed] [Google Scholar]; Seminal review that helped advance the Brenner Hypothesis that nephron loss contributes to hypertension development.
- 81.Habib YH, Gowayed MA, Abdelhady SA, El-Deeb NM, Darwish IE, El-Mas MM. Modulation by antenatal therapies of cardiovascular and renal programming in male and female offspring of preeclamptic rats. Naunyn-Schmiedeberg’s Archives of Pharmacology 2021. doi: 10.1007/s00210-021-02146-7. [DOI] [PubMed] [Google Scholar]
- 82.Anderson CM, Lopez F, Zimmer A, Benoit JN. Placental insufficiency leads to developmental hypertension and mesenteric artery dysfunction in two generations of Sprague-Dawley rat offspring. Biol Reprod 2006;74(3):538–44. doi: 10.1095/biolreprod.105.045807. [DOI] [PubMed] [Google Scholar]
- 83.Alexander BT. Placental insufficiency leads to development of hypertension in growth-restricted offspring. Hypertension 2003;41(3):457–62. doi: 10.1161/01.Hyp.0000053448.95913.3d. [DOI] [PubMed] [Google Scholar]
- 84.Baserga M, Hale MA, Wang ZM, Yu X, Callaway CW, McKnight RA, et al. Uteroplacental insufficiency alters nephrogenesis and downregulates cyclooxygenase-2 expression in a model of IUGR with adult-onset hypertension. Am J Physiol Regul Integr Comp Physiol 2007;292(5):R1943–R55. doi: 10.1152/ajpregu.00558.2006. [DOI] [PubMed] [Google Scholar]
- 85.Alexander BT, Hendon AE, Ferril G, Dwyer TM. Renal denervation abolishes hypertension in low-birth-weight offspring from pregnant rats with reduced uterine perfusion. Hypertension 2005;45(4):754–8. doi: 10.1161/01.HYP.0000153319.20340.2a. [DOI] [PubMed] [Google Scholar]
- 86.Master JS, Zimanyi MA, Yin KV, Moritz KM, Gallo LA, Tran M, et al. Transgenerational left ventricular hypertrophy and hypertension in offspring after uteroplacental insufficiency in male rats. Clinical and Experimental Pharmacology & Physiology 2014;41(11):884–90. doi: 10.1111/1440-1681.12303. [DOI] [PubMed] [Google Scholar]
- 87.Yeung KR, Sunderland N, Lind JM, Heffernan S, Pears S, Xu B, et al. Increased salt sensitivity in offspring of pregnancies complicated by experimental preeclampsia. Clinical and Experimental Pharmacology & Physiology 2018;45(12):1302–8. doi: 10.1111/1440-1681.13008. [DOI] [PubMed] [Google Scholar]
- 88.Nehiri T, Duong Van Huyen JP, Viltard M, Fassot C, Heudes D, Freund N, et al. Exposure to maternal diabetes induces salt-sensitive hypertension and impairs renal function in adult rat offspring. Diabetes 2008;57(8):2167–75. doi: 10.2337/db07-0780. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Amri K, Freund N, Vilar J, Merlet-Bénichou C, Lelièvre-Pégorier M. Adverse effects of hyperglycemia on kidney development in rats: in vivo and in vitro studies. Diabetes 1999;48(11):2240–5. doi: 10.2337/diabetes.48.11.2240. [DOI] [PubMed] [Google Scholar]
- 90.Langley SC, Jackson AA. Increased systolic blood pressure in adult rats induced by fetal exposure to maternal low protein diets. Clin Sci 1994;86(2):217–22; discussion 121. doi: 10.1042/cs0860217. [DOI] [PubMed] [Google Scholar]
- 91.Mizuno M, Siddique K, Baum M, Smith SA. Prenatal programming of hypertension induces sympathetic overactivity in response to physical stress. Hypertension 2013;61(1):180–6. doi: 10.1161/hypertensionaha.112.199356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Pladys P, Sennlaub F, Brault S, Checchin D, Lahaie I, Lê NLO, et al. Microvascular rarefaction and decreased angiogenesis in rats with fetal programming of hypertension associated with exposure to a low-protein diet in utero. American Journal of Physiology-Regulatory, Integrative and Comparative Physiology 2005;289(6):R1580–R8. doi: 10.1152/ajpregu.00031.2005. [DOI] [PubMed] [Google Scholar]
- 93.Corstius HB, Zimanyi MA, Maka N, Herath T, Thomas W, van der Laarse A, et al. Effect of intrauterine growth restriction on the number of cardiomyocytes in rat hearts. Pediatr Res 2005;57(6):796–800. doi: 10.1203/01.Pdr.0000157726.65492.Cd. [DOI] [PubMed] [Google Scholar]
- 94.Amer MG, Mohamed NM, Shaalan AAM. Gestational protein restriction: study of the probable effects on cardiac muscle structure and function in adult rats. Histol Histopathol 2017;32(12):1293–303. doi: 10.14670/hh-11-883. [DOI] [PubMed] [Google Scholar]
- 95.Chou HC, Wang LF, Lu KS, Chen CM. Effects of maternal undernutrition on renal angiotensin II and chymase in hypertensive offspring. Acta Histochem 2008;110(6):497–504. doi: 10.1016/j.acthis.2008.01.002. [DOI] [PubMed] [Google Scholar]
- 96.Benediktsson R, Lindsay RS, Noble J, Seckl JR, Edwards CRW. Glucocorticoid exposure in utero: new model for adult hypertension. Lancet 1993;341(8841):339–41. doi: 10.1016/0140-6736(93)90138-7. [DOI] [PubMed] [Google Scholar]
- 97.Dagan A, Kwon HM, Dwarakanath V, Baum M. Effect of renal denervation on prenatal programming of hypertension and renal tubular transporter abundance. Am J Physiol Renal Physiol 2008;295(1):F29–34. doi: 10.1152/ajprenal.00123.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Figueroa JP, Rose JC, Massmann GA, Zhang J, Acuña G. Alterations in fetal kidney development and elevations in arterial blood pressure in young adult sheep after clinical doses of antenatal glucocorticoids. Pediatr Res 2005;58(3):510–5. doi: 10.1203/01.Pdr.0000179410.57947.88. [DOI] [PubMed] [Google Scholar]
- 99.Zhang J, Massmann GA, Rose JC, Figueroa JP. Differential effects of clinical doses of antenatal betamethasone on nephron endowment and glomerular filtration rate in adult sheep. Reprod Sci 2010;17(2):186–95. doi: 10.1177/1933719109351098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Eckman DM, Kerr BA, Fuloria M, Simandle SA, Watt SE, Rose JC, et al. Antenatal betamethasone alters vascular reactivity in adult female ovine cerebral arteries. Pediatr Res 2010;68(4):344–8. doi: 10.1203/PDR.0b013e3181edb9fa. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Pulgar VM, Figueroa JP. Antenatal betamethasone administration has a dual effect on adult sheep vascular reactivity. Pediatr Res 2006;60(6):705–10. doi: 10.1203/01.pdr.0000246481.05231.17. [DOI] [PubMed] [Google Scholar]
- 102.Lee J-H, Zhang J, Flores L, Rose JC, Massmann GA, Figueroa JP. Antenatal betamethasone has a sex-dependent effect on the in vivo response to endothelin in adult sheep. Am J Physiol Regul Integr Comp Physiol 2013;304(8):R581–R7. doi: 10.1152/ajpregu.00579.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Moritz KM, Mazzuca MQ, Siebel AL, Mibus A, Arena D, Tare M, et al. Uteroplacental insufficiency causes a nephron deficit, modest renal insufficiency but no hypertension with ageing in female rats. J Physiol 2009;587(Pt 11):2635–46. doi: 10.1113/jphysiol.2009.170407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Baum M. Role of renal sympathetic nerve activity in prenatal programming of hypertension. Pediatr Nephrol 2018;33(3):409–19. doi: 10.1007/s00467-016-3359-8. [DOI] [PubMed] [Google Scholar]
- 105.Van Abeelen AF, Veenendaal MV, Painter RC, De Rooij SR, Thangaratinam S, Van Der Post JA, et al. The fetal origins of hypertension: a systematic review and meta-analysis of the evidence from animal experiments of maternal undernutrition. J Hypertens 2012;30(12):2255–67. doi: 10.1097/HJH.0b013e3283588e0f. [DOI] [PubMed] [Google Scholar]
- 106.Roghair RD, Aldape G. Naturally occurring perinatal growth restriction in mice programs cardiovascular and endocrine function in a sex- and strain-dependent manner. Pediatr Res 2007;62(4):399–404. doi: 10.1203/PDR.0b013e31813cbf16. [DOI] [PubMed] [Google Scholar]
- 107.Vehaskari VM, Woods LL. Prenatal programming of hypertension: lessons from experimental models. J Am Soc Nephrol 2005;16(9):2545–56. doi: 10.1681/asn.2005030300. [DOI] [PubMed] [Google Scholar]
- 108.Woods LL, Weeks DA, Rasch R. Programming of adult blood pressure by maternal protein restriction: role of nephrogenesis. Kidney Int 2004;65(4):1339–48. doi: 10.1111/j.1523-1755.2004.00511.x. [DOI] [PubMed] [Google Scholar]
- 109.Boubred F, Daniel L, Buffat C, Tsimaratos M, Oliver C, Lelièvre-Pégorier M, et al. The magnitude of nephron number reduction mediates intrauterine growth-restriction-induced long term chronic renal disease in the rat. A comparative study in two experimental models. J Transl Med 2016;14(1):331. doi: 10.1186/s12967-016-1086-3. [DOI] [PMC free article] [PubMed] [Google Scholar]