

Abstract
Background and Objective
Drug–drug interactions between direct oral anticoagulants (DOAC) and antiseizure medications via the cytochrome P450 (CYP) or the P-glycoprotein (P-gp) systems may lead to under-anticoagulation. The clinical relevance of these interactions is unclear. We aimed to elucidate the risk of thromboembolism with concurrent DOAC and CYP/P-gp modulating antiseizure medications.
Methods
In this propensity score-weighted population-based retrospective cohort study, we used competing risk regression analyses to determine the risks of ischemic stroke, venous thromboembolism, and death in DOAC recipients taking CYP/P-gp-modulating antiseizure medications (phenytoin, valproate, levetiracetam, carbamazepine, or phenobarbital) versus those taking CYP/P-gp-neutral antiseizure medications (pregabalin, gabapentin, or clobazam). We also performed secondary analyses for the epilepsy and atrial fibrillation subgroups.
Results
Among DOAC users, CYP/P-gp-modulating antiseizure medications were collectively associated with an increased risk of ischemic stroke (adjusted hazard ratio 1.28, 95% confidence interval 1.05–1.57, p = 0.017). In addition, phenytoin (adjusted hazard ratio 1.34, 95% confidence interval 1.07–1.68, p = 0.011) and valproate (adjusted hazard ratio 1.38, 95% confidence interval 1.10–1.74, p = 0.006) were associated with increased mortality. In the epilepsy subgroup, the risk of ischemic stroke and venous thromboembolism did not differ between CYP/P-gp-modulating and CYP/P-gp-neutral antiseizure medications.
Conclusions
Although CYP/P-gp-modulating antiseizure medications were associated with an increased risk of ischemic stroke when paired with DOAC in the primary analysis, such a phenomenon was not found among patients with epilepsy who took phenytoin, valproate, or levetiracetam with DOAC. Therefore, these antiseizure medication options among patients with epilepsy with concurrent DOAC should not be restricted solely based on their potential drug–drug interactions. Yet, the increased mortality during concurrent use of DOAC with phenytoin or valproate might call for caution.
Supplementary Information
The online version contains supplementary material available at 10.1007/s40263-022-00971-9.
Key Points
| Pharmacokinetic studies reported lower direct oral anticoagulant (DOAC) levels with the co-use of cytochrome P450 or P-glycoprotein-inducing antiseizure medications. Whether the drug–drug interactions confer a clinical risk of thromboembolism was unclear. |
| In our population-based cohort study involving 8746 patients taking concurrent antiseizure medications and DOAC over a 5-year period, cytochrome P450/ P-glycoprotein-modulating antiseizure medications were associated with a 28% increase in the odds of ischemic stroke compared with DOAC users taking cytochrome P450/P-glycoprotein-neutral antiseizure medications. Among which, phenytoin users had a 39% increase in the odds of developing ischemic stroke. |
| In a subgroup analysis of 2173 patients with epilepsy taking DOAC, the use of phenytoin, valproate, or levetiracetam was not associated with ischemic stroke or venous thromboembolism. |
| While phenytoin or valproate use with DOAC was associated with increased mortality, levetiracetam in combination with DOAC was not associated with an increased risk of death. |
Introduction
Direct oral anticoagulants (DOAC) are safe and efficacious ischemic stroke prophylaxis for atrial fibrillation (AF) [1]. Despite fewer potential drug–drug interactions compared with vitamin K antagonists, DOAC metabolism may still be altered by cytochrome P450 (CYP) or P-glycoprotein (P-gp) inducers, potentially leading to thromboembolic events [2]. International anticoagulation guidelines therefore cautioned against concomitant use of certain CYP/P-gp-modulating drugs with DOAC [3]. For instance, common antiseizure medications (ASMs) such as phenytoin, valproate, levetiracetam, carbamazepine, and phenobarbital were either contraindicated or recommended to be used with caution when co-prescribed with DOAC. These recommendations limited the therapeutic choices of important medical conditions such as post-stroke epilepsy that frequently mandate anticoagulation because of coexisting AF [4].
Of note, current guideline recommendations were predominantly based on animal models, pharmacokinetic studies, and case reports [5–7]. Pharmacokinetically, phenytoin, levetiracetam, valproate, and carbamazepine may reduce the half-lives of DOAC [8]. However, emerging evidence challenged the clinical relevance of these studies. For instance, a hospital-based retrospective study did not detect an increased ischemic stroke risk among DOAC users with concurrent CYP/P-gp-inducing ASMs [9]. Risk evaluation of ASM-DOAC combinations in the real-world practice may reveal appropriate treatment options balancing the efficacy of DOAC and control of epilepsy, which are both fundamental to survival and cerebrovascular health. In this population-based study, we aimed to determine the thromboembolic risks in DOAC recipients taking CYP/P-gp-modulating ASMs.
Methods
Study Design and Data Source
We performed a territory-wide, propensity score-weighted, retrospective cohort study that included all patients taking concurrent ASMs and DOAC in public hospitals or clinics in Hong Kong from 1 January, 2015 to 31 December, 2020. We retrieved clinical parameters from the Clinical Data Analysis and Reporting System of the Hospital Authority, a territory-wide electronic healthcare database that records clinical information of over 90% of the 7.5-million population. The coding accuracy of the Clinical Data Analysis and Reporting System was reported to be 85.4–100% [10–12]. The institutional review board approved the study (Joint CUHK-NTEC CREC Ref No. 2021.349) and waived the need for a written informed consent given the deidentification of data. The study was academic initiated with no funding from the pharmaceutical industry or other sources.
Study Subjects
We identified all patients taking apixaban, dabigatran, edoxaban, or rivaroxaban concurrently prescribed an oral ASM (pregabalin, gabapentin, phenytoin, valproate, carbamazepine, oxcarbazepine, levetiracetam, lamotrigine, phenobarbital, clobazam, or topiramate) within the study period. We recorded the baseline demographics and laboratory results (see Covariates section). Based on International Classification of Diseases, Ninth Revision, Clinical Modification (ICD-9-CM), we retrieved the following medical comorbidities: AF (ICD-9-CM 427.31), epilepsy (ICD-9-CM 345), hyperlipidemia (ICD-9-CM 272), congestive heart failure (ICD-9-CM 428), hypertension (ICD-9-CM 401), diabetes mellitus (ICD-9-CM 250), ischemic heart disease (ICD-9-CM 410-414), peripheral vascular disease (ICD-9-CM 443), ischemic stroke (ICD-9-CM 433, 434, 436), and venous thromboembolism (pulmonary embolism: ICD-9-CM 415.1, deep vein thrombosis: ICD-9-CM 453). Major bleeding was defined as a composite of intraocular hemorrhage (ICD-9-CM 379.23), intracranial hemorrhage (ICD-9-CM 430-432), retroperitoneal hemorrhage (ICD-9-CM 568.81), and hemarthrosis (ICD-9-CM 719.10) according to international guidelines [13]. We also recorded concomitant use of antiplatelet agents, statins, CYP/P-gp modulators other than ASMs, non-steroidal anti-inflammatory drugs, and cyclo-oxygenase-2 inhibitors (see Covariates section). CHADS2-Vasc and HAS-BLED scores were calculated [14]. We excluded patients with mitral stenosis (ICD-9-CM 394), valvular replacement (ICD-9-CM V42.2, V43.3), antiphospholipid syndrome (ICD-9-CM 289.81), or familial thrombophilia (ICD-9-CM 289.81).
Outcome
The primary outcome was the first ischemic stroke episode during combination use of DOAC and ASM. The secondary endpoints were venous thromboembolism and death.
Covariates
For the primary and secondary analyses in our propensity score weighting model, we included four demographic variables (age, sex, smoking, drinking habit), 13 background medical comorbidities (congestive heart failure, ischemic heart disease, ischemic stroke, venous thromboembolism, diabetes mellitus, hypertension, hyperlipidemia, peripheral vascular disease, AF, major bleeding, epilepsy, chronic kidney disease, chronic liver disease), nine background laboratory parameters (low-density and high-density lipoprotein cholesterol, total cholesterol, triglycerides, creatinine, alanine transaminase, hemoglobin, platelet, glycated hemoglobin A1c), and 15 background medications at the commencement of the DOAC and ASM combination (amiodarone, dronedarone, rifampicin, ibuprofen, indomethacin, naproxen, celecoxib, etorixocib, aspirin, clopidogrel, cilostazol, ticagrelor, simvastatin, atorvastatin, rosuvastatin).
Statistical Analyses
We expressed normally distributed continuous variables as mean (standard deviation), non-normally distributed continuous variables as median (interquartile range), and categorical variables as number (percentage). Time to event was defined as the months between the date of the first outcome event and the start date of ASM-DOAC combination therapy. For patients who did not develop outcome events, time to event was months between the commencement date of the DOAC-ASM combination and (1) end date of the ASM-DOAC combination, (2) death date, or (3) study-end date, whichever was earlier. Time-to-event analyses were performed by a Fine-Gray model putting death as a competing risk. Cox regression was used to analyze all-cause death. The cumulative incidence of the primary and secondary endpoints was estimated by Gray’s method, while that of death was estimated by the Kaplan–Meier method. Multiple comparisons were adjusted by Bonferroni correction. A two-sided test with a p-value <0.05 was considered statistically significant.
Primary Analysis
In the primary analysis, we first compared the outcomes in patients taking DOAC using CYP/P-gp-modulating ASMs (phenytoin, valproate, levetiracetam, carbamazepine, phenobarbital) to those taking CYP/P-gp-neutral ASMs (gabapentin, pregabalin, clobazam [3]). We then performed per-ASM analyses if the sample size permitted. We estimated the propensity score of each patient by generalized boosted models incorporating age, sex, baseline laboratory results, background comorbidities, and co-medications mentioned in the covariates section [15]. After estimating the propensity score, propensity score weighting based on inverse probability of treatment weighting was used to attempt an unbiased estimate on the average treatment effects of the four treatments (CYP/P-gp-neutral ASMs, phenytoin, valproate, levetiracetam) [16]. Inverse probability of treatment weighting uses weights based on the propensity score to create a synthetic sample in which the distribution of measured baseline covariates is independent of treatment assignment. We minimized the mean and maximum of the absolute standardized mean difference and Kolmogorov–Smirnov statistic as the four stopping rules to determine the corresponding optimal iteration of generalized boosted models. The stopping rule with overall the best subgroup balance and effective sample size was adopted. Generalized boosted models for propensity score have been proven to have a lower prediction error and provide more stable weights than logistic regression [15]. An absolute standardized mean difference of < 0.1 indicated a good balance. Covariates that failed to achieve an absolute standardized mean difference of < 0.1 were adjusted in a doubly robust model.
Assuming missing baseline data were random, we substituted them with values from multiple imputation with chained equations to create 20 complete data sets after the first 20 iterations. The imputed variables, in descending order of missingness, were glycated hemoglobin A1c (9.4%), lipid profile (9.0%), alanine transaminase (9.0%), clotting profile (8.0%), platelet (3.0%), hemoglobin (1.7%), and creatinine (0.7%). Imputed values were constrained within plausible ranges. Sensitivity analyses were performed using patients with complete data only. All statistical analyses were performed with R software version 3.5.1.
Secondary Analyses
By the same propensity score weighting principles, we performed two planned subgroup analyses based on the prescribing indications of ASM or DOAC: (1) epilepsy may confer a higher intrinsic risk of cerebrovascular events compared with non-epileptic conditions [17]. To avoid this bias in favor of CYP/P-gp-neutral ASMs (gabapentin or pregabalin), which are frequently prescribed in non-epileptic conditions such as neuralgia, we performed a subgroup analysis for patients with epilepsy alone. (2) As patients with AF have a higher risk of cerebrovascular events than those with venous thromboembolism [14], we conducted a subgroup analysis for patients with AF.
Results
From 1 January, 2015 to 31 December, 2020, among the total 72,581 patients taking DOAC attending public hospitals or clinics in Hong Kong, 9595 (13.2%) patients concurrently received ASMs. We excluded patients taking lamotrigine, topiramate, and oxcarbazepine because of the small sample size (Fig. 1). The patients in the final analyses (n = 8746) were taking apixaban (n = 3657), dabigatran (n = 2351), edoxaban (n = 493), or rivaroxaban (n = 2245). The indications for DOAC were AF (n = 5931) and venous thromboembolism (n = 1501). In terms of ASMs, patients were taking gabapentin (n = 4067), pregabalin (n = 1223), clobazam (n = 290), phenytoin (n = 1167), valproate (n = 1161), levetiracetam (n = 643), carbamazepine (n = 146), and phenobarbital (n = 49). The indications for ASMs were epilepsy (n = 2173), neuralgia (n = 3156), and psychiatric disorders (n = 2224). Daily dosages of ASM, medical comorbidities, comedications, and laboratory parameters are listed in Table 1.
Fig. 1.

Study flow diagram
Table 1.
Imputed patient characteristics after balancing with the inverse probability of treatment weighting method (grouped by ASMs)
| CYP/P-gp neutral ASMs | CYP/P-gp modulating ASMs | AMSD | Phenytoin | AMSD | Valproate | AMSD | Levetiracetam | AMSD | |
|---|---|---|---|---|---|---|---|---|---|
| n | 5580 | 3166 | 1167 | 1161 | 643 | ||||
| Daily dosage, mg, mean (SD) |
GBP: 516.6 (346.4) PGB: 113.6 (106.0) Clobazam: 18.2 (8.0) |
Phenytoin: 150.7 (94.5) Valproate: 568 (258.6) Levetiracetam: 1029 (270.3) Carbamazepine: 471 (148.1) Phenobarbital: 105 (52.2) |
150.7 (94.5) | 567.6 (258.6) | 1028.6 (270.3) | ||||
| Demographics | |||||||||
| Age, years, mean (SD) | 75.6 (11.7) | 73.2 (13.4) | 0.026 | 75.1 (12.4) | 0.035 | 73.4 (13.4) | 0.130 | 70.1 (14.1) | 0.303 |
| Female, n (%) | 2464 (44.2) | 1634 (51.6) | 0.026 | 594 (50.9) | 0.034 | 603 (51.9) | 0.064 | 363 (56.5) | 0.112 |
| Smoker, n (%) | 95 (1.7) | 44 (1.4) | 0.002 | 19 (1.6) | 0.008 | 14 (1.2) | 0.003 | 10 (1.6) | 0.001 |
| Drinker, n (%) | 81(1.5) | 64(2.0) | 0.005 | 23 (2.0) | 0.001 | 20 (1.7) | 0.003 | 17 (2.6) | 0.015 |
| Medical co-morbidities, n (%) | |||||||||
| CHF | 1888 (33.8) | 821 (25.9) | 0.025 | 329 (28.2) | 0.050 | 312 (26.9) | 0.057 | 142 (22.1) | 0.094 |
| IHD | 1462 (26.2) | 709 (22.4) | 0.014 | 280 (24.0) | 0.005 | 246 (21.2) | 0.048 | 139 (21.6) | 0.030 |
| IS | 1782 (31.9) | 1820 (57.5) | 0.039 | 747 (64.0) | 0.059 | 674 (58.1) | 0.191 | 344 (53.5) | 0.135 |
| ATE | 97 (1.7) | 29 (0.9) | 0.006 | 8 (0.7) | 0.010 | 15 (1.3) | 0.006 | 6 (0.9) | 0.009 |
| VTE | 864 (15.5) | 637 (20.1) | 0.011 | 233 (20.0) | 0.021 | 200 (17.2) | 0.010 | 154 (24.0) | 0.067 |
| DM | 1702 (30.5) | 967 (30.5) | 0.017 | 385 (33.0) | 0.046 | 359 (30.9) | 0.005 | 188 (29.2) | 0.010 |
| Hypertension | 3349 (60.0) | 1892 (59.8) | 0.013 | 727 (62.3) | 0.026 | 702 (60.5) | 0.001 | 348 (54.1) | 0.047 |
| PVD | 269 (4.8) | 157 (5.0) | 0.009 | 61 (5.2) | 0.007 | 62 (5.3) | 0.004 | 29 (4.5) | 0.006 |
| Hyperlipidemia | 1660 (29.7) | 998 (31.5) | 0.010 | 388 (33.2) | 0.016 | 363 (31.3) | 0.012 | 200 (31.1) | 0.016 |
| AF | 3789 (67.9) | 2142 (67.7) | 0.002 | 817 (70.0) | 0.008 | 816 (70.3) | 0.027 | 394 (61.3) | 0.051 |
| Epilepsy | 300 (5.4) | 1873 (59.2) | 0.068 | 756 (64.8) | 0.110 | 609 (52.5) | 0.213 | 417 (64.9) | 0.143 |
| Major bleeding | 324 (5.8) | 138 (4.4) | 0.004 | 47 (4.0) | 0.019 | 51 (4.4) | 0.011 | 33 (5.1) | 0.003 |
| CLD | 38 (0.7) | 22 (0.7) | 0.001 | 9 (0.8) | 0.005 | 2 (0.2) | 0.005 | 9 (1.4) | 0.008 |
| CKD | 315 (5.6) | 137 (4.3) | 0.004 | 55 (4.7) | 0.015 | 56 (4.8) | 0.006 | 22 (3.4) | 0.018 |
| CHADS2Vasc, mean (SD) | 4.2 (2.0) | 4.3 (2.1) | NA | 4.7 (2.0) | NA | 4.4 (2.1) | NA | 3.9 (2.2) | NA |
| HASBLED, mean (SD) | 2.7 (1.3) | 3.2 (1.4) | NA | 3.4 (1.3) | NA | 3.2 (1.4) | NA | 3(1.5) | NA |
| Background medications, n (%) | |||||||||
| Apixaban | 2345 (42.0) | 1311 (41.4) | 0.006 | 476 (40.8) | 0.025 | 489 (42.1) | 0.018 | 275 (42.8) | 0.046 |
| Dabigatran | 1518 (27.2) | 912 (28.8) | 0.004 | 306 (26.2) | 0.014 | 307 (26.4) | 0.000 | 167 (26.0) | 0.015 |
| Edoxaban | 352 (6.3) | 143 (4.5) | 0.010 | 47 (4.0) | 0.015 | 55 (4.7) | 0.013 | 37 (5.8) | 0.005 |
| Rivaroxaban | 1365 (24.5) | 782 (24.7) | 0.013 | 338 (29.0) | 0.031 | 310 (26.7) | 0.018 | 164 (25.5) | 0.000 |
| Aspirin | 1550 (27.8) | 1007 (31.8) | 0.016 | 387 (33.2) | 0.051 | 377 (32.5) | 0.047 | 191 (29.7) | 0.020 |
| Cilostazol | 23 (0.4) | 4 (0.1) | 0.010 | 1 (0.1) | 0.001 | 3 (0.3) | 0.002 | 0 (0.0) | 0.004 |
| Clopidogrel | 410 (7.3) | 183 (5.8) | 0.002 | 75 (6.4) | 0.017 | 61 (5.3) | 0.024 | 35 (5.4) | 0.019 |
| Ticagrelor | 31 (0.6) | 7 (0.2) | 0.003 | 3 (0.3) | 0.004 | 1 (0.1) | 0.005 | 3 (0.5) | 0.001 |
| Simvastatin | 2099 (37.6) | 1291 (40.8) | 0.014 | 517 (44.3) | 0.064 | 489 (42.1) | 0.039 | 217 (33.7) | 0.047 |
| Atorvastatin | 0 (0.0) | 0 (0.0) | 0.006 | 0 (0.0) | NA | 0 (0.0) | NA | 0 (0.0) | NA |
| Rosuvastatin | 234 (4.2) | 97 (3.1) | 0.003 | 35 (3.0) | 0.009 | 32 (2.8) | 0.016 | 24 (3.7) | 0.002 |
| Amiodarone | 159 (2.8) | 111 (3.5) | 0.003 | 42 (3.6) | 0.003 | 38 (3.3) | 0.005 | 28 (4.4) | 0.016 |
| Dronedarone | 58 (1.0) | 11 (0.3) | 0.001 | 2 (0.2) | 0.009 | 6 (0.5) | 0.004 | 1 (0.2) | 0.007 |
| Rifampicin | 6 (0.1) | 7 (0.2) | 0.014 | 2 (0.2) | 0.001 | 1 (0.1) | 0.000 | 4 (0.6) | 0.005 |
| NSAID/COX2i | 377 (6.8) | 97 (3.1) | 0.006 | 25 (2.1) | 0.029 | 39 (3.4) | 0.027 | 23 (3.6) | 0.024 |
| Laboratory parameters | |||||||||
| APTT(s), median (IQR) | 34.3 (7.8) | 31.7 (7.9) | NA | 32.8 (7.0) | NA | 33.5 (7.5) | NA | 33.4 (7.6) | NA |
| PT(s), median (IQR) | 24.3 (5.1) | 22.4 (5.4) | NA | 23.5 (4.9) | NA | 23.8 (4.8) | NA | 23.9 (5.2) | NA |
| Hemoglobin (g/dL), mean (SD) | 12 (1.9) | 12.1 (1.8) | 0.004 | 12.1 (1.8) | 0.040 | 12.3 (1.8) | 0.109 | 12.1 (1.9) | 0.019 |
| Platelet (10^9/L), mean (SD) | 233.7 (79.4) | 228.4 (80.2) | 0.041 | 232 (79.2) | 0.022 | 223.1 (77.8) | 0.134 | 231 (84.3) | 0.057 |
| Creatinine (umol/L), mean (SD) | 95.5 (52.9) | 91.0 (55.2) | 0.006 | 92.6 (60.0) | 0.003 | 91.8 (51.3) | 0.061 | 90.6 (58.5) | 0.054 |
| ALT (IU/L), mean (SD) | 33 (43.5) | 36.2 (42.5) | 0.004 | 36.8 (47.6) | 0.018 | 31.3 (35.2) | 0.001 | 44.7 (47.4) | 0.049 |
| HbA1c (%), mean (SD) | 6.3 (1.1) | 6.2 (1.1) | 0.025 | 6.2 (1.1) | 0.019 | 6.2 (1.1) | 0.077 | 6.2 (1.0) | 0.107 |
| TC (mmol/L), mean (SD) | 4 (0.9) | 1.3 (0.4) | 0.000 | 4 (0.9) | 0.032 | 4 (0.9) | 0.016 | 4 (1.0) | 0.007 |
| LDL-C (mmol/L), mean (SD) | 2.1 (0.8) | 2.1 (0.8) | 0.008 | 2.2 (0.8) | 0.040 | 2.1 (0.8) | 0.065 | 2.1 (0.8) | 0.043 |
| HDL-C (mmol/L), mean (SD) | 1.3 (0.4) | 4.0 (0.9) | 0.003 | 1.3 (0.4) | 0.006 | 1.3 (0.4) | 0.081 | 1.3 (0.4) | 0.105 |
| Triglyceride (mmol/L), mean (SD) | 1.3 (0.7) | 1.3 (0.7) | 0.009 | 1.3 (0.7) | 0.032 | 1.3 (0.7) | 0.016 | 1.3 (0.7) | 0.013 |
| Annual incidence rates of outcomes (%) | |||||||||
| IS | 3.9% | 5.5% | NA | 7.1% | NA | 5.1% | NA | 6.0% | NA |
| VTE | 3.0% | 4.2% | NA | 3.6% | NA | 3.7% | NA | 3.8% | NA |
| Mortality | 17.8% | 22.4% | NA | 23.6% | NA | 21.3% | NA | 18.4% | NA |
CYP/P-gp-modulating ASMs collectively included phenytoin (n = 1167), valproate (n = 1161), levetiracetam (n = 643), carbamazepine (n = 146), and phenobarbital (n = 49)
ASMs antiseizure medications, AF atrial fibrillation, ALT alanine aminotransferase, AMSD absolute mean standardized difference, APTT activated partial thromboplastin time, ATE non-ischemic stroke arterial thromboembolism, CHF congestive heart failure, CKD chronic kidney disease, CLD chronic liver disease, COX2i cyclo-oxygenase-2 inhibitor, CYP/P-gp cytochrome P450/P-glycoprotein, DM diabetes mellitus, GBP gabapentin, HbA1c glycated hemoglobin A1c, HDL-C high-density lipoprotein cholesterol, ICH intracranial hemorrhage, IHD ischemic heart disease, IQR interquartile range, IS ischemic stroke, LDL-C low-density lipoprotein cholesterol, NA not applicable, NSAID non-steroidal anti-inflammatory drugs, PGB pregabalin, PT prothrombin time, PVD peripheral vascular disease, SD standard deviation, TC total cholesterol, VTE venous thromboembolism
Primary Analysis
Compared with CYP/P-gp-neutral ASMs, CYP/P-gp-modulating ASMs were collectively associated with an increased risk of ischemic stroke (annual incidence rate [AIR] 5.5% vs 3.9%, adjusted hazard ratio [aHR] 1.28, 95% confidence interval [CI] 1.05–1.57, p = 0.017). Per-ASM analysis revealed that phenytoin was associated with ischemic stroke (AIR: 7.1% vs 3.9%, aHR 1.39 (1.00–1.94), p = 0.049). Phenytoin (AIR: 23.6% vs 17.8%, aHR 1.43, 95% CI 1.24–1.64, p < 0.001) and valproate (AIR: 21.3% vs 17.8%, aHR 1.21, 95% CI 1.00–1.48, p = 0.040) were associated with increased mortality. Annual incidence rates of ischemic stroke (valproate: 5.1%; levetiracetam: 6.0%; neutral: 3.9%) and venous thromboembolism (valproate: 3.7%; levetiracetam: 3.8%; neutral: 3.0%) were higher among valproate and levetiracetam users but not statistically different from CYP/P-gp-neutral ASMs in the weighted comparisons (Tables 1, 2). We excluded carbamazepine and phenobarbital users from the per-ASM analysis because of the small sample size. Figure 2A–C illustrates the cumulative incidences of study endpoints. Table S1 of the Electronic Supplementary Material (ESM) lists the annual stroke incidence of each ASM-DOAC combination.
Table 2.
Outcome comparisons between ASMs
| Primary analysis (n = 8746) | Epilepsy subgroup (n = 2173) | Atrial fibrillation subgroup (n = 5816) | |||||||
|---|---|---|---|---|---|---|---|---|---|
| Median follow-up (months) | aHR (95% CI) | P-value | Median follow-up (months) | aHR (95% CI) | P-value | Median follow-up (months) | aHR (95% CI) | P-value | |
| Ischemic stroke | |||||||||
| CYP/P-gp-modulating vs CYP/P-gp-neutral ASM | 15 | 1.28 (1.05–1.57) | 0.017 | 15 | 0.91 (0.66–1.27) | 0.592 | 15 | 1.33 (1.07–1.66) | 0.012 |
| Phenytoin vs neutral ASM | 15 | 1.39 (1.00–1.94) | 0.049 | 15 | 1.17 (0.71–1.93) | 0.536 | 15 | 1.56 (1.11–2.21) | 0.010 |
| Valproate vs neutral ASM | 14 | 1.29 (0.85–1.95) | 0.227 | 14 | 1.19 (0.76–1.86) | 0.454 | 14 | 1.11 (0.66–1.89) | 0.686 |
| Levetiracetam vs neutral ASM | 13 | 1.27 (0.34–4.68) | 0.717 | 13 | 0.74 (0.61–1.55) | 0.926 | 13 | 1.02 (0.56–1.86) | 0.940 |
| Venous thromboembolism | |||||||||
| CYP/P-gp-modulating vs CYP/P-gp-neutral ASM | 16 | 1.41 (1.11–1.80) | 0.005 | 16 | 1.06 (0.67–1.68) | 0.791 | 16 | 1.11 (0.66–1.87) | 0.696 |
| Phenytoin vs neutral ASM | 16 | 1.46 (0.93–2.30) | 0.098 | 16 | 0.87 (0.53–1.44) | 0.560 | 16 | 1.19 (0.63–2.28) | 0.580 |
| Valproate vs neutral ASM | 15 | 1.71 (0.94–3.09) | 0.076 | 15 | 0.79 (0.45–1.32) | 0.340 | 15 | 0.79 (0.18–3.42) | 0.749 |
| Levetiracetam vs neutral ASM | 14 | 2.56 (0.56–11.7) | 0.221 | 14 | 1.12 (0.67–2.12) | 0.540 | 14 | 1.89 (0.10–34.4) | 0.659 |
| Death | |||||||||
| CYP/P-gp-modulating vs CYP/P-gp-neutral ASM | 18 | 1.26 (1.15–1.39) | <0.001 | 18 | 1.38 (1.11–1.71) | 0.003 | 18 | 1.46 (1.30–1.63) | < 0.001 |
| Phenytoin vs neutral ASM | 18 | 1.43 (1.24–1.64) | <0.001 | 18 | 1.34 (1.07–1.68) | 0.011 | 18 | 1.81 (1.56–2.09) | < 0.001 |
| Valproate vs neutral ASM | 17 | 1.21 (1.00–1.48) | 0.040 | 17 | 1.38 (1.10–1.74) | 0.006 | 17 | 1.59 (1.29–1.99) | < 0.001 |
| Levetiracetam vs neutral ASM | 16 | 1.06 (0.59–1.91) | 0.839 | 16 | 1.07 (0.78–1.47) | 0.669 | 16 | 1.76 (0.61–5.11) | 0.511 |
aHR adjusted hazard ratio, ASMs antiseizure medications, CI confidence interval, NA not applicable
CYP/P-gp-modulating ASMs collectively included phenytoin, valproate, levetiracetam, carbamazepine, and phenobarbital
Fig. 2.
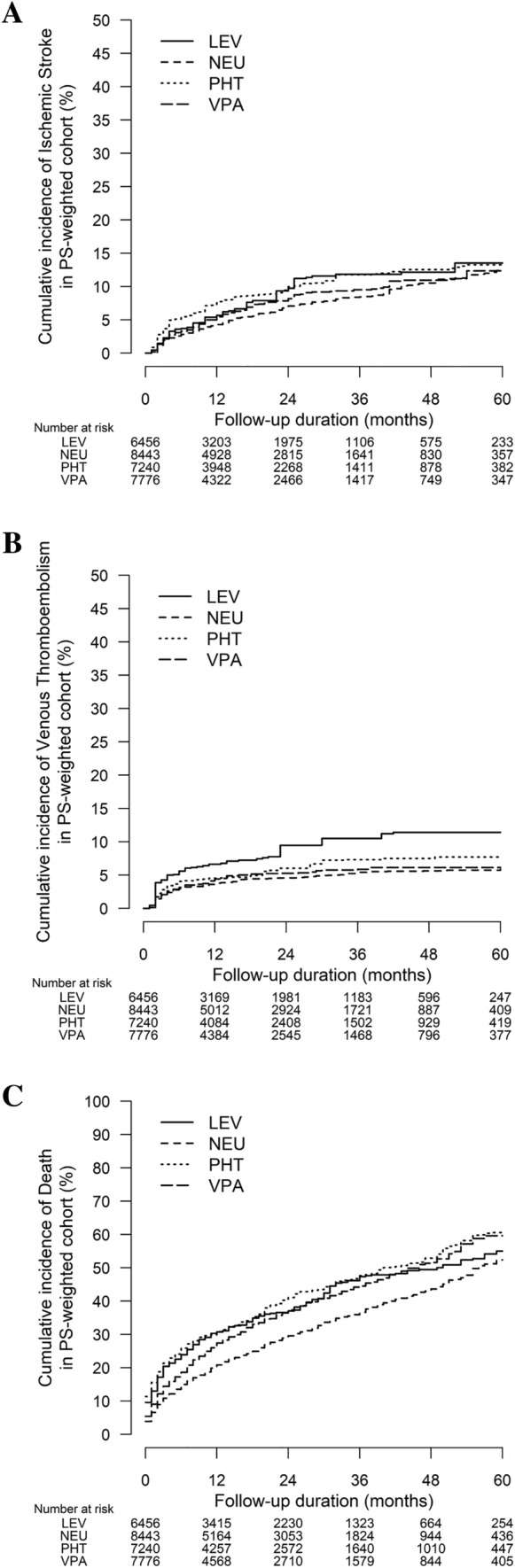
Cumulative incidences of (A) ischemic stroke, (B) venous thromboembolism, and (C) death among patients taking concurrent antiseizure medications and direct oral anticoagulants. LEV levetiracetam, NEU cytochrome P450/P-glycoprotein neutral antiseizure medications, PHT phenytoin, PS propensity score, VPA valproate
Secondary Analyses
Epilepsy Subgroup
In contrast to the primary analysis, CYP/P-gp-modulating ASMs among patients with epilepsy (n = 1873) were not associated with an increased risk of thromboembolism. Per-ASM analysis of phenytoin, valproate, and levetiracetam did not find an increased risk of thromboembolism compared to CYP/P-gp-neutral ASMs (Table 2, Table S2 of the ESM). Phenytoin (AIR: 18.1% vs 13.2%, aHR 1.34, 95% CI 1.07–1.68, p = 0.011) and valproate (AIR: 19.8% vs 13.2%, aHR 1.38; 95% CI 1.10–1.74, p = 0.006) remained associated with increased mortality (Table 2). Figure 3A–C shows the cumulative incidences of study endpoints. Table S3 of the ESM listed the annual stroke incidence of each ASM-DOAC combination in the epilepsy subgroup.
Fig. 3.
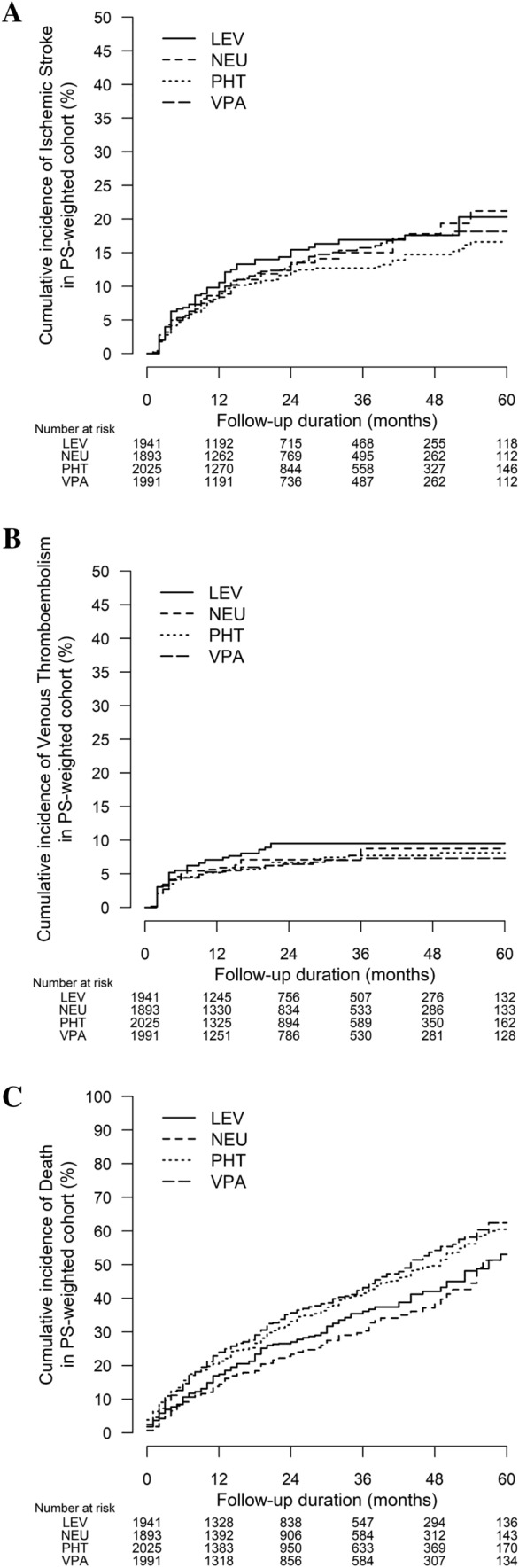
Cumulative incidences of (A) ischemic stroke, (B) venous thromboembolism, and (C) death among patients taking concurrent antiseizure medications and direct oral anticoagulants, the epilepsy subgroup. LEV levetiracetam, NEU cytochrome P450/P-glycoprotein neutral antiseizure medications, PHT phenytoin, PS propensity score, VPA valproate
AF Subgroup
Among patients in the AF subgroup (n = 5931) [Table S4 of the ESM], CYP/P-gp-modulating ASMs were associated with an increased risk of ischemic stroke (AIR: 3.0% vs 5.6%, aHR 1.33, 95% CI 1.07–1.66, p = 0.012). Per-ASM analysis showed that phenytoin was again associated with an increased risk of ischemic stroke (Table 2). Table S5 of the ESM listed the annual stroke incidence of each ASM-DOAC combination in the AF subgroup.
Sensitivity Analyses
Sensitivity analyses of 7796 patients without any missing data yielded similar findings. Phenytoin was associated with an increased risk of ischemic stroke in the overall analysis. Phenytoin and valproate were associated with a higher risk of death. In the epilepsy subgroup, phenytoin, valproate, and levetiracetam were not associated with an increased risk of thromboembolism (Tables S6–8 of the ESM).
Discussion
In this population-based study on patients with a co-prescription of DOAC and ASM: (1) CYP/P-gp-modulating ASMs were collectively associated with a higher risk of thromboembolism compared with CYP/P-gp-neutral ASMs. (2) Phenytoin was associated with ischemic stroke in the primary analysis. Yet, in the epilepsy subgroup, phenytoin, valproate, and levetiracetam were not associated with an increased risk of ischemic stroke compared to CYP/P-gp-neutral ASMs. (3) Phenytoin and valproate were associated with increased mortality across all analyses.
To our knowledge, this is the first population-based cohort study that evaluated thromboembolic risks during concurrent use of DOAC and ASMs. Pharmacokinetic studies have shown that CYP/P-gp-inducing ASMs were associated with lower DOAC levels [5, 18, 19]. Two small cohort studies descriptively reported a 4–6% annual incidence of thromboembolism among DOAC recipients who took CYP/P-gp-inducing ASMs [19, 20], which was higher than historical DOAC cohorts [21]. Our population-based cohort involving 8746 patients revealed a significant increase in ischemic stroke with phenytoin use and a non-statistically significant increase in ischemic stroke with valproate or levetiracetam use among DOAC recipients, which suggested such drug–drug interactions may be clinically relevant. The prominent effect of phenytoin on ischemic stroke, apart from lowering the DOAC level, could be in part secondary to its pro-inflammatory and atherogenic effects [22]. However, the indications of ASMs in the primary analysis were heterogenous, thus representing patients with vastly different background cerebrovascular risks.
Because of the discrepancy in the prevalence of epilepsy between patients with CYP/P-gp-neutral and CYP/P-gp-modulating ASMs (5.6% vs 58.3%) in the primary analysis, there could be bias in favor of CYP/P-gp-neutral ASMs owing to the high intrinsic stroke risk among patients with epilepsy [17]. Therefore, we performed a subgroup analysis for patients with epilepsy. Surprisingly, the associations of ischemic stroke with phenytoin, valproate, or levetiracetam among DOAC users were abolished. This finding was consistent with a hospital-based cohort study of 320 DOAC users with a history of ischemic stroke and AF, which concluded that CYP/P-gp-modulating ASMs were collectively not associated with recurrent ischemic stroke [9]. We postulate that such observations could be mediated by a complex interplay between optimal seizure control and effective anticoagulation. Among patients with epilepsy, optimal seizure control may prevent the overproduction of reactive oxygen species, which is a key mediator of atherosclerosis and subsequent ischemic stroke [17]. On the other hand, the degree of reduction in the DOAC level by CYP/P-gp-inducing ASMs that can result in ischemic strokes among patients with epilepsy remained uncertain. Further studies supported by serum DOAC level measurements and the degree of seizure control are warranted to provide mechanistic explanations for our observations. Our findings suggest that among patients with epilepsy who require DOAC, the choice of ASM should not be solely based on their potential pharmacokinetic interactions with DOAC, but also requires considerations on the potential merits that adequate seizure control may contribute to cerebrovascular health. However, it should be noted that with a median follow-up period of 16 months, our study was only able to determine the short-term cerebrovascular risks in patients with epilepsy who required concurrent ASM and DOAC. As an increased cardiovascular risk was observed among phenytoin users following 10 years of exposure [23], long-term ischemic stroke risk of ASM-DOAC combinations should be subjected to further research.
Levetiracetam use with DOAC had been controversial yet of paramount clinical importance because of the highly restricted therapeutic options in patients with epilepsy who require concurrent DOAC [24]. A nested case-control study suggested levetiracetam might increase the risk of systemic embolism among DOAC users [25]. The analysis was limited by the small number of patients using levetiracetam with no specification on prescribing indications. Previous guidelines even suggested that levetiracetam should be contraindicated among DOAC users [26]. This view was challenged as levetiracetam was only a substrate of mouse P-gp but not human P-gp [6], human liver microsomes experiments also found that levetiracetam had no pharmacokinetic interactions through the CYP pathway [27]. Our finding that levetiracetam was not associated with ischemic stroke in the epilepsy subgroup was consistent with these laboratory observations. Based on our findings in patients with epilepsy taking levetiracetam with a high prevalence of stroke (> 60%), as well as the proven efficacy of levetiracetam on post-stroke epilepsy [28], levetiracetam may be a reasonable choice in treating patients with post-stroke epilepsy who require concurrent DOAC. Nevertheless, caution should be exercised in the combined use of levetiracetam and DOAC in non-epileptic conditions because of the non-statistically significant increase in ischemic stroke.
Although phenytoin or valproate use with DOAC was not associated with an increased risk of ischemic stroke among patients with epilepsy, the higher risk of death with these two ASMs among DOAC users was consistent with other studies that involved patients without anticoagulation [29]. Possible explanations include higher rates of adverse events related to older ASMs such as hepatotoxicity, cardiac arrhythmia, and allergic reactions. Alternatively, the use of phenytoin or valproate might have implied more severe epilepsy that might inherently compromise survival. Given our study findings, close monitoring of adverse events or switching to alternative ASMs among phenytoin or valproate users with concurrent DOAC, if possible, should be considered.
Our study had several limitations. First, DOAC-specific coagulation assays were not available to ascertain the degree of interaction between DOAC and ASMs. Second, unmeasured confounding factors could be present because of the retrospective study design. We attempted to minimize confounding by including critical covariates in our propensity score weighting algorithm. Third, our analyses did not include short-term use of intravenous ASMs. Fourth, as a retrospective study, causative effects of ASM-DOAC combinations on study outcomes cannot be confirmed. Fifth, although we provided the AIRs of each ASM-DOAC combination, our study design did not allow multiple comparisons across them. Further studies targeting specific ASM-DOAC combinations with clinical-laboratory correlations are therefore warranted. Sixth, the control of epilepsy, which is also related to survival [30], was not ascertained in our study. Last, the study findings remained to be confirmed in other ethnicities because of potential inter-ethnic variations in cerebrovascular risks [31].
Conclusion
In conclusion, although the combined use of DOAC and CYP/P-gp-modulating ASM, especially phenytoin, may increase the risk of ischemic stroke, these findings were not observed in patients with epilepsy. As phenytoin, valproate, or levetiracetam use with DOAC did not increase the risk of cerebrovascular events among patients with epilepsy, these ASM options among DOAC recipients who have epilepsy should not be restricted solely based on their potential drug–drug interactions. Yet, the increased mortality during concurrent use of DOAC with phenytoin or valproate might call for caution. Further studies with DOAC level measurements are warranted to elucidate the mechanisms that resulted in such observations.
Supplementary Information
Below is the link to the electronic supplementary material.
Acknowledgments
The authors acknowledge the contribution from Dr. and Mrs. Robert Dan, Dr. Wendy Lau, Ms. Anki Miu, and Kwok Tak Seng Centre for Stroke Research and Intervention.
Declarations
Authors’ Contributions
BYI conceived and designed this research; BYI, HK, LHL, AYL, XL, HL, HHC, HYC, and YOS participated in data collection and interpretation; BYI, TCY, and GLW performed the statistical analyses; and BYI, HK, VCM, and TWL wrote the paper and participated in the revisions of it. All authors read and approved the final manuscript.
Funding
No sources of funding were received for the preparation of this article. The open access fee was supported by funding from the Chinese University of Hong Kong.
Availability of Data and Material
The data underlying this article will be shared on reasonable request to the corresponding author.
Code Availability
Not applicable.
Conflicts of Interest
Bonaventure Y. Ip, Ho Ko, Grace LH Wong, Terry CF Yip, Louis HS Lau, Alexander YL Lau, Xinyi Leng, Howan Leung, Howard HW Chan, Helen YF Chan, Vincent CT Mok, Yannie OY Soo, and Thomas W Leung, have no conflicts of interest that are directly relevant to the content of this article.
Ethics Approval
The Joint Chinese University of Hong Kong-New Territories East Cluster Clinical Research Ethics Committee approved the study (CREC Ref No. 2021.349).
Consent to Participate
Not applicable.
Consent for Publication
Not applicable.
Footnotes
Bonaventure Y. Ip and Ho Ko contributed equally as co-first authors of the article.
References
- 1.Menichelli D, Del Sole F, Di Rocco A, Farcomeni A, Vestri A, Violi F, et al. Real-world safety and efficacy of direct oral anticoagulants in atrial fibrillation: a systematic review and meta-analysis of 605 771 patients. Eur Heart J Cardiovasc Pharmacother. 2021;7(FI1):f11–f19. doi: 10.1093/ehjcvp/pvab002. [DOI] [PubMed] [Google Scholar]
- 2.Taha M, Li W, Schmidt CM, Gonzalez-Castellon M, Taraschenko O. The interactions between anticonvulsants and non-vitamin K antagonist oral anticoagulant agents: a systematic review. Epilepsy Res. 2020;162:106304. doi: 10.1016/j.eplepsyres.2020.106304. [DOI] [PubMed] [Google Scholar]
- 3.Steffel J, Collins R, Antz M, Cornu P, Desteghe L, Haeusler KG, et al. 2021 European Heart Rhythm Association practical guide on the use of non-vitamin K antagonist oral anticoagulants in patients with atrial fibrillation. Europace. 2021;23(10):1612–1676. doi: 10.1093/europace/euab065. [DOI] [PubMed] [Google Scholar]
- 4.Leung T, Leung H, Soo YO, Mok VC, Wong KS. The prognosis of acute symptomatic seizures after ischaemic stroke. J Neurol Neurosurg Psychiatry. 2017;88(1):86–94. doi: 10.1136/jnnp-2015-311849. [DOI] [PubMed] [Google Scholar]
- 5.Galgani A, Palleria C, Iannone LF, De Sarro G, Giorgi FS, Maschio M, et al. Pharmacokinetic interactions of clinical interest between direct oral anticoagulants and antiepileptic drugs. Front Neurol. 2018;9:1067. doi: 10.3389/fneur.2018.01067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Baltes S, Gastens AM, Fedrowitz M, Potschka H, Kaever V, Loscher W. Differences in the transport of the antiepileptic drugs phenytoin, levetiracetam and carbamazepine by human and mouse P-glycoprotein. Neuropharmacology. 2007;52(2):333–346. doi: 10.1016/j.neuropharm.2006.07.038. [DOI] [PubMed] [Google Scholar]
- 7.Grewal GK, Kukal S, Kanojia N, Madan K, Saso L, Kukreti R. In vitro assessment of the effect of antiepileptic drugs on expression and function of ABC transporters and their interactions with ABCC2. Molecules. 2017;22(10):1484. doi: 10.3390/molecules22101484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Stollberger C, Finsterer J. Interactions between non-vitamin K oral anticoagulants and antiepileptic drugs. Epilepsy Res. 2016;126:98–101. doi: 10.1016/j.eplepsyres.2016.06.003. [DOI] [PubMed] [Google Scholar]
- 9.Ho CJ, Chen SH, Lin CH, Lu YT, Hsu CW, Tsai MH. Non-vitamin K oral anticoagulants and anti-seizure medications: a retrospective cohort study. Front Neurol. 2020;11:588053. doi: 10.3389/fneur.2020.588053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Lau LH, Guo CL, Yip TC, Mak JW, Wong SH, Lam KL, et al. Risks of post-colonoscopic polypectomy bleeding and thromboembolism with warfarin and direct oral anticoagulants: a population-based analysis. Gut. 2022;71(1):100–110. doi: 10.1136/gutjnl-2020-323600. [DOI] [PubMed] [Google Scholar]
- 11.Yip TC, Wong VW, Chan HL, Tse YK, Lui GC, Wong GL. Tenofovir is associated with lower risk of hepatocellular carcinoma than entecavir in patients with chronic HBV infection in China. Gastroenterology. 2020;158(1):215–25.e6. doi: 10.1053/j.gastro.2019.09.025. [DOI] [PubMed] [Google Scholar]
- 12.Wong AY, Root A, Douglas IJ, Chui CS, Chan EW, Ghebremichael-Weldeselassie Y, et al. Cardiovascular outcomes associated with use of clarithromycin: population based study. BMJ. 2016;352:h6926. doi: 10.1136/bmj.h6926. [DOI] [PubMed] [Google Scholar]
- 13.Schulman S, Kearon C, Subcommittee on Control of Anticoagulation of the Scientific and Standardization Committee of the International Society on Thrombosis and Haemostasis Definition of major bleeding in clinical investigations of antihemostatic medicinal products in non-surgical patients. J Thromb Haemost. 2005;3(4):692–694. doi: 10.1111/j.1538-7836.2005.01204.x. [DOI] [PubMed] [Google Scholar]
- 14.Lane DA, Lip GY. Use of the CHA(2)DS(2)-VASc and HAS-BLED scores to aid decision making for thromboprophylaxis in nonvalvular atrial fibrillation. Circulation. 2012;126(7):860–865. doi: 10.1161/CIRCULATIONAHA.111.060061. [DOI] [PubMed] [Google Scholar]
- 15.McCaffrey DF, Griffin BA, Almirall D, Slaughter ME, Ramchand R, Burgette LF. A tutorial on propensity score estimation for multiple treatments using generalized boosted models. Stat Med. 2013;32(19):3388–3414. doi: 10.1002/sim.5753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Austin PC. An introduction to propensity score methods for reducing the effects of confounding in observational studies. Multivariate Behav Res. 2011;46(3):399–424. doi: 10.1080/00273171.2011.568786. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Chang CS, Liao CH, Lin CC, Lane HY, Sung FC, Kao CH. Patients with epilepsy are at an increased risk of subsequent stroke: a population-based cohort study. Seizure. 2014;23(5):377–381. doi: 10.1016/j.seizure.2014.02.007. [DOI] [PubMed] [Google Scholar]
- 18.Perlman A, Goldstein R, Choshen Cohen L, Hirsh-Raccah B, Hakimian D, Matok I, et al. Effect of enzyme-inducing antiseizure medications on the risk of sub-therapeutic concentrations of direct oral anticoagulants: a retrospective cohort study. CNS Drugs. 2021;35(3):305–316. doi: 10.1007/s40263-021-00795-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Candeloro M, Eikelboom JW, Chan N, Bhagirath V, Douketis JD, Schulman S. Carbamazepine, phenytoin, and oral anticoagulants: drug-drug interaction and clinical events in a retrospective cohort. Res Pract Thromb Haemost. 2022;6(2):e12650. doi: 10.1002/rth2.12650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Giustozzi M, Mazzetti M, Paciaroni M, Agnelli G, Becattini C, Vedovati MC. Concomitant use of direct oral anticoagulants and antiepileptic erugs: a prospective cohort study in patients with atrial fibrillation. Clin Drug Investig. 2021;41(1):43–51. doi: 10.1007/s40261-020-00982-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Zhu W, Ye Z, Chen S, Wu D, He J, Dong Y, et al. Comparative effectiveness and safety of non-vitamin K antagonist oral anticoagulants in atrial fibrillation patients. Stroke. 2021;52(4):1225–1233. doi: 10.1161/STROKEAHA.120.031007. [DOI] [PubMed] [Google Scholar]
- 22.Hamed SA. Atherosclerosis in epilepsy: its causes and implications. Epilepsy Behav. 2014;41:290–296. doi: 10.1016/j.yebeh.2014.07.003. [DOI] [PubMed] [Google Scholar]
- 23.Josephson CB, Wiebe S, Delgado-Garcia G, Gonzalez-Izquierdo A, Denaxas S, Sajobi TT, et al. Association of enzyme-inducing antiseizure drug use with long-term cardiovascular disease. JAMA Neurol. 2021;78(11):1367–1374. doi: 10.1001/jamaneurol.2021.3424. [DOI] [PubMed] [Google Scholar]
- 24.von Oertzen TJ, Trinka E, Bornstein NM. Levetiracetam and non-vitamin K antagonist oral anticoagulants in patients with atrial fibrillation and epilepsy: a reasonable combination. Eur Heart J. 2019;40(46):3800–3801. doi: 10.1093/eurheartj/ehz657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Gronich N, Stein N, Muszkat M. Association between use of pharmacokinetic-interacting drugs and effectiveness and safety of direct acting oral anticoagulants: nested case–control study. Clin Pharmacol Ther. 2021;110(6):1526–1536. doi: 10.1002/cpt.2369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Steffel J, Verhamme P, Potpara TS, Albaladejo P, Antz M, Desteghe L, et al. The 2018 European Heart Rhythm Association practical guide on the use of non-vitamin K antagonist oral anticoagulants in patients with atrial fibrillation. Eur Heart J. 2018;39(16):1330–1393. doi: 10.1093/eurheartj/ehy136. [DOI] [PubMed] [Google Scholar]
- 27.Nicolas JM, Collart P, Gerin B, Mather G, Trager W, Levy R, et al. In vitro evaluation of potential drug interactions with levetiracetam, a new antiepileptic agent. Drug Metab Dispos. 1999;27(2):250–254. [PubMed] [Google Scholar]
- 28.Werhahn KJ, Trinka E, Dobesberger J, Unterberger I, Baum P, Deckert-Schmitz M, et al. A randomized, double-blind comparison of antiepileptic drug treatment in the elderly with new-onset focal epilepsy. Epilepsia. 2015;56(3):450–459. doi: 10.1111/epi.12926. [DOI] [PubMed] [Google Scholar]
- 29.Sarycheva T, Lavikainen P, Taipale H, Tiihonen J, Tanskanen A, Hartikainen S, et al. Antiepileptic drug use and mortality among community-dwelling persons with Alzheimer disease. Neurology. 2020;94(20):e2099–e2108. doi: 10.1212/WNL.0000000000009435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Sperling MR, Feldman H, Kinman J, Liporace JD, O'Connor MJ. Seizure control and mortality in epilepsy. Ann Neurol. 1999;46(1):45–50. doi: 10.1002/1531-8249(199907)46:1<45::AID-ANA8>3.0.CO;2-I. [DOI] [PubMed] [Google Scholar]
- 31.Estol CJ, Bath PM, Gorelick PB, Cotton D, Martin RH. Differences in ischemic and hemorrhagic recurrence rates among race-ethnic groups in the PRoFESS secondary stroke prevention trial. Int J Stroke. 2014;9(100):43–47. doi: 10.1111/ijs.12269. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.