

Abstract
Without clinical efficacy data, vaccine protective effect may be extrapolated from animals to humans using an immunologic marker that correlates with protection in animals. This immunobridging approach was used for the two-dose Ebola vaccine regimen Ad26.ZEBOV, MVA-BN-Filo. Ebola virus (EBOV) glycoprotein binding antibody data obtained from 764 vaccinated healthy adults in five clinical studies (NCT02416453, NCT02564523, NCT02509494, NCT02543567, NCT02543268) were used to calculate mean predicted survival probability (with preplanned 95% confidence interval [CI]). We used a logistic regression model based on EBOV glycoprotein binding antibody responses in vaccinated non-human primates (NHPs) and NHP survival after EBOV challenge. While the protective effect of the vaccine regimen in humans can be inferred in this fashion, the extrapolated survival probability cannot be directly translated into vaccine efficacy. The primary immunobridging analysis evaluated the lower limit of the CI against predefined success criterion of 20% and passed with mean predicted survival probability of 53.4% (95% CI: 36.7–67.4).
Subject terms: Viral infection, Biomarkers
Introduction
Since the discovery of the Ebola virus (EBOV) in 19761, the number of outbreaks and fatalities have been increasing over time, with the devastating 2014 to 2016 outbreak in West Africa causing more cases and deaths than all previous outbreaks combined2. Since then, six EBOV disease (EVD) outbreaks have been declared in the Democratic Republic of the Congo (DRC) and Guinea3.
Reactive use of rVSV-ZEBOV-GP (Ervebo®), a recombinant replicating vector-based vaccine that demonstrated high efficacy during the outbreaks in Guinea4 and DRC5, is recommended by the World Health Organization (WHO) Strategic Advisory Group of Experts on Immunization (SAGE) during outbreak control for people at high risk of EVD6. Despite the demonstrated efficacy of this vaccine in a reactive setting, the long-lasting 2018 to 2020 outbreak highlighted the importance of complementary prophylactic vaccines. Indeed, the WHO SAGE recommended vaccination of lower-risk populations with the two-dose Ad26.ZEBOV, MVA-BN-Filo regimen (Janssen Vaccines and Prevention, B. V.) in the 2018 to 2020 outbreak6. Ad26.ZEBOV is a monovalent, recombinant, replication-incompetent, Ad26-vectored vaccine encoding the glycoprotein (GP) of the EBOV Mayinga variant. MVA-BN-Filo (Janssen Vaccines and Prevention, B.V., produced in collaboration with Bavarian Nordic) is a recombinant, non-replicating, modified vaccinia Ankara (MVA)–vectored vaccine encoding the EBOV Mayinga variant GP, the Sudan virus Gulu GP, the Marburg virus Musoke GP, and the Taï Forest virus nucleoprotein. Furthermore, conditional approval under an exceptional emergency situation was granted by the Rwanda Food and Drug Authority, and the regimen was used in a large vaccination campaign7. In July 2020, the Ad26.ZEBOV, MVA-BN-Filo regimen received approval for prophylactic use in adults and children aged ≥1 year in the European Union (Zabdeno®, Mvabea®)8,9.
Specific regulatory guidelines, including the U.S. Food and Drug Administration (FDA) Animal Rule10 and European Medicines Agency (EMA) conditional approval11 or approval under exceptional circumstances12, allow for licensure when human efficacy studies are not feasible. These guidelines specify that an immunologic marker that correlates with protection in a suitable animal model can be used to demonstrate the likelihood of clinical benefit, a concept called immunobridging, as a basis for licensure, with additional post-licensure commitments10–12. Evaluation of the protective effect of Ad26.ZEBOV, MVA-BN-Filo for European licensure under exceptional circumstances was based on such an immunobridging approach. A non-human primate (NHP) challenge model was used in which disease symptoms after EBOV challenge are similar to, but more stringent than, human EVD13,14. The NHP model is considered stringent as it is fully lethal, compared to an average human case fatality rate of 50%, and NHPs have a shorter time to onset of symptoms (average of 5.4 days compared to 6.2-9.7 days in humans) and an extremely rapid disease progression with a shorter time to death (after symptom onset, mean survival time in NHPs is 1.4 days relative to 5.8-14.4 days to death for lethal human cases)15–24. It has long been recognized that EBOV GP binding antibody levels correlate with vaccine-induced protection against EBOV25,26, which we confirmed for the Ad26.ZEBOV, MVA-BN-Filo vaccine regimen in Roozendaal et al.24. Based on this, Roozendaal et al. constructed a logistic regression model that relates binding antibody levels measured in NHPs to their survival probability24. For the current immunobridging analysis, we utilized a “one lab, one assay” approach and rebuilt the logistic regression model using GP binding antibody concentrations that were measured by the same accredited laboratory where the clinical samples were analyzed. We describe the modeling of antibody levels after vaccination in participants from five clinical studies to calculate a mean predicted survival probability with an associated confidence interval (CI), of which the lower limit had a pre-defined success criterion. Due to the stringency of the NHP model, the calculated mean predicted survival probability is expected to be an underestimation of clinical vaccine efficacy. Hence, immunobridging provides support for the vaccine protective effect in humans, yet the inferred mean predicted survival probability cannot be directly translated into actual vaccine efficacy.
Results
Protective effect in healthy adults (aged 18-50 years)
The primary immunobridging analysis was based on the pooled per-protocol immunogenicity (PPI) dataset of 764 healthy adults from five studies (EBL2001 [France, UK]27,28, EBL2002 [Burkina Faso, Côte d’Ivoire, Kenya, Uganda]29–31, EBL3001 [Sierra Leone]32–34, EBL3002 [USA]35,36, and EBL3003 [USA]36,37). The baseline and demographic characteristics are summarized in Supplementary Table 1 in the Supplementary Appendix. The mean age was 30.6 years, and most participants were male (65%) and from either the USA (51%) or African countries (43% in total; 28% from Sierra Leone). At 21 days post-Dose 2, the EBOV GP binding antibody geometric mean concentration (GMC) was 3918 enzyme-linked immunosorbent assay (ELISA) units (EU)/mL for study EBL3001 conducted in Sierra Leone and ranged from 8109 EU/mL to 11,054 EU/mL in the other four clinical studies (conducted in Burkina Faso, Côte d’Ivoire, France, Kenya, Uganda, UK, and USA); regardless of GMC, responder rates were consistent in all five studies, ranging from 98% to 100% (Supplementary Table 2 in the Supplementary Appendix).
The primary immunobridging analysis demonstrated a mean predicted survival probability of 53.4% with a lower limit of the pre-planned 95% CI of 36.7% and a post-hoc 98.7% CI (a conservative threshold regularly used in interim analyses) with a lower limit of 33.8% (Table 1). Both lower limits were well above 20%, thereby passing the pre-defined success criterion of the primary immunobridging analysis. Although the mean predicted survival probability cannot be interpreted as the actual vaccine efficacy, the analysis provides support for a protective effect of the vaccine regimen in humans.
Table 1.
Immunobridging analysis using a logistic regression model based on data from NHPs vaccinated with the Ad26.ZEBOV, MVA-BN-Filo vaccine regimen in a 56-day interval; PPI analysis set.
| Participants Vaccinated, N | 764 |
|---|---|
| Pre-planned Immunobridging Analysis | |
| Mean Predicted Survival Probability, % (95% CI) | 53.4 (36.7–67.4) |
| Post-hoc Analysis | |
| Mean Predicted Survival Probability, % (98.7% CI) | 53.4 (33.8–70.9) |
| O’Brien-Fleming Adjustment (One-sided Alpha of 0.0066) |
CI confidence interval, NHP non-human primate, PPI per-protocol immunogenicity.
This analysis was based on the pooled data of healthy adults (aged 18-50 years) vaccinated with Ad26.ZEBOV, MVA-BN-Filo in a 56-day interval in five clinical studies (EBL2001, EBL2002, EBL3001, EBL3002, and EBL3003) using a logistic regression model based on NHP data from the Ad26.ZEBOV, MVA-BN-Filo vaccine regimen in a 56-day interval.
Sensitivity to potential influencing factors is shown in Fig. 1. Given the inherent variability of subgroup analyses, and that some subgroups were very small and therefore not sufficiently powered, the lower limit of the CI was not formally compared against the success criterion defined for the primary analysis. The pre-specified sensitivity analyses were consistent overall with the primary analysis, and for the subgroup analyses by region, only a lower mean predicted survival probability was observed in the West African subgroup (36.4%), containing pooled data from Burkina Faso, Côte d’Ivoire, and Sierra Leone. A post-hoc analysis performed by country (Fig. 2) indicated that, in line with the lower immunogenicity observed in study EBL3001 (Supplementary Table 2 in the Supplementary Appendix), a lower country-specific mean predicted survival probability was obtained for Sierra Leone (30.9%), but not for the other West African countries Burkina Faso (61.2%) and Côte d’Ivoire (54.2%).
Fig. 1. Forest plot of mean predicted survival probability and 95% CI – prespecified subgroup analyses by baseline EBOV GP binding antibody concentration, sex, age category, race, and region; PPI analysis set.

This analysis was based on the pooled data of healthy adults (aged 18-50 years) vaccinated with Ad26.ZEBOV, MVA-BN-Filo in a 56-day interval in five clinical studies (EBL2001, EBL2002, EBL3001, EBL3002, and EBL3003) using a logistic regression model based on NHP data from the Ad26.ZEBOV, MVA-BN-Filo vaccine regimen in a 56-day interval. Mean predicted survival probability and the 95% bootstrapped CI are reported. CI confidence interval, EBOV Ebola virus, ELISA enzyme-linked immunosorbent assay, EU enzyme-linked immunosorbent assay units, GMC geometric mean concentration, GP glycoprotein, LLOQ lower limit of quantification (36.11 EU/mL), N number of participants with data, NHP non-human primate, PPI per-protocol immunogenicity, vertical dashed line = mean predicted survival probability from primary analysis including all participants. *The subgroup analyses excluding the participants from clinical study EBL3001 and the stratification by baseline EBOV GP ELISA level are described in more detail in the Supplementary Results.
Fig. 2. Forest plot of mean predicted survival probability and 95% CI – post hoc subgroup analyses by country.

This analysis was based on the pooled data of healthy adults (aged 18-50 years) vaccinated with Ad26.ZEBOV, MVA-BN-Filo in a 56-day interval in five clinical studies (EBL2001, EBL2002, EBL3001, EBL3002, and EBL3003) using a logistic regression model based on NHP data from the Ad26.ZEBOV, MVA-BN-Filo vaccine regimen in a 56-day interval. Mean predicted survival probability and the 95% bootstrapped CI are reported. CI confidence interval, EU enzyme-linked immunosorbent assay units, GMC geometric mean concentration, N number of participants with data, NHP non-human primate; vertical dashed line = mean predicted survival probability from primary analysis including all participants.
Protective effect in specific subpopulations: healthy adults (aged > 50 years), people living with human immunodeficiency virus (PLWH; aged 18-50 years), and children (aged 1-17 years)
Subgroup analyses were performed to assess the protective effects in older healthy adults, adult PLWH, and children because immune responses may vary in different populations and age groups. Among the 53 older participants aged >50 years, the mean age was 57.1 years (standard deviation, 4.6) and ages ranged from 51 to 69 years. The antibody GMC at 21 days post-Dose 2 was 7700 EU/mL in older adults, 5283 EU/mL in PLWH, and 13,509 EU/mL in children aged 1-17 years, with corresponding responder rates of 98%, 100%, and 98.5%, respectively (Fig. 3 and Supplementary Table 3 in the Supplementary Appendix). The mean predicted survival probability was 53% in older adults, 42.0% in PLWH, and 70.1% in children aged 1-17 years (Fig. 3). Among children, the mean predicted survival probability increased with decreasing age groups: 65.0% in children aged 12-17 years, 66.9% in children aged 4-11 years, and 82.6% in children aged 1-3 years (Fig. 3). Overall, these analyses, as well as similar analyses conducted in the full analysis set (FAS; Supplementary Table 4 in the Supplementary Appendix) indicate that Ad26.ZEBOV, MVA-BN-Filo is likely to confer protection against EVD in elderly healthy adults, PLWH, and children.
Fig. 3. Forest plot of mean predicted survival probability and 95% CI for healthy adults aged >50 years, PLWH (aged 18-50 years), and children (aged 1-17 years); PPI analysis set.

This analysis was based on the pooled data of participants vaccinated with Ad26.ZEBOV, MVA-BN-Filo in a 56-day interval in five clinical studies (EBL2001, EBL2002, EBL3001, EBL3002, and EBL3003) using a logistic regression model based on data from NHPs vaccinated with the Ad26.ZEBOV, MVA-BN-Filo vaccine regimen in a 56-day interval. CI confidence interval, EU enzyme-linked immunosorbent assay units, NHP non-human primate, PLWH people living with human immunodeficiency virus, PPI per-protocol immunogenicity. *PLWH were on a stable antiretroviral therapy regimen, had a CD4 + cell count >350 cells/μL, and were considered to be in otherwise reasonably good medical condition.
Discussion
The EMA and FDA guidelines on the clinical evaluation of vaccines state that when it is not possible to conduct a traditional clinical efficacy study, other approaches to estimate vaccine efficacy can be considered, including extrapolation of vaccine efficacy from animals to humans10–12. While this assumes that the protective mechanism is conserved between the animal model and humans, the immunologic parameter selected for immunobridging only needs to correlate with survival and is not necessarily the main mechanistic contributor to protection. We previously identified EBOV GP binding antibodies as an immune parameter strongly correlating with survival after EBOV challenge in vaccinated NHPs24. Vaccination-elicited immune responses are similar between NHPs and humans, and the same assay in the same laboratory was used to measure the vaccine-elicited binding antibody responses in NHPs and humans24. An immunobridging approach based on EBOV GP binding antibody responses was used to infer the protective effect of the vaccine regimen in people. A 3-week post–second vaccination time point was selected based on the kinetics of the antibody response observed in NHPs and confirmed in humans38–40; it was the final immunogenicity sampling time point before the transfer of NHPs to the BSL4 facility for challenge one week later. At this time point and at the clinical dose, human antibody response levels are typically two- to four-fold lower than antibody responses in NHPs28,30,33,36,38–40. While NHPs are protected at this dose level, the predicted survival probability based on human immune response levels is lower. However, as noted previously, the survival rate for EVD in humans is higher than that of NHPs. As no adjustments were made for this in the immunobridging, lower antibody levels do not necessarily indicate lower survival.
Prior to unblinding of the five clinical studies, it was agreed with the EMA that the vaccine would be considered protective if the lower limit of the 95% CI around the mean predicted survival probability was above a pre-specified success criterion of 20%. This success criterion was selected considering that, in unvaccinated NHPs, the observed survival was 0% (0/13 unvaccinated NHPs survived). A lower limit of 20% for the 95% CI provides a sufficiently large margin to indicate a true protective effect, and is similar to thresholds employed in vaccine field efficacy studies supporting regulatory approval41. Due to the stringency of the NHP challenge model, the calculated mean predicted survival probability is difficult to interpret and the inferred likelihood of protection is likely an underestimation of clinical vaccine efficacy. The primary immunobridging analysis demonstrated a mean predicted survival probability of 53.4%. Lower limits of the pre-planned 95% CI and post-hoc 98.7% CI, 36.7% and 33.8%, respectively, were well above the pre-specified success criterion of 20%, thereby confirming the likelihood of protection in healthy adults (aged 18-50 years). While immunobridging provides support for the vaccine protective effect, the mean predicted survival probability derived from the model cannot be directly translated into the actual level of clinical vaccine efficacy, which will need to be determined in a field study. In view of the severity and lethal risk posed by EVD, the benefit of likely protection together with the absence of evidence of a safety concern27–37 outweighs the current uncertainty on the exact clinical vaccine efficacy.
A substantial number of healthy adults (120 out of 899) included in the FAS had received a delayed MVA-BN-Filo vaccination (with an interval up to 454 days between the two vaccines) due to a study pause. As reported previously, EBOV GP binding antibody responses in participants who received a delayed second dose were at least as high as those who received the two vaccines in the per-protocol–defined 56-day interval28,30,31, demonstrating that the vaccine regimen is at least as immunogenic if the second vaccination is administered later than planned. In line with this observation, no notable differences were observed between the immunobridging analyses based on the FAS (see Supplementary Notes) and the primary (PPI) analysis set. The predictive vaccine protective effect was inferred in healthy adults aged >50 years, PLWH, and children aged ≥1 year, with the highest survival estimate (82.6%) observed in the youngest children (aged 1-3 years) in a post-hoc analysis.
The lower country-specific mean predicted survival probability calculated for Sierra Leone (30.9%) was expected based on the lower EBOV GP binding antibody GMC observed 21 days post-MVA-BN-Filo in study EBL3001 (Supplementary Table 2 in the Supplementary Appendix). This lower immunogenicity was not related to pre-existing immunity against the Ad26 vector, nor to demographic or logistic factors (shipment and storage of vaccines and/or sera). The lower immunogenicity in Sierra Leone may be explained by a combination of factors specific to the region as the study was conducted in the Kambia district, one of the most rural areas of Sierra Leone32,33. Individuals living in rural areas compared to more urban areas have lower life expectancy and worse health status42, which may contribute to lower immune responses to vaccination. Other geographic differences, such as nutritional deficiencies or genetic variability, could also affect vaccine immunogenicity43,44. Even in the worst-case scenario of a one-to-one translation of mean predicted survival probability to vaccine efficacy, epidemiologic modeling predicts that, at a population level, 30% vaccine efficacy would still result in a significant reduction in deaths caused by EVD (dependent on vaccination coverage)45. Upon administration of an Ad26.ZEBOV booster two years after the primary regimen, participants from Sierra Leone showed a strong anamnestic response within seven days33, demonstrating that Ad26.ZEBOV, MVA-BN-Filo had induced immunologic memory that likely contributes to protection upon exposure to EBOV.
This immunobridging analysis uses a logistic regression curve based only on the level of virus-specific antibodies present in the circulation shortly after vaccination and, while this does not take waning immunity after vaccination into account, it also does not allow vaccination-induced immunologic memory to contribute to the calculated mean predicted survival probability. In addition, due to the short time to death in NHPs, the contribution of immune memory to the durability of vaccine-elicited protection cannot be measured. Assessing the durability of the vaccine protective effect would require an animal model in which the disease course is slower and more reflective of human disease24, allowing the vaccination-induced immunologic memory to contribute to protection. However, such a model is currently not available and, in the absence of evidence for durability of protection, an Ad26.ZEBOV booster after the primary Ad26.ZEBOV, MVA-BN-Filo vaccination regimen is currently recommended as a precautionary measure in the situation of imminent risk of exposure to maximize the protective effect8,9. The research described here supports a new approach to demonstrate the probability of vaccine-mediated protection. Immunobridging has been used to support regulatory approval of the anthrax vaccine (BioThrax®)46 and will likely be used more often for other emerging infectious diseases, like Marburg virus disease, Crimean-Congo hemorrhagic fever, or Nipah47, for which evaluation of clinical efficacy testing will be as challenging.
Methods
Regression model
The logistic regression model used for this immunobridging study was previously developed using data from NHP challenge studies24. Seven NHP challenge studies were conducted with a single 0.5 mL intramuscular administration of the well-characterized EBOV Kikwit virus recommended by the Filovirus Animal Non-Clinical Group (FANG)24,48 at a fully lethal target dose of 100 plaque-forming units. A penalized logistic regression model based on NHP data was developed using Firth’s method49, with survival outcome as a dependent variable and EBOV GP binding antibody concentrations (EU/mL, log10) measured 21 days post-Dose 2 as an independent variable. For the model described in Roozendaal et al.24, EBOV GP binding concentrations were analyzed by the Battelle Biomedical Research Center (Columbus, OH, USA). For the current immunobridging analysis, all NHP samples were reanalyzed by Q2 Solutions Vaccine Testing Laboratory (San Juan Capistrano, CA, USA) to enable a direct comparison to the human immunogenicity data. More information on the selection of the immune parameter and the model development can be found in Roozendaal et al.24.
The immunogenicity data of participants vaccinated with the two-dose, heterologous Ad26.ZEBOV, MVA-BN-Filo regimen in a 56-day interval were obtained from two phase 2 and three phase 3 randomized, observer-blind, and placebo-controlled studies in healthy adults, PLWH, and pediatric (aged 1-17 years) participants: EBL2001 (France, UK)27,28, EBL2002 (Burkina Faso, Côte d’Ivoire, Kenya, Uganda)29–31, EBL3001 (Sierra Leone)32–34, EBL3002 (USA)35,36, and EBL3003 (USA)36,37. All participants received an intramuscular injection (0.5 mL) with 5 × 1010 virus particles of Ad26.ZEBOV as Dose 1, followed 56 days later by 1 × 108 Infectious Units of MVA-BN-Filo as Dose 2 (heterologous two-dose Ad26.ZEBOV, MVA-BN-Filo vaccine regimen). The protocol-defined window around Dose 2 was ±3 days for all studies, except EBL2001 (±1 day) and EBL3001 (±7 days). In all five clinical studies, EBOV GP binding antibody concentrations were measured by Q2 Solutions Laboratory. Study details are available online27,29,32,35,37 and are previously published28,30,31,33,34,36.
For immunogenicity assessments in the previous NHP challenge studies and clinical studies, serum samples were obtained from all NHPs and clinical study participants immediately before the first vaccination and three weeks after the second vaccination. Immunoglobulin G responses against EBOV GP were analyzed in all NHP and clinical samples using the same EBOV GP (Kikwit) FANG ELISA28,50, validated for both human and NHP serum, in the same Q2 Solutions Laboratory. Responses were summarized as group GMCs with 95% CI. For human samples, all values below the lower limit of quantification (LLOQ [36.11 EU/mL]) were imputed with half of the LLOQ value (ie, LLOQ/2). Clinical study participants were considered responders if the post-vaccination concentration was >2.5-fold the LLOQ in baseline seronegative individuals or >2.5-fold the baseline value in baseline seropositive participants.
Two logistic regression models were constructed for the immunobridging analysis. The primary analysis model shown in Fig. 4 contained only data from NHPs vaccinated with Ad26.ZEBOV, MVA-BN-Filo in a 56-day interval (N = 66). The second model was based on all available data of NHPs (N = 108) vaccinated with either Ad26.ZEBOV or Ad26.Filo and MVA-BN-Filo in different vaccine sequences and intervals between doses. The second model was only used in a sensitivity analysis to evaluate the robustness of the primary immunobridging result. The data that were used for the calculation of the logistic regression models are shown in Supplementary Table 5 in the Supplementary Appendix, and the result is shown in Supplementary Table 6.
Fig. 4. Logistic regression model for main regimen.
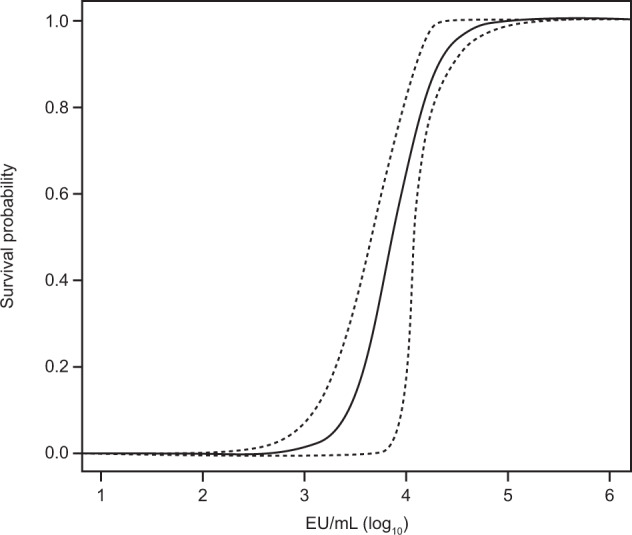
Figure depicts the logistic regression model and its 95% confidence band (bootstrap-derived using 10,000 bootstraps of the NHP data of the main regimen). EU enzyme-linked immunosorbent assay units, NHP non-human primate.
The fitted logistic regression model was used to predict a survival probability for each human ELISA value measured at 21 days post-Dose 2. The individual predicted human survival probabilities were averaged to calculate the mean predicted survival probability. First, a 95% CI was calculated using a non-parametric double bootstrap method. This method consisted of resampling the NHP and human datasets 10,000 times with replacement, repeating the fitting of a logistic model on the re-sampled NHP data, and calculating a mean predicted survival probability by inserting the re-sampled human ELISA data into the logistic model and averaging the predicted individual survival probabilities. As a result, 10,000 mean predicted survival probabilities were obtained. The 95% CI was derived as the 2.5th and 97.5th percentiles of the distribution of the mean predicted survival probabilities.
The immunobridging analysis using data from the five clinical studies was originally pre-planned as an interim futility analysis, with no foreseen Type I error rate adjustment. In view of the 2018 to 2020 outbreak and the persisting public health need, this analysis was used as the basis for marketing authorization approval in Europe. To adjust the CI for alpha spending post-hoc, a 98.7% CI was calculated. The 98.7% CI was based on the O’Brien-Fleming alpha spending rules, as this approach is conservative and regularly used in interim analyses (approximately 65% of the pre-planned data available, resulting in an O’Brien-Fleming adjusted one-sided alpha of 0.0066 [obtained using Wang-Tsiatis bounds where Δ = 0]). The same non-parametric double bootstrap procedure was used to calculate the 98.7% CI but was based on 100,000 bootstraps to ensure sufficient resolution in the extreme regions of the distribution.
Statistical analyses
The primary analysis aimed to evaluate whether the lower limit of the CI was above a pre-defined success criterion of 20%, a cutoff agreed upon with the EMA. Immunogenicity data from healthy adult participants (aged 18-50 years) vaccinated with Ad26.ZEBOV, MVA-BN-Filo in the five contributing clinical studies were pooled. The FAS comprised all participants who were randomized (and non-randomized, open-label stage 1 of study EBL3001) and received ≥1 dose of study vaccine, regardless of the occurrence of protocol deviations. The PPI analysis set represented the primary analysis set and included all randomized (and non-randomized, open-label stage 1 of study EBL3001) and vaccinated participants who received Dose 1 and Dose 2 within the protocol-defined windows, had ≥1 evaluable post-vaccination immunogenicity sample, and had no major protocol deviations influencing the immune response. Only participants with an available 21 days post-Dose 2 ELISA result were included in the immunogenicity and immunobridging analyses. Immunogenicity data were analyzed descriptively, and immunobridging was performed on the PPI analysis set (primary analysis) and on the FAS.
Immunobridging sensitivity analyses
Pre-specified immunobridging sensitivity analyses were conducted to evaluate the robustness of the primary analysis and assess potential influencing factors, such as baseline positivity in the ELISA, sex, age, race, and geographic region (Supplementary Tables 1, 7, and 8 in the Supplementary Appendix). Firstly, because Sierra Leone was the only country included in the five clinical studies that was previously affected by an EBOV outbreak, the analysis was repeated including only participants from study EBL3001 (which was conducted in Sierra Leone), stratified per baseline EBOV GP FANG ELISA levels (<LLOQ [36.11 EU/mL], LLOQ-100, >100-1000, >1000 EU/mL), to assess the impact of pre-existing EBOV GP binding antibody levels on the immunobridging analysis (Supplementary Fig. 1 in the Supplementary Appendix). Secondly, to further assess the effect of possible pre-exposure to EBOV, the analysis was repeated, excluding the participants of the Sierra Leone study (EBL3001; Supplementary Fig. 2)32. Thirdly, demographic subanalyses were conducted, stratified by age (18-30 and 31-50 years of age), sex (female and male), race (Asian, Black or African American, White, and other), and geographic region (East Africa [Kenya, Uganda], West Africa [Burkina Faso, Côte d’Ivoire, Sierra Leone], Europe [France, UK], and North America [USA]; Fig. 1 and Supplementary Fig. 2). Mean predicted survival probabilities with a 95% CI were also calculated based on the pooled data of healthy elderly participants (aged > 50 years), PLWH (aged 18-50 years), and children (1-17 years, and in three age categories: 1-3 years, 4-11 years, and 12-17 years) using the primary analysis model (Fig. 3). PLWH in this analysis were on a stable antiretroviral therapy regimen, had a CD4 + cell count >350 cells/μL, and were considered to be in otherwise reasonably good medical condition without an acquired immunodeficiency syndrome–defined diagnosis or a clinically significant disease. For all sensitivity analyses, the 95% CI were calculated based on the non-parametric double bootstrap method. Fourthly, post-hoc, a subgroup immunobridging analysis was conducted by country (Burkina Faso, Côte d’Ivoire, France, Kenya, Sierra Leone, Uganda, UK, and USA), with the 95% CI calculated based on the non-parametric double bootstrap method (Fig. 2). Finally, as a prespecified sensitivity analysis, the primary analysis was repeated using the second model based on all available ELISA data of NHPs (N = 108) vaccinated with either Ad26.ZEBOV or Ad26.Filo and MVA-BN-Filo in different vaccine sequences and intervals between doses (Supplementary Table 6)33. Results from the pre-specified sensitivity analyses are described in Section 1 (Supplementary Notes) in the Supplementary Appendix.
Reporting summary
Further information on research design is available in the Nature Research Reporting Summary linked to this article.
Supplementary information
Acknowledgements
The authors wish to acknowledge the contribution of all participants who have taken part in the clinical studies. The authors thank Chelsea McLean, PhD (Janssen Vaccines and Prevention B.V., Leiden, The Netherlands) for her review and helpful contributions to the manuscript. This work was supported/funded by: Janssen Vaccines and Prevention B.V.; the Biomedical Advanced Research and Development Authority (BARDA; Contracts: HHSO100201500008C and HHSO100201700013C); The National Institute of Allergy and Infectious Diseases (NIAID), part of the National Institutes of Health (NIH; Contracts: HHSN272200800056C, HHSN272201000006I, and HHSN272201200003I) and the Innovative Medicines Initiative 2 Joint Undertaking (grant agreement numbers 115854 [EBOVAC1] and 115861 [EBOVAC2]). Medical writing support for the development of this manuscript, under the direction of the authors, and publication coordination were provided by Vikki Clayton of Ashfield MedComms, an Ashfield Health company, and funded by Janssen Vaccines and Prevention B.V. Additional editorial assistance was provided by Courtney St. Amour, PhD, of Lumanity Communications Inc., and was funded by Janssen Vaccines and Prevention B.V., and additional publication coordination was provided by Sónia Silva (Janssen Vaccines & Prevention B.V., Leiden, The Netherlands). The initial version of the manuscript was drafted by Dr Bockstal.
Author contributions
Conception or design of the work: V.B., B.S., C.R., R.R., A.G., R.Z., B.C., J.H., K.L., M.D., H.S., and J.V.H. Acquisition of the data: V.B., M.L., J.N.S., G.A.V.R., L.S., and J.H. Analysis or interpretation of the data: V.B., M.L., D.H., B.S., C.R., R.R., T.V.E., A.G., G.A.V.R., L.S., R.Z., J.H., K.L., M.D., H.S., and J.V.H. Drafted the work: V.B. Critically reviewed the manuscript: all authors. Both V.B. and M.L. contributed equally to this article.
Data availability
Janssen has an agreement with the Yale Open Data Access (YODA) Project to serve as the independent review panel for the evaluation of requests for clinical study reports and participant-level data from investigators and physicians for scientific research that will advance medical knowledge and public health. Data will be made available following publication and approval by YODA of any formal requests with a defined analysis plan. For more information on this process or to make a request, please visit the Yoda Project site at http://yoda.yale.edu. The data sharing policy of Janssen Pharmaceutical Companies of Johnson & Johnson is available at https://www.janssen.com/clinical-trials/transparency.
Competing interests
Janssen had a role in the conception or design of the work, acquisition of the data, analysis or interpretation of the data, preparation of the manuscript, and decision to publish. All authors were full-time employees of Janssen Pharmaceutical Companies of Johnson & Johnson at the time of the study and may own shares in Janssen Pharmaceutical Companies of Johnson & Johnson. Dr Van Effelterre reports ownership of shares in GlaxoSmithKline. All authors also report receiving grant support, paid to their institution, from the following: Biomedical Advanced Research and Development Authority (BARDA; Contract: HHSO100201700013C); the National Institute of Allergy and Infectious Diseases (NIAID), part of the National Institutes of Health (NIH; Contracts: HHSN272200800056C, HHSN272201000006I, and HHSN272201200003I); and the Innovative Medicines Initiative 2 Joint Undertaking (grant agreement number 115854 [EBOVAC1]). Additionally, this project has received funding from the Innovative Medicines Initiative 2 (www.imi.europa.eu) Joint Undertaking under EBOVAC2 grant agreement No 115861; this Joint Undertaking receives support from the European Union’s Horizon 2020 research and innovation programme and the European Federation of Pharmaceutical Industries and Association (EFPIA). Dr Callendret is listed as an inventor on an international patent application (number PCT/US2015/048357) with publication number WO 2016/036955, entitled “Methods and compositions for inducing protective immunity against filovirus infection.” Dr Solforosi has participated in advisory boards for Abilita Bio, Inc. No other disclosures are reported.
Footnotes
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
These authors contributed equally: Viki Bockstal, Maarten Leyssen.
Supplementary information
The online version contains supplementary material available at 10.1038/s41541-022-00564-z.
References
- 1.WHO/International Study Team. Ebola haemorrhagic fever in Sudan, 1976. Bull. World Health Organ. 1978;56:270. [PMC free article] [PubMed] [Google Scholar]
- 2.Centers for Disease Control and Prevention. 2014–2016 Ebola outbreak in West Africa. https://www.cdc.gov/vhf/ebola/history/2014-2016-outbreak/index.html (2019).
- 3.World Health Organization. 10th Ebola outbreak in the Democratic Republic of the Congo declared over; vigilance against flare-ups and support for survivors must continue. https://www.who.int/news/item/25-06-2020-10th-ebola-outbreak-in-the-democratic-republic-of-the-congo-declared-over-vigilance-against-flare-ups-and-support-for-survivors-must-continue (2020).
- 4.Henao-Restrepo AM, et al. Efficacy and effectiveness of an rVSV-vectored vaccine in preventing Ebola virus disease: final results from the Guinea ring vaccination, open-label, cluster-randomised trial (Ebola Ca Suffit!) Lancet. 2017;389:505–518. doi: 10.1016/S0140-6736(16)32621-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.World Health Organization. Preliminary results on the efficacy of rVSV-ZEBOV-GP Ebola vaccine using the ring vaccination strategy in the control of an Ebola outbreak in the Democratic Republic of the Congo: an example of integration of research into epidemic response. https://www.who.int/publications/m/item/preliminary-results-on-the-efficacy-of-rvsv-zebov-gp-ebola-vaccine-using-the-strategy-in-the-control-of-an-ebola-outbreak (2019).
- 6.World Health Organization. Strategic Advisory Group of Experts (SAGE) on immunization interim recommendations on vaccination against Ebola virus disease (EVD). https://www.who.int/immunization/policy/position_papers/interim_ebola_recommendations_may_2019.pdf (2019).
- 7.Rwanda Biomedical Centre. The Ministry of Health launched UMURINZI Ebola vaccine program campaign. https://rbc.gov.rw/index.php?id=100&tx_news_pi1%5Bnews%5D=530&tx_news_pi1%5Bday%5D=14&tx_news_pi1%5Bmonth%5D=12&tx_news_pi1%5Byear%5D=2019&cHash=9bcec711608382d977905e5f68b0e56a (2019).
- 8.European Medicines Agency. Zabdeno. https://www.ema.europa.eu/en/medicines/human/EPAR/zabdeno (2020).
- 9.European Medicines Agency. Mvabea. https://www.ema.europa.eu/en/medicines/human/EPAR/mvabea (2020).
- 10.U.S. Food and Drug Administration. Product development under the animal rule: guidance for industry. https://www.fda.gov/files/drugs/published/Product-Development-Under-the-Animal-Rule.pdf (2015).
- 11.European Medicines Agency. Conditional marketing authorisation. https://www.ema.europa.eu/en/human-regulatory/marketing-authorisation/conditional-marketing-authorisation (2004).
- 12.European Medicines Agency. Guideline on procedures for the granting of a marketing authorization under exceptional circumstances, pursuant to Article 14 (8) of regulation (EC) no. 726/2004. https://www.ema.europa.eu/en/documents/regulatory-procedural-guideline/guideline-procedures-granting-marketing-authorisation-under-exceptional-circumstances-pursuant/2004_en.pdf (2005).
- 13.St Claire MC, Ragland DR, Bollinger L, Jahrling PB. Animal models of ebolavirus infection. Comp. Med. 2017;67:253–262. [PMC free article] [PubMed] [Google Scholar]
- 14.Siragam V, Wong G, Qiu XG. Animal models for filovirus infections. Zool. Res. 2018;39:15–24. doi: 10.24272/j.issn.2095-8137.2017.053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Ajelli M, et al. The 2014 Ebola virus disease outbreak in Pujehun, Sierra Leone: epidemiology and impact of interventions. BMC Med. 2015;13:281. doi: 10.1186/s12916-015-0524-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Bwaka MA, et al. Ebola hemorrhagic fever in Kikwit, Democratic Republic of the Congo: clinical observations in 103 patients. J. Infect. Dis. 1999;179(Suppl 1):S1–S7. doi: 10.1086/514308. [DOI] [PubMed] [Google Scholar]
- 17.Fitzpatrick G, et al. The contribution of Ebola viral load at admission and other patient characteristics to mortality in a Medecins Sans Frontieres Ebola Case Management Centre, Kailahun, Sierra Leone, June-October 2014. J. Infect. Dis. 2015;212:1752–1758. doi: 10.1093/infdis/jiv304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Hunt L, et al. Clinical presentation, biochemical, and haematological parameters and their association with outcome in patients with Ebola virus disease: an observational cohort study. Lancet Infect. Dis. 2015;15:1292–1299. doi: 10.1016/S1473-3099(15)00144-9. [DOI] [PubMed] [Google Scholar]
- 19.Khan AS, et al. The reemergence of Ebola hemorrhagic fever, Democratic Republic of the Congo, 1995. Commission de Lutte contre les Epidemies a Kikwit. J. Infect. Dis. 1999;179(Suppl 1):S76–S86. doi: 10.1086/514306. [DOI] [PubMed] [Google Scholar]
- 20.Ndambi R, et al. Epidemiologic and clinical aspects of the Ebola virus epidemic in Mosango, Democratic Republic of the Congo, 1995. J. Infect. Dis. 1999;179(Suppl 1):S8–S10. doi: 10.1086/514297. [DOI] [PubMed] [Google Scholar]
- 21.Van Kerkhove MD, Bento AI, Mills HL, Ferguson NM, Donnelly CA. A review of epidemiological parameters from Ebola outbreaks to inform early public health decision-making. Sci. Data. 2015;2:150019. doi: 10.1038/sdata.2015.19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Xu Z, et al. Epidemiologic characteristics, clinical manifestations, and risk factors of 139 patients with Ebola virus disease in western Sierra Leone. Am. J. Infect. Control. 2016;44:1285–1290. doi: 10.1016/j.ajic.2016.04.216. [DOI] [PubMed] [Google Scholar]
- 23.Yan T, et al. Clinical characteristics of 154 patients suspected of having Ebola virus disease in the Ebola holding center of Jui Government Hospital in Sierra Leone during the 2014 Ebola outbreak. Eur. J. Clin. Microbiol Infect. Dis. 2015;34:2089–2095. doi: 10.1007/s10096-015-2457-z. [DOI] [PubMed] [Google Scholar]
- 24.Roozendaal R, et al. Nonhuman primate to human immunobridging to infer the protective effect of an Ebola virus vaccine candidate. npj Vaccines. 2020;5:112. doi: 10.1038/s41541-020-00261-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Sullivan NJ, Martin JE, Graham BS, Nabel GJ. Correlates of protective immunity for Ebola vaccines: implications for regulatory approval by the animal rule. Nat. Rev. Microbiol. 2009;7:393–400. doi: 10.1038/nrmicro2129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Wong G, et al. Immune parameters correlate with protection against Ebola virus infection in rodents and nonhuman primates. Sci. Transl. Med. 2012;4:158ra146. doi: 10.1126/scitranslmed.3004582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Janssen Vaccines & Prevention B.V. A study to assess safety, tolerability, and immunogenicity of three heterologus 2-dose regimens of the candidate prophylactic vaccines for Ebola in healthy adults. ClinicalTrials.gov. NCT02416453. https://clinicaltrials.gov/ct2/show/NCT02416453 (2021).
- 28.Pollard AJ, et al. Safety and immunogenicity of a two-dose heterologous Ad26.ZEBOV and MVA-BN-Filo Ebola vaccine regimen in adults in Europe (EBOVAC2): a randomised, observer-blind, participant-blind, placebo-controlled, phase 2 trial. Lancet Infect. Dis. 2021;21:493–506. doi: 10.1016/S1473-3099(20)30476-X. [DOI] [PubMed] [Google Scholar]
- 29.Janssen Vaccines & Prevention B.V. Safety, tolerability and immunogenicity study of 3 prime-boost regimens for Ebola vaccines Ad26.ZEBOV/MVA-BN-Filo in healthy adults, children and human immunodeficiency virus positive (HIV+) adults. ClinicalTrials.gov. NCT02564523. https://clinicaltrials.gov/ct2/show/NCT02564523 (2022)
- 30.Barry H, et al. Safety and immunogenicity of 2-dose heterologous Ad26.ZEBOV, MVA-BN-Filo Ebola vaccination in healthy and HIV-infected adults: a randomised, placebo-controlled phase II clinical trial in Africa. PLoS Med. 2021;18:e1003813. doi: 10.1371/journal.pmed.1003813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Anywaine Z, et al. Safety and immunogenicity of 2-dose heterologous Ad26.ZEBOV, MVA-BN-Filo Ebola vaccination in children and adolescents in Africa: a randomised, placebo-controlled, multicentre phase II clinical trial. PLoS Med. 2022;19:e1003865. doi: 10.1371/journal.pmed.1003865. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Janssen Vaccines & Prevention B.V. Staged phase 3 study to assess the safety and immunogenicity of Ebola candidate vaccines Ad26.ZEBOV and MVA-BN-Filo (EBOVAC-Salone). ClinicalTrials.gov. NCT02509494. https://clinicaltrials.gov/ct2/show/NCT02509494 (2022).
- 33.Ishola D, et al. Safety and long-term immunogenicity of the two-dose heterologous Ad26.ZEBOV and MVA-BN-Filo Ebola vaccine regimen in adults in Sierra Leone: a combined open-label, non-randomised stage 1, and a randomised, double-blind, controlled stage 2 trial. Lancet Infect. Dis. 2022;22:97–109. doi: 10.1016/S1473-3099(21)00125-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Afolabi MO, et al. Safety and immunogenicity of the two-dose heterologous Ad26.ZEBOV and MVA-BN-Filo Ebola vaccine regimen in children in Sierra Leone: a randomised, double-blind, controlled trial. Lancet Infect. Dis. 2022;22:110–122. doi: 10.1016/S1473-3099(21)00128-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Janssen Vaccines & Prevention B.V. A study to evaluate a range of dose levels of Ad26.ZEBOV and MVA-BN-Filo in healthy adult participants. ClinicalTrials.gov. NCT02543567. https://clinicaltrials.gov/ct2/show/NCT02543567 (2017).
- 36.Bockstal V, et al. Assessments of different batches and dose levels of a heterologous two-dose Ad26.ZEBOV, MVA-BN-Filo vaccine regimen: two phase 3, randomized, placebo-controlled studies. npj Vaccines. 2021;6:157. doi: 10.1038/s41541-021-00402-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Crucell Holland BV. A study to evaluate the immunogenicity, safety and tolerability of Ad26.ZEBOV and MVA-BN-Filo in healthy adult participants. ClinicalTrials.gov. NCT02543268. https://clinicaltrials.gov/ct2/show/NCT02543268 (2016).
- 38.Milligan ID, et al. Safety and immunogenicity of novel adenovirus type 26- and modified vaccinia ankara-vectored Ebola vaccines: a randomized clinical trial. JAMA. 2016;315:1610–1623. doi: 10.1001/jama.2016.4218. [DOI] [PubMed] [Google Scholar]
- 39.Mutua G, et al. Safety and immunogenicity of a 2-dose heterologous vaccine regimen with Ad26.ZEBOV and MVA-BN-Filo Ebola vaccines: 12-month data from a phase 1 randomized clinical trial in Nairobi, Kenya. J. Infect. Dis. 2019;220:57–67. doi: 10.1093/infdis/jiz071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Anywaine Z, et al. Safety and immunogenicity of a 2-dose heterologous vaccination regimen with Ad26.ZEBOV and MVA-BN-Filo Ebola vaccines: 12-month data from a phase 1 randomized clinical trial in Uganda and Tanzania. J. Infect. Dis. 2019;220:46–56. doi: 10.1093/infdis/jiz070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.European Medicines Agency. Assessment report: Dengvaxia. https://www.ema.europa.eu/en/documents/assessment-report/dengvaxia-epar-public-assessment-report_en.pdf (2018).
- 42.Strasser R, Kam SM, Regalado SM. Rural health care access and policy in developing countries. Annu Rev. Public Health. 2016;37:395–412. doi: 10.1146/annurev-publhealth-032315-021507. [DOI] [PubMed] [Google Scholar]
- 43.Valdez Y, Brown EM, Finlay BB. Influence of the microbiota on vaccine effectiveness. Trends Immunol. 2014;35:526–537. doi: 10.1016/j.it.2014.07.003. [DOI] [PubMed] [Google Scholar]
- 44.Mentzer, A. J., O’Connor, D., Pollard, A. J. & Hill, A. V. Searching for the human genetic factors standing in the way of universally effective vaccines. Philos. Trans. R Soc. Lond. B Biol. Sci.370, 20140341 (2015). [DOI] [PMC free article] [PubMed]
- 45.Potluri R, et al. Impact of prophylactic vaccination strategies on Ebola virus transmission: a modeling analysis. PLoS One. 2020;15:e0230406. doi: 10.1371/journal.pone.0230406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Fay MP, et al. Anthrax vaccine-induced antibodies provide cross-species prediction of survival to aerosol challenge. Sci. Transl. Med. 2012;4:151ra126. doi: 10.1126/scitranslmed.3004073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.GAVI, the Vaccine Alliance. 10 infectious diseases that could be the next pandemic. https://www.gavi.org/vaccineswork/10-infectious-diseases-could-be-next-pandemic (2020).
- 48.Hirschberg R, et al. Challenges, progress, and opportunities: proceedings of the Filovirus Medical Countermeasures Workshop. Viruses. 2014;6:2673–2697. doi: 10.3390/v6072673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Firth D. Bias reduction of maximum likelihood estimates. Biometrika. 1993;80:27–38. doi: 10.1093/biomet/80.1.27. [DOI] [Google Scholar]
- 50.Logue J, et al. Use of the Filovirus Animal Non-Clinical Group (FANG) Ebola virus immuno-assay requires fewer study participants to power a study than the Alpha Diagnostic International assay. J. Virol. Methods. 2018;255:84–90. doi: 10.1016/j.jviromet.2018.02.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Janssen has an agreement with the Yale Open Data Access (YODA) Project to serve as the independent review panel for the evaluation of requests for clinical study reports and participant-level data from investigators and physicians for scientific research that will advance medical knowledge and public health. Data will be made available following publication and approval by YODA of any formal requests with a defined analysis plan. For more information on this process or to make a request, please visit the Yoda Project site at http://yoda.yale.edu. The data sharing policy of Janssen Pharmaceutical Companies of Johnson & Johnson is available at https://www.janssen.com/clinical-trials/transparency.