

Abstract
The lack of knowledge about the effect of inspiratory hyperoxia on the lung-specific tumour microenvironment and progression of lung cancer has attracted considerable attention. This study proposes that inspiratory hyperoxia has special significance for the malignant phenotype of lung cancer cells. The effects of different oxygenation parameters on the proliferation, apoptosis, invasion and migration of lung cancer cells were systematically evaluated in vitro and in vivo. Our results reveal that inspiratory hyperoxia treatment (60% oxygen, 6 h·day−1) not only has no tumour progression-promoting effects, but also suppresses lung cancer metastasis and promotes long-term survival. In addition, we combined transcriptome, proteome and metabolome analysis and found that hyperoxia treatment induced significant intracellular metabolic changes in lung cancer cells. Overall, we established that MYC/SLC1A5-induced metabolic reprogramming and glutamine addiction is a new mechanism that drives lung cancer metastasis, which can be significantly suppressed by inspiratory hyperoxia treatment. These findings are relevant to the debate on the perils, promises and antitumour effect of inspiratory hyperoxia, especially for patients with lung cancer.
Short abstract
Inspiratory hyperoxia is beneficial to lung cancer patients https://bit.ly/3wLDami
Introduction
The tissue of tumour origin is a crucial factor determining the intrinsic malignant phenotype of a tumour and the characteristics of external tumour microenvironment (TME) [1–3]. Tumours originating in the same organ usually have similar gene expression signatures, since they retain the transcriptional features of the parental tissue. The tissue of origin can also create an organ-specific TME for cancer cells, including epigenetically regulated gene expression, cellular composition and tissue architecture [4, 5]. The TME with organ-specific characteristics will impose a variety of non-cell independent pressures on cancer cells and participate in the regulation of tumour initiation, progression, and metastasis. Notably, the TME of lung tumours is extremely unique due to distinct environment created by the gas exchange in which the oxygen (O2) concentration is constantly changing [6, 7]. However, although this must be an acute challenge to lung tumours, few studies have been conducted to explain the impact of this lung-specific TME on the progression of lung cancer.
The prognosis of patients with lung cancer remains dismal due to its aggressive malignancy and profound resistance to various forms of therapy. Thus, there is a strong impetus to identify new therapeutic targets for lung cancer. Inspiratory hyperoxia has been widely used as an adjuvant therapy in many clinical settings [8]. The effect of inspiratory hyperoxia on cancer growth and metastasis remains controversial, especially in lung cancer. Existing studies have reported that, due to its ability to improve the oxygenation status of tumours, inspiratory hyperoxia can be used in conjunction with radiotherapy or chemotherapy, but this application also raises concerns that an increased O2 pressure may stimulate tumour growth via re-oxygenation of hypoxic tumour cells and increased neovascularisation [9–11]. Hatfield et al. [12] suggested that supplemental oxygenation (60%) can weaken the hypoxia-driven and adenosine A2A receptor or adenosine A2B receptor (A2AR/A2BR)-mediated (hypoxia-A2-adenosinergic) immunosuppression in the TME and serve as an immunological co-adjuvant in combination with current lung cancer immunotherapies. Notably, the effect of inspiratory hyperoxia on the TME of lung tumours is comprehensive and subversive. Not only immune cells, but also mixed cells, including cancer cells, endothelial cells and stromal cells, are under stress from lung-specific TME. Therefore, whether inspiratory hyperoxia has anti- or pro-tumourigenic effects on lung tumours needs to be further studied.
In recent decades, the advanced metabolic analysis of cancer cells has expanded the understanding of the consequences and mechanisms of cancer metabolism in different stages of tumourigenesis [13, 14]. The classical oncogenic drivers, such as MYC, KRAS, etc., can induce metabolic reprogramming in cancer cells, which allows them to actively assist tumour growth and dissemination in nutrient- and O2-limited environments [15–17]. Glucose and glutamine are major anabolic carbon sources supporting cancer cell survival and biosynthesis in most tumours. In some malignant tumours, aberrantly activated oncogenes drive cancer cells into a state of nutritional addiction or to use unconventional nutritional catabolism pathways, such as the Warburg effect and glutamine addiction [18]. Recent studies have shown that the metabolic phenotype of tumours is also affected by the organ of origin and TME. An excellent example is that MYC activates glutamine catabolism in lung tumours, but in liver tumours it activates glutamine synthesis [19].
Here, we hypothesised that the change in O2 concentration is of unique significance to the TME and the malignant phenotype of lung tumour cells. To confirm this hypothesis, the effects of different oxygenation parameters on the proliferation, apoptosis, invasion and migration of lung cancer cells were systematically evaluated in vitro and in vivo. Transcriptome, proteome and metabolome analysis were combined to determine the effect of supplemental O2 on the malignant phenotype of lung cancer and the detailed underlying mechanism. The findings of this study will provide an adequate theoretical basis to clarify the safety of oxygenation therapy for lung cancer patients and ascertain whether it has the potential to be used as an adjuvant in combination with current lung cancer treatment methods.
Materials and methods
Cell lines
The immortalised human lung epithelial cell line BEAS-2b, Lewis lung carcinoma (LLC) cells and the human lung cancer cell lines H1299 and A549 were obtained from FuHeng Biology (Shanghai, China). Cells were cultured in DMEM or RPMI 1640 medium (Gibco/Thermo Fisher Scientific, Waltham, MA, USA) supplemented with 10% fetal bovine serum and 1% penicillin/streptomycin solution, in an incubator at 37°C, with a humidified atmosphere containing 5% carbon dioxide (CO2).
Mice
Balb/c nude mice (4–6 weeks old) were purchased from Beijing Weitong Lihua Laboratory Animal Technology (Beijing, China). All animal experiments were approved by the animal protection and utilization committee of Xuzhou Medical University (Xuzhou, China). No preference in mouse sex was given for any of the studies.
Clinical samples
A series of tissue samples from patients with nonsmall cell lung cancer (NSCLC) who did not receive radiotherapy or chemotherapy were obtained from the Department of Pathology of Shanghai Pulmonary Hospital (Shanghai, China). All specimens were obtained under the guidance of the United States Health Insurance Portability and Accountability Act protocol and supervised by the ethics committee of the hospital, and were confirmed pathologically as NSCLC.
Establishment of lung cancer model
An orthotopic lung tumour model was established as reported previously [20]. Briefly, Balb/c nude mice were anaesthetised with an intraperitoneal injection of sodium pentobarbital (60 mg·kg−1). After confirming that mice had been adequately anesthetised, LLC cells (1.0×106) with 50% Matrigel matrix (Corning, Corning, NY, USA) were injected orthotopically into the left lung. Mice were examined daily for infection, bleeding, weight loss, lethargy and changes in food and/or water consumption.
For the lung metastasis model, luciferase-labelled LLC and H1299 cells (4×106) were injected into mice through the tail vein. The fluorescence expression was detected after 1 month, and then the lungs were harvested.
Hyperoxic exposure
Mice were placed in an ATTENDOR animal gas control system (China Innovation Instrument, Ningbo, China), and exposed to a well-controlled gas composition to mimic protocols of supplemental O2 delivery to humans. This system provides an independent environment with a controllable O2 concentration for animals. The gas monitoring module can effectively control CO2 emissions and supply gas of excellent quality for the breathing of the mice. O2 concentrations of 21%, 30%, 60%, 90% and 98% were selected and supplied to mice for 3, 6 and 12 h per day for 30 days.
A Smartor-118-three gas incubator (China Innovation Instrument) was used to mimic protocols of supplemental O2 delivery in vitro. BEAS-2b, LLC, H1299, A549 and H226 cells were exposed to hyperoxia (60% O2/5% CO2/35% nitrogen (N2)) for 4, 8, 12, 24, 48 and 72 h.
Tumour xenograft study
H1299 cells (5×106) were injected subcutaneously into the flanks of the mice. Then, mice in the hyperoxia group were treated with inspiratory hyperoxia (60% O2) for 21 days, and the mice in the control group were routinely fed. Tumour volume (V) was monitored every 4 days by measuring the long axis (L) and the short axis (W) of xenograft tumours and calculated using the following formula: V=(L×W2)/2.
RNA-sequencing analysis
Total RNA was isolated from the H1299 cells exposed to hyperoxia (60% O2, 24 h) and control cells, quantified and its integrity checked. The high-throughput RNA-sequencing (seq) analysis was performed by Shanghai Aksomics Biotech (Shanghai, China). Briefly, the ribosomal RNA (rRNA) was removed from the total RNA using the NEBNext rRNA Depletion Kit (New England Biolabs, Ipswich, MA, USA) following the manufacturer's instructions. RNA libraries were constructed using the NEBNext Ultra II Directional RNA Library Prep Kit (New England Biolabs) according to the manufacturer's instructions. Then, the HTSeq software (v0.9.1) was used to obtain the transcript level raw count as the expression profiling, and the edgeR software (v3.16.5) was used to perform normalisation, and differentially expressed genes were identified by their p-value and expression fold change.
Liquid chromatography tandem mass spectrometry for proteome analysis
H1299 cells (1.0×107) were lysed in 1000 µL RIPA buffer at 4°C. The total protein content of each sample was then determined using a bicinchoninic acid protein assay. Then, after performing acetone precipitation and trypsin digestion, the samples were labelled with tandem mass tagging (TMT). Subsequently, after removing the sodium deoxycholate from the samples, the peptides were desalinated. After reverse-phase high-performance liquid chromatography (RP-HPLC), peptides were fractionated into 120 fractions using high pH RP-RP-HPLC, and then combined into eight fractions. For each fraction, ∼2 µg peptide were separated and analysed using a nano-ultra-preformance liquid chromatography (Easy-nLC 1200) coupled to Q-Exactive mass spectrometry (Thermo Fisher Scientific).
The original data obtained by liquid chromatography-tandem mass spectrometry (LC-MS/MS) were searched and quantified using the Max Quant software (version 1.5.6.0; Max-Planck-Institute of Biochemistry, Martinsried, Germany). The protein sequence database (Uniprot_organism_2016_09) was downloaded from UNIPROT. This database and its reverse decoy were then searched using the Max Quant software. Both peptide and protein false discovery rate (FDR) should be <0.01. Only unmodified unique peptides were used for quantification. All the other parameters were kept as default.
Metabolomics
The 1260 infinity HPLC system (Agilent Technologies, Santa Clara, CA, USA) coupled with Q-Exactive MS/MS (Thermo Fisher Scientific) was used for metabolomics analysis in this study. Cells were washed twice with ammonium carbonate (75 mM) at pH 7.4 and snap-frozen in liquid nitrogen. Metabolites were extracted with methanol:acetonitrile (ACN) (1:1, v/v). The samples were incubated for 1 h at −20°C, followed by 15 min centrifugation at 20 000 ×g and 4°C to precipitate proteins. After drying and reconstituting with ACN:H2O (1:1, v/v), the extracts were centrifuged for 15 min at 19 676×g to remove insoluble debris. Samples were stored at −80°C until LC/MS analysis.
For LC/MS analysis, samples were separated on an amide column with a mobile phase A consisting of water mixed with ammonium acetate (25 mM) and ammonium hydroxide (25 mM) and a mobile phase B consisting of ACN. The MS/MS analysis coupled with HPLC was performed on a Q-Exactive MS/MS system (Thermo Fisher Scientific). The different data-dependent analysis methods were performed as follows: full scan range: 60–900 (m/z); resolution for MS1 and ddMS2: 70 000 and 17 500, respectively; maximum injection time for MS1 and ddMS2: 100 and 45 ms, respectively; automatic gain control (AGC) for MS1 and ddMS2: 3×106 and 2×105, respectively; isolation window: 1.6 m/z; normalised collision energies: 10, 17, 25 or 30, 40, 50. The full scan method was performed as follows: the full scan range was 60–900 m/z; the AGC was 3×106 ions; resolution was 140 000; and the maximum injection time was 100 ms. Univariate analysis was used to analysis the metabolomics dataset. The relative difference coefficient (d-value) between groups was first calculated.
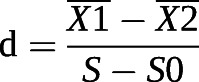 |
(X1: the average content of a metabolite in H1299 cells cultured in normoxia; X2: the average content of a metabolite in H1299 cells with 60% O2 treatment for 24 h; S: the sum of intraclass variance; S0: correction value of S.) Subsequently, significance analysis of microarrays was used to identify deferentially expressed metabolites and to estimate the FDR.
Preparation of plasmids, lentivirus and stable cells
Short hairpin RNAs (shRNAs) were designed and synthesised by GeneChem (Shanghai, China). The shRNA-control vector and shRNA-SLC1A5 or shRNA-MYC were transfected into H1299 and A549 cells using Lipofectamine 2000 transfection reagent (Invitrogen, Carlsbad, CA, USA). Cells were infected with lentivirus for 48 h and then selected with 2 ng·mL−1 puromycin for 2 weeks, refreshing the medium every 3 days.
Cell proliferation
Cell proliferation was measured by the cell counting kit-8 (CCK-8) and 5-ethynyl-2′-deoxyuridine (EdU) incorporation. Cells (5×103) were seeded in 96-well plates, and the recommended volume of CCK-8 solution was added at 24, 48 and 72 h, respectively. The EdU incorporation assay was performed using an EdU incorporation kit, following the instructions. Briefly, cells were inoculated in 96-well plates (3×103), cultured in 60% O2 for 24 h, and incubated in EdU at a concentration of 50 μM for 2 h. Cells were then fixed and stained with Apollo dye (which reacts with EdU to detect it). Ultimately, cell nuclei were stained with Hoechst33342.
Cell invasion, migration and wound healing assays
For the migration assay, H1299, A549, H226 and LLC cells (2∼4×104) were seeded in serum-free medium in the upper chamber and cultured in 60% O2 for 24 h. For the invasion assay, the Transwell filter inserts were coated with Matrigel, and 1×105 cells were seeded in serum-free medium in the upper chamber and in 60% O2 for 24 h. Cancer cells that traversed the membrane were stained with crystal violet (0.04%) and counted.
Analysis of cell apoptosis
The annexin V fluorescein isothiocyanate/propidium iodide apoptosis detection kit (KeyGen Biotech, Nanjing, China) was used for the detection of apoptotic cells with corresponding treatment.
RNA extraction, reverse transcription and quantitative reverse transcriptase PCR analysis
RNA was extracted with TRIzol (Invitrogen) and cDNA was synthesised according to the method described in the HiScript First Strand cDNA synthesis kit (Vazyme Biotech, Nanjing, China). The quantitative reverse transcriptase (qRT)-PCR analysis was performed according to the procedure described in the UltraSYBR one-step qRT-PCR kit (CWBIO, Beijing, China). The primers used for qRT-PCR analysis are listed in supplementary table S1.
Western blot analysis
Cells were lysed using the Cell Total Protein Extraction Kit (Sangon Biotech, Shanghai, China) for whole-cell lysate. Total protein was separated by SDS-PAGE and transferred to 0.45 μm polyvinylidene fluoride membranes (MilliporeSigma, Burlington, MA, USA). After blocking with 5% nonfat milk for 1 h, primary antibodies against SLC1A4, SLC1A5, c-MYC, TBX4, TBX15, BACH1, HK2, PFK2, MCT1, GPI, HIF1α, GAPDH and tubulin were separately incubated with the membranes at 4°C overnight. Subsequently, the membranes were incubated with the corresponding secondary antibody for 1 h at room temperature. Eventually, the immunoreacted protein bands were visualised. The antibodies used in this study are listed in supplementary table S4.
ELISA
To detect intracellular glutamine and glutathione, cells were incubated overnight under normal conditions, followed by a change to fresh medium and continued incubation at 21% O2 and 60% O2 for 12, 24 and 48 h. The culture medium was removed and cells were rinsed with PBS before adding the appropriate amount of lysis buffer to obtain the cell lysate. Then, the cell lysate was centrifuged at 10 000–14 000 ×g for 3–5 min and the supernatant was collected and used in subsequent measurements as described in the kit.
Glutamine uptake
The day before 60% O2 exposure, H1299 and A549 cells were seeded in 24-well plates at a density of 5×104 cells per well and incubated in a normal incubator (21% O2) or a 60% hyperoxia incubator for 24 h. H1299 and A549 cells with or without shSlc1a5 treatment were seeded in 24-well plates at a density of 5×104 cells per well and incubated in a normal incubator for 24 h. Cells were then washed twice with PBS and fresh medium containing [14C]-glutamine (glutamine concentration of 4 mM) was added for 15 min. Background cells as well as counting wells (normalised) were added with normal medium and test wells were added with radiolabelled medium. Then, cells were lysed with 1% SDS and lysis products were counted using a scintillation counter.
Extracellular flux measurements
The extracellular acidification rate (ECAR) and oxygen consumption rate (OCR) were determined using an Agilent Seahorse XFe96 extracellular flux analyser (Agilent Technologies) as described in the manufacturer's protocol. Cells were seeded in 96-well microplates the day before the experiment and incubated in a normal incubator or a 60% hyperoxia incubator for 12 h. The hydrated probe plates were placed in a CO2-free incubator overnight. Glucose (10 mM), oligomycin (1 mM) and 2-deoxy-d-glucose (50 mM) were added successively to conduct the glycolytic stress test. Oligomycin (1.5 mM), carbonyl cyanide-p-trifluoromethoxyphenylhydrazone (FCCP; 1 mM) and rotenone and antimycin A (0.5 mM) were added successively to conduct the mitochondrial stress test.
Immunohistochemical staining
The tissue microarrays were blocked with goat serum for 30 min before adding the primary antibody. Specimens were incubated with primary anti-SLC1A5 antibody for 12 h at 4°C. Eventually, the microarrays were stained with 3,3-diaminobenzidine solution and haematoxylin. The slides were photographed using an inverted microscope (Olympus Corporation, Tokyo, Japan).
Two pathologists separately assessed the specimens under blinded experimental conditions. The expression levels of SLC1A5 were evaluated by combining the percentage of cells with the staining intensity. The intensity of immunoreacted SLC1A5 was scored using a 0–3 scale (0: negative; 1: weak; 2: moderate; 3: strong); the percentage of immunoreactivity cells was graded as 1 (0–25%), 2 (26–50%), 3 (51–75%) and 4 (76–100%). The staining index value (values, 0–12) was determined by multiplying the score for staining intensity by the score for the positive area. For statistical analysis, the level of SLC1A5 expression was categorised as low (0–4), middle (4–8) and high (8–12) expression.
Chromatin immunoprecipitation analysis
Cells were cultured under normal conditions for 24 h and chromatin immunoprecipitation (ChIP) assay was performed using a ChIP assay kit (Beyotime, Shanghai, China) using the procedure described by the kit manufacturer. Cells (1×106) were fixed in 1% formaldehyde for 10 min. Nuclei extracts were sonicated to fragment DNA to 200–1000 bp. The fragmented chromatin obtained was divided into two ChIP reactions (C-MYC or control IgG). DNA was enriched by immunoprecipitation using 4 mg of anti-C-MYC or normal rabbit IgG control antibody complexed with protein A agarose beads. Protein K treatment removed the protein and unenriched DNA samples were treated in the same way as control input. PCR was performed on the ChIP and input DNA using primers for the SLC1A5 promoter region containing the MYC binding site.
Luciferase reporter assay
The luciferase reporter assay was performed according to a prior report [21]. Briefly, the luciferase reporter vector was constructed using 0.5 μg of pGL3 plasmid containing the putative MYC binding site or response element or its mutant. The constructed luciferase reporter vector was transiently co-transfected in triplicate into 293 T cells. The Dual Luciferase Kit (Promega, Madison, WI, USA) was used to determine the luciferase activity. Firefly luciferase activity was normalised to the Renilla luciferase control value and shown as the average of triplicate measurements.
Quantification and statistical analysis
The statistical analyses were performed using GraphPad Prism 8.0.1 (GraphPad Software, San Diego, CA, USA). Values are presented as the mean±sem unless stated otherwise. Details of the specific statistical analysis are indicated in the figure captions.
Results
Inspiratory hyperoxia suppresses lung cancer metastasis
To determine the effects of long-term inspiratory hyperoxia on lung cancer cells, we established a lung cancer model in immunodeficient mice. Using a gas control delivery system, mice were exposed to various concentrations of O2 (21%, 30%, 60%, 90% and 98% O2) to mimic protocols of supplemental O2 delivery to humans (figure 1a). Tumour-free mice exposed to 98% O2 (6 h·day−1) had a mortality rate >50% within 30 days. Such mortality rate may be the result of O2 toxicity, nonspecific inflammatory responses and extensive alveolar haemorrhage (supplementary figure S1a, c). The survival of tumour-free mice exposed to 60% O2 for <6 h was not affected. Histological examination revealed that ∼14% of the mice exposed to 60% O2 had nonfatal emphysema or pulmonary bulla (supplementary figure S1b, d). In addition, mice exposed to 60% O2 showed no significant difference in general physical signs (heart rate, body temperature, respiration rate) except for weight loss (supplementary figure S1e). In general, the exposure to 60% O2 (<6 h·day−1) proved to be safe in long-term treatment.
FIGURE 1.

Inspiratory hyperoxia (IH) suppresses lung cancer metastasis. a) Schematic of the workflow for the establishment of lung cancer models and IH treatment. b) Effects of IH treatment (30% oxygen (O2), 60% O2, 90% O2) on long-term survival in the orthotopic lung tumour models (n=15). Statistical significance was calculated using a log-rank test. c) Representative lung sections; arrows indicate lung tumours. d) Percentage of mice with lymph node metastasis and distant metastasis and representative images; Chi-squared test. e) 60% O2 treatment leads to long-term survival in the lung metastasis models with Lewis lung carcinoma (LLC) cells intravenous injection (n=15); log-rank test. f) 60% O2 treatment leads to long-term survival in the lung metastasis models with H1299 cells i.v. injection (n=15); log-rank test. g) Representative lung photos and lung sections of tumour-bearing mice. h) IH decreases lung tumour burden in mice 4 weeks after i.v. injection of H1299 cells (n=10); green dot indicates mice with distant metastasis. i) Bioluminescence imaging 4 weeks after injection into mice via tail vein and box plot (n=5). *: p<0.05, ***: p<0.001 versus the indicated group, two-tailed t-test.
Tumour-bearing mice exposed to 60% O2 (6 h·day−1) showed significantly improved survival and suppressed lung tumour progression (figure 1b, c). The incidence of lymph node metastasis and distant metastasis in the control group was 2.5-fold and four-fold higher than in the inspiratory hyperoxia exposed mice, respectively (figure 1d). Immunohistochemical staining showed that exposure to inspiratory hyperoxia downregulated the expression of high mobility group AT-hook 2 (HMGA2) protein, which has been reported to be a marker of tumour metastasis (supplementary figure S1f) [22].
In Balb/c nude mice, inspiratory hyperoxia also prevented LLC cells and H1299 cells from metastasising to the lung and reduced the primary tumour burden and improved survival after intravenous injection (figure 1e–h). The results of a luciferase reporter assay using H1299 cells stably expressing firefly luciferase implanted into Balb/c nude mice by i.v. injection further confirmed that inspiratory hyperoxia has antimetastatic effect (figure 1i).
Effects of inspiratory hyperoxia on the proliferation, invasiveness and apoptosis of lung cancer cells
To further evaluate the effects of inspiratory hyperoxia on lung tissues and tumours, BEAS-2b, H1299, A549, H226 and LLC cells were cultured under high O2 concentration (60% O2, 5% CO2, 35% N2) to simulate inspiratory hyperoxia in vitro. The measurement of the proliferation rate using the CCK-8 assay and EdU incorporation showed that exposure to 60% O2 for 24 h significantly reduced the proliferation rate of human normal lung epithelial cells (BEAS-2b cell line), human lung cancer cells (H1299, A549 and H226 cell lines) and LLC cells (supplementary figure S2a–f). However, it did not alter the growth of primary tumours when H1299 cells were injected subcutaneously into the mice (supplementary figure S2g, h). Experiments using hypoxia reagent to label hypoxia area in primary tumours revealed that inspiratory hyperoxia did not effectively reduce or reverse the hypoxic area in tumour xenografts (supplementary figure S2i). These data suggest that the inhibition of lung tumour growth by inspiratory hyperoxia may be closely related to lung-specific TME.
In addition, exposure to hyperoxia for <24 h had no effect on apoptosis and necrosis in BEAS-2b and lung cancer cell lines, but in BEAS-2b, LLC, H1299, H226 and A549 cells exposed to 60% O2 for >48 h induced significant apoptosis and necrosis (supplementary figure S3). Cell migration, invasion and scratch assays showed that lung cancer cells exposed to 60% O2 had lower invasive and migratory abilities (supplementary figure S4).
Transcriptomic and proteomic profiling of hyperoxia-exposed H1299 cells
RNA-seq (Gene Expression Omnibus identifier GSE192839) and TMT-based quantitative proteomics analyses (iProX data licence: IPX0003908000) on controls and 60% O2 (24 h) exposed H1299 cells were performed, and 59 577 RNAs and 5970 proteins were identified, respectively (figure 2a,b). The expression levels of 5460 identified proteins and their corresponding mRNAs showed a poor correlation (R2=0.0312; figure 2c, d). For mRNA and protein differential expression analyses, a false discovery rate (FDR) cut-off of 5% and a two-fold change threshold were used. When integrating these datasets, we identified 43 overlapping differentially expressed genes and proteins (figure 2e).
FIGURE 2.

Multi-omics data of H1299 cells treated with hyperoxia. a) Schematic diagram of the workflow for the analysis of the transcriptome, proteome and metabolome. b) Dynamic range of protein expression. c) Scatter plot of the correlation between genes quantified in both transcriptomic and proteomic datasets. d) Venn diagram showing the number of identified mRNAs and proteins by RNA-sequencing (seq) and liquid chromatography-tandem mass spectrometry (LC-MS/MS). e) Venn diagram showing significantly enriched mRNAs (fold change ≥2; false discovery rate (FDR) <0.05) and proteins (fold change ≥2; FDR <0.05), and their overlap. f) Gene ontology (GO) enrichment analysis is used to classify the significant differentially expressed mRNAs and proteins. g) Kyoto Encyclopedia of Genes and Genomes (KEGG) enrichment analysis of the significant differentially expressed mRNAs and proteins. h) Upregulation of Human Cancer Metastasis Database (HCMDB) genes in 60% oxygen (O2) (24 h) treated H1299 cells versus controls. i, j) Gene set enrichment analysis of significant differentially expressed mRNAs with normalised enrichment score (NES) and FDR Q-value. TMT: tandem mass tagging; RP-HPLC: reverse-phase high-performance liquid chromatography.
Gene Ontology enrichment analysis revealed that these significantly differentially expressed genes and proteins were enriched in “carbohydrate metabolic process”, “transepithelial transport”, “ADP metabolic process” and cell motility and migration regulation process (figure 2f). Kyoto Encyclopedia of Genes and Genomes (KEGG) enrichment analysis revealed a marked enrichment in cell metabolism as well as pathways associated with cancer development (figure 2g). To identify cellular pathways involved in lung cancer metastasis and metabolism, Gene Set Enrichment Analysis (GSEA) performed on the RNA-seq data determined that, compared with control cells, cells exposed to hyperoxia showed reduced enrichment of metastasis-related genes reported in the Human Cancer Metastasis Database (figure 2h) [23]. Furthermore, GSEA also revealed a significant decrease in metabolic pathways, including mitochondrial inner membrane, oxidative phosphorylation, glucose metabolism and glutamine metabolism genes, upon hyperoxia exposure (figure 2i, j, supplementary figure S5). In general, the multi-omics data suggested that exposure to 60% O2 (24 h) may lead to significant metabolic reprogramming of lung cancer cells, which may explain its antitumour metastasis activity.
Metabolomic changes following hyperoxia treatment
Our analysis of the cellular metabolome of normoxia- and hyperoxia-exposed H1299 cells using untargeted metabolomics to establish a holistic overview of cancer metabolism mechanisms underlying hyperoxia treatment identified a total of 319 structurally named metabolites across both treatment groups. Compared with the normoxia group, in hyperoxia-treated H1299 cells, 13 metabolites, including 6-methylquinoline, hippuric acid and pyridoxal, were significantly increased, and 15 metabolites, including 4-hydroxybenzaldehyde, l-glutamine, asparagine and pyruvic acid, were significantly decreased (figure 3a–g). Additionally, analysis of the KEGG metabolic library using the MetaboAnalyst (3.0) software revealed that altered metabolites were enriched in “alanine, aspartate and glutamate metabolism”, “arginine biosynthesis”, “glyoxylate and dicarboxylate metabolism”, “tricarboxylic acid cycle” and “d-glutamine and d-glutamate metabolism” processes (figure 3h). It is known that glucose and glutamine are the main anabolic carbon sources that support tumour cell proliferation and invasion. Therefore, we hypothesise that hyperoxia exposure may cause changes in the metabolism of glutamine and glucose, thereby regulating the metastasis of lung cancer.
FIGURE 3.

Metabolic profiles with metabolite compositions and metabolic pathways. a) Heat map analysis of metabolite compositions between 60% oxygen (O2) (24 h) and normoxia groups. b–g) Quantitation of the levels of b) 4-hydroxybenzaldehyde, c) l-glutamine, d) argininosuccinic acid, e) dl-4-hydroxyphenyllactic acid, f) 6-methylquinoline and g) pyruvic acid; two-tailed t-test. h) The metabolic pathways identified using MetaboAnalyst 3.0 software. TCA: tricarboxylic acid.
Inspiratory hyperoxia decreases the glutamine uptake of lung tumours
The assessment of the effect of hyperoxia on glutamine metabolism in lung cancer cells through measuring the level of intracellular glutamine by ELISA revealed that hyperoxia exposure caused a continuous decrease in glutamine in H1299 and A549 cells (figure 4a–c). Heat map and gene enrichment analysis indicated that the metabolic pathway of glutamine was inhibited in H1299 cells exposed to hyperoxia (figure 4d–f). qRT-PCR and immunoblotting analyses were used to confirm the regulatory effects of hyperoxia on the expression of genes largely involved in the transport, synthesis and catabolism of glutamine. The results indicated that solute carrier family 1 member 5 (SLC1A5), also known as ASCT2, a major glutamine transporter, which was highly expressed in normoxia H1299, A549, H226 and H520 cells, was downregulated by 60% O2 treatment (figure 4g–j, supplementary figure S6).
FIGURE 4.

Inspiratory hyperoxia decreases the glutamine uptake in lung tumours. a) Schematic diagram of the workflow for the measurement of intracellular glutamine levels. The red line indicates that the cells that were cultured in 60% oxygen (O2), and the black line indicates that the cells that were cultured in 21% O2. b, c) Hyperoxia treatment decreases the level of glutamine in H1299 and A549 cells (n=5); one-way ANOVA followed by Tukey's post hoc test. d) Heat map depicting changes in the expression of genes involved in “glutamine metabolism” between 60% O2 (24 h) and the normoxia group. e, f) Gene Set Enrichment Analysis (GSEA) results indicate that genes related to e) “glutamine metabolic process” and f) “glutathione metabolism” are differentially enriched in H1299 cells cultured in normoxia and 60% O2 (24 h). g, h) The levels of glutamine metabolism related genes are determined by quantitative reverse transcriptase PCR in g) H1299 and h) A549 cells (n=3). i, j) Western blot analysis for the expressions of SLC1A4 and SLC1A5 in i) H1299 and j) A549 cells (n=3); one-way ANOVA followed by Tukey's post hoc test. k) Inspiratory hyperoxia treatment reduces the expression of SLC1A5 in lung tumour in lung metastasis models; scale bars 200 μm (left), 50 μm (right). l) Immunohistochemistry staining for the expression of SLC1A5 in xenograft tumours; scale bars=50 μm. m, n) Effect of 60% O2 (24 h) treatment on the relative glutamine uptake in m) H1299 and n) A549 cells (n=5). NES: normalised enrichment score; FDR: false discovery rate. *: p<0.05, **: p<0.01, ***: p<0.001, ns: p>0.05 versus the normoxia or indicated group; two-tailed t-test.
In an in vivo lung metastasis mouse model, we found that SLC1A5 expression was significantly reduced in inspiratory hyperoxia exposed lung tumours compared with that in the control group (figure 4k). However, 60% O2 treatment had no effect on the expression of SLC1A5 in subcutaneously transplanted tumours in mice (figure 4l). We inferred that the inspiratory hyperoxia-induced decrease in SLC1A5 expression in lung tumour led to reduced glutamine uptake and metabolic demands to regulate tumour metastasis. The measurement of glutamine uptake further confirmed that hyperoxia significantly decreased the intracellular transport of glutamine in lung cancer cells (figure 4m, n).
Furthermore, the level of glutathione was significantly increased in BEAS-2b cells treated with 60% O2, suggesting that hyperoxia may play a role in the maintenance of cellular redox homeostasis. However, the opposite effect on intracellular glutathione was observed in H1299 and A549 cells (supplementary figure S7a–c). We speculate that the hyperoxia induced decrease of the glutamine level may lead to the obstruction of glutathione synthesis in lung cancer cells. Hyperoxia caused a more significant increase in reactive oxygen species (ROS) levels in lung cancer cells compared with BEAS-2b cells (supplementary figure S7d–f). Remarkably, the treatment with the antioxidant N-acetylcysteine could prevent the apoptosis caused by exposure to hyperoxia for >48 h, but promoted cancer progression (supplementary figure S7g–i).
The progression of lung cancer depends on glucose and glutamine catabolism
NSCLC cells have been reported to use glycolysis as the main pathway of glucose catabolism, even in an O2-enriched environment, that is, the Warburg effect [24]. Our metabolomic and transcriptomics data showed that exposure to hyperoxia leads to an increase in the heterogeneity of carbohydrate small molecule compounds and gene expression levels involved in glucose metabolism in lung cancer cells (supplementary figure S8a, b). We analysed the protein expression levels of glycolysis-related enzymes and found lower levels of HK2, PFK2 and MCT1 in H1299 and A549 cells exposed to 60% O2 (24 h) compared to controls (supplementary figure S8c). The measurement of the OCR and ECAR in BEAS-2b, H1299 and A549 cells revealed that H1299 and A549 cells exposed to hyperoxia showed decreased basal and maximum ECAR and OCR compared to the control group (supplementary figure S8d–i). The glycolytic capacity of lung cancer cells exposed to 60% O2 for 12 h was 40–50% lower than that in controls. However, hyperoxia did not significantly affect the glycolytic capacity and mitochondrial respiratory capacity of BEAS-2b cells (supplementary figure S8j–o).
In addition, we found that the proliferation and survival of both lung cancer cells and BEAS-2b cells are heavily dependent on glucose. Compared with glucose deprivation, glutamine deprivation is more associated with reducing the aggressiveness of tumour cells, but less with inducing proliferation arrest and apoptosis of BEAS-2b, H1299 and A549 cells (supplementary figure S9). These results suggest that antitumour strategies that target glutamine catabolism have greater safety and feasibility.
SLC1A5 mediates the hyperoxia-induced inhibition of glutamine catabolism
Functional analysis of SLC1A5 showed that shRNA-mediated knockdown of SLC1A5 expression significantly decreased the proliferation of H1299, A549 and H226 cells (figure 5a–c, supplementary figure S10) and reduced the migration and invasion abilities in H1299 cells (figure 5d, e). In in vivo metastasis assays, SLC1A5 knockdown significantly inhibited metastasis of H1299 cells to the lung and reduced the primary tumour burden (figure 5f–h). Further investigation of the role of SLC1A5 in the growth of lung tumour in a mouse xenograft model indicated that a low expression level of SLC1A5 could reduce tumour weight and volume (figure 5i–l). Moreover, SLC1A5 overexpression increased the level of intracellular glutamine in lung cancer cells and partially reversed the hyperoxia-induced inhibition of invasion (supplementary figure S11).
FIGURE 5.

SLC1A5 mediates the hyperoxia-induced suppression of glutamine catabolism. a, b) Knockdown of SLC1A5 is confirmed at the a) mRNA and b) protein level in H1299 and A549 cells (n=5). c) Determination of the relative glutamine uptake in H1299 and A549 cells with shSLC1A5 treatment (n=5). d, e) The d) migration and e) invasion of H1299 cells (12 h) with short hairpin (sh)control and shSLC1A5 treatment (n=5); scale bar=50 μm. f) Representative bioluminescence images of mice 4 weeks after intravenous injection of H1299 cells. g) Lung tumour burden in mice 4 weeks after i.v. injection of H1299 cells and h) representative lung section (n=5). i) Photographs of Matrigel plugs excised from mice after 21 days of growth in vivo and j, k) quantitative analysis of the j) tumour volume and k) tumour weight (n=10). l) Detection of SLC1A5 expressions in xenograft models by Western blot analysis (n=6). *: p<0.05, **: p<0.01, ***: p<0.001 versus the indicated group; two-tailed t-test.
MYC directly activates SLC1A5 transcription
The possible mechanism of the downregulation of SLC1A5 was further investigated by determining the expression of RNA-seq-identified differentially expressed transcription factors by qRT-PCR and Western blotting analyses. The results showed that 60% O2 exposure significantly reduced the level of MYC in H1299 and H226 cells (figure 6a–c, supplementary figure S6). We proposed that the SLC1A5 gene may be a target of oncogenic MYC. The inactivation of C-MYC by JQ1 consistently reduced expression of SLC1A5 in H1299 and A549 cells. Similarly, MYC depletion led to the decrease of SLC1A5 expression in H1299 cells (figure 6d–g). Among the 441 hyperoxia-induced differentially expressed genes, we identified a MYC-binding motif in the proximal promoters of 82 genes, including SLC1A5 (figure 6h, i). Analysis of a publicly available ChIP sequencing dataset (GSE80151) further suggested that SLC1A5 may be a MYC target gene. To determine whether the MYC-binding sequence participates in SLC1A5 transcriptional activation, we cloned the 400- to 500-bp promoter region containing the putative (wild type (wt)) MYC binding sequence and a mutated (mut) version into the luciferase reporter vector (figure 6j). As expected, the luciferase activity from the SLC1A5 promoter was 2.5-fold higher in MYC-REwt than in RE-mut in 293T cells (figure 6k). The results of the ChIP-qPCR assay further confirmed the decreased MYC occupancy on the SLC1A5 promoter in H1299 and A549 cells treated with JQ1 or MYC knockdown (figure 6l–o). Thus, the oncogene MYC activates transcription of SLC1A5, which raises the possibility that MYC regulates glutamine uptake.
FIGURE 6.

MYC directly activates SLC1A5 transcription. a) Quantitative reverse-transcriptase (qRT)-PCR analysis is used to determine the levels of transcription factors with significant differences identified by RNA-sequencing (seq) in H1299 cellls (n=3). b, c) Western blots analysis showing that exposure to 60% oxygen (O2) for 12 h and 24 h results in a decrease in MYC protein expression in H1299 cells (n=5). d–f) Analysis of MYC and SLC1A5 expressions in H1299 and A549 cells treated with JQ1. g) Effect of MYC knockdown on the expression level of SLC1A5 protein in H1299 cells (n=3). h) Venn diagram of the RNA-seq data showing the number of genes differentially expressed in H1299 cells with 60% O2 treatment versus the control cells (blue). Small circle show genes with MYC binding motifs in their proximal promoter. i) Predicted binding site MYC-binding sites of SLC1A5. Yellow arrows show primers used for chromatin immunoprecipitation (ChIP)-qPCR analysis. j) Schematic of the MYC binding site, the MYC response elements (MYC REwt), and its mutants (REmut) within the promoter of SLC1A5. k) Luciferase activity of 293T cell with and without ectopically expressed MYC (n=3). l–o) Binding of MYC to SLC1A5 is analysed by ChIP assays in H1299 and A549 cells with and without JQ1 and MYC knockdown treatment (n=3). *: p<0.05, **: p<0.01, ***: p<0.001 versus the normoxia or indicated group; two-tailed t-test.
SLC1A5 expression level is associated with tumour stage, lymph node metastases and poor survival in human NSCLC
We performed microarray analysis using the Gene Expression Profiling Interactive Analysis online platform and database to identify the expressions of SLC1A5 and MYC in NSCLC. The results indicated that the SLC1A5 level was higher in NSCLC than in the adjacent tissues. Notably, MYC was only significantly expressed in lung squamous cell carcinoma (LUSC), and did not appear to play a significant role in lung adenocarcinoma (LUAD) (figure 7a–d). Similar findings were also observed in cervical squamous cell carcinoma, cholangiocarcinoma, pancreatic cancer and other malignant tumours (supplementary figure S12a, b). In addition, there was a significant positive correlation between MYC and SLC1A5 mRNA levels in NSCLC (figure 7e, supplementary figure S12c, d). Analysis of The Cancer Genome Atlas datasets also showed that the levels of SLC1A5 and MYC were upregulated in LUAD with lymph node or distant metastasis (figure 7f–i, supplementary figure S12e–l).
FIGURE 7.

SLC1A5 is associated with the tumour stage, lymph node metastases and poor survival in human nonsmall cell lung cancer (NSCLC). a–d) SLC1A5 and MYC expressions were analysed in NSCLC tissues (n=969) and their adjacent noncancerous tissues in the Gene Expression Profiling Interactive Analysis database. e) Correlation between mRNA levels of SLC1A5 versus MYC in NSCLC tissues according to The Cancer Genome Atlas (TCGA) database; r=0.430, p<0.001. f–i) Data of TCGA datasets showing that SLC1A5 is closely related to lymph node metastasis and distant metastasis in lung adenocarcinoma (LUAD). j) Representative images from human NSCLC tumours stained with SLC1A5; scale bars=100 μm. k–n) Correlation of SLC1A5 expression with tumour grade, lymph node metastasis, distant metastasis and epidermal growth factor receptor (EGFR) mutation in 219 NSCLC patients; Chi-squared test. o) Kaplan–Meier survival curve analysis was performed to examine the effects of the genes on the survival rate in NSCLC; p<0.001, log-rank test. LUSC: lung squamous cell carcinoma; ns: nonsignificant. *: p<0.05, **: p<0.01, ns: p>0.05 versus indicated group.
In addition, we constructed a tissue microarray of 219 human NSCLC specimens, and used it for immunohistochemical analysis (figure 7j). We found that the level of SLC1A5 was significantly associated with clinical tumour stage and lymph node metastasis in both LUAD and LUSC (figure 7k–n, supplementary tables S2 and S3). Additionally, Kaplan–Meier analysis indicated that NSCLC patients with high levels of SLC1A5 have poor survival prognosis (figure 7o).
Discussion
In this study, we hypothesised that the change in O2 concentration has a unique significance for the lung-specific TME and the malignant phenotype of lung tumour cells. To confirm this hypothesis, the effects of different oxygenation parameters on the proliferation, apoptosis and invasion of lung cancer cells were systematically evaluated in vitro and in vivo. In addition, transcriptome, proteome and metabolome analyses were combined to determine the effect of supplemental O2 on the malignant phenotype of lung cancer and the detailed mechanism mediating this effect. The findings of this study will provide a satisfactory theoretical basis to clarify the safety of oxygenation therapy for lung cancer patients and ascertain whether it has the potential to be used as an adjuvant in combination with current lung cancer treatment methods.
Effectively reducing tumour burden and metastasis risk is the main challenge in the clinical response and management of advanced lung cancer [25, 26]. This study demonstrated that MYC/SLC1A5-induced metabolic reprogramming and glutamine addiction serves as a new mechanism that drives lung cancer metastasis, which can be significantly suppressed by inspiratory hyperoxia (60% O2). These findings are relevant to the debate on the perils, promises and antitumour effect of inspiratory hyperoxia, especially for patients with lung cancer. The clinical protocol of respiratory hyperoxia is widely used in patients with oxygenation deficiency caused by various diseases, including lung cancer. Previous studies have shown that respiratory hyperoxia has a confirmed stimulatory effect on the proliferation of endothelial and epithelial cells in wound healing [27–30]. However, the unknown impact of inspiratory hyperoxia on local lung cancer progression has raised some concerns. Over the past decade, studies have strongly suggested that respiratory hyperoxia did not have a stimulatory effect on tumour growth, recurrence and metastasis in most cancers [31]. Evidence from clinical and animal studies has shown that hyperbaric oxygen can enhance the effects of chemotherapy and radiotherapy in cervical and breast cancer [32, 33]. A possible explanation is that hyperbaric oxygen overcomes the hypoxia and insufficient vascular development inside the tumour, thereby increasing the transport of drug molecules and therapeutic sensitivity [34]. Kim et al. [9] intravenously injected mice with LLC cells and exposed them to a 24-h normobaric (95% O2)/normoxia cycle for 2 weeks, and found that respiratory hyperoxia inhibited the progression of lung cancer by inducing apoptosis. Additionally, studies by Hatfield and co-workers [12, 35, 36] revealed the antitumour effects of respiratory hyperoxia by reversing the hypoxia-adenosinergic immunosuppression in the TME. In this study, our data indicated that exposure to 60% O2 (6 h·day−1) inhibited lung cancer metastasis and increased the survival time of tumour-bearing mice. We emphasise that it is very important to take the proper time of application, duration, pressure and dose to achieve the overall benefit for mice with lung tumours. It is not recommended to use >60% O2 and a duration of >12 h, to avoid serious complications, including pulmonary bullae, alveolar haemorrhage, oxygen toxicity and even death.
Glutamine is an essential amino acid for NSCLC growth in vitro and in vivo. Recent studies have proposed that glutamine supports lung cancer growth through a KRAS-regulated metabolic pathway under hypoxia [37, 38]. Momcilovic et al. [39] also showed that LUSC could circumvent inhibition of mTOR and glycolysis by upregulation of glutamine metabolism. Another major finding of our untargeted metabolomics study is that inspiratory hyperoxia had a profound effect on intracellular metabolism of lung cancer cells. We found that hyperoxia exposure significantly decreased the expression of SLC1A5 and the level of intracellular glutamine in lung cancer cells. Additional data confirmed that hyperoxia-induced suppression of the aggressiveness of lung cancer is mediated by inhibition of glutamine uptake, in a manner similar to glutamine deprivation. In vitro, hyperoxia-induced suppression of glycolysis may be the main contributor to its inhibition of the proliferation of lung cancer cells and BEAS-2b cells. However, hyperoxia did not significantly affect the growth of tumour xenografts in vivo. We believe that there may be two reasons for this inconsistency. First, O2 supplementation is not enough to counteract the limitations of tumour volume increase and vascular development defects. Second, the inhibitory effect of inspiratory hyperoxia on lung tumour growth may depend on lung-specific TME. In fact, tumour cells cultured in vitro and early primary lung tumours do not experience significant hypoxia, which suggests that reversing hypoxia is not the main mechanism of hyperoxia-induced inhibition of the proliferation of lung cancer cells and reduction of the lung tumour burden.
The transcription factor MYC is one of the most common somatically mutated oncogenes in human cancer. High level of MYC is found in both high-grade pre-malignancy and invasive tumours and is associated with poor outcome in different human tumour types [40, 41]. Recent studies suggest that MYC is specifically necessary for the metastasis of tumour cells independent of its effects on proliferation and survival [42]. Our data indicated that exposure to 60% O2 for 12 h significantly downregulated MYC expression at both mRNA and protein levels. Additionally, we found that MYC directly activates SLC1A5 transcription, leading to the reprogramming of glutamine metabolism. In summary, we discovered a MYC/SLC1A5 signalling pathway regulated by hyperoxia, which is the basis for glutamine uptake and lung cancer metastasis (figure 8).
FIGURE 8.

Summary of the mechanism of inspiratory hyperoxia supresses lung cancer metastasis through a MYC/SLC1A5-dependent metabolic pathway. Hyperoxia treatment (60% oxygen (O2)) reduces the expression level of MYC, resulting in downregulation of the MYC target gene SLC1A5 in NSCLC cells. Low levels of SLC1A5 lead to disturbance in glutamine uptake and catabolism, ultimately leading to a reduction in lung cancer cell invasiveness and inhibition of metastasis. PR: promoter region.
Moreover, our preliminarily investigation of the role of inspiratory hyperoxia-induced oxidative stress in the progression of lung cancer revealed that hyperoxia exposure caused a decrease in the glutathione level and a significant increase in intracellular ROS in BEAS-2b, H1299 and A549 cells. Recent studies provide compelling evidence that ROS have antitumourigenic roles with the potential to become a novel anticancer target [43–46]. Our study also confirmed that exogenous antioxidants promote the metastasis of lung cancer under inspiratory hyperoxia, which is consistent with previous reports [16]. However, exposure to hyperoxia for more than 48 h could significantly increase intracellular ROS to toxic levels and induced severe apoptosis and necrosis in both lung cancer cells and BEAS-2b cells. This reminds us that balancing the antitumour activity and toxic effect of ROS needs to be cautiously considered when using inspiratory hyperoxia.
In conclusion, our study reveals that inspiratory hyperoxia exposure not only has no tumour progression-inducing effects, but also suppresses lung cancer metastasis. We also propose that intracellular metabolic reprogramming driven by the oncogene MYC/SLC1A5 axis is the main mechanism by which inspiratory hyperoxia inhibits cancer metastasis. These findings will have implications for future therapeutic approaches as inspiratory hyperoxia can potentially synergise with therapies that target glutamine metabolism, glycolysis or ROS, such as chemotherapy and radiation.
Supplementary material
Please note: supplementary material is not edited by the Editorial Office, and is uploaded as it has been supplied by the author.
Supplementary material ERJ-00062-2022.Supplement (3.1MB, pdf)
Shareable PDF
Acknowledgements
We thank Shanghai Aksomics Biotech (Shanghai, China) for assistance with the analysis of multiomics data.
Footnotes
Author contributions: Hao Zhang and Chang Chen conceived the project. Xiucheng Liu, Hao Qin and Zheng Li performed all experiments on cell lines and mice. Wei Zhuang, Shoujie Feng, Chen Guo and Yin Lv helped establish lung cancer models. Xiucheng Liu and Xiaoyu Quan wrote the manuscript. All authors contributed to experimental design and data analysis. Xiucheng Liu composed the manuscript. All authors reviewed the manuscript and discussed the work.
This article has an editorial commentary: https://doi.org/10.1183/13993003.01357-2022
Conflict of interest: The authors declare that they have no competing interests.
Support statement: This work was supported by funds from the Social Development Projects of Key R&D Programs in Jiangsu Province (BE2019643 to Hao Zhang), from the Social Development Projects of Key R&D Programs in Xuzhou Ciy (2022 to Hao Zhang), from the National Natural Science Foundation of Jiangsu Province (BK20171178 to Hao Zhang), from General Program of Jiangsu Commission of Health (H2017083 to Hao Zhang), and from the Project of Invigorating Health Care through Science, Technology and Education, Jiangsu Provincial Medical Youth Talent (QNRC2016778 to Hao Zhang). Funding information for this article has been deposited with the Crossref Funder Registry.
References
- 1.Hoadley KA, Yau C, Hinoue T, et al. Cell-of-origin patterns dominate the molecular classification of 10,000 tumors from 33 types of cancer. Cell 2018; 173: 291–304. doi: 10.1016/j.cell.2018.03.022 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Klemm F, Maas RR, Bowman RL, et al. Interrogation of the microenvironmental landscape in brain tumors reveals disease-specific alterations of immune cells. Cell 2020; 181: 1643–1660. doi: 10.1016/j.cell.2020.05.007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Faubert B, Solmonson A, DeBerardinis RJ. Metabolic reprogramming and cancer progression. Science 2020; 368: eaaw5473. doi: 10.1126/science.aaw5473 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Guha P, Gardell J, Rabinowitz B, et al. Monocytic and granulocytic myeloid-derived suppressor cell plasticity and differentiation are organ-specific. Oncogene 2021; 40: 693–704. doi: 10.1038/s41388-020-01559-7 [DOI] [PubMed] [Google Scholar]
- 5.Zhang C, Yu D. Suppressing immunotherapy by organ-specific tumor microenvironments: what is in the brain? Cell Biosci 2019; 9: 82. doi: 10.1186/s13578-019-0349-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Schulz M, Salamero-Boix A, Niesel K, et al. Microenvironmental regulation of tumor progression and therapeutic response in brain metastasis. Front Immunol 2019; 10: 1713. doi: 10.3389/fimmu.2019.01713 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Quail DF, Joyce JA. Microenvironmental regulation of tumor progression and metastasis. Nat Med 2013; 19: 1423–1437. doi: 10.1038/nm.3394 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Schwarte LA, Schober P, Loer SA. Benefits and harms of increased inspiratory oxygen concentrations. Curr Opin Anaesthesiol 2019; 32: 783–791. doi: 10.1097/ACO.0000000000000791 [DOI] [PubMed] [Google Scholar]
- 9.Kim SW, Kim IK, Ha JH, et al. Normobaric hyperoxia inhibits the progression of lung cancer by inducing apoptosis. Exp Biol Med 2018; 243: 739–748. doi: 10.1177/1535370218774737 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Schumann M, Schulz H, Hackney AC, et al. Feasibility of high-intensity interval training with hyperoxia vs. intermittent hyperoxia and hypoxia in cancer patients undergoing chemotherapy – study protocol of a randomized controlled trial. Contemp Clin Trials Commun 2017; 8: 213–217. doi: 10.1016/j.conctc.2017.11.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Al-Waili NS, Butler GJ. Effects of hyperbaric oxygen on inflammatory response to wound and trauma: possible mechanism of action. ScientificWorldJournal 2006; 6: 425–441. doi: 10.1100/tsw.2006.78 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Hatfield SM, Kjaergaard J, Lukashev D, et al. Immunological mechanisms of the antitumor effects of supplemental oxygenation. Sci Transl Med 2015; 7: 277ra230. doi: 10.1126/scitranslmed.aaa1260 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Pavlova NN, Thompson CB. The emerging hallmarks of cancer metabolism. Cell Metab 2016; 23: 27–47. doi: 10.1016/j.cmet.2015.12.006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.DeBerardinis RJ, Chandel NS. Fundamentals of cancer metabolism. Sci Adv 2016; 2: e1600200. doi: 10.1126/sciadv.1600200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Wise DR, DeBerardinis RJ, Mancuso A, et al. Myc regulates a transcriptional program that stimulates mitochondrial glutaminolysis and leads to glutamine addiction. Proc Natl Acad Sci USA 2008; 105: 18782–18787. doi: 10.1073/pnas.0810199105 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Wiel C, Le Gal K, Ibrahim MX, et al. BACH1 stabilization by antioxidants stimulates lung cancer metastasis. Cell 2019; 178: 330–345. doi: 10.1016/j.cell.2019.06.005 [DOI] [PubMed] [Google Scholar]
- 17.Ou Y, Wang SJ, Li D, et al. Activation of SAT1 engages polyamine metabolism with p53-mediated ferroptotic responses. Proc Natl Acad Sci USA 2016; 113: E6806–E6812. doi: 10.1073/pnas.1607152113 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Ma T, Patel H, Babapoor-Farrokhran S, et al. KSHV induces aerobic glycolysis and angiogenesis through HIF-1-dependent upregulation of pyruvate kinase 2 in Kaposi's sarcoma. Angiogenesis 2015; 18: 477–488. doi: 10.1007/s10456-015-9475-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Yuneva MO, Fan TW, Allen TD, et al. The metabolic profile of tumors depends on both the responsible genetic lesion and tissue type. Cell Metab 2012; 15: 157–170. doi: 10.1016/j.cmet.2011.12.015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Justilien V, Fields AP. Utility and applications of orthotopic models of human non-small cell lung cancer (NSCLC) for the evaluation of novel and emerging cancer therapeutics. Curr Protoc Pharmacol 2013; 62: 14.27.1–14.27.17. doi: 10.1002/0471141755.ph1427s62 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Yue M, Jiang J, Gao P, et al. Oncogenic MYC activates a feedforward regulatory loop promoting essential amino acid metabolism and tumorigenesis. Cell Rep 2017; 21: 3819–3832. doi: 10.1016/j.celrep.2017.12.002 [DOI] [PubMed] [Google Scholar]
- 22.Morishita A, Zaidi MR, Mitoro A, et al. HMGA2 is a driver of tumor metastasis. Cancer Res 2013; 73: 4289–4299. doi: 10.1158/0008-5472.CAN-12-3848 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Zheng G, Ma Y, Zou Y, et al. HCMDB: the Human Cancer Metastasis Database. Nucleic Acids Res 2018; 46: D950–D955. doi: 10.1093/nar/gkx1008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Feng S, Zhang L, Liu X, et al. Low levels of AMPK promote epithelial-mesenchymal transition in lung cancer primarily through HDAC4- and HDAC5-mediated metabolic reprogramming. J Cell Mol Med 2020; 24: 7789–7801. doi: 10.1111/jcmm.15410 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Kim TH, Kim E, Yoon D, et al. Recombinant human prothrombin kringles have potent anti-angiogenic activities and inhibit Lewis lung carcinoma tumor growth and metastases. Angiogenesis 2002; 5: 191–201. doi: 10.1023/A:1023835102832 [DOI] [PubMed] [Google Scholar]
- 26.Wang X, Adjei AA. Lung cancer and metastasis: new opportunities and challenges. Cancer Metastasis Rev 2015; 34: 169–171. doi: 10.1007/s10555-015-9562-4 [DOI] [PubMed] [Google Scholar]
- 27.Tompach PC, Lew D, Stoll JL. Cell response to hyperbaric oxygen treatment. Int J Oral Maxillofac Surg 1997; 26: 82–86. doi: 10.1016/S0901-5027(05)80632-0 [DOI] [PubMed] [Google Scholar]
- 28.Villacampa P, Liyanage SE, Klaska IP, et al. Stabilization of myeloid-derived HIFs promotes vascular regeneration in retinal ischemia. Angiogenesis 2020; 23: 83–90. doi: 10.1007/s10456-019-09681-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Liu ZJ, Velazquez OC. Hyperoxia, endothelial progenitor cell mobilization, and diabetic wound healing. Antioxid Redox Signal 2008; 10: 1869–1882. doi: 10.1089/ars.2008.2121 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Fosen KM, Thom SR. Hyperbaric oxygen, vasculogenic stem cells, and wound healing. Antioxid Redox Signal 2014; 21: 1634–1647. doi: 10.1089/ars.2014.5940 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Wenwu L, Xuejun S, Hengyi T, et al. Hyperbaric oxygen and cancer: more complex than we expected. Target Oncol 2013; 8: 79–81. doi: 10.1007/s11523-013-0259-8 [DOI] [PubMed] [Google Scholar]
- 32.Bennett M, Feldmeier J, Smee R, et al. Hyperbaric oxygenation for tumour sensitisation to radiotherapy. Cochrane Database Syst Rev 2005; 4: CD005007. doi: 10.1002/14651858.CD005007.pub2 [DOI] [PubMed] [Google Scholar]
- 33.Heys SD, Smith IC, Ross JA, et al. A pilot study with long term follow up of hyperbaric oxygen pretreatment in patients with locally advanced breast cancer undergoing neo-adjuvant chemotherapy. Undersea Hyperb Med 2006; 33: 33–43. [PubMed] [Google Scholar]
- 34.Baish JW, Gazit Y, Berk DA, et al. Role of tumor vascular architecture in nutrient and drug delivery: an invasion percolation-based network model. Microvasc Res 1996; 51: 327–346. doi: 10.1006/mvre.1996.0031 [DOI] [PubMed] [Google Scholar]
- 35.Steingold JM, Hatfield SM. Targeting hypoxia-A2A adenosinergic immunosuppression of antitumor T cells during cancer immunotherapy. Front Immunol 2020; 11: 570041. doi: 10.3389/fimmu.2020.570041 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Halpin-Veszeleiova K, Hatfield SM. Oxygenation and A2AR blockade to eliminate hypoxia/HIF-1alpha-adenosinergic immunosuppressive axis and improve cancer immunotherapy. Curr Opin Pharmacol 2020; 53: 84–90. doi: 10.1016/j.coph.2020.07.005 [DOI] [PubMed] [Google Scholar]
- 37.Son J, Lyssiotis CA, Ying H, et al. Glutamine supports pancreatic cancer growth through a KRAS-regulated metabolic pathway. Nature 2013; 496: 101–105. doi: 10.1038/nature12040 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Yang S, Hwang S, Kim M, et al. Mitochondrial glutamine metabolism via GOT2 supports pancreatic cancer growth through senescence inhibition. Cell Death Dis 2018; 9: 55. doi: 10.1038/s41419-017-0089-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Momcilovic M, Bailey ST, Lee JT, et al. The GSK3 signaling axis regulates adaptive glutamine metabolism in lung squamous cell carcinoma. Cancer Cell 2018; 33: 905–921. doi: 10.1016/j.ccell.2018.04.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Wolfer A, Ramaswamy S. MYC and metastasis. Cancer Res 2011; 71: 2034–2037. doi: 10.1158/0008-5472.CAN-10-3776 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Romero OA, Torres-Diz M, Pros E, et al. MAX inactivation in small cell lung cancer disrupts MYC-SWI/SNF programs and is synthetic lethal with BRG1. Cancer Discov 2014; 4: 292–303. doi: 10.1158/2159-8290.CD-13-0799 [DOI] [PubMed] [Google Scholar]
- 42.Wolfer A, Wittner BS, Irimia D, et al. MYC regulation of a “poor-prognosis” metastatic cancer cell state. Proc Natl Acad Sci USA 2010; 107: 3698–3703. doi: 10.1073/pnas.0914203107 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Le Gal K, Ibrahim MX, Wiel C, et al. Antioxidants can increase melanoma metastasis in mice. Sci Transl Med 2015; 7: 308re8. doi: 10.1126/scitranslmed.aad3740 [DOI] [PubMed] [Google Scholar]
- 44.Wang H, Liu X, Long M, et al. NRF2 activation by antioxidant antidiabetic agents accelerates tumor metastasis. Sci Transl Med 2016; 8: 334ra351. doi: 10.1126/scitranslmed.aad6095 [DOI] [PubMed] [Google Scholar]
- 45.Piskounova E, Agathocleous M, Murphy MM, et al. Oxidative stress inhibits distant metastasis by human melanoma cells. Nature 2015; 527: 186–191. doi: 10.1038/nature15726 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Lee HY, Kim IK, Lee HI, et al. Combination of carboplatin and intermittent normobaric hyperoxia synergistically suppresses benzo[a]pyrene-induced lung cancer. Korean J Intern Med 2018; 33: 541–551. doi: 10.3904/kjim.2016.334 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Please note: supplementary material is not edited by the Editorial Office, and is uploaded as it has been supplied by the author.
Supplementary material ERJ-00062-2022.Supplement (3.1MB, pdf)
This one-page PDF can be shared freely online.
Shareable PDF ERJ-00062-2022.Shareable (229.9KB, pdf)