

Summary
Mass spectrometry-based proteomic technology has greatly improved and has been widely applied in various biological science fields. However, proteome-wide accurate quantification of proteins in signaling pathways remains challenging. Here, we report a genome-wide amino acid coding-decoding quantitative proteomic (GwAAP) system to facilitate precise proteome quantification. For each protein, a unique code peptide was assigned and incorporated into the N-terminus of the targeted protein and used for identification and quantification. As a proof of principle, we systematically tagged 40 yeast proteins with codes and employed mass spectrometry to decode. We successfully recovered all 40 code peptides with a large and consistent quantitative dynamic range (CV slope <10%, R2 > 0.8). We further verified the alteration of the glucose and galactose metabolism pathways in yeast under different carbon source conditions. The GwAAP system could potentially provide a strategy to achieve absolute quantification of the entire yeast proteome without bias.
Subject areas: Biochemistry methods, Biological sciences research methodologies, Proteomics
Graphical abstract
Highlights
-
•
Unique code peptides were tagged for quantifying proteins on a genome-wide scale
-
•
Code peptides were enriched by an HA antibody to increase the MS detection sensitivity
-
•
Code peptides representing different proteins shared comparable MS response curve
-
•
GwAAP has the potential for intermolecular quantification
Biochemistry methods; Biological sciences research methodologies; Proteomics
Introduction
A systematic understanding of complex biological processes depends on the identification and quantification of critical proteins in multiple pathways. Over the last few decades, mass spectrometry-based proteomic technologies have been developed.1 However, there are still obstacles to impeding the application of proteomics to precisely identify and quantify large-scale proteins.2 For instance, (1) the dynamic ranges of the proteome span ten orders of magnitude, which exceeds the detection range of proteomic technology. Thus, all protein components in a certain pathway might not be fully covered in one experiment. (2) For a certain protein, the peptides detected by mass spectrometry (MS) can vary among different experiments, which results in MS-based protein quantification diversity from batch to batch. (3) For some proteins, unique quantitative peptides are lacking and it is difficult to distinguish based on their proteotypic peptides. For instance, the protein sequence similarity of paralogous ribosomal proteins in the yeast proteome is nearly 100%.3 Taking Rpb13a and Rpb13b as an example, only two amino acids are different between them. The abundance and the stoichiometry ratio between the two proteins are hardly accessed based on the peptides digested from their protein sequences.
To obtain full coverage and accurate quantification, many methods and algorithms have been developed. The protein amount could be calculated as the sum of intensities of all peptide peaks divided by the number of theoretically observable tryptic peptides (intensity-based absolute quantification, iBAQ).4 In addition, to quantitatively compare the protein abundances across different experiments, the iBAQ values were normalized into fraction of total (FOT) values. The FOT value of a protein was defined as the iBAQ value of the protein divided by the total iBAQ values of all identified proteins within one sample. Another reported strategy was to reduce the deviation of large-scale proteins.5 The ‘quantotypic peptides’, which share consistent signal response linearity, can be chosen to quantify each protein.6 Accordingly, a SCRIPPT-MAP database based on the best responder (BR) peptides was established, and the quantified proteins exhibited a consistent linear MS response curve. However, these strategies are highly time consuming and depend on massive accumulation of detected samples. Therefore, global absolute quantification of the proteome remains challenging.
Currently, methods based on chemical labeling, such as TMT, enables proteome-wide relatively quantification across many samples and absolute quantification could be available by adding standard peptides. However, because of the complexity of samples, the preferential detection of high-abundance proteins is hard to avoid and the coverage depth is limited.7 Data-independent acquisition (DIA)-based technologies, including sequential window acquisition of all theoretical spectra (SWATH), multiple reaction monitoring (MRM) and parallel reaction monitoring (PRM), coupled with targeted data analysis are independent of the composition of precursor ions for fragmentation, overcome the limitations of inherent irreproducibility and undersampling of data-dependent acquisition (DDA) and are widely applied in proteomics research.8 However, DIA inevitably results in false positives and negatives, and the high dynamic range of the proteome still limits the sensitivity of DIA.
To increase the sensitivity and signal response linearity of proteomics, affinity purification-based MS was applied.9,10 This method could increase the relative abundance of certain proteins or peptides, but only a few targeted proteins could be detected. In addition, when affinity enrichments are performed, different antibodies corresponding to different proteins are needed. The use of antibodies for different proteins may introduce various affinity efficiencies and loss of specificity from cross-reactivity.11 Recently, a genetically engineered sperm cell line and a model animal library were constructed in which each cell line was labeled by an artificial GST tag on the N-terminus of a certain protein.12,13,14,15 This method could precisely provide the expression information of all proteins. However, the project required a large number of stable cell lines to be constructed, and only one protein could be identified in a cell line at a time. Nevertheless, these strategies provide us with the idea that it is possible to artificially label proteins on a whole-genome scale.
Saccharomyces cerevisiae is the first single-celled eukaryote to be whole-genome sequenced and has a genome of approximately 12 Mbp organized on 16 chromosomes, and the proteome is currently estimated at 5,858 proteins (Saccharomyces Genome Database, www.yeastgenome.org). A previous comprehensive proteome-wide abundance study suggested that protein tags did not significantly influence protein abundance.16 In addition, the Synthetic Yeast Genome Project (Sc2.0), whose attempt was to synthesize a eukaryotic genome, indicated that it is possible to recode genes on a chromosome-wide scale.17,18,19,20,21 In addition, contributing to the capacity to take up and recombine DNA fragments,22 it is able to assemble numerous cellular pathways into a neochromosome to study their function and regulation. Thus, S. cerevisiae is an ideal model to prove our coding-decoding quantification proteomic system.
Here, we report a genome-wide amino acid coding-decoding quantification proteomic system. A four digital amino acid library system was designed in which each protein could be assigned a distinct code. Using synthetic biology technology, the amino acid codes were coded in the N-terminus of the corresponding proteins in the yeast (S. cerevisiae) genome so that the copy numbers of the code peptides were identical to those of the targeted proteins in yeast. The code peptides were enriched by an anti-HA antibody and decoded and quantified by an MS strategy. In general, this method increased the detection sensitivity by peptide enrichment, which reduced the dynamic range and complexity of proteomics. In addition, the code peptides with similar physical and chemical properties resulted in comparable linear signal responses in the mass spectrum, which significantly improved the accuracy of proteomic quantification.
Results
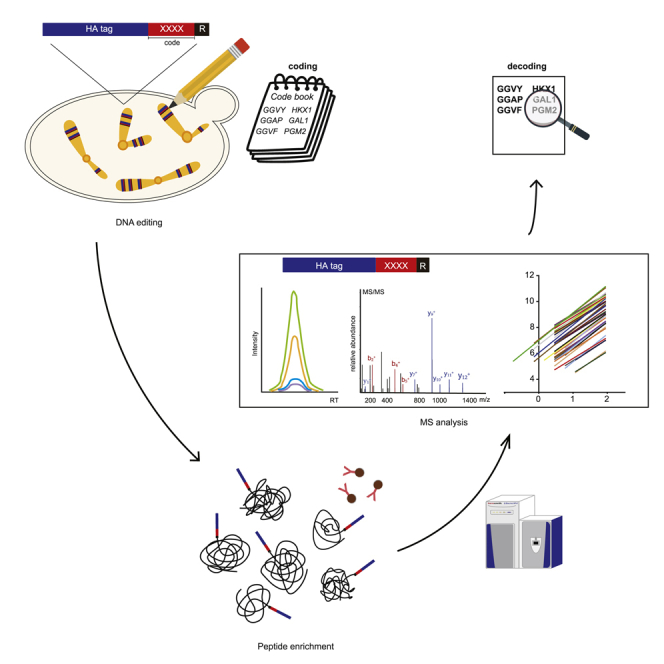
The workflow for the genome-wide amino acid coding-decoding proteomic quantification system
To establish a genome-wide amino acid coding-decoding proteomic quantification system, we created a library of four digital amino acids according to the following two principles: (1) the code sequence should contain only one of the leucine (L) and isoleucine (I) as their m/z (mass to charge ratio) were identical and could not be distinguished by MS; (2) the code sequence could not contain lysine (K) or arginine (R) because they are the cleavage sites of trypsin. Next, a synthetic biology approach was applied to construct a yeast strain in which each gene contained a 14-AA code sequence at the N-terminus. The first 9 of the 14-AA sequences were an HA tag, and the following 4 AAs were code sequences randomly assigned from a library we established. The last AA was arginine (R), the cleavage site of trypsin. Here, we defined the 14-AA sequence at the N-terminus of a gene as its “code peptide”. As the code peptides were expressed with the corresponding proteins, they were present in equimolar amounts to the corresponding tagged proteins. Whole yeast protein extract was prepared and digested by trypsin, and the HA codes were enriched using an antibody against HA. Subsequently, the expression of each gene was identified by detecting the code peptides by MS (Figure 1).
Figure 1.

The GwAAP workflow
Schematic of the GwAAP workflow. A library of code peptides was established, and synthetic chromosomes were constructed using a synthetic biology technique. The N-terminus of genes on each chromosome contained 14-AA code peptides. The samples were lysed, and proteins were extracted and proteolyzed using trypsin. The peptide mixtures were further enriched by anti-HA antibodies and identified and quantified by LC-MS/MS in the DDA or PRM data acquisition modes.
Writing the HA codes into 40 yeast proteins by synthetic biology
As ‘proof of principle’, 40 yeast proteins in three metabolic pathways, i.e., the glycolysis pathway, galactose metabolism pathway and ergosterol biosynthesis pathway, were chosen and constructed by a plasmid system into S. cerevisiae. These proteins represent an ideal example to demonstrate the dynamic range of the technique because they contain proteins that are from extremely high concentrations (FBA1/YKL060C, 1.0 × 106 copies/cell, one of the most abundant proteins in S. cerevisiae) to very low concentrations (GAL80/YML051W, approximately 24 copies/cell) and contain proteins whose abundance could not be measured by other MS approaches (e.g., TDH1/YJL052W).16
To tag the 40 proteins with unique codes, two amino acids out of the 4 digital AA codes with random selection could produce 289 AA combinations, theoretically. In addition, as glycine residue can increase the ionization efficiency,23 we used two glycine amino acids in all of the 40 codes (GGXX). When engineering on a genome scale, all the 4 amino acids can be randomly selected. We synthesized the 40 protein-coding genes as 12 DNA chunks, each of which contained three or four genes (Figure S1A and Table S1). Each gene on the plasmid contained 500 bp upstream and 200 bp downstream sequences with the 14-AA code sequence in the N-terminus at its open reading frame (ORF) region. Every two genes were divided by one 100 bp random sequence. A 250 bp random sequence was inserted as homologous arms to assemble the synthetic gene array, and 200 bp DNA sequences were included in chunk A and chunk L, which were homologous to any pRS plasmid.24 We assembled the 12 chunks into one vector using transformation-associated recombination (TAR)25 (Figure S1A). Following transformation, yeast cells were plated on selective medium and screened by PCR. The amplicons with the indicated sizes demonstrated proper in vivo assembly of all 12 chunks and the vector (Figure S1B). To determine the abundances of the 40 genes, proteins were extracted and resolved by SDS-PAGE and visualized by Coomassie staining (Figure S1C, left panel) and western blot (Figure S1C, right panel), which illustrated that the genes on the plasmid could be expressed as expected.
Decoding and quantifying the tagged proteins by proteomic approaches
To establish an MS/MS spectral library covering the target code peptides representing the 40 conditionally expressed proteins, yeast cells cultivated in glucose or galactose medium were analyzed by MS. All the code peptides representing the 40 target proteins were successfully identified as listed in Table 1. For example, YPYDVPDYAGGVYR with an m/z of 817.8779, represented the HA code of HXK1 (Figure 2A). Nine fragment ions, y1, y7, y9, y10, y11, y12, b2, b4 and b5, were covered in the YPYDVPDYAGGVYR MS/MS spectrum, demonstrating that fragment ions containing four digital specific AAs were identified by the MS/MS spectrum (Figure 2A). Overall, these results indicate that the code sequences of peptides used in the GwAAP strategy could be specifically identified by MS.
Table 1.
The isolation list of the 40 code peptides
| NO. | Gene | Tag sequence | Parent ion M/z | RT (Min) |
|---|---|---|---|---|
| 1 | GLK1 | YPYDVPDYAGGVWR | 829.3859 | 43.36–53.36 |
| 2 | HXK1 | YPYDVPDYAGGVYR | 817.9779 | 37.99–47.99 |
| 3 | HXK2 | YPYDVPDYAGGVDR | 793.8597 | 35.21–45.21 |
| 4 | GAL80 | YPYDVPDYAGGASR | 765.8466 | 31.50–41.50 |
| 5 | GAL4 | YPYDVPDYAGGATR | 772.8544 | 35.66–45.66 |
| 6 | GAL3 | YPYDVPDYAGGACR | 802.3459 | 28.74–38.74 |
| 7 | GAL1 | YPYDVPDYAGGAPR | 770.8570 | 28.29–38.29 |
| 8 | GAL10 | YPYDVPDYAGGVGR | 764.8570 | 30.56–40.56 |
| 9 | GAL7 | YPYDVPDYAGGVAR | 771.8648 | 31.39–41.39 |
| 10 | GAL2 | YPYDVPDYAGGVVR | 785.8805 | 37.03–47.03 |
| 11 | PGM1 | YPYDVPDYAGGVLR | 792.8883 | 42.27–52.27 |
| 12 | PGM2 | YPYDVPDYAGGVFR | 809.8805 | 46.57–56.57 |
| 13 | PGI1 | YPYDVPDYAGGGCR | 795.3381 | 26.61–36.63 |
| 14 | PFK1 | YPYDVPDYAGGGPR | 763.8492 | 26.95–36.95 |
| 15 | FBA1 | YPYDVPDYAGGAAR | 757.8492 | 26.41–36.41 |
| 16 | PFK2 | YPYDVPDYAGGAGR | 750.8413 | 30.74–40.74 |
| 17 | TPI1 | YPYDVPDYAGGAVR | 771.8648 | 31.49–41.49 |
| 18 | TDH1 | YPYDVPDYAGGALR | 778.8276 | 38.62–48.62 |
| 19 | TDH2 | YPYDVPDYAGGAFR | 795.8648 | 41.88–51.88 |
| 20 | TDH3 | YPYDVPDYAGGAWR | 815.3703 | 42.44–52.44 |
| 21 | PGK1 | YPYDVPDYAGGAYR | 803.8623 | 32.52–42.52 |
| 22 | GPM1 | YPYDVPDYAGGADR | 779.8441 | 24.55–34.55 |
| 23 | ENO1 | YPYDVPDYAGGAHR | 790.8601 | 20.60–30.60 |
| 24 | ENO2 | YPYDVPDYAGGANR | 779.3521 | 24.94–34.94 |
| 25 | PYK2 | YPYDVPDYAGGAER | 786.8519 | 32.19–42.19 |
| 26 | CDC19 | YPYDVPDYAGGAQR | 786.3599 | 25.25–35.25 |
| 27 | ERG1 | YPYDVPDYAGGGGR | 743.8335 | 23.63–33.63 |
| 28 | ERG2 | YPYDVPDYAGGGAR | 750.8413 | 33.25–43.25 |
| 29 | ERG3 | YPYDVPDYAGGGVR | 764.8570 | 30.03–40.03 |
| 30 | ERG4 | YPYDVPDYAGGGLR | 771.8649 | 31.37–41.37 |
| 31 | ERG5 | YPYDVPDYAGGGFR | 788.8570 | 38.84–48.84 |
| 32 | ERG6 | YPYDVPDYAGGGWR | 808.3624 | 39.10–49.10 |
| 33 | ERG7 | YPYDVPDYAGGGYR | 796.8544 | 39.00–49.00 |
| 34 | ERG9 | YPYDVPDYAGGGDR | 772.8362 | 24.45–34.45 |
| 35 | ERG11 | YPYDVPDYAGGGHR | 522.9039 | 21.09–31.09 |
| 36 | ERG20 | YPYDVPDYAGGGNR | 772.3442 | 25.97–35.97 |
| 37 | ERG24 | YPYDVPDYAGGGER | 779.8441 | 24.78–34.78 |
| 38 | ERG25 | YPYDVPDYAGGGQR | 779.3521 | 26.02–36.02 |
| 39 | ERG26 | YPYDVPDYAGGGSR | 758.8388 | 24.08–34.08 |
| 40 | ERG27 | YPYDVPDYAGGGTR | 765.8466 | 34.47–44.47 |
Figure 2.

The decoding process of GwAAP
(A) MS and MS/MS spectra of the code peptide YPYDVPDYAGGVYR (Hkx1p, m/z 817.9779, 2+) in the DDA mode.
(B) A heatmap of 40 peptides containing their precursor and corresponding fragment ions.
(C) YPYDVPDYAGGVYR was targeted in PRM analyses, showing the dominant y9 fragment ion by cleavage at the N-terminal bond of the proline residue.
(D) Peak area contributions of the individual fragment ions of the peptide. The relative contribution of each fragment ion to each peptide peak is displayed as different colors.
To accurately quantify the 40 peptides, we applied PRM mode to target identical precursors. The DDA raw dataset containing identification information (MS1/MS2) of the 40 code peptides was applied to set up spectral libraries, covering a total of 61 precursors and 548 transitions (Table S2). The digested yeast peptides were enriched by an anti-HA antibody and detected by PRM. For each code peptide, up to 6 fragment ions were selected and quantified using Skyline software.26 As a result, all 40 code peptides were accurately quantified by 202 transitions (Figure 2B). MS and MS/MS spectrum and the ion chromatograms of HXK1 derived from PRM detection were showed as an example (Figures 2C and 2D). The MS/MS spectra of the YPYDVPDYAGGVYR code sequence indicated that fragmentation at the N-terminal side of the proline residue in a mass spectrometer generated an intense y9 fragment ion containing the 4 digital AA code, and the fragment ion peak was dramatically overrepresented in the MS/MS spectrum, which facilitated more sensitive quantification of the 40 code peptides.
The ‘proteotypic’ peptides used in GwAAP maintained a higher consistent signal response
In general MS analysis, the peptide amounts were determined by the intensities of randomly identified peptide peaks. Andrew, B. S.27proposed a definition of ‘proteotypic’ peptides, which exhibited good response signals on the mass spectrum and the fragmentation pattern with salient features for accurate detection and quantification. Ideally, it would be more reproducible and linear if specific ‘proteotypic’ peptides were used for the identification and quantification of certain proteins in each experiment. Consequently, in the GwAAP strategy, the code peptides were designed to quantify the corresponding proteins, which could potentially be ‘proteotypic’ peptides.
To determine the stability of GwAAP, we firstly assessed the correlation across three independent experiments. Protein abundance from different experiments were pair-wise analyzed. The Pearson correlations were all above 0.95, indicating high degrees of reproducibility (Figures 3A and 3B, Tables S3A and S3B). To test the feasibility of GwAAP in quantifying the protein abundance in yeast, a range of serially diluted yeast lysates (from 0.3125 μg to 160 μg) were collected and analyzed. The R2 (correlation between total area fragment against loading amounts) and slope values were measured to determine the linear performance of the code peptides. The slope range of the response curve for all 40 code peptides was 1.60–2.14, and the R2 values were all above 0.8 (Figure 3C and Table S4). We also surveyed the linear response curve of the corresponding proteins (functioning in the glycolysis and galactose metabolism pathways) annotated in Script-Map. As a result, 12 out of 40 proteins were detected, in which the slope of the BR peptide ranged from −0.081-61.66. The dynamic range of the linear curve slope observed by the GwAAP approach (1.60–2.14) was significantly smaller than that obtained by the Script-Map method (−0.08-61.66), indicating that comparing with the existing peptides digested from proteins, the code peptides used in GwAAP maintained a higher consistent signal response of each protein (Figure 3D). In summary, the results demonstrated that GwAAP was feasible and reliable for quantitatively analyzing the protein abundance in S. cerevisiae.
Figure 3.

The reproducibility and linearity of GwAAP
(A) Pearson correlation coefficients within the three independent experiments of trypsin digestion (top right). The samples were digested by trypsin and detected directly by MS to assess the enzymatic efficiency. The 40 protein abundances were log2-transformed in each pairwise comparison, which was plotted as a scatterplot in the bottom left. The green lines represented the least-squares best fit for each pairwise comparison.
(B) Pearson correlation coefficients within the three independent experiments of peptide enrichment (top right). The samples were digested by trypsin, enriched by an HA antibody and detected by MS to assess the enrichment efficiency. The 40 protein abundances were log2-transformed in each pairwise comparison, which was plotted as a scatterplot in the bottom left. The green lines represented the least-squares best fit for each pairwise comparison.
(C) Comparison of the linearity range of dilution curves with the GwAAP and Script-Map approaches. The dilution curves containing 40 code peptides obtained from PRM analysis with the GwAAP approach (left panel). Total area fragment meant the sum of the area under the curve (AUC) of the 6 most intense fragment ions. In contrast, Script-Map measured the linearity range of 11 out of 40 proteins (right panel), except ALDOB, of which the curve slope was −0.081. The AUC was the peak area for the precursor.
(D) Slope comparison of GwAAP (colored in blue) and Script-Map (colored in red). The linearity and coverage of GwAAP are more consistent than those of Script-Map.
Writing the HA codes into yeast genome using a CRISPR-Cas12a-based genome editing system
As the expression pattern of genes on plasmid may be different from those on genome, we established a CRISPR-Cas12a-based genome editing system to realize the code sequence in vivo knocked into the yeast genome28 (Figure 4A). Taking 26 genes from the glycolysis and galactose metabolism pathways as an example (Figure 4B), we investigated the effect of the HA codes knock-in. In addition, in the experiment of quantification of 40 code peptides, we found that it was difficult to distinguish the peptides with the similar m/z and RT range in MS1 level. The unique transitions in MS2 level were needed to identify and distinguish these peptides. Therefore, two codes shared the similar m/z were changed to simplify the identification. They were codes of GAL7 and GPM1, from GGVA to GGGE and GGAD to GGGG, respectively. The growth of engineered strains (JDY6086) was largely indistinguishable from that of the wild-type strain in glucose media, but showed modest defects in galactose media (Figure 4C), which indicated that the HA codes had little influence on the function of the 17 proteins in the glycolysis pathway. The abundance of the overall proteome and the targeted 26 proteins showed high correlation between JDY6086 and BY4741 when they were cultured in galactose media (Figure 4D and Table S3C).
Figure 4.

Construction of yeast strains with HA code sequences knock-in in vivo
(A) The workflow of a CRISPR-Cas12a-based genome editing system.
(B) PCR analysis of JDY6086, the strain with 26 HA codes knock-in. The forward primer was the same for all the genes and the size of the amplicon for each gene was list in Table S5.
(C) Fitness analysis of BY4741 with or without HA code knock-in on glucose (YPD) and galactose (YPGal) media. All the photos were taken at 48 h JDY6086, strains with 26 HA codes knock-in, 17 glycolytic genes and 9 galactose metabolic genes; JDY6090, strains with 9 HA codes knock-in, 9 galactose metabolic genes.
(D) The correlation of the protein expression levels between BY4741 and JDY6086 cultured in galactose-containing media. The overall proteome (left panel) and the targeted 26 proteins (right panel) were analyzed by DDA mode.
To clarify the function of which gene(s) was influenced by the added code, several strains with different gene editing in the galactose metabolism pathway were constructed. Only when the HA codes of PGM1 and PGM2 were knocked in at the same time, growth defects were detected (Figures 4C, S2A, and S2B), which suggested that the HA code at the N-terminus had impact on the activation of the phosphohexose mutase, both on Pgm1p and Pgm2p. In addition, the expressions of GAL genes were correctly induced when galactose was existed (Figure S2C) and the strain with HA codes of GAL genes displayed similar growth rate compared with wild-type strain (Figures S2A and S2B). These results suggested that the HA codes added did not affect the function of 7 GAL genes, including a transcription factor, Gal4p.
Monitoring the dynamic stoichiometry of the glycolysis and galactose metabolism pathways in glucose and galactose culture conditions by GwAAP
The carbon source gave rise to proteomic alteration in S. cerevisiae,29 and therefore, we monitored the dynamic stoichiometry of the glycolysis and galactose metabolism pathways in yeast cultured in glucose and galactose media using GwAAP. Cells were cultured in glucose- or galactose-containing media to mid-log phase and harvested for subsequent analysis. The 26 code peptides were analyzed by GwAAP (Figure 5A). To demonstrate the accuracy of GwAAP, a TMT experiment was performed at the same time (Figure 5A). We successfully identified and quantified the expression levels of all the 26 proteins. As an illustration, the YPYDVPDYAGGASR peptide, representing Gal80p, was identified in all 3 groups (galactose, glucose, and raffinose). The precursor and fragment ions (y5, b4, y7, y9, y11, y12) were detected with PRM mode. The protein abundance of Gal80p was quantified by the peak areas derived from 6 individual fragment ions (Figure 5B). Subsequently, we compared the abundance of all 26 enzymes between the two groups (Figures S2A and 5C). All of the enzymes in the galactose metabolism pathway (Gal3p, Gal80p, Gal4p, Gal2p, Gal1p, Gal7p, Gal10p) were upregulated in response to galactose culture condition, which was consistent with previous report.29 For instance, the abundances of Gal1p, Gal7p and Gal10p were 4- to 3100-fold higher in the galactose group than in the glucose group. Comparing with GwAAP, 4 proteins were not identified by TMT detection (Figure S3B), demonstrating that the GwAAP strategy is superior to TMT methods in terms of low-abundance protein identification. In addition, the GwAAP quantification results revealed that the GAL genes were induced in the present of galactose whereas the glycolytic genes showed similar expression levels, which was consistent with the TMT results (Figure S3C and Table S3D). These results suggested that GwAAP was a reliable quantification method.
Figure 5.

GwAAP was applied to determine stoichiometry of the glycolysis and galactose metabolism pathways in yeast
(A) Workflow of the GwAAP analysis of S. cerevisiae grown in three different carbon sources.
(B) Examples of MS and MS/MS spectra of YPYDVPDYAGGASR (Gal80p) in two different carbon sources.
(C) The 26 proteins comprising glycolysis and galactose metabolism pathways were quantified. The colored bars with fold-change values indicate the relative abundance of proteins in the galactose- or glucose-containing medium groups (glucose group as control; red, upregulated; blue, downregulated). The number above the colored bars represents the protein stoichiometry, which is depicted as the log10 transformed abundance ratio of each protein versus the minimum protein.
To further demonstrate the accuracy and sensitivity of GwAAP, we compared the protein levels of key enzymes in galactose metabolism pathway and glycolysis pathway detected by both GwAAP and TMT. Pfk1p/Pfk2p and Gal10p are the rate-limiting enzymes of the glycolysis and galactose metabolic pathways, respectively. As expected, when galactose was carbon source, the abundance of Gal10p substantially increased, which was 22.1-fold increased detected by GwAAP and 6.3-fold by TMT (Figure S3D). Pfk1p and Pfk2p were of slightly decreased abundance in responded to galactose, of which the fold-changes were 0.96 by GwAAP and 0.72 by TMT for Pfk1p and 0.87 by GwAAP and 0.83 by TMT for Pfk2p (Figure S3E). These results suggested that glucose was the preferred carbon source as galactose was needed to converted into a glycolytic metabolite, glucose 6-phosphate, to ferment (Figure 5C).
Furthermore, GwAAP enabled us to demonstrate the stoichiometry of isomerases in glycolysis pathway. Both Eno1p and Eno2p function as phosphohydrolases. The expression of the two genes varies in yeast because of the different transcriptional regulatory elements.30 The relative changes of Eno1p and Eno2p in glucose- and galactose-containing media were assessed by GwAAP and TMT, which were 2.48-fold and 1.73-fold for Eno1p and 0.60-fold and 0.68-fold for Eno2p, respectively (Figure S3F). In addition, the stoichiometry ratio of Eno1p and Eno2p can be determined by GwAAP, which was 1:3.1 in yeast under galactose culture condition and decreased to 1:12.6 in yeast grown in glucose medium (Figure 5C and Table S3E). Similarly, we also observed that the expression level of Cdc19p was significantly higher than that of the isomerase Pyk2p (stoichiometry ratio 87.1:1 in galactose culture condition and 81.3:1 in glucose condition) by GwAAP, consistent with a previous study showing that Cdc19p was the dominant pyruvate kinase31 (Figure 5C and Table S3E).These results demonstrate that the stoichiometric measurement of different proteins using GwAAP could precisely quantitate the dynamics of the proteome in various life processes.
Discussion
Here, we provided a GwAAP strategy that tagged genes with a series of synthetic sequences of unique identifier peptides and enabled MS identification and quantification on a genome-wide scale. Our study proved that the GwAAP strategy integrated with synthetic biology techniques is a powerful tool, enabling genome-wide labeling with artificial coding tags as ‘proteotypic’ peptides, reducing the sample complexity and dynamic range and increasing the coverage depth of low-abundance proteins. Our code peptides shared similar physical and chemical properties so that each single protein maintained a relatively consistent signal response curve. Moreover, the fragmentation of each code peptide at the N-terminal side of proline generated an intense fragment ion (y9) containing the four digital AA codes. These fragmentation patterns with specific features are conducive to accurate quantification.
By way of its ease of genetic operation and relatively simple genetic diversity, the quantitative proteome of S. cerevisiae has been well characterized by several independent approaches, which are either base on immunoblot analyzes with tandem affinity purification (TAP),32 flow cytometry analyzes with GFP tagging33 or MS.34 The protein abundance information was collected from the corresponding tagged strains and thousands of individual strains were needed to be analyzed simultaneously. In the GwAAP strategy, the tags of all the proteins were inserted in one strain and a 4 amino acid sequence was used as a unique barcode to assign tags to their corresponding proteins. Thus, all the quantification information was obtained in one experiment by MS. On the other hand, the collection strains have been used for the fitness measurements,32,35which provide information to assess the impact of tag insertion. Compared with the traditional proteotypic peptide-based MS strategy, the GwAAP strategy offers several advantages. First, GwAAP provide an alternative solution for proteins which lack suitable endogenous quantitative peptide for MS detection36 (e.g., YDR544C and YNL338W). Second, all the proteins were quantified by the code peptides, which contained 4 unique amino acids and a fixed affinity tag sequence and shared similar physical and chemical properties. Third, the complexity and dynamic range of the peptides in the GwAAP samples could be reduced by peptide purification as all peptides contained a fixed affinity tag. Fourthly, code peptides could be used to distinguish the proteins lacking unique peptides, such as paralogous proteins (e.g., Rpl13a and Rpl13b). At last, GwAAP has the potential for intermolecular quantification with its stoichiometric measurement ability.
In addition, when the strain with genome-widely recoded is constructed, it can be applied to any MS detection approaches, such as TMT and DIA. The TMT reagents can label code peptides across multiplexing samples and improve accurate quantification. Besides, the code peptides we tagged can be used to create a mass-spectrometric map of entire proteome of yeast as the prior knowledge of DIA. It benefits the high quality of fragment ion spectra and improves the accurate quantification by reducing stochastic and irreproducible precursor ion selection. With the whole genomic recoded strain, we can develop an entire proteome map of yeast with a high coverage, similar to DIA, and a high accuracy, similar to PRM.
To make the GwAAP strategy generally applicable, there are also some limitations to be overcome, one of which is that cells with recoded genes are not easily available. Multiplex genome editing by CRISPR/Cas systems provides a solution to tag genes in certain metabolic pathways of interest.37 The developed synthetic genome technology makes it possible to tag genes on the chromosomal scale in S. cerevisiae, and the Genome Project-write (GP-write) is working on the whole-genome engineering of human cell lines and other eukaryotic organisms.38 We anticipate that an increasing number of eukaryotic organisms could be redesigned and reconstructed on the whole-genome level, similarly to S. cerevisiae.
Another challenge is that when it comes in a genome-wide tagging, the complexity of the code peptides would be dramatically increased and how to design the code peptides and make sure that they can be specifically identified is a key issue. In our design, four amino acids were used as unique ID for each protein (Figure 1), resulting in about 80,000 random sequences which is met the requirement of the yeast genome. Before tagging into the yeast genome, a priori assessments could be done. Some peptide sequence-based retention time prediction tools, such as CharmeRT,39 AutoRT40 and Prosit,41 could assess the properties of code peptides. In addition, peptide libraries either from SPOT synthesis,36 phage display42 or bacteria expressed43 could be used to generate reference fragment ion spectra which enable optimal detection of code peptides by MS. Benefiting with these two ways, rational design of code peptides for whole genome is feasible and reliable. Moreover, to simplify the identification and achieve the accuracy of quantification of the GwAAP system, the coding sequences should be avoided the codes peptides with the same m/z.
In summary, with engineered yeast labeled genome-wide with ‘proteotypic’ code peptides, the GwAAP strategy can precisely quantitate the dynamics of the proteome with deep coverage in various life processes.
Limitations of the study
There are several limitations of this study. First, the immunogen presentation efficiency should be specifically calculated in absolute proteomic quantification as antibody sensitivity and specificity can vary between batches. A heavy labeled synthetic peptide could be used as an internal standard for efficiency calculation. Second, incorporation of code peptides into the terminus (N- terminus or C-terminus) of some proteins may alter three-dimensional structure/stability and affect biological functions; careful assessment is required when performing genome-wide tagging. Third, GwAAP is not yet easily applied to other species, especially eukaryotic organisms. More high-efficiency gene tagging methods may be required in the future. Fourth, only up to 40 code peptides were incorporated and detected simultaneously in this study; a strain containing more code peptide tags is to be constructed in the next study.
STAR★Methods
Key resources table
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Pierce™ Anti-HA Agarose | Thermo Scientific | Cat#26181; RRID: AB_2537081 |
| Mouse mono-cloned α-HA | Sigma | Cat#H3663; RRID:AB_262051 |
| Goat Anti-Mouse IgG (H&L)-HRP Conjugated | Easybio | Cat#BE0102-100; RRID:AB_2923205 |
| Chemicals, peptides and recombinant proteins | ||
| Easytaq DNA polymerase | Transgen | Cat#AP111-01 |
| Not I | NEB | Cat#R3189S |
| Urea | Sigma | Cat#U5128 |
| Bradford protein assay kit | SanGon Biotech | Cat#C503041 |
| Ammonium bicarbonate | Sigma-Aldrich | Cat#A6141 |
| Fomic acid | Thermo Scientific | Cat#85171 |
| Acetonitrile | Thermo Scientific | Cat#51101 |
| 10-plex TMT Label Reagents | Thermo Scientific | Cat#A34808 |
| Trypsin | Promega | Cat#V5280 |
| HPLC-grade H2O | J.T.Baker | Cat#4218-03 |
| Critical commercial assays | ||
| Tanon™ High-sig ECL Western Blotting Substrate | Tanon | Cat#180-5001 |
| Pierce™ Quantitative Fluorescent Peptide Assay | Thermo Scientific | Cat#23290 |
| Deposited data | ||
| Mass spectrometry proteomics data | iProX (https://www.iprox.org.cn) | IPX0002656000 |
| Experimental models: Organisms/strains | ||
| S. cerevisiae: Strain background: BY4741 | EUROSCARF | http://www.euroscarf.de/index.php?name = News |
| Oligonucleotides | ||
| For all PCR primers, see Table S5 | This paper | N/A |
| Recombinant DNA | ||
| For all plasmids, see Table S1 | This paper | N/A |
| Software and algorithms | ||
| Thermo Proteome Discoverer 2.3 | Thermo Scientific | |
| Skyline (version 19.1.1.309) | Open Source | https://skyline.ms/project/home/software/skyline/begin.view |
| Xcalibur | Thermo Fisher Scientific | https://www.thermofisher.com/us/en/home/industrial/mass-spectrometry/liquid-chromatography-mass-spectrometry-lc-ms/lc-ms-software/multi-omics-data-analysis |
| GraphPad Prism8 (version 8.0.1) | Graphpad | GraphPad Prism; RRID: SCR_002798 |
| R | R code | https://www.r-project.org |
| Other | ||
| Saccharomyces Genome Database (SGD) | SGD community | https://yeastgenome.org/ |
Resource availability
Lead contact
Further information and request for resources should be directed to and will be fulfilled by the lead contact, Chen Ding (chend@fudan.edu.cn).
Materials availability
This study did not generate any new unique reagents. All the requests for the generated plasmids and strains should be directed to the lead contact and will be made available on request after completion of a Materials Transfer Agreement.
Experimental model and subjectdetails
Strains and growth media
The yeast strains used in this study were derivates of BY4741(MATa his3Δ1 leu2Δ0 ura3Δ0 met15Δ0). Standard methods for yeast culture, gene disruption and transformation were applied. The strains used in this study were list in Table S6.
Method details
Construction of yeast strains
For the plasmid model strain construction, the 40 genes were synthesized into 12 chunks, each of which contained three or four genes (BGI). Each chunk was flanked by two recognition sites of restriction enzymes (Not I). All chunks and the vector pRS413 were digested by Not I, purified in gel and used for yeast transformation. The yeast transformation was carried out using the lithium acetate transformation protocol. 30 ng purified synthetic chunk fragments and 10 ng linearized vector were co-transformed into BY4741 and plated onto SC-His plate (1.75 g/L Difco yeast nitrogen base w/o amino acids, 5 g/L ammonium sulfate, 2 g/L amino acid drop-out mix, 2% glucose and 2% agar). Correct assembled colonies were verified by junction PCR. Primers used in this study were listed at Table S5.
The genome edited strains were obtained according to previously reported by Swiat MA, et al.28 Briefly, plasmids carrying double or quadruple Cas12a crRNAs were constructed by standard Golden Gate Cloning. The crRNAs were flanked by short direct repeats of 19 bp. Double-stranded repair DNA fragments containing about 200 bp homolog arms were obtained by PCR, using the synthesized chunks with corresponding code sequence as templates. For each round editing, 100 ng crRNA array expression plasmid and 1 μg corresponding repair DNA fragments were co-transformed into yeast cells expressing Cas12a. Correct editing colonies were verified by diagnose PCR and SANGER sequencing and used for next round editing. Primers used in this study were listed at Table S5.
Yeast colony PCR for rapid screening
The colonies appeared on the selective plates were resuspended in 20 μL 20 mM NaOH and lysed in a thermocycler (98°C/3 min, 4°C/2 min, 5 cycles, kept at 15°C). After short centrifugation, 1 μL of supernatant was used as template in 10 μL PCR reaction.
Genomic DNA preparation for PCR analysis
Yeast cells were collected and washed with sterile water. After centrifugation, the pellet was resuspended into 100 μL breaking buffer (10 mM Tris-Cl, pH 8.0, 100 mM NaCl, 1 mM EDTA, pH 8.0, 2% (v/v) Triton X-100, 1% (w/v) SDS). 100 μL of 0.5 mm Glass Beads (Biospec, 11079105) and 200 μL of phenol/chloroform/isoamyl alcohol (25:24:1) were added into the tube and disrupted by Beadbeater at 2000 rpm for 5 min. Then 100 μL sterile water were added into the tube, and mix briefly. Centrifuge the tube at 13,000 rpm for 10 min. Transfer the top layer (about 180 μL) into a new tube with 500 μL 100% ethanol and chill the tube at −20°C for 15 min. Centrifuge the tube at 13,000 rpm for 5 min. The pellet was washed with 500 μL 75% ethanol and dried in vacuum pump (SpeedVac, Eppendorf, 45°C, 5 min). The genomic DNA was dissolved in 100 μL sterile water and stored at −20°C.
Immunoblotting
Undigested protein extract from each sample was boiled in 1× LDS buffer (Invitrogen) and separated on a 10% bis-Tris denaturing and reducing SDS-PAGE. Gels were then transferred onto a nitrocellulose membrane (Bio-Rad) for immunoblotting. Membranes were blocked with 5% non-fat dry milk in TBS-tween buffer and probed for HA tag. All primary incubations were done at 4°C overnight using a 1:4,000 dilution (Sigma). Secondary incubations were performed in 5% non-fat dry milk in TBS-tween using 1:10,000 diluted peroxidase-conjugated goat anti-mouse IgG (H + L) (Easybio). Membranes were visualized using an ECL plus western blotting kit (Tanon) and detected with radiographic film (Thermo).
Growth assay
Single colonies were cultured at 30°C in YP media (2% bacto-peptone, 1% yeast extract) supplemented with 2% glucose or 2% galactose for overnight at 220 rpm. Each strain was diluted to an OD600 of 0.1 in the same media. Distribute the cells into a 96-well plate containing 200 μL of YPD or YPGal media. Plates were covered with a Breathe-Easy membrane (Sigma), placed in a BioTek plate reader and incubated with continuous shaking. Measurements were made every 15 min. At the same time, these overnight cultures were adjusted to an OD600 of 1.0, series diluted by 10-fold and spotted onto YPD and YPGal plates.
Protein digestion and peptide Immunoprecipitation
Yeast cells were cultured in glucose or galactose medium. Cell pellet were resuspended in lysis buffer (8 M urea, 100 mM Tris, pH 8), and homogenized. The lysates were centrifuged at 12,000 g for 30 min, and the supernatants were further transferred in another clean 1.5 mL tube. The protein concentration was determined in the lysate using the bradford protein assay kit (SanGon Biotech). 200 μg of cell extract were loaded into 10 kDa Microcon filtration devices (Millipore), and the clear supernatants were washed three times with 50 mM ammonium bicarbonate. Then the samples were digested using trypsin at an enzyme to protein mass ratio of 1:50 overnight at 37°C. Peptides were extracted and dried (SpeedVac, Eppendorf).44 The dried peptides were re-dissolved with IP buffer, containing 50 mM MOPS/NaOH, 10 mM Na2HPO4, 50 mM NaCl, and incubated beads conjugated with an anti-HA antibody (Pierce, 26181) at 4°C for 16 h. The beads then rinsed with IP buffer, and eluted with 0.2% TFA for 15 min twice. Peptides were dissolved in the supernatant by centrifugation and desalted using C18 SpinTips (Protea) then lyophilized and stored at −80°C.
LC-MS/MS HCD DDA method
EASY-nLC 1200 nanoflow liquid chromatography system (Thermo Fisher Scientific) coupled to a Q-Exactive HF-X Mass Spectrometer (Thermo Fisher Scientific, Rockford, IL, USA) was used for all of the LC-MS/MS analyses. Dried peptides were resuspended in 0.1% fomic acid (FA) and loaded onto a C18 trap column (100 μm × 2 cm, homemade; particle size, 3 μm; pore size, 120 Å; SumChrom, USA) with a max pressure of 280 bar using Solvent A (100% H2O containing 0.1% (v/v) formic acid), then separated on a home-made silica microcolumn (150 μm × 12 cm, homemade; particle size, 1.9 μm, pore size, 120 Å; SumChrom, USA) with a gradient of 5-50% mile phase B (80% acetonitrile containing 0.1% (v/v) formic acid) at a flow rate of 600 nL/min for 75 min. A precursor MS scan (m/z 300–1,400) of intact peptides was acquired in the Orbitrap at the resolution of 120,000 at 200 m/z; a minimum MS signal threshold of 13,000 was used for triggering data-dependent fragmentation events. The most intense ions selected under top-speed mode were isolated in Quadrupole with a 1.6 m/z and fragmented by higher energy collisional dissociation (HCD) at a normalized collision energy of 27%. For a full mass spectrometry scan, the automatic gain control (AGC) value was 3 × 106 with a maximum inject time of 80 ms. Then the MS/MS spectra were acquired in the Orbitrap with an AGC target of 5 × 104 and a maximum inject time of 20 ms. A 12 s dynamic exclusion window was used to prevent repeated analysis of the same components. Finally, data were acquired by Xcalibur software (Thermo Fisher Scientific).
DDA data analysis and library generation
The DDA data processing was performed with minor changes, implemented on PD 2.3 software using Mascot as previously described. The yeast RefSeq protein database (updated on 04-07-2013) was downloaded in the National Center for Biotechnology Information (NCBI), in whichthe 40 metabolic enzymes containing distinct 14-AA code peptides were incorporated as novel proteins to form a new yeast database named ‘yeast RefSeq 40 plus’. MS raw files were processed against the ‘yeast RefSeq 40 plus’ protein database. In all cases the precursor mass tolerance was set to 20 ppm and fragment ion mass tolerance to 50 milli mass unit (mmu). The maximum number of missed cleavage sites was set to 2. Acetylation on N terminal and methionine oxidation were used as a variable modification. All spectra were validated by Percolator Node with the false discovery rate of 1%. Then for each identified peptide, the XIC (extracted ion Chromatograph) was extracted by searching against the MS1 based on its identification information, and the abundance was estimated by calculating the area under the extracted XIC curve (AUC). To generate the spectral libraries, the acquired DDA result files (Thermo’s Mass Spec Format, pdresult) were imported to Skyline. Peptide and transition settings were edited in the Skyline, and 40 peptides and their corresponding fragment ions were imported into Skyline to create background proteome. The following parameters were exported in the isolation list (in.csv format): Mass [m/z], CS [z], polarity, Start [min], End [min], NCE, comment.
LC-MS/MS PRM method
Code peptide-enriched samples were resuspended in 0.1% formic acid and subjected to liquid chromatography on an EASY-nLC 1200 system. All separations were performed using a gradient 12%–24% ACN in 0.1% formic acid over 40 min (75 min total) at a flow rate of 600 nL/min. The HPLC was coupled directly with a Q-Exactive mass spectrometer. The PRM method consisted of a full MS scan configured as above followed by up to 20 targeted MS/MS scans as defined by a time-scheduled inclusion list (30 k resolution, 2e5 AGC target, 100 ms maximum injection time, 1.6 m/z isolation window, 27% normalized collision energy, centroid mode). For DDA, AGC targets were optimized for speed with a goal of reaching the target before reaching maximum injection time. However, for PRM, the AGC target was selected for enhanced sensitivity and dynamic range with a goal of reaching the maximum injection time before reaching the target. PRM assay scheduling was performed within Skyline (version 19.1.1.309). To confirm the schedule, a scheduled isolation list with refined 10 min windows was exported from Skyline as a.csv file and imported directly into the instrument PRM method as an inclusion list.
PRM data analysis
For spectrum-centric analysis of PRM mass spectrometry results, Peptide-centric analysis was performed using Skyline. Signal extraction was performed on +2, +3, +4 precursors and +1, +2 b and y fragment ions. Full MS resolving power was set to 120,000 and MS/MS resolving power set to 30,000. Peptide identifications were either represented by Total Area Fragment, derived from Skyline, or refined by manual interpretation using several criteria including fragment ion mass accuracy, correlation of precursor and fragment ion peak shapes, and signal-to-noise ratios. The manual interpretation standard is consistent with the Skyline quantification method. The 6 most intense fragment ions are quantified by integrating the area under the curve (AUC) or maximum feature intensity. Specifically, we required at least three highly-resolved fragment ions without interference to consider a peptide identified.
TMT labeling
100 μg of JDY6086 yeast cell extract were loaded into 10 kDa Microcon filtration devices, and the clear supernatants were washed three times with 0.05 M Triethylammonium bicarbonate (TEAB). Then the samples were digested by trypsin at a 50:1 protein-to-trypsin ratio at 37°C for 12 h. Immediately before use, the TMT Label Reagents (Thermo Scientific, 90,110) were equilibrated to room temperature. For the 0.8 mg vials, 41 μL of anhydrous acetonitrile was added to each tube. Measure protein digest concentration using Thermo Scientific Pierce Quantitative Fluorescent Peptide Assay (Product No. 23290). We selected six of 10-plex TMT Label Reagents,126, 127T, 127N, 128N, 129N, 130N, 131, and carefully add 41 L of the TMT Label Reagent to each 100 μL sample (25 μg protein digest) in turn. Following incubation at room temperature for 1 h, the samples were quenched with 8 μL of 5% hydroxylamine for 15 min. The TMT-labeled samples were combined in a new microcentrifuge tube at a 1: 1: 1: 1: 1: 1 ratio and speedvaced to dry labeled peptide sample. The sample was separated in a home-made reverse-phase C18 column in a pipet tip. The TMT-labeled peptides were eluted and separated into nine fractions using a stepwise gradient of increasing acetonitrile (6%, 9%, 12%, 15%, 18%, 21%, 25%, 30%, and 35%) at pH 10. The nine fractions were combined to three fractions, dried in a vacuum concentrator (Thermo Scientific), and then analyzed by liquid chromatography-tandem mass spectrometry (LC-MS/MS).
TMT methods
The samples were resuspended in 0.1% formic acid and subjected to liquid chromatography on an EASY-nLC 1200 system. Samples were separated with a gradient of 8-50% mobile phase B (acetonitrile and 0.1% formic acid) at a flow rate of 600 nL/min for 75 min. The isolation window of the quadrupole was set to 0.7 m/z. The MS analysis for Q Exactive HFX were set with one full scan (300–1400 m/z, R = 60,000) at automatic gain control target (AGC target) of 3e6 ions, followed by to 20 MS/MS scan with higher-energy collision dissociation (AGC target 2e5 ions, max injection time 20 ms, isolation window 0.7 m/z, normalized collision energy of 35%), detected in the Orbitrap (R = 45,000). Dynamic exclusion time was performed as 25 s.
TMT data analysis
Mass spectra was processed using PD 2.3 software using Mascot as previously described. “Yeast RefSeq 40 plus” as a database was performed using a 20-ppm precursor ion tolerance and the product ion tolerance was set to 50 mmu. TMT tags on lysine residues and peptide N termini (+229.163 Da) and carbamidomethyl cysteine was searched as a fixed modification. Oxidized methionine, protein N-term acetylation were set as variable modifications. The false discovery rates (FDR) of peptide were <0.01.
Quantification and statistical analysis
Statistical analysis and graphing were performed using R (version 3.4.3) and GraphPad Prism (version 8.0.1). Comparison of two groups was performed using Student’s t test. The p values less than 0.05, 0.01, 0.001,0.00001 were marked with ∗, ∗∗, ∗∗∗, ∗∗∗∗ respectively.
Acknowledgments
This work was supported by National Key R&D Program of China (2022YFA1303200, 2022YFA1303201, 2017YFA0505102, 2018YFE0201603, 2017YFA0505101, 2020YFE0201600, 2016YFA0502500, 2018YFA0507501, 2017YFA0505103 and 2017YFC0908404), National Natural Science Foundation of China (31770886, 31972933, 31700682, 31725002, 31800069 and 32001042), Science and Technology Commission of Shanghai Municipality (2017SHZDZX01), Major Project of Special Development Funds of Zhangjiang National Independent Innovation Demonstration Zone (ZJ2019-ZD-004), Shuguang Program of Shanghai Education Development Foundation and Shanghai Municipal Education Commission: 19SG02, Program of Shanghai Academic/Technology Research Leader, CAMS Innovation Fund for Medical Sciences (CIFMS, 2019-12M-5-063), Fudan original research personalized support project, Shenzhen Science and Technology Program (KQTD20180413181837372), Guangdong Provincial Key Laboratory of Synthetic Genomics (2019B030301006) and Shenzhen Outstanding Talents Training Fund.
Author contributions
C.D., J.D., and F.H. conceived the project and designed the experiment. L.C. and X.Y. conducted the experiments. L.C., X.Y., and Z.Q. performed data analysis. L.C., X.Y., Z.Q., and X.S. visualized the data. X.Y. processed the data. C.D. and J.D. provided the Funding support. L.C., X.Y., and C.D. wrote the manuscript. H.Z., X.Y., J.D., and C.D. edited the manuscript.
Declaration of interests
The authors declare no competing interests.
Published: December 22, 2022
Footnotes
Supplemental information can be found online at https://doi.org/10.1016/j.isci.2022.105471.
Contributor Information
Fuchu He, Email: hefc@nic.bmi.ac.cn.
Junbiao Dai, Email: junbiao.dai@siat.ac.cn.
Chen Ding, Email: chend@fudan.edu.cn.
Supplemental information
Table S3A. The forty tagged protein expression in three biological repeats of trypsin digestion experiments, related to Figure 3. The samples were digested by trypsin and detected directly by PRM to assess the enzymatic efficiency. The abundance of code peptide represented corresponding protein abundances.
Table S3B. The forty tagged protein expression in three independent experiments of peptide enrichment, related to Figure 3. The samples were digested by trypsin, enriched by HA antibody and detected by PRM to assess the enrichment efficiency. The abundance of code peptide represented corresponding protein abundances.
Table S3C. The protein expression levels by profiling between BY4741 and JDY6086, related to Figure 4.
Table S3D. Twenty-six proteins of galactose and glycolysis pathway in JPY6086 yeast cultured in glucose and glucose medium using TMT methods, related to Figure 5. The data was processed in normalization and scaling mode on PD 2.3 software.
Table S3E. The intensities of twenty-six proteins in JPY6086 yeast cultured in glucose and glucose media quantified by GWAAP methods, related to Figure 5. The abundance of code peptide represented corresponding protein abundances.
Table S4A. The total area fragments of forty tagged protein in the serially diluted yeast lysates.
Table S4B. The curves of the best response peptides for 12 proteins detected by SCRIPT-MAP.
Data and code availability
-
•
The dataset supporting the findings of this study have been deposited at iProX repository (https://www.iprox.org.cn) and are publicly available as of the date of publication. Accession numbers are listed in the key resources table.
-
•
This paper does not report any original code.
-
•
Any additional information required to reanalyze the data reported in this paper is available form the lead contact upon request.
References
- 1.Pappireddi N., Martin L., Wühr M. A review on quantitative multiplexed proteomics. Chembiochem. 2019;20:1210–1224. doi: 10.1002/cbic.201800650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Mallick P., Kuster B. Proteomics: a pragmatic perspective. Nat. Biotechnol. 2010;28:695–709. doi: 10.1038/nbt.1658. [DOI] [PubMed] [Google Scholar]
- 3.Planta R.J., Mager W.H. The list of cytoplasmic ribosomal proteins of Saccharomyces cerevisiae. Yeast. 1998;14:471–477. doi: 10.1002/(SICI)1097-0061(19980330)14:5<471::AID-YEA241>3.0.CO;2-U. [DOI] [PubMed] [Google Scholar]
- 4.Schwanhäusser B., Busse D., Li N., Dittmar G., Schuchhardt J., Wolf J., Chen W., Selbach M. Global quantification of mammalian gene expression control. Nature. 2011;473:337–342. doi: 10.1038/nature10098. [DOI] [PubMed] [Google Scholar]
- 5.Liu W., Wei L., Sun J., Feng J., Guo G., Liang L., Fu T., Liu M., Li K., Huang Y., et al. A reference peptide database for proteome quantification based on experimental mass spectrum response curves. Bioinformatics. 2018;34:2766–2772. doi: 10.1093/bioinformatics/bty201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Rauniyar N. Parallel reaction monitoring: atargeted experiment performed using high resolution and high mass accuracy mass spectrometry. Int. J. Mol. Sci. 2015;16:28566–28581. doi: 10.3390/ijms161226120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Muntel J., Kirkpatrick J., Bruderer R., Huang T., Vitek O., Ori A., Reiter L. Comparison of protein quantification in a complex background by DIA and TMT workflows with fixed instrument time. J. Proteome Res. 2019;18:1340–1351. doi: 10.1021/acs.jproteome.8b00898. [DOI] [PubMed] [Google Scholar]
- 8.Zhang F., Ge W., Ruan G., Cai X., Guo T. Data-independent acquisition mass spectrometry-based proteomics and software tools: a glimpse in 2020. Proteomics. 2020;20:1900276. doi: 10.1002/pmic.201900276. [DOI] [PubMed] [Google Scholar]
- 9.Udeshi N.D., Pedram K., Svinkina T., Fereshetian S., Myers S.A., Aygun O., Krug K., Clauser K., Ryan D., Ast T., et al. Antibodies to biotin enable large-scale detection of biotinylation sites on proteins. Nat. Methods. 2017;14:1167–1170. doi: 10.1038/nmeth.4465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Arul A.B., Robinson R.A.S. Sample multiplexing strategies in quantitative proteomics. Anal. Chem. 2019;91:178–189. doi: 10.1021/acs.analchem.8b05626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Whiteaker J.R., Zhao L., Lin C., Yan P., Wang P., Paulovich A.G. Sequential multiplexed analyte quantification using peptide immunoaffinity enrichment coupled to mass spectrometry. Mol. Cell. Proteomics. 2012;11 doi: 10.1074/mcp.M111.015347. M111.015347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Youngren K.K., Coveney D., Peng X., Bhattacharya C., Schmidt L.S., Nickerson M.L., Lamb B.T., Deng J.M., Behringer R.R., Capel B., et al. The Ter mutation in the dead end gene causes germ cell loss and testicular germ cell tumours. Nature. 2005;435:360–364. doi: 10.1038/nature03595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Yang H., Shi L., Wang B.A., Liang D., Zhong C., Liu W., Nie Y., Liu J., Zhao J., Gao X., et al. Generation of genetically modified mice by oocyte injection of androgenetic haploid embryonic stem cells. Cell. 2012;149:605–617. doi: 10.1016/j.cell.2012.04.002. [DOI] [PubMed] [Google Scholar]
- 14.Zhong C., Yin Q., Xie Z., Bai M., Dong R., Tang W., Xing Y.H., Zhang H., Yang S., Chen L.L., et al. CRISPR-Cas9-Mediated genetic screening in mice with haploid embryonic stem cells carrying a guide RNA library. Cell Stem Cell. 2015;17:221–232. doi: 10.1016/j.stem.2015.06.005. [DOI] [PubMed] [Google Scholar]
- 15.Komor A.C., Kim Y.B., Packer M.S., Zuris J.A., Liu D.R. Programmable editing of a target base in genomic DNA without double-stranded DNA cleavage. Nature. 2016;533:420–424. doi: 10.1038/nature17946. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Ho B., Baryshnikova A., Brown G.W. Unification of protein abundance datasets yields a quantitative Saccharomyces cerevisiae proteome. Cell Syst. 2018;6:192–205.e3. doi: 10.1016/j.cels.2017.12.004. [DOI] [PubMed] [Google Scholar]
- 17.Mitchell L.A., Wang A., Stracquadanio G., Kuang Z., Wang X., Yang K., Richardson S., Martin J.A., Zhao Y., Walker R., et al. Synthesis, debugging, and effects of synthetic chromosome consolidation: synVI and beyond. Science. 2017;355:eaaf4831. doi: 10.1126/science.aaf4831. [DOI] [PubMed] [Google Scholar]
- 18.Shen Y., Wang Y., Chen T., Gao F., Gong J., Abramczyk D., Walker R., Zhao H., Chen S., Liu W., et al. Deep functional analysis of synII, a 770-kilobase synthetic yeast chromosome. Science. 2017;355:eaaf4791. doi: 10.1126/science.aaf4791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Wu Y., Li B.Z., Zhao M., Mitchell L.A., Xie Z.X., Lin Q.H., Wang X., Xiao W.H., Wang Y., Zhou X., et al. Bug mapping and fitness testing of chemically synthesized chromosome X. Science. 2017;355:eaaf4706. doi: 10.1126/science.aaf4706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Xie Z.X., Li B.Z., Mitchell L.A., Wu Y., Qi X., Jin Z., Jia B., Wang X., Zeng B.X., Liu H.M., et al. "Perfect" designer chromosome V and behavior of a ring derivative. Science. 2017;355:eaaf4704. doi: 10.1126/science.aaf4704. [DOI] [PubMed] [Google Scholar]
- 21.Zhang W., Zhao G., Luo Z., Lin Y., Wang L., Guo Y., Wang A., Jiang S., Jiang Q., Gong J., et al. Engineering the ribosomal DNA in a megabase synthetic chromosome. Science. 2017;355:eaaf3981. doi: 10.1126/science.aaf3981. [DOI] [PubMed] [Google Scholar]
- 22.Gibson D.G. Synthesis of DNA fragments in yeast by one-step assembly of overlapping oligonucleotides. Nucleic Acids Res. 2009;37:6984–6990. doi: 10.1093/nar/gkp687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Miyamoto K., Aoki W., Ohtani Y., Miura N., Aburaya S., Matsuzaki Y., Kajiwara K., Kitagawa Y., Ueda M. Peptide barcoding for establishment of new types of genotype-phenotype linkages. PLoS One. 2019;14:e0215993. doi: 10.1371/journal.pone.0215993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Brachmann C.B., Davies A., Cost G.J., Caputo E., Li J., Hieter P., Boeke J.D. Designer deletion strains derived from Saccharomyces cerevisiae S288C: a useful set of strains and plasmids for PCR-mediated gene disruption and other applications. Yeast. 1998;14:115–132. doi: 10.1002/(SICI)1097-0061. [DOI] [PubMed] [Google Scholar]
- 25.Gibson D.G., Benders G.A., Axelrod K.C., Zaveri J., Algire M.A., Moodie M., Montague M.G., Venter J.C., Smith H.O., Hutchison C.A., 3rd One-step assembly in yeast of 25 overlapping DNA fragments to form a complete synthetic Mycoplasma genitalium genome. Proc. Natl. Acad. Sci. USA. 2008;105:20404–20409. doi: 10.1073/pnas.0811011106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.MacLean B., Tomazela D.M., Shulman N., Chambers M., Finney G.L., Frewen B., Kern R., Tabb D.L., Liebler D.C., MacCoss M.J. Skyline: an open source document editor for creating and analyzing targeted proteomics experiments. Bioinformatics. 2010;26:966–968. doi: 10.1093/bioinformatics/btq054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Stergachis A.B., MacLean B., Lee K., Stamatoyannopoulos J.A., MacCoss M.J. Rapid empirical discovery of optimal peptides for targeted proteomics. Nat. Methods. 2011;8:1041–1043. doi: 10.1038/nmeth.1770. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Swiat M.A., Dashko S., den Ridder M., Wijsman M., van der Oost J., Daran J.M., Daran-Lapujade P. FnCpf1: a novel and efficient genome editing tool for Saccharomyces cerevisiae. Nucleic Acids Res. 2017;45:12585–12598. doi: 10.1093/nar/gkx1007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Paulo J.A., O'Connell J.D., Gaun A., Gygi S.P. Proteome-wide quantitative multiplexed profiling of protein expression: carbon-source dependency in Saccharomyces cerevisiae. Mol. Biol. Cell. 2015;26:4063–4074. doi: 10.1091/mbc.E15-07-0499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Cohen R., Yokoi T., Holland J.P., Pepper A.E., Holland M.J. Transcription of the constitutively expressed yeast enolase gene ENO1 is mediated by positive and negative cis-acting regulatory sequences. Mol. Cell Biol. 1987;7:2753–2761. doi: 10.1128/mcb.7.8.2753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Boles E., Schulte F., Miosga T., Freidel K., Schlüter E., Zimmermann F.K., Hollenberg C.P., Heinisch J.J. Characterization of a glucose-repressed pyruvate kinase (Pyk2p) in Saccharomyces cerevisiae that is catalytically insensitive to fructose-1, 6-bisphosphate. J. Bacteriol. 1997;179:2987–2993. doi: 10.1128/jb.179.9.2987-2993.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Huh W.K., Falvo J.V., Gerke L.C., Carroll A.S., Howson R.W., Weissman J.S., O'Shea E.K. Global analysis of protein localization in budding yeast. Nature. 2003;425:686–691. doi: 10.1038/nature02026. [DOI] [PubMed] [Google Scholar]
- 33.Newman J.R.S., Ghaemmaghami S., Ihmels J., Breslow D.K., Noble M., DeRisi J.L., Weissman J.S. Single-cell proteomic analysis of S. cerevisiae reveals the architecture of biological noise. Nature. 2006;441:840–846. doi: 10.1038/nature04785. [DOI] [PubMed] [Google Scholar]
- 34.Picotti P., Bodenmiller B., Mueller L.N., Domon B., Aebersold R. Full dynamic range proteome analysis of S. cerevisiae by targeted proteomics. Cell. 2009;138:795–806. doi: 10.1016/j.cell.2009.05.051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Breslow D.K., Cameron D.M., Collins S.R., Schuldiner M., Stewart-Ornstein J., Newman H.W., Braun S., Madhani H.D., Krogan N.J., Weissman J.S. A comprehensive strategy enabling high-resolution functional analysis of the yeast genome. Nat. Methods. 2008;5:711–718. doi: 10.1038/nmeth.1234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Picotti P., Clément-Ziza M., Lam H., Campbell D.S., Schmidt A., Deutsch E.W., Röst H., Sun Z., Rinner O., Reiter L., et al. A complete mass-spectrometric map of the yeast proteome applied to quantitative trait analysis. Nature. 2013;494:266–270. doi: 10.1038/nature11835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.McCarty N.S., Graham A.E., Studená L., Ledesma-Amaro R. Multiplexed CRISPR technologies for gene editing and transcriptional regulation. Nat. Commun. 2020;11:1281. doi: 10.1038/s41467-020-15053-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Boeke J.D., Church G., Hessel A., Kelley N.J., Arkin A., Cai Y., Carlson R., Chakravarti A., Cornish V.W., Holt L., et al. GENOME ENGINEERING.The genome project-write. Science. 2016;353:126–127. doi: 10.1126/science.aaf6850. [DOI] [PubMed] [Google Scholar]
- 39.Dorfer V., Maltsev S., Winkler S., Mechtler K. CharmeRT: boosting peptide identifications by chimeric spectra identification and retention time prediction. J. Proteome Res. 2018;17:2581–2589. doi: 10.1021/acs.jproteome.7b00836. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Wen B., Li K., Zhang Y., Zhang B. Cancer neoantigen prioritization through sensitive and reliable proteogenomics analysis. Nat. Commun. 2020;11:1759. doi: 10.1038/s41467-020-15456-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Gessulat S., Schmidt T., Zolg D.P., Samaras P., Schnatbaum K., Zerweck J., Knaute T., Rechenberger J., Delanghe B., Huhmer A., et al. Prosit: proteome-wide prediction of peptide tandem mass spectra by deep learning. Nat. Methods. 2019;16:509–518. doi: 10.1038/s41592-019-0426-7. [DOI] [PubMed] [Google Scholar]
- 42.Li W., Caberoy N.B. New perspective for phage display as an efficient and versatile technology of functional proteomics. Appl. Microbiol. Biotechnol. 2010;85:909–919. doi: 10.1007/s00253-009-2277-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Egloff P., Zimmermann I., Arnold F.M., Hutter C.A.J., Morger D., Opitz L., Poveda L., Keserue H.A., Panse C., Roschitzki B., Seeger M.A. Engineered peptide barcodes for in-depth analyses of binding protein libraries. Nat. Methods. 2019;16:421–428. doi: 10.1038/s41592-019-0389-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Zhu Y., Weiss T., Zhang Q., Sun R., Wang B., Yi X., Wu Z., Gao H., Cai X., Ruan G., et al. High-throughput proteomic analysis of FFPE tissue samples facilitates tumor stratification. Mol. Oncol. 2019;13:2305–2328. doi: 10.1002/1878-0261.12570. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Table S3A. The forty tagged protein expression in three biological repeats of trypsin digestion experiments, related to Figure 3. The samples were digested by trypsin and detected directly by PRM to assess the enzymatic efficiency. The abundance of code peptide represented corresponding protein abundances.
Table S3B. The forty tagged protein expression in three independent experiments of peptide enrichment, related to Figure 3. The samples were digested by trypsin, enriched by HA antibody and detected by PRM to assess the enrichment efficiency. The abundance of code peptide represented corresponding protein abundances.
Table S3C. The protein expression levels by profiling between BY4741 and JDY6086, related to Figure 4.
Table S3D. Twenty-six proteins of galactose and glycolysis pathway in JPY6086 yeast cultured in glucose and glucose medium using TMT methods, related to Figure 5. The data was processed in normalization and scaling mode on PD 2.3 software.
Table S3E. The intensities of twenty-six proteins in JPY6086 yeast cultured in glucose and glucose media quantified by GWAAP methods, related to Figure 5. The abundance of code peptide represented corresponding protein abundances.
Table S4A. The total area fragments of forty tagged protein in the serially diluted yeast lysates.
Table S4B. The curves of the best response peptides for 12 proteins detected by SCRIPT-MAP.
Data Availability Statement
-
•
The dataset supporting the findings of this study have been deposited at iProX repository (https://www.iprox.org.cn) and are publicly available as of the date of publication. Accession numbers are listed in the key resources table.
-
•
This paper does not report any original code.
-
•
Any additional information required to reanalyze the data reported in this paper is available form the lead contact upon request.