

Abstract
The genome of the basidiomycete yeast Dioszegia hungarica strain PDD-24b-2 isolated from cloud water at the summit of puy de Dôme (France) was sequenced using a hybrid PacBio and Illumina sequencing strategy. The obtained assembled genome of 20.98 Mb and a GC content of 57% is structured in 16 large-scale contigs ranging from 90 kb to 5.56 Mb, and another 27.2 kb contig representing the complete circular mitochondrial genome. In total, 8,234 proteins were predicted from the genome sequence. The mitochondrial genome shows 16.2% cgu codon usage for arginine but has no canonical cognate tRNA to translate this codon. Detected transposable element (TE)-related sequences account for about 0.63% of the assembled genome. A dataset of 2,068 hand-picked public environmental metagenomes, representing over 20 Tbp of raw reads, was probed for D. hungarica related ITS sequences, and revealed worldwide distribution of this species, particularly in aerial habitats. Growth experiments suggested a psychrophilic phenotype and the ability to disperse by producing ballistospores. The high-quality assembled genome obtained for this D. hungarica strain will help investigate the behavior and ecological functions of this species in the environment.
Keywords: Tremellaceae, mitochondrial genome, de novo sequencing, fungi, airborne microorganisms, transposable elements, Dioszegia hungarica strain PDD-24b-2, aeromicrobiology, fungal spore, cold environment
Introduction
There is increasing evidence that airborne microorganisms participate in chemical transformations and physical processes in the atmosphere (Šantl-Temkiv et al. 2022). In particular, microorganisms found in clouds play a central role in reactions of carbon-containing compounds at night, whereas during the day, photochemistry is dominant (Vaïtilingom et al. 2012, 2013). Both prokaryotic and eukaryotic microorganisms can be found in clouds (Delort et al. 2017). Regarding eukaryotes, 1–3% of sequenced 18S rDNA amplicons belong to the class Tremellomycetes of basidiomycete yeasts (Amato et al. 2017), which include the genus Dioszegia (Order: Tremellales; Family: Tremellaceae/Bulleribasidiaceae) (Liu et al. 2015). Dioszegia hungarica strain PDD-24b-2 was isolated from cloud water collected at the summit of the puy de Dôme, France (Vaïtilingom et al. 2012) (Supplementary Fig. 1) (backward trajectory calculated according to Stein et al. 2015). Strains identified as Dioszegia sp. are frequently isolated from cloud water sampled at this site (in 70% of studied samples; Vaïtilingom et al. 2012). This fungal taxon was repeatedly identified in various cold environments, such as snow and glacial meltwater rivers (De García et al. 2007), and is also associated with plants in Antarctica (Ferreira et al. 2019). The D. hungarica type strain CBS 4214T was isolated from soil in Külsó-tó, Hungary as described in Takashima et al. (2001). Also found in warmer environments, it is part of the core fungal community of the wheat phyllosphere (the aerial parts of plants) (Karlsson et al. 2017; Sapkota et al. 2017). Dioszegia hungarica was identified as one of the few “microbial hub taxa” that, when influenced by plant host and abiotic factors, act on the plant microbiome. For example, it directly inhibits the growth of specific bacterial taxa on Arabidopsis thaliana seedlings, thus decreasing the phyllosphere bacterial community diversity (Agler et al. 2016). The atmospheric environment in which airborne microbes are found represents both a source (immigration) and a sink (emigration) for the phyllosphere microbiome (Kinkel 1997). Examining the genome of D. hungarica may provide valuable information to better understand the dynamics of fungal diversity, especially at the plant/atmosphere interface, and its role in climate change-relevant ecosystems (e.g. clouds, cold environments, phyllosphere).
Dioszegia hungarica, formerly classified as Cryptococcus hungaricus and Bullera armeniaca (Takashima et al. 2001), is one of the 23 species of Dioszegia identified so far (Li et al. 2020). To date, genomes of 3 other Dioszegia species have been sequenced: D. aurantiaca strain JCM 2956 and D. crocea strain JCM 2961, isolated from overwintered nettle stems of Urtica sp. and strawberry phyllosphere, respectively (Takashima et al. 2019), and D. cryoxerica strain ANT03-071 (https://mycocosm.jgi.doe.gov/Diocr1), isolated from moss in Antarctica (Connell et al. 2010). Previous analyses of the internal transcribed spacer (ITS) and D1/D2 regions of the large subunit rRNA gene showed that D. hungarica is phylogenetically distant from these genome-sequenced representatives of the genus (Trochine et al. 2017; Li et al. 2020). This makes the species D. hungarica a good candidate to further investigate fungal genetic diversity. In this study, we describe the high-quality assembled genome sequence of D. hungarica strain PDD-24b-2 obtained by a hybrid PacBio and Illumina sequencing strategy. The assembled genome features 17 contigs, 16 large-scale linear contigs and a smaller contig representing the complete circular mitochondrial genome.
Materials and methods
Strain and growth conditions
Dioszegia hungarica strain PDD-24b-2 was isolated from cloud water collected at the summit of puy de Dôme, France on 17 January 2008 (Vaïtilingom et al. 2012). R2A liquid medium was prepared as described previously (Reasoner and Geldreich 1985). Commercial dehydrated R2A agar (Oxoid, Hampshire, UK) was used as solid medium. Yeast mold (YM) medium (pH 6.2) contained per liter 3 g yeast extract, 3 g malt extract, 5 g peptone (pancreatic digest gelatin), 10 g d-glucose, and was supplemented with 20 g agar for solid medium. Liquid cultures were grown at 17°C with agitation (Sanyo MIR 254 refrigerated incubator, MA, USA). The ability to produce ballistospores was assessed on R2A solid medium, placing an inoculated Petri dish above a sterile one as described previously (Ianiri et al. 2014).
DNA extraction and PCR amplification
Total DNA was extracted from a 4-day aerobic culture (OD at 600 nm of 0.97) in 200 mL R2A medium incubated at 17°C, using the MasterPure complete DNA and RNA purification kit as described by the manufacturer (Lucigen, WI, USA). The 18S rRNA gene was PCR-amplified from total DNA (25 ng) using primers Dios20F (5′-GTGCGTCTGATTCTTGACTCC-3′) and Dios11R (5′-CCCGACCGTCCCTATTAATCA-3′) and DreamTaq DNA polymerase, as recommended by the manufacturer (Thermo Fisher Scientific Baltics, Vilnius, Lithuania). The PCR program (Biometra TOne thermocycler, Analytik Jena, Jena, Germany) involved DNA denaturation at 95°C for 5 min, 30 cycles of 45 s at 93°C, 20 s at 56°C and 1 min at 72°C, and a final 10 min extension at 72°C. The amplified 1,080 bp PCR fragment was sequenced by the Sanger method (Microsynth France, Vaulx-en-Velin, France).
Genome sequencing, assembly, and automatic annotation
Illumina library preparation (Nextera XT kit), PacBio library preparation (SMRTbell express template prep kit 2.0) and high throughput sequencing of D. hungarica PDD-24b-2 were performed by GenoScreen (Lille, France). Libraries were sequenced using the MiSeq Illumina platform and the PacBio platform (SMRT cell Pacbio Sequel). Illumina and PacBio reads were quality checked with FastQC v0.11.9 (Andrews, Braham Bioinformatics).
Illumina adapter sequences were removed with CutAdapt v2.10 (Martin 2011) and paired-end reads cleaned with Prinseq v0.20.4 (Schmieder and Edwards 2011): the first 15 nucleotides of each read were cut, nucleotides with a Phred score under 30 were cut from the read 3′ end, reads shorter than 60 nucleotides were discarded, reads with a mean Phred score under 30 were discarded, as well as those containing undetermined nucleotides. Only paired reads were conserved. After these processing steps 8,383,275 read pairs were obtained.
PacBio subreads were assembled with Flye v2.8.2 (Kolmogorov et al. 2019). The cleaned Illumina read pairs were used to correct the PacBio assembly using BOWTIE2 v2.4.1 (Langmead and Salzberg 2012; Langmead et al. 2019) and Pilon v1.23 (Walker et al. 2014). Contigs were aligned to each other using BLASTn v2.10.1 (Camacho et al. 2009) to resolve alternative haplotypes. Telomeric repeats of the sequence T2AG3–5, akin to those of Cryptococcus neoformans (Edman 1992) were searched and visualized in this assembly using IGV v 2.12.0 (Robinson et al. 2011). Completeness of the D. hungarica PDD-24b-2 genome assembly was assessed with BUSCO v5.2.2 (Manni et al. 2021) against tremellomycetes_odb10, and compared with the 3 previously released genomes for the Dioszegia genus.
The nuclear genome was deposited in the MycoCosm platform (Grigoriev et al. 2014) and automatically annotated, as previously described (Kuo et al. 2014) using the JGI Annotation Pipeline. The mitochondrial genome annotation pipeline combined ab initio predictions, homology-based predictions with a curated mitochondrial protein set, and Hidden Markov models (HMM) based predictions, as described in Haridas et al. (2018). The EuKaryotic Orthologous Groups (KOG) classification scheme was used to evaluate the number of genes associated with predicted processes with detailed gene ID available at https://mycocosm.jgi.doe.gov/cgi-bin/kogBrowser?type=KOG&db=Diohu1. The Kyoto Encyclopedia of Genes and Genomes (KEGG) pathway database was used to identify metabolic pathway genes (https://mycocosm.jgi.doe.gov/cgi-bin/metapathways?db=Diohu1).
Identification of TEs
Putative TEs were searched in the D. hungarica PDD-24b-2 genome sequence using 2 de novo approaches: the RepeatModeler v2.0.3 pipeline (Flynn et al. 2020) with its long-terminal repeat (LTR) pipeline extensions, and the Extensive de novo TE Annotator pipeline (Bell et al. 2022). TEs were also identified by protein homology with transposon sequences from other fungi (Saitozyma podzolica, Cryptococcus neoformans, Cryptococcus gattii, Rhodotorula toruloides, Candida glabrata) using BLAST + tblastx v2.11.0 (Camacho et al. 2009). Detected sequences were manually curated using CD Search (Marchler-Bauer and Bryant 2004; Marchler-Bauer et al. 2017) to predict conserved protein domains. Target site duplications (TSD) were identified by manually checking for direct repeats in sequences adjacent to identified TEs, and confirmed by surveying several copies. Detected putative TEs were classified into Orders and Superfamilies, as previously described (Wicker et al. 2007). Unique candidate TEs for each family, with typical TE domains (e.g. transposase, reverse transcriptase, RNase H, integrase, aspartic protease, gag domains) were then used to build a putative TE library (Supplementary File 1) to screen the genome sequence of D. hungarica PDD-24b-2 using RepeatMasker v4.1.2-p1 and estimate putative TE copy number, including full-length and truncated copies. The RepeatMasker.out file was parsed with the tool “One code to find them all” (Bailly-Bechet et al. 2014) to assemble detected TE fragments. More information on the TE-mining process can be found in the following GitHub repository: https://github.com/JarrigeD/Dioszegia_hungarica_sequencing.
Phylogenetic analysis
The ITS region (ITS1, 5.8S ribosomal RNA gene, ITS2 and large subunit ribosomal RNA gene partial sequence) phylogenetic tree of the Dioszegia genus was constructed entirely with MEGA11 (Tamura et al. 2021). An alignment of the ITS region was generated using MUSCLE (Edgar 2004) on a total of 462 positions for D. hungarica PDD-24b-2, reference strains of the 23 Dioszegia species described to date, and 2 Hannaella type strains used as outgroup (Supplementary Table 2). The MEGA11 “find best fit substitution model” tool was used to choose the substitution model for tree building. The General Time Reversible model with rate heterogeneity across sites (GTR + G) (Tavaré 1986; Yang 1996) had the lowest Bayesian information criterion score and corrected Akaike information criterion score and was used to calculate the matrix of pairwise distances. A discrete Gamma distribution was used to model evolutionary rate differences among sites [5 categories (+G, parameter = 0.1890)]. A total of 500 replicate trees were built with the Maximum Likelihood method to calculate bootstrap support values, and the best tree topology with the highest log likelihood (−2379.73) was selected.
Geographical distribution
A total of 2,068 whole-genome shotgun (WGS) raw read metagenomic datasets were hand-picked to maximize geographic and environmental variety and retrieved from the Sequence Read Archive (SRA) using sra-tools. The presence of D. hungarica sequences was tested using sra-tools blastn_vdb megablast, with strain PDD-24-2b-2 ITS region as query and a minimum percentage identity threshold of 97%. The resulting BLAST hits were filtered to target members of the genus Dioszegia (≥45 nt with ≥99% identity to the PDD-24b-2 5.8S rRNA gene sequence) and of the species D. hungarica (≥15 nt to ITS1 or ITS2 sequences, E-value ≤10e−10). These thresholds were defined using alignments of ITS regions of D. hungarica PDD-24b-2 to those of fungal type strains in the NCBI ITS_RefSeq_Fungi database. Maps of WGS datasets with D. hungarica or Dioszegia sp. hits were plotted in Python v3.10.2 using Matplotlib v3.5.1 and GeoPandas v0.10.2. Details of metadata and dataset accessions, homolog search scripts, filtering parameters, and mapping processes are available at https://github.com/JarrigeD/Dioszegia_hungarica_sequencing.
The GlobalFungi database (accessible at https://globalfungi.com/, last accessed on 2022-09-12) (17,000 ITS amplicons environmental samples) (Větrovský et al. 2020), the MarDB database (accessible at https://mmp.sfb.uit.no/blast/, last accessed on 2021-11-23) (14,600 marine microbial genomes) (Klemetsen et al. 2018; Priyam et al. 2019) and TARA Ocean Gene Atlas databases (Villar et al. 2018) (accessible at https://tara-oceans.mio.osupytheas.fr/ocean-gene-atlas, last accessed on 2022-09-12) EUK_SMAGs (713 eukaryotic plankton Metagenome Assembled Genomes: MAGs) (Delmont et al. 2022), MATOUv1_metaG (116.8 million eukaryotic expressed genes + 530 Arctic Ocean MAGs) (Carradec et al. 2018), and OM-RGC_v2_metaG (370 marine metagenomes + 530 MAGs) (Salazar et al. 2019) were also searched for close homologs of the ITS region of Dioszegia hungarica PDD-24b-2.
Results and discussion
Cell morphology and growth characteristics
Single ovoid cells of ∼4 µm in length, dividing by polar budding, were observed by optical microscopy (Fig. 1a). Dioszegia hungarica strain PDD-24b-2 grows in R2A and YM media with the characteristic deep orange color typical of this genus (Inacio et al. 2005), which becomes more pronounced at higher cell density (Fig. 1b and c). Growth in YM was faster than in R2A (Fig. 1d), with incubation temperature strongly affecting growth (Fig. 1d). The shortest doubling times were observed at 17°C in both YM (297 ± 5 min) and R2A (381 ± 33 min) media. Incubation at 4°C and 25°C resulted in 3- and 2-times longer doubling times, respectively. No growth was observed at 30°C and 37°C after 2 weeks under all tested conditions (data not shown). This is in line with the temperatures of low-altitude clouds at the French site from which the strain was isolated, i.e. 5°C mean and 17°C maximal temperature, respectively (Vaïtilingom et al. 2010). In addition, strain PDD-24b-2 was able to produce ballistospores, a launched spore type specific to basidiomycetes, at 17°C on R2A solid medium after 6 days of culture. Ballistospores are able to launch from an inoculated plate to a neighboring sterile one, on which colonies will grow following incubation, forming a “mirror” image of the inoculated plate (Fig. 1e). Ballistosporic basidiospores have been proposed to act as giant cloud condensation nuclei that could increase precipitation by coalescing smaller droplets (Hassett et al. 2015). Unlike the strain studied in this work, however, one of the D. hungarica strains isolated from terrestrial habitats was unable to produce ballistospores (Takashima et al. 2001), suggesting that this trait is not conserved within D. hungarica.
Fig. 1.

Morphological and growth characteristics of D. hungarica strain PDD-24b-2. a) Cellular morphology observed with a Leica DM4000 B microscope at ×1,000 magnification after growth in YM broth at 17°C. b) Colonies on R2A solid medium at 17°C. c) Liquid cultures after growth at different temperatures for 4 days. Highest cell density was observed at 17°C. No growth was observed above 25°C or in sterile YM medium. d) Effect of culture incubation temperatures on growth rate (µ). For each medium (YM or R2A), the mean growth rate was the mean of 2 biological replicates. e) Ballistospore production on solid R2A medium. The inoculated plate (left) was placed on top of the uninoculated one (right). After 6 days of incubation, colonies also appeared on the bottom plate as a partial mirror image of the top inoculated plate.
Genome sequencing, assembly, and completeness
The genome of D. hungarica PDD-24b-2 was sequenced by a hybrid strategy using a combination of PacBio (average coverage of 97×, median subread size of 3,584 bp, and 474,621 subreads in total) and Illumina (average coverage of 101×; 9,901,968 read pairs of 151 bp) sequencing, yielding a high-quality assembly. The 28 contigs assembled from PacBio subreads were corrected with the Illumina pair-end reads. BLASTn alignment analysis identified 2 identical contigs which were merged, and 9 small contigs nearly identical to larger ones (between 99.96% and 100% identity) which could represent alternative haplotypes and were thus discarded from the final assembly. This yielded a final genome assembly of 18 contigs, with 17 linear contigs corresponding to the nuclear genome. One contig was circular, as evidenced by more than 500 Illumina reads bridging its ends (data not shown) and corresponded to the mitochondrial genome. Its size of 27,226 bp was in close agreement with that estimated for the D. hungarica strain CBS 4214T from average contour-length on electron micrographs 20 years ago (27.3 kb, Gácser et al. 2002).
After assembly, the beginning 5′ third and the remaining 3′ parts of the 18S rRNA gene were located at the termini of 2 nuclear contigs, i.e. the smallest contig of 2 kb (contig20) and the contig of 1.11 Mbp (contig11, which also contained the remainder of the rRNA-encoding region). To confirm the linkage between contig20 and contig11, PCR amplification of the 18S rRNA gene was performed using primers targeting contig20 and contig11. The full 18S rRNA gene sequence including the 19 nt gap initially left out of the assembly was sequenced. Accordingly, contig11 was merged with the smaller contig20 to restore a complete 18S rRNA gene within the reunited rRNA region composed of 5S rRNA, 18S rRNA, 5.8S rRNA, and 25S rRNA genes. Genome regions with rRNA genes are notoriously difficult to resolve in eukaryotic genomic assemblies, as rRNA genes can be found in tens to thousands tandem copies (Nelson et al. 2019). For instance, Cryptococcus neoformans, another basidiomycetous yeast, contains around 55 tandem repeats of a single rRNA gene region (Loftus et al. 2005; Ganley and Kobayashi 2007). The rRNA gene copy number is usually estimated by relative read coverage (Lofgren et al. 2019). For strain PDD-24b-2, Illumina read depth coverage of contig20 was 35 times higher than for the rest of the genome (Supplementary Fig. 2). As expected, a similar increased coverage was also observed for the terminal part of contig11 in which the rRNA gene cluster is located. This suggests that ribosomal RNA genes are present in about 35 copies in D. hungarica PDD-24b-2, although the precise number of tandem repeats remains unknown. To estimate the length of the whole region containing copies of the rRNA gene cluster, we multiplied the rRNA unit length (10.29 kb) by its relative coverage of 35. The estimated length of the complete contig11 would thus be ∼1.46 Mbp (Fig. 2).
Fig. 2.
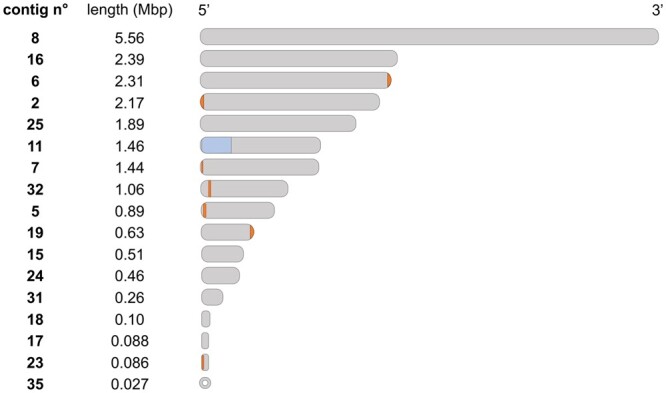
Telomeric sequences distribution in assembled contigs of D. hungarica PDD-24b-2. Detected telomeric T2AG3–5 repeats are indicated as dark bars (not drawn to scale). The complete ribosomal RNA gene region (with an estimated 35 copies of the ribosomal RNA gene cluster) is highlighted on contig11. Identification labels for assembled contigs are given as in MycoCosm (https://mycocosm.jgi.doe.gov/Diohu1). Contig 35 corresponds to the circular mitochondrial genome.
For 7 contigs, T2AG3-5 telomeric repeats were detected at one of the ends only (Fig. 2), suggesting incomplete resolution of the nuclear genome. Nevertheless, the statistics and characteristics of the obtained final genome assembly of D. hungarica strain PDD-24b-2, with 16 large-scale contigs and a complete mitochondrial genome contig, compare favorably with previously reported genomes for the genus Dioszegia (Table 1). Specifically, and with an L50 value of 4 and a N50 length of 2.17 Mbp, the genome assembly of D. hungarica PDD-24b-2 contains no gaps, unlike the 3 previously sequenced genomes of Dioszegia strains (Table 1). In particular, the assembly of the D. cryoxerica ANT 3-071T genome was strongly fragmented, with 111 scaffolds under 2 kb, a L50 of 96 and a N50 of 0.12 Mbp. In the case of D. aurantiaca JCM 2956T and D. crocea JCM 2961T, some large and a few small (<2 kb) scaffolds were reported.
Table 1.
Genome assembly and predicted annotation data of D. hungarica PDD-24b-2 compared with the 3 other genome-sequenced Dioszegia sp.
Annotation statisticsBUSCO v5.2.2 assembly completeness assessment (%)cComplete
| Content | Dioszegia hungarica PDD-24b-2 | Dioszegia aurantiaca JCM 2956T | Dioszegia crocea JCM 2961T | Dioszegia cryoxerica ANT 03-071T |
|---|---|---|---|---|
| BioProject accession no. | PRJNA809585 | PRJDB3721 | PRJDB3718 | PRJNA196046 |
| Reference | This study | Takashima et al. (2019) | Takashima et al. (2019) | L.B. Connell (personal communication) |
| Sequencing and assemblya statistics | ||||
| Sequencing read coverage depth (technology) | 97× (PacBio Sequel) + 101× (Illumina MiSeq) | 112× (Illumina HiSeq 2500) | 176× (Illumina HiSeq 2500) | 98.5× (Illumina HiSeq 2500) |
| Assembly size (Mbp) | 20.96 | 19.34 | 20.60 | 39.52 |
| Scaffolds/contigs | 18/18 | 52/139 | 26/86 | 865/1,318 |
| Longest scaffold (Mbp) | 5.56 | 4.12 | 3.59 | 0.43 |
| L50 scaffold value | 4 | 5 | 10 | 96 |
| N50 scaffold length (Mbp) | 2.17 | 1.28 | 1.95 | 0.12 |
| Scaffolds over 2 kb | 17b | 44 | 21 | 754 |
| GC content (%) | 57.2 | 53.6 | 53.2 | 56.9 |
| Gaps (%) | 0.00 | 0.82 | 0.59 | 1.3 |
| Linear contigs | 16b | 139 | 86 | 1.318 |
| Mitochondrial genome (kb) | 27 (circular) | NA | NA | 36 |
| Gene models | 8,219 | 8,106 | 8,753 | 15,948 |
| Average transcript length (bp) | 1,538 | 1,817 | 1,801 | 1,415 |
| Average exon/intron length (bp) | 247/67 | 258/59 | 259/61 | 264/61 |
| Average exons per gene | 6.23 | 5.90 | 5.80 | 5.36 |
| Average protein length (aa) | 513 | 507 | 500 | 429 |
| Genes with GO annotations | 3,925 | NA | NA | 6,951 |
| 89.3 | 89.0 | 89.3 | 90.0 | |
| Single | 88.8 | 88.9 | 88.7 | 36.6 |
| Duplicated | 0.5 | 0.1 | 0.6 | 53.4 |
| Fragmented | 3.3 | 3.9 | 3.9 | 3.2 |
| Missing | 7.4 | 7.1 | 6.8 | 6.8 |
Haploid assemblies except for D. cryoxerica ANT 03-071T which is probably diploid.
Scaffolds and linear contigs left after merging contig20 and contig11.
BUSCO reference dataset: tremellomycetes_odb10 (2021 June 28).
NA, not available.
The GC content of D. hungarica is about 57%, similar to that reported for D. cryoxerica, and higher than about 53% for D. aurantiaca and D. crocea. A detailed comparative assembly assessment of the 4 genomes using BUSCO v5.2.2 (Manni et al. 2021) was performed using the tremellomycetes_odb10 database. The estimated occurrence of complete genes was similar for the 4 compared Dioszegia genomes, yet at about 89.5% instead of the expected 100%. This suggests a lineage specific bias for the Dioszegia genus of the reference tremellomycetes_odb10 database. A notable difference between Dioszegia genomes is the high percentage of duplicated genes reported for D. cryoxerica (53.4%), possibly reflecting unresolved haplotypes in the diploid assembly of its genome (https://mycocosm.jgi.doe.gov/Diocr1). This would also be consistent with the twice larger length of its assembly (39.5 Mpb) compared with the 3 other reported genomes including D. hungarica.
Genome annotation for protein-coding genes and predicted metabolic pathways
The obtained number of 8,219 predicted protein-coding genes is close to that reported for D. aurantiaca JCM 2956T and D. crocea JCM 2961T (Table 1) and also to the total number of unique protein-coding genes of D. cryoxerica ANT 3-071T. The KOG classification scheme was used to evaluate the number of genes involved in cellular processes and signaling (1,301), information storage and processing (1,067), metabolism (1,460), and genes with unknown functions (1,056) (detailed gene ID available at https://mycocosm.jgi.doe.gov/cgi-bin/kogBrowser?type=KOG&db=Diohu1). The largest gene families include transporters from the Major Facilitator Superfamily (138) and sugar transporters (66), protein kinases (102), and clusters of genes with WD domain (100) and helicase-domain (75). Secondary metabolism is represented by 3 NRPS-like gene clusters and a single PKS-like gene cluster.
Gene predictions were analyzed in the light of experimentally characterized metabolic traits in D. hungarica (Takashima et al. 2001). Genes for glycolysis/gluconeogenesis (39 genes), the TCA cycle (19 genes), starch utilization and production (61 genes), and nitrite utilization (1 nitrite reductase-encoding gene) (KOG classification within the MycoCosm plateform) reflect the previously reported utilization by D. hungarica of glucose, succinic and citric acid, starch, and nitrite, respectively. Conversely, no genes were predicted for methanol or nitrate utilization, or for thiamine biosynthesis, confirming the reported inability of D. hungarica to use methanol or nitrate, and its thiamine auxotrophy. Identified genes for carotenoid biosynthesis (36 putative genes, KEGG annotations, JGI Annotation Pipeline) are in line with previous reports of carotenoids in Dioszegia strains (Madhour et al. 2005; Amaretti et al. 2014; Villarreal et al. 2016) and also with the bright orange color culture observed for D. hungarica PDD-24b-2 (Fig. 1). Carotenoids prevent oxidative stress (Madhour et al. 2005) and act as photo-protectants (Moliné et al. 2009) and cryo-protectants (Dieser et al. 2010), and may thus favor survival under the harsh conditions of clouds (Šantl-Temkiv et al. 2022). In this context, strain PDD-24b-2 also encodes a putative antifreeze protein (protein ID: 32937), with a predicted ice-binding protein domain (InterPro entry: IPR021884) and a predicted secretion signal. Secreted antifreeze proteins impair ice crystal formation and protect cell integrity under cold conditions (Hashim et al. 2013), suggesting a role of this protein in cold protection of D. hungarica in the cloud environment that remains to be experimentally validated.
Transposable elements
A total of 311 putative sequences related to TEs were detected and classified in 16 TE families (Supplementary Table 1 and File 1). TEs are dominated by Class I elements representing 12 families. Of those, 7 families of Copia and 1 family of Gypsy LTR TE were found. Class I non-LTR elements putative families were distributed in 3 LINE families and one DIRS family. Four families of Class II terminal inverted repeat (TIR) elements were also detected. Only 1 family encodes a transposase gene carrying a cl24015 domain attributed to MULE TE DDE transposases (Babu et al. 2006). The 10 bp long TSD supports an assignation to the Mutator Superfamily. Four families of nonautonomous Miniature Inverted-Repeat Transposable Elements (MITE) were also detected. One of them is related to the aforementioned Mutator element (same TIR and 10 bp long TSD). The others may be related to the hAT superfamily, according to their TSD length of 8 bp. However, we could not detect the corresponding autonomous copies encoding the transposases to confirm their annotation. In total, putative transposon-related sequences (around 130 kb) represent 0.63% of the D. hungarica PDD-24b-2 genome, among the lowest so far for basidiomycete fungi (Castanera et al. 2017). However, reported TE contents are highly variable (ranging between 0.1% and 42%), possibly also reflecting in part differences in sequence assembly and TE annotation protocols (Castanera et al. 2017).
Circular mitochondrial genome
This study provides the first complete and circular mitochondrial genome for D. hungarica. Organization of the mitochondrial genome of strain PDD-24b-2 differs from that of other D. hungarica strains basing on previously reported physical maps (Gácser et al. 2002). This is not unexpected as mitochondrial genome maps differed between D. hungarica strains.
The mitochondrial genome of strain PDD-24b-2 is smaller (27 kb) than those of D. changbaiensis (35 kb; Tan and Wang 2021) and D. cryoxerica ANT 03-071T (36 kb; L. B. Connell, personal communication) but of similar GC content (40–42%). The PDD-24b-2 mitochondrial genome contains all 15 known core protein-coding genes of mitochondria in Basidiomycetes, 23 tRNAs, and 2 rRNAs (Table 2).
Table 2.
Comparison of the mitochondrial genomes of D. hungarica and D. changbaiensis.
General statisticsMitochondrial genome contents
| Content | Dioszegia hungarica PDD-24b-2 | Dioszegia changbaiensis CGMCC AS 2.2309T |
|---|---|---|
| Accession no. | JAKWFO000000000 | MT755637 |
| Reference | This study | Tan and Wang (2021) |
| Size (bp) | 27,226 | 34,853 |
| GC content (%) | 40.6 | 41.9 |
| Protein-coding genes | atp6, atp8, atp9, cob, cox1, cox2, cox3, nad1, nad2, nad3, nad4, nad4L, nad5, nad6, rps3 | atp6, atp8, atp9, cob, cox1, cox2, cox3, nad1, nad2, nad3, nad4, nad4L, nad5, nad6, rps3 |
| tRNA | trnA(ugc), trnD(guc), trnE(uuc), trnF(gaa), trnG(ucc), trnH(gug), trnI(gau), trnK(uuu), trnL(uaa), trnL(uag), trnM(cau), trnM(cau), trnN(guu), trnP(ugg), trnQ(uug), trnR(ucg), trnR(ucu), trnS(gcu), trnS(uga), trnT(ugu), trnV(uac), trnW(cca), trnY(gua) | trnA(ugc), trnD(guc), trnE(uuc), trnF(gaa), trnG(ucc), trnH(gug), trnI(gau), trnK(uuu), trnL(uaa), trnL(uag), trnM(cau), trnM(cau) trnN(guu), trnP(ugg), trnQ(uug), trnR(ucg), trnS(gcu), trnS(uga), trnT(ugu), trnV(uac), trnW(cca), trnY(gua) |
| rRNA | rns, rnl | rns, rnl |
| Arg codons in mitochondrial CDS | aga: 0, agg: 1, cga: 72, | aga: 1, agg: 0, cga: 66, |
| cgc: 0, cgg: 10, cgu: 16 | cgc : 1, cgg: 8, cgu: 20 |
The additional tRNA gene in D. hungarica is underlined.
One major difference between the mitochondrial genome of D. hungarica and that of D. changbaiensis, the only other Dioszegia annotated mitochondrial genome to date, is the presence in D. hungarica of an additional tRNA gene, trnR(ucu) for arginine (Table 2). Although similar arginine codon usages are found in both strains, this is not the case for the aga and agg codons that are exclusively found in one of the mitochondrial genome. It is possible that this additional tRNA-Arg(ucu) in D. hungarica is used to translate the agg codon (Agris et al. 2007). On the other hand, in the absence of tRNA-Arg(ucu) the translation of the aga codon in D. changbaiensis remains unexplained.
A noticeable similarity between the mitochondrial genome of D. hungarica and that of D. changbaiensis is a high cgu codon usage for arginine (16.2% of arginine codons for D. hungarica and 20.8% for D. changbaiensis) (Table 2). Thus, as this cgu codon cannot be canonically translated by either tRNA-Arg(ucg) or tRNA-Arg(ucu) without post-transcriptional modifications (Phizicky and Hopper 2010), experiments are needed to identify yet unknown modification processes and their roles in translation in D. hungarica and D. changbaiensis mitochondria.
Phylogenetic analysis and environmental distribution
A phylogenetic tree based on the analysis of the ITS region was constructed for 24 strains of the genus Dioszegia, including strain PDD-24b-2, with 2 strains of the genus Hannaella, as outgroups, using the Maximum Likelihood method (ITS sequence information in Supplementary Table 2). In this tree, D. hungarica PDD-24b-2 and the D. hungarica type strain are clustered together and distinct from genome-sequenced strains of other Dioszegia species (Fig. 3), in accordance with previous taxonomical studies (Trochine et al. 2017; Li et al. 2020).
Fig. 3.

Phylogenetic analysis of the Dioszegia genus based on the ITS region. The tree was obtained from a sequence alignment of 462 nt of the ITS region with the Maximum Likelihood method and General Time Reversible model. Branch lengths (number of substitutions per site) are indicated under each branch, bootstrap support values (percentage of replicate trees in which the associated taxa clustered together in the bootstrap test 500 replicates) are indicated in larger fontsize. Sequence accession numbers are indicated in brackets. Diamonds (◆) indicate strains for which a draft genome is available. Hannaella sinensis and Hannaella luteola were used as outgroups.
Geographical distribution and potential habitat specificity of D. hungarica were investigated with a large set of public metagenomes selected to represent a wide diversity of environments, using the ITS region of strain PDD-24b-2 as a query (Fig. 4a). Dioszegia hungarica was detected at diverse latitudes around the world (Fig. 4b), and mostly in aerial biomes. In contrast, representatives of the Dioszegia genus were found to be more diversely distributed (Fig. 4c). Strikingly, ITS sequences specific of D. hungarica were not detected in marine samples in our dataset of selected metagenomes, nor in the Mar and TARA Ocean Gene Atlas databases (no fungal hits with over 97% identity were found). This suggests that D. hungarica is scarce in open sea environments. On the other hand, D. hungarica sequences were not detected in soil metagenomes either (Fig. 4c). This was surprising since the Dioszegia hungarica type strain was isolated from soil (Takashima et al. 2001). However, when using the GlobalFungi database, which is a terrestrial soil-focused database, ITS sequences of D. hungarica were detected in soil samples. Considering the significant differences in types of sequences between metagenomes (WGS, short raw reads) and the GlobalFungi database (targeted amplification of longer ITS sequences), the stringency of search parameters used in our analysis may contribute to explain this discrepancy, especially in environments with low abundance of D. hungarica communities. Nevertheless, the low occurrence of D. hungarica in oceans is somewhat paradoxical considering that strain PDD-24b-2 was isolated from a cloud of oceanic origin (Supplementary Fig. 1). We thus hypothesize that D. hungarica was picked up during air mass travel across France through the puy de Dôme sampling site. As such, the detection of D. hungarica in cloud water could serve as an indicator of air mass contact with terrestrial surfaces in future studies where detailed characterization of investigated cloud microbiomes is of interest.
Fig. 4.

Geographical and environmental distribution of the Dioszegia genus and D. hungarica species. a) Environmental metagenome exploration pipeline. The megablast hits filtering process is indicated with unfiltered hits (left), filtered hits representing D. hungarica (right, light hits), or Dioszegia sp. sequences (right, dark hits) mapped on D. hungarica PDD-24b-2 ITS region. b) Geographical distribution of sequences assigned to D hungarica or Dioszegia sp. in environmental metagenomic datasets (see Supplementary Table 3 for detailed information). c) Environmental distribution of D hungarica and Dioszegia sp. compared with that of the complete metagenomic dataset.
In conclusion, the obtained high-quality assembled and annotated genome of the orange-pigmented psychrotrophic yeast D. hungarica PDD-24b-2, a major representative of the cloud microbiome, now provides a blueprint for future functional genomics analyses of this environmentally relevant fungus. This will help characterize its mechanisms of resistance to UV radiation (Inacio et al. 2005) and of survival in cold environments (Dalluge et al. 2019), contribute to develop yeast enzymatic processes at low temperatures (Vaz et al. 2011), and help to identify and characterize the biotic factors that play a role in cloud chemistry.
Supplementary Material
Acknowledgments
We thank Léa Eck for her help with DNA extraction and Amandine Moreno for physiological tests and microscopy photographs, Dr Laurie B Connell for giving access to the D. cryoxerica ANT 03-071T genome prior to publication, Dr Joseph Schacherer for discussion of sequencing strategy and Prof. Hubert Becker for his help with tRNA analysis. This article is dedicated to Dr Anne-Marie Delort, pioneer of investigations on the role of microorganisms in atmospheric chemistry, on the occasion of her retirement.
Funding
This study and the postdoctoral grant to DJ were funded by the French National Agency, grant ANR-19-CE01-0004-02, project METACLOUD. Genome annotation was performed by the U.S. Department of Energy Joint Genome Institute, a DOE Office of Science User Facility, supported by the Office of Science of the U.S. Department of Energy under contract no. DE-AC02-05CH11231.
Conflicts of interest
None declared.
Author contributions
FB initiated the study. FB and DJ wrote the manuscript. FB, DJ, and SV revised the manuscript. MJ and MS cultivated the strain under supervision of PA. FB performed wet lab experiments. SH performed genome annotation on the MycoCosm platform under supervision of IG. DJ performed the phylogenetic analysis and geographic distribution study. DJ performed the transposable element search under the guidance of CBG. TN contributed to discussion. All authors have read, edited, and approved the final manuscript.
Contributor Information
Domitille Jarrige, Génétique Moléculaire, Génomique, Microbiologie (GMGM), Université de Strasbourg, UMR 7156 CNRS, Strasbourg, France.
Sajeet Haridas, Lawrence Berkeley National Laboratory, U.S. Department of Energy Joint Genome Institute, Berkeley, CA 94720, USA.
Claudine Bleykasten-Grosshans, Génétique Moléculaire, Génomique, Microbiologie (GMGM), Université de Strasbourg, UMR 7156 CNRS, Strasbourg, France.
Muriel Joly, Université Clermont Auvergne, Clermont Auvergne Institut National Polytechnique (INP), Centre National de la Recherche Scientifique (CNRS), Institut de Chimie de Clermont-Ferrand (ICCF), 63000 Clermont-Ferrand, France.
Thierry Nadalig, Génétique Moléculaire, Génomique, Microbiologie (GMGM), Université de Strasbourg, UMR 7156 CNRS, Strasbourg, France.
Martine Sancelme, Université Clermont Auvergne, Clermont Auvergne Institut National Polytechnique (INP), Centre National de la Recherche Scientifique (CNRS), Institut de Chimie de Clermont-Ferrand (ICCF), 63000 Clermont-Ferrand, France.
Stéphane Vuilleumier, Génétique Moléculaire, Génomique, Microbiologie (GMGM), Université de Strasbourg, UMR 7156 CNRS, Strasbourg, France.
Igor V Grigoriev, Lawrence Berkeley National Laboratory, U.S. Department of Energy Joint Genome Institute, Berkeley, CA 94720, USA; Department of Plant and Microbial Biology, University of California Berkeley, Berkeley, CA 94720, USA.
Pierre Amato, Université Clermont Auvergne, Clermont Auvergne Institut National Polytechnique (INP), Centre National de la Recherche Scientifique (CNRS), Institut de Chimie de Clermont-Ferrand (ICCF), 63000 Clermont-Ferrand, France.
Françoise Bringel, Génétique Moléculaire, Génomique, Microbiologie (GMGM), Université de Strasbourg, UMR 7156 CNRS, Strasbourg, France.
Data Availability
The Dioszegia hungarica PDD-24b-2 Whole Genome Shotgun project was deposited at DDBJ/ENA/GenBank under accession number JAKWFO000000000. The genome version used in this report is JAKWFO010000000. The raw Illumina and PacBio reads were deposited at the Sequence Read Archive under accessions numbers SRR18177991 and SRR18177990, respectively. Details on genome assembly and gene model properties are provided on the MycoCosm genome portal (https://mycocosm.jgi.doe.gov/Diohu1). Strain Dioszegia hungarica PDD-24b-2 is available upon request to Dr Pierre Amato or Dr Françoise Bringel. Representative sequences of putative D. hungarica PDD-24b-2 TE families are in Supplementary File 1. Putative TEs detected in D. hungarica PDD-24b-2 are in Supplementary Table 1. ITS sequences used to construct the phylogenetic tree of the Dioszegia genus are in Supplementary Table 2. Environmental samples in which D. hungarica and Dioszegia species were searched are provided in Supplementary Table 3. The air mass trajectory of the cloud from which D. hungarica strain PDD-24b-2 was isolated is shown in Supplementary Fig. 1. A close-up of the Illumina read depth coverage of D. hungarica PDD-24b-2 rRNA gene region is provided in Supplementary Fig. 2. The homolog search scripts, environmental metagenome dataset, as well as more information on biogeographic analyses and TE mining are available at https://github.com/JarrigeD/Dioszegia_hungarica_sequencing.
Supplemental material is available at G3 online.
Literature cited
- Agler MT, Ruhe J, Kroll S, Morhenn C, Kim S-T, Weigel D, Kemen EM. Microbial hub taxa link host and abiotic factors to plant microbiome variation. PLoS Biol. 2016;14(1):e1002352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Agris PF, Vendeix FAP, Graham WD. tRNA’s wobble decoding of the genome: 40 years of modification. J Mol Biol. 2007;366(1):1–13. [DOI] [PubMed] [Google Scholar]
- Amaretti A, Simone M, Quartieri A, Masino F, Raimondi S, Leonardi A, Rossi M. Isolation of carotenoid-producing yeasts from an alpine glacier. Chem Eng Trans. 2014;38:217–222. [Google Scholar]
- Amato P, Joly M, Besaury L, Oudart A, Taib N, Moné AI, Deguillaume L, Delort A-M, Debroas D. Active microorganisms thrive among extremely diverse communities in cloud water. PLoS One. 2017;12(8):e0182869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Andrews S. FastQC. Babraham Bioinformatics. https://www.bioinformatics.babraham.ac.uk/projects/fastqc/
- Babu MM, Iyer LM, Balaji S, Aravind L. The natural history of the WRKY–GCM1 zinc fingers and the relationship between transcription factors and transposons. Nucleic Acids Res. 2006;34(22):6505–6520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bailly-Bechet M, Haudry A, Lerat E. “One code to find them all”: a perl tool to conveniently parse RepeatMasker output files. Mob DNA. 2014;5:13. [Google Scholar]
- Bell EA, Butler CL, Oliveira C, Marburger S, Yant L, Taylor MI. Transposable element annotation in non‐model species: the benefits of species‐specific repeat libraries using semi‐automated EDTA and DeepTE de novo pipelines. Mol Ecol Resour. 2022;22(2):823–833. [DOI] [PubMed] [Google Scholar]
- Camacho C, Coulouris G, Avagyan V, Ma N, Papadopoulos J, Bealer K, Madden TL. BLAST+: architecture and applications. BMC Bioinformatics. 2009;10(1):421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carradec Q, Pelletier E, Da Silva C, Alberti A, Seeleuthner Y, Blanc-Mathieu R, Lima-Mendez G, Rocha F, Tirichine L, Labadie K, et al. ; Tara Oceans Coordinators. A global Ocean Atlas of eukaryotic genes. Nat Commun. 2018;9(1):373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Castanera R, Borgognone A, Pisabarro AG, Ramírez L. Biology, dynamics, and applications of transposable elements in basidiomycete fungi. Appl Microbiol Biotechnol. 2017;101(4):1337–1350. [DOI] [PubMed] [Google Scholar]
- Connell LB, Redman R, Rodriguez R, Barrett A, Iszard M, Fonseca Á. Dioszegia antarctica sp. nov. and Dioszegia cryoxerica sp. nov., psychrophilic basidiomycetous yeasts from polar desert soils in Antarctica. Int J Syst Evol Microbiol. 2010;60(Pt 6):1466–1472. [DOI] [PubMed] [Google Scholar]
- Dalluge JJ, Brown EC, Connell LB. Toward a rapid method for the study of biodiversity in cold environments: the characterization of psychrophilic yeasts by MALDI-TOF mass spectrometry. Extremophiles. 2019;23(4):461–466. [DOI] [PubMed] [Google Scholar]
- Delmont TO, Gaia M, Hinsinger DD, Frémont P, Vanni C, Fernandez-Guerra A, Eren AM, Kourlaiev A, d'Agata L, Clayssen Q, et al. Functional repertoire convergence of distantly related eukaryotic plankton lineages abundant in the sunlit ocean. Cell Genomics. 2022;2(5):100123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Delort AM, Vaïtilingom M, Joly M, Amato P, Wirgot N, Lallement A, Sancelme M, Matulova M, Deguillaume L. Clouds: a transient and stressing habitat for microorganisms. In: Chénard C, Lauro FM, editors. Microbial Ecology of Extreme Environments. Cham: Springer International Publishing; 2017. p. 215–245. [Google Scholar]
- Dieser M, Greenwood M, Foreman CM. Carotenoid pigmentation in Antarctic heterotrophic bacteria as a strategy to withstand environmental stresses. Arct Antarct Alp Res. 2010;42(4):396–405. [Google Scholar]
- Edgar RC. MUSCLE: multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Res. 2004;32(5):1792–1797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Edman JC. Isolation of telomerelike sequences from Cryptococcus neoformans and their use in high-efficiency transformation. Mol Cell Biol. 1992;12:7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferreira EMS, de Sousa FMP, Rosa LH, Pimenta RS. Taxonomy and richness of yeasts associated with angiosperms, bryophytes, and meltwater biofilms collected in the Antarctic peninsula. Extremophiles. 2019;23(1):151–159. [DOI] [PubMed] [Google Scholar]
- Flynn JM, Hubley R, Goubert C, Rosen J, Clark AG, Feschotte C, Smit AF. RepeatModeler2 for automated genomic discovery of transposable element families. Proc Natl Acad Sci USA. 2020;117(17):9451–9457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gácser A, Hamari Z, Pfeiffer I, Litter J, Kevei F, Kucsera J. Organization of mitochondrial DNA in the basidiomycetous Dioszegia hungarica (Cryptococcus hungaricus) species. FEMS Microbiol Lett. 2002;212(1):1–6. [DOI] [PubMed] [Google Scholar]
- Ganley ARD, Kobayashi T. Highly efficient concerted evolution in the ribosomal DNA repeats: total rDNA repeat variation revealed by whole-genome shotgun sequence data. Genome Res. 2007;17(2):184–191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De García V, Brizzio S, Libkind D, Buzzini P, Van Broock M. Biodiversity of cold-adapted yeasts from glacial meltwater rivers in Patagonia, Argentina: yeasts from Patagonian glacial waters. FEMS Microbiol Ecol. 2007;59(2):331–341. [DOI] [PubMed] [Google Scholar]
- Grigoriev IV, Nikitin R, Haridas S, Kuo A, Ohm R, Otillar R, Riley R, Salamov A, Zhao X, Korzeniewski F, et al. MycoCosm portal: gearing up for 1000 fungal genomes. Nucleic Acids Res. 2014;42(Database Issue):D699–D704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haridas S, Salamov A, Grigoriev IV. Fungal genome annotation. In: de Vries RP, Tsang A, Grigoriev IV, editors. Fungal Genomics. Methods in Molecular Biology. New York (NY: ): Springer; 2018. p. 171–184. [DOI] [PubMed] [Google Scholar]
- Hashim NHF, Bharudin I, Nguong DLS, Higa S, Bakar FDA, Nathan S, Rabu A, Kawahara H, Illias RM, Najimudin N, et al. Characterization of Afp1, an antifreeze protein from the psychrophilic yeast Glaciozyma antarctica PI12. Extremophiles. 2013;17(1):63–73. [DOI] [PubMed] [Google Scholar]
- Hassett MO, Fischer MWF, Money NP. Mushrooms as rainmakers: how spores act as nuclei for raindrops. PLoS One. 2015;10(10):e0140407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ianiri G, Abhyankar R, Kihara A, Idnurm A. Phs1 and the synthesis of very long chain fatty acids are required for ballistospore formation. PLoS One. 2014;9(8):e105147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Inacio J, Portugal L, Spencer-Martins I, Fonseca A. Phylloplane yeasts from Portugal: seven novel anamorphic species in the Tremellales lineage of the Hymenomycetes (Basidiomycota) producing orange-coloured colonies. FEMS Yeast Res. 2005;5(12):1167–1183. [DOI] [PubMed] [Google Scholar]
- Karlsson I, Friberg H, Kolseth A-K, Steinberg C, Persson P. Organic farming increases richness of fungal taxa in the wheat phyllosphere. Mol Ecol. 2017;26(13):3424–3436. [DOI] [PubMed] [Google Scholar]
- Kinkel LL. Microbial population dynamics on leaves. Annu Rev Phytopathol. 1997;35:327–347. [DOI] [PubMed] [Google Scholar]
- Klemetsen T, Raknes IA, Fu J, Agafonov A, Balasundaram SV, Tartari G, Robertsen E, Willassen NP. The MAR databases: development and implementation of databases specific for marine metagenomics. Nucleic Acids Res. 2018;46(D1):D692–D699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kolmogorov M, Yuan J, Lin Y, Pevzner PA. Assembly of long, error-prone reads using repeat graphs. Nat Biotechnol. 2019;37(5):540–546. [DOI] [PubMed] [Google Scholar]
- Kuo A, Bushnell B, Grigoriev IV. Fungal genomics: Sequencing and annotation. In: Martin FM, editor. Fungi. Advances in Botanical Research. Cambridge (UK): Elsevier Academic Press; 2014. p. 1–52. [Google Scholar]
- Langmead B, Salzberg SL. Fast gapped-read alignment with Bowtie 2. Nat Methods. 2012;9(4):357–359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Langmead B, Wilks C, Antonescu V, Charles R. Scaling read aligners to hundreds of threads on general-purpose processors. Bioinformatics. 2019;35(3):421–432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li A-H, Yuan F-X, Groenewald M, Bensch K, Yurkov AM, Li K, Han P-J, Guo L-D, Aime MC, Sampaio JP, et al. Diversity and phylogeny of basidiomycetous yeasts from plant leaves and soil: proposal of two new orders, three new families, eight new genera and one hundred and seven new species. Stud Mycol. 2020;96:17–140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu X-Z, Wang Q-M, Göker M, Groenewald M, Kachalkin AV, Lumbsch HT, Millanes AM, Wedin M, Yurkov AM, Boekhout T, et al. Towards an integrated phylogenetic classification of the Tremellomycetes. Stud Mycol. 2015;82:1–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lofgren LA, Uehling JK, Branco S, Bruns TD, Martin F, Kennedy PG. Genome‐based estimates of fungal rDNA copy number variation across phylogenetic scales and ecological lifestyles. Mol Ecol. 2019;28(4):721–730. [DOI] [PubMed] [Google Scholar]
- Loftus BJ, Fung E, Roncaglia P, Rowley D, Amedeo P, Bruno D, Vamathevan J, Miranda M, Anderson IJ, Fraser JA, et al. The genome of the basidiomycetous yeast and human pathogen Cryptococcus neoformans. Science. 2005;307(5713):1321–1324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Madhour A, Anke H, Mucci A, Davoli P, Weber RWS. Biosynthesis of the xanthophyll plectaniaxanthin as a stress response in the red yeast Dioszegia (Tremellales, Heterobasidiomycetes, Fungi). Phytochemistry. 2005;66(22):2617–2626. [DOI] [PubMed] [Google Scholar]
- Manni M, Berkeley MR, Seppey M, Simão FA, Zdobnov EM. BUSCO update: novel and streamlined workflows along with broader and deeper phylogenetic coverage for scoring of eukaryotic, prokaryotic, and viral genomes. Mol Biol Evol. 2021;38:4647–4654. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marchler-Bauer A, , BoY, , HanL, , HeJ, , LanczyckiCJ, , LuS, , ChitsazF, , DerbyshireMK, , GeerRC, , Gonzales NR, et al. CDD/SPARCLE: functional classification of proteins via subfamily domain architectures. Nucleic Acids Res. 2017;45(D1):D200–D203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marchler-Bauer A, , Bryant SH. CD-Search: protein domain annotations on the fly. Nucleic Acids Res. 2004;32(Web Server issue):W327–W331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martin M. Cutadapt removes adapter sequences from high-throughput sequencing reads. EMBnet J. 2011;17(1):10–12. [Google Scholar]
- Moliné M, Libkind D, del Carmen Diéguez M, van Broock M. Photoprotective role of carotenoids in yeasts: response to UV-B of pigmented and naturally-occurring albino strains. J Photochem Photobiol B. 2009;95(3):156–161. [DOI] [PubMed] [Google Scholar]
- Nelson JO, Watase GJ, Warsinger-Pepe N, Yamashita YM. Mechanisms of rDNA copy number maintenance. Trends Genet. 2019;35(10):734–742. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Phizicky EM, Hopper AK. tRNA biology charges to the front. Genes Dev. 2010;24(17):1832–1860. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Priyam A, Woodcroft BJ, Rai V, Moghul I, Munagala A, Ter F, Chowdhary H, Pieniak I, Maynard LJ, Gibbins MA, et al. Sequenceserver: a modern graphical user interface for custom BLAST databases. Mol Biol Evol. 2019;36(12):2922–2924. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reasoner DJ, Geldreich EE. A new medium for the enumeration and subculture of bacteria from potable water. Appl Environ Microbiol. 1985;49(1):1–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robinson JT, Thorvaldsdóttir H, Winckler W, Guttman M, Lander ES, Getz G, Mesirov JP. Integrative genomics viewer. Nat Biotechnol. 2011;29(1):24–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Salazar G, Paoli L, Alberti A, Huerta-Cepas J, Ruscheweyh H-J, Cuenca M, Field CM, Coelho LP, Cruaud C, Engelen S, et al. ; Tara Oceans Coordinators. Gene expression changes and community turnover differentially shape the global ocean metatranscriptome. Cell. 2019;179(5):1068–1083.e21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Šantl-Temkiv T, Amato P, Casamayor EO, Lee PKH, Pointing SB. Microbial ecology of the atmosphere. FEMS Microbiol Rev. 2022;46:fuac009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sapkota R, Jørgensen LN, Nicolaisen M. Spatiotemporal variation and networks in the mycobiome of the wheat canopy. Front Plant Sci. 2017;8:1357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmieder R, Edwards R. Quality control and preprocessing of metagenomic datasets. Bioinformatics. 2011;27(6):863–864. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stein AF, , DraxlerRR, , RolphGD, , StunderBJB, , CohenMD, , Ngan F. NOAA’s HYSPLIT atmospheric transport and dispersion modeling system. Bull Am Meteorol Soc. 2015;96(12):2059–2077. [Google Scholar]
- Takashima M, Deak T, Nakase T. Emendation of Dioszegia with redescription of Dioszegia hungarica and two new combinations, Dioszegia aurantiaca and Dioszegia crocea. J Gen Appl Microbiol. 2001;47(2):75–84. [DOI] [PubMed] [Google Scholar]
- Takashima M, Manabe R, Ohkuma M. Draft genome sequences of basidiomycetous epiphytic phylloplane yeast type strains Dioszegia crocea JCM 2961 and Dioszegia aurantiaca JCM 2956. Microbiol Resour Announc. 2019;8:e01727-18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tamura K, Stecher G, Kumar S. MEGA11: Molecular Evolutionary Genetics Analysis version 11. Mol Biol Evol. 2021;38:3022–3027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tan M, Wang Q. Characterization of the complete mitochondrial genome of Dioszegia changbaiensis (Tremellales: bulleribasidiaceae) with phylogenetic implications. Mitochondrial DNA B Resour. 2021;6(12):3315–3317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tavaré S. Some probabilistic and statistical problems in the analysis of DNA sequences. Lect Math Life Sci. 1986;17:57–86. [Google Scholar]
- Trochine A, Turchetti B, Vaz ABM, Brandao L, Rosa LH, Buzzini P, Rosa C, Libkind D. Description of Dioszegia patagonica sp. nov., a novel carotenogenic yeast isolated from cold environments. Int J Syst Evol Microbiol. 2017;67(11):4332–4339. [DOI] [PubMed] [Google Scholar]
- Vaïtilingom M, Amato P, Sancelme M, Laj P, Leriche M, Delort A-M. Contribution of microbial activity to carbon chemistry in clouds. Appl Environ Microbiol. 2010;76(1):23–29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vaïtilingom M, Attard E, Gaiani N, Sancelme M, Deguillaume L, Flossmann AI, Amato P, Delort A-M. Long-term features of cloud microbiology at the puy de Dôme (France). Atmos Environ. 2012;56:88–100. [Google Scholar]
- Vaïtilingom M, Deguillaume L, Vinatier V, Sancelme M, Amato P, Chaumerliac N, Delort A-M. Potential impact of microbial activity on the oxidant capacity and organic carbon budget in clouds. Proc Natl Acad Sci USA. 2013;110(2):559–564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vaz ABM, Rosa LH, Vieira MLA, de Garcia V, Brandão LR, Teixeira LCRS, Moliné M, Libkind D, van Broock M, Rosa CA, et al. The diversity, extracellular enzymatic activities and photoprotective compounds of yeasts isolated in Antarctica. Braz J Microbiol. 2011;42(3):937–947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Větrovský T, Morais D, Kohout P, Lepinay C, Algora C, Awokunle Hollá S, Bahnmann BD, Bílohnědá K, Brabcová V, D'Alò F, et al. GlobalFungi, a global database of fungal occurrences from high-throughput-sequencing metabarcoding studies. Sci Data. 2020;7(1):228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Villar E, Vannier T, Vernette C, Lescot M, Cuenca M, Alexandre A, Bachelerie P, Rosnet T, Pelletier E, Sunagawa S, et al. The Ocean Gene Atlas: exploring the biogeography of plankton genes online. Nucleic Acids Res. 2018;46(W1):W289–W295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Villarreal P, Carrasco M, Barahona S, Alcaíno J, Cifuentes V, Baeza M. Tolerance to ultraviolet radiation of psychrotolerant yeasts and analysis of their carotenoid, mycosporine, and ergosterol content. Curr Microbiol. 2016;72(1):94–101. [DOI] [PubMed] [Google Scholar]
- Walker BJ, Abeel T, Shea T, Priest M, Abouelliel A, Sakthikumar S, Cuomo CA, Zeng Q, Wortman J, Young SK, et al. Pilon: an integrated tool for comprehensive microbial variant detection and genome assembly improvement. PLoS One. 2014;9(11):e112963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wicker T, Sabot F, Hua-Van A, Bennetzen JL, Capy P, Chalhoub B, Flavell A, Leroy P, Morgante M, Panaud O, et al. A unified classification system for eukaryotic transposable elements. Nat Rev Genet. 2007;8(12):973–982. [DOI] [PubMed] [Google Scholar]
- Yang Z. Among-site rate variation and its impact on phylogenetic analyses. Trends Ecol Evol. 1996;11(9):367–372. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The Dioszegia hungarica PDD-24b-2 Whole Genome Shotgun project was deposited at DDBJ/ENA/GenBank under accession number JAKWFO000000000. The genome version used in this report is JAKWFO010000000. The raw Illumina and PacBio reads were deposited at the Sequence Read Archive under accessions numbers SRR18177991 and SRR18177990, respectively. Details on genome assembly and gene model properties are provided on the MycoCosm genome portal (https://mycocosm.jgi.doe.gov/Diohu1). Strain Dioszegia hungarica PDD-24b-2 is available upon request to Dr Pierre Amato or Dr Françoise Bringel. Representative sequences of putative D. hungarica PDD-24b-2 TE families are in Supplementary File 1. Putative TEs detected in D. hungarica PDD-24b-2 are in Supplementary Table 1. ITS sequences used to construct the phylogenetic tree of the Dioszegia genus are in Supplementary Table 2. Environmental samples in which D. hungarica and Dioszegia species were searched are provided in Supplementary Table 3. The air mass trajectory of the cloud from which D. hungarica strain PDD-24b-2 was isolated is shown in Supplementary Fig. 1. A close-up of the Illumina read depth coverage of D. hungarica PDD-24b-2 rRNA gene region is provided in Supplementary Fig. 2. The homolog search scripts, environmental metagenome dataset, as well as more information on biogeographic analyses and TE mining are available at https://github.com/JarrigeD/Dioszegia_hungarica_sequencing.
Supplemental material is available at G3 online.