

Abstract
Background
We previously report that yes-associated protein (YAP), the core downstream effector of Hippo pathway, promotes the malignant progression of glioblastoma (GBM). However, although classical regulatory mechanisms of YAP are well explored, how YAP is modulated by the Hippo-independent manner remains poorly understood. Meanwhile, the nonreceptor tyrosine kinase Fyn-related kinase (FRK), which exhibits low expression and possesses tumor suppressor effects in GBM, is reported to be involved in regulation of protein phosphorylation. Here, we examined whether FRK could impede tumor progression by modulating YAP activities.
Methods
Human GBM cells and intracranial GBM model were used to assess the effects of FRK and YAP on the malignant biological behaviors of GBM. Immunoblotting and immunohistochemistry were used to detect the expression of core proteins in GBM tissues. Co-immunoprecipitation, proximity ligation assay, luciferase assay and ubiquitination assay were utilized to determine the protein–protein interactions and related molecular mechanisms.
Results
The expression levels of FRK and YAP were inversely correlated with each other in glioma tissues. In addition, FRK promoted the ubiquitination and degradation of YAP, leading to tumor suppression in vitro and in vivo. Mechanistically, FRK interacted with and phosphorylated YAP on Tyr391/407/444, which recruited the classical E3 ubiquitin ligase Siah1 to catalyze ubiquitination and eventually degradation of YAP. Siah1 is required for YAP destabilization initiated by FRK.
Conclusions
We identify a novel mechanism by which FRK orchestrates tumor-suppression effect through phosphorylating YAP and inducing its ubiquitination by Siah1. FRK-Siah1-YAP signaling axis may serve as a potential therapeutic target for GBM treatment.
Keywords: GBM, FRK, YAP, Siah1, ubiquitination
Key Points.
YAP is a new substrate of FRK and Siah1 is a new E3 ubiquitin ligase of YAP.
Phosphorylation of YAP by FRK is required for recruiting E3 ubiquitin ligase Siah1.
FRK inhibits GBM progression via inducing YAP phosphorylation and degradation.
Importance of the Study.
In this study, we reported that FRK, a member of SRC family kinase, interacted with and phosphorylated YAP on Tyr391/407/444, which recruited the ubiquitin ligase Siah1 for binding to and destabilizing YAP, leading to tumor suppression in vitro and in vivo. In human glioma specimens, the level of FRK and Siah1 were downregulated and negatively correlated with that of YAP, indicating that YAP increase may be related to the decrease of FRK and Siah1. Our study identifies that YAP is a substrate of FRK and Siah1 is a new E3 ubiquitin ligase of YAP. FRK inhibits GBM progression via phosphorylating YAP and inducing its ubiquitylation by Siah1. FRK-Siah1-YAP signaling axis may serve as a potential therapeutic target for the treatment of GBM.
Glioblastoma (GBM) is the most common and aggressive human primary brain malignancy. Despite recent advancement in comprehensive treatments, the prognosis for GBM patients remains undesirable and the median survival is less than 15 months.1,2 Therefore, it is urgent to explore potential molecular mechanism and new therapy strategies against GBM.
The Hippo pathway is a conserved tumor suppressor signaling involved in regulating organ size and cell proliferation in drosophila and mammals.3,4 Dysregulation of Hippo pathway components is often associated with abnormal cell growth and tumorigenesis.5 The transcriptional co-activator Yes-associated protein (YAP) is a key downstream effector of Hippo pathway and negatively regulated by a kinase cascade, including mammalian sterile-20-like 1/2 (MST1/2) kinases and large tumor suppressor 1/2 (LATS1/2) kinases.3 YAP is finally phosphorylated at Ser127 and Ser381 by LATS1/2, which leads to its cytoplasmic retention by binding to 14-3-3 and subsequent proteasomal degradation by β-Trcp.6,7 YAP is frequently activated in various human cancers, including liver cancer, ovarian cancer, esophageal cancer, small cell lung cancer, prostate cancer, and melanoma.5 Moreover, we also report that YAP is up-regulated in gliomas and promotes GBM progression by inhibiting GSK3β and then activating β-catenin.8 Recently, we find that, by promoting the translation of fibroblast growth factor 2 and DNA repair, YAP is involved in radioresistance of gliomas.9 However, why YAP protein is up-regulated and activated in GBM remains elusive.
YAP is firstly identified by its ability to bind to the nonreceptor tyrosine kinase YES. Therefore, it is called Yes-associated protein. YAP phosphorylation by SRC/YES promotes YAP/Runx2 interaction that inhibits expression of osteocalcin in bone.10 Actually, several SRC nonreceptor tyrosine kinase family members, such as SRC, YES, LCK, have been reported to be able to phosphorylate and activate YAP directly. For example, LCK regulates YAP tyrosine phosphorylation and nuclear localization in cholangiocarcinoma cells independent of LATS activity.11 Fyn-related kinase (FRK), also known as protein tyrosine kinase 5 (PTK5) or Rak, belongs to the SRC family kinases.12 However, FRK differs from the other SRC family members in many structural features, including the presence of a putative bipartite nuclear localization signal and the lack of a consensus myristoylation motif.13 FRK inhibits cell proliferation and epidermal growth factor receptor (EGFR) signaling in a variety of cancer cells. FRK-mediated tyrosine phosphorylation of PTEN and BRCA1 is crucial for their protein stability and function.14,15 In fact, we have also previously reported that FRK is down-regulated in gliomas and FRK suppresses the proliferation of human GBM cells by inhibiting cyclin D1 nuclear accumulation.16–20 However, whether FRK can regulate YAP, like other SRC family kinases, remains little explored.
In this study, we identified that FRK inhibits GBM progression via phosphorylating YAP on Tyr391/407/444 and inducing its ubiquitination by seven in absentia homolog1 (Siah1), a highly conserved member of the RING family E3 ubiquitin ligases and involved in the regulation of multiple signal pathways in GBM.21–24 YAP is a new substrate of FRK and Siah1 is a new E3 ubiquitin ligase of YAP. Consistently, the protein levels of FRK and Siah1 are negatively associated with that of YAP in glioma tissues, indicating that YAP increase may be related to the decrease of FRK and Siah1. Our study identifies that the novel FRK-Siah1-YAP signaling axis may be a potential molecular therapeutic target for GBM.
Materials and Methods
Glioma Specimens
Human glioma specimens were obtained from the Affiliated Hospital of Xuzhou Medical University and the patients were followed up under a protocol approved by the Institutional Review Board. All tissue samples were collected in compliance with informed consent policy. Fifty paraffin embedded samples were constructed to tissue microarray (TMA) as described in our previous study9 and the detailed information was described in Supplemental Table S1.
Cell Culture
Human U251 cell and human embryonic kidney 293T (HEK293T) cell were obtained from the Cell Bank of China Science Academy (Shanghai, China).The U87 cell was purchased from American Type Culture Collection. Low passage primary GBM cells (GBM 1120D and TBD0220) were kindly donated by Prof. Chunsheng Kang in Department of Neurosurgery, Tianjin Medical University General Hospital.25 U251, U87, and HEK293T cell lines were maintained in DMEM (Gibco) supplemented with 10% fetal bovine serum (Gibco) at 37°C, in 5% CO2 humid atmosphere. GBM 1120D was maintained in DMEM (Gibco) supplemented with 15% fetal bovine serum (Gibco). TBD0220 was maintained in DMEM/F-12 (Corning) supplemented with 10% fetal bovine serum (Gibco).
Antibodies and Reagents
Primary antibodies used were as follows: Anti-HA (#3724), anti-p-YAPSer127 (#13008), anti-LATS1 (#3477), anti-p-LATS1Ser909 (#9157), and anti-β-Trcp (#4394) were from Cell Signaling Technology. Anti-YAP (WH0010413M1), anti-Flag (F1804), anti-PCNA (05-347), and anti-Siah1 (AV38212) were from Sigma. Anti-p-YAPTyr357 (ab62751), anti-YAP (ab52771), and anti-SKP1 (ab76502) were from abcam. Anti-Phosphotyrosine (#05-321) was from Millipore. Anti-FRK (sc-166478), anti-CYR61 (sc-374129), and anti-CTGF (sc-101586) were from Santa Cruz Biotechnology. Anti-LATS2 (A16249), anti-p-LATS1T1079/LATS2T1041 (AP0912), anti-β-actin (AC026), and anti-Siah1 (A2494) were from ABclonal. Cycloheximide (CHX) and MG132 were from TargetMol. Puromycin, Protein A/G agarose and verteporfin (VP) were from MedChemExpress. PolyJet was from Signagen. Polybrene was from Sigma.
Plasmids and Stable Cell Line Generation
The Flag-FRK was kindly gifted by Dr. Shiaw-Yih Lin in MD Anderson Cancer Center.14 The lentivirus-based FRK construct for in vivo experiment was generated in our previous study.16 The pCDH-CMV-EF1-YAP was kindly gifted by Prof. Hongbin Ji in the Institute of Biochemistry and Cell Biology, Shanghai Institutes for Biological Sciences.26 The Flag tagged YAPWT, YAPY188F, YAPY247/8F, YAPY391F, YAPY407F, and YAPY444F were kindly donated by Dr. Jianmin Zhang in Roswell Park Cancer Institute.27 The Siah1WT and Siah1C44S were provided by Dr. Hengliang Shi in Xuzhou Medical University. The shSiah1s were constructed by Shanghai Genechem Company and the sequences (5′–3′) were as follows:
shSiah1#1: 5′-CTGATAGGAACACGCAAGCAA-3′
shSiah1#2: 5′-CCCTGTAAATATGCGTCTTCT-3′
shSiah1#3: 5′-ATGGCAATTTAGGCATCAATG-3′
Plasmids transfection and stable cell line generation were performed according to the manufacturer’s instruction and our previous study.9
Immunoprecipitation and Immunoblotting
Immunoprecipitation (IP) and immunoblotting analyses were performed as we previously described.28 In brief, cells were lysed in mild IP lysis buffer (Beyotime) with 1% cocktail protease inhibitors (CWBIO) and centrifuged at 12,000 rpm for 15 min at 4°C. Then, the supernatants were incubated with 1 μg of indicated antibodies overnight at 4°C and followed by addition of 40 ml of protein A/G magnetic beads (MedChemExpress) for an additional 3 h. The precipitates were washed four times with mild lysis buffer, resuspended in 1* Loading buffer, and resolved by SDS-PAGE followed by immunoblotting analysis.
Immunohistochemistry and Immunofluorescence
Immunohistochemistry and immunofluorescence were performed according to our previous study.28 To quantify FRK, YAP, Siah1, and PCNA expression, we measured the immunostaining scores of FRK, YAP, and PCNA according to the percentage of positive staining cells and the staining intensity in glioma tissues.
Cell Growth and Death Assays
The cell growth was estimated by EdU incorporation assay (RiboBio) and colony formation assay according to the manufacturer’s instruction and our pervious study.9 The cell death was estimated by Calcein-AM·PI staining assay (KeyGEN BioTECH) according to the manufacturer’s instruction.
Cell Luciferase Assay
The 8xGTIIC luciferase reporter (Addgene) which harbors TEAD-binding sites was utilized to indicate the activation of YAP. Luciferase reporter assay was performed following the manufacturer’s instructions (Promega E1910). After transfection with plasmids for 24 h, the corresponding luminescence value was obtained and a construct containing Renilla luciferase was used as internal control. The ratio of firefly/renilla was normalized to that of the empty vector controls.
Proximity Ligation Assay
Proximity ligation assay (PLA) was performed by using a commercial kit (Duolink PLA) according to the manufacturer’s instructions. Briefly, after being washed three times with cold dulbecco’s phosphate buffered saline (DPBS), the cells were fixed with 4% paraformaldehyde for 15 min, blocked with 3% BSA for 1 h to reduce background binding, and incubated with primary antibodies of FRK and YAP for 12 h at 4°C. Thereafter, the cells were incubated with probes for 30 min at 37°C. Then, the cells were washed three times with cold DPBS, mixed with backbone, connector, DNA ligase, and buffer in 100 μL of reaction solution for 1 h. Subsequently, the cells were incubated with DNA polymerase, dNTP, and buffer for 90 min in 37°C. Later, the cells were washed three times with cold DPBS, and the cell nuclei were stained with 4′,6-diamidino-2-phenylindole, dihydrochloride (DAPI) for 5 min. Finally, the fluorescence was observed under a laser confocal microscope.
Ubiquitination Assay
Cells were co-transfected with the indicated plasmids and HA-ubiquitin for 48 h and treated with the proteasome inhibitor MG132 (20 µM) for 6 h. Cell lysates were immunoprecipitated using the indicated antibodies and analyzed by immunoblotting using an anti-HA antibody.
Quantitative PCR and Reverse-Transcription PCR
After RNA isolation, cDNA was synthesized using the FastQuant Kit (TIANGEN) according to the manufacturer’s instructions.The cDNA products were used for reverse-transcription PCR or quantitative PCR analysis using SuperReal PreMix Plus (TIANGEN). The PCR primers were as follows:
YAP Forward 5′-CACAGCTCAGCATCTTCGAC-3′
Reverse 5′-TATTCTGCTGCACTGGTGGA-3′
FRK Forward 5′-ACCGCAACTCCATACAGC-3′
Reverse 5′-TTCCGAGACTCCAGATAGGC-3′
Siah1 Forward 5′-CTGGTGCTGTTGACTGGGTGATG-3′
Reverse 5′-TACGATTGCGAAGAACTGCTGGTG-3′
β-actin Forward 5′-CCAACCGCGAGAAGATGA-3′
Reverse 5′-CCAGAGGCGTACAGGGATAG-3′
GBM Intracranial Mouse Model
All experimental protocols were carried out according to the guidelines of Xuzhou Medical University for animal research and were approved by Institutional Animal Use Committee. Five-week-old male nude mice, purchased from the GemPharmatech Co., Ltd., were used to establish intracranial GBM xenografts. A total of 6 × 105 U87 cells with luciferase were injected into the right striatum of each mouse. Bioluminescence imaging was taken on day 9, 16, 23, and 30 after implantation to monitor intracranial tumor growth. Last, the mice were sacrificed when they exhibited hemiplegia, listlessness, cachexia, and other neurological symptoms. Kaplan–Meier survival curves were plotted to show the survival time.
Public Available Clinical Data Analysis
The clinical data of FRK, Siah1, YAP, and target genes (CTGF and CYR61) were downloaded from the Cancer Genome Atlas (TCGA, https://portal.gdc.cancer.gov) and Chinese glioma Genome Atlas (CGGA, http://www.cgga.org.cn/).
Statistical Analysis
All of the experiments were repeated independently at least three times. The data were presented as mean ± SD, as indicated in each figure caption. The P values were determined using two-sided unpaired t-test or one- or two-way analysis of variance (ANOVA) with GraphPad Prism 9 software. Survival analysis was conducted using the Kaplan–Meier model with a two-sided log-rank test. The values were considered statistically significant at P < 0.05.
Results
The Expression Level of FRK is Negatively Correlated with that of YAP in Glioma Samples
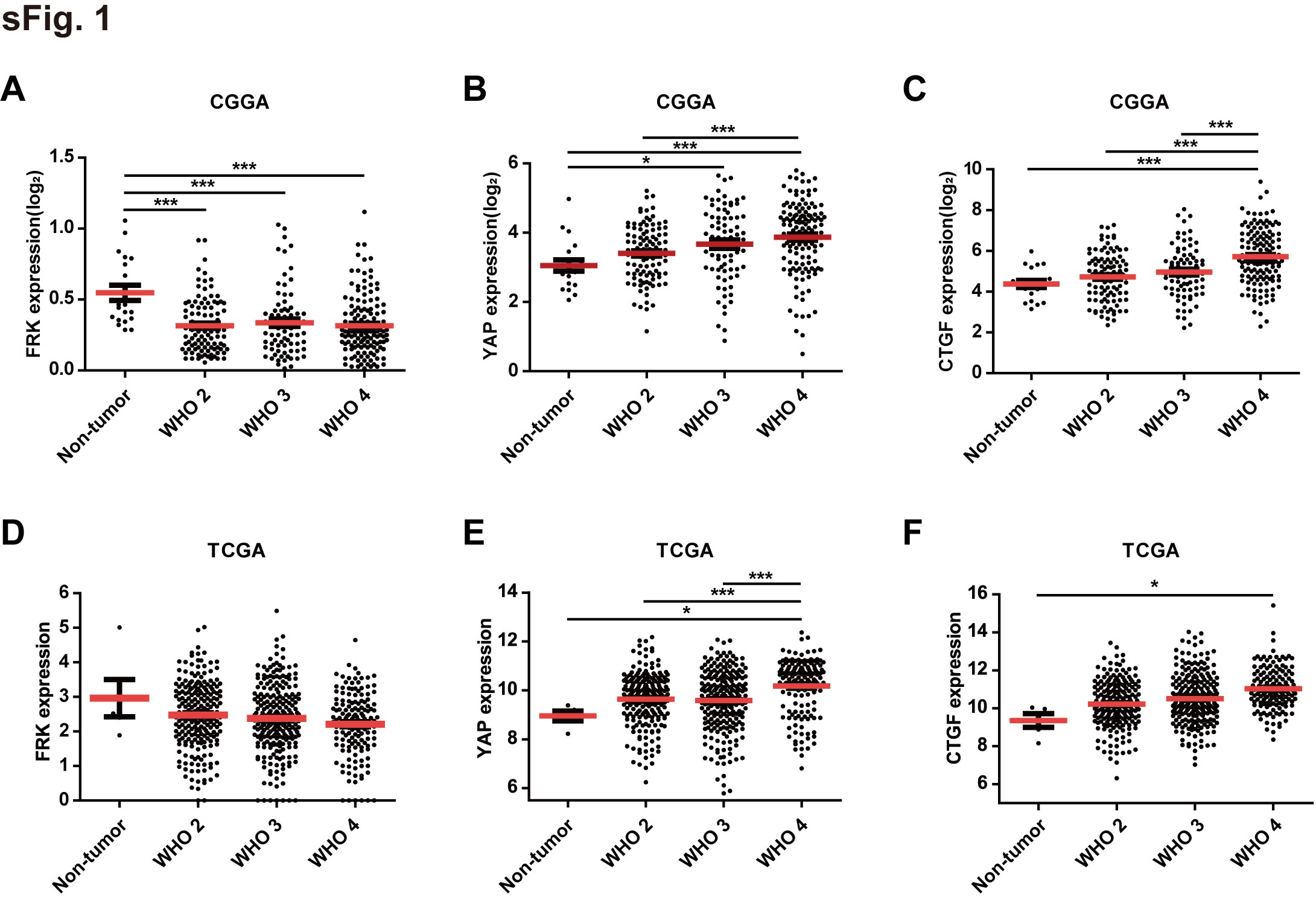
To determine the potential clinical relevance of FRK and YAP, we detected the expression of FRK and YAP in 5 nontumor brain tissues and 45 human glioma specimens using immunohistochemistry staining (Figure 1A–E). As a result, the level of FRK in the high-grade gliomas was significantly lower than that in the low-grade gliomas and nontumor brain tissues, while YAP exhibited opposite tendency (Figure 1A–D), in line with outcomes of TCGA and CGGA analyzing (Supplemental Figure 1). Furthermore, the level of FRK and YAP exhibited negative correlation (Figure 1E, r = −0.8718, P < 0.001, n = 50). Notably, the subcellular localization of FRK and YAP showed exclusive distribution in the same region. As shown in Figure 1A, where the FRK level was high, the YAP level was low, and vice versa. In addition, according to our clinical follow-up data of high-grade glioma samples (n = 25), patients with high expression of FRK had longer survival time than those with low expression (Figure 1F). In contrast, patients with high expression of YAP had significantly shorter survival time than those with low YAP (Figure 1G). Most interestingly, patients with high YAP and low FRK showed poorer prognosis (Figure 1H).
Fig. 1.

The expression level of FRK is negatively correlated with that of YAP in gliomas. (A&B) Representative images of immunohistochemical staining (A) and positive cell percentage (B) of FRK and YAP in nontumor brain tissues (n = 5) and human glioma specimens (n = 45). Scale bar: 20 μm. (C&D) Immunohistochemical scores (+: 0–15%; ++: 15%–30%; +++: 30%–65%; ++++: >65%) of FRK (C) and YAP (D) in gliomas with different grades. (E) The positive staining percentage of FRK and YAP exhibited negative correlation among different specimens (n = 50, r = −0.8718, P < 0.0001). (F–H) Kaplan–Meier survival analysis of high-grade glioma patients expressing indicated proteins (n = 25). The cut-off to define high or low expression was the median value. #, *P < 0.05, ##, **P < 0.01, ###, ***P < 0.001.
FRK Promotes YAP Ubiquitination and Degradation
Previous studies reported that YAP has been regulated by SRC family kinases (SFKs), such as YES,29 SRC,10,30,31 and LCK.11 In this context, we therefore asked whether FRK, a member of SFKs, could regulate YAP. First, we co-expressed Flag-FRK and YAP in HEK293T cells and found that the protein abundance of YAP and p-YAPS127 decreased after FRK over-expression (Supplemental Figure 2A, 2B). Similarly, the protein level of YAP also decreased after FRK over-expression in low-passage human primary GBM cells (GBM 1120D, Figure 2A). Next, we generated GFP-FRK over-expression U251 and U87 GBM cells via lentivirus system (Supplemental Figure 2C, 2D) and found that exogenous FRK decreased endogenous YAP protein level (Figure 2B, 2C), but not its mRNA level (Supplemental Figure 2E). Interestingly, FRK upregulated LATS1 protein level without affecting the phosphorylation either on LATS1Ser909 and LATS1T1079 or on LATS2T1041 (Figure 2B, 2C), indicating that the regulation of FRK on YAP was independent of Hippo pathway.
Fig. 2.

FRK promotes YAP ubiquitination and degradation. (A) Lysates of low passage primary GBM cell (GBM 1120D) transfected with Flag-YAP with or without FRK were examined by immunoblotting. (B&C) Representative immunoblots (B) and statistical results (C) of indicated proteins in FRK over-expression U251 and U87 GBM cells. (D-F) Lysates of FRK over-expression U251 and U87 GBM cells treated with 10 µg/ml cycloheximide (CHX) for the indicated times were examined by immunoblotting (D) and YAP levels were quantified (E, F). (G&H) Lysates of FRK over-expression U251 and U87 GBM cells treated with MG132 (20 μmol/L) or CQ (20 μmol/L) were immunoblotted as indicated antibodies. (I) HEK293T and U251 cells, as well as GBM 1120D cells were transfected with the indicated plasmids and treated with MG132. Cell lysates were immunoprecipitated using anti-YAP antibody and subjected to immunoblotting. *P < 0.05, **P < 0.01, ***P < 0.001.
To determine whether the protein stability of YAP is controlled by FRK, we blocked de novo protein synthesis using cycloheximide (CHX) and found that the half-life of YAP was much shorter in FRK over-expression cells (Figure 2D–F). Furthermore, the proteasomal inhibitor MG132 completely reversed YAP destabilization (Figure 2G), whereas the lysosomal inhibitor chloroquine (CQ) had no such effect (Figure 2H). As expected, over-expression of FRK greatly promoted YAP ubiquitylation in the presence of MG132 in HEK293T and U251, as well as in GBM 1120D cells (Figure 2I). Taken together, these results validated that FRK promotes the ubiquitylation and degradation of YAP in GBM.
Over-expression of YAP Partially Abolishes the Inhibition Effect of FRK on GBM Progression In Vitro and In Vivo
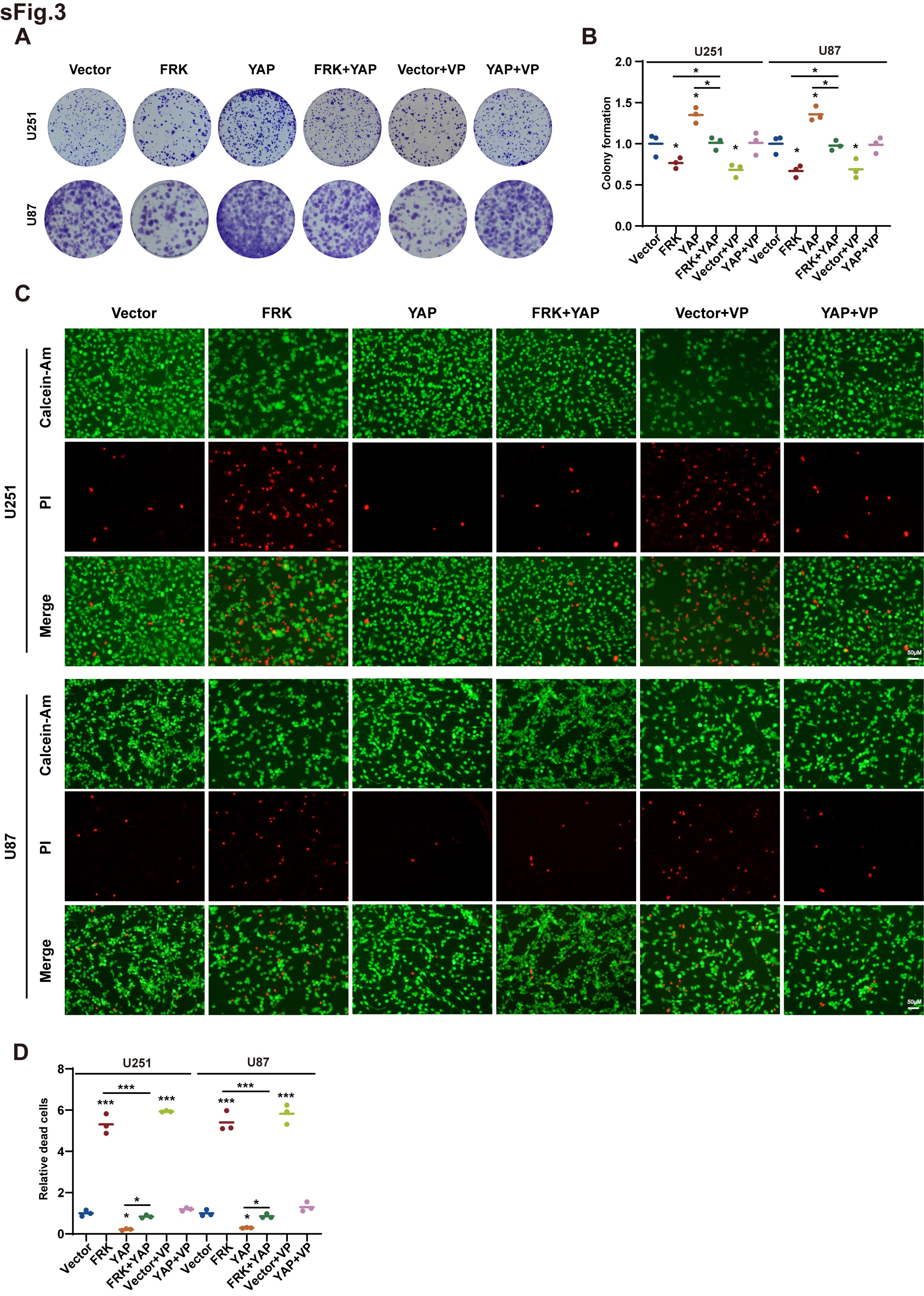
To investigate the biological relevance of FRK-induced degradation of YAP, we first assessed the properties of FRK in regulation of cell proliferation. As is shown in Figure 3A,3B and Supplemental Figure 3A, 3B, FRK decreased cell proliferation and colony-formation ability, which were attenuated by over-expression of YAP. In addition, cell growth inhibition after over-expression of FRK was similar to that of the commonly used inhibitor verteporfin (VP) administration, and the promotion effect of YAP on cell growth was inhibited by VP, indicating that YAP is a main player of tumor growth (Figure 3A,3B and Supplemental Figure 3A, 3B). On the contrary, cell death assayed by Calcein-AM·PI staining showed the opposite trend (Supplemental Figure 3C,3D).
Fig. 3.

Over-expression of YAP partially abolishes the inhibition effect of FRK on GBM progression in vitro and in vivo. (A&B) Cell proliferation was assessed using EdU assay in GBM cells stably over-expressing FRK or/and YAP with or without VP (5 μg/mL) treatment. Representative images were shown (A) and EdU-positive cells were quantified (B). Scale bar: 100 µm. (C) Schematic illustration of the in vivo experimental workflow. (D&E) Representative bioluminescence images (D) and the photon flux was analyzed quantitatively (E). P value was calculated on day 30. (F&G) Representative HE staining images of consecutive brain sections (F, n = 3 per group) and the quantified results (G). (H) Kaplan–Meier curve showed the overall survival time of GBM-bearing mice of each group (n = 7 per group). (I&J) Representative immunostaining images of PCNA in the GBM xenografts (I, n = 3 per group) and the quantified results were shown (J). Scale bar: 50 µm. (K) Representative immunoblots of the extracts from tumors derived from each group probed with indicated antibodies. *P < 0.05, **P < 0.01, ***P < 0.001.
Motivated by the above results, we investigated whether YAP acts as a critical factor in the inhibition effect of FRK on GBM progression in vivo. Orthotopic GBM mouse models were constructed by intracranially injecting the luciferase labeled U87 cells and tumor growth was monitored every 7 days using bioluminescence values (Figure 3C). As is shown in Figure 3D–G, examined by bioluminescence imaging analysis and HE staining, we found that FRK over-expression inhibited tumor growth, while YAP over-expression promoted it. Notably, the inhibitory effect of FRK on GBM growth could be largely blocked by YAP over-expression (Figure 3D–G). In addition, the median survival time of mice injected with FRK over-expression cells prolonged, which could be reversed by the simultaneously over-expressed YAP (Figure 3H). Consistently, the intensity of PCNA-positive proliferative cells in FRK-over-expression tumors decreased, whereas it was attenuated by concomitantly over-expressed YAP (Figure 3I,3J). Furthermore, the protein levels of FRK, YAP, and LATS1/2 in the xenograft tissues were similar to those in GBM cells (Figure 3K), suggesting that the regulation relationship between these proteins also existed in the xenograft. Collectively, these results suggest that FRK-induced GBM growth inhibition is partially abolished by over-expression of YAP.
Phosphorylation of YAP on Tyr 391/407/444 by FRK is Required for its Ubiquitylation and Degradation
We then asked whether FRK modulates YAP by interacting with it. The forward or reverse Co-IP assay showed that exogenous FRK interacted with endogenous YAP (Figure 4A,4B). Besides, FRK and YAP co-localized in the perinuclear and nuclear region (Supplemental Figure 4A). Interestingly, where the expression of FRK is higher, the expression of YAP is lower, consistent with the results of our tissue immunohistochemistry (Figure 1A). In addition, to further elucidate the interaction between endogenous FRK and YAP, we detected the positive signals via PLA both in GBM cells (U251, U87) and in low-passage primary GBM cells (GBM 1120D, TBD0220) (Figure 4C). The results strongly showed that endogenous FRK and YAP generally interacted with each other. We found that the positive signals were distributed both in cytosol and in nuclear compartments, which is consistent with the results of our immunofluorescence experiment (Supplemental Figure 4A). Because the results of immunofluorescence and PLA showed that FRK and YAP interacted with each other both in cytosol and in nucleus, we therefore performed the ubiquitylation assay again by using cytosol and nuclear fraction, respectively. We found that FRK could promote the ubiquitylation of YAP both in cytosol and in nucleus (Figure 4D).
Fig. 4.

Phosphorylation of YAP on Tyr391/407/444 by FRK is required for its ubiquitination and degradation. (A&B) Reciprocal Co-IP of Flag-FRK with endogenous YAP. U251 cell extracts were immunoprecipitated using antibodies against YAP and FRK, and subjected to immunoblotting analysis. IgG was used as the isotype control. (C) Duolink PLA was performed with anti-FRK and anti-YAP primary antibodies in GBM cells (U251, U87) and low-passage primary GBM cells (GBM 1120D, TBD 0220). The red signal showed the interaction of FRK and YAP. (D) HEK293T cells were transfected with the indicated plasmids and treated with MG132. The cytoplasm and nuclear fraction were immunoprecipitated using anti-YAP antibody and subjected to immunoblotting using anti-HA antibody respectively. (E&F) Effect of FRK over-expression on YAP tyrosine phosphorylation by immunoblotting using p-Tyr in U251 cells. (G) Amino acid sequence conservation in different species of the six tyrosine residues in YAP. (H) Effects of YAPY188F, YAPY247/8F, YAPY391F, YAPY407F, and YAPY444F on YAP tyrosine phosphorylation in HEK293T cells co-transfected with Flag-FRK. (I) Cell lysates were analyzed by immunoblotting using p-YAPY407 in FRK-over-expression U251 cell. (J) Protein levels of YAPWT, YAPY188F, YAPY247/8F, YAPY391F, YAPY407F, and YAPY444F mutant in HEK293T cells co-transfected with Flag-FRK. (K) HEK293T cells were transfected with HA-Ub, Flag-FRK and Flag-YAPWT or YAPY391F, YAPY407F, and YAPY444F mutant, and then treated with 20 µM MG132. Cell lysates were immunoprecipitated using anti-YAP antibody and analyzed by immunoblotting using anti-HA antibody.
Next, we sought to investigate whether YAP is a substrate of FRK kinase. Using a specific phosphotyrosine antibody (p-Tyr), we found that over-expressing FRK promoted YAP tyrosine phosphorylation (Figure 4E,4F). The sequences of short and long isoforms of YAP1 (YAP1-1β and YAP1-2γ) harbor six conserved tyrosine sites (Figure 4G). To identify the FRK-dependent phosphorylation site(s) in YAP, we used Flag-tagged Y-to-F YAP mutants, which could not be phosphorylated, of each potential site (YAPY188F, YAPY247/8F, YAPY391F, YAPY407F, or YAPY444F) to analyze their tyrosine phosphorylation in HEK293T cells overexpressing FRK. We found that FRK-mediated tyrosine phosphorylation of YAP decreased significantly after YAPY391F, YAPY407F, or YAPY444F transfection (Figure 4H). Moreover, using a specific antibody against p-YAPY407, we detected the increase of p-YAPY407 level in HEK293T cells transfected with FRK and wild-type YAP (YAPWT) (Figure 4I, Supplemental Figure 4B).
To analyze the potential role of Y391/407/444 phosphorylation in the regulation of YAP protein degradation by FRK, we examined the protein level of YAP mutants after FRK over-expression. As expected, the level of YAPWT, YAPY188F and YAPY247/8F mutants decreased after FRK over-expression, but those of YAPY391F, YAPY407F and YAPY444F mutants did not (Figure 4J). Moreover, as shown in Figure 4K, YAPY391F, YAPY407F, and YAPY444F mutants rendered YAP less responsive to FRK-mediated ubiquitylation. Taken together, our data indicate that FRK may interact with and phosphorylates YAP at Y391/407/444, which is critical for ubiquitylation and degradation of YAP.
Siah1 is an E3 Ubiquitin Ligase of YAP
As Tyr391/407/444 phosphorylation by FRK promoted YAP ubiquitylation, we therefore wondered which is the E3 ubiquitin ligase for YAP in cooperation with FRK. We tried to identify this de novo E3 ubiquitin ligase from YAP binding proteins. According to the literature, three Labs32–34 used YAP as baits to identify interacting proteins by affinity purification and mass spectrometry. All three interactomes revealed 21 shared proteins, most of which are known core members of the Hippo pathway and only the E3-ubiquitin-ligase-associated CACYBP (SIP1) and SKP1 are novel interactors35 (Figure 5A). We notified that both CACYBP (SIP1) and SKP1 are the core of Siah1 E3 ubiquitin ligase complex.36 Cullin-1-like adaptor protein SIP1 provides a physical link between the RING domain of Siah1 and the substrate engaged by the SKP1/Ebi F-Box protein.36 Structural-functional analysis of various Siah1 substrates has identified a consensus Siah1-binding motif (VxP) among Siah1 substrates.37 We closely examined the protein sequence of YAP and revealed a potential Siah1 substrate-binding motif (VxP), which is highly conserved in YAP proteins from different species (Figure 5B). Thus, we performed Co-IP assay and revealed that either known-Siah1 binding proteins (β-Trcp and SKP1) or Siah1 could be readily detected in YAP immunoprecipitates in GBM cells (Figure 5C,5D). Consistently, immunofluorescence assay showed that Siah1 and YAP co-localized in U87 cells (Figure 5E).
Fig. 5.

Siah1 is an E3 ubiquitin ligase of YAP. (A) Venn diagram indicating the shared YAP interacting proteins reported with high confidence in the three proteomic studies. (B) Comparison of putative Siah1 binding motif in YAP with the Siah1-binding consensus motif defined in the known Siah1 substrates. (C&D) U251 cell extracts were immunoprecipitated using antibodies against YAP or Flag, and subjected to immunoblotting analysis. IgG was used as the isotype control. (E) Representative images of co-localization of Siah1 with YAP in U87 cells. Scale bar: 5 μm. (F) HEK293T cells were transfected with Siah1 WT or C44S mutant and YAP levels were assessed by immunoblotting. (G) qRT-PCR examined the effects of Siah1 WT or C44S mutant on YAP mRNA levels in U251 cells. (H) U251 cells were transfected with Siah1 WT or C44S mutant and treated with or without 20 µM MG132. Cell lysates were analyzed by immunoblotting. (I) HEK293T cells were transiently transfected with indicated plasmids along with the luciferase reporter. Luciferase activity was detected at 24 h after transfection. (J) HEK293T and U251 cells were transfected with the indicated plasmids and treated with MG132. Cell lysates were immunoprecipitated by anti-YAP antibody and analyzed by immunoblotting. ***P < 0.001, ns: no significance.
To investigate whether Siah1 was involved in the degradation of YAP, we overexpressed wide-type (WT) or RING domain mutant (C44S) of Siah1, which is the ubiquitin-ligase-activity-dead mutant, in U251 cells. We found that YAP protein levels decreased after over-expression of Siah1WT, but not Siah1C44S mutant, suggesting that Siah1- modulated YAP depends on its ubiquitin ligase activity (Figure 5F). However, both WT and C44S mutant of Siah1 could not alter YAP mRNA levels (Figure 5G).
In addition, we also found that Siah1WT over-expression markedly decreased the expression levels of YAP, as well as CTGF and CYR61, two known target genes of YAP. By contrast, MG132 treatment abated the reduction of YAP, CTGF and CYR61 levels, which is caused by Siah1WT over-expression (Figure 5H). Consistently, we found that Siah1WT, but not Siah1C44S mutant, inhibited the transcriptional activity of YAP via cell luciferase assay (Figure 5I). Taken together, these findings confirmed that Siah1 modulates YAP protein levels and its downstream activity in a proteasome-dependent manner.
To investigate whether Siah1 actually catalyzes the ubiquitination of YAP, we co-expressed HA-ubiquitin with either WT or the C44S mutant of Siah1 in HEK293 and U251 cells. After immunoprecipitating YAP, we observed that Siah1WT significantly promoted YAP ubiquitination, while the Siah1C44S mutant could not (Figure 5J). Together, these results demonstrate that Siah1 is a bona fide E3 ubiquitin ligase targeting YAP protein for ubiquitination.
Siah1 Expression is Negatively Correlated with YAP in Glioma Samples
To further determine the potential clinical relevance of our findings, we analyzed the TCGA and CGGA database and observed that Siah1 shows opposite prognostic trend with YAP in glioma patients (Supplemental Figure 5A-E), consistent with the IHC results of our glioma samples (Figure 1). Furthermore, an apparent negative correlation was detected between the level of Siah1 and that of YAP target genes (CTGF and CYR61) in glioma patients (Supplemental Figure 5F, 5G).
Next, we examined the expression of Siah1 and YAP proteins in serial sections of 5 nontumor brain tissues and 45 human glioma specimens and found that expression levels of Siah1 were negatively correlated with those of YAP (Supplemental Figure 5H, 5I, r = −0.4501, P = 0.0010). In addition, according to the data of high-grade glioma patients with follow-up information (n = 25), we found that patients with low Siah1 showed poorer prognosis (Supplemental Figure 5J). Importantly, patients with Siah1 low and YAP high have poorer prognosis (Supplemental Figure 5K), similar to that of FRK low and YAP high patients (Figure 1H).
Siah1 is Required for FRK-induced YAP Ubiquitination and Degradation
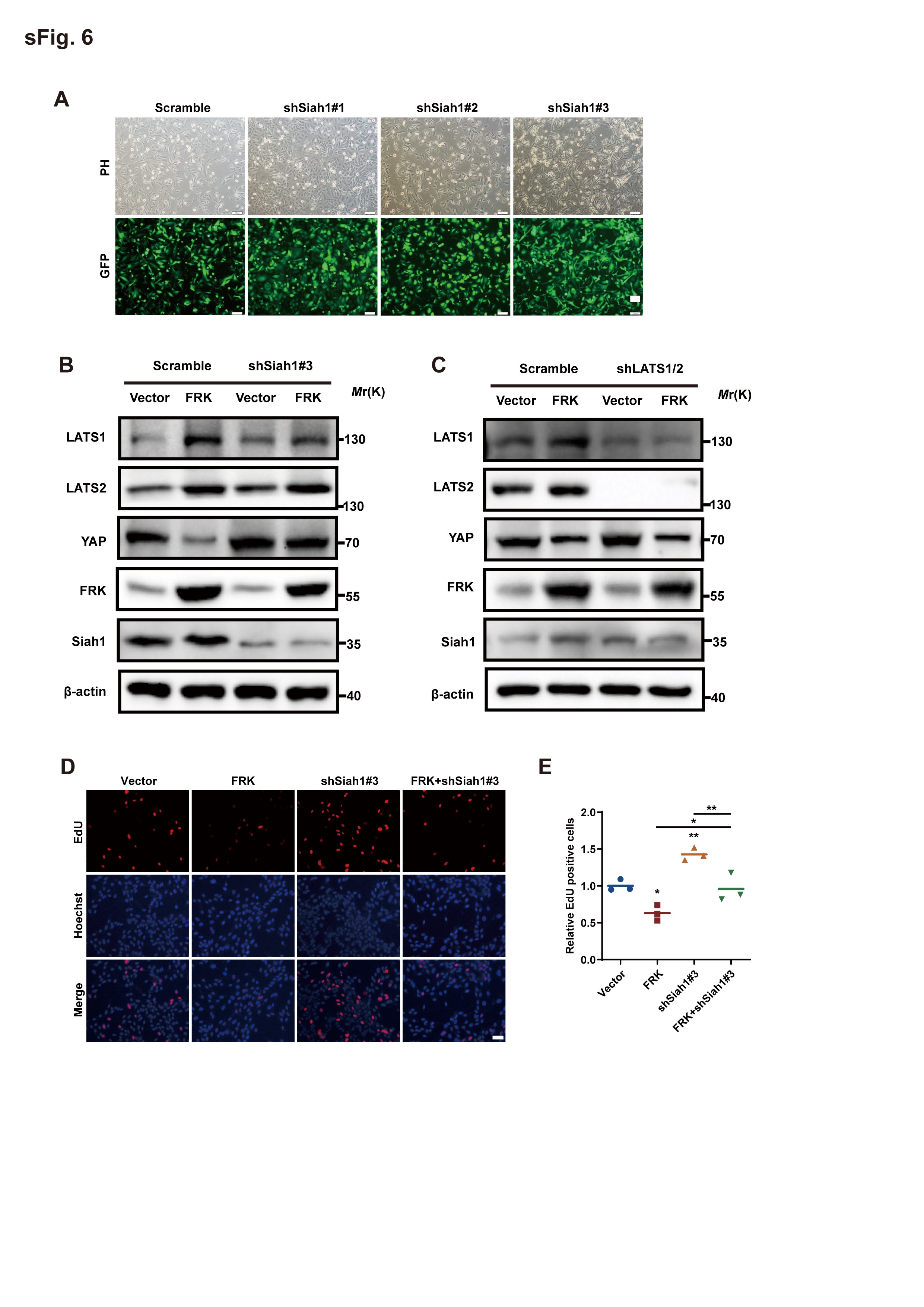
To further determine whether the inhibitory effect of FRK on YAP is mediated by Siah1, we expressed Siah1 shRNA in GBM cells (Supplemental Figure 6A) and found that YAP protein levels increased significantly in shSiah1#3 cells which exhibited better downregulation efficiency than other two shSiah1 (Figure 6A). Consistently, down-regulation of Siah1 dramatically decreased YAP poly-ubiquitylation (Figure 6B).
Fig. 6.

Siah1 is required for FRK-induced YAP ubiquitination and protein degradation. (A) GBM cells were transfected with three independent shRNAs targeting Siah1, and levels of Siah1 and YAP were detected by immunoblotting. (B) HEK293T and U251 cells were transfected with the indicated plasmids and treated with MG132. Cell lysates were immunoprecipitated using an anti-YAP antibody and subjected to immunoblotting. (C) U251 cells expressing Siah1 shRNA#3 (shSiah1#3) and exogenous FRK, and YAP levels were assessed by immunoblotting. (D) HEK293T cells were transfected with HA-Ub, Flag-FRK and shSiah1#3, and then treated with 20 µM MG132. Cell lysates were immunoprecipitated using an anti-YAP antibody. (E) HEK293T cells were transfected with HA-YAP, Flag-Siah1 and upgrade volume of Flag-FRK. Cell lysates were immunoprecipitated using an antibody against YAP. (F) HEK293T cells were transfected with Flag-FRK and YAPWT or YAP mutants. Cell lysates were immunoprecipitated using an antibody against YAP. (G) Working model: In physiological conditions, FRK phosphorylates YAP at Y391/407/444 and enhances its binding to Siah1, leading to YAP protein degradation both in cytoplasm and in nucleus, which in turn inhibits GBM progression. Conversely, the expression of FRK decreased in GBM, leading to the opposite effect. * P < 0.05, ** P < 0.01.
Intriguingly, Siah1 down-regulation also partially abolished FRK-induced YAP degradation (Figure 6C), but did not affect LATS1/2 level (Supplemental Figure 6B). In addition, knockdown of LATS1/2 did not affect the modulation of YAP via FRK (Supplemental Figure 6C). The above results further indicated that the modulation of YAP via FRK and Siah1 is independent of Hippo signal pathway. Consistently, down-regulation of Siah1 inhibited FRK-induced YAP ubiquitylation (Figure 6D), indicating that Siah1 is required for FRK-induced YAP ubiquitination and degradation.
In support of a role for Siah1 in FRK-induced YAP poly-ubiquitination, we deduced that FRK altered the binding of YAP with Siah1. To validate our hypothesis, we performed Co-IP experiments and found that the binding of Siah1 and YAP dramatically increased along with the increase of FRK level (Figure 6E), indicating that FRK promotes YAP binding to Siah1. Furthermore, the increase of FRK-induced interaction between Siah1 and YAP was abolished by YAPY391F, YAPY407F, and YAPY444F mutant (Figure 6F). These results demonstrate that FRK-dependent phosphorylation of YAP is critical for the binding of YAP with Siah1 and its subsequence ubiquitylation. In line with above results, the growth-inhibitory effect of FRK on GBM cells was reversed by Siah1 down-regulation (Supplemental Figure 6D, 6E).
Discussion
Previous studies have demonstrated that YAP frequently amplified and activated in several types of human cancers.5 However, although classical regulatory mechanisms of YAP are well explored, the Hippo-independent manners of how YAP is modulated remains poorly understood. Here, we found that FRK negatively regulates YAP protein stability through phosphorylation of YAP on Tyr391/407/444, which promotes YAP ubiquitination and degradation through recruiting Siah1 (Figure 6G).
Several studies have shown that phosphorylation may be a common mechanism for regulation of protein stability. It is reported that FRK positively regulates PTEN and BRCA1 stability by tyrosine phosphorylation in breast cancer.14,15 In our studies, although only Y407 phosphorylation by FRK has been verified through phospho-YAP-Tyr407, we found that FRK may phosphorylate YAP on Tyr391/407/444 by a serial experiment. Unfortunately, since there are no commercial antibodies against phospho-YAP-Tyr391 and phospho-YAP-Tyr444, we could not verify these two sites now. In addition, our present study identified that YAP phosphorylation by FRK promotes its ubiquitination and degradation. However, several SRC nonreceptor tyrosine kinase family members, such as SRC, YES, LCK, were shown to directly phosphorylate YAP at Y391/407/444 and promoted YAP nuclear translocation and enhanced its activation.10,11,29–31 This may be the reason why FRK and other SFKs play opposite roles in cancer progression. It is interesting that FRK and SRC phosphorylates YAP at the similar sites but exhibits different effect on YAP activity. We have no answer for this question now and deduce it may due to the different subcellular location of FRK and SRC. This question deserved further study in the future.
It has been reported that YAPS381 phosphorylation modulates β-Trcp-mediated ubiquitination and degradation.7 Because FRK has not affected the activity of LATS, and knockdown of LATS1/2 has not affected the modulation of YAP via FRK, we therefore deduce that FRK-induced YAP ubiquitination and degradation was independent of Hippo signal pathway and was not performed by β-Trcp. Besides β-Trcp, FBXW7, SOCS5/6, SKP2, STUB1, DCAF12, RNF187, PARK2, and TRAF6 has also been reported to be the E3 ligases of YAP.38–45 In this study, we identified and validated that Siah1 is a novel E3 ubiquitin ligase of YAP. Accumulated data have shown that Siah1 is a member of RING family E3 ubiquitin ligases and a few proteins including NcoR, TRAF, β-catenin, c-Myb, APC, and Kid have been identified as Siah1 substrates.46–49 In the current study, we found that YAP phosphorylation by FRK increases the association of YAP with Siah1 and then ubiquitylation, indicating that YAP is a new Siah1 substrates.
In recent years, small-molecule-induced protein degradation has emerged as a powerful therapeutic strategy, such as proteolysis-targeting chimeras (PROTACs), which has been developed to degrade a wide range of clinically relevant target proteins. These small-molecule degraders engage both the E3 ligase and the target protein to promote the formation of a substrate–drug–ligase ternary complex. BI-3802 has been reported to induce specific degradation of BCL6 by E3 ligase Siah1.50 In the future, we would like to find a similar small molecule compound that can induce the specific degradation of YAP by Siah1.
In conclusion, we identify that FRK phosphorylates and destabilizes YAP by promoting recruitment of Siah1, leading to GBM growth inhibition. YAP is a substrate of FRK and Siah1 is a new E3 ubiquitin ligase of YAP. Lack of FRK and Siah1 in GBM causes YAP accumulation and activation to promote tumor progression. Our present study has strongly suggested that the FRK-Siah1-YAP axis is critical for GBM malignant progression and targeting this axis is a potential therapeutic strategy against GBM.
Supplementary Material
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Acknowledgments
We thank Dr. Jianmin Zhang (Roswell Park Cancer Institute) and Prof. Hongbin Ji (Institute of Biochemistry and Cell Biology, Chinese Academy of Sciences) for providing various plasmids. We thank Prof. Chunsheng Kang (Department of Neurosurgery, Tianjin Medical University General Hospital) for providing the low passage primary GBM cells. We thank Prof. Chunmei Zhu at Department of English in Xuzhou Medical University for English writing assistance.
Contributor Information
Yan Wang, Institute of Nervous System Diseases, Xuzhou Medical University, Xuzhou, Jiangsu, China; Department of Neurosurgery, the Affiliated Hospital of Xuzhou Medical University, Xuzhou, Jiangsu, China; The Graduate School, Xuzhou Medical University, Xuzhou, Jiangsu, China.
Kai Wang, Institute of Nervous System Diseases, Xuzhou Medical University, Xuzhou, Jiangsu, China; Department of Neurosurgery, the Affiliated Hospital of Xuzhou Medical University, Xuzhou, Jiangsu, China; The Graduate School, Xuzhou Medical University, Xuzhou, Jiangsu, China.
Jiale Fu, Institute of Nervous System Diseases, Xuzhou Medical University, Xuzhou, Jiangsu, China; Department of Neurosurgery, the Affiliated Hospital of Xuzhou Medical University, Xuzhou, Jiangsu, China; The Graduate School, Xuzhou Medical University, Xuzhou, Jiangsu, China.
Yu Zhang, Institute of Nervous System Diseases, Xuzhou Medical University, Xuzhou, Jiangsu, China; Department of Neurosurgery, the Affiliated Hospital of Xuzhou Medical University, Xuzhou, Jiangsu, China.
Yufei Mao, Institute of Nervous System Diseases, Xuzhou Medical University, Xuzhou, Jiangsu, China; Department of Neurosurgery, the Affiliated Hospital of Xuzhou Medical University, Xuzhou, Jiangsu, China; The Graduate School, Xuzhou Medical University, Xuzhou, Jiangsu, China.
Xu Wang, Institute of Nervous System Diseases, Xuzhou Medical University, Xuzhou, Jiangsu, China; Department of Neurosurgery, the Affiliated Hospital of Xuzhou Medical University, Xuzhou, Jiangsu, China; The Graduate School, Xuzhou Medical University, Xuzhou, Jiangsu, China.
Xiang Wang, Institute of Nervous System Diseases, Xuzhou Medical University, Xuzhou, Jiangsu, China; Department of Neurosurgery, the Affiliated Hospital of Xuzhou Medical University, Xuzhou, Jiangsu, China; The Graduate School, Xuzhou Medical University, Xuzhou, Jiangsu, China.
Rutong Yu, Institute of Nervous System Diseases, Xuzhou Medical University, Xuzhou, Jiangsu, China; Department of Neurosurgery, the Affiliated Hospital of Xuzhou Medical University, Xuzhou, Jiangsu, China.
Xiuping Zhou, Institute of Nervous System Diseases, Xuzhou Medical University, Xuzhou, Jiangsu, China; Department of Neurosurgery, the Affiliated Hospital of Xuzhou Medical University, Xuzhou, Jiangsu, China.
Funding
This study was supported by the National Natural Science Foundation of China (81872053 and 82072770 to X.Z.; 81902526 to Y.W.); Natural Science Foundation of Jiangsu province (BK20201458 to X.Z.).
Conflict of interest statement. The authors declare that there is no conflict of interest in this work.
Authorship statement. XZ and RY were responsible for the conception and design and study supervision. XZ, YW, KW and JF performed the experiments and wrote the manuscript. YZ and YM analyzed the data. XW and XW collected clinical specimens and followed up. All authors read and approved the final version of this paper.
Ethics Statement
All the tissue microarrays used obtained the informed consent of the patients and the study was approved by the institutional Ethics Committee. Clinical histories were recorded at screening. All animal experimental protocols were carried out according to the guidelines of Xuzhou Medical University for animal research and were approved by Institutional Animal Use Committee.
References
- 1. Jiang T, Nam DH, Ram Z, Poon WS, Wang Q. Clinical practice guidelines for the management of adult diffuse gliomas. Cancer Lett. 2020;499:60–72. [DOI] [PubMed] [Google Scholar]
- 2. Hu H, Mu Q, Bao Z, et al. . Mutational landscape of secondary glioblastoma guides MET-targeted trial in brain tumor. Cell. 2018;175(6):1665–1678.e18. [DOI] [PubMed] [Google Scholar]
- 3. Meng Z, Moroishi T, Guan KL. Mechanisms of Hippo pathway regulation. Genes Dev. 2016;30(1):1–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Ma S, Meng Z, Chen R, Guan KL. The hippo pathway: biology and pathophysiology. Annu Rev Biochem. 2019;88:577–604. [DOI] [PubMed] [Google Scholar]
- 5. Pan D. The hippo signaling pathway in development and cancer. Dev Cell. 2010;19(4):491–505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Zhao B, Wei X, Li W, et al. . Inactivation of YAP oncoprotein by the Hippo pathway is involved in cell contact inhibition and tissue growth control. Genes Dev. 2007;21(21):2747–2761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Zhao B, Li L, Tumaneng K, Wang CY, Guan KL. A coordinated phosphorylation by Lats and CK1 regulates YAP stability through SCF(beta-TRCP). Genes Dev. 2010;24(1):72–85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Wang Y, Pan P, Wang Z, et al. . beta-catenin-mediated YAP signaling promotes human glioma growth. J Exp Clin Cancer Res. 2017;36(1):136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Zhang Y, Wang Y, Zhou D, et al. . Radiation-induced YAP activation confers glioma radioresistance via promoting FGF2 transcription and DNA damage repair. Oncogene. 2021;40(27):4580–4591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Zaidi SK, Sullivan AJ, Medina R, et al. . Tyrosine phosphorylation controls Runx2-mediated subnuclear targeting of YAP to repress transcription. EMBO J. 2004;23(4):790–799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Sugihara T, Werneburg NW, Hernandez MC, et al. . YAP tyrosine phosphorylation and nuclear localization in cholangiocarcinoma cells are regulated by LCK and independent of LATS activity. Mol Cancer Res. 2018;16(10):1556–1567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Goel RK, Lukong KE. Understanding the cellular roles of Fyn-related kinase (FRK): implications in cancer biology. Cancer Metastasis Rev. 2016;35(2):179–199. [DOI] [PubMed] [Google Scholar]
- 13. Chandrasekharan S, Qiu TH, Alkharouf N, et al. . Characterization of mice deficient in the Src family nonreceptor tyrosine kinase Frk/rak. Mol Cell Biol. 2002;22(14):5235–5247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Yim EK, Peng G, Hui D, et al. . Rak functions as a tumor suppressor by regulating PTEN protein stability and function. Cancer Cell. 2009;15(4):304–314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Kim JL, Ha GH, Campo L, et al. . The role of Rak in the regulation of stability and function of BRCA1. Oncotarget. 2017;8(49):86799–86815. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Hua L, Wang G, Wang Z, et al. . Activation of STAT1 by the FRK tyrosine kinase is associated with human glioma growth. J Neuro Oncol. 2019;143(1):35–47. [DOI] [PubMed] [Google Scholar]
- 17. Hua L, Zhu M, Song X, et al. . FRK suppresses the proliferation of human glioma cells by inhibiting cyclin D1 nuclear accumulation. J Neurooncol. 2014;119(1):49–58. [DOI] [PubMed] [Google Scholar]
- 18. Shi Q, Song X, Wang J, et al. . FRK inhibits migration and invasion of human glioma cells by promoting N-cadherin/beta-catenin complex formation. J Mol Neurosci. 2015;55(1):32–41. [DOI] [PubMed] [Google Scholar]
- 19. Wang J, Cai C, Nie D, et al. . FRK suppresses human glioma growth by inhibiting ITGB1/FAK signaling. Biochem Biophys Res Commun. 2019;517(4):588–595. [DOI] [PubMed] [Google Scholar]
- 20. Zhou X, Hua L, Zhang W, et al. . FRK controls migration and invasion of human glioma cells by regulating JNK/c-Jun signaling. J Neurooncol. 2012;110(1):9–19. [DOI] [PubMed] [Google Scholar]
- 21. Yan S, Li A, Liu Y. CacyBP/SIP inhibits the migration and invasion behaviors of glioblastoma cells through activating Siah1 mediated ubiquitination and degradation of cytoplasmic p27. Cell Biol Int. 2018;42(2):216–226. [DOI] [PubMed] [Google Scholar]
- 22. Zhu Y, Qiu Z, Zhang X, et al. . Jab1 promotes glioma cell proliferation by regulating Siah1/beta-catenin pathway. J Neurooncol. 2017;131(1):31–39. [DOI] [PubMed] [Google Scholar]
- 23. Shi H, Zheng B, Wu Y, et al. . Ubiquitin ligase Siah1 promotes the migration and invasion of human glioma cells by regulating HIF-1alpha signaling under hypoxia. Oncol Rep. 2015;33(3):1185–1190. [DOI] [PubMed] [Google Scholar]
- 24. He Y, Roos WP, Wu Q, Hofmann T, Kaina B. The SIAH1-HIPK2-p53ser46 damage response pathway is involved in temozolomide-induced glioblastoma cell death. Mol Cancer Res. 2019;17(5):1129–1141. [DOI] [PubMed] [Google Scholar]
- 25. Yi K, Zhan Q, Wang Q, et al. . PTRF/cavin-1 remodels phospholipid metabolism to promote tumor proliferation and suppress immune responses in glioblastoma by stabilizing cPLA2. Neuro Oncol. 2021;23(3):387–399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Huang H, Zhang W, Pan Y, et al. . YAP suppresses lung squamous cell carcinoma progression via deregulation of the DNp63-GPX2 axis and ROS accumulation. Cancer Res. 2017;77(21):5769–5781. [DOI] [PubMed] [Google Scholar]
- 27. Li YW, Guo J, Shen H, et al. . Phosphorylation of Tyr188 in the WW domain of YAP1 plays an essential role in YAP1-induced cellular transformation. Cell Cycle. 2016;15(18):2497–2505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Zhou X, Xie S, Wu S, et al. . Golgi phosphoprotein 3 promotes glioma progression via inhibiting Rab5-mediated endocytosis and degradation of epidermal growth factor receptor. Neuro Oncol. 2017;19(12):1628–1639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Rosenbluh J, Nijhawan D, Cox AG, et al. . beta-Catenin-driven cancers require a YAP1 transcriptional complex for survival and tumorigenesis. Cell. 2012;151(7):1457–1473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Calvo F, Ege N, Grande-Garcia A, et al. . Mechanotransduction and YAP-dependent matrix remodelling is required for the generation and maintenance of cancer-associated fibroblasts. Nat Cell Biol. 2013;15(6):637–646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Li P, Silvis MR, Honaker Y, et al. . alpha E-catenin inhibits a Src-YAP1 oncogenic module that couples tyrosine kinases and the effector of Hippo signaling pathway. Genes Develop. 2016;30(7):798–811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Kwon Y, Vinayagam A, Sun X, et al. . The Hippo signaling pathway interactome. Science. 2013;342(6159):737–740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Wang W, Li X, Huang J, et al. . Defining the protein-protein interaction network of the human hippo pathway. Mol Cell Proteomics. 2014;13(1):119–131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Couzens AL, Knight JD, Kean MJ, et al. . Protein interaction network of the mammalian Hippo pathway reveals mechanisms of kinase-phosphatase interactions. Sci Signal. 2013;6(302):rs15. [DOI] [PubMed] [Google Scholar]
- 35. Moya IM, Halder G. Discovering the Hippo pathway protein-protein interactome. Cell Res. 2014;24(2):137–138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Bhattacharya S, Lee YT, Michowski W, et al. . The modular structure of SIP facilitates its role in stabilizing multiprotein assemblies. Biochemistry. 2005;44(27):9462–9471. [DOI] [PubMed] [Google Scholar]
- 37. House CM, Frew IJ, Huang HL, et al. . A binding motif for Siah ubiquitin ligase. Proc Natl Acad Sci USA. 2003;100(6):3101–3106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Tu K, Yang W, Li C, et al. . Fbxw7 is an independent prognostic marker and induces apoptosis and growth arrest by regulating YAP abundance in hepatocellular carcinoma. Mol Cancer. 2014;13:110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Hong X, Nguyen HT, Chen Q, et al. . Opposing activities of the Ras and Hippo pathways converge on regulation of YAP protein turnover. EMBO J. 2014;33(21):2447–2457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Yao F, Zhou Z, Kim J, et al. . SKP2- and OTUD1-regulated non-proteolytic ubiquitination of YAP promotes YAP nuclear localization and activity. Nat Commun. 2018;9(1):2269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Tang DE, Dai Y, Lin LW, et al. . STUB1 suppresseses tumorigenesis and chemoresistance through antagonizing YAP1 signaling. Cancer Sci. 2019;110(10):3145–3156. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 42. Cho YS, Li S, Wang X, et al. . CDK7 regulates organ size and tumor growth by safeguarding the Hippo pathway effector Yki/Yap/Taz in the nucleus. Genes Dev. 2020;34(1-2):53–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Wang Z, Kong Q, Su P, et al. . Regulation of Hippo signaling and triple negative breast cancer progression by an ubiquitin ligase RNF187. Oncogenesis. 2020;9(3):36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Zhou X, Li Y, Wang W, et al. . Regulation of Hippo/YAP signaling and esophageal squamous carcinoma progression by an E3 ubiquitin ligase PARK2. Theranostics. 2020;10(21):9443–9457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Lv Y, Kim K, Sheng Y, et al. . YAP controls endothelial activation and vascular inflammation through TRAF6. Circ Res. 2018;123(1):43–56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Gwak J, Song T, Song JY, et al. . Isoreserpine promotes β-catenin degradation via Siah-1 up-regulation in HCT116 colon cancer cells. Biochem Biophys Res Commun. 2009;387(3):444–449. [DOI] [PubMed] [Google Scholar]
- 47. Hu G, Fearon ER. Siah-1 N-Terminal RING domain is required for proteolysis function, and C-terminal sequences regulate oligomerization and binding to target proteins. Mol Cell Biol. 1999;19(1):724–732. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Xiao JH, Ghosn C, Hinchman C, et al. . Adenomatous Polyposis Coli (APC)-independent regulation of β-catenin degradation via a retinoid X receptor-mediated pathway. J Biol Chem. 2003;278(32):29954–29962. [DOI] [PubMed] [Google Scholar]
- 49. Bruzzoni-Giovanelli H, Fernandez P, Veiga L, et al. . Distinct expression patterns of the E3 ligase SIAH-1 and its partner Kid/KIF22 in normal tissues and in the breast tumoral processes. J Exp Clin Cancer Res. 2010;29(1):10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Slabicki M, Yoon H, Koeppel J, et al. . Small-molecule-induced polymerization triggers degradation of BCL6. Nature. 2020;588(7836):164–168. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.