

Abstract
A defining characteristic of mammalian prions is their capacity for self‐sustained propagation. Theoretical considerations and experimental evidence suggest that prion propagation is modulated by cell‐autonomous and non‐autonomous modifiers. Using a novel quantitative phospholipase protection assay (QUIPPER) for high‐throughput prion measurements, we performed an arrayed genome‐wide RNA interference (RNAi) screen aimed at detecting cellular host‐factors that can modify prion propagation. We exposed prion‐infected cells in high‐density microplates to 35,364 ternary pools of 52,746 siRNAs targeting 17,582 genes representing the majority of the mouse protein‐coding transcriptome. We identified 1,191 modulators of prion propagation. While 1,151 modified the expression of both the pathological prion protein, PrPSc, and its cellular counterpart, PrPC, 40 genes selectively affected PrPSc. Of the latter 40 genes, 20 augmented prion production when suppressed. A prominent limiter of prion propagation was the heterogeneous nuclear ribonucleoprotein Hnrnpk. Psammaplysene A (PSA), which binds Hnrnpk, reduced prion levels in cultured cells and protected them from cytotoxicity. PSA also reduced prion levels in infected cerebellar organotypic slices and alleviated locomotor deficits in prion‐infected Drosophila melanogaster expressing ovine PrPC. Hence, genome‐wide QUIPPER‐based perturbations can discover actionable cellular pathways involved in prion propagation. Further, the unexpected identification of a prion‐controlling ribonucleoprotein suggests a role for RNA in the generation of infectious prions.
Keywords: High‐throughput screen, Hnrnpk, Neurodegeneration, Prion, Protein aggregation
Subject Categories: Molecular Biology of Disease, RNA Biology
A novel quantitative phospholipase protection assay in combination with a genome‐wide RNA interference screen leads to the identification of host factors involved in prion propagation.

Introduction
The life cycle of mammalian prions entails the misfolding and aggregation of the cellular protein PrPC and its incorporation into a nucleated higher‐order isoform called PrPSc (Aguzzi & Calella, 2009). Once the PrPSc aggregates reach a critical size, they break and elongate again by recruiting additional monomers (Knowles et al, 2009). This cyclic sequence of events is the basis for the increase in prion infectivity (Nuvolone et al, 2009). However, it is still unknown whether this process occurs autonomously akin to crystal growth, or if it necessitates auxiliary cofactors (Deleault et al, 2012a, 2012b). The latter is suggested by the observation that propagation of prions in a cell‐free system is inefficient and necessitates extreme conditions such as cyclic high‐energy sonication, shaking, or partial chemical denaturation (Saborio et al, 2001; Atarashi et al, 2011). In contrast, infection of animals or cultured cells with prions can yield titer increases by several orders of magnitude under physiological conditions (Prusiner et al, 1982; Klöhn et al, 2003). This suggests that living systems contain important cofactors that enable prion propagation, e.g., by lowering the thresholds of rate‐limiting reactions.
How could one possibly identify such cofactors? In the case of other neurodegenerative diseases, crucial insights were derived from human genetics. The study of families afflicted by inherited forms of Alzheimer's and Parkinson's disease have yielded a plethora of genes encoding proteins directly linked to the offending aggregates (van Rheenen et al, 2016; Jansen et al, 2019; Nalls et al, 2019). However, this approach has not been as successful in the case of prion diseases, partly because of their rarity which precludes large genome‐wide association studies (Lloyd et al, 2013). As a result, the only modifiers robustly associated with predisposition to prion diseases are genetic polymorphisms within the PrPC‐encoding PRNP gene itself (Mead et al, 2009, 2012; Sanchez‐Juan et al, 2014; Jones et al, 2020).
A possible approach to this conundrum consists of investigating candidate genes which may be inferred from existing reports or from their role in phenomena pertinent to prion propagation. For example, transcription factors involved in PRNP mRNA expression (Rybner et al, 2002; Bellingham et al, 2009; Vincent et al, 2009; Dery et al, 2013), or proteins involved in its degradation (Shyu et al, 2002; Parkyn et al, 2008; Vincent et al, 2009), may represent such candidates. However, this approach has major limitations. Any potential candidates, in order to be identified as such, must have been described previously in similar contexts. Consequently, any fundamentally novel mechanisms cannot be discovered because they would not exist as priors.
Forward genetic screens, in which each protein‐coding gene is being modified and hits are identified by their effect on the phenotype of interest, represent a less biased and more inclusive approach with the potential of yielding wholly unpredicted hits. Moreover, the identification of relationships between hits, e.g., because they fall within a single pathway, or because they encode individual components of a single physical complex, can fortify the confidence in the validity of the results. In the past, such screens have been most effectively performed in unicellular organisms that undergo a haploid phase, such as yeast (Derkatch et al, 2001; Kryndushkin & Wickner, 2007). However, more recent technologies such as RNA interference (RNAi) and CRISPR have enabled the deployment of forward genetic screens in diploid mammalian cells (Mohr et al, 2010; Kampmann, 2018; Heinzer et al, 2021).
In this work, we have used arrayed RNAi to interrogate all genes of the mouse genome for their influence on prion propagation. We have discovered 40 such genes. Twenty of these were found to reduce prion propagation when suppressed, but 20 genes enhanced prion propagation when silenced. Some of these modifiers fell within pathways expected to control prion propagation (Marbiah et al, 2014). However, others were entirely surprising, including the small heteronuclear RNA binding protein, Hnrnpk.
Results
Establishment of a genome‐wide high‐throughput screen for identification of prion modulators
Scalable, reproducible high‐throughput assays should consist of only a few steps and should not require analyte transfers to different reaction containers. Immunochemical prion detection (by Western blotting, enzyme‐linked immunoassay, or other methods) is typically preceded by limited proteolysis using proteinase K (PK), eliminates PrPC and ensures that any residual signal is specific to PrPSc (Bolton et al, 1982). However, digestion with PK requires fastidious titration and accurate timing, which may introduce confounders (McKinley et al, 1983).
To solve these issues, we took advantage of phosphatidylinositol‐specific phospholipase C (PIPLC), an enzyme that cleaves proteins attached to the membrane via a glycophosphatidylinositol (GPI) anchor from the surface of cells (Heinz et al, 1995). While PrPC is GPI‐anchored (Stahl et al, 1990), prion aggregates appear to associate in cells independently of the anchoring. PIPLC treatment of intact cells leads to the release of most of the PrPC into the supernatant, while prions remain cell associated (Stahl et al, 1990; Borchelt et al, 1993) and in endocytic compartments (Taraboulos et al, 1992).
Prion assemblies were disaggregated using sodium hydroxide (NaOH, pH = 14, 66.6 mM; Peretz et al, 2001), neutralized with NaH2PO4 buffer (pH = 4.5, 83.3 mM) to near‐neutral pH (Li, 2016), and a Förster energy transfer donor‐acceptor antibody pair (Allophycocyanin‐POM1 (APC‐POM1) and Europium‐POM19 (EU‐POM19)) was added (Polymenidou et al, 2008; Ballmer et al, 2017; Pease et al, 2019). PrP was then detected by time resolved (TR) FRET (Ballmer et al, 2017; Pease et al, 2019; Heinzer et al, 2021). We termed the resulting assay QUantItative Prion PhospholipasE pRotection assay (QUIPPER, Fig 1A).
Figure 1. A cell‐based high‐throughput prion detection assay for an arrayed whole genome RNAi screen.
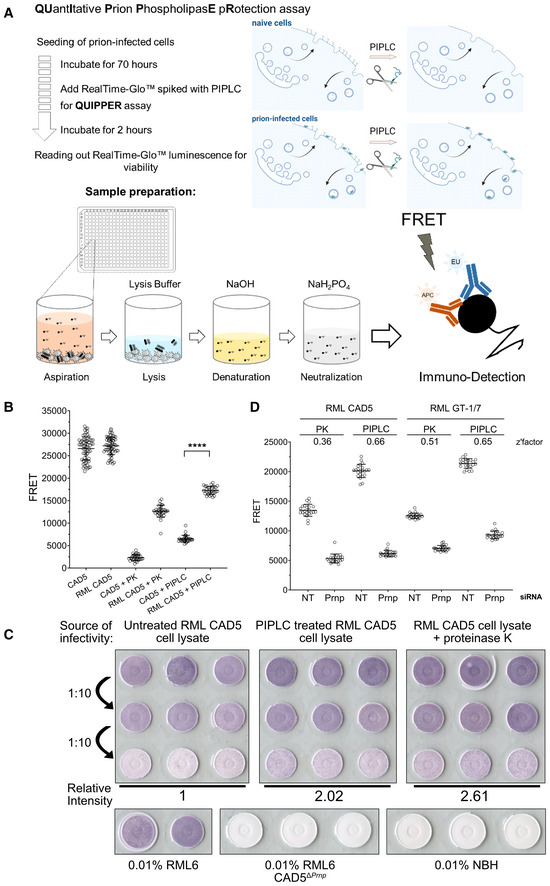
- Workflow of the QUIPPER assay.
- PK vs. PIPLC treatment to determine prion loads in infected cells in 384‐well plates. Both treatments discriminate between RML prion infected CAD5 and non‐infectious brain homogenate (NBH)‐treated CAD5 cells. ****P‐value < 0.0001 (Student's t‐test). Shown are mean ± SD, n ≤ 30 individually cultured wells.
- The infectivity of PIPLC and PK‐treated cell lysates was determined by infecting CAD5 cells with lysates as indicated. The signal intensity of the highest dilution was measured and compared with untreated RML CAD5 cell lysate. PIPLC‐treated cells and PK‐treated lysates showed similar infectivity titers. Prion‐infected and NBH, as well as RML on CAD5ΔPrnp were used for control.
- RML‐infected CAD5 and GT‐1/7 cells were transfected with non‐targeting (NT) or Prnp targeting siRNAs in a 384‐well plate and subjected to PK or PIPLC treatment. Z'‐factors were calculated for each condition. Shown are mean ± SD, n = 22 individually cultured wells.
Source data are available online for this figure.
We then tested our assumption that PIPLC‐resistant PrP (henceforth termed “PrPPLC”) is a plausible surrogate for prion infectivity. Therefore, we generated chronically RML6‐prion infected CAD5 cells (RML CAD5; Fig EV1A) by treatment with mouse brain homogenate containing the Rocky Mountain Laboratory (RML) strain of prions (Solassol et al, 2003; Avar et al, 2020). For control, we used cells inoculated with non‐infectious brain homogenate (NBH CAD5). We then measured PrP by QUIPPER and by PK digestion in 384‐well microtiter plates. The readout yielded a clear separation between infected and non‐infected cells (Fig 1B). We conclude that QUIPPER can reliably detect prion infection.
Figure EV1. QUIPPER assay, example plate maps.
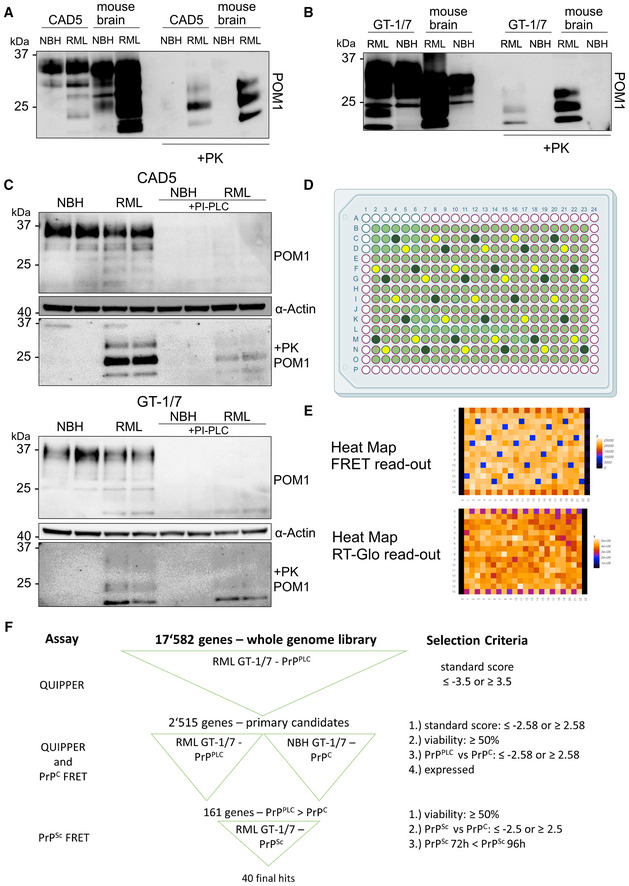
- Western Blot analysis of chronically RML6‐infected CAD5 cells following PK digestion. Anti‐PrP antibody POM1 is used for probing the membrane. Brain homogenates were used as controls.
- Western Blot analysis of chronically RML6‐infected GT‐1/7 cells after PK treatment. Membrane is probed with anti‐PrP antibody POM1. Brain homogenates were used as controls.
- Western blot analysis of infected and uninfected CAD5 and GT‐1/7 cells following PIPLC treatment and subsequent PK digestion. Membranes are probed with anti‐PrP antibody POM1. ⍺: anti. Anti‐actin antibody is used to probe the membrane as loading control.
- Plate map used in the screen depicting controls and samples. Light green represents wells containing siRNAs from the library, dark green represents wells containing Prnp‐targeting control siRNAs and yellow represents wells containing non‐targeting control siRNAs.
- Examples of heat maps for FRET as well as viability read‐out from the primary screen.
- Schematic of the hit‐selection process over all the screens performed in this study with the corresponding criteria.
Source data are available online for this figure.
Previous reports suggested that PIPLC treatment of chronically infected cells reduces the amount of PrPSc (Enari et al, 2001). In order to ensure that PrPPLC can be used as a surrogate for prion infectivity, we performed a scrapie cell assay in endpoint format (SCEPA; Mahal et al, 2008), arguably the most precise method to determine infectious prion titers in cellula. We inoculated naïve CAD5 cells with three decadic dilutions from lysates of RML‐infected CAD5 cells treated with PIPLC just prior to lysis. For control, we used untreated RML CAD5 lysate and PK‐treated lysate, as well as naïve and CAD5ΔPrnp cells inoculated with NBH or RML. After three passages, cells were spotted onto EliSPOT membranes and digested with PK to selectively detect infected cells (Fig 1C). Image analysis of the optical density of the membranes showed that PrPPLC retained full infectivity associated with prions. Furthermore, retainment of PrPSc was confirmed using western blotting following PIPLC treatment, which yielded a positive signal following PK digestion in prion infected GT‐1/7 (Fig EV1B) and CAD5 cells (Fig EV1C).
We then compared the discriminatory power of QUIPPER vs. PK digestion for identifying modulators of prion propagation. Chronically infected RML CAD5 and RML GT‐1/7 cells were treated with Prnp‐targeting siRNAs or non‐targeting (NT) siRNA controls in 384‐well plates. Computation of the Z'‐factor, a measure of the separation between positive and negative controls (Zhang et al, 1999), showed that QUIPPER outperformed PK digestion in both RML GT‐1/7 and RML CAD5 cell lines (Fig 1D). We opted to use RML GT‐1/7 cells for the genome‐wide screen because of their strong adherence to tissue culture plates, which facilitated their handling in 384‐well microplates.
Genome‐wide screen for prion modifiers
We used a genome‐wide murine siRNA library containing a pool of three distinct siRNAs per target transcript. Each siRNA mixture was dispensed in duplicate to a final concentration of 20 nM using an acoustic dispenser. Each 384‐well plate was loaded with 264 gene‐targeting siRNA triplets, 22 NT siRNA, and 22 Prnp‐targeting siRNAs. The outermost wells were left blank as it was found to be prone to evaporation (Fig EV1D). Controls and duplicates were strategically positioned for identifying and correcting any artifactual plate gradients, dispensing errors, or hotspots (Pease et al, 2019; Heinzer et al, 2021). Such gradients can arise from problems with the dispensing and aspiration steps or from temperature/humidity inhomogeneities during the incubation. After 3 days of culture, RealTime‐Glo (RT‐Glo), a reagent for cell‐viability readout, and PIPLC were added and incubated at 37°C for 2 h. Subsequently, RT‐Glo luminescence was measured, medium was aspirated, cells were lysed, and PrPPLC was disaggregated and denatured. Finally, the antibody pairs were added to each well, and TR‐FRET was measured after a 24 h incubation (Fig 1A).
We screened a total of 136 plates entailing 17,582 in duplicates using siRNA triplets as well as 2,992 negative and positive controls, respectively. For each plate, heatmaps of TR‐FRET and RT‐Glo values (Fig EV1E) were generated to detect any artifactual signal gradients or hotspots, which may have occurred during the screening process. 15,548 genes were assayed in duplicates and 2,030 genes in single measurements; four genes were not assayed during the primary screening. Z'‐factors (Zhang et al, 1999) were > 0.5 for 125 plates and 0–0.5 for 11 plates, confirming the robustness of the screen (Fig 2A). A plot of all TR‐FRET values obtained from the screen showed two non‐overlapping populations corresponding to Prnp‐targeting and NT controls, whereas the majority of the genes interrogated by the library had no effect on prion levels (Fig 2B). The determination coefficient r 2 between duplicates was 0.38, indicating a correlation sufficient to enable candidate selection (Taylor, 1990; Fig 2C).
Figure 2. Whole‐genome screen for prion modulators.
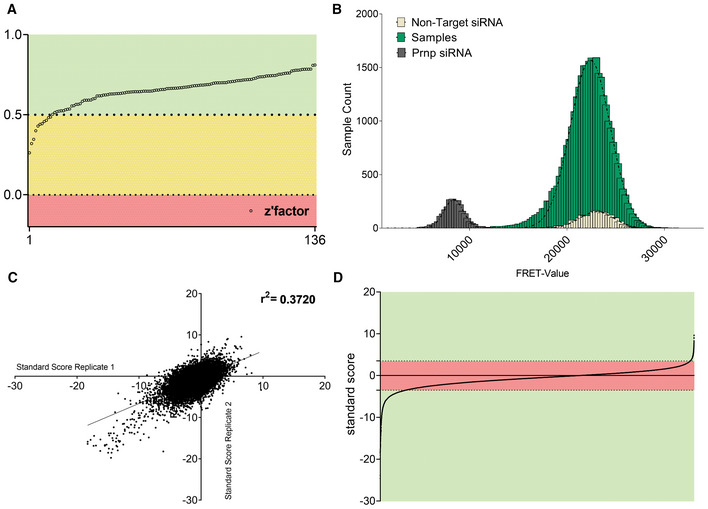
- Z'‐factor for each plate of the whole genome screen representing the robustness of the screen based on the separability of the positive (Prnp targeting) and negative (non‐targeting) controls.
- Histogram representing the influence of each protein‐coding gene as well as NT and Prnp targeting controls, on prion levels. Abscissa: prion levels measured by FRET. Ordinate: number of genes yielding a given FRET‐range. Controls showed a strong separation, allowing for confident hit‐selection. Only a few genes affected prion levels.
- Correlation of standard scores for all genes that were assayed in duplicates in the primary screen. r 2: coefficient of determination.
- All individual data points from the primary screening. Genes reaching a z‐score of [< −3.5] ∪ [> 3.5] in one or both duplicates were considered as hits (green area).
We then computed the standard score (z‐score) for each candidate (Birmingham et al, 2009), and selected the top scoring 2,515 candidates (z‐score = [<−3.5] ∪ [>3.5]) for a confirmatory screen (Fig 2D). Of these candidates, 2,154 had a negative z‐score, whereas only 361 genes had a positive z‐score. Hence, 86% of modifiers, when suppressed, reduced PrPPLC levels, whereas modifiers whose suppression enhanced PrPPLC levels were rarer (14%; for candidate selection process, also see Fig EV1F).
Confirmatory screens on prion modulators
Since PrPC is necessary for prion propagation, some prion modifiers may act by changing PrPC expression or localization, whereas others may act selectively on PrPSc. We therefore performed two secondary screens. In the first screen, all top‐scoring 2,515 hits were tested for the modulation of PrPC. To exclude any potential confounders, GT‐1/7 cells were exposed to NBH and passaged identically to the prion‐infected cells, and the assays were performed as in the previous screen except for the omission of PIPLC treatment. In a second screen, we performed QUIPPER on the same 2,515 hits and measured PrPPLC.
All plates passed quality control (Fig EV2A) and the reproducibility of duplicates was high (r 2 for the PrPC and PrPPLC subsets: 0.77 and 0.76 for QUIPPER and 0.8 and 0.88 for viability, respectively; Fig EV2B and C). The TR‐FRET scores of duplicates were averaged and a z‐score for measuring the effect size of the manipulation of each gene was computed (Dataset EV1). The PrPC and PrPPLC subsets were strongly correlated (r 2: 0.62), suggesting that selective PrPPLC regulators are rare (Fig 3A). Moreover, there was a strong correlation between the RT‐Glo measurements for NBH and RML infected GT‐1/7 cells (Fig EV2D), implying that no gene knockdown resulted in synthetic lethality with prion infection under these conditions.
Figure EV2. Secondary screen quality metrics and viability readout.
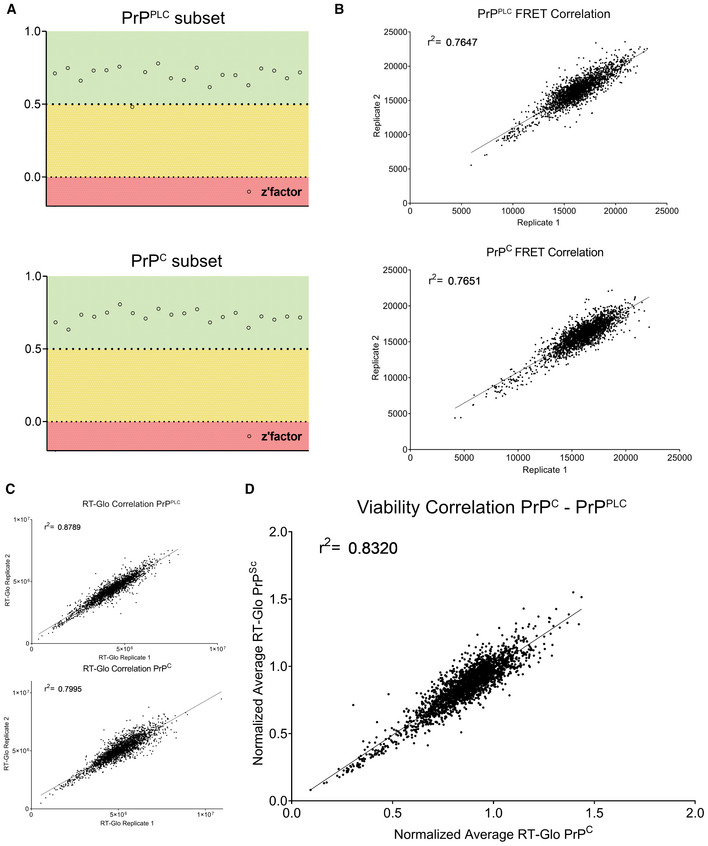
- Z'‐factor for each plate of both deconvolution screens (for regulators of PrPPLC, top panel, for regulators of PrPC, bottom panel) representing the robustness of the screens based on the separability of the positive (Prnp targeting) and negative (non‐targeting) control. 0.5–1 (green) = excellent assay, 0–0.5 (yellow) = acceptable assay, 0 ≥ (red) = unacceptable assay.
- Duplicate correlation of FRET‐data for each screen. Coefficient of determination (r 2‐value) is depicted in the graph.
- Duplicate correlation of viability‐data measured using RealTime‐Glo™ luminescence readout for each screen. Coefficient of determination (r 2‐value) is depicted in the graph.
- Duplicates from (C) were averaged and normalized for each screen, and the two normalized values for each gene is correlated to assess if any gene regulates viability dependent on prion infection. The high r 2‐value, as well as the lack of outliers demonstrates the lack of synthetic lethal genes in the subset assessed in the deconvolution screen.
Figure 3. Secondary screens and shortlisting of 40 candidates.
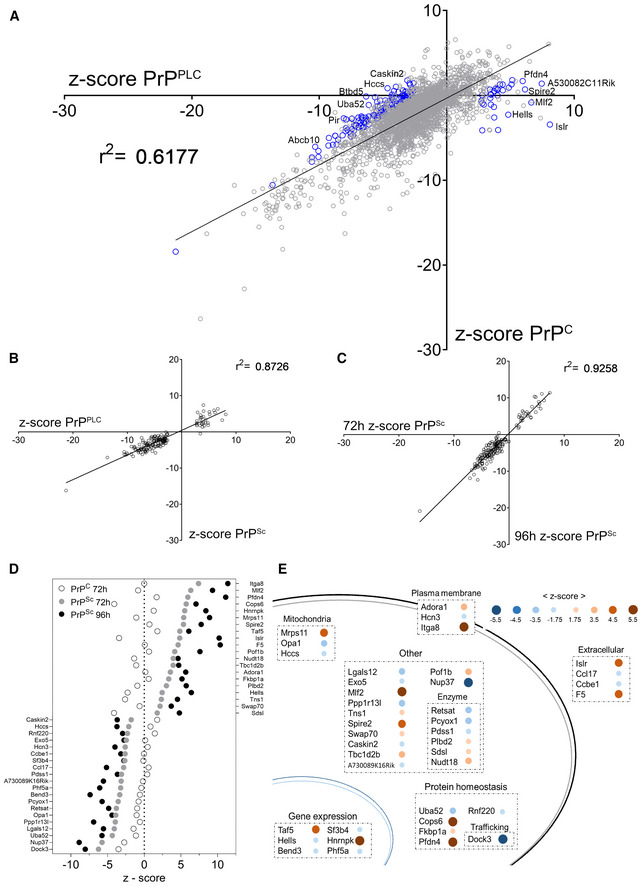
- Regression of the values of the confirmatory screen for prion specific modulators in mock‐ and prion infected GT‐1/7 cells. Z‐scores of genes from the two independent screens, assessing either regulation of PrPC or PrPPLC, yield a coefficient of determination (r 2‐value) of 0.62 indicating that most genes are modulating prion levels via regulating PrPC. Blue circles indicate 161 prion specific hits selected for downstream counter screens. The most conspicuous modulators were labeled.
- Correlation of z‐scores obtained from a secondary screen to assess the effect of PK digestion on 161 PrPPLC modulators in RML GT‐1/7 cells after 72 h of RNAi treatment. Z‐scores of genes from the two independent screens (PIPLC or PK for two different sample preparation approaches) yield a coefficient of determination (r 2) of 0.87, indicating that the candidates regulate PK‐resistant prions.
- Correlation of the z‐scores obtained from the counter‐screenings to assess the effect of PK digestion on the prion modulators in RML GT‐1/7 cells after 72 and 96 h of RNAi treatment. The coefficient of determination (r 2‐value) of 0.93 and the increase in effect size for the prolonged treatment condition indicate a robust effect of the candidates on prion levels.
- Summary of the effect of the 40 shortlisted hits on PrPC (after 72 h) and PrPSc (after 72 and 96 h), assayed using PK digestion, given as z‐scores.
- Function and topology of the 40 hits. Blue dots: prion stabilizers; brown dots: prion limiters. Size and color saturation represent the effect size of each hit based on its z‐score (72 h RNAi treatment; PK readout).
We then applied layered criteria to identify genes modulating specifically PrPPLC. Firstly, in the repetition of the QUIPPER assay, only genes with a z‐score [<−2.58] ∪ [>2.58], corresponding to a P value of 0.01, were considered hits. Secondly, in order to exclude any genes with a strong impact on viability, we limited the hit calling to samples in which the raw RT‐Glo signal was not below 50% of the plate‐specific NT control. Thirdly, we considered only genes whose effect size was ≥ 2.58 standard deviations higher for PrPPLC than for PrPC in uninfected cells. The latter criterion led to the exclusion of 99% of the overlap in two datasets. Lastly, all genes fulfilling these criteria were filtered according to their expression levels in RML GT‐1/7 cells (Dataset EV1), thereby excluding genes that were not expressed (also see Fig EV1E). This led to a list of 161 genes whose expression has a stabilizing (n = 131) or limiting (n = 30) effect on the amount of PrPPLC (Fig 3A, blue circles).
Role for prion specific modulators in sporadic Creutzfeldt‐Jakob disease (sCJD) susceptibility
We wondered whether any of the 161 modulators of PrPPLC may have a role in genetic susceptibility to sCJD (Heinzer et al, 2021). No gene passed the threshold for multiple hypothesis testing. Despite lack of statistical significance, one associated gene was DOCK3 (Multi‐marker Analysis of GenoMic Annotation (MAGMA) unadjusted P = 0.00063), a brain‐resident guanine exchange factor serving as a binding partner to presenilin, which is implicated in Alzheimer's disease and neurodegeneration (Chen et al, 2002, 2009; Tachi et al, 2012; Bai et al, 2013; Dataset EV1).
Validation of prion‐specific regulators
Our observations suggest that QUIPPER may detect perturbations of prion propagation more sensitively than PK digestion. However, because QUIPPER is a new assay that has not yet been validated extensively by multiple laboratories, we subjected prion‐infected GT‐1/7 cells to PK digestion upon treatment with siRNA triplets corresponding to each of the 161 hits after 72 and 96 h of siRNA treatment. We found a remarkable convergence between the QUIPPER and PK assays over the entire collection of genes (Fig 3B), represented by the high r 2 of 0.87, bolstering our confidence in the robustness of the targets identified. Prolonging the treatment with siRNA enhanced the effects observed (Fig 3C). We then asked whether the hits were specific to a particular prion strain. For that, we infected GT‐1/7 cells with the 22L strain of prions (Fig EV3A) and treated them with siRNA triplets corresponding to 97 randomly selected hits, filling a 384‐well plate. Most genes showed similar effects on RML and 22L‐infected cells at both timepoints tested (Fig EV3B).
Figure EV3. Prion strain dependence of the 97 candidates.
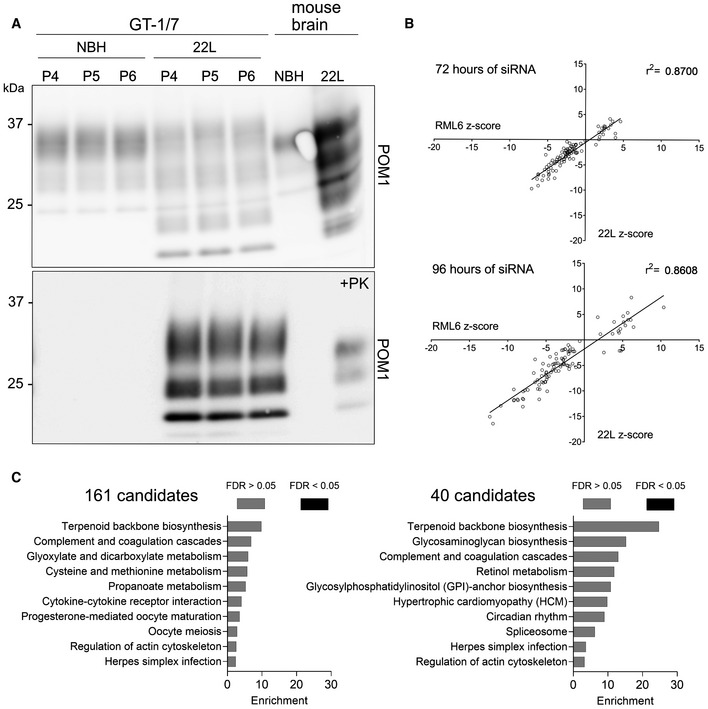
- Western Blot analysis of chronically 22L‐infected GT‐1/7 cells after PK treatment. Membrane is probed with anti‐PrP antibody POM1. P4, P5 and P6 corresponds to the amount of passaging after exposure to 22L brain homogenate. Brain homogenates were used as controls.
- Correlation of the effect of 97 candidates on their effect on PrPSc of two different prion strains (RML6 and 22L). Next to RML6 infected GT‐1/7, 97 hits were assessed for their effect on 22L prion infected GT‐1/7 for 72 h as well as 96 h‐long treatment duration. The high coefficient of determination (r 2‐value) of the effect observed for both prion strains indicates that the candidates do not show strain‐specificity.
- Gene set overrepresentation analysis for 161 hits and 40 hits from the prion modifier screen. No pathway passed the threshold for significance.
Source data are available online for this figure.
In summary, 40 out of the 161 candidates showed a robust and consistent prion modulation across all detection methods (see Fig EV1E). Of these 40 candidates, 20 reduce prion propagation upon silencing, and 20 candidates enhanced prion propagation upon silencing, and henceforward are called stabilizers or limiters, respectively (Fig 3D and E; Dataset EV1). When these genes were interrogated for an overrepresentation in a pathway, we did not see a statistically significant enrichment (Fig EV3C).
Hnrnpk expression limits prion propagation in mouse and human cells
Intriguingly, the suppression of Hnrnpk, an essential gene whose ablation causes cell death (Tsherniak et al, 2017), strongly enhanced prion levels while changing PrPC levels only slightly. Hence, Hnrnpk acts as a limiter of prion propagation (Figs 3D and 4A). To broaden our validation efforts, we treated cells with Psammaplysene A (PSA), which had been described to bind Hnrnpk (Boccitto et al, 2017). PSA treatment led to a strong dose‐dependent decrease of PrPSc in prion‐infected GT‐1/7 cells (Fig 4B and D), whereas it only led to a slight change in PrPC levels. We then assessed mRNA levels via bulk RNA‐Sequencing (RNA‐Seq) of Prnp upon Hnrnpk downregulation and PSA treatment and found that Hnrnpk siRNA treatment of GT‐1/7 cells lead to an efficient downregulation of Hnrnpk as well as a slight increase in Prnp levels, corroborating the screening efforts and PSA had no effect on either Hnrnpk or Prnp mRNA levels (Dataset EV1; Fig 4C).
Figure 4. Hnrnpk and PSA limits prion levels in chronically infected cells.
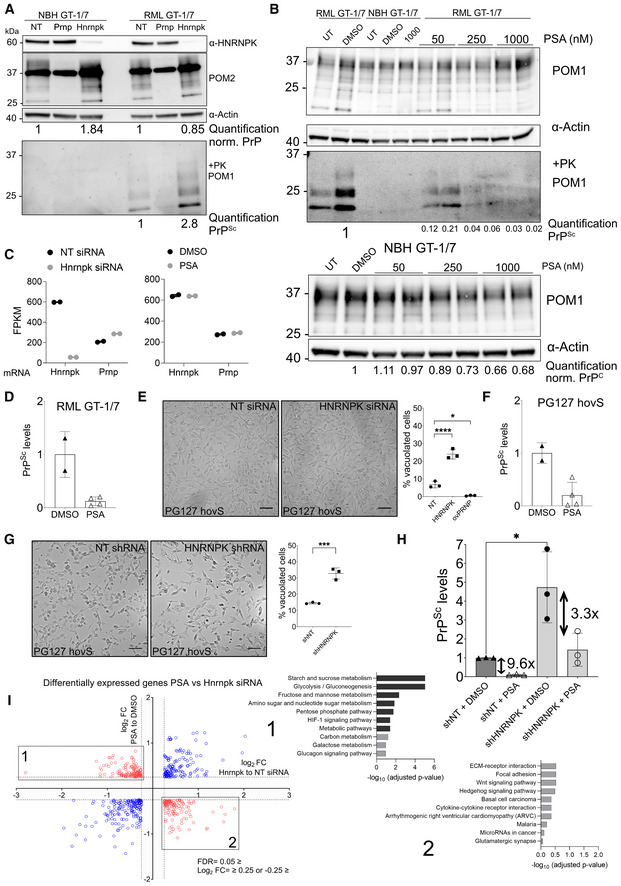
- Western blot showing Hnrnpk siRNA transfection (96 h.) decreases Hnrnpk protein levels while increases PrPSc in RML prion infected GT‐1/7. Prnp siRNAs suppressed both PrPC and PrPSc as expected. ⍺: anti Quantifications are reported as normalized to Actin and in comparison, to NT. PK‐western blot is quantified relative to NT.
- Western Blot of PSA‐treated uninfected and infected GT‐1/7 cells. Increasing concentration of PSA leads to a more prominent reduction of PrPSc in mouse cells. PK‐western blot is quantified relative to DMSO.
- mRNA levels of Hnrnpk and Prnp following siRNA and PSA treatment. FPKM: Fragments per kilobase of transcript per million mapped reads. Hnrnpk siRNAs lead to a decrease in Hnrnpk mRNA levels as well as an increase in Prnp mRNA levels. No difference is seen between DMSO‐treated and PSA‐treated cells for either Hnrnpk levels of Prnp levels. n = 2 per treatment group.
- Quantification of PrPSc levels in RML GT‐1/7 cells following treatment with 1 μM PSA in comparison to DMSO. Each dot represents an experiment. Shown are mean ± SD.
- Brightfield microscopy images of the effect of siRNA mediated HNRNPK downregulation on prion‐induced vacuolation in PG127 hovS cells. Downregulation of HNRNPK leads to an enhanced cytopathological vacuolation phenotype when compared with NT siRNA. ovPRNP siRNA transfected, as well as uninfected cells were used as controls (Fig 4B). Downregulation of ovPRNP in the infected hovS eliminates the vacuoles; Right panel: Quantification of vacuoles of NT, HNRNPK and ovPRNP siRNA‐treated PG127 hovS. Cells from pictures at three different positions in the well were manually counted and the amount of vacuolated cells was normalized to the total amount of cells. Values represent mean ± SD. *P = 0.0113, ****P ≤ 0.0001 (Dunnett's multiple comparisons test). Scale bar = 100 μm. n = 3 technical replicates.
- Quantification of PrPSc levels in PG127 hovS cells following treatment with 1 μM PSA in comparison to DMSO. Each dot represents an experiment. Shown are mean ± SD.
- Same as in E, using shRNAs instead of siRNA. Downregulation of HNRNPK leads to an enhanced cytopathological vacuolation phenotype when compared with NT shRNA. Uninfected cells were used as controls (Fig EV4E). Right panel: Quantification of vacuoles of NT and HNRNPK shRNA‐treated PG127 hovS. Cells from pictures at three different positions in the well were manually counted and the amount of vacuolated cells was normalized to the total amount of cells. Values represent mean ± SD. ***P ≤ 0.0009 (Dunnett's multiple comparisons test). Scale bar = 100 μm. n = 3 technical replicates.
- Quantification of PrPSc levels in PG127 hovS cells following treatment with shRNA against HNRNPK and 1 μM PSA in comparison to DMSO and NT. Each dot represents an experiment. *P‐value < 0.027 (Student's t‐test). Shown are mean ± SD.
- Gene set overrepresentation analysis of differentially expressed genes (log2FC ‐0.25 ≥ or 0.25 ≤ and FDR ≤ 0.05) for siRNA mediated Hnrnpk downregulation or PSA treatment in RML GT‐1/7 cells analyzed by RNAseq. Differentially regulated genes (up in siRNA treatment and down in PSA or vice versa) were overlapped and used for pathway analysis. No significantly enriched pathways are detected for upregulated genes in Hnrnpk and downregulated in PSA treatment. For the opposing direction, an enrichment of genes involved in glucose metabolism was detected.
Source data are available online for this figure.
We repeated these experiments in hovS, a human cell line expressing ovine but not human PrPC, which is readily infectible with the PG127 strain of ovine prions (Avar et al, 2020). Again, the downregulation of HNRNPK in prion‐infected hovS cells led to an increase in PrPSc (Fig EV4A). Prion infection of hovS cells induces a prominent cytopathology consisting of cytosolic vacuolation. HNRNPK suppression exacerbated vacuolation, whereas ovine PRNP (ovPRNP) suppression completely abolished it, in line with the notion that prion levels determine the extent of hovS cytopathology (Fig 4E). PSA treatment of infected hovS also led to a decrease in prion levels (Figs EV4C and F). To validate HNRNPK as a limiter of prion propagation independent of siRNA transfection, the experiment was additionally performed using shRNAs through lentiviral transduction in hovS. The results obtained with a shRNA targeting HNRNPK was congruent to the results obtained via siRNA transfection (Figs 4G and EV4D), again highlighting the validity of HNRNPK as a modulator of prion formation.
Figure EV4. Phenotypic response of PG127prion infected hovS upon HNRNPK downregulation using siRNA or shRNA.
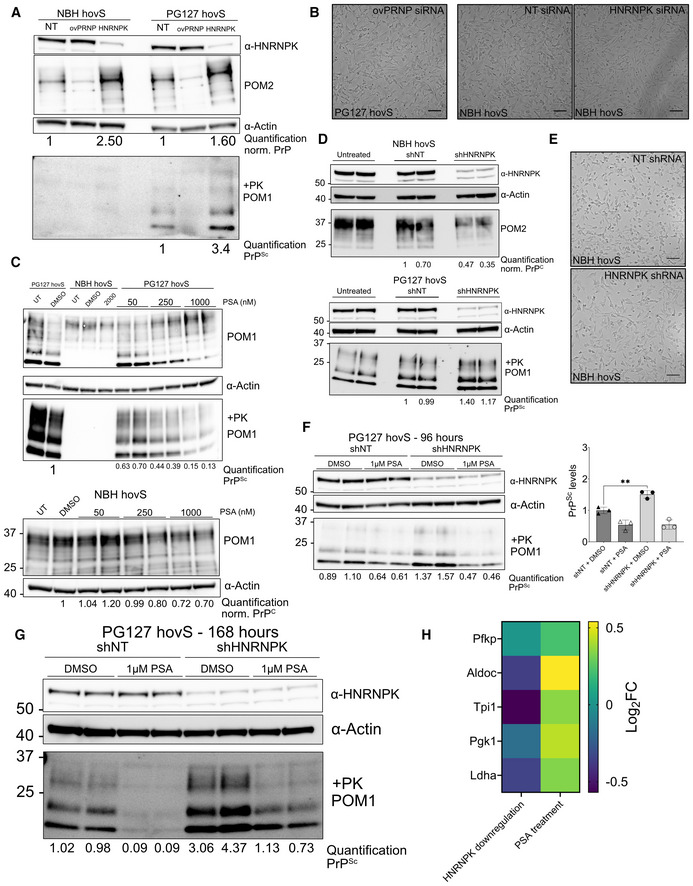
- Western blot showing HNRNPK siRNA transfection (96 h) decreases HNRNPK protein levels while it increases PrPSc in prion infected hovS cells. PRNP siRNAs suppressed both PrPC and PrPSc as expected. ⍺: anti Quantifications are reported as normalized to Actin and in comparison, to NT. PK‐western blot is quantified relative to NT.
- Brightfield microscopy images of the effect of siRNA mediated HNRNPK downregulation on vacuolation in PG127 and NBH hovS cells. Downregulation of ovPRNP in the infected hovS eliminates the vacuoles; HNRNPK downregulation in uninfected cells does not yield a vacuolation phenotype. Scale bar = 100 μm.
- Western Blot of PSA‐treated uninfected and PG127 infected hovS cells. Increasing concentration of PSA leads to a more prominent reduction of PrPSc . PK‐western blot is quantified relative to DMSO.
- Western blot showing HNRNPK shRNA transduction (96 h) decreases HNRNPK protein levels while it increases PrPSc in PG127 prion infected hovS cells. ⍺: anti. Quantifications are reported as normalized to Actin and in comparison, to NT. PK‐western blot is quantified relative to NT.
- Brightfield microscopy images of the effect of shRNA mediated HNRNPK downregulation on vacuolation in NBH hovS cells. HNRNPK downregulation in uninfected cells does not yield a vacuolation phenotype. Scale bar = 100 μm.
- Representative Western Blot analysis of the effect of 2 days of PSA treatment on PrPSc and HNRNPK levels in PG127 prion infected hovS cells following 96 h after transduction of control (NT) or HNRNPK targeting shRNAs. PSA treatment does not affect the levels of HNRNPK (upper panel) when compared with DMSO‐treated cells. HNRNPK downregulation significantly increases PrPSc levels. ⍺, anti. PK‐western blot is quantified relative to DMSO NT samples. Data points represent individual experiments. **P = 0.0051 (unpaired t‐test).
- Representative Western Blot analysis of the effect of 5 days of PSA treatment on PrPSc and HNRNPK levels in PG127 prion infected hovS cells following 168 h after transduction of control (NT) or HNRNPK targeting shRNAs. ⍺: anti. PK‐western blot is quantified relative to DMSO NT samples.
- Heat map of the log2 fold‐change of functional targets of Hnrnpk upon siRNA or PSA treatment.
Source data are available online for this figure.
Next, we asked whether PSA indeed works on regulating PrPSc levels through its interaction with HNRNPK. As a knockout was not possible, due to the cell‐essential nature of HNRNPK (Tsherniak et al, 2017) for long‐term suppression, we used the shRNA constructs (HNRNPK‐targeting and NT) and applied PSA at a concentration of 1 μM and started treatment 2 days after lentiviral transduction of shRNAs to allow time for HNRNPK downregulation to take place, and continued PSA treatment for either 2 or 5 days. PK‐digested western blots (Figs EV4F and G) first confirmed that HNRNPK downregulation does significantly increase prion levels and second, PSA's effect after 5 days does seem to be limited when HNRNPK shRNAs are applied, with a three‐fold reduction, upon comparison to NT‐shRNAs (Fig 4H). Moreover, we found that PSA does not alter HNRNPK levels (Fig EV4F and G) and thus its antiprion effect potentially arises through enhancing the activity of HNRNPK.
We then further analyzed the RNAseq data of RML GT‐1/7 cells treated either with siRNAs against Hnrnpk or with PSA. As siRNA treatment increases prion levels and treatment with PSA leads to a decrease in prion levels, we intersected differentially expressed genes in opposing directions for both treatments. No significant enrichment was detected for genes that are upregulated in the siRNA treatment and downregulated in the PSA treatment. However, when genes that are downregulated in the siRNA treatment condition and upregulated in the PSA treatment condition were taken into account, genes related to glucose metabolism showed a significant enrichment (Figs 4I and EV4H), a function previously reported for Hnrnpk (Sun et al, 2018).
Psammaplysene A treatment leads to decrease in prion levels ex vivo and in vivo
We then asked whether a reduction of prions would be possible in a more physiological model system for prion infection and propagation. We found cerebellar organotypic cultured slice (COCS; Falsig & Aguzzi, 2008) to be an ideal model to assess the efficacy of PSA treatment, as it represents a primary cell culture system with all relevant cell types represented as they are in vivo, while allowing easy experimental manipulation in a dish system. Therefore, we infected the cultures with prions using RML and started treatment of 1 μM PSA diluted in culture medium 2 weeks post‐infection. Subsequently the COCS were homogenized and PK‐resistant PrPSc amount was assessed with immunoblotting. We found that PSA treatment following prion infection significantly reduced prion levels (Fig 5A), without altering PrPC levels (Fig 5B) further highlighting Hnrnpk as a limiter of prion propagation. Furthermore, we took advantage of a Drosophila model of prion propagation for the testing of in vivo efficacy of PSA. The model takes advantage of the ectopic overexpression of ovine PrPC in Drosophila melanogaster (Thackray et al, 2018). Following prion infection, which is achieved through feeding the larvae with prion‐containing food, flies produce bona fide prions and suffer from neurotoxicity associated with prions, which can be assessed with a negative geotaxis climbing assay. The experiment was performed as previously described (Thackray et al, 2018) and constant PSA treatment was achieved through feeding flies with the compound over the time course of 40 days. During this period, flies were subjected to a negative geotaxis climbing assay to assess whether locomotor deficits upon prion infection showed any improvement by treatment with PSA. Strikingly, a dose‐dependent improvement of the performance of the animals was observed (Fig 5C). Moreover, to address if the amelioration of the locomotor function corresponds to a reduction of prions, an RT‐QuIC assay using hamster PrP (haPrP) as a substrate was performed to assess the amount of seeding active prions in the fly brains. Therefore, whole‐head homogenates from 20 animals per treatment group were prepared and the samples were diluted 1:20 in PBS prior to their introduction into the RT‐QuIC. Although flies treated with NBH and subsequently fed DMSO as controls yielded the same outcome as the prion‐negative sample, an early peak in ThT signal were observed for flies that have been prion‐infected and were later control fed using DMSO (Fig 5D). Remarkably, a dose‐dependent reduction of seeding active prions was observed in response to PSA treatment of flies infected with prions, as quantified based on the lag time (Shi et al, 2013) corresponding to the negative geotaxis climbing assay, demonstrating that PSA can alter prion levels in vivo in Drosophila.
Figure 5. PSA reduces prion formation ex vivo and in vivo .
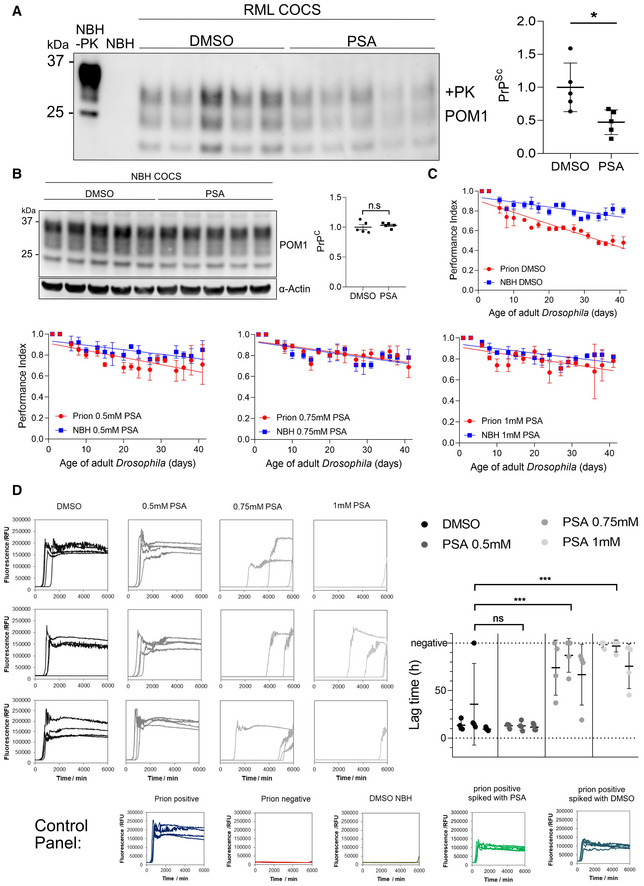
- Western Blot of RML prion infected COCS treated with 1 μM PSA or DMSO. PSA treatment was started 2 weeks after infection and continued until lysis. PSA reduced the amount of PrPSc. Right panel: Quantification of the Western Blot; n = 5 biological replicates. Values: mean ± SD. *P = 0.0211 (Student's t‐test).
- Western Blot analysis of NBH‐treated COCS. PSA treatment was identical to the samples of A. PSA did not affect PrPC expression. Right panel: Quantification of the Western Blot; n = 5 biological replicates. Values represent mean ± SD. n.s., not significant (Student's t‐test). ⍺, anti.
- Negative geotaxis climbing assay in prion infected Drosophila. Flies were treated with DMSO, 0.5 mM PSA, 0.75 mM PSA, or 1 mM PSA at the larval stage and during adulthood for the duration of the assay. Climbing ability was assessed on groups of flies (n = 3 × 15) three times a week and expressed as a performance index. Statistical analysis on the difference between PG127 prion infected versus control prion‐free treatment group data in each graph was performed by an unpaired t‐test: DMSO: P = 0.0002; PSA: 0.5 mM: P = 0.0186; 0.75 mM: P = n.s.; 1 mM: P = n.s.; n.s., not significant. Shown are mean ± SD.
- RT‐QuIC analysis of whole‐head homogenates of prion infected Drosophila. For each sample, 10 male and 10 female heads from the same treatment group were homogenized, 1:20 diluted and applied to the RT‐QuIC. Shown are the RT‐QuIC reactions of three independent homogenates per treatment group, each assessed in quadruplicates. For quantification, the lag‐time of each reaction was calculated and plotted in a graph. Shown are mean ± SD. The assays were performed for 100 h, samples not yielding a positive reaction are considered negative. ***P ≤ 0.0006; n.s., not significant, flies fed with NBH and treated with DMSO were used as a negative control, a standard prion‐free and a prion‐containing sample were used as assay controls. To control for potential interference of DMSO or PSA with the RT‐QuIC reaction, prion positive sample was spiked with 1 μM PSA.
Source data are available online for this figure.
Discussion
Most previous attempts to identify prion modifiers were designed to test individual, biologically plausible candidate genes, which have been successful for example by uncovering the essential role of the B‐cell receptor (Klein et al, 1997) and the CXCR5 chemokine receptor (Prinz et al, 2003) in prion propagation. Here, we attempted a radically different approach by performing a functional genomic screen to identify prion modifiers in cultured cells, with the only ideological constraint that all tested genes are protein‐coding. Classically, prion detection relies on limited proteolysis, e.g. using proteinase K (Bendheim et al, 1984). As this approach is complex and imprecise, and therefore unsuitable to high‐throughput screens, we have developed the QUIPPER, a robust method for the quantitative, sensitive, and scalable detection of prion propagation in cultured cells. The principle of QUIPPER relies on the finding that at steady state most PrPC resides at the cell membrane and can be released by digesting with PIPLC, whereas prions remain associated with the cells (Stahl et al, 1990; Borchelt et al, 1993). We termed the identified PrP species PrPPLC as it represents a mixture of prions and PrPC from the endocytic compartments. To then allow the detection of PrPPLC, sodium hydroxide was added to induce disaggregation, enabling the recognition of the monomers by FRET donor‐acceptor antibodies upon pH neutralization (Pease et al, 2019). Notably, the entire QUIPPER procedure is carried out as a one‐pot assay, which greatly enhances its throughput and allows for extensive automation with programmable liquid handlers. Accordingly, QUIPPER enabled us to perform a whole‐genome functional genomics screen using RNAi in chronically prion‐infected mouse cells. Although we chose to employ QUIPPER in the context of a genetic interference screen, which in the future can be complemented by a gene activation screen and expanded to further cell lines, we believe that it will also be indispensable for future high‐throughput drug discovery campaigns in search of antiprion compounds.
Following testing of the whole genome, a secondary screen consisting of 2,515 candidates was performed and additionally assessed for changes in the levels of PrPC. Unsurprisingly, most of the primary candidates were regulating PrPPLC by affecting the biosynthesis or degradation of PrPC and PrPPLC specific regulators were rare. In addition, regulators of PrPC demonstrated the strongest effects on prion levels, arguing for modulation of PrPC as a potent and valid target in prion diseases (Raymond et al, 2019; Vallabh et al, 2020). Nevertheless, we selected 161 candidates showing an enhanced effect on PrPPLC and assessed those with classical PK digestion to identify modulators of PrPSc, as some of the candidates might affect PrPPLC levels by changing the localization of PrPC, rendering it insensitive to PIPLC digestion. Thereby, we identified 40 hits that modulated prion levels, withstanding different biochemical prion detection methods. Among those, we reassuringly found hits that have been previously associated with prion disease (Marbiah et al, 2014; Wu et al, 2019), confirming the validity of our approach. When interrogated, these 40 hits did not reveal any significant associations with each other, such as working in concert on a known pathway or as interaction partners. However, it did not escape out attention that a group of hits have been previously associated with other neurodegenerative diseases (Chen et al, 2001; Stone et al, 2009; Takano et al, 2014; Banerjee et al, 2017; Schludi et al, 2017; Caminati & Procacci, 2020; Bampton et al, 2021), implying that there might be common host factors responsible for the progression of these ailments. One of them is Pfdn4, a member of the prefoldin complex (Takano et al, 2014), responsible for the folding of native peptides into their functional form. Although the occurrence of cytosolic PrPC and its involvement in the formation of prions remains controversial (Ma & Lindquist, 2001, 2002; Ma et al, 2002), Prefoldin might be shielding cytosolic PrPC from being accessed by prions, thereby preventing its incorporation, as upon downregulation of Pfdn4 we observed a strong increase in PrPSc levels. In addition, we identified several targets (Pof1b, Spire2, Swap70, Tns1, Dock3 and Itga8) that were independently described to be involved in actin binding or dynamics. Such a high prevalence might demonstrate the importance of the host–actin network in the cellular propagation of misfolded proteins. Indeed, actin has been shown to influence prion propagation in yeast (Dorweiler et al, 2020) and might be applicable as well in mammalian cells (preprint: See et al, 2021; Victoria & Zurzolo, 2017). Based on our data, the actin network might be a worthwhile target to further investigate. In addition, previous studies have shown promising results in modulating the propagation of pathological aggregates by interfering with the actin network (Rostami et al, 2017), in which actin may be a major component for cell‐to‐cell transmission of aggregates, through regulating processes such as synaptic vesicle exocytosis and others (Oliveira da Silva & Liz, 2020).
We put efforts forth in investigating Hnrnpk as a prion modulator, due to its strong effect size, ubiquitous expression (Uhlén et al, 2015), and its recent implications in several protein misfolding diseases (Moujalled et al, 2015; Bampton et al, 2021; Sidhu et al, 2022). As Hnrnpk is a cell‐essential gene (Tsherniak et al, 2017), we followed several lines of validation in human and mouse cells as well as different modes of genetic perturbation. In all instances tested, HNRNPK downregulation led to an increase in prion levels, leading to the conclusion that HNRNPK is a host‐factor regulating prion formation, irrespective of prion strain and even species in question. Moreover, in our human cell model for prion propagation, we found that the application of siRNAs and shRNAs targeting HNRNPK, led to a robust increase in prion‐induced cytopathological vacuolation, and targeting PrPC completely abolished it. In addition, a neuroprotective compound, Psammaplysene A, reported to bind Hnrnpk (Boccitto et al, 2017), allowed us to investigate the target pharmacologically. Psammaplysene A (PSA) application in vitro, ex vivo and in vivo, relieved prion propagation and ameliorated prion toxicity in a dose‐dependent manner and therefore acts as a potent inhibitor of prion formation, repeatedly, independent of species in question and prion strain, in contrast to most other anti‐prion drugs, which have been limited in their effect for different prion strains (Ghaemmaghami et al, 2010). The conclusions of this are twofold: First, it demonstrates the possibility and effectiveness of targeting a host‐resident factor in prion diseases. Second, it suggests that different prion strains rely on the same host‐factors for effective propagation, implying that host factors might provide efficient anti‐prion targets independent of prion strains. Although the mode of action of PSA and its effect on Hnrnpk could so far not be clarified, we posit that Hnrnpk's endogenous anti‐prion function is enhanced upon PSA treatment, as we found no difference in Hnrnpk levels following PSA treatment. One limitation of our study is showing the dependence of the effect of PSA on Hnrnpk, which has proven to be difficult due to the essentiality of Hnrnpk. However, considering PSA only binds to Hnrnpk in the presence of RNA (Boccitto et al, 2017), therefore, potentially interfering with its RNA binding properties, RNA‐binding of Hnrnpk might be crucial for limiting prion propagation by limiting the amount of free RNA which has been described as a potential scaffolding factor for the formation of prions (Deleault et al, 2003). Alternatively, as Hnrnpk is predominantly localized to the nucleus, it may be acting specifically as a limiting factor for nuclear PrPSc formation (Mangé et al, 2004). In addition, our transcriptomics data point towards an involvement of the glucose metabolism, for which Hnrnpk has been associated with (Sun et al, 2018). Further investigation of the function of or mimicry of the effect of PSA might provide a novel therapeutic approach for prion diseases and potentially other neurodegenerative diseases, which is of great interest. Moreover, we are confident that other prion modifiers discovered in this study will be of great use to investigate cellular mechanisms of prion propagation, and perhaps represent general cellular host‐factors of protein aggregation.
Materials and Methods
Cell culture
GT‐1/7 cells (Accession ID: CVCL_0281, Mellon et al, 1990), CAD5 cells (CATH‐A‐Differentiated, Accession ID: CVCL_0199, Mahal et al, 2008) and hovS cells (ovinized SH‐SY5Y cells, produced in house) were cultured in T75 or T150 tissue culture flasks (TPP, Trasadingen, Switzerland) in OptiMEM containing Phenol unless stated otherwise (Gibco, Thermo Fisher Scientific, Waltham, MA, USA). As supplements 10% of FBS (Takata, Göteborg, Sweden), 1% of Non‐Essential Amino Acids (NEAA, Gibco), 1% GlutaMaX (Gibco) and 1% of Penicillin/Streptomycin mix (P/S, Gibco) were used. During the screening process phenol was omitted from the media using a no‐Phenol formulation for OptiMEM (Gibco), to eliminate a potential interference with the TR‐FRET readout. During harvesting of the cells Accutase (Gibco) was used and collection of the detached cells was done using 1× phosphate‐buffered saline (PBS, Kantonsapotheke Zurich, Switzerland or Gibco) followed by centrifugation at 330 g (Sorvall Legend XT, Thermo Fisher Scientific) for 4 min with the aim of eliminating dead cells. Later, cell counting was done using trypan blue (Gibco). For culturing hovS cells, 400 μg/ml geneticin (G418 sulfate, Life Technologies, Gibco) was added to the media. For freezing of the cell culture stocks, cells were resuspended in Bambanker Freezing Medium (LubioScience, Zurich, Switzerland) or DMSO (Sigma Aldrich, St. Louis, MO, USA) containing 10% FBS and stored in −80°C or a liquid nitrogen tank, respectively.
Prion infection of cells
For infection of cells with different strains of prions, a previously established protocol was followed (Avar et al, 2020). Cells were seeded in 6‐well plates and the infection occurred for all prion strains (22L, RML, PG127) at the same weight/volume ratio of 0.25% brain homogenate containing prions or non‐infectious brain homogenate (NBH) in a total culture media volume of 1.5 ml. Cells were incubated together with the infectious material for 3 days, followed by continuous splitting for eight passages to ensure a state of persistent prion infection was achieved.
siRNA library preparation and screening
The whole genome Silencer Mouse siRNA library Version 3, consisting of three unique siRNAs targeting each annotated 17,582 mouse gene (52,746 siRNAs) was purchased from Thermo Fisher Scientific in a lyophilized format amounting to 0.25 nmol/siRNA. The library was resuspended, pooled and aliquoted in house as described (Heinzer et al, 2021) to a final concentration of 5 μM (1.67 μM each). For the screening procedure, the aliquoted pooled library was reformatted into the destination plates (384‐well Culture Plate, Perkin Elmer, Beaconsfield, UK) along with the Prnp targeting (Thermo Fisher Scientific) positive controls, NT controls (Thermo Fisher Scientific) serving as negative control and a cell death‐inducting control (Qiagen, Hilden, Germany) to make up a final concentration of 20 nM. Plates were frozen at −40°C until further use. On the assay day, plates were thawed and 5 μl of RNAiMAX (Invitrogen, Carlsbad, CA, USA) diluted in culture media without antibiotics (3% v/v) to a final concentration of 0.5% was dispensed with a Biotek MultiFlo FX multi‐drop dispenser (Vinooski, VT, USA), followed by centrifugation (1,000 rpm, 1 min, Eppendorf 5804R, Hamburg, Germany) and subsequently the plates were incubated for 30 min at room temperature (RT). Later, cells (RML GT‐1/7 or 22L GT‐1/7, NBH GT‐1/7, RML CAD5, NBH CAD5) were seeded on top of the siRNA‐RNAiMAX mixture at a density of 12,500 cells/well (3,000 cells/well for RML CAD5 and NBH CAD5) in a total volume of 25 μl to achieve reverse transfection. Cells were incubated in a rotating tower incubator (LiCONiC StoreX STX, Schaanwald, Liechtenstein) for 70 h and were removed for viability measurements. To measure the viability of cells, 10 μl of 4× concentrated Realtime‐Glo (RT‐Glo) reagent (Promega, Madison, WI, USA) diluted in antibiotic‐free medium was dispensed. In addition, for the screening rounds involving a PIPLC readout, the enzyme was spiked into the media containing the viability reagent at a dilution of 1:800 and plates were returned to the rotating incubator for further incubation for another 2 h. Subsequently, the luminescent signal was read out in an EnVision multimode plate reader (Perkin Elmer) at 37°C. For the sample preparation with the PIPLC readout, culture media was aspirated from the plates using a Biotek EL406 plate washer and lysed with 13 μl of lysis buffer containing 0.5% Na‐deoxycholate (Sigma Aldrich), 0.5% Triton‐X (Sigma Aldrich) and EDTA‐free cOmplete Mini protease inhibitor cocktail (Roche, Basel, Switzerland). The lysis of the contents of the wells was performed on a plate shaker (Eppendorf Thermo Mixer Comfort) for 10 min at 4°C at 500 rpm shaking, followed by an additional incubation step at 4°C in still standing position for 1 h. B. cereus PIPLC was produced in house as described previously (Ryan et al, 1996; Hornemann et al, 2004) in E. coli. For the PK readout, the protease inhibitor was omitted from the lysis buffer contents. Instead, the lysis buffer was spiked with a 2.5 μg/ml final concentration of PK, dispensed 10 μl, incubated for 30 min on a plate shaker at 37°C with shaking, followed by addition of 3 μl of final 2.3 mM phenylmethylsulfonyl fluoride (PMSF, a serine hydrolase inhibitor; Sigma Aldrich) diluted in PBS/isopropanol (Sigma Aldrich) mixture and incubated on a plate shaker for 10 min at RT and 500 rpm shaking conditions. For both the PIPLC and the PK readouts, the following steps involving denaturation and neutralization for sample preparation as well as the TR‐FRET procedure for the final readout were identical. Denaturation was performed by dispensing 2 μl of 0.5 M NaOH to a final concentration of 66.6 mM and plates were shaken with 500 rpm at RT for a total of 2 min followed by 8 min more of incubation time in still standing position. Denaturation of the fibrils was followed by a neutralization step to adjust the pH of the contents of the wells prior to the addition of the antibodies. 3 μl of 0.5 M NaH2PO4, was added to make up a final concentration of 83.3 mM and plates were shaken once more with 500 rpm at RT for a total of 2 min followed by 7 min more of incubation time in still standing position. For the read‐out of PrPC levels in the deconvolution screen, the usage of PIPLC, as well as the subsequent sample preparation steps were omitted. Instead, cells were lysed in 18 μl of lysis buffer. Finally, for all the approaches, 9 μl of TR‐FRET antibody pairs, POM1 (Polymenidou et al, 2008) coupled to Allophycocyanin (APC) and POM19 coupled to Europium (EU) as previously described (Ballmer et al, 2017; Heinzer et al, 2021) were added to a final concentration of 5 and 2.5 nM, respectively. After each dispensing step during sample preparation and TR‐FRET, plates were centrifuged at 1,000 rpm for 1 min. Plates were returned to the fridge for incubation overnight and read out on the following day on the EnVision (Perkin Elmer) plate reader with the previously described parameters (Ballmer et al, 2017).
Data analysis
Sample size was estimated, and screening data was analyzed based on a previous study (Heinzer et al, 2021). Data quality and robustness was assessed at several steps during the analysis pipeline. Heatmaps were visually inspected for any occurrence of gradients, arising through dispensing errors. Wells with prominent dispensing errors were excluded from further analyses during the primary screen. Z'‐Factor (Zhang et al, 1999; Zhang, 2011) was computed for each plate as a metric for separability of the positive (Prnp targeting siRNAs) and negative (NT siRNAs) controls. Net‐FRET and z‐score values for each candidate were calculated and regressed onto each other for assessing the reproducibility of the duplicates. Hit selection was based on the z‐scores of each gene, as explained in the results section of this manuscript. Results were depicted using GraphPad Prism.
RNA‐Seq experiments
RNeasy Mini Kit (Qiagen) was used for RNA‐extraction according to the manufacturer's guidelines. Libraries were prepped with the Illumina TruSeq stranded mRNA protocol (Illumina, San Diego, CA, USA) and quality control (QC) was assessed on the Agilent 4200 TapeStation System (Agilent Technologies, Santa Clara, CA, USA). Subsequently, libraries were pooled equimolecular and sequenced on the Illumina NovaSeq6000 platform with single‐end 100 bp reads. Sequencing depth was around 20 million reads per sample. Experiments were run in biological duplicates, unless otherwise stated. Data analysis was done using a previously established pipeline (Hatakeyama et al, 2016; Dataset EV1). Pathway analysis for significantly enriched cellular processes or components were done using WEB‐based GEne SeT AnaLysis Toolkit (WebGestalt; Wang et al, 2017) using the KEGG functional category version 88.2 dated January 11, 2018.
siRNA transfections and PSA treatment in large‐well format
Cells were seeded at a density of 500,000 cells/well for RML GT‐1/7 and 450,000 cells/well for PG127 hovS in 1.5 ml of culture medium in 6‐well plates (TPP). Next day, media was exchanged with optiMEM (Gibco) with no antibiotics. To enable efficient transfection, RNAiMAX (Thermo Fisher Scientific) with the same concentration as in the screening process was added (final conc. For hovS: 0.3%). Next day, siRNAs diluted in water were mixed with RNAiMAX and a final concentration of 20 μM (10 μM for hovS) was administered to the culture media in a dropwise manner to achieve forward transfection. Incubation lasted 72 or 96 h, as indicated per experiment. Media containing siRNAs was aspirated and cells were washed once with 1× PBS (Kantonsapotheke) and lysed for downstream analysis. Imaging of the cells was performed using a Nikon T2 Eclipse (Nikon, Tokyo, Japan) microscope and images were processed using ImageJ (Schneider et al, 2012). PSA (Aobious Inc., Gloucester, MA, USA) was freshly prepared at the beginning of any experiment by serial dilution in DMSO. For each concentration, one aliquot per day of treatment was stored at −20°C. The desired final concentrations of PSA were prepared daily by diluting 1 to 1,000 the DMSO stock aliquot in culture media. In the PSA experiments, the cells were seeded in a total volume of 1.5 ml media in six‐well plates at a density of 300,000 PG127 and NBH hovS cells 450,000 and RML and NBH GT‐1/7 cells/well. From the following day for the next 5 days, daily medium changes were performed with freshly prepared media at differing concentrations of PSA.
Immunoblotting
Lysed cells were centrifuged at 2,000 g for 5 min. BCA assay (Pierce, Thermo Fisher Scientific) was used to measure the total protein content of each sample and for all downstream analysis involving proteins, sample volume was adapted to contain the same amount. For immunoblotting, samples were prepared, diluted to achieve the same total protein concentration, and digested using PK (final concentration 2.5 μg/ml). Digestion was stopped with boiling the samples after addition of 1 mM final Dithiothreitol (DTT, Bio‐Rad, Hercules, CA, USA) in NuPAGE 4× LDS loading buffer (Thermo Fisher Scientific). Samples were then loaded onto a NuPAGE 4–12% Bis‐Tris gradient gel (Invitrogen, Thermo Fisher Scientific) and blotted onto a nitrocellulose membrane using the iBlot dry transfer system (Invitrogen, Thermo Fisher Scientific). Membrane was blocked using 5% SureBlock (LubioScience) diluted in 1× PBS containing 0.1% Tween‐20 (PBST, Sigma Aldrich) for 30 min. Membranes were then incubated with primary antibodies diluted in 1% SureBlock‐PBST (POM1, POM2 as anti‐PrP antibody, 300 ng/ml final concentration; Polymenidou et al, 2008), anti‐Hnrnpk antibody (ab70492, 1:5,000 diluted, Abcam, Cambridge, UK), anti‐Actin antibody (MAB1501, 1:10,000 diluted, Millipore Sigma, Darmstadt, Germany) overnight at 4°C under shaking conditions. For detection, anti‐mouse HRP or anti‐rabbit HRP (Bio‐Rad) was diluted 1:5,000 in 1% SureBlock‐PBST. Imaging was done on LAS‐3000 System (Fujifilm, Tokyo, Japan).
Cerebellar slice culture experiments
Animal experiments were performed in accordance with the Swiss Animal Protection law and under a permit issued by the Canton of Zurich (Nr: 236/2019). Cerebellar slices were prepared from 12‐day‐old Tga20 mouse pups according to our published protocol (Falsig & Aguzzi, 2008). Briefly, acutely dissected cerebella were embedded in 2% low melting point agarose and cut into 350‐μm thick sections with a Leica vibratome in ice‐cold Gey's balanced salt solution (GBSS) supplemented with kynurenic acid and glucose. For prion infection, slices were exposed to 0.001% brain homogenate derived from terminally sick RML6 prion inoculated mice (or normal brain homogenate as control) for 1 h at 4°C on a shaker. After several washes, six to eight slices were put onto a Millicell‐CM Biopore PTFE membrane insert (Merck Millipore) and cultured in an incubator on top of slice culture medium containing 50% vol/vol MEM, 25% vol/vol basal medium Eagle, 25% vol/vol horse serum, 0.65% w/vol glucose, 1% vol/vol penicillin/streptomycin and 1% Glutamax. Culture medium was changed three times per week. To treat the cultured slices with PSA, stock PSA solution prepared in DMSO was diluted into the culture medium with a final concentration of 1 μM. Culture medium with the same concentration of DMSO was used as control. Fresh PSA was supplied to the media and media was changed every 2–3 days for a total duration of 1 month. At the end of the experiments, cultured slices were collected into the RIPA buffer and homogenized. Protein concentrations in the lysates were quantified using the BCA method. To detect prions by western blotting, lysates containing 50 μg total proteins were mixed with proteinase K (with a final concentration of 20 μg/ml) in a reaction volume of 30 μl and incubated at 37°C for 30 min. After mixing each digested sample with 10 μl 4× loading buffer and boiled at 95°C for 5 min, 18 μl of each sample was loaded onto the gel for western blotting. Quantification was done using ImageJ software and results were plotted using GraphPad Prism8. To assess statistical significance an unpaired Student's t‐test was performed.
SCEPA for detection of infectivity in PK‐ and PIPLC‐treated cells
For assessing whether prion infectivity is intact after treatment with PK (Roche) or PIPLC (Thermo Fisher Scientific) 100.000 RML CAD5 cells were seeded in 2 ml of full culture media and grown to confluency for 4 days. Later, samples for PIPLC were treated with 0.1 U/ml of the enzyme which was spiked into the media and placed back in the incubator for 1 h. Cells were then harvested in 1× PBS, centrifuged at 1,500 g for 5 min for pelleting and finally the PBS was aspirated. 40 μl 1× PBS was added to the cell pellet and lysis of the contents of the tube was followed with 5 freeze–thaw cycles in liquid nitrogen with vortexing. Subsequently, the lysate was centrifuged at 1,000 g for 3 min and supernatant amounting to 50 μl was transferred to a new Eppendorf tube. For PK digestion, 10 μl of 15 μg/ml PK was added to the cells and digestion followed for 30 min at 37°C. Digestion was stopped using 5 μl of PMSF (Sigma Aldrich; final concentration 2.3 mM) under the same conditions described in the screening section of the manuscript. Untreated lysates as well as PIPLC‐treated samples were harvested as described above and 15 μl PBS was added to the samples to make up the same volume. SCEPA was performed as previously described (Sorce et al, 2020).
shRNA design, cloning, lentiviral vector preparation and transduction
Using NEBuilder HiFi DNA Assembly Cloning Kit (NEB #E5520), eGFP coding sequence (Addgene # 17397) was cloned into our Auto‐TDP‐43‐HA LV transfer vector (preprint: Hruska‐Plochan et al, 2021), replacing TDP‐43‐HA, making Auto‐EGFP. Custom MHP_shRNA cassette was synthetized by GenScript so that human U6 promoter is directly followed by random sequence of 54 bp flanked by HindIII and PacI restriction sites. TTTTTTT was used for efficient PolIII termination (Gao et al, 2018) and TGTGCTT for loop (Jensen et al, 2012). This cassette was then cloned into Auto‐EGFP via HiFi assembly, upstream from TRE promoter, generating pSHE LV transfer vector.
HNRNPK transcript variant 5 (NM_001318186.2) was then used as an input sequence for shRNA design using the following algorithms: Broad Institute GPP Web Portal, Invitrogen BLOCK‐iT™ RNAi Designer, Kay Lab siRNA/shRNA/Oligo Optimal Design tool and RNAinverse server of ViennaRNA Web Services. Five highest‐scoring target sequences per algorithm were kept and 2 final target sequences were selected using rational design following the standard rules for pre‐miRNA‐like shRNA design (Bofill‐De Ros & Gu, 2016), including Dicer loop‐counting rule (Gu et al, 2012). Q5 site directed mutagenesis (NEB #E0554S) was used to clone the designed shRNAs into pSHE, substituting the random sequence of the original cassette, generating pSHE‐shHNRNPKa and pSHE‐shHNRNPKb LV transfer vectors. shRNA targeting the HaloTag sequence was then designed as a non‐targeting shRNA control and was cloned into pSHE as described above, generating pSHE‐shHaloTag.
pSHE vectors were then packaged into lentivirus (LV) as described previously (preprint: Hruska‐Plochan et al, 2021). The resulting lentiviral pellets were then resuspended in PBS to achieve 10× concentrated LV preparations, which were titrated using Lenti‐X™ GoStix™ Plus (Takara #631280). 10× concentrate of pSHE LVs was then used at 300 ng (of lentiviral p24 protein as per GoStix Value (GV)) per well of a 6 well plate of hovS cells pipetting the LV concentrate directly onto the culture (dropwise). Spent hovS media was then added to reach 1,000 μl total. Medium was exchanged completely the following day.
HNRNPK knockdown efficacy of pSHE‐shHNRNPKa and pSHE‐shHNRNPKb was assessed in SH‐SY5Y cells and pSHE‐shHNRNPKa was selected for hovS experiments. For immunoblotting of transduced cells, see above.
Primer list: shHNRNPKa_F.
5′‐TGCTTAAGCATTCCACAGCATCTTTTTTTAATTAACATGGTCCCAGC‐3′.
shHNRNPKa_R.
5′‐AGCACAGCTTAAGCATTCCACAGCATCAAGCTTTCGTCCTTTCCAC‐3′.
shHNRNPKb_F.
5′‐TGCTTAAACCACCAACAATAACTTTTTTTAATTAACATGGTCCCAGC‐3′.
shHNRNPKb_R.
5′‐AGCACAGCTTAAACCACCAACAATAACAAGCTTTCGTCCTTTCCAC‐3′.
shHaloTag_F.
5′‐TGCTAAATGCAATACCTTTGACTTTTTTTAATTAACATGGTCCCAGC‐3′.
shHaloTag_R.
5′‐AGCACAGCTAAATGCAATACCTTTGACAAGCTTTCGTCCTTTCCAC‐3′.
RT‐QuIC assay
The reaction buffer of the RT‐QuIC consisted of 1 mM EDTA (Life Technologies), 10 μM thioflavin T, 170 mM NaCl, and 1× PBS (incl. 130 mM NaCl) and HaPrP23‐231 filtered using 100‐kD centrifugal filters (Pall Nanosep OD100C34) at a concentration of 0.1 mg/ml. Fly brain homogenates were diluted in PBS and 2 μl of the diluted homogenates were used to assess seeding activity, resuspended in 98ul of assay buffer in a 96‐well plate format. The experimenter was blinded. The plate was loaded into a FLUOstar Omega plate reader (BMG Labtech) and the shaking cycles were set as follows: 7× (90 s shaking; 900 rpm [double orbital]; 30 s rest) and 60 s reading. Reading was carried out with excitation at 450 nm and emission at 480 nm every 15 min. The amplification was performed at 42°C for 105 h. Four replicates per sample were measured. Lag time was determined as timepoint at which the sample reached 20,000 RFU.
Fly stocks
The UAS‐PrP fly line w; M{VRQ‐PrP(GPI), 3xP3‐RFP.attP}ZH‐51D transgenic for V136R154Q171 (VRQ) ovine PrP, expressed with an N‐terminal leader peptide and C‐terminal GPI signal sequence, was generated by PhiC31 site‐specific transformation (Thackray et al, 2012) with Cre‐mediated removal of the red fluorescent protein (RFP) as described previously (Thackray et al, 2014). The Elav‐GAL4(P{w[+mW.hs] = GawB}elav[C155]) pan neuronal driver fly line was obtained from the Department of Genetics, University of Cambridge, UK. Drosophila were raised on standard cornmeal media at 25°C and maintained at low to medium density.
PSA treatment and prion inoculation of Drosophila
PSA was prepared in DMSO to give 0, 0.5, 0.75 or 1 mM final concentration of drug. Two hundred microliters of the relevant dilution of PSA or DMSO alone were added to the top of fly feed in 3‐inch plastic vials and the feed allowed to dry for 24 h prior to use. A cross between the UAS‐PrP fly line and the Elav‐GAL4 driver fly line was set up in each of the drug‐treated or control fly food vials described above. The parental flies were removed from the vials once first instar larvae were evident. For prion inoculation, 250 μl of 1% (w/v) PG127 scrapie‐positive sheep brain homogenate (Andréoletti et al, 2011) prepared in PBS pH 7.4, were added to drug‐treated third instar VRQ ovine PrP Drosophila larvae. Following eclosion (i.e. hatching), flies were collected that were transgenic for VRQ ovine PrP expressed pan neuronally and were transferred to fresh drug‐treated vials every other day for the duration of the study (40 days). The locomotor ability of flies was assessed in a negative geotaxis climbing assay as previously described (Thackray et al, 2018). Drosophila head homogenate was prepared from 5 and 40 day old flies as previously described (Thackray et al, 2018) and subjected to RT‐QuIC analysis as described in the materials and methods. Statistical analysis of the negative geotaxis climbing assay data was performed by the unpaired t‐test, using Prism (GraphPad Software Inc., San Diego, USA).
Author contributions
Merve Avar: Conceptualization; data curation; formal analysis; validation; investigation; visualization; methodology; writing – original draft; project administration; writing – review and editing. Daniel Heinzer: Conceptualization; data curation; formal analysis; validation; investigation; visualization; methodology; writing – original draft; project administration; writing – review and editing. Alana M Thackray: Data curation; formal analysis; visualization; methodology; writing – review and editing. Yingjun Liu: Data curation; formal analysis; visualization; methodology; writing – review and editing. Marian Hruska‐Plochan: Data curation; formal analysis; visualization; methodology; writing – review and editing. Stefano Sellitto: Data curation; formal analysis; validation; visualization; methodology; writing – review and editing. Elke Schaper: Software. Daniel Patrick Pease: Methodology. Jiang‐An Yin: Resources. Asvin KK Lakkaraju: Conceptualization. Marc Emmenegger: Resources; methodology. Marco Losa: Resources. Andra Chincisan: Software. Simone Hornemann: Resources; writing – review and editing. Magdalini Polymenidou: Conceptualization; resources; writing – review and editing. Raymond Bujdoso: Conceptualization; formal analysis; visualization; writing – review and editing. Adriano Aguzzi: Conceptualization; supervision; funding acquisition; writing – original draft; project administration; writing – review and editing.
Disclosure and competing interests statement
The authors declare that they have no conflict of interest.
Supporting information
Expanded View Figures PDF
Dataset EV1
Source Data for Expanded View
Source Data for Figure 1
Source Data for Figure 4
Source Data for Figure 5
PDF+
Acknowledgements
A.A. is the recipient of an Advanced Grant of the European Research Council (ERC Prion2020, 670958) and grants from the Swiss National Research Foundation (179040), the Nomis Foundation, the Swiss Personalized Health Network (SPHN, 2017DRI17), and a donation from the estate of Dr. Hans Salvisberg. We would like to thank Dr. Emilio Yangüez and Dr. Maria Domenica Moccia and the Functional Genomics Center Zurich (FGCZ) for their help with the RNA‐Sequencing experiment. We thank Dr. M. Ryan and Dr. O.H. Griffith for providing us with the expression system for B. cereus PIPLC. We thank Prof. Simon Mead for help with the analysis of the sCJD dataset. We thank Prof. Robert Kalb and Dr. Marco Boccitto for insightful discussions. In addition, we wholeheartedly thank Irina Abakumova and Rita Moos for their technical help. Summary Fig 1A depicting the QUIPPER assay and Fig EV1C have been created with BioRender.
The EMBO Journal (2022) 41: e112338
Data availability
All the data presented are available in this study and no data were deposited in external repositories. The analysis pipeline for the screening can be accessed online at https://github.com/elkeschaper/hts.
References
- Aguzzi A, Calella AM (2009) Prions: protein aggregation and infectious diseases. Physiol Rev 89: 1105–1152 [DOI] [PubMed] [Google Scholar]
- Andréoletti O, Orge L, Benestad SL, Beringue V, Litaise C, Simon S, Le Dur A, Laude H, Simmons H, Lugan S et al (2011) Atypical/Nor98 scrapie infectivity in sheep peripheral tissues. PLoS Pathog 7: e1001285 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Atarashi R, Sano K, Satoh K, Nishida N (2011) Real‐time quaking‐induced conversion: a highly sensitive assay for prion detection. Prion 5: 150–153 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Avar M, Heinzer D, Steinke N, Doğançay B, Moos R, Lugan S, Cosenza C, Hornemann S, Andréoletti O, Aguzzi A (2020) Prion infection, transmission, and cytopathology modeled in a low‐biohazard human cell line. Life Sci Alliance 3: e202000814 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bai N, Hayashi H, Aida T, Namekata K, Harada T, Mishina M, Tanaka K (2013) Dock3 interaction with a glutamate‐receptor NR2D subunit protects neurons from excitotoxicity. Mol Brain 6: 22 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ballmer BA, Moos R, Liberali P, Pelkmans L, Hornemann S, Aguzzi A (2017) Modifiers of prion protein biogenesis and recycling identified by a highly parallel endocytosis kinetics assay. J Biol Chem 292: 8356–8368 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bampton A, Gatt A, Humphrey J, Cappelli S, Bhattacharya D, Foti S, Brown AL, Asi Y, Low YH, Foiani M et al (2021) HnRNP K mislocalisation is a novel protein pathology of frontotemporal lobar degeneration and ageing and leads to cryptic splicing. Acta Neuropathol 142: 609–627 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Banerjee M, Datta M, Bhattacharyya NP (2017) Modulation of mutant huntingtin aggregates and toxicity by human myeloid leukemia factors. Int J Biochem Cell Biol 82: 1–9 [DOI] [PubMed] [Google Scholar]
- Bellingham SA, Coleman LA, Masters CL, Camakaris J, Hill AF (2009) Regulation of prion gene expression by transcription factors SP1 and metal transcription factor‐1. J Biol Chem 284: 1291–1301 [DOI] [PubMed] [Google Scholar]
- Bendheim PE, Barry RA, DeArmond SJ, Stites DP, Prusiner SB (1984) Antibodies to a scrapie prion protein. Nature 310: 418–421 [DOI] [PubMed] [Google Scholar]
- Birmingham A, Selfors LM, Forster T, Wrobel D, Kennedy CJ, Shanks E, Santoyo‐Lopez J, Dunican DJ, Long A, Kelleher D et al (2009) Statistical methods for analysis of high‐throughput RNA interference screens. Nat Methods 6: 569–575 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boccitto M, Lee N, Sakamoto S, Spruce LA, Handa H, Clardy J, Seeholzer SH, Kalb RG (2017) The neuroprotective marine compound Psammaplysene a binds the RNA‐binding protein HNRNPK. Mar Drugs 15: 246 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bofill‐De Ros X, Gu S (2016) Guidelines for the optimal design of miRNA‐based shRNAs. Methods 103: 157–166 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bolton DC, McKinley MP, Prusiner SB (1982) Identification of a protein that purifies with the scrapie prion. Science 218: 1309–1311 [DOI] [PubMed] [Google Scholar]
- Borchelt DR, Rogers M, Stahl N, Telling G, Prusiner SB (1993) Release of the cellular prion protein from cultured cells after loss of its glycoinositol phospholipid anchor. Glycobiology 3: 319–329 [DOI] [PubMed] [Google Scholar]
- Caminati G, Procacci P (2020) Mounting evidence of FKBP12 implication in neurodegeneration. Neural Regen Res 15: 2195–2202 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen Q, Yoshida H, Schubert D, Maher P, Mallory M, Masliah E (2001) Presenilin binding protein is associated with neurofibrillary alterations in Alzheimer's disease and stimulates tau phosphorylation. Am J Pathol 159: 1597–1602 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen Q, Kimura H, Schubert D (2002) A novel mechanism for the regulation of amyloid precursor protein metabolism. J Cell Biol 158: 79–89 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen Q, Peto CA, Shelton GD, Mizisin A, Sawchenko PE, Schubert D (2009) Loss of modifier of cell adhesion reveals a pathway leading to axonal degeneration. J Neurosci 29: 118–130 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deleault NR, Lucassen RW, Supattapone S (2003) RNA molecules stimulate prion protein conversion. Nature 425: 717–720 [DOI] [PubMed] [Google Scholar]
- Deleault NR, Piro JR, Walsh DJ, Wang F, Ma J, Geoghegan JC, Supattapone S (2012a) Isolation of phosphatidylethanolamine as a solitary cofactor for prion formation in the absence of nucleic acids. Proc Natl Acad Sci U S A 109: 8546–8551 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deleault NR, Walsh DJ, Piro JR, Wang F, Wang X, Ma J, Rees JR, Supattapone S (2012b) Cofactor molecules maintain infectious conformation and restrict strain properties in purified prions. Proc Natl Acad Sci U S A 109: E1938–E1946 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Derkatch IL, Bradley ME, Hong JY, Liebman SW (2001) Prions affect the appearance of other prions: the story of [PIN(+)]. Cell 106: 171–182 [DOI] [PubMed] [Google Scholar]
- Dery MA, Jodoin J, Ursini‐Siegel J, Aleynikova O, Ferrario C, Hassan S, Basik M, LeBlanc AC (2013) Endoplasmic reticulum stress induces PRNP prion protein gene expression in breast cancer. Breast Cancer Res 15: R22 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dorweiler JE, Oddo MJ, Lyke DR, Reilly JA, Wisniewski BT, Davis EE, Kuborn AM, Merrill SJ, Manogaran AL (2020) The actin cytoskeletal network plays a role in yeast prion transmission and contributes to prion stability. Mol Microbiol 114: 480–494 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Enari M, Flechsig E, Weissmann C (2001) Scrapie prion protein accumulation by scrapie‐infected neuroblastoma cells abrogated by exposure to a prion protein antibody. Proc Natl Acad Sci U S A 98: 9295–9299 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Falsig J, Aguzzi A (2008) The prion organotypic slice culture assay‐‐POSCA. Nat Protoc 3: 555–562 [DOI] [PubMed] [Google Scholar]
- Gao Z, Herrera‐Carrillo E, Berkhout B (2018) Delineation of the exact transcription termination signal for type 3 polymerase III. Mol Ther Nucleic Acids 10: 36–44 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ghaemmaghami S, May BC, Renslo AR, Prusiner SB (2010) Discovery of 2‐aminothiazoles as potent antiprion compounds. J Virol 84: 3408–3412 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gu S, Jin L, Zhang Y, Huang Y, Zhang F, Valdmanis PN, Kay MA (2012) The loop position of shRNAs and pre‐miRNAs is critical for the accuracy of dicer processing in vivo . Cell 151: 900–911 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hatakeyama M, Opitz L, Russo G, Qi W, Schlapbach R, Rehrauer H (2016) SUSHI: an exquisite recipe for fully documented, reproducible and reusable NGS data analysis. BMC Bioinformatics 17: 228 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heinz DW, Ryan M, Bullock TL, Griffith OH (1995) Crystal structure of the phosphatidylinositol‐specific phospholipase C from Bacillus cereus in complex with myo‐inositol. EMBO J 14: 3855–3863 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heinzer D, Avar M, Pease DP, Dhingra A, Yin JA, Schaper E, Doğançay B, Emmenegger M, Spinelli A, Maggi K et al (2021) Novel regulators of PrPC biosynthesis revealed by genome‐wide RNA interference. PLoS Pathog 17: e1010013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hornemann S, Schorn C, Wüthrich K (2004) NMR structure of the bovine prion protein isolated from healthy calf brains. EMBO Rep 5: 1159–1164 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hruska‐Plochan M, Betz KM, Ronchi S, Wiersma VI, Maniecka Z, Hock E‐M, Laferriere F, Sahadevan S, Hoop V, Delvendahl I et al (2021) Human neural networks with sparse TDP‐43 pathology reveal NPTX2 misregulation in ALS/FTLD. bioRxiv 10.1101/2021.12.08.471089 [PREPRINT] [DOI] [Google Scholar]
- Jansen IE, Savage JE, Watanabe K, Bryois J, Williams DM, Steinberg S, Sealock J, Karlsson IK, Hägg S, Athanasiu L et al (2019) Genome‐wide meta‐analysis identifies new loci and functional pathways influencing Alzheimer's disease risk. Nat Genet 51: 404–413 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jensen SM, Schmitz A, Pedersen FS, Kjems J, Bramsen JB (2012) Functional selection of shRNA loops from randomized retroviral libraries. PLoS One 7: e43095 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jones E, Hummerich H, Viré E, Uphill J, Dimitriadis A, Speedy H, Campbell T, Norsworthy P, Quinn L, Whitfield J et al (2020) Identification of novel risk loci and causal insights for sporadic Creutzfeldt‐Jakob disease: a genome‐wide association study. Lancet Neurol 19: 840–848 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kampmann M (2018) CRISPRi and CRISPRa screens in mammalian cells for precision biology and medicine. ACS Chem Biol 13: 406–416 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klein MA, Frigg R, Flechsig E, Raeber AJ, Kalinke U, Bluethmann H, Bootz F, Suter M, Zinkernagel RM, Aguzzi A (1997) A crucial role for B cells in neuroinvasive scrapie. Nature 390: 687–690 [DOI] [PubMed] [Google Scholar]
- Klöhn PC, Stoltze L, Flechsig E, Enari M, Weissmann C (2003) A quantitative, highly sensitive cell‐based infectivity assay for mouse scrapie prions. Proc Natl Acad Sci U S A 100: 11666–11671 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Knowles TPJ, Waudby CA, Devlin GL, Cohen SIA, Aguzzi A, Vendruscolo M, Terentjev EM, Welland ME, Dobson CM (2009) An analytical solution to the kinetics of breakable filament assembly. Science 326: 1533–1537 [DOI] [PubMed] [Google Scholar]
- Kryndushkin D, Wickner RB (2007) Nucleotide exchange factors for Hsp70s are required for [URE3] prion propagation in Saccharomyces cerevisiae. Mol Biol Cell 18: 2149–2154 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li B (2016) Establishment of an automated digital prion infectivity cell assay and PrP‐HPFRET based high‐throughput siRNA screening platform. Doctoral Dissertation, University of Zurich, Zurich, Switzerland. [Google Scholar]
- Lloyd SE, Mead S, Collinge J (2013) Genetics of prion diseases. Curr Opin Genet Dev 23: 345–351 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma J, Lindquist S (2001) Wild‐type PrP and a mutant associated with prion disease are subject to retrograde transport and proteasome degradation. Proc Natl Acad Sci U S A 98: 14955–14960 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma JY, Lindquist S (2002) Conversion of PrP to a self‐perpetuating PrPSc‐like conformation in the cytosol. Science 298: 1785–1788 [DOI] [PubMed] [Google Scholar]
- Ma JY, Wollmann R, Lindquist S (2002) Neurotoxicity and neurodegeneration when PrP accumulates in the cytosol. Science 298: 1781–1785 [DOI] [PubMed] [Google Scholar]
- Mahal SP, Demczyk CA, Smith EW, Klohn PC, Weissmann C (2008) Assaying prions in cell culture: the standard scrapie cell assay (SSCA) and the scrapie cell assay in end point format (SCEPA). Methods Mol Biol 459: 49–68 [DOI] [PubMed] [Google Scholar]
- Mangé A, Crozet C, Lehmann S, Béranger F (2004) Scrapie‐like prion protein is translocated to the nuclei of infected cells independently of proteasome inhibition and interacts with chromatin. J Cell Sci 117: 2411–2416 [DOI] [PubMed] [Google Scholar]
- Marbiah MM, Harvey A, West BT, Louzolo A, Banerjee P, Alden J, Grigoriadis A, Hummerich H, Kan HM, Cai Y et al (2014) Identification of a gene regulatory network associated with prion replication. EMBO J 33: 1527–1547 [DOI] [PMC free article] [PubMed] [Google Scholar]
- McKinley MP, Bolton DC, Prusiner SB (1983) A protease‐resistant protein is a structural component of the scrapie prion. Cell 35: 57–62 [DOI] [PubMed] [Google Scholar]
- Mead S, Poulter M, Uphill J, Beck J, Whitfield J, Webb TE, Campbell T, Adamson G, Deriziotis P, Tabrizi SJ et al (2009) Genetic risk factors for variant Creutzfeldt‐Jakob disease: a genome‐wide association study. Lancet Neurol 8: 57–66 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mead S, Uphill J, Beck J, Poulter M, Campbell T, Lowe J, Adamson G, Hummerich H, Klopp N, Rückert IM et al (2012) Genome‐wide association study in multiple human prion diseases suggests genetic risk factors additional to PRNP. Hum Mol Genet 21: 1897–1906 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mellon PL, Windle JJ, Goldsmith PC, Padula CA, Roberts JL, Weiner RI (1990) Immortalization of hypothalamic GnRH neurons by genetically targeted tumorigenesis. Neuron 5: 1–10 [DOI] [PubMed] [Google Scholar]
- Mohr S, Bakal C, Perrimon N (2010) Genomic screening with RNAi: results and challenges. Annu Rev Biochem 79: 37–64 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moujalled D, James JL, Yang S, Zhang K, Duncan C, Moujalled DM, Parker SJ, Caragounis A, Lidgerwood G, Turner BJ et al (2015) Phosphorylation of hnRNP K by cyclin‐dependent kinase 2 controls cytosolic accumulation of TDP‐43. Hum Mol Genet 24: 1655–1669 [DOI] [PubMed] [Google Scholar]
- Nalls MA, Blauwendraat C, Vallerga CL, Heilbron K, Bandres‐Ciga S, Chang D, Tan M, Kia DA, Noyce AJ, Xue A et al (2019) Identification of novel risk loci, causal insights, and heritable risk for Parkinson's disease: a meta‐analysis of genome‐wide association studies. Lancet Neurol 18: 1091–1102 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nuvolone M, Aguzzi A, Heikenwalder M (2009) Cells and prions: a license to replicate. FEBS Lett 583: 2674–2684 [DOI] [PubMed] [Google Scholar]
- Oliveira da Silva MI, Liz MA (2020) Linking alpha‐synuclein to the actin cytoskeleton: consequences to neuronal function. Front Cell Dev Biol 8: 787 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parkyn CJ, Vermeulen EG, Mootoosamy RC, Sunyach C, Jacobsen C, Oxvig C, Moestrup S, Liu Q, Bu G, Jen A et al (2008) LRP1 controls biosynthetic and endocytic trafficking of neuronal prion protein. J Cell Sci 121: 773–783 [DOI] [PubMed] [Google Scholar]
- Pease D, Scheckel C, Schaper E, Eckhardt V, Emmenegger M, Xenarios I, Aguzzi A (2019) Genome‐wide identification of microRNAs regulating the human prion protein. Brain Pathol 29: 232–244 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peretz D, Scott MR, Groth D, Williamson RA, Burton DR, Cohen FE, Prusiner SB (2001) Strain‐specified relative conformational stability of the scrapie prion protein. Protein Sci 10: 854–863 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Polymenidou M, Moos R, Scott M, Sigurdson C, Shi YZ, Yajima B, Hafner‐Bratkovic I, Jerala R, Hornemann S, Wuthrich K et al (2008) The POM monoclonals: a comprehensive set of antibodies to non‐overlapping prion protein epitopes. PLoS One 3: e3872 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prinz M, Heikenwalder M, Junt T, Schwarz P, Glatzel M, Heppner FL, Fu YX, Lipp M, Aguzzi A (2003) Positioning of follicular dendritic cells within the spleen controls prion neuroinvasion. Nature 425: 957–962 [DOI] [PubMed] [Google Scholar]
- Prusiner SB, Cochran SP, Groth DF, Downey DE, Bowman KA, Martinez HM (1982) Measurement of the scrapie agent using an incubation time interval assay. Ann Neurol 11: 353–358 [DOI] [PubMed] [Google Scholar]
- Raymond GJ, Zhao HT, Race B, Raymond LD, Williams K, Swayze EE, Graffam S, Le J, Caron T, Stathopoulos J et al (2019) Antisense oligonucleotides extend survival of prion‐infected mice. JCI Insight 5: e131175 [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Rheenen W, Shatunov A, Dekker AM, McLaughlin RL, Diekstra FP, Pulit SL, van der Spek RA, Võsa U, de Jong S, Robinson MR et al (2016) Genome‐wide association analyses identify new risk variants and the genetic architecture of amyotrophic lateral sclerosis. Nat Genet 48: 1043–1048 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rostami J, Holmqvist S, Lindström V, Sigvardson J, Westermark GT, Ingelsson M, Bergström J, Roybon L, Erlandsson A (2017) Human astrocytes transfer aggregated alpha‐synuclein via tunneling nanotubes. J Neurosci 37: 11835–11853 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ryan M, Smith MP, Vinod TK, Lau WL, Keana JF, Griffith OH (1996) Synthesis, structure‐activity relationships, and the effect of polyethylene glycol on inhibitors of phosphatidylinositol‐specific phospholipase C from Bacillus cereus. J Med Chem 39: 4366–4376 [DOI] [PubMed] [Google Scholar]
- Rybner C, Hillion J, Sahraoui T, Lanotte M, Botti J (2002) All‐trans retinoic acid down‐regulates prion protein expression independently of granulocyte maturation. Leukemia 16: 940–948 [DOI] [PubMed] [Google Scholar]
- Saborio GP, Permanne B, Soto C (2001) Sensitive detection of pathological prion protein by cyclic amplification of protein misfolding. Nature 411: 810–813 [DOI] [PubMed] [Google Scholar]
- Sanchez‐Juan P, Bishop MT, Kovacs GG, Calero M, Aulchenko YS, Ladogana A, Boyd A, Lewis V, Ponto C, Calero O et al (2014) A genome wide association study links glutamate receptor pathway to sporadic Creutzfeldt‐Jakob disease risk. PLoS One 10: e0123654 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schludi MH, Becker L, Garrett L, Gendron TF, Zhou Q, Schreiber F, Popper B, Dimou L, Strom TM, Winkelmann J et al (2017) Spinal poly‐GA inclusions in a C9orf72 mouse model trigger motor deficits and inflammation without neuron loss. Acta Neuropathol 134: 241–254 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schneider CA, Rasband WS, Eliceiri KW (2012) NIH image to ImageJ: 25 years of image analysis. Nat Methods 9: 671–675 [DOI] [PMC free article] [PubMed] [Google Scholar]
- See SK, Chen M, Bax S, Tian R, Woerman A, Tse E, Johnson IE, Nowotny C, Muñoz EN, Sengstack J et al (2021) PIKfyve inhibition blocks endolysosomal escape of α‐synuclein fibrils and spread of α‐synuclein aggregation. bioRxiv 10.1101/2021.01.21.427704 [PREPRINT] [DOI] [Google Scholar]
- Shi S, Mitteregger‐Kretzschmar G, Giese A, Kretzschmar HA (2013) Establishing quantitative real‐time quaking‐induced conversion (qRT‐QuIC) for highly sensitive detection and quantification of PrPSc in prion‐infected tissues. Acta Neuropathol Commun 1: 44 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shyu WC, Harn HJ, Saeki K, Kubosaki A, Matsumoto Y, Onodera T, Chen CJ, Hsu YD, Chiang YH (2002) Molecular modulation of expression of prion protein by heat shock. Mol Neurobiol 26: 1–12 [DOI] [PubMed] [Google Scholar]
- Sidhu R, Gatt A, Fratta P, Lashley T, Bampton A (2022) HnRNP K mislocalisation in neurons of the dentate nucleus is a novel neuropathological feature of neurodegenerative disease and ageing. Neuropathol Appl Neurobiol 48: e12793 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Solassol J, Crozet C, Lehmann S (2003) Prion propagation in cultured cells. Br Med Bull 66: 87–97 [DOI] [PubMed] [Google Scholar]
- Sorce S, Nuvolone M, Russo G, Chincisan A, Heinzer D, Avar M, Pfammatter M, Schwarz P, Delic M, Müller M et al (2020) Genome‐wide transcriptomics identifies an early preclinical signature of prion infection. PLoS Pathog 16: e1008653 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stahl N, Borchelt DR, Prusiner SB (1990) Differential release of cellular and scrapie prion proteins from cellular membranes by phosphatidylinositol‐specific phospholipase C. Biochemistry 29: 5405–5412 [DOI] [PubMed] [Google Scholar]
- Stone TW, Ceruti S, Abbracchio MP (2009) Adenosine receptors and neurological disease: neuroprotection and neurodegeneration. Handb Exp Pharmacol: 535–587 10.1007/978-3-540-89615-9_17 [DOI] [PubMed] [Google Scholar]
- Sun Z, Zhu M, Lv P, Cheng L, Wang Q, Tian P, Yan Z, Wen B (2018) The Long noncoding RNA Lncenc1 maintains naive states of mouse ESCs by promoting the glycolysis pathway. Stem Cell Reports 11: 741–755 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tachi N, Hashimoto Y, Matsuoka M (2012) MOCA is an integrator of the neuronal death signals that are activated by familial Alzheimer's disease‐related mutants of amyloid β precursor protein and presenilins. Biochem J 442: 413–422 [DOI] [PubMed] [Google Scholar]
- Takano M, Tashiro E, Kitamura A, Maita H, Iguchi‐Ariga SM, Kinjo M, Ariga H (2014) Prefoldin prevents aggregation of α‐synuclein. Brain Res 1542: 186–194 [PubMed] [Google Scholar]
- Taraboulos A, Raeber AJ, Borchelt DR, Serban D, Prusiner SB (1992) Synthesis and trafficking of prion proteins in cultured cells. Mol Biol Cell 3: 851–863 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taylor R (1990) Interpretation of the correlation coefficient: a basic review. J Diagn Med Sonogr 6: 35–39 [Google Scholar]
- Thackray AM, Muhammad F, Zhang C, Di Y, Jahn TR, Landgraf M, Crowther DC, Evers JF, Bujdoso R (2012) Ovine PrP transgenic Drosophila show reduced locomotor activity and decreased survival. Biochem J 444: 487–495 [DOI] [PubMed] [Google Scholar]
- Thackray AM, Di Y, Zhang C, Wolf H, Pradl L, Vorberg I, Andréoletti O, Bujdoso R (2014) Prion‐induced and spontaneous formation of transmissible toxicity in PrP transgenic Drosophila . Biochem J 463: 31–40 [DOI] [PubMed] [Google Scholar]
- Thackray AM, Andréoletti O, Bujdoso R (2018) Mammalian prion propagation in PrP transgenic Drosophila . Brain 141: 2700–2710 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsherniak A, Vazquez F, Montgomery PG, Weir BA, Kryukov G, Cowley GS, Gill S, Harrington WF, Pantel S, Krill‐Burger JM et al (2017) Defining a cancer dependency map. Cell 170: 564–576.e16 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Uhlén M, Fagerberg L, Hallström BM, Lindskog C, Oksvold P, Mardinoglu A, Sivertsson Å, Kampf C, Sjöstedt E, Asplund A et al (2015) Proteomics. Tissue‐based map of the human proteome. Science 347: 1260419 [DOI] [PubMed] [Google Scholar]
- Vallabh SM, Minikel EV, Schreiber SL, Lander ES (2020) Towards a treatment for genetic prion disease: trials and biomarkers. Lancet Neurol 19: 361–368 [DOI] [PubMed] [Google Scholar]
- Victoria GS, Zurzolo C (2017) The spread of prion‐like proteins by lysosomes and tunneling nanotubes: implications for neurodegenerative diseases. J Cell Biol 216: 2633–2644 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vincent B, Sunyach C, Orzechowski HD, St George‐Hyslop P, Checler F (2009) p53‐dependent transcriptional control of cellular prion by presenilins. J Neurosci 29: 6752–6760 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang J, Vasaikar S, Shi Z, Greer M, Zhang B (2017) WebGestalt 2017: a more comprehensive, powerful, flexible and interactive gene set enrichment analysis toolkit. Nucleic Acids Res 45: W130–W137 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu W, Zhao D, Shah SZA, Zhang X, Lai M, Yang D, Wu X, Guan Z, Li J, Zhao H et al (2019) OPA1 overexpression ameliorates mitochondrial cristae remodeling, mitochondrial dysfunction, and neuronal apoptosis in prion diseases. Cell Death Dis 10: 710 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang XD (2011) Illustration of SSMD, z score, SSMD*, z* score, and t statistic for hit selection in RNAi high‐throughput screens. J Biomol Screen 16: 775–785 [DOI] [PubMed] [Google Scholar]
- Zhang JH, Chung TD, Oldenburg KR (1999) A simple statistical parameter for use in evaluation and validation of high throughput screening assays. J Biomol Screen 4: 67–73 [DOI] [PubMed] [Google Scholar]