

Abstract

P-glycoprotein (P-gp) is found to be of considerable interest for the design of drugs capable of treating chemoresistant tumors. This transporter is an interesting target for which an efficient approach has not yet been developed in terms of computer simulation. In this work, we use a combination of docking, molecular dynamics, and metadynamics to fully explore the states that occur during the capture of a ligand and subsequent efflux by P-gp. The proposed approach allowed us to substantiate a number of experimentally established facts, as well as to develop a new criterion for identifying potential P-gp inhibitors.
1. Introduction
The important role that P-glycoprotein (P-gp) plays in the human body’s functioning has drawn the attention of researchers and is reflected in a number of papers published annually on this topic. The most characteristic P-gp function is the protection of organs and individual cells due to the release of various xenobiotics through the membrane. This transporter possesses a low specificity; its substrates include various endogenous substrates, dyes, and drugs.1−3
P-gp (∼170 kDa) consists of two pseudosymmetrical parts, and each includes a cytoplasmic ATP-binding domain and six hydrophobic transmembrane alpha-helices that form the substrate translocation pathway4−6 (Figure 1). The efflux is provided by the binding and subsequent hydrolysis of two ATP molecules within the nucleotide-binding domains (NBDs). The binding of ATP molecules promotes rearrangements in the transmembrane domains (TMDs) and changes in the protein conformation required for the transport of substances across the membrane.7
Figure 1.

At the moment, there is no single concept of substance transfer; vacuum cleaner and flippase model are among the key models.8,9 However, NBD dimerization and switch are clearly the key steps.10 It is worth noting that the transport cycle is usually considered from the apo state with ligand capture, followed by the binding of two ATP molecules.9,10 There are also diverging views on the role of ATP. Although most researchers discuss about the need to bind and convert ATP to ADP for conformational rearrangements,7,11 a number of works consider ATP hydrolysis as an energy source.9,12
Many experimental data show that some small molecules are able to suppress the efflux of classical P-gp substrates, which is of interest from the point of view, for example, of tumor chemoresistance suppression, as it is the activity of this mechanism that is the key way to reduce the sensitivity of tumor cells to drugs.13,14
At the moment, a number of effective P-gp inhibitors have been proposed; all of them were obtained by the comprehensive optimization (HTS, QSAR, and combinatorial chemistry) of compounds that showed the ability to compete for P-gp binding in addition to the main biological effect.15,16 However, failures in clinical trials clearly indicate the need to create the next generation of inhibitors, which requires a transition to the rational design of compounds and the development of a set of criteria to identify potential inhibitors.
From the point of view of computational methods, almost the only approach is to substantiate the experimentally established activity using docking.17,18 For example, the authors9 substantiate the ability of tyrosine kinase inhibitors to suppress doxorubicin efflux by the fact that they bind to the same site according to the docking results.
It should be noted that in the case of rigid targets, such as Mdm219−21 or AMPK,22−24 docking clearly proves its effectiveness and high predictive power. However, the structure of P-gp has a number of fundamental differences. Binding sites for several classical substrates were historically identified within the structure of the transporter: R-, H-, and so forth.25−27 At the same time, almost the entire inner surface of the protein has the ability to interact with the ligand, which makes it extremely difficult to clearly identify a specific affinity site for docking. Moreover, the transport activity of the protein is determined by significant changes in the tertiary structure that accompany the transfer of small molecule compounds through the membrane, which also complicates the choice of the most relevant fixed state required for docking.
Through the implementation of a comprehensive approach to the study of P-gp, we propose a new look at the process of small molecule compound transfer and inhibitor identification, which allows not only to substantiate previously obtained practical results but also to implement rational drug development approaches starting from the stage of molecular modeling.
2. Results and Discussion
2.1. Identification of Inhibitors by Docking
Small molecule ligands can bind to P-gp over a large surface, entering P-gp both directly from the membrane and from the cytosol.8,28−30 Although three main sites are known (H site, R site,25,31 and modulator M site4), various binding options have been proposed even for high-affinity P-gp substrates.32 The location of the H-site is more controversial; fluorescence resonance energy transfer investigations suggested a location of Hoechst 33342 in the cytoplasmic/inner leaflet side of the transporter,33 while the use of a pharmacophore pattern for the same dye suggested a location within the outer leaflet.34
We chose two areas for docking, which include the sites mentioned in various works, and also first considered the area formed at the final stage of the transported substance efflux on the extracellular side of the transporter (Figure 2). Two structures were used, respectively: the most cited murine P-gp crystallographic structure at the inward-facing unbound (apo) conformation (PDB ID: 4Q9H)35 and human P-gp in the ATP-bound, outward-facing conformation (PDB ID: 6C0V).12
Figure 2.

Three docking areas as exemplified by bis-benzimide.
We used two groups of substances (Figure 3), expecting to see differences between them: high-affinity P-gp substrates (bis-benzimide, rhodamine 123, and doxorubicin) and advanced P-gp inhibitors (tariquidar, zosuquidar, and elacridar).
Figure 3.

Considered compounds: high-affinity P-gp substrates (A); third generation P-gp inhibitors (B).
According to the calculations, both bis-benzimide and rhodamine 123 tend to bind to site II, which is weakly consistent with the literature data.32,36 Similarly, the calculations do not explain the highest efficiency of zosuquidar observed in vitro (IC50 = 10 μM vs 28 and 29 μM for other inhibitors),37 as there is no difference in docking at site II, and at sites I and III, zosuquidar shows weak results. In addition, the scatter of the obtained binding energy values for the studied compounds at the inward sites I and II does not exceed 2.5 kcal/mol (Figure 4).
Figure 4.

Docking results. Energetics of ligand interactions with different docking areas (A); ligand interactions with P-gp alpha helices, as exemplified by bis-benzimide (B).
Thus, none of the sites, taken separately, allows one to demonstrate an unambiguous advantage of inhibitors over substrates. However, analyzing the set of results, it can be assumed that inhibitors have a greater affinity to the P-gp cavity and are weakly bound to P-gp from the extracellular side compared with substrates. Such a comprehensive approach, despite the absence of a pronounced difference in the binding energy, can be applied during the design of P-gp modulators. The formulated criterion for the selection of small molecule compounds as P-gp inhibitors and substrates is in line with the conclusions on the fact that inhibitors and substrates differ in the ratio of rates of elimination and reuptake by the cell membrane, made in the work.38
Despite the formulated criterion obtained based on the docking results, the analysis of binding poses of the studied compounds showed that the binding occurs simultaneously with several TMD alpha-helices (Figure 4B). This fact can ultimately significantly affect the obtained docking results given the high conformational mobility of P-gp.
2.2. Simulation of P-gp Transport Cycle Dynamics
In the case of such a conformationally labile target as the transport protein that provides the transfer of substances due to changes in its spatial structure, it becomes practically unescapable to use methods that consider its dynamics. The analysis of conformational rearrangements of a target protein in the presence of a small molecule ligand often allows one to formulate the criteria for the design of targeted molecules.39 Hence, we performed a computational study of P-gp by the methods of molecular dynamics and metadynamics.
2.2.1. Role of Nucleotides in P-gp Structuring and Flexibility
Efflux requires the binding of ATP molecules to P-gp. Although there is a sufficient amount of ATP in the cell, it is assumed that ATP uptake occurs after the binding of a substrate.9,10 Nevertheless, the structures of P-gp presented in PDB are in the inward-facing conformation, which do not contain ATP due to the specifics of their construction.
To analyze the role of nucleotides in P-gp structuring and flexibility, we simulated the dynamics of the entire system in the membrane in the presence and absence of ATP using our own completed protein model (see Materials and Methods).
The results unambiguously demonstrate that ATP stabilizes a sandwich consisting of two NBDs with a layer of two ATP molecules (Figures 5 and 6), while in this state, the transporter can successfully capture substrates by maintaining the dimensions of a fairly spacious internal cavity (Figure 6, green, view II). Thus, when modeling, it is necessary to take into account that, in the cell, where a sufficient amount of ATP is always present, P-gp predominantly exists in this form.
Figure 5.

Flexibility and distance between P-gp NBDs: yellow, apo form of P-gp (efflux requires the binding of two ATP molecules); green, each NBD is bound by one ATP molecule (corresponds to the active form ready for substrate transfer).
Figure 6.

Spatial structure of P-gp: red, XRD data collected in the absence of ATP (PDB ID: 4Q9H); green, dynamic snapshot of homologously extended P-gp in the presence of ATP (ATP not shown); light green, cryo-electron microscopy data collected in the presence of 9 mM Mg2+/ATP (PDB ID: 6C0V, ATP not shown). View I, location of NBDs; view II, formation of a cavity between TMDs.
We managed to reconstruct the processes that occur in the transport protein during the efflux process using metadynamics: we obtained a panel of states through which the protein passes in the presence of ATP (Figures 7 and 8). The free-energy surfaces are presented in the Supporting Information; the analysis of the results is shown in Figure 8.
Figure 7.

P-gp metadynamics in the absence of a substrate. Amino acids used in the construction of the free-energy surface of the system are indicated: red ○—S430, orange ○—K1208, blue ○—E93.
Figure 8.

Energy profiles of P-gp conformational changes depending on the bound nucleotides: dark gray—Apo-P-gp; yellow—P-gp-ATP/ADP; light gray—P-gp-ADP/ADP; green—P-gp-ATP/ATP.
The obtained results again indicate that in the cell the protein is most likely associated with nucleotides, as the apo form is frankly unfavorable (Figure 8). In addition, the computational experiment shows that the binding of two ADP molecules is energetically very close to the binding of two ATP molecules. However, the ATP concentration is significantly higher than that of ADP in the real biological system, and the ATP-bound state should dominate.
In the resulting protein conformations, held together by ATP molecules, there is still enough space to capture a ligand from the cytoplasm or directly from the membrane (Figure 9, green). In this state, a catalytically active P-gp-ATP/ATP complex is formed, and hydrolysis of one ATP molecule, accompanied by a large-scale rearrangement of the transporter, occurs: in the P-gp-ADP/ATP complex (Figure 9, yellow), the substrate-binding cavity contracts, but a channel on the extracellular side of the membrane, previously closed in the P-gp-ATP/ATP state (Figure 9, green), through which efflux is possible, opens.
Figure 9.

Energetically favorable states of the transporter in the presence of the nucleotides: green—P-gp-ATP/ATP; yellow—P-gp-ATP/ADP.
The sandwich held together by ATP/ADP formed after the first hydrolysis turns out to be even more energetically favorable (Figure 8) than P-gp-ATP/ATP, which allows it to hydrolyze the second ATP molecule in the second catalytic center. In turn, the resulting P-gp-ADP/ADP complex is less strengthened, and NBDs diverge, after which ADP molecules are replaced by new ATP molecules to repeat the transport cycle (Figure 10).
Figure 10.

Alternative transport cycles of P-gp.
The data obtained also suggest that a single nucleotide substitution may be sufficient for efflux at the stage of the formation of the P-gp-ADP/ADP complex (Figure 10, dashed arrow), as the resulting P-gp-ATP/ADP complex both energetically and spatially allows ligand binding in the substrate-binding cavity (Figure 9). This assumption explains the phenomenon noted in the course of studying the P-gp interaction with its classical substrate:40 at a concentration of 1.5 mM ATP, 1 mol of triphosphate is consumed for every 0.57 mol of the substrate, while at a lower ATP level (0.3 mM), hydrolysis of 1 mol of ATP releases 0.83 mol of the substrate.
Our results suggest that at a high ATP content, a rapid change of both nucleotides in NBDs is possible, while in a situation of energy deficiency or a significant excess of the transported substrate, the substrate capture without a complete renewal of NBDs with a shift in the ATP/substrate ratio from 2:1 to 1:1 is more likely (Figure 10). This view is consistent with the concepts of alternating catalytic sites scheme41 and unidirectional transport mechanism.42
2.2.2. Role of Ligands in P-gp Conformational Rearrangement
P-gp possesses the basal activity: in a real system, the cycle that we simulated above is realized even in the absence of potential substrates. However, binding to small molecule compounds can significantly change the hydrolysis activity toward both stimulation and suppression.
Analyzing the conformations of the transporter realized in the presence of ligands (Figure 11) that bind to the substrate-binding cavity of P-gp (high-affinity P-gp substrate bis-benzimide and inhibitor tariquidar), we found a significant difference in the energy profile of P-gp interactions with the substrate and inhibitor (Figure 12).
Figure 11.

P-gp metadynamics in the presence of a substrate.
Figure 12.

Energy profiles of P-gp conformational changes depending on the bound nucleotides and compounds: green—P-gp-ATP/ATP; blue—P-gp-ATP/ATP + bis-benzimide; orange—P-gp-ATP/ATP + tariquidar.
The energy profile for tariquidar (an effective P-gp inhibitor) practically corresponds to the profile of the P-gp-ATP/ATP complex (Figure 12): the binding of the inhibitor contributes to a smooth decrease in the free energy of the system in each intermediate state, and the range of energetically favorable states in this case is sufficiently wide and does not show a tendency to collapse (Figure 13A).
Figure 13.

Inhibitor binding evenly increases the energy advantage of all intermediate states (A), while substrate binding promotes the transition to the outward-facing state (B).
The presence of a substrate (bis-benzimide) leads to a significant change in the curve (Figure 12): the energy minimum shifts toward the most collapsed states (Figure 13B). Such a profile is typical for the asymmetric state of P-gp-ATP/ADP (Figure 8), in which the release of the substrate molecule into the extracellular space is possible.
Thus, the system tends to accelerate the rearrangement into the outward-facing state in the presence of a substrate, while the inhibitor contributes to the fact that all states, both outward- and inward-facing, become equally stable (Figure 13).
Thus, the state of the transporter remains as close as possible to the initial P-gp-ATP/ATP in the presence of an inhibitor, and the inhibitor will not be transferred to the extracellular space in the case of ATP hydrolysis in this state (Figure 14, orange). At the same time, the binding of a proper substrate facilitates the processes required for the efflux: the transporter collapses with the opening of a channel for the release of the substance into the extracellular space (Figure 14, blue). The obtained data support the previously stated hypothesis that the inhibitor can act as a spacer between the branches of P-gp, preventing the conformational rearrangements required for the release of a substance.27
Figure 14.
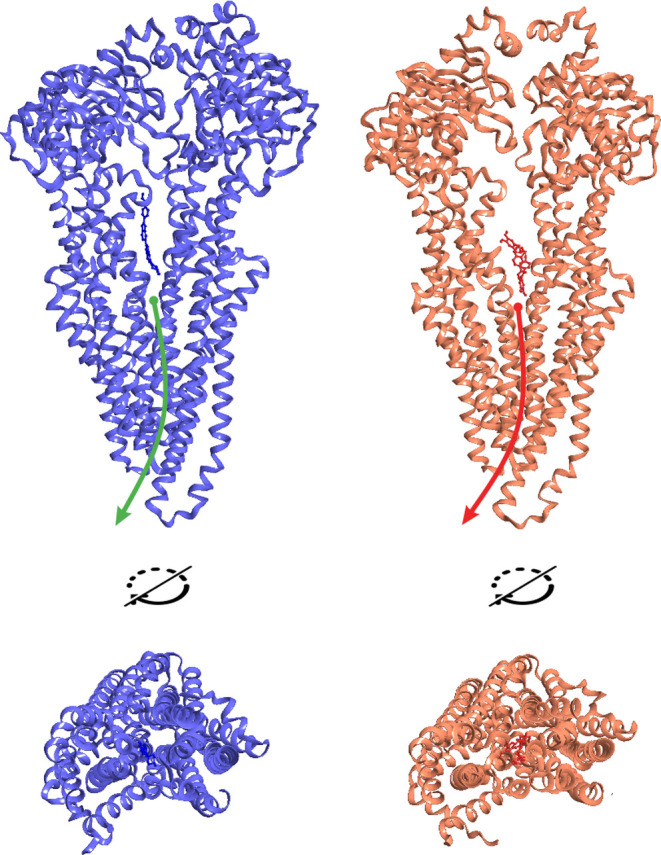
Energetically favorable states of the P-gp-ATP/ATP complex in the presence of bis-benzimide (blue) and tariquidar (orange). In the case of bis-benzimidine, a channel for the release of the substance is formed, while the release of tariquidar is not possible due to the absence of a channel.
3. Conclusions
P-gp has proven to be a difficult target for in silico studies due to its high flexibility. P-gp is able to bind a wide range of compounds, and the very fact of binding does not allow us to conclude about their future fate: the substance can turn out to be both a substrate, which is quickly eliminated from the cell, and an inhibitor, which hinders the conformational rearrangements of P-gp. In this case, classical docking as a part of screening of large libraries has a low predictive power. However, a thorough multiparameter analysis of the docking results can bear fruit. Trends can be judged by the binding difference at several sites or by the presence of specific interactions, such as the appearance of an energetically favorable brace at site III.
We identified characteristic energy profiles that differ significantly in the case of P-gp inhibitors and substrates, which allows one to unambiguously identify them, using metadynamics. It can be expected that metadynamics will soon become a routine screening method as docking, which will open up the possibility for the rational approach to design efflux inhibitors using the methods of computer simulation.
Despite many years of research on efflux transporters, there is currently no unified concept of the transport mechanism. In the course of studying the transporter dynamics, we proposed a scheme that is in good agreement with both the results of crystallography and modeling of other transporters and with the biochemical data on the multifaceted activity of P-gp. In particular, we managed to explain the experimentally established regularity on the nonconstant ratio of the number of hydrolysable ATP molecules in one efflux cycle. Our data also confirm that P-gp practically does not occur in the apo form but is usually associated with nucleotides, and this must be considered when modeling various kinds of interactions.
4. Materials and Methods
4.1. Structure Preparation
Small molecule structures were drawn in ChemBioDraw (PerkinElmer). The generation of 3D structures of small molecules was performed using the LigPrep tool implemented in Schrodinger Suite 2020-4 in the OPLS3e force field, with the generation of tautomers and stereoisomers.43
RCSB Protein Data Bank was used as a source of protein structures.44,45 All the used protein structures were preliminarily prepared using the Protein Preparation Wizard tools implemented in Schrodinger Suite 2020-4.43 Missing hydrogen atoms were added; bond multiplicities, side chains of amino acid residues, hydrogen-bond networks (for models obtained by XRD analysis), and desolvation energies (for models with a solvent from which it was removed) were corrected. The structures PDB ID 4Q9H(35) and 6C0V(12) were used.
In order to eliminate the breaks in the P-gp chains, 6QEX,464M1M,47 and 6FN1(48) were used for homological modeling. Discontinuities were repaired using a combination of homologous and de novo modeling (the Prime tool, Schrodinger Suite 2020-4).49
The membrane model is a phospholipid bilayer (POPE) calculated for a temperature of 310 K (37 °C). The positioning coordinates of the transmembrane part of P-gp were established in accordance with the data presented in the OPM database of transmembrane proteins.50
4.2. Ligand–Protein Docking
To parameterize the binding of small molecules considered as P-gp interactants, we used the method of flexible molecular docking implemented in the Glide program.51
The size of the docking area is consistent with the size of the ligand; the upper size limit is 15 Å. Up to 20 poses were generated for each structure. The optimality of posing was determined based on the GlideScore and Emodel indicators, as well as on the clustering ability of docking solutions.
Docking of small molecule compounds was performed with reference to the following amino acids: Inward site I—M982 and F299; Inward site II—F979, M982, M945 (PDB ID: 4Q9H); Outward site—F336, F739, F971 (PDB ID: 6C0V).
4.3. Molecular Dynamics and Metadynamics
Molecular dynamics and metadynamics of the studied structures were simulated using the Schrodinger Suite 2020-4 software package, the Desmond module;52 molecular systems with an explicitly specified solvent were used.
The protein model in the membrane was placed in an orthorhombic region with a buffer zone of 20 Å from the protein surface. The simulation area was filled with a model solvent simulating saline (water, SPC model53). The charge of the system was neutralized by adding additional sodium or chloride ions. The OPLS3e force field was used.54
All molecular systems were preminimized and equilibrated. Simulation parameters: time, 100 ns; NPT environment; temperature, 310 K; and registration step, 0.02 ns. In the case of metadynamics, the bias potential was set as an increase in energy by 0.03 kcal/mol in 0.09 ps.
Collective variables required to construct the free-energy surface of the system were set as follows: the distance between Cα of selected amino acids S430 and K1208, as well as the angle between Cα of amino acids S430, E93, and K1208. The choice of binding sites is associated with their high rigidity, which reduces the likelihood of conformational noise that leads to blurring of free-energy indicators. S430 and K1208 are located in rigidly structured fragments essential for ATP binding, while E93 reflects the rotation axis, around which NBD-containing half-molecules of P-gp are displaced during the transport cycle.
For each considered system based on P-gp, a free-energy surface was constructed. Points corresponding to the local and absolute free-energy minima of the system, as well as points describing ultimate indicators of the system, were selected for the analysis.
Acknowledgments
The work was supported by the Russian Science Foundation (project no. 19-73-10150). The authors are also grateful to Gureev Maxim (https://orcid.org/0000-0002-0385-922X) for the technical support during computational studies.
Glossary
Abbreviations
- P-gp
P-glycoprotein, MDR1
- TMD
transmembrane domain
- NBD
nucleotide-binding domain
- PDB
Protein Data Bank
- HTS
high-throughput screening
- QSAR
quantitative structure–activity relationship
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acsomega.2c04768.
Energy profiles of conformational changes for P-gp and P-gp complexes with nucleotides and small molecules (PDF)
The authors declare no competing financial interest.
Supplementary Material
References
- Zhou S. F. Structure, function and regulation of P-glycoprotein and its clinical relevance in drug disposition. Xenobiotica 2008, 38, 802–832. 10.1080/00498250701867889. [DOI] [PubMed] [Google Scholar]
- Grigoreva T.; Sagaidak A.; Romanova A.; Novikova D.; Garabadzhiu A.; Tribulovich V. Establishment of drug-resistant cell lines under the treatment with chemicals acting through different mechanisms. Chem.-Biol. Interact. 2021, 344, 109510. 10.1016/j.cbi.2021.109510. [DOI] [PubMed] [Google Scholar]
- Hodges L. M.; Markova S. M.; Chinn L. W.; Gow J. M.; Kroetz D. L.; Klein T. E.; Altman R. B. Very important pharmacogene summary: ABCB1 (MDR1, P-glycoprotein). Pharmacogenet. Genomics 2011, 21, 152–161. 10.1097/fpc.0b013e3283385a1c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferreira R. J.; Ferreira M. J. U.; dos Santos D. J. V. A. Molecular docking characterizes substrate-binding sites and efflux modulation mechanisms within P-glycoprotein. J. Chem. Inf. Model. 2013, 53, 1747–1760. 10.1021/ci400195v. [DOI] [PubMed] [Google Scholar]
- Javed W.; Vallet S.; Clement M. P.; Le Roy A.; Moulin M.; Härtlein M.; Breyton C.; Burlet-Schiltz O.; Marcoux J.; Orelle C.; Ebel C.; Martel A.; Jault J. M. Structural insights into the catalytic cycle of a bacterial multidrug ABC efflux pump. J. Mol. Biol. 2022, 434, 167541. 10.1016/j.jmb.2022.167541. [DOI] [PubMed] [Google Scholar]
- Grigoreva T.; Sagaidak A.; Novikova D.; Tribulovich V. Implication of ABC transporters in non-proliferative diseases. Eur. J. Pharm. 2022, 935, 175327. 10.1016/j.ejphar.2022.175327. [DOI] [PubMed] [Google Scholar]
- Cole S. P. C. Multidrug resistance protein 1 (MRP1, ABCC1), a ″multitasking″ ATP-binding cassette (ABC) transporter. J. Biol. Chem. 2014, 289, 30880–30888. 10.1074/jbc.r114.609248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clay A. T.; Sharom F. J. Lipid bilayer properties control membrane partitioning, binding, and transport of P-glycoprotein substrates. Biochemistry 2013, 52, 343–354. 10.1021/bi301532c. [DOI] [PubMed] [Google Scholar]
- Bender J.; Fang J.; Simon R. A computational study of the inhibition mechanisms of P-glycoprotein mediated paclitaxel efflux by kinase inhibitors. BMC Syst. Biol. 2017, 11, 108. 10.1186/s12918-017-0498-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dei S.; Braconi L.; Romanelli M. N.; Teodori E. Recent advances in the search of BCRP- and dual P-gp/BCRP-based multidrug resistance modulators. Cancer Drug Resist. 2019, 2, 710–743. 10.20517/cdr.2019.31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prieß M.; Göddeke H.; Groenhof G.; Schäfer L. V. Molecular mechanism of ATP hydrolysis in an ABC transporter. ACS Cent. Sci. 2018, 4, 1334–1343. 10.1021/acscentsci.8b00369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim Y.; Chen J. Molecular structure of human P-glycoprotein in the ATP-bound, outward-facing conformation. Science 2018, 359, 915–919. 10.1126/science.aar7389. [DOI] [PubMed] [Google Scholar]
- Lehne G. P-glycoprotein as a drug target in the treatment of multidrug resistant cancer. Curr. Drug Targets 2000, 1, 85–99. 10.2174/1389450003349443. [DOI] [PubMed] [Google Scholar]
- Nanayakkara A. K.; Follit C. A.; Chen G.; Williams N. S.; Vogel P. D.; Wise J. G. Targeted inhibitors of P-glycoprotein increase chemotherapeutic-induced mortality of multidrug resistant tumor cells. Sci. Rep. 2018, 8, 967. 10.1038/s41598-018-19325-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Palmeira A.; Sousa E.; H Vasconcelos M. H.; M Pinto M. M. Three decades of P-gp inhibitors: skimming through several generations and scaffolds. Curr. Med. Chem. 2012, 19, 1946–2025. 10.2174/092986712800167392. [DOI] [PubMed] [Google Scholar]
- Srivalli K. M. R.; Lakshmi P. K. Overview of P-glycoprotein inhibitors: a rational outlook. Braz. J. Pharm. Sci. 2012, 48, 353–367. 10.1590/s1984-82502012000300002. [DOI] [Google Scholar]
- Zahra R.; Furqan M.; Ullah R.; Mithani A.; Saleem R. S. Z.; Faisal A. A cell-based high-throughput screen identifies inhibitors that overcome P-glycoprotein (Pgp)-mediated multidrug resistance. PLoS One 2020, 15, e0233993 10.1371/journal.pone.0233993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Braconi L.; Teodori E.; Contino M.; Riganti C.; Bartolucci G.; Manetti D.; Romanelli M.; Perrone M.; Colabufo N.; Guglielmo S.; Dei S. Overcoming multidrug resistance (MDR): design, biological evaluation and molecular modelling studies of 2,4-substituted quinazoline derivatives. ChemMedChem 2022, 17, e202200027 10.1002/cmdc.202200027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grigoreva T. A.; Novikova D. S.; Gureev M. A.; Garabadzhiu A. V.; Tribulovich V. G. Amino acids as chiral derivatizing agents for antiproliferative substituted N-benzyl isoindolinones. Chirality 2018, 30, 785–797. 10.1002/chir.22854. [DOI] [PubMed] [Google Scholar]
- Grigoreva T. A.; Novikova D. S.; Petukhov A. V.; Gureev M. A.; Garabadzhiu A. V.; Melino G.; Barlev N. A.; Tribulovich V. G. Proapoptotic modification of substituted isoindolinones as MDM2-p53 inhibitors. Bioorg. Med. Chem. Lett. 2017, 27, 5197–5202. 10.1016/j.bmcl.2017.10.049. [DOI] [PubMed] [Google Scholar]
- Krasavin M.; Gureyev M. A.; Dar’in D.; Bakulina O.; Chizhova M.; Lepikhina A.; Novikova D.; Grigoreva T.; Ivanov G.; Zhumagalieva A.; Garabadzhiu A. V.; Tribulovich V. G. Design, in silico prioritization and biological profiling of apoptosis-inducing lactams amenable by the Castagnoli-Cushman reaction. Bioorg. Med. Chem. 2018, 26, 2651–2673. 10.1016/j.bmc.2018.04.036. [DOI] [PubMed] [Google Scholar]
- Novikova D. S.; Garabadzhiu A. V.; Melino G.; Barlev N. A.; Tribulovich V. G. Small-molecule activators of AMP-activated protein kinase as modulators of energy metabolism. Russ. Chem. Bull. 2015, 64, 1497–1517. 10.1007/s11172-015-1036-x. [DOI] [Google Scholar]
- Novikova D. S.; Grigoreva T. A.; Ivanov G. S.; Melino G.; Barlev N. A.; Tribulovich V. G. Activating effect of 3-benzylidene oxindoles on AMPK: from computer simulation to high-content screening. ChemMedChem 2020, 15, 2521–2529. 10.1002/cmdc.202000579. [DOI] [PubMed] [Google Scholar]
- Novikova D. S.; Grigoreva T. A.; Zolotarev A. A.; Garabadzhiu A.; Tribulovich V. G. Advanced palladium free approach to the synthesis of substituted alkene oxindoles via aluminum-promoted Knoevenagel reaction. RSC Adv. 2018, 8, 34543–34551. 10.1039/c8ra07576j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shapiro A. B.; Ling V. Positively cooperative sites for drug transport by P-glycoprotein with distinct drug specificities. Eur. J. Biochem. 1997, 250, 130–137. 10.1111/j.1432-1033.1997.00130.x. [DOI] [PubMed] [Google Scholar]
- Aller S. G.; Yu J.; Ward A.; Weng Y.; Chittaboina S.; Zhuo R. P.; Harrell P. M.; Trinh Y. T.; Zhang Q. H.; Urbatsch I. L.; Chang G. Structure of P-glycoprotein reveals a molecular basis for poly-specific drug binding. Science 2009, 323, 1718–1722. 10.1126/science.1168750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grigoreva T.; Romanova A.; Sagaidak A.; Vorona S.; Novikova D.; Tribulovich V. Mdm2 inhibitors as a platform for the design of P-glycoprotein inhibitors. Bioorg. Med. Chem. Lett. 2020, 30, 127424. 10.1016/j.bmcl.2020.127424. [DOI] [PubMed] [Google Scholar]
- Shapiro A. B.; Corder A. B.; Ling V. P-glycoprotein-mediated Hoechst 33342 transport out of the lipid bilayer. Eur. J. Biochem. 1997, 250, 115–121. 10.1111/j.1432-1033.1997.00115.x. [DOI] [PubMed] [Google Scholar]
- Stein W. D. Kinetics of the P-glycoprotein, the multidrug transporter. Exp. Physiol. 1998, 83, 221–232. 10.1113/expphysiol.1998.sp004106. [DOI] [PubMed] [Google Scholar]
- Homolya L.; Orbán T. I.; Csanády L.; Sarkadi B. Mitoxantrone is expelled by the ABCG2 multidrug transporter directly from the plasma membrane. Biochim. Biophys. Acta, Biomembr. 2011, 1808, 154–163. 10.1016/j.bbamem.2010.07.031. [DOI] [PubMed] [Google Scholar]
- Loo T. W.; Clarke D. M. Location of the rhodamine-binding site in the human multidrug resistance P-glycoprotein. J. Biol. Chem. 2002, 277, 44332–44338. 10.1074/jbc.m208433200. [DOI] [PubMed] [Google Scholar]
- Martinez L.; Arnaud O.; Henin E.; Tao H. C.; Chaptal V.; Doshi R.; Andrieu T.; Dussurgey S.; Tod M.; Di Pietro A.; Zhang Q. H.; Chang G.; Falson P. Understanding polyspecificity within the substrate-binding cavity of the human multidrug resistance P-glycoprotein. FEBS J. 2014, 281, 673–682. 10.1111/febs.12613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qu Q.; Sharom F. J. Proximity of bound Hoechst 33342 to the ATPase catalytic sites places the drug binding site of P-glycoprotein within the cytoplasmic membrane leaflet. Biochemistry 2002, 41, 4744–4752. 10.1021/bi0120897. [DOI] [PubMed] [Google Scholar]
- Pajeva I. K.; Globisch C.; Wiese M. Structure-function relationships of multidrug resistance P-glycoprotein. J. Med. Chem. 2004, 47, 2523–2533. 10.1021/jm031009p. [DOI] [PubMed] [Google Scholar]
- Szewczyk P.; Tao H.; McGrath A. P.; Villaluz M.; Rees S. D.; Lee S. C.; Doshi R.; Urbatsch I. L.; Zhang Q.; Chang G. Snapshots of ligand entry, malleable binding and induced helical movement in P-glycoprotein. Acta Crystallogr., Sect. D: Biol. Crystallogr. 2015, 71, 732–741. 10.1107/s1399004715000978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mamizadeh R.; Hosseinzadeh Z.; Razzaghi-Asl N.; Ramazani A. In silico analysis of a few dietary phytochemicals as potential tumor chemo-sensitizers. Struct. Chem. 2018, 29, 1139–1151. 10.1007/s11224-018-1098-0. [DOI] [Google Scholar]
- Chufan E. E.; Kapoor K.; Ambudkar S. V. Drug-protein hydrogen bonds govern the inhibition of the ATP hydrolysis of the multidrug transporter P-glycoprotein. Biochem. Pharmacol. 2016, 101, 40–53. 10.1016/j.bcp.2015.12.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eytan G. D.; Regev R.; Oren G.; Assaraf Y. G. The role of passive transbilayer drug movement in multidrug resistance and its modulation. J. Biol. Chem. 1996, 271, 12897–12902. 10.1074/jbc.271.22.12897. [DOI] [PubMed] [Google Scholar]
- Gureev M.; Novikova D.; Grigoreva T.; Vorona S.; Garabadzhiu A.; Tribulovich V. Simulation of MDM2 N-terminal domain conformational lability in the presence of imidazoline based inhibitors of MDM2-p53 protein-protein interaction. J. Comput.-Aided Mol. Des. 2020, 34, 55–70. 10.1007/s10822-019-00260-6. [DOI] [PubMed] [Google Scholar]
- Shapiro A. B.; Ling V. Stoichiometry of coupling of rhodamine 123 transport to ATP hydrolysis by P-glycoprotein. Eur. J. Biochem. 1998, 254, 189–193. 10.1046/j.1432-1327.1998.2540189.x. [DOI] [PubMed] [Google Scholar]
- Senior A. E.; al-Shawi M. K.; Urbatsch I. L. The catalytic cycle of P-glycoprotein. FEBS Lett. 1995, 377, 285–289. 10.1016/0014-5793(95)01345-8. [DOI] [PubMed] [Google Scholar]
- Xu Y. Y.; Seelig A.; Bernèche S. Unidirectional transport mechanism in an ATP dependent exporter. ACS Cent. Sci. 2017, 3, 250–258. 10.1021/acscentsci.7b00068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sastry G. M.; Adzhigirey M.; Day T.; Annabhimoju R.; Sherman W. Protein and ligand preparation: parameters, protocols, and influence on virtual screening enrichments. J. Comput.-Aided Mol. Des. 2013, 27, 221–234. 10.1007/s10822-013-9644-8. [DOI] [PubMed] [Google Scholar]
- Berman H. M.; Westbrook J.; Feng Z.; Gilliland G.; Bhat T. N.; Weissig H.; Shindyalov I. N.; Bourne P. E. The Protein Data Bank. Nucleic Acids Res. 2000, 28, 235–242. 10.1093/nar/28.1.235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burley S. K.; Bhikadiya C.; Bi C.; Bittrich S.; Chen L.; Crichlow G. V.; Christie C. H.; Dalenberg K.; Di Costanzo L.; Duarte J. M.; Dutta S.; Feng Z.; Ganesan S.; Goodsell D. S.; Ghosh S.; Green R. K.; Guranović V.; Guzenko D.; Hudson B. P.; Lawson C. L.; Liang Y.; Lowe R.; Namkoong H.; Peisach E.; Persikova I.; Randle C.; Rose A.; Rose Y.; Sali A.; Segura J.; Sekharan M.; Shao C.; Tao Y.-P.; Voigt M.; Westbrook J. D.; Young J. Y.; Zardecki C.; Zhuravleva M. RCSB Protein Data Bank: powerful new tools for exploring 3D structures of biological macromolecules for basic and applied research and education in fundamental biology, biomedicine, biotechnology, bioengineering and energy sciences. Nucleic Acids Res. 2021, 49, D437–D451. 10.1093/nar/gkaa1038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alam A.; Kowal J.; Broude E.; Roninson I.; Locher K. P. Structural insight into substrate and inhibitor discrimination by human P-glycoprotein. Science 2019, 363, 753–756. 10.1126/science.aav7102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li J. Z.; Jaimes K. F.; Aller S. G. Refined structures of mouse P-glycoprotein. Protein Sci. 2014, 23, 34–46. 10.1002/pro.2387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alam A.; Kung R.; Kowal J.; McLeod R. A.; Tremp N.; Broude E. V.; Roninson I. B.; Stahlberg H.; Locher K. P. Structure of a zosuquidar and UIC2-bound human-mouse chimeric ABCB1. Proc. Natl. Acad. Sci. U.S.A. 2018, 115, E1973–E1982. 10.1073/pnas.1717044115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jacobson M. P.; Pincus D. L.; Rapp C. S.; Day T. J.; Honig B.; Shaw D. E.; Friesner R. A. A hierarchical approach to all-atom protein loop prediction. Proteins 2004, 55, 351–367. 10.1002/prot.10613. [DOI] [PubMed] [Google Scholar]
- Lomize M. A.; Pogozheva I. D.; Joo H.; Mosberg H. I.; Lomize A. L. OPM database and PPM web server: resources for positioning of proteins in membranes. Nucleic Acids Res. 2012, 40, D370. 10.1093/nar/gkr703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Repasky M. P.; Shelley M.; Friesner R. A.. Flexible ligand docking with Glide. Current Protocols in Bioinformatics; Wiley Online Library, 2007; Chapter 8, Unit 8 12. [DOI] [PubMed] [Google Scholar]
- Bowers K. J.; Chow E.; Xu H.; Dror R. O.; Eastwood M. P.; Gregersen B. A.; Klepeis J. L.; Kolossvary I.; Moraes M. A.; Sacerdoti F. D.; Salmon J. K.; Shan Y.; Shaw D. E.. Scalable algorithms for molecular dynamics simulations on commodity clusters. Proceedings of the 2006 ACM/IEEE conference on Supercomputing; Association for Computing Machinery: 84: Tampa, Florida, 2006.
- Toukan K.; Rahman A. Molecular-dynamics study of atomic motions in water. Phys. Rev. B: Condens. Matter Mater. Phys. 1985, 31, 2643–2648. 10.1103/physrevb.31.2643. [DOI] [PubMed] [Google Scholar]
- Roos K.; Wu C.; Damm W.; Reboul M.; Stevenson J. M.; Lu C.; Dahlgren M. K.; Mondal S.; Chen W.; Wang L.; Abel R.; Friesner R. A.; Harder E. D. OPLS3e: extending force field coverage for drug-like small molecules. J. Chem. Theory Comput. 2019, 15, 1863–1874. 10.1021/acs.jctc.8b01026. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.