

Abstract
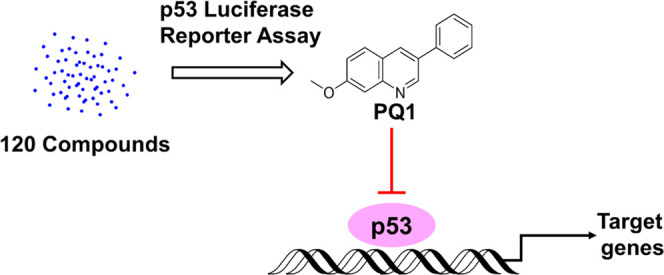
Transcription factor p53 regulates cellular responses to environmental perturbations via the transcriptional activation of downstream target genes. Inappropriate p53 activation can trigger abnormal cellular responses, therefore leading to acute or chronic tissue damage, human developmental syndromes, and neurodegenerative diseases. Antagonists of p53 transcriptional activity provide prospective therapeutic applications and molecular probes. In this article, we identified five 3-phenylquinoline derivatives as potential p53 inhibitors through screening a chemical library consisting of 120 compounds, in which PQ1 was the most active compound. PQ1 had no effect on p53 protein levels and decreased the expression of p53 target gene p21. PQ1 thermally stabilizes the wild-type p53 protein. Further, transcriptomics confirmed that PQ1 exposure generated a similar regulatory effect to transcription profiles with a reported p53 transcriptional inhibitor pifithrin-α. However, compared to pifithrin-α, PQ1 increased the sensitivity of SW480 cells to 5FU. Taken together, PQ1 was a novel antagonist of p53 transcriptional activity. We propose that PQ1 could be developed as a chemical tool to pinpoint the physiological functions of p53 and a novel lead compound for targeting dysfunctional p53 activation.
1. Introduction
The p53 tumor suppressor protein is best known as a transcription factor involved in the cellular response to environmental perturbations.1 Since its discovery over 40 years ago, p53 has been considerably explored and heads the list of 10 most studied genes of all time.2 Given that p53 is the most frequently mutated gene in cancer and the p53 signaling pathway is frequently perturbed in almost all cancers, the most popular investigations about p53 are focused on the development and treatment of tumors.3−5 Mutation of p53 leads to the malfunction of its tumor suppression, making that reactivation of mutant p53 is a promising strategy for efficient cancer therapy.6 Besides, too little p53 activity can result in tumor development, and activation of wild-type p53 is also an attractive strategy for anticancer therapy.7
Counterintuitively, increased p53 activity in normal tissues during cancer therapy causes organ damage and dose-limiting toxicity.8 More importantly, inappropriate p53 activation can trigger cellular responses, such as excessive apoptosis or decreased proliferation, and therefore drives tissue-specific developmental defects, including hematopoietic, neuronal, craniofacial, cardiovascular, and pigmentation defects.9 Besides, p53 activation is also implicated in ischemia/oxidative stress-related tissue damage and neurodegenerative diseases, such as Parkinson, amyotrophic lateral sclerosis, and frontotemporal dementia.10,11 Therefore, inhibition of p53 activation has clinical therapeutic significance for acute or chronic tissue damage, human developmental syndromes, and neurodegenerative diseases.
Until now, only four p53 inhibitors have been developed. Pifithrin-α (PFTα), pifithrin-β, and NSC194598 are antagonists (Figure 1) of p53 transcriptional activity,8,12,13 and pifithrin-μ disrupts p53 function via inhibiting p53 binding to mitochondria.14 In this regard, such an inhibitor has not yet been studied in clinic. Hence, investigations that develop potent and specific p53 inhibitors are crucial for the drug for diseases with inappropriate p53 activation. In this article, we first performed p53 promoter-based luciferase reporter assays to screen compounds that modulate p53 activity. Fortunately, five phenylquinoline derivatives were identified as potent p53 inhibitors from 120 compounds and the most active compound named PQ1. Follow-up experiments revealed that PQ1 and PFTα shared similar regulation of p21 expression, p53 thermal stability, and cellular transcription profiles.
Figure 1.

Structures of antagonists against p53 transcriptional activity in literature studies.
2. Results
2.1. Generation of p53 Inhibitors
In this study, a p53 promoter-based luciferase reporter assay was applied to screen small-molecule inhibitors of p53. For the assay, we established two luciferase reporter cell lines, which express luciferase reporter gene under the transcriptional control of the p53 response element in SW480 and HCT116 cells. SW480 cells express the R273H/P309S mutation of p53 (mutp53 R273H/P309S), and HCT116 cells express wild-type p53 (wtp53). A compound library encompassing 120 small molecules in our laboratory was screened on the basis of luciferase activity (Figure 2A). We successively performed chemical screenings in SW480, HCT116, and 5FU-treated HCT116 cells to check the assay reproducibility and exclude false-positive compounds (Figure 2A). 5FU was used to activate the activity of p53 by inducing its expression in HCT116 cells. These screenings identified 5 top hits (#001, 003, 005, 010, 064) that showed ∼2-fold decreased luciferase activity compared with the DMSO control in the three screenings (Figure 2A). These five compounds shared similar structural scaffolds to phenylquinoline (Figure 2B). In consonance with the well-known p53 inhibitor PFTα, all compounds inhibited p53 activity in a dose-dependent manner (Figure 2C). Compound 001, named PQ1, was more potent than the positive compound PFTα and the other four compounds (Figure 2C). To explore whether these compounds directly decrease the activity of luciferase, NH2 cells expressing HIV LTR-dependent luciferase were applied. The results showed that 001 (PQ1), 003, 005, 010, 064, and PFTα inceased the activity of HIV LTR-dependent luciferase, revealing that these compounds did not inhibit the activity of luciferase (Figure S1). Hence, PQ1 is a potent p53 inhibitor.
Figure 2.

Screening p53 inhibitors. (A) Screening data for 120 compounds in HCT116 and SW480 cells. Five top hits are individually labeled with a black box. (B) Structures of five top hits. (C) Concentration-dependent inhibition of p53 activity of five top hits in HCT116 and SW480 cells. The data are presented as the mean ± SD (n = 3).
2.2. PQ1 Inhibits the Intracellular p53 Transcriptional Activity
It has been demonstrated that both decreasing p53 expression levels and inhibiting p53 transcriptional activity reduced the luciferase activity in p53-dependent luciferase reporter-based assays; we therefore investigated the effect of PQ1 on the expression of p53 and its target gene p21. Given that p53 was expressed at a low level in HCT116 cells without stress stimulation, 5FU was used to increase the expression of p53 and p21 in HCT116 cells via the induction of DNA damage (Figure 3). PQ1 and PFTα inhibited the expression of p21 in 5FU-treated HCT116 cells (Figure 3). Similar downregulation of p21 expression was also observed in SW480 cells in the presence of PQ1 and PFTα (Figure 3). Meanwhile, PQ1 and PFTα had no effect on the expression of p53 in both HCT116 and SW480 cells (Figure 3). Based on these results, in accordance with PFTα, PQ1 inhibited the transcriptional activity of p53 but did not decrease p53 expression levels.
Figure 3.

PQ1 inhibits the intracellular p53 transcriptional activity. (A) Western blot analysis of indicated protein expression in HCT116 and SW480 cells with PQ1 and PFTα treatment. Cells were treated with indicated treatment for 24 h and subjected to western blotting analysis. (B) Relative quantification of western blotting bands in panel (A). The bands of western blotting were subjected to qualitative analysis via Image Lab software. The data are presented as the mean ± SD (n = 3).
In addition, PQ1 induced an increase of γH2AX levels (a protein marker for DNA damage), whereas the positive drug PFTα had no effect (Figure 3). To explore the distinct effect of PQ1 and PFTα on γH2AX in depth, the effects of p53 function deficiency on the γH2AX levels were detected in wild-type p53 harboring cells (HCT116 and LoVo cells) and p53 function-deficient cells (SW480 and HCT116 p53–/– cells). Compared to HCT116 and LoVo cells, SW480 and HCT116 p53–/– cells showed higher expression levels of γH2AX (Figure S2), indicating that p53 function deficiency induced DNA damage. Owing to the fact that PQ1 was more potent than the positive compound PFTα, PQ1 more potently abolished p53 function than PFTα. Thus, it is easy to speculate that PQ1 induced more severe DNA damage than PFTα, which resulted in the distinct effect of PQ1 and PFTα on γH2AX.
2.3. PQ1 Thermally Stabilizes Wild-Type p53
To further investigate whether PQ1 binds directly to the p53 protein, a cellular thermal shift assay (CETSA) was performed to assay for drug cellular uptake and target engagement in vivo. PQ1 and PFTα were analyzed for thermal stabilization of the p53 protein in HCT116 and SW480 cells (Figure 4). The expression levels of wild-type p53 in HCT116 cells were low and undetectable in the condition without stress stimulation. Thus, we applied 5FU to induce the increased expression of wild-type p53, enabling p53 detectability. PQ1 and PFTα stabilized the p53 protein against thermally induced protein aggregation at denaturation temperatures ranging from 41 to 44 °C in HCT116 cells. However, thermostabilization of the p53 protein by PQ1 and PFTα was not observed in SW480 cells. In conclusion, PQ1 and PFTα thermally stabilize the wtp53 protein but not the p53 R275H/P309S mutant protein, indicating that PQ1 directly bound to the p53 protein as well as PFTα in HCT116 cells. Whether PQ1 has an impact on the p53 protein of SW480 cells needs further study.
Figure 4.

CETSA showed that PQ1 and PFTα thermally stabilize wtp53 but not mutp53 R275H/P309S. HCT116 and SW480 cells were treated with PQ1 and PFTα for 3 h and then subjected to CETSA. 5FU was used so that the p53 expression of HCT116 cells could be detected by western blot.
2.4. Effect of PQ1 on the Transcription Profiles of SW480 Cells
To further evaluate the effect of PQ1 on the intracellular p53 in SW480 cells, we performed RNA sequencing to evaluate the effect of PQ1 on the transcription profiles. A total of 459 and 385 DEGs were identified in PQ1- and PFTα-treated SW480 cells, respectively (Figure 5A). Overall, PQ1 and PFTα individually induced 276 and 245 downregulated DEGs and triggered 183 and 140 upregulated DEGs in SW480 cells (Figure 5A). We further identified overlapping genes among PQ1-decreased DEGs, PFTα-decreased DEGs, and p53 target genes using Venn diagram analysis. Venn diagram revealed that more than 80% of these decreased genes were p53 target genes in SW480 cells (Figure 5B), revealing that PQ1 and PFTα showed similar regulation of p53 target genes in SW480 cells.
Figure 5.

Overview of the effect of PQ1 and PFTα on the transcription profiles in SW480 cells. (A) Venn diagram of the number of DEGs induced by PQ1 and PFTα. PQ1- and PFTα-induced total, upregulated, and downregulated DEGs were subjected to Venn diagram analysis. (B) Venn diagram of the number of p53 target genes and DEGs decreased by PQ1 and PFTα. PQ1- and PFTα-decreased DEGs and p53 target genes were subjected to Venn diagram analysis. (C) Cluster heatmap based on the DEGs induced by PQ1. The transcriptional data were subjected to hierarchical clustering analysis.
Indeed, we applied other two cell lines, including HCT116 and HCT116 p53–/– (p53 deletion). Hierarchical clustering and heatmap revealed that similar regulation of gene expression was observed in PQ1- and PFTα-treated cells (Figures 5C and S3). Especially, PQ1 and PFTα distinctly changed the transcription profiles of SW480 and HCT116 cells but have no obvious effect on gene expression profiles in HCT116 p53–/– cells (Figure S4), revealing that the effects of PQ1 and PFTα on cellular gene expression were dependent on p53 levels.
In addition, KEGG enrichment analysis was performed to annotate the total, upregulated, and downregulated DEGs in SW480 cells. The top 20 enriched pathways of PQ1- and PFT-induced DEGs were fairly aggregated together in a grouped graph (Figure 6). The sum number of pathways enriched from total DEGs was 27, 82.5% of which are regulated by both PQ1 and PFTα (Figure 6), revealing that PQ1 and PFTα shared a similar effect on the cellular pathways of SW480 cells.
Figure 6.

KEGG pathways enriched from DEGs induced by PQ1 and PFTα in SW480 cells. DEGs were subjected to KEGG enrichment analysis, and the top 20 enriched pathways of each treatment were fairly aggregated in a grouped graph. P ≤ 0.05 is considered statistically significant.
Taken together, PQ1 was similar to PFTα in regulating cellular transcription profiles, firmly indicating that PQ1 was an antagonist of p53 transcriptional activity as well as PFTα.
2.5. Modeling of the Interaction between p53 and PQ1
To further enhance the understanding of the interaction between p53 and PQ1, we identified the binding site of PQ1 in p53 by molecular docking. PQ1 was docked into the binding site of the p53 DNA binding domain, and the result is shown in Figure 7. The maximum binding affinity between PQ1 and the p53 DNA binding domain was predicted to be −5.0 kcal/mol. The compound PQ1 adopted a compact conformation to bind at the binding pocket of the p53 DNA binding domain. PQ1 was located at the hydrophobic site, surrounded by the residues Leu-114, Val-122, and Pro-142, forming a stable hydrophobic binding. Importantly, one key hydrogen bond interaction was observed between PQ1 and the residue Thr-123, with the bond length of 2.2 Å, which was the main interaction between PQ1 and the p53 DNA binding domain. All of these interactions helped PQ1 to anchor in the binding site of p53 DNA binding domain. In summary, the above molecular simulations give us a rational explanation of the interactions between PQ1 and p53.
Figure 7.

Molecular docking complex between PQ1 and p53 in 3D view: (A) top view and (B) detailed view.
2.6. PQ1 Increased the Sensitivity of SW480 Cells to 5FU
Given that mutant p53 transcriptional effects have been shown to lead to increased cell proliferation and cell death resistance,15 we evaluate the effect of PQ1 on the cell growth and drug sensitivity of SW480 cells. The results showed that PFTα had no obvious cytotoxicity against HCT116 and SW480 cells in the concentration range of 1–50 μM (Figure 8A). However, PQ1 showed weak cytotoxicity against SW480 cells, which was more potent than HCT116 cells (Figure 8A). Then, the antitumor potency of PQ1 or PFTα in combination with 5FU was assessed to evaluate the effect of PQ1 on drug sensitivity. Compared to HCT116 cells, SW480 cells showed resistance to 5FU (Figure 8B). PFTα protected HCT116 cells from the cytotoxicity of 5FU (Figure 8B). At the same time, PQ1 enhanced the cell growth inhibitory activity of 5FU against SW480 cells rather than HCT116 cells (Figure 8B), indicating that PQ1 increased the sensitivity of SW480 cells to 5FU.
Figure 8.

PQ1 increased the sensitivity of SW480 cells to 5FU. (A) Effect of PQ1 on the cell growth of SW480 and HCT116 cells. (B) Effect of PQ1, PFTα, 5FU, and their combination on the cell growth of SW480 and HCT116 cells. Cells were treated with indicated drugs for 72 h and subjected to in vitro antitumor assays. The data were presented as the mean ± SD (n = 3).
3. Discussion
PQ1 belongs to 3-phenylquinoline compounds, a structurally distinct inhibitor of p53 transcriptional activity from reported p53 inhibitors. PFTα is the first identified chemical inhibitor of p53 transcriptional activity and belongs to thiazole imide derivatives (Figure 1). Pifithrin-β (Figure 1) derives from PFTα via cyclization and dehydration and is more potent than PFTα.13 NSC194598 is identified as an inhibitor of p53 DNA binding through a cell-free high-throughput screening assay and is a fluorene derivative (Figure 1). Hence, our research provides a novel scaffold for lead compounds to inhibit p53 transcriptional activity.
In luciferase reporter assays, western blot analysis, and transcriptome sequencing, the effects of PQ1 on HCT116 cells were basically in accordance with those on SW480 cells. However, PQ1 displayed distinct effects on the thermostability of the p53 protein in HCT116 and SW480 cells. In cellular thermal shift assays of this article, HCT116 cells expressed wild-type phosphorylated p53, which was activated by 5FU-induced DNA damage, and SW480 cells expressed R273H/P309S mutant p53. R273H/P309S mutant p53 eliminates direct DNA contacts, preserves the wild-type DNA binding domain structure, and shares a not significantly changed conformation with wild-type p53.16−18 Thus, it is proposed that the R273H/P309S mutation of p53 is impossible to trigger the distinct effects of PQ1 on the thermostability of p53 proteins in HCT116 and SW480 cells. Meanwhile, reversible protein phosphorylation regulates the thermostability of a protein.19 The phosphorylation of Ser392 increases the thermostability of the p53 protein,19 indicating that the protein conformational flexibility of p53 is different between unmodified and phosphorylated proteins. The thermostability of proteins is related to protein conformational flexibility;20 therefore, the distinct effects of PQ1 on thermostability of the p53 protein in HCT116 and SW480 cells may be due to the different phosphorylation status of proteins.
Due to the fact that p53 confers its functions primarily through regulating the expression of downstream target genes, underlying p53 target genes helps us to understand precisely how p53 modulates a stress response in cells.21 Although genetic strategies such as knockdown and knockout have been used most frequently to mine p53 target genes and understand their functions, such methods have limitations that long-term p53 deficiency usually induces gene expressions and reprograms cell signaling pathways. Compared to these methods, smal-molecule modulators are an ideal tool for transiently modulating p53 function in a dose- and time-dependent manner without altering p53 levels. Through RNA sequencing, we successfully identified 32 genes overlapped from PQ1 and PFTα treatment (Figure 5B). These candidates are commonly downregulated by PQ1 and PFTα and are potentially novel target genes of the p53 R273H/309S protein. However, we cannot exclude the probability that decreased gene expression indirectly results from the inhibition of p53 transcriptional activity. Therefore, future experiments should aim to confirm the roles of p53 as pivotal modulators of these newly identified candidates.
Reported p53 inhibitors, including PFTα, pifithrin-β, and NSC194598, have been subjected to pharmacodynamic studies. PFTα shows protective effects against radiation-induced damage, epileptiform-like activity, diabetic cardiomyopathy, mitochondrial compromise, nonalcoholic fatty liver disease, extensive testicular injury, and progressive hearing loss in specific experimental models.12,22−27 Pifithrin-β protects against neuronal death in models of stroke and neurodegenerative disorders.28 NSC194598 protects mice from radiation toxicity via inhibiting transcriptional output.8 Although antagonists of p53 transcriptional activity provide prospective therapeutic applications, p53 inhibitors have not been applied in clinic.29 Thus, discovering small-molecule inhibitors of p53 is an important research goal for generating drug leads of therapeutic strategies targeting p53 function. Our research provides a new choice for exploring the clinical significance of inhibiting p53 function. More studies are needed to understand the therapeutic applications of PQ1 in depth.
PFTα and NSC194598 protect cells harboring wild-type p53 from p53-mediated cytotoxicity,8,12 whereas PQ1 does not display this effect. We found that PQ1 showed weak cytotoxicity against SW480 cells and increased the sensitivity of SW480 cells to 5FU, which is distinct from the biological activity of PFTα. These results are partly in line with the results reported in the literature, in which the disruption of mutant p53 is beneficial for cancer treatment. Knockdown of mutant p53 sensitizes SW480 cells to growth suppression by various chemotherapeutic drugs.30 SAHA shows preferential cytotoxicity in mutant p53 cancer cells by destabilizing mutant p53.31 BCL7C protein suppresses mutant p53-mediated gene transcription, consequently leading to inhibition of ovarian cancer cell proliferation.32 Taken together with these results, PQ1 is an antagonist of p53 transcriptional activity, which is distinct from PFTα.
4. Conclusions
In conclusion, we identify PQ1 as a novel antagonist of p53 transcriptional activity and dissect the intracellular engagement of p53 inhibition. PQ1 inhibits the activity of p53-induced luciferase and the expression of p53 target gene p21, supporting the fact that PQ1 inhibits p53 transcriptional activity but not its expression. PQ1 directly bound to the p53 protein as well as the well-known p53 transcriptional inhibitor PFTα in HCT116 cells. Further, transcriptomics shows that PQ1 exposure generates a similar regulatory effect to transcription profiles with PFTα, firmly confirming that PQ1 is an inhibitor of p53 transcriptional activity. However, compared to PFTα, PQ1 increased the sensitivity of SW480 cells to 5FU, indicating that PQ1 is distinct from PFTα. Our findings present a chemical tool to pinpoint the physiological functions of p53 and provide a novel lead compound for targeting dysfunctional p53 activation.
5. Materials and Methods
5.1. Reagents
The compound library containing 120 small molecules was from the laboratory of Zhenyu Li. The phenylquinoline derivatives were synthesized through the Suzuki coupling reaction as in our previous description.33 PFTα (Cat#: T2707) was from Targetmol (China), 5FU (Cat#: F6627-1G) was obtained from Sigma-Aldrich. Anti-p21 antibody (Cat#: 103551-1-AP) and anti-p53 antibody (Cat#: 103551-1-AP) were from Proteintech (China). Anti-γH2AX antibody (Cat#: ab11174) and anti-GAPDH antibody (Cat#: ab181602) were from Abcam. Seven Color Protein Marker II (Cat#: SW175-02) was obtained from Sevenbio (China). DMEM medium (Cat#: 01-052-1ACS) and RPMI-1640 medium (Cat#: 01-100-1ACS) were from Biological Industries (Israel). The fetal bovine serum (FBS) of Sijiqing (Cat#: 13011-8611) was purchased from Zhejiang Tianhang Biotechnology Co., Ltd. (China).
5.2. Cell Culture
HCT116, HCT116 p53–/–, and NH2 cells were maintained in DMEM complete medium, and SW480 and LoVo cells were maintained in RPMI-1640 complete medium. All complete media were supplemented with 10% FBS, 50 μg/mL penicillin, and 50 μg/mL streptomycin. Cells were cultured at 37 °C in a humidified atmosphere containing 5% CO2. All cells were provided by the Cell Bank of the Shanghai Institute for Biological Sciences, Chinese Academy of Sciences.
5.3. Chemical Screening with Luciferase Reporter Assays
The chemical screening was performed in 96-well plates. HCT116 and SW480 cells stably expressing p53 promoter-based luciferase reporter gene were generated from our previous literature.34 Cells were seeded in plates at a density of 10 000–20 000 cells per well according to cell types and incubated overnight for cell attachment. Cells were exposed to 50 μM drugs for 24 h. Then, after removing cell culture medium, cells were cautiously washed three times with cold PBS. After that, the luciferase activity was analyzed with a Firefly luciferase reporter gene assay kit (RG006, Beyotime) according to the product manual.
5.4. Western Blot Analysis
Cell lysates were sampled using 4× protein loading buffer and heated at 95 °C for 5 min. The sampled proteins were separated by SDS-PAGE. Following electrophoresis, the resolved proteins were transferred to the PVDF membrane using a tank blotting system. The block was performed by incubating the membrane for 1 h in blocking solution (5% bovine serum albumin solution formulated in TBST) at room temperature. The membrane was incubated overnight with specific primary antibodies at 4 °C for the binding of primary antibodies and then incubated with horseradish peroxidase-conjugated secondary antibodies for 1 h at room temperature for the binding of secondary antibodies. After incubating with antibodies, the membrane was washed three times with TBST at room temperature. Finally, signals of bots were detected by measuring chemiluminescence intensity using electrochemiluminescence reagent (P0018, Beyotime) on a ChemiDoc XRS+ imaging system (Bio-Rad).
5.5. Cellular Thermal Shift Assay
Cells were treated with detected compounds for 3 h and collected with a cell scraper. The collected cells were washed with PBS and resuspended in PBS containing protease inhibitors. The cell suspension was divided into smaller (50 μL) aliquots and heated individually at different temperatures for 3 min followed by cooling in ice. Then, the heated cells were freeze-thawed three times using liquid nitrogen and subjected to centrifugation at 20 000g for 20 min at 4 °C. The supernatant was analyzed by western blot.
5.6. RNA Preparation and Sequencing
SW480 cells were treated with PQ1 (50 μM) or PFTα (50 μM) for 24 h and collected into TRIzol reagent (Invitrogen) by a cell scraper. The total RNA was isolated from the collected cells according to the user manual and sequentially subjected to quality inspection, mRNA fragmentation, and cDNA synthesis. The library was constructed by an Illumina HiSeq sequencing platform for sequencing cDNA to generate the raw data in FASTQ format (raw data). The RNA-seq data were evaluated for data filtering, base content distribution, base quality distribution, and distribution of mean sequence quality. Filtered RNA-seq data were mapped to the genome sequence on the HISAT2 platform for the annotation of reads. Then, the expression levels of all transcripts were normalized by the FPKM (fragments per kilo bases per million fragments) method. Through the comparison of gene expression levels between two samples, differentially expressed genes (DEGs) were identified according to P-adjust < 0.05 and |log 2 FC| ≥ 1.
5.7. Bioinformatics Analysis
The total target genes of p53 were obtained from the hTFtarget database.35 Venn diagram analysis was performed by the MajorBio Biochip system. Cluster analysis and KEGG pathway enrichment analysis were conducted by GENESCLOUD of Shanghai Personalbio Technology Co., Ltd (https://www.genescloud.cn/login).
5.8. Molecular Docking
A molecular docking study was performed to investigate the binding mode between PQ1 and p53 using Autodock vina 1.1.2.36 The three-dimensional (3D) structure of the p53 DNA binding domain (PDB ID: 6ZNC) was downloaded from the RCSB Protein Data Bank (www.rcsb.org). The 2D structure of PQ1 was drawn by ChemBioDraw Ultra 14.0 and converted to the 3D structure by ChemBio3D Ultra 14.0 software. The AutoDockTools 1.5.6 package was employed to generate the docking input files.37,38 The ligand was prepared for docking by merging nonpolar hydrogen atoms and defining rotatable bonds. The search grid of the p53 DNA binding domain was identified as center_x: 155.496, center_y: −4.397, and center_z: 35.68 with dimension size_x: 15, size_y: 15, and size_z: 15. To increase the docking accuracy, the value of exhaustiveness was set to 16. For Vina docking, the default parameters were used if it was not mentioned. The best-scoring pose as judged by the Vina docking score was chosen and visually analyzed using PyMoL 1.7.6 software (www.pymol.org).
5.9. Cell Growth Inhibition Assay
The sulforhodamine B (SRB) assay was used to measure the inhibition of cell growth. Cells were cultured at a seeding density of 3000–5000 cells per well in 96-well plates. After cell attachment, the cells were exposed to the tested compounds for 72 h. After removal of the cell culture medium, cold 10% (wt/vol) trichloroacetic acid (TCA) was gently added to each well, and the plate was fixed for at least 1 h at 4 °C. The plate was washed five times with slow-running tap water, and the excess water was removed using paper towels. The fixed cells were stained for 15 min with 0.4% (wt/vol) SRB solution at room temperature. After staining, the plate was immediately rinsed five times with 1% (vol/vol) acetic acid to remove the unbound dye and dried at room temperature. Lastly, the protein-bound dye was dissolved in 10 mM Tris base solution (pH 10.5), and the absorbance of the sample was measured at 570 nm in a microplate reader. Cell growth inhibition rate (%) was calculated by (ODcontrol – ODgroup)/(ODcontrol – ODblank) × 100, where ODcontrol is the OD value in the presence of 0.1% DMSO, ODgroup is the OD value in the presence of drugs, and ODblank is the OD value in the absence of cells.
5.10. Statistical Analysis
GraphPad Prism 8.0 software was applied to perform data analysis. IC50 value for inhibition of p53 activity was calculated with an equation of [inhibitor] vs normalized response – variable slope. Significant differences of all pairwise combinations were evaluated using the two-way ANOVA multiple comparison test. Values of P < 0.05 were considered significant. Significance levels are indicated by *P < 0.05, **P < 0.01, and ***P < 0.001.
Acknowledgments
This work was supported by the financial support from Shandong Provincial Natural Science Foundation (ZR2020MH399), Shanxi Key Laboratory of Redevelopment of Famous Local Traditional Chinese Medicines (202104010910001), and Shanxi Provincial Education Reform Project for Postgraduate (2022YJJG017).
Glossary
Abbreviations
- PFTα
pifithrin-α
- 5FU
5-fluorouracil
- FBS
fetal bovine serum
- CETSA
cellular thermal shift assay
- SDS
sodium dodecyl sulfate
- PAGE
polyacrylamide gel electrophoresis
- FPKM
fragments per kilo bases per million fragments
- DEG
differentially expressed gene
- KEGG
Kyoto Encyclopedia of Genes and Genomes
- FC
fold of change
- ANOVA
analysis of variance
- IC50
50% inhibitory concentration
- SD
standard deviation
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acsomega.2c05891.
Assays for the tested compounds against HIV LTR-dependent luciferase (Figure S1); relationship between p53 deficiency and DNA damage response (Figure S2); overview of the effect PQ1 and PFTα on the transcription profiles (Figure S3) (PDF)
Author Contributions
X.W.: funding acquisition, data curation, formal analysis, investigation, methodology, writing—original draft, writing—review and editing. L.W.: data curation, formal analysis, investigation, methodology, visualization. Z.L.: funding acquisition, data curation, formal analysis, investigation, methodology, writing—review and editing.
The authors declare no competing financial interest.
Supplementary Material
References
- Kastenhuber E. R.; Lowe S. W. Putting p53 in context. Cell 2017, 170, 1062–1078. 10.1016/j.cell.2017.08.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dolgin E. The most popular genes in the human genome. Nature 2017, 551, 427–431. 10.1038/d41586-017-07291-9. [DOI] [PubMed] [Google Scholar]
- Liu Y.; Leslie P. L.; Zhang Y. Life and death decision-making by p53 and implications for cancer immunotherapy. Trends Cancer 2021, 7, 226–239. 10.1016/j.trecan.2020.10.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lieschke E.; Wang Z.; Kelly G. L.; Strasser A. Discussion of some ’knowns’ and some ’unknowns’ about the tumour suppressor p53. J. Mol. Cell Biol. 2019, 11, 212–223. 10.1093/jmcb/mjy077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miller J. J.; Gaiddon C.; Storr T. A balancing act: using small molecules for therapeutic intervention of the p53 pathway in cancer. Chem. Soc. Rev. 2020, 49, 6995–7014. 10.1039/D0CS00163E. [DOI] [PubMed] [Google Scholar]
- Bykov V. J. N.; Eriksson S. E.; Bianchi J.; Wiman K. G. Targeting mutant p53 for efficient cancer therapy. Nat. Rev. Cancer 2018, 18, 89–102. 10.1038/nrc.2017.109. [DOI] [PubMed] [Google Scholar]
- Ladds M. J. G. W.; Lain S. Small molecule activators of the p53 response. J. Mol. Cell Biol. 2019, 11, 245–254. 10.1093/jmcb/mjz006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Q.; Karim R. M.; Cheng M.; Das M.; Chen L.; Zhang C.; Lawrence H. R.; Daughdrill G. W.; Schonbrunn E.; Ji H.; Chen J. Inhibition of p53 DNA binding by a small molecule protects mice from radiation toxicity. Oncogene 2020, 39, 5187–5200. 10.1038/s41388-020-1344-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bowen M. E.; Attardi L. D. The role of p53 in developmental syndromes. J. Mol. Cell Biol. 2019, 11, 200–211. 10.1093/jmcb/mjy087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Checler F.; da Costa C. A. p53 in neurodegenerative diseases and brain cancers. Pharmacol. Ther. 2014, 142, 99–113. 10.1016/j.pharmthera.2013.11.009. [DOI] [PubMed] [Google Scholar]
- Maor-Nof M.; Shipony Z.; Lopez-Gonzalez R.; Nakayama L.; Zhang Y. J.; Couthouis J.; Blum J. A.; Castruita P. A.; Linares G. R.; Ruan K.; Ramaswami G.; Simon D. J.; Nof A.; Santana M.; Han K.; Sinnott-Armstrong N.; Bassik M. C.; Geschwind D. H.; Tessier-Lavigne M.; Attardi L. D.; Lloyd T. E.; Ichida J. K.; Gao F. B.; Greenleaf W. J.; Yokoyama J. S.; Petrucelli L.; Gitler A. D. p53 is a central regulator driving neurodegeneration caused by C9orf72 poly(PR). Cell 2021, 184, 689–708 e20. 10.1016/j.cell.2020.12.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Komarov P. G.; Komarova E. A.; Kondratov R. V.; Christov-Tselkov K.; Coon J. S.; Chernov M. V.; Gudkov A. V. A chemical inhibitor of p53 that protects mice from the side effects of cancer therapy. Science 1999, 285, 1733–1737. 10.1126/science.285.5434.1733. [DOI] [PubMed] [Google Scholar]
- Pietrancosta N.; Moumen A.; Dono R.; Lingor P.; Planchamp V.; Lamballe F.; Bahr M.; Kraus J. L.; Maina F. Imino-tetrahydro-benzothiazole derivatives as p53 inhibitors: Discovery of a highly potent in vivo inhibitor and its action mechanism. J. Med. Chem. 2006, 49, 3645–3652. 10.1021/jm060318n. [DOI] [PubMed] [Google Scholar]
- Strom E.; Sathe S.; Komarov P. G.; Chernova O. B.; Pavlovska I.; Shyshynova I.; Bosykh D. A.; Burdelya L. G.; Macklis R. M.; Skaliter R.; Komarova E. A.; Gudkov A. V. Small-molecule inhibitor of p53 binding to mitochondria protects mice from gamma radiation. Nat. Chem. Biol. 2006, 2, 474–479. 10.1038/nchembio809. [DOI] [PubMed] [Google Scholar]
- Pfister N. T.; Prives C. Transcriptional regulation by wild-type and cancer-related mutant forms of p53. Cold Spring Harbor Perspect. Med. 2017, 7, a026054 10.1101/cshperspect.a026054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen S.; Wu J. L.; Liang Y.; Tang Y. G.; Song H. X.; Wu L. L.; Xing Y. F.; Yan N.; Li Y. T.; Wang Z. Y.; Xiao S. J.; Lu X.; Chen S. J.; Lu M. Arsenic trioxide rescues structural p53 mutations through a cryptic allosteric site. Cancer Cell 2021, 39, 225–239. 10.1016/j.ccell.2020.11.013. [DOI] [PubMed] [Google Scholar]
- Bullock A. N.; Henckel J.; Fersht A. R. Quantitative analysis of residual folding and DNA binding in mutant p53 core domain: definition of mutant states for rescue in cancer therapy. Oncogene 2000, 19, 1245–1256. 10.1038/sj.onc.1203434. [DOI] [PubMed] [Google Scholar]
- Joerger A. C.; Ang H. C.; Veprintsev D. B.; Blair C. M.; Fersht A. R. Structures of p53 cancer mutants and mechanism of rescue by second-site suppressor mutations. J. Biol. Chem. 2005, 280, 16030–16037. 10.1074/jbc.M500179200. [DOI] [PubMed] [Google Scholar]
- Huang J. X.; Lee G.; Cavanaugh K. E.; Chang J. W.; Gardel M. L.; Moellering R. E. High throughput discovery of functional protein modifications by Hotspot Thermal Profiling. Nat. Methods 2019, 16, 894–901. 10.1038/s41592-019-0499-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vihinen M. Relationship of protein flexibility to thermostability. Protein Eng. Des. Sel. 1987, 1, 477–480. 10.1093/protein/1.6.477. [DOI] [PubMed] [Google Scholar]
- Moyer S. M.; Wasylishen A. R.; Qi Y.; Fowlkes N.; Su X.; Lozano G. p53 drives a transcriptional program that elicits a non-cell-autonomous response and alters cell state in vivo. Proc. Natl. Acad. Sci. U.S.A. 2020, 117, 23663–23673. 10.1073/pnas.2008474117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Samarasinghe R. A.; Miranda O. A.; Buth J. E.; Mitchell S.; Ferando I.; Watanabe M.; Allison T. F.; Kurdian A.; Fotion N. N.; Gandal M. J.; Golshani P.; Plath K.; Lowry W. E.; Parent J. M.; Mody I.; Novitch B. G. Identification of neural oscillations and epileptiform changes in human brain organoids. Nat. Neurosci. 2021, 24, 1488–1500. 10.1038/s41593-021-00906-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Si R.; Zhang Q.; Tsuji-Hosokawa A.; Watanabe M.; Willson C.; Lai N.; Wang J.; Dai A.; Scott B. T.; Dillmann W. H.; Yuan J. X.; Makino A. Overexpression of p53 due to excess protein O-GlcNAcylation is associated with coronary microvascular disease in type 2 diabetes. Cardiovasc. Res. 2020, 116, 1186–1198. 10.1093/cvr/cvz216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gu J.; Wang S.; Guo H.; Tan Y.; Liang Y.; Feng A.; Liu Q.; Damodaran C.; Zhang Z.; Keller B. B.; Zhang C.; Cai L. Inhibition of p53 prevents diabetic cardiomyopathy by preventing early-stage apoptosis and cell senescence, reduced glycolysis, and impaired angiogenesis. Cell Death Dis. 2018, 9, 82. 10.1038/s41419-017-0093-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoshino A.; Ariyoshi M.; Okawa Y.; Kaimoto S.; Uchihashi M.; Fukai K.; Iwai-Kanai E.; Ikeda K.; Ueyama T.; Ogata T.; Matoba S. Inhibition of p53 preserves Parkin-mediated mitophagy and pancreatic beta-cell function in diabetes. Proc. Natl. Acad. Sci. U.S.A. 2014, 111, 3116–3121. 10.1073/pnas.1318951111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang J. K.; Zhao T. X.; Chen J. D.; Kang L.; Ei Y. X.; Wu Y. H.; Han L. D.; Shen L. J.; Long C. L.; Wu S. D.; Wei G. H. Multiple transcriptomic profiling: p53 signaling pathway is involved in DEHP-induced prepubertal testicular injury via promoting cell apoptosis and inhibiting cell proliferation of Leydig cells. J. Hazard. Mater. 2021, 406, 124316 10.1016/j.jhazmat.2020.124316. [DOI] [PubMed] [Google Scholar]
- Zhang L.; Gao Y.; Zhang R.; Sun F.; Cheng C.; Qian F.; Duan X.; Wei G.; Sun C.; Pang X.; Chen P.; Chai R.; Yang T.; Wu H.; Liu D. THOC1 deficiency leads to late-onset nonsyndromic hearing loss through p53-mediated hair cell apoptosis. PLoS Genet. 2020, 16, e1008953 10.1371/journal.pgen.1008953. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Da Pozzo E.; La Pietra V.; Cosimelli B.; Da Settimo F.; Giacomelli C.; Marinelli L.; Martini C.; Novellino E.; Taliani S.; Greco G. p53 functional inhibitors behaving like pifithrin-beta counteract the Alzheimer peptide non-beta-amyloid component effects in human SH-SY5Y cells. ACS Chem. Neurosci. 2014, 5, 390–399. 10.1021/cn4002208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsai Y. Y.; Su C. H.; Tarn W. Y. p53 activation in genetic disorders: different routes to the same destination. Int. J. Mol. Sci. 2021, 22, 9307. 10.3390/ijms22179307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yan W.; Liu G.; Scoumanne A.; Chen X. Suppression of inhibitor of differentiation 2, a target of mutant p53, is required for gain-of-function mutations. Cancer Res. 2008, 68, 6789–6796. 10.1158/0008-5472.CAN-08-0810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li D.; Marchenko N. D.; Moll U. M. SAHA shows preferential cytotoxicity in mutant p53 cancer cells by destabilizing mutant p53 through inhibition of the HDAC6-Hsp90 chaperone axis. Cell Death Differ. 2011, 18, 1904–1913. 10.1038/cdd.2011.71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang C.; Hao Q.; Shi G.; Zhou X.; Zhang Y. BCL7C suppresses ovarian cancer growth by inactivating mutant p53. J. Mol. Cell Biol. 2021, 13, 141–150. 10.1093/jmcb/mjaa065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu Y.; Li Z.; Liu Z.; et al. Antitumor and Topoisomerase IIα Inhibitory Activities of 3-Aryl-7-hydroxyquinolines. Chin. J. Org. Chem. 2019, 39, 3230–3236. 10.6023/cjoc201905013. [DOI] [Google Scholar]
- Lu Y.; Wu X.; Qin X. Exploring the antitumor activities of arylquinolines aginst colorectal cancer based on p53 signaling pathway. J. Shanxi Univ. (Nat. Sci. Ed.) 2022, 45, 1107–1116. [Google Scholar]
- Zhang Q.; Liu W.; Zhang H. M.; Xie G. Y.; Miao Y. R.; Xia M.; Guo A. Y. hTFtarget: a comprehensive database for regulations of human transcription factors and their targets. Genomics, Proteomics Bioinform. 2020, 18, 120–128. 10.1016/j.gpb.2019.09.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trott O.; Olson A. J. AutoDock Vina: improving the speed and accuracy of docking with a new scoring function, efficient optimization, and multithreading. J. Comput. Chem. 2010, 31, 455–461. 10.1002/jcc.21334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanner M. F. Python: a programming language for software integration and development. J. Mol. Graphics Model. 1999, 17, 57–61. [PubMed] [Google Scholar]
- Morris G. M.; Huey R.; Lindstrom W.; Sanner M. F.; Belew R. K.; Goodsell D. S.; Olson A. J. AutoDock4 and AutoDockTools4: Automated docking with selective receptor flexibility. J. Comput. Chem. 2009, 30, 2785–2791. 10.1002/jcc.21256. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.