

Abstract

The production of aromatic hydrocarbons from the waste tire pyrolysis attracts more and more attention because of its tremendous potential. Based on styrene-butadiene rubber (SBR), which is the main rubber in the waste passenger car tires, this work studies the temperature influence on primary pyrolysis product distribution by experimental techniques (Py-GC/MS, TG–MS), and then, the formation mechanism of monocyclic aromatic hydrocarbons (MAHs) observed in the experiment was analyzed by first-principles calculations. The experimental results show that the MAHs during the pyrolysis mainly include styrene, toluene, and xylene, and subsequent calculations showed that these compounds were formed through a series of primary and secondary reactions. The formation pathways of these typical MAHs were studied via the reaction energy barrier analysis, respectively. It shows that the MAHs were not only derived from the benzene ring in the SBR chain but also generated from short-chain alkenes through the Diels–Alder reaction. The obtained pyrolysis reaction mechanism provides theoretical guidance for the regulation of the pyrolysis product distribution of MAHs.
1. Introduction
The recycling and treatment of waste passenger car tires are of great significance1 to deal with environmental problems caused by the disposal of waste tires. Pyrolysis is considered to be an effective way for the utilization of waste passenger car tires.2,3 The waste tires were puts into a high-temperature pyrolysis reaction kettle in the absence of oxygen, and the gas, char, oil, and steel components were obtained under controlled conditions.4,5 In view of the complex composition and molecular structure in tires, many chemical reactions are involved in the pyrolysis process, and the corresponding reaction mechanisms are also very complicated.6 Nevertheless, in-depth analysis of the product formation mechanism is still the theoretical basis for realizing the efficient cascade utilization of waste tire pyrolysis product.
The waste tire pyrolysis oil (WTPO) is the main product of tire pyrolysis, and the composition of the oil mainly includes aliphatic, aromatic, and impurities (sulfur or nitrogen compounds, polycyclic aromatic hydrocarbons).7 Aromatic hydrocarbons (MAHs) are important high-value components in WTPO,8 which are mainly generated from the pyrolysis of styrene butadiene rubber (SBR)9 in passenger car tires.10 Compared with the gas and char components, the study of the formation mechanism of the oil component is more challenging due to various reaction pathways. Although the reaction mechanism involved in the formation of MAHs in the oil is extremely complicated, researchers are still studying the molecular mechanism of the pyrolysis process by various methods. Experimentally, it is possible to determine the type and distribution of products at different pyrolysis temperatures through the method of temperature programming combined with synchronous detection and then to infer the formation mechanism of some pyrolysis products.11 In addition, the formation mechanism of different components can also be studied by real-time tracking of essential elements (such as S and N) during the pyrolysis.12 However, the complex molecular structure and composition of the waste tire system bring an enormous amount of work to the experimental study of the reaction mechanism.
Apart from the direct experimental characterization, an efficient way to understand the pyrolysis reaction mechanisms entails an in-depth analysis of experimental kinetics and thermodynamics data by the combination of the model-free and -fitting methods. For example, the third-order reaction model was found to efficiently describe the entire pyrolysis of waste tires as a single process according to the master-plots method.13 The devolatilization stages including the corresponding sub-stages of waste rubber and polyurethane tires were best described by the reaction mechanism models.14 Notably, the artificial neural network-based data analysis was adopted for the operation optimization.15,16 The theoretical models combined with experimental data can facilitate a better understanding and optimization of the pyrolysis of waste tires.17,18 However, sufficient experimental data are also required for building best-fit functions. In addition, the lack of key intermediate product data may also bring difficulties to the analysis of the reaction path.
Computer simulation has always played an indispensable role in the fields of materials and chemical engineering, especially for complex systems or processes that are difficult to carry out in experiment.19,20 The calculation work related to the pyrolysis process and reaction mechanism of the waste tire system has reported in recent years, and most of the work is reactive force field (ReaxFF) molecular dynamics (MD) simulation.21−24 The ReaxFF MD can efficiently study the product distribution of the pyrolysis process under different reaction conditions and provide theoretical guidance on the experiment.21 However, due to the lack of a set of dedicated ReaxFF parameters for the waste tire system, it is difficult to use this method to effectively study the reaction pathway of specific products. In terms of reaction mechanism research, the density functional theory (DFT) method can accurately calculate the activation energy of the chemical reaction in the pyrolysis process and analyze the reaction kinetics in combination with the corresponding model.25 Moreover, ab-initio (or first-principles) MD (AIMD) is formulated in which the potential energy surface is generated “on the fly” from the instantaneous ground state of the electrons within DFT. This method can study the dynamic reaction process within a short, sub-nanosecond physical timescale. However, there is a lack of systematic first-principles calculations work on the formation mechanism of key products in the pyrolysis process of SBR or other polymers in waste tires.
In-depth study of the formation mechanism will provide theoretical guidance for the modulation of pyrolysis products. Therefore, this work focuses on the formation mechanism of MAHs in the pyrolysis of SBR. The SBR is derived from two monomers, styrene and butadiene. Therefore, we choose styrene and alkenes as starting reactants for the formation of MAHs, which are also the primary pyrolysis products observed in the experiments.26 Generally, hydrogenation, methylation and dissociation of styrene are the main pathways to generate subsequent pyrolysis products, while the conversion from alkenes to MAHs can be via the Diels–Alder reaction pathway. The SBR was pyrolyzed by Py-GC/MS to obtain the distribution of pyrolysis products at different temperatures. On the basis of experiment results, the reaction pathways for the formation of MAHs were calculated using DFT, and the energy barriers and reaction energies of these pathways were compared to determine the difficulty and feasibility of the reaction.
2. Methods
2.1. Experimental Details
2.1.1. Materials
The SBR was randomly sampled from Qingdao ECOSTAR Co., Ltd in Qingdao of Shandong province. The sample was first cleaned with deionized water and then was air-dried in the oven at 423 K for 4 h to eliminate the moisture remaining in the sample. After then, the rubber sample was ground and sieved through a 16-mesh screen to gather 1 mm particles, and finally, sample particles were sealed and stored in a dry environment for subsequent experiments.
2.1.2. Experimental Method
TG-MS experiments were performed in a nitrogen atmosphere within the TGA analyzer (TG 209 Libra, NETZSCH, Germany) coupling with mass spectrum analyzer (QMS 403 Aëolos Quadro, NETZSCH, Germany). 15.0 mg of sample particles was put into the Al2O3 crucible and heated from room temperature to the desired temperature (738 and 758 K) with the heating rate of 10 K/min. Nitrogen gas (40 mL min–1) was used as the carried gas to provide an inert atmosphere during the pyrolytic procedure, and swept gaseous released from the sample pyrolysis in TG into the MS through a capillary tube heated to about 533 K. The main products were recorded by MS analyzer. Each experiment was repeated three times to ensure reproducibility of the result.
Py–GC/MS experiments were carried on in the nitrogen atmosphere within self-designed pyrolytic instrument (see Figure S1) coupling with the detection system composed of GC (gas chromatography, Shimadzu GC-2030, Japan) with a capillary column (25 m × 0.25 mm, 3 μm film thickness) and aforementioned MS (scanning range from 1 to 300). The GC method had been presented in Table S1 in the Supporting Information. The 50 mL min–1 carrier gas (N2) was first introduced into the system for 2 h to eliminate O2 resided in the pipeline; subsequently, it was adjusted to 40 mL min–1. Moreover, 10.0 g of rubber particles was placed in the fixed bed preheated to the desired temperature (575, 625, 675, 725, 775, and 825 K); then, the pyrolytic products pass through the cooling device and the sampler, successively. It is noted that the sample maintained at a constant temperature of 453 K comprised a six-way and an eight-way valve. The former has two mode-sampling and injection, and the latter contains eight quantification rings, meaning that eight samples can be taken during the pyrolytic experiment. The cracking oil was collected from the cooling device and used for subsequent tests, and the gases in the quantitative loop were analyzed by the GC–MS system. All the experiments were repeated three times to ensure the repeatability of the results.
2.2. Simulation Details
All DFT simulations were carried out for the formation mechanism of aromatic compounds during pyrolysis of SBR within the framework of the generalized gradient approximation with the Perdew–Burke–Ernzerh27 of the functional in the VASP code.28,29 Due to the magnetic properties of radical species, the spin-polarized effect was considered in our calculations. The cutoff energy of plane-wave basis expansion was set to 400 eV. Electronic convergence was set to 10–4 eV, and geometries were converged to less than 0.05 eV/Å. The effect of vdW interaction was significant for the reaction mechanism in our precious study.30−35 Therefore, the DFT-D3 method of Grimme et al.36,37 was utilized to calculate all the energetics and structures of the intermediates and transition states (TSs). The PAW was carried out to describe the interaction between the core-electron and valence electron. The structure optimization was calculated using the conjugate-gradient algorithm. To avoid interactions between system images, a (15 Å × 15 Å × 15 Å) unit cell was constructed with 1 × 1 × 1 Monkhorst–Pack k-point mesh sampling.38 The TSs were searched using the method called a constrained optimization scheme.39−41 The geometries of TSs were converged to less than 0.05 eV/Å. The TSs were confirmed by two rules: (i) all forces on atoms vanish; (ii) the total energy is a maximum along the reaction coordinate but a minimum with respect to the rest of the degrees of freedom. Vibrational frequency analyses42 were performed to confirm the integrity of TSs.
The AIMD simulation was carried out in a canonical (NVT) ensemble. The temperature of the systems was controlled using a Nose–Hoover thermostat at 2000 K. The time steps are 0.5 fs and total steps are 2000 steps.
3. Results and Discussion
3.1. Analysis of Pyrolysis Product Distribution
The distribution of pyrolysis products was the basis for the analysis of subsequent reaction pathways and formation mechanisms. Therefore, the Py-GC/MS experiments for SBR were performed to analyze the distribution of products at different temperatures (Figure S2), we selected the result of 775 K which was favorable for MAH generation from the pyrolysis experiment (see Figure S3), and the results are shown in Figure 1. The main components of gaseous were composed of alkanes (C2H6, C3H8, and C4H10) and alkenes (C2H4, C3H6, C4H8, and C4H6). The most of the alkenes were from scission of β bond and side chains attached to aromatic hydrocarbon.43,44 Notably, under the high temperature, these alkenes were unstable and would transform to the corresponding saturated hydrocarbon by combining with the dissociative H.5 Compared to the gaseous, the liquid components were more complex, which comprised single ring aromatics and a small amount of alkenes. Figure 1 indicated that the main aromatic components were toluene (10#), xylene (12&14#), styrene (11#) and α-methylstyrene (17#), and 2,5-dimethylstyrene (23#). Furthermore, these aromatics were generated from Diels–Alder reactions among alkenes or secondary reactions of aromatic hydrocarbons.45−47 The detailed reaction mechanisms of alkanes, alkenes, and aromatics would be discussed later.
Figure 1.

Chromatogram of pyrolytic products of SBR at 775 K (about 10.0 g of 1 mm samples were cracked with a gas flow rate of 40 mL min–1 in a N2 atmosphere).
3.2. Primary Pyrolysis Mechanism of SBR
The SBR was formed by the polymerization of styrene and butadiene monomers. Various compounds were obtained during the pyrolysis of SBR such as aliphatic, aromatic, and others. However, prior to studies on the cracking mechanism of SBR, several pyrolytic experiments were performed to fully understand the influence of reside residence time on pyrolyzates. As shown in Figure S4, the residence time can only change the content of the products, but not its composition. It also indicates that residence time has no influence on the formation mechanism of pyrolyzates. To study the formation mechanisms of MAHs in the pyrolysis processes, the primary pyrolysis mechanism of the SBR chain was examined by DFT calculations. Limited by the computing power, two kinds of short-chain segments derived from the SBR chain were considered in the calculation, one has a benzene ring and the other is alkene (Figure 2).
Figure 2.

Pyrolysis mechanism and reaction energy barrier of chain segments with the benzene ring (a) and alkene (b).
3.2.1. Pyrolysis of SBR
The pyrolysis of SBR was the initial step of the pyrolysis process, which provided the possibility for the subsequent formation of aromatic compounds and aliphatic long chains. Therefore, we first studied the C–C bond breaking mechanism by DFT calculations. The bond energy of the single bond was much lower than that of the double bond. As for the first kind of segment (Figure 2a), four possible reaction pathways were C1–C2, C2–C3, C3–C4, and C5–C6 bond breaking, respectively. The bond breaking through these pathways provided reasonable reaction sites for the subsequent products (aromatic compounds and long aliphatic chains). The reaction energy barriers for different pathways were also calculated, it showed that the C–C bond breaking in the chain segment with benzene ring was a thermodynamically endothermic process. Moreover, the reaction energies of the four possible pathways were between 3 and 4 eV. The reaction energy barriers of C2–C3 (Ea = 3.72 eV) and C3–C4 bond breaking (Ea = 3.95 eV) were significantly lower than those of C1–C2 (Ea = 4.56 eV) and C4–C5 bond breaking (Ea = 5.07 eV). Besides, the reaction energies of C2–C3 (ΔE = 3.15 eV) and C3–C4 bond breaking (ΔE = 3.81 eV) were also lower than those of C1–C2 (ΔE = 4.55 eV) and C4–C5 (ΔE = 4.75 eV). Through the four reactions pathways described above, the chain segment with the benzene ring was cleaved into an aromatic compound with free radicals and an aromatic straight-chain compound with free radicals. The C2–C3 bond was relatively easy to break, and the corresponding styrene and butadiene would be preferentially generated and exist in large quantities. Our previous work by ReaxFF MD simulation also found that the single bond connected with a double bond or benzene was easy to break.22
The other short-chain segment derived from the SBR chain was the alkene-like segment. This kind of segment was also unstable at high temperatures and would be further hydrogenated to short-chain structures, such as ethylene, propylene, and 1,3-butadiene et al.47 We adopted C9H12 as the alkene-like segment model to simulate the breaking of the alkene chain. Three possible reaction pathways are C5–C6, C6–C7, and C2–C3 bond breaking (Figure 2b), respectively. The C–C bond breaking in C9H12 was an endothermic reaction process with high reaction energy barrier. Among these three pathways, the reaction energies were quite close, and the lowest reaction energy barrier was 5.35 eV in pathway 3, which was slightly lower than those of pathway 1 and pathway 2. Therefore, pathway 3 is more likely to occur and results in the product of 1,3-butadiene. However, three pathways have similar reaction energies and are all likely to occur to form corresponding pyrolysis products.
3.2.2. Hydrogenation of Pyrolysis Intermediates
The free radicals of short chain alkenes and side chains attached to the aromatic hydrocarbon were unstable and preferred to react with dissociative H+ generated from the pyrolysis process. The DFT calculation was applied to study the reaction mechanism between the H+ and unsaturated hydrocarbons.
According to the pyrolysis mechanism of the chain segment with the benzene ring, the optimal bond-breaking routes were pathway 2 and pathway 3 in Figure 2a, which form α-methylstyrene and styrene intermediates, respectively. As shown in Figure 3a, pathway 1 was the hydrogenation of the styrene intermediate, where the reaction energy was −2.49 eV and the activation energy barrier was 2.37 eV. The energy barrier for the hydrogenation of α-methylstyrene (pathway 2) was 2.66 eV and the reaction energy was −1.65 eV. Both pathways were thermodynamically exothermic reactions, in which the energy barrier for the hydrogenation of the styrene intermediate was lower than that of the α-methylstyrene intermediate because the energy barrier for the hydrogenation of unsaturated double bonds was lower than that for unsaturated single bonds.
Figure 3.

Hydrogenation process and reaction energy barrier of (a) styrene and α-methylstyrene; (b) vinyl (pathway 1), propylene (pathway 2), and butadiene (pathway 3).
After the aliphatic long chain was cracked, six kinds of alkene intermediates would be formed, which would also be further hydrogenated to obtain alkene products.48 We simulated the hydrogenation of shorter-chain product intermediates (ethylene, propylene, and 1,3-butadiene), and the reaction pathways and energy barriers are shown in Figure 3b. The hydrogenation reaction energy barriers of forming butadienyl (pathway 2) and ethylene (pathway 3) were 2.30 and 2.32 eV, and the reaction energies were −2.63 and −2.65 eV. The lowest reaction energy barrier occurred in the propylene hydrogenation reaction (Pathway 1), and the reaction energy barrier and reaction energy were 2.23 and −2.71 eV, respectively. These three pathways were all exothermic in thermodynamics, the hydrogenation reaction energy barriers of ethylene, propylene, and butadienyl intermediates were quite close, so all three pathways could occur in the reaction process.
3.3. Conversion of Aromatic Products from Rubber Pyrolysis
In addition to styrene and α-methylstyrene, other aromatic products, such as cumene and phenylethane, would be detected during the SBR pyrolysis process. Therefore, we simulated the subsequent reactions starting from α-methylstyrene or styrene, which were the main products of SBR pyrolysis.
3.3.1. Hydrogenation of α-Methylstyrene
The hydrogenation of α-methylstyrene was first studied by DFT calculations. As shown in Figure S5, by comparing the reaction energy barrier, it was found that the first step of hydrogenation occurs at the C1 site (Ea1 = 2.38 eV), and the second step occurs at the C2 site. The calculated energy barriers for α-methylstyrene to generate isopropylbenzene through two-step hydrogenation reactions were 2.38 and 2.87 eV, respectively. This result indicated that the second hydrogenation step had a higher reaction energy barrier, so it was the decisive step in this reaction path. In addition, the reaction energy of this step was −1.42 eV, which was an exothermic reaction. Finally, we concluded the hydrogenation reaction mechanism of α-methylstyrene through the comparison of energy barrier. Because of the conjugate structure formed by the double bond and the benzene ring, the first step of hydrogenation would occur at the C1 position near the benzene ring to break the conjugate structure, followed by the second step of hydrogenation at the C2 position.
3.3.2. Hydrogenation, Decomposition, and Methylation of Styrene
For the subsequent reaction starting from the styrene, as shown in Figure 4a, we considered three possible reactions pathways. The styrene could be hydrogenated, decomposed, and methylated to obtain ethylbenzene, benzene, and β-methylstyrene, respectively. For the hydrogenation reaction (pathway 1), the calculated reaction potential energy is shown in Figure 4b. The second hydrogenation step has a higher reaction energy barrier (Ea2 = 2.82 eV) than the first hydrogenation step (Ea1 = 2.32 eV), which was the rate-determining step in this reaction path. The reaction energy of this path was −1.42 eV, which was an exothermic process. Consistent with the reaction results of the formation of isopropylbenzene from α-methylstyrene, the first step of hydrogenation was also carried out on C1 (Ea1 = 2.32 eV), which further confirmed the hypothesis that the C near the benzene ring was easier to be hydrogenated.
Figure 4.

Hydrogenation, decomposition, and methylation of styrene. (a) Mechanism; (b) energy barrier.
For the reaction in which styrene was decomposed to obtain a benzene ring (pathway2), there were mainly two possible decomposition mechanisms: (1) first hydrogenation and then breaking the C–C bond; (2) first breaking the C–C bond and then hydrogenation. We compared the energy barrier results of these two mechanisms, which showed that the energy barrier of C–C bond breaking is higher than the first step hydrogenation, and then, the hydrogenation was more likely to occur (Ea1 = 2.46 eV). The styrene was difficult to be destroyed due to the conjugated large π structure. If a hydrogen radical was introduced to attack the styrene first, the large π bond could be easily broken. Further analysis of the potential energy diagram showed that the reaction energy of the decomposition was 2.35 eV, which was an endothermic reaction. The energy barrier of the reverse reaction was lower than that of the forward reaction and the reverse reaction was exothermic. Therefore, the reaction tended to occur in the reverse direction, that is, styrene was formed from the reaction between benzene and ethylene.
Large amounts of methyl radicals provided a favorable environment for methylation reactions. The methyl radicals and hydrogen radicals were also among the main products during the pyrolysis process by reaction MD.49 For the methylation reaction (pathway 3), we also simulated two possible reaction mechanisms: (1) addition of methyl first and then dehydrogenation; (2) dehydrogenation first and then addition of methyl. The reaction energy barrier of the first step in mechanism 1 was 0.27 eV, which was much lower than that of the first step in mechanism 2, indicating that the reaction pathway of adding methyl first was more feasible. This was because the introduction of methyl radicals led to the opening of double bonds and then free radicals were formed at the C2 position to combine with CH3, so the free radicals at C1 position formed a double bond structure with C2 after the removal of an H from C2.
3.4. Synthesis of Aromatic Compounds from Aliphatic Chain Products
Apart from the formation of aromatics, a series of alkenes were generated in the pyrolysis processes of SBR. Short-chain alkenes among them would be further formed into the cycloalkenes through the Diels–Alder reactions, and then, cycloalkenes were formed into aromatics through dehydrogenation. To explore these reaction processes, the SBR sample was pyrolyzed at 557, 625, 675, 725, 775, and 825 K, respectively. The normalized peak-area of the GC profile can represent the relative content of the product components.50 Moreover, the pyrolytic residual from low temperature was further pyrolyzed at a higher temperature to eliminate influence from previous pyrolysis, and these results are displayed in Figure 5.
Figure 5.

Contents of product components at different temperatures: (a) <C5; (b) C6–C7; and (c) C8–C9 (10.0 g of 1 mm sample was cracked under a N2 atmosphere with 40 mL min–1 flow rate).
Figure 5a–c indicted that the content of short-chain alkenes (<5) presents a decline trend from 575 to 650 K and reaches its maximum at 725 K and then declines in the period between 725 and 775 K. However, the increment of cyclohexene, 4-methylcyclohexene, benzene, toluene, and styrene appear a secondary peak and peak shoulders at the same temperature range. Combined with previous experimental studies and previous reports,44 it can be inferred that the decreasing amounts of alkenes at 575–650 and 725–750 K are mainly transformed into benzene, toluene, and styrene through Diels–Alder reactions. Therefore, DFT was used to calculate the possible reaction pathways to explore the complex alkene conversion processes. The detailed processes are as follows.
3.4.1. Synthesis of Cycloalkene by Addition of Alkene
Three reactions to synthesize cycloalkenes from short-chain alkenes were calculated by DFT. As shown in Figure 6, cyclohexene, 4-methylcyclohexene, and 4-vinylcyclohexene were synthesized in three reactions, respectively. The highest reaction energy barrier of 2.50 eV for reaction 2 occurred in the first reaction step, which was lower than that of reaction 1 and reaction 3. Among the three reactions, only reaction 3 is endothermic. The highest energy barrier of 2.93 eV occurred in reaction 1. In these reactions, the trans-1,3-butadiene always transformed to the cis configuration first and then connected with the C at one end of the double bond of the alkene to form a long-chain intermediate. After that, the intermediate bonded to the other end of the double bond C to form the cycloalkene. This phenomenon also consisted with the published work that the trans conformation of 1,3-butadiene reacted with alkene was more difficult than the cis conformation in the Diels–Alder reaction.51,52 Among these three reactions, reaction 2 had the lowest reaction energy barrier, the highest exothermic heat, and the best reactivity. Therefore, the content of 4-methylcyclohexene was the highest in the cycloalkene product of alkene synthesis.
Figure 6.
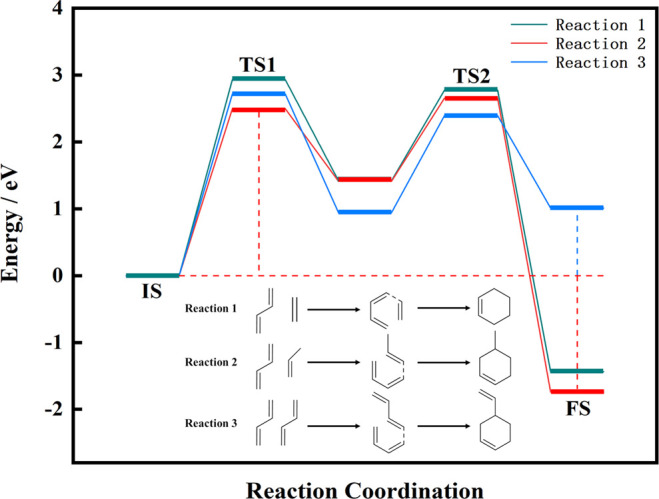
Reaction energy barrier diagram of three alkene synthesis of cycloalkene.
3.4.2. Dehydrogenation of Cyclic Alkenes to Synthesize Aromatic Hydrocarbons
The cycloalkene product obtained by alkene addition needs to go through four steps of dehydrogenation to obtain the aromatic product, so the DFT calculations were carried out for the cycloalkene dehydrogenation reaction. For the cyclohexene, 4-methylcyclohexene, and 4-vinylcyclohexene (Figure 6), we calculated the energy barriers of each step of the dehydrogenation reaction, respectively.
As shown in Figure 7 and Table S2, for cyclohexene dehydrogenation (reaction 1), the first dehydrogenation site always occurred at the C closest to the double bond. The second dehydrogenation formed a double bond and thus a stable conjugated structure was obtained. Finally, a benzene ring is formed after these four steps, and the highest energy barrier was 3.41 eV occurred in the first step. For 4-methylcyclohexene dehydrogenation (reaction 2), there were four possible sites for the first dehydrogenation step. The simulation result showed that the optimal dehydrogenation site for the first step is C1 with an energy barrier of 4.15 eV. Using the same method, the optimal dehydrogenation sites for the rest three steps are C2, C4, and C3 in order, with energy barriers of 2.13, 3.43, and 1.28 eV, respectively. For the dehydrogenation of 4-vinylcyclohexene (reaction 3), the optimal dehydrogenation sites for the four steps are C2, C1, C4, and C3 in order, and the highest energy barrier was 3.36 eV. According to the energy barrier analysis, the most likely reaction among the three routes was reaction 3, and the final product is styrene.
Figure 7.

(a) Transition, initial, and final state structure of aromatics synthesized by cyclic alkene dehydrogenation; (b) reaction energy barrier diagram of cyclic alkenes dehydrogenation to aromatic hydrocarbons.
Besides, we also calculated the reaction route of benzene ring methylation to toluene. The methylation reaction of benzene had a very low energy barrier (Ea1 = 0.21 eV, Ea2 = 1.19 eV) and a large exotherm (ΔE = −1.87 eV), it means that the benzylation reaction is easy to carry out to form toluene, and toluene was also the final product of reaction 2. Therefore, we judged that the content of styrene and toluene are both higher than that of benzene in the aromatic products formed from aliphatic chains. This conclusion was also consistent with the experimental results (Figure 1).
3.4.3. Toluene Methylation Reaction
In addition to toluene, products such as xylene and trimethylbenzene were monitored in the pyrolysis experiment of SBR, and we also concerned about the formation of xylene and trimethylbenzene. From Figure 5b,c, we can see that the increment of toluene decreases rapidly at 765–785 K, while the increment of xylene and trimethylbenzene have a peak shoulder in the corresponding temperature range. Comprehensive analysis showed that toluene was converted into xylene and trimethylbenzene through methylation at this temperature, and xylene was also converted into trimethylbenzene when it was generated. The reaction pathway was simulated theoretically by means of DFT and AIMD.
First, we carried out a theoretical simulation of the methylation reaction of toluene, which included four reaction pathways and five different products. As shown in Figure 8a. The first methylation reaction of toluene yields three different xylene products (o-xylene, m-xylene, and p-xylene). The o-xylene was further methylated to synthesize 1,2,3-trimethylbenzene, while the m-xylene was methylated to form 1,2,3-trimethylbenzene and 1,2,5-trimethylbenzene via two different reaction pathways. It can be seen from Figure 8b that the four pathways were all exothermic reactions. The highest energy barrier of two-step methylation of pathway 3 was 1.19 eV, which was lower than that of other pathways. Moreover, the reaction energies of the two-step methylation of pathway 3 were −1.86 and −1.87 eV, respectively. Therefore, toluene was easily transformed into m-xylene through the first step of methylation and then into mesitylene. For the toluene methylation reaction, the reaction mechanism is consistent with the previous methylation reactions in Section 3.3.2, that is, the C site was first attacked by the methyl radical followed by the dehydrogenation reaction.
Figure 8.

(a) Reaction pathway and reaction rate of methylation reaction of toluene; (b) energy barrier diagram of the toluene methylation reaction.
To have a deep understanding of toluene methylation, we also calculated the reaction kinetics of toluene methylation and obtained the reaction rate of each step. As shown in Figure 8a, the reaction rate of toluene in the first step was on the order of the sixth power of ten, and the reaction rate of trimethylbenzene formation in the second step was on the order of the fourth power of ten. According to the energy barrier and kinetic analysis, the methylation reaction rate of toluene in the first step was higher than that in the second step, which results in high content of xylene in the final products. Among them, pathway 3 has the fastest reaction rate, it indicated that the content of mesitylenes would be higher than that of 1,2,3-trimethylbenzene. Li et al. also discussed that methyl benzenes and mesitylenes had a high proportion in aromatic hydrocarbons.44
AIMD simulations were able to present an intuitive dynamic process of toluene methylation reaction. As shown in Figure 9, AIMD simulations were performed for the first step methylation reactions in pathway 1 and pathway 3 (Figure 8). The methylation reaction of toluene is achieved by three steps. First, the free methyl radical in the environment approached C on the benzene ring; second, the H in the toluene gradually falls off; and third, the H freely combines with the free methyl group to form methane. The AIMD results verify that this simulation pathway is reasonable.
Figure 9.

AIMD simulation results of toluene methylation reaction to form (a) 1,3-dimethylbenzene and (b) 1,2-dimethylbenzene.
4. Conclusions
To understand the formation mechanism of aromatic hydrocarbons is important for WTPO upgrading into an alternative fuel, which is the main strategy for the application of this oil up until now. Starting from the SBR chain, this work uses experimental methods to explore the product distribution at different temperatures and studies the formation mechanism of important MAHs by DFT and AIMD calculations. The theoretical calculations are in good agreement with experimental results. The important conclusions are as follows:
-
1
The styrene, toluene, and xylene are the main MAHs produced by the pyrolysis of SBR in the experiment. This result was also confirmed in the following theoretical calculations due to the different bond breaking sites and secondary reactions.
-
2
Aromatic products in rubber pyrolysis are obtained not only directly from SBR pyrolysis but also from short-chain alkenes through the Diels–Alder reaction to synthesize cyclic alkenes, followed by dehydrogenation. In the case of cyclic olefin dehydrogenation, it always occurs preferentially on the C adjacent to the double bond, followed by further dehydrogenation to form the double bond, and thus, the main dehydrogenation product is toluene.
-
3
The aromatic products are possibly further converted into other aromatic products through methylation and hydrogenation reactions. For the mechanism of the methylation reaction, we conclude from the energy barrier analysis obtained by DFT and AIMD calculations that the C atom is always attacked by the methyl radical first, and then, the H atom is gradually dissociated. The hydrogenation reaction always hydrogenates the C at the α position first and then the β position.
Acknowledgments
The authors would like to express appreciation for the support of National Key R&D Program of China [Grant no. 2018YFC1902601].
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acsomega.2c04994.
Experimental equipment for rubber pyrolysis; GC program settings; component of pyrolytic products at different flow rates; yield of pyrolytic products at different temperature; Py-GC/MS experiments for SBR were performed to analyze the distribution of SBR pyrolytic products at 575, 625, 675, 725, 775, and 825 K, respectively; reaction energy barrier diagram of α-methylstyrene two-step hydrogenation; and dehydrogenation energy barrier and reaction energy of cyclic alkene (PDF)
Author Contributions
∥ J.L. and D.Z. contributed equally to this work.
The authors declare no competing financial interest.
Supplementary Material
References
- Torretta V.; Rada E. C.; Ragazzi M.; Trulli E.; Istrate I. A.; Cioca L. I. Treatment and disposal of tyres: Two EU approaches. A review. Waste Manage. 2015, 45, 152–160. 10.1016/j.wasman.2015.04.018. [DOI] [PubMed] [Google Scholar]
- Chen B.; Zheng D.; Xu R.; Leng S.; Han L.; Zhang Q.; Liu N.; Dai C.; Wu B.; Yu G.; Cheng J. Disposal methods for used passenger car tires: One of the fastest growing solid wastes in China. Green Energy Environ. 2022, 7, 1298. 10.1016/j.gee.2021.02.003. [DOI] [Google Scholar]
- Martínez J. D.; Puy N.; Murillo R.; García T.; Navarro M. V.; Mastral A. M. Waste tyre pyrolysis – A review. Renewable Sustainable Energy Rev. 2013, 23, 179–213. 10.1016/j.rser.2013.02.038. [DOI] [Google Scholar]
- Osorio-Vargas P.; Campos C. H.; Torres C. C.; Herrera C.; Shanmugaraj K.; Bustamante T. M.; Diaz de Leon J. N.; Medina F.; Arteaga-Pérez L. E. Catalytic pyrolysis of used tires on noble-metal-based catalysts to obtain high-value chemicals: Reaction pathways. Catal. Today 2022, 394, 475. 10.1016/j.cattod.2021.06.029. [DOI] [Google Scholar]
- Tang X.; Chen X.; He Y.; Evrendilek F.; Chen Z.; Liu J. Co-pyrolytic performances, mechanisms, gases, oils, and chars of textile dyeing sludge and waste shared bike tires under varying conditions. Chem. Eng. J. 2022, 428, 131053. 10.1016/j.cej.2021.131053. [DOI] [Google Scholar]
- Osorio-Vargas P.; Lick I. D.; Sobrevía F.; Correa-Muriel D.; Menares T.; Manrique R.; Casella M. L.; Arteaga-Pérez L. E. Thermal Behavior, Reaction Pathways and Kinetic Implications of Using a Ni/SiO2 Catalyst for Waste Tire Pyrolysis. Waste Biomass Valorization 2021, 12, 6465–6479. 10.1007/s12649-021-01494-y. [DOI] [Google Scholar]
- Pan Y.; Yang D.; Sun K.; Wang X.; Zhou Y.; Huang Q. Pyrolytic transformation behavior of hydrocarbons and heteroatom compounds of scrap tire volatiles. Fuel 2020, 276, 118095. 10.1016/j.fuel.2020.118095. [DOI] [Google Scholar]
- Pan Y.; Sima J.; Wang X.; Zhou Y.; Huang Q. BTEX recovery from waste rubbers by catalytic pyrolysis over Zn loaded tire derived char. Waste Manage. 2021, 131, 214–225. 10.1016/j.wasman.2021.06.013. [DOI] [PubMed] [Google Scholar]
- Liu S.; Yu J.; Bikane K.; Chen T.; Ma C.; Wang B.; Sun L. Rubber pyrolysis: Kinetic modeling and vulcanization effects. Energy 2018, 155, 215–225. 10.1016/j.energy.2018.04.146. [DOI] [Google Scholar]
- Labaki M.; Jeguirim M. Thermochemical conversion of waste tyres-a review. Environ. Sci. Pollut. Res. Int. 2017, 24, 9962–9992. 10.1007/s11356-016-7780-0. [DOI] [PubMed] [Google Scholar]
- Ding Z.; Chen Z.; Liu J.; Evrendilek F.; He Y.; Xie W. Co-combustion, life-cycle circularity, and artificial intelligence-based multi-objective optimization of two plastics and textile dyeing sludge. J. Hazard. Mater. 2022, 426, 128069. 10.1016/j.jhazmat.2021.128069. [DOI] [PubMed] [Google Scholar]
- Chen G.; Sun B.; Li J.; Lin F.; Xiang L.; Yan B. Products distribution and pollutants releasing characteristics during pyrolysis of waste tires under different thermal process. J. Hazard. Mater. 2022, 424, 127351. 10.1016/j.jhazmat.2021.127351. [DOI] [PubMed] [Google Scholar]
- Aslan D. I.; Parthasarathy P.; Goldfarb J. L.; Ceylan S. J. W. M. Pyrolysis reaction models of waste tires: Application of Master-Plots method for energy conversion via devolatilization. Waste Manage. 2017, 68, 405–411. 10.1016/j.wasman.2017.06.006. [DOI] [PubMed] [Google Scholar]
- Tang X.; Chen Z.; Liu J.; Chen Z.; Xie W.; Evrendilek F.; Buyukada M. J. J. o. H. M. Dynamic pyrolysis behaviors, products, and mechanisms of waste rubber and polyurethane bicycle tires. J. Hazard. Mater. 2021, 402, 123516. 10.1016/j.jhazmat.2020.123516. [DOI] [PubMed] [Google Scholar]
- Chen Z.; Liu J.; Chen H.; Ding Z.; Tang X.; Evrendilek F. J. R. E. Oxy-fuel and air atmosphere combustions of Chinese medicine residues: Performances, mechanisms, flue gas emission, and ash properties. Renewable Energy 2022, 182, 102–118. 10.1016/j.renene.2021.10.010. [DOI] [Google Scholar]
- Ding Z.; Chen Z.; Liu J.; Evrendilek F.; He Y.; Xie W. J. J. o. H. M. Co-combustion, life-cycle circularity, and artificial intelligence-based multi-objective optimization of two plastics and textile dyeing sludge. J. Hazard. Mater. 2022, 426, 128069. 10.1016/j.jhazmat.2021.128069. [DOI] [PubMed] [Google Scholar]
- Tang X.; Chen X.; He Y.; Evrendilek F.; Chen Z.; Liu J. J. C. E. J. Co-pyrolytic performances, mechanisms, gases, oils, and chars of textile dyeing sludge and waste shared bike tires under varying conditions. Chem. Eng. J. 2022, 428, 131053. 10.1016/j.cej.2021.131053. [DOI] [Google Scholar]
- Fu J.; Liu J.; Xu W.; Chen Z.; Evrendilek F.; Sun S. J. B. T. Torrefaction, temperature, and heating rate dependencies of pyrolysis of coffee grounds: Its performances, bio-oils, and emissions. Bioresour. Technol. 2022, 345, 126346. 10.1016/j.biortech.2021.126346. [DOI] [PubMed] [Google Scholar]
- Shi S. Advances in modeling hydrocarbon cracking kinetic predictions by quantum chemical theory: A review. Int. J. Energy Res. 2018, 42, 3164–3181. 10.1002/er.4049. [DOI] [Google Scholar]
- Li X.; Zheng M.; Ren C.; Guo L. ReaxFF Molecular Dynamics Simulations of Thermal Reactivity of Various Fuels in Pyrolysis and Combustion. Energy Fuels 2021, 35, 11707–11739. 10.1021/acs.energyfuels.1c01266. [DOI] [Google Scholar]
- Wei X.; Yu J.; Du J.; Sun L. A ReaxFF molecular dynamic study on pyrolysis behavior and sulfur transfer during pyrolysis of vulcanized natural rubber. Waste Manage. 2022, 139, 39–49. 10.1016/j.wasman.2021.12.022. [DOI] [PubMed] [Google Scholar]
- Deng S.; Zhuo H.; Wang Y.; Leng S.; Zhuang G.; Zhong X.; Wei Z.; Yao Z.; Wang A. J.-G. Multiscale Simulation on Product Distribution from Pyrolysis of Styrene-Butadiene Rubber. Polymers 2019, 11, 1967. 10.3390/polym11121967. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yan S.; Xia D.; Zhang X.; Jiang B. A complete depolymerization of scrap tire with supercritical water participation: A molecular dynamic simulation study. Waste Manage. 2019, 93, 83–90. 10.1016/j.wasman.2019.05.030. [DOI] [PubMed] [Google Scholar]
- Bei L.; Han Y.; Qiao L.; Bikane K.; Yu J.; Sun L. The in-situ effect of H2S on the decomposition of natural rubber and catalyst activity. Chemosphere 2021, 283, 131252. 10.1016/j.chemosphere.2021.131252. [DOI] [PubMed] [Google Scholar]
- Liu J.; Fan X. R.; Zhao W.; Yang S. W.; Hu B.; Yang S. G.; Lu Q. Mechanical insight into the formation of H2S from thiophene pyrolysis: The influence of H2O. Chemosphere 2021, 279, 130628. 10.1016/j.chemosphere.2021.130628. [DOI] [PubMed] [Google Scholar]
- Zheng D.; Cheng J.; Dai C.; Xu R.; Wang X.; Liu N.; Wang N.; Yu G.; Chen B. Study of passenger-car-waste-tire pyrolysis: Behavior and mechanism under kinetical regime. Waste Manage. 2022, 148, 71–82. 10.1016/j.wasman.2022.05.024. [DOI] [PubMed] [Google Scholar]
- Perdew J. P.; Burke K.; Ernzerhof M. Generalized Gradient Approximation Made Simple. Phys. Rev. Lett. 1996, 77, 3865–3868. 10.1103/physrevlett.77.3865. [DOI] [PubMed] [Google Scholar]
- Kresse G.; Furthmüller J. Efficiency of ab-initio total energy calculations for metals and semiconductors using a plane-wave basis set. Comput. Mater. Sci. 1996, 6, 15–50. 10.1016/0927-0256(96)00008-0. [DOI] [PubMed] [Google Scholar]
- Kresse G.; Hafner J. Ab initio molecular-dynamics simulation of the liquid-metal–amorphous-semiconductor transition in germanium. Phys. Rev. B: Condens. Matter Mater. Phys. 1994, 49, 14251–14269. 10.1103/physrevb.49.14251. [DOI] [PubMed] [Google Scholar]
- Cheng J.; Song T.; Hu P.; Lok C. M.; Ellis P.; French S. A density functional theory study of the α-olefin selectivity in Fischer–Tropsch synthesis. J. Catal. 2008, 255, 20–28. 10.1016/j.jcat.2008.01.027. [DOI] [Google Scholar]
- Yao Z. H.; Guo C. X.; Mao Y.; Hu P. Quantitative Determination of C-C Coupling Mechanisms and Detailed Analyses on the Activity and Selectivity for Fischer-Tropsch Synthesis on Co(0001): Microkinetic Modeling with Coverage Effects. ACS Catal. 2019, 9, 5957–5973. 10.1021/acscatal.9b01150. [DOI] [Google Scholar]
- Wei Z. Z.; Yao Z. H.; Zhou Q.; Zhuang G. L.; Zhong X.; Deng S. W.; Li X. N.; Wang J. G. Optimizing Alkyne Hydrogenation Performance of Pd on Carbon in Situ Decorated with Oxygen-Deficient TiO2 by Integrating the Reaction and Diffusion. ACS Catal. 2019, 9, 10656–10667. 10.1021/acscatal.9b03300. [DOI] [Google Scholar]
- Mao Y.; Hu P. Identification of the active sites and mechanism for partial methane oxidation to methanol over copper-exchanged CHA zeolites. Sci. China: Chem. 2020, 63, 850–859. 10.1007/s11426-019-9695-9. [DOI] [Google Scholar]
- Yao Z.; Zhao J.; Zhao C.; Deng S.; Zhuang G.; Zhong X.; Wei Z.; Li Y.; Wang S.; Wang J. A first-principles study of reaction mechanism over carbon decorated oxygen-deficient TiO2 supported Pd catalyst in direct synthesis of H2O2. Chin. J. Chem. Eng. 2021, 31, 126–134. 10.1016/j.cjche.2020.11.016. [DOI] [Google Scholar]
- Yao Z.; Zhao J.; Bunting R. J.; Zhao C.; Hu P.; Wang J. Quantitative Insights into the Reaction Mechanism for the Direct Synthesis of H2O2 over Transition Metals: Coverage-Dependent Microkinetic Modeling. ACS Catal. 2021, 11, 1202–1221. 10.1021/acscatal.0c04125. [DOI] [Google Scholar]
- Grimme S.; Ehrlich S.; Goerigk L. Effect of the damping function in dispersion corrected density functional theory. J. Comput. Chem. 2011, 32, 1456–1465. 10.1002/jcc.21759. [DOI] [PubMed] [Google Scholar]
- Grimme S.; Antony J.; Ehrlich S.; Krieg H. A consistent and accurate ab initio parametrization of density functional dispersion correction (DFT-D) for the 94 elements H-Pu. J. Chem. Phys. 2010, 132, 154104. 10.1063/1.3382344. [DOI] [PubMed] [Google Scholar]
- Monkhorst H. J.; Pack J. D. Special points for Brillouin-zone integrations. Phys. Rev. B: Solid State 1976, 13, 5188–5192. 10.1103/physrevb.13.5188. [DOI] [Google Scholar]
- Liu Z.-P.; Hu P. General Rules for Predicting Where a Catalytic Reaction Should Occur on Metal Surfaces: A Density Functional Theory Study of C–H and C–O Bond Breaking/Making on Flat, Stepped, and Kinked Metal Surfaces. J. Am. Chem. Soc. 2003, 125, 1958–1967. 10.1021/ja0207551. [DOI] [PubMed] [Google Scholar]
- Zhang C.; Hu P.; Alavi A. A General Mechanism for CO Oxidation on Close-Packed Transition Metal Surfaces. J. Am. Chem. Soc. 1999, 121, 7931–7932. 10.1021/ja991475x. [DOI] [Google Scholar]
- Alavi A.; Hu P.; Deutsch T.; Silvestrelli P. L.; Hutter J. CO Oxidation on Pt(111): An Ab Initio Density Functional Theory Study. Phys. Rev. Lett. 1998, 80, 3650–3653. 10.1103/physrevlett.80.3650. [DOI] [Google Scholar]
- Cortright R. D.; Dumesic J. A. Kinetics of heterogeneous catalytic reactions: Analysis of reaction schemes. Adv. Catal. 2001, 46, 161–264. 10.1016/s0360-0564(02)46023-3. [DOI] [Google Scholar]
- Hita I.; Arabiourrutia M.; Olazar M.; Bilbao J.; Arandes J. M.; Castaño P. Opportunities and barriers for producing high quality fuels from the pyrolysis of scrap tires. Renewable Sustainable Energy Rev. 2016, 56, 745–759. 10.1016/j.rser.2015.11.081. [DOI] [Google Scholar]
- Li D.; Lei S.; Lin F.; Zhong L.; Ma W.; Chen G. Study of scrap tires pyrolysis – Products distribution and mechanism. Energy 2020, 213, 119038. 10.1016/j.energy.2020.119038. [DOI] [Google Scholar]
- Chao L.; Zhang C.; Zhang L.; Gholizadeh M.; Hu X. Catalytic pyrolysis of tire waste: Impacts of biochar catalyst on product evolution. Waste Manage. 2020, 116, 9–21. 10.1016/j.wasman.2020.07.045. [DOI] [PubMed] [Google Scholar]
- Ding K.; Zhong Z.; Zhang B.; Song Z.; Qian X. Pyrolysis Characteristics of Waste Tire in an Analytical Pyrolyzer Coupled with Gas Chromatography/Mass Spectrometry. Energy Fuels 2015, 29, 3181–3187. 10.1021/acs.energyfuels.5b00247. [DOI] [Google Scholar]
- Xu F.; Wang B.; Yang D.; Ming X.; Jiang Y.; Hao J.; Qiao Y.; Tian Y. TG-FTIR and Py-GC/MS study on pyrolysis mechanism and products distribution of waste bicycle tire. Energy Convers. Manage. 2018, 175, 288–297. 10.1016/j.enconman.2018.09.013. [DOI] [Google Scholar]
- Kaminsky W.; Sinn H., Pyrolysis of Plastic Waste and Scrap Tires Using a Fluidized-Bed Process. Thermal Conversion of Solid Wastes and Biomass; U.S. Department of Energy Office of Scientific and Technical Information, 1980; Vol. 130, pp 423–439. [Google Scholar]
- Ye W.; Xu X.; Zhan M.; Huang Q.; Li X.; Jiao W.; Yin Y. Formation behavior of PAHs during pyrolysis of waste tires. J. Hazard. Mater. 2022, 435, 128997. 10.1016/j.jhazmat.2022.128997. [DOI] [PubMed] [Google Scholar]
- Song Z.; Liu L.; Yang Y.; Sun J.; Zhao X.; Wang W.; Mao Y.; Yuan X.; Wang Q. Characteristics of limonene formation during microwave pyrolysis of scrap tires and quantitative analysis. Energy 2018, 142, 953–961. 10.1016/j.energy.2017.10.101. [DOI] [Google Scholar]
- Cui C.-X.; Liu Y.-J. A DFT study on Diels-Alder cycloadditions oftrans-1,3-butadiene to C60and C70. J. Phys. Org. Chem. 2015, 28, 281–289. 10.1002/poc.3408. [DOI] [Google Scholar]
- Cui C.-X.; Liu Y.-J. A thorough understanding of the Diels-Alder reaction of 1,3-butadiene and ethylene. J. Phys. Org. Chem. 2014, 27, 652–660. 10.1002/poc.3313. [DOI] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.