

Abstract
Lipoarabinomannan (LAM) is a component of the mycobacterial surface which has been associated with a variety of deleterious effects on immune system function. Despite the importance of LAM to the pathogenesis of mycobacterial infection, there is no information available on its fate in vivo. In this study, we determined the pharmacokinetics and tissue distribution of exogenously administered LAM in mice. For measurements of serum and tissue LAM concentrations, we developed an enzyme-linked immunosorbent assay which used monoclonal antibodies of different isotypes to capture and detect LAM at concentrations of ≥0.4 μg/ml. Intravenous administration of LAM to mice resulted in transient serum levels with organ deposition in the spleen and in the liver. Immunohistochemical studies localized LAM to the spleen marginal zone macrophages and, to a lesser degree, to liver macrophages. When LAM was administered to mice previously given a LAM-binding immunoglobulin M (IgM), LAM was very rapidly cleared from circulation. In those mice, deposition of LAM in the spleen was significantly reduced while LAM deposition in the liver increased. Administration of LAM-binding IgM resulted in significant levels of IgM to LAM in bile consistent with an increased hepatobiliary excretion of LAM in the presence of specific antibody. Bile, liver extracts, and bile salts were found to rapidly inactivate the immunoreactivity of LAM. The results indicate that serum clearance and organ deposition of LAM in mice are affected by the presence of LAM-binding antibody and suggest a mechanism by which antibody could modify the course of mycobacterial infection.
Lipoarabinomannan (LAM) is a mycobacterial envelope glycolipid (3) that has been implicated in the virulence and pathogenesis of Mycobacterium tuberculosis infection. In vitro LAM has been shown to inhibit the proliferation of human T cells (20), scavenge oxygen free radicals, block the transcriptional activation of gamma interferon-inducible genes, inhibit protein kinase C activity (4), and alter cytokine expression by macrophages (2). LAM has also been implicated in fever induction, tissue necrosis (24), and lung cavity formation (5). Despite considerable evidence that LAM can function as an immunomodulator, there is no information available on the fate of LAM in vivo. Another unexplored question is the role, if any, of LAM-binding antibody in altering the fate and tissue distribution in vivo. LAM is immunogenic and elicits antibody responses during natural mycobacterial infection (14, 29). There is evidence that some antibodies to M. tuberculosis may contribute to host defense (9). In this regard, a serum immunoglobulin G (IgG) response to LAM has been associated with reduced likelihood of M. tuberculosis dissemination in children (8).
The liver and the spleen are important organs in the clearance of microbial products, and the presence of specific antibody can have profound effects on antigen clearance in vivo. For example, Cryptococcus neoformans glucuronoxylomannan (GXM) is deposited primarily in the spleen in naive rodents, but antibody administration results in rapid serum clearance with deposition in liver (11, 18). Pneumococcal type III capsular polysaccharide and C carbohydrate are cleared by hepatobiliary transport when specific IgA is present (25). In this study, we evaluated the fate and organ deposition of LAM in mice in the presence and absence of LAM-binding IgM. The results indicate a role for LAM-binding antibody and the hepatobiliary system in the elimination of this mycobacterial glycolipid.
(This work was presented in part at the 36th IDSA annual meeting in Denver, Colo., November 1998, and at the 4th International Conference on the Pathogenesis of Mycobacterial Infections, Stockholm, Sweden, July 1999.)
MATERIALS AND METHODS
Lipoarabinomannan.
LAM from M. tuberculosis H37Rv was kindly provided by J. T. Belisle and P. J. Brennan (Department of Microbiology, Colorado State University, Fort Collins) and was suspended in sterile phosphate-buffered saline (PBS) prior to injection.
MAbs.
The murine LAM-binding monoclonal antibody (MAb) 5c11 (IgM) was previously described (10). MAb 12A1, an IgM to Cryptococcus neoformans GXM (21), was used as an irrelevant isotype-matched control. MAb CS-40, a LAM-binding IgG1 (6), was kindly provided by J. T. Belisle and P. J. Brennan. MAbs injected into mice (5c11 and 12A1) were in ascites. MAbs used for enzyme-linked immunosorbent assay (ELISA) were in cell supernatant (5c11 and 12A1) or in a purified form suspended in PBS (CS-40).
Mice.
Adult female BALB/c mice aged 7 to 30 weeks were purchased from either the Jackson Laboratories (Bar Harbor, Maine) or the National Cancer Institute (Frederick, Md.).
Capture ELISA for the detection of LAM.
Microtiter plate wells (Corning, Corning, N.Y.) were coated with 50 μl of goat anti-mouse IgG1 (Southern Biotechnology Associates, Inc., Birmingham, Ala.) at 1 μg/ml in Tris-buffered saline (TBS). The plates were incubated for 1.5 h at 37°C and blocked with 1% bovine serum albumin (BSA) in TBS. After incubation at 37°C for 1.5 h, the plates were washed three times with 0.05% Tween 20 in TBS. MAb CS-40 was added to the wells at a concentration of 1 μg/ml in 1% BSA in TBS and incubated for 1 h. The wells were then washed, and a sample of body fluid or organ homogenate supernatant (described below) was added to the wells and serially diluted in 1% BSA in TBS. After incubation for 1.5 h at 37°C and washing, 1 μg of MAb 5c11/per ml in 1% BSA was added and the plates were incubated for 1 h, followed by washing. Alkaline phosphatase-conjugated goat anti-mouse IgM (Southern Biotechnology Associates, Inc.) was then added at 1 μg/ml in 1% BSA and incubated for 1 h at 37°C. After washing five times, p-nitrophenylphosphate (Southern Biotechnology Associates, Inc.) at 1 mg/ml and in substrate buffer (0.001 M MgCl2, 0.05 M Na2Co3 [pH 9.8]) was added to the plates, and the absorbance at 405 nm was read with a Ceres 900 Hdi reader (Bio-Tek Instruments Inc., Winooski, Vt.). LAM concentrations were calculated relative to a standard solution in which a known amount of LAM was dissolved in 1% BSA in TBS and serially diluted as above.
Preparation of samples for the detection of LAM.
The methodology for preparing tissue samples for LAM detection was adopted from protocols used to detect C. neoformans polysaccharide in vivo. Serum and bile were diluted in PBS with proteinase K (1.25 mg/ml) (Boehringer Mannheim, Indianapolis, Ind.) and incubated overnight at 37°C. The samples were then boiled for 15 min to inactivate proteinase K, diluted in 1% BSA, and used in the capture ELISA for LAM detection. Organs were homogenized for 2 min (Ultra Turrax T25 homogenizer; Janke and Kunkel, Staufen, Germany), and the homogenate was digested with proteinase K (1 mg/ml) overnight at 37°C. After proteinase K digestion, the samples were boiled, cell debris was removed by centrifugation, and the organ supernatants were analyzed for LAM by capture ELISA. This process was validated by demonstrating that there was no reduction in the recovery of LAM from serum containing MAb 5c11 and LAM processed with proteinase K and subjected to boiling relative to that of serum containing LAM without MAb 5c11. Hence, proteinase K treatment and boiling dissociated LAM-protein complexes.
Immunohistochemistry.
Localization of LAM in tissue was done by immunohistochemistry. Organs were fixed in 10% buffered formalin, embedded in paraffin, sectioned into 4-μm slices, and placed on glass slides. Tissue sections were treated with 3% H2O2 for 30 min and blocked with 2% BSA in PBS for 1 h. MAb to LAM 5c11 (IgM) or CS-40 (IgG1) was added as the primary antibody at 1 μg/ml, and the slide was incubated for 2 h at room temperature or overnight at 4°C. After washing, the slides were incubated with horseradish peroxidase-conjugated goat anti-mouse IgM or IgG1 (Southern Biotechnology Associates, Inc.) diluted in 2% BSA at a ratio of 1:200 for 2 h at room temperature. After additional washing, color was developed with diaminobenzidine. Negative controls consisted of tissue sections from a mouse that received no LAM that was treated as described above, as well as tissue sections from a LAM-treated mouse incubated with MAb 12A1 (isotype-matched control). To confirm the localization of LAM in spleen cells, 10-μm frozen sections of splenic tissue from a mouse treated with LAM were incubated with MAb ER-TR9, a rat anti-mouse IgM (32) specific for marginal zone macrophages (Accurate Chemical and Scientific Corp., Westbury, N.Y.), followed by horseradish peroxidase-conjugated goat anti-rat IgM (Southern Biotechnology Associates, Inc.). Color was developed with diaminobenzidine. The frozen sections were further incubated with MAb CS-40 at 37°C for 1 h, followed by fluorescein isothiocyanate (FITC)-labeled anti-mouse IgG (Southern Biotechnology Associates, Inc.) for 45 min at room temperature. Each step was followed by washing with PBS. Negative controls consisted of frozen sections of splenic tissue treated as described above, with the exception of MAb CS-40. Additional controls consisted of frozen sections of spleen from a mouse receiving no LAM treated as described above (with and without MAb CS-40).
Pharmacokinetics studies.
To determine the kinetics of LAM in mice, 50 μg of LAM was dissolved in sterile PBS and injected into the tail vein. Blood was obtained from the retroorbital plexus at various times after injection, and serum was separated by centrifugation and stored at −20°C. The mice were killed by cervical dislocation, and the liver, spleen, kidney, lung, brain, and gallbladder were removed. Organs were weighed, and a small portion of each was placed in 10% buffered formalin for immunohistochemical analysis. The bulk of the organs as well as the serum and bile were used in the LAM capture ELISA as described above. To determine the effects of MAbs on the kinetics of LAM, 1 mg of MAb 5c11 or MAb 12A1 was administered to mice intraperitoneally (i.p.) 2 h prior to the injection of LAM.
Measurement of IgM to LAM in biological fluids.
The titers of LAM-binding IgM in serum, gallbladder bile, and urine were determined by ELISA. Serum was obtained as described above. The gallbladder was removed, placed in a 1-ml tube, perforated with a 23-gauge needle, and centrifuged to separate bile from the gallbladder sac, and the bile was transferred into a clean tube. Urine was collected by placing the mice in a plastic dish. Microtiter plate wells were coated with 1 μg of LAM solution in carbonate buffer (pH 9.6) per ml, incubated at 37°C for 1.5 h, and blocked with 3% BSA in TBS. After washing, serum, bile, and urine samples were diluted in 1% BSA in TBS and added to the plates, and the plates were incubated for 1 h at 37°C or overnight at 4°C, followed by additional washing. Alkaline phosphatase-conjugated goat anti-mouse IgM (Southern Biotechnology Associates, Inc.) was added to each well at a concentration of 1 μg/ml in 1% BSA, and the plates were incubated for 1 h at 37°C. The wells were then washed five times, p-nitrophenylphosphate (Southern Biotechnology Associates, Inc.) was added at a concentration of 1 mg/ml in substrate buffer (0.001 M MgCl2, 0.05 M Na2Co3 [pH 9.8]), and the absorbance at 405 nm was read with a Ceres 900 Hdi reader (Bio-Tek Instruments Inc.). The titers of LAM-binding IgM were calculated.
Stability of LAM in liver extract.
Liver extract was prepared by homogenizing the liver from a naive BALB/c mouse. The homogenate was filtered through a 2-μm membrane to remove cell debris. LAM was added to the extract and to a PBS control, and the mixtures were incubated for 1 h at 37°C. After proteinase K digestion, the samples were boiled, cell debris was removed by centrifugation, and LAM concentration in the organ supernatant was measured by ELISA.
Stability of LAM in bile and bile salts.
LAM was added to gallbladder bile (collected as described above) or to a bile salts solution consisting of 11 mM taurocholic acid sodium salt (Calbiochem, La Jolla, Calif.) and 2 mM taurodeoxycholic acid sodium salt (Calbiochem). LAM in PBS was used as a control. Bile was incubated for 1 h, bile salts solutions were incubated for various times at 37°C, and the LAM concentration was measured by ELISA.
Statistical analysis.
Results were analyzed by the Kruskal-Wallis test. A P value of ≤0.05 was considered significant.
RESULTS
Development of a capture ELISA for the detection of LAM.
Using MAbs of different isotypes to capture and detect LAM, we were able to measure LAM immunoreactivity in serum, bile, and organ homogenates. LAM suspended in 1% BSA in TBS was used as the standard and was detected at concentrations of ≥0.4 μg/ml (Fig. 1). The detectability of LAM by capture ELISA was not affected by the dilution of LAM in serum or spleen homogenate samples.
FIG. 1.

Capture ELISA for the detection of LAM with two LAM-binding MAbs of different isotypes. Shown is the absorbance dose-response curve for the ELISA used in this study. LAM was diluted in 1% BSA in TBS. Symbols indicate average of four measurements. Error bars indicate standard deviation from the mean. LAM was detected by ELISA at concentrations of ≥0.4 μg/ml. Application of this ELISA to the determination of LAM concentration after dilution in serum or spleen homogenate resulted in similar curves.
Pharmacokinetics of LAM in serum and organs by capture ELISA.
Mice were injected with 50 μg of LAM, and serum LAM concentrations were determined at various times. LAM was detected in a serum sample obtained 10 min after its administration, decreased rapidly, and was not detectable in serum samples 2 to 3 h later. To establish the site of LAM tissue localization, various organs were examined 6.5 h after LAM administration. LAM was detected primarily in the spleen, with a smaller amount detected in the liver. No LAM was detected in kidney, lung, or brain. In a mouse studied on day 13 after LAM injection, small amounts of LAM were detected only in the spleen. Hence, LAM injected intravenously (i.v.) into mice is rapidly cleared from serum and deposited in spleen and liver tissues.
Tissue deposition by immunohistochemistry.
Immunohistochemical stains for LAM demonstrated LAM in the marginal zone of the spleen 6.5 h after injection of LAM (Fig. 2). Within the marginal zone, LAM appeared to localize within large cells. LAM was also detected in the liver, mainly in macrophages. No staining was observed in kidney, lung, brain, or control tissue sections.
FIG. 2.
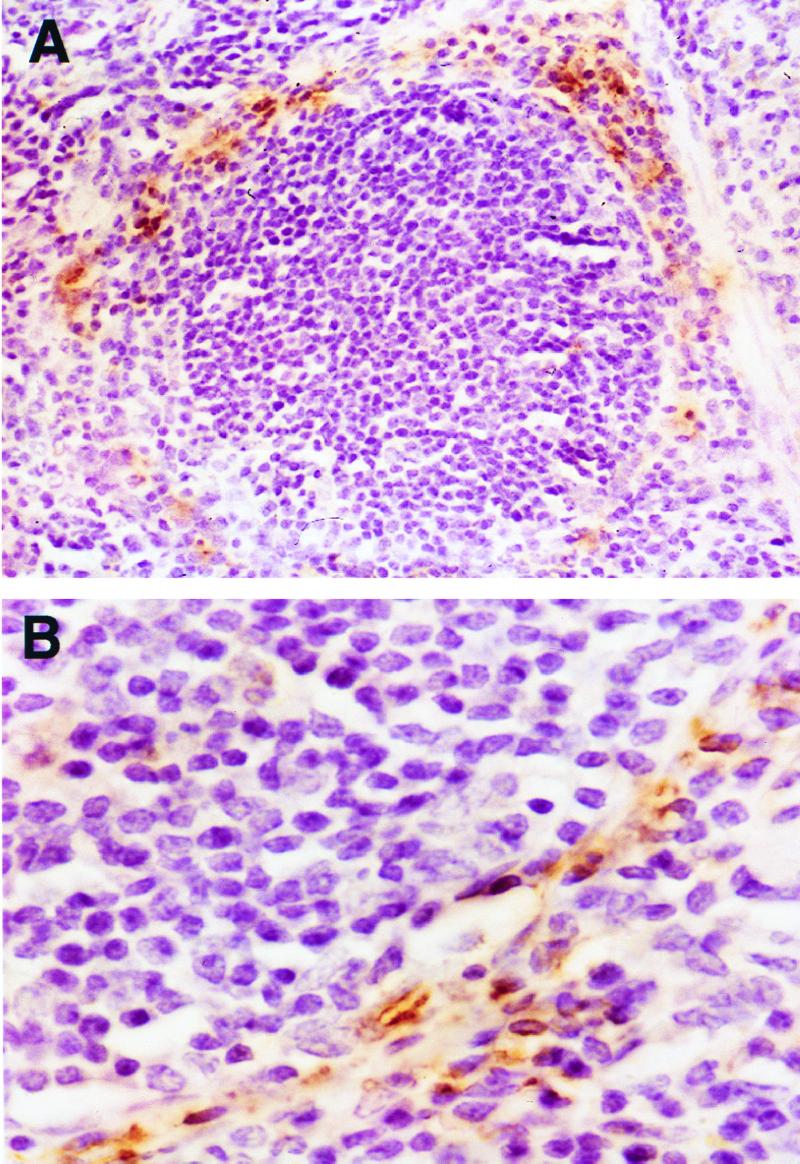
Detection of LAM in mouse spleen by immunohistochemistry 6.5 h after administration of LAM. A follicle with staining of the marginal zone (magnification, ×150) (A); staining is localized to macrophage-like cells in the marginal zone (magnification, ×800) (B).
To further identify the target cells for LAM deposition in the spleen, a double staining procedure was performed with MAb ER-TR9, which is specific for mouse marginal zone macrophages, and MAb CS-40, which is specific for LAM. Colocalization of staining was observed in the marginal zone of the spleen (Fig. 3), indicating that LAM was associated with marginal zone macrophages. Control mice demonstrated staining of marginal zone cells but no staining for LAM.
FIG. 3.
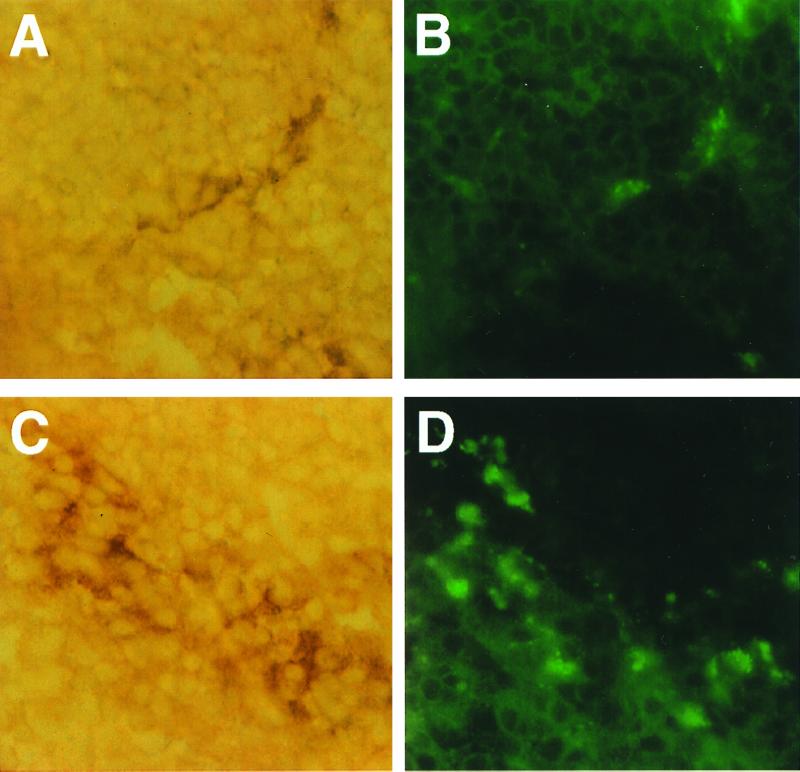
Double staining of frozen spleen sections with MAb ER-TR9 specific for mouse marginal zone macrophages (A and C) and MAb CS-40 specific for LAM (B and D). Shown is an immunofluorescence image of LAM (B and D) colocalized to cells stained by immunohistochemistry with MAb ER-TR9 (A and C). The picture was generated with a Kodak RFS 2035 scanner and Adobe Photoshop 6.5 for Macintosh. Magnification, ×250.
Pharmacokinetics of LAM in the presence and absence of LAM-binding antibody.
Administration of MAb 5c11 2 h prior to injection of LAM altered the distribution of LAM. Mice given MAb 5c11 had significantly lower LAM serum levels than mice given the control IgM, MAb 12A1 (Fig. 4). The difference was statistically significant at 10 min and at 30 min (P < 0.01) and 1.5 h (P < 0.05) (Fig. 4).
FIG. 4.

Serum LAM concentration in the presence of LAM-binding MAb (5c11) or control. Mice were given 50 μg of LAM 2 h after administration of 1 mg of MAb. Symbols indicate means of LAM concentration, and error bars represent 1 standard deviation from the mean. Numbers of mice receiving MAb 5c11 or control MAb 12A1 were 5, 8, 5, 3, and 5 for 10-min, 30-min, 1.5-h, 2.5-h, and 5-h times, respectively. P values were calculated with the Kruskal-Wallis test. ∗, P < 0.01; ∗∗, P < 0.05.
Organ distribution in the presence and absence of LAM-binding antibody as measured by capture ELISA.
Administration of MAb 5c11 also affected the organ distribution of LAM. Significantly lower levels of LAM were detected at 6.5 h in the spleens of mice that received MAb 5c11 (P < 0.01) relative to those in mice that received the control, MAb 12A1 (Fig. 5). The level of LAM in the liver was greater in mice receiving MAb 5c11 than in controls (P = 0.045). (Mice were killed 6.5 to 9.5 h after LAM administration, alternating control and experimental mice to avoid timing bias.) LAM was not detected in other organs nor was it detected in gallbladder bile.
FIG. 5.

LAM concentrations in organs of mice given MAb 5c11 (LAM binding) or control mice 6.5 h after injection of LAM. No LAM was detected in liver, brain, kidney, or lung. Error bars represent one standard deviation of the mean. Control mice received MAb 12A1 (n = 5) or no MAb (n = 1). Error bars denote standard deviation from the mean. P values were calculated with the Kruskal-Wallis test. ∗, P < 0.01; ∗∗, P < 0.05.
Organ distribution in the presence and absence of LAM-binding antibody as detected by immunohistochemistry.
Immunohistochemistry confirmed the results obtained by capture ELISA in tissues of mice sacrificed at 6.5 h, demonstrating a reduced staining in the spleens of mice receiving MAb 5c11 relative to those of controls receiving MAb 12A1. In the liver, LAM was detected mainly in macrophages from both experimental and control groups. LAM was not detected in other organs.
Pharmacokinetics of MAb 5c11.
To further substantiate that MAb 5c11 may be transporting LAM to the hepatobiliary system as an antigen-antibody complex, the titer of LAM-binding IgM was measured in serum, bile, and urine following i.p. administration of 1 mg of MAb 5c11. LAM-binding IgM was detected in serum 30 min after its administration, and high titers in serum were measured up to 24 h later. High titers of LAM-binding IgM were also detected in bile 30 min after i.p. administration of MAb 5c11. Serial measurements demonstrated LAM-binding IgM in gallbladder bile up to 14 h after its administration. LAM-binding IgM was not detected in the urine except for a titer of 1:18 at 30 min, which may reflect excretion of IgM light chains. To exclude the possibility that LAM-binding IgM detected in the gallbladder was the result of contamination after i.p. administration, we measured LAM-binding IgM in bile following i.v. injection of 0.5 mg of MAb 5c11. LAM-binding IgM was detected in gallbladder bile 10 min after i.v. administration, increased rapidly, and reached high titers 30 min after administration (Fig. 6).
FIG. 6.

Titer of LAM-binding IgM in gallbladder bile after i.v. administration of 0.5 mg of MAb 5c11. Symbols denote antibody titers and horizontal lines represent the median for each time.
Effect of liver extract, bile, and bile salts on LAM immunoreactivity.
Failure to detect LAM in bile and the smaller magnitude of difference in liver LAM concentrations between MAb-treated and control groups as compared to those in the spleen (Fig. 5) led us to hypothesize that hepatobiliary substances may reduce the immunoreactivity of LAM. A solution of LAM was mixed with liver extract, bile, and bile salts and incubated for various times. Incubation of LAM with liver extract, bile, or bile salts resulted in a rapid loss of immunoreactivity relative to that of untreated LAM (Fig. 7). The reduced reactivity of LAM was not due to the interference of bile salts with the ELISA, since the reactivity of MAb 5c11 and MAb CS-40 was unaffected by the addition of bile salts. Bile salts reduced the immunoreactivity of LAM at various concentrations (ranging from 2 to 50 μg/ml), and their effect was very rapid. For example, approximately one-half of the original concentration was detected after incubation for 5 min. Longer incubations resulted in even greater reductions in reactivity (Fig. 7C).
FIG. 7.

LAM immunoreactivity determined by ELISA after incubation with liver extract (A), bile (B), or bile salts (C). In each experiment, the initial concentration of LAM was 25 μg/ml. In each group, open symbols correspond to the treated group. Each point was calculated based on four (A) or two (B and C) measurements and expressed as absorbance (A and B) or as a percentage of the absorbance of LAM in PBS (C).
DISCUSSION
Despite many studies on the in vitro effects of LAM on immune cells, the fate of LAM in vivo has not been studied. This information is important because LAM is a major constituent of the mycobacterial cell surface (approximately 15 mg are generated from 1 g of bacteria) (4) and has been shown to cause biological effects by interacting with immune system cells. The ability of the body to eliminate LAM may determine how long LAM can exert its effects on the immune system. Our studies show that in the absence of a LAM-binding MAb, LAM is cleared from the serum within 3 h and is taken up by the spleen, where it localizes to the marginal zone, and by the liver, where it is found in macrophages. The marginal zone in mice is composed of reticular cells, macrophages, and B lymphocytes (16). Staining with antibody specific to marginal zone macrophages demonstrates that the distribution of LAM within the marginal zone is consistent with uptake by macrophages (Fig. 3). Some uptake of LAM by B lymphocytes, however, could not be ruled out. Previous studies have reported that marginal zone macrophages avidly take up microbial antigens, especially T-cell-independent polysaccharides (11, 17), presumably as a result of specialized carbohydrate receptors on marginal zone macrophages. The uptake of LAM by macrophages has been shown to occur through mannose receptors (23, 28). Furthermore, LAM from virulent M. tuberculosis has been demonstrated to serve as a ligand to the mannose receptor (27). Localization of LAM to the phagocytic cells may result in immunomodulatory effects. It is also of interest that mannose receptor-mediated uptake of mycobacteria by human macrophages (1, 26) has been recently reported to result in evasion of macrophage bactericidal response (1).
LAM was cleared from serum significantly faster in mice given LAM-binding IgM. Our data further suggests that the administration of LAM-specific IgM also affected organ distribution by reducing LAM deposition in the spleen and directing it to the liver. Immune complexes have been described to be eliminated via the liver (15, 19). To further explore the role of MAbs in directing LAM to the hepatobiliary system, we studied the pharmacokinetics of MAb 5c11 in serum, gallbladder bile, and urine. High titers of MAb 5c11 were detected in gallbladder bile shortly after administration, suggesting a rapid passage of this IgM MAb from serum to bile. These results are consistent with a previous report that passive administration of IgM resulted in its detection in rat bile within 30 min (13) and suggest that 5c11-LAM complexes may be eliminated via the bile in addition to the deposition of LAM in liver cells. However, we were unable to detect LAM in bile after MAb 5c11 administration, and we noticed that the differences in liver LAM levels between the treatment and control groups were smaller than the differences observed in the spleen LAM levels (P < 0.05 vs. P < 0.01) (Fig. 5). We hypothesized that these observations were due to inactivation or chemical modification by bile salts. A precedent for this has been reported for lipopolysaccharide (LPS) which is inactivated by bile (31). To explore this possibility, we incubated LAM with liver extract and gallbladder bile and demonstrated with capture ELISA that both rapidly reduced the reactivity of LAM. Furthermore, LAM was mixed with bile salts in concentrations similar to those found in rodent bile (7) and at a ratio previously described by others (30). Like bile and liver extract, bile salts rapidly reduced the reactivity of LAM by ELISA. This result strongly suggests a mechanism for the rapid disappearance of LAM from serum whereby IgM promotes liver uptake and biliary excretion with inactivation of LAM by bile salts. Similar (antibody-independent) biliary effects have been reported for LPS (31). LAM is structurally similar to LPS from gram-negative bacteria inasmuch as both are composed of sugar and lipid components. LPS is also involved in virulence, causing the hemodynamic, hematologic, and metabolic changes that are seen in sepsis caused by gram-negative bacteria (33). Our results suggest that MAb 5c11 prevents the uptake of LAM by spleen cells and shunts it toward the hepatobiliary system, where it may undergo inactivation by bile salts and elimination via the bile system. Bile salts-LAM interaction may also result in the masking of the epitope recognized by MAb 5c11. Either mechanism could prevent the recognition of LAM by the capture ELISA and by receptors on spleen cells. The ability of MAb 5c11 to modify the serum clearance and tissue distribution of LAM is similar to observations made previously with C. neoformans GXM, which showed that IgM promoted liver uptake (11, 18). However, GXM-binding IgM promoted spleen uptake as opposed to MAb 5c11, which prevented uptake of LAM by the spleen. Hence, LAM-binding antibody can alter the pharmacokinetics and tissue distribution of LAM, and this suggests a mechanism by which antibody to LAM could affect the course of infection.
In summary, in the absence of LAM-binding antibody, LAM is taken up by marginal zone macrophages and liver cells. In the presence of LAM-binding antibody, uptake of LAM by the spleen is reduced and LAM appears to be rapidly shunted to the hepatobiliary system for uptake, clearance, and inactivation by bile. The exact mechanism by which the liver participates in the MAb-mediated clearance of LAM and the nature of the alteration of LAM by liver extract, bile, and bile salts remain to be determined. It is of interest that bacilli in the liver grow at a lower rate than those in the spleen (22) and that BCG was originally selected after repeated passages of virulent Mycobacterium bovis in media supplemented with ox bile (12).
This study is a first step in understanding the kinetics of LAM in vivo. Additional studies are necessary to determine the clearance and organ distribution of LAM during mycobacterial infection and the role, if any, of MAb 5c11 during infection.
ACKNOWLEDGMENTS
A. Glatman-Freedman has been an Aaron Diamond Young Investigator Awardee, and this work was supported in part by a grant from the Aaron Diamond Foundation. A. Glatman-Freedman is currently supported by NIH grant 1K08AI01691. Arturo Casadevall is supported by NIH grants AI-33142, AI-33774, and HL-59842 and by a Burroughs-Wellcome Fund Scholar Award in Experimental Therapeutics. This work was also supported in part by an NIH training grant in HIV, AIDS, and Opportunistic Infections (1 T32 AI07501-01).
We thank John T. Belisle and Patrick J. Brennan for supplying LAM as part of NIH contract NO1-AI-75320, “Tuberculosis Research Materials and Vaccine Testing.” We further thank David Cohen from the liver center at the Albert Einstein College of Medicine for his valuable input and Marta Feldmesser and FengYing Chen for their assistance.
REFERENCES
- 1.Astarie-Dequeker C, N'Diaye E-N, Le Cabec V, Rittig M G, Prandi J, Maridonneau-Parini I. The mannose receptor mediates uptake of pathogenic and nonpathogenic mycobacteria and bypasses bactericidal responses in human macrophages. Infect Immun. 1999;67:469–477. doi: 10.1128/iai.67.2.469-477.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Barnes P F, Chatterjee D, Abrams J S, Lu S, Wang E, Yamamura M, Brennan P J, Modlin R L. Cytokine production induced by Mycobacterium tuberculosis lipoarabinomannan. J Immunol. 1992;149:541–547. [PubMed] [Google Scholar]
- 3.Besra G S, Chatterjee D. Lipids and carbohydrates of Mycobacterium tuberculosis. In: Bloom B R, editor. Tuberculosis: pathogenesis, protection, and control. Washington, D.C.: ASM Press; 1994. pp. 285–306. [Google Scholar]
- 4.Chan J, Fan X, Hunter S W, Brennan P J, Bloom B R. Lipoarabinomannan, a possible virulence factor involved in persistence of Mycobacterium tuberculosis within macrophages. Infect Immun. 1991;59:1755–1761. doi: 10.1128/iai.59.5.1755-1761.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Chang J C, Wysocki A, Tchou-Wong K, Moskowitz N, Zhang Y, Rom W N. Effect of Mycobacterium tuberculosis and its components on macrophages and the release of matrix metalloproteinases. Thorax. 1996;51:306–311. doi: 10.1136/thx.51.3.306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Chatterjee D, Lowell K, Rivoire B, McNeil M R, Brennan P J. Lipoarabinomannan of Mycobacterium tuberculosis. Capping with mannosyl residues in some strains. J Biol Chem. 1992;267:6234–6239. [PubMed] [Google Scholar]
- 7.Coleman R, Iqbal S, Godfrey P P, Billington D. Membrane and bile formation: composition of several mammalian biles and their membrane-damaging properties. Biochem J. 1979;178:201–208. doi: 10.1042/bj1780201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Costello A M, Kumar A, Narayan V, Akbar M S, Ahmed S, Abou-Zeid C, Rook G A W, Stanford J, Moreno C. Does antibody to mycobacterial antigens, including lipoarabinomannan, limit dissemination in childhood tuberculosis? Trans R Soc Trop Med Hyg. 1992;86:686–692. doi: 10.1016/0035-9203(92)90192-f. [DOI] [PubMed] [Google Scholar]
- 9.Glatman-Freedman A, Casadevall A. Serum therapy for tuberculosis revisited: reappraisal of the role of antibody-mediated immunity against Mycobacterium tuberculosis. Clin Microbiol Rev. 1998;11:514–532. doi: 10.1128/cmr.11.3.514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Glatman-Freedman A, Martin J M, Riska P F, Bloom B R, Casadevall A. Monoclonal antibodies to surface antigens of Mycobacterium tuberculosis and their use in a modified enzyme-linked immunosorbent spot assay for detection of mycobacteria. J Clin Microbiol. 1996;34:2795–2802. doi: 10.1128/jcm.34.11.2795-2802.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Goldman D L, Lee S C, Casadevall A. Tissue localization of Cryptococcus neoformans glucuronoxylomannan in the presence and absence of specific antibody. Infect Immun. 1995;63:3448–3453. doi: 10.1128/iai.63.9.3448-3453.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Guerin C. The history of BCG. In: Rosenthal S R, editor. BCG vaccination against tuberculosis. Boston, Mass: Little, Brown & Co.; 1957. pp. 48–53. [Google Scholar]
- 13.Hansen P G C, Jackson G D F. Movement of IgM antibody from blood to bile in rats. Hepatology. 1991;14:518–522. [PubMed] [Google Scholar]
- 14.Hetland G, Wiker H G, Hogasen K, Hamasur B, Svenson S B, Harbon M. Involvement of antilipoarabinomannan antibodies in classical complement activation in tuberculosis. Clin Diagn Lab Immunol. 1998;5:211–218. doi: 10.1128/cdli.5.2.211-218.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Hopf U, Schaefer H E, Hess G, Buschenfelde K H M Z. In vivo uptake of immune complexes by parenchymal and nonparenchymal liver cells in mice. Gastroenterology. 1981;80:250–259. [PubMed] [Google Scholar]
- 16.Kraal G. Cells in the marginal zone of the spleen. Int Rev Cytol. 1992;132:31–74. doi: 10.1016/s0074-7696(08)62453-5. [DOI] [PubMed] [Google Scholar]
- 17.Kraal G, Ter Hart H, Meelhuizen C, Venneker G, Claassen E. Marginal zone macrophages and their role in the immune response against T-independent type 2 antigens: modulation of the cells with specific antibody. Eur J Immunol. 1989;19:675–680. doi: 10.1002/eji.1830190416. [DOI] [PubMed] [Google Scholar]
- 18.Lendvai N, Casadevall A, Liang Z, Goldman D L, Mukherjee J, Zuckier L. Effect of immune mechanisms on the pharmacokinetics and organ distribution of cryptococcal polysaccharide. J Infect Dis. 1998;177:1647–1659. doi: 10.1086/515329. [DOI] [PubMed] [Google Scholar]
- 19.Ljunghusen O, Johansson A, Skogh T. Hepatic immune complex elimination studied with FITC-labelled antigen. J Immunol Methods. 1990;128:1–7. doi: 10.1016/0022-1759(90)90457-7. [DOI] [PubMed] [Google Scholar]
- 20.Moreno C, Mehlert A, Lamb J. The inhibitory effects of mycobacterial lipoarabinomannan and polysaccharides upon polyclonal and monoclonal human T cell proliferation. Clin Exp Immunol. 1988;74:206–210. [PMC free article] [PubMed] [Google Scholar]
- 21.Mukherjee J, Casadevall A, Scharff M D. Molecular characterization of the humoral responses to Cryptococcus neoformans infection and glucuronoxylomannan-tetanus toxoid conjugate immunization. J Exp Med. 1993;177:1105–1116. doi: 10.1084/jem.177.4.1105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Orme I M, Collins F M. Mouse model of tuberculosis. In: Bloom B R, editor. Tuberculosis: pathogenesis, protection, and control. Washington, D.C.: ASM Press; 1994. pp. 113–134. [Google Scholar]
- 23.Prigozy T I, Sieling P A, Clemens D, Stewart P L, Behar S M, Porcelli S A, Brenner M B, Modlin R L, Kronnenberg M. The mannose receptor delivers lipoglycan antigens to endosomes for presentation to T cells by CD1b molecules. Immunity. 1997;6:187–197. doi: 10.1016/s1074-7613(00)80425-2. [DOI] [PubMed] [Google Scholar]
- 24.Rook G A W, Al Attiyah R, Foley N. The role of cytokines in the immunopathology of tuberculosis and the regulation of agalactosyl IgG. Lymphokine Res. 1989;8:323–328. [PubMed] [Google Scholar]
- 25.Russell M W, Brown T A, Claflin L J, Schroer K, Mestecky J. Immunoglobulin A-mediated hepatobiliary transport constitutes a natural pathway for disposing of bacterial antigens. Infect Immun. 1983;42:1041–1048. doi: 10.1128/iai.42.3.1041-1048.1983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Schlesinger L S. Macrophage phagocytosis of virulent but not attenuated strains of Mycobacterium tuberculosis is mediated by mannose receptors in addition to complement receptors. J Immunol. 1993;150:2920–2930. [PubMed] [Google Scholar]
- 27.Schlesinger L S, Hull S R, Kaufman T M. Binding of the terminal mannosyl units of lipoarabinomannan from a virulent strain of Mycobacterium tuberculosis to human macrophages. J Immunol. 1994;152:4070–4079. [PubMed] [Google Scholar]
- 28.Schlesinger L S, Kaufman T M, Iyer S, Hull S R, Marchiando L K. Differences in mannose receptor-mediated uptake of lipoarabinomannan from virulent and attenuated strains of Mycobacterium tuberculosis by human macrophages. J Immunol. 1996;157:4568–4575. [PubMed] [Google Scholar]
- 29.Sousa A O, Henry S, Maroja F M, Lee F K, Brum L, Singh M, Lagrange P H, Aucouturier P. IgG subclass distribution of antibody responses to protein and polysaccharide mycobacterial antigens in leprosy and tuberculosis patients. Clin Exp Immunol. 1998;111:48–55. doi: 10.1046/j.1365-2249.1998.00452.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Van Bossuyt H, Desmaretz C, Gaeta G B, Wisse E. The role of bile acids in the reduction in lipopolysaccharide uptake by cultured rat Kupffer cells. Virchows Arch B. 1989;57:141–147. doi: 10.1007/BF02899075. [DOI] [PubMed] [Google Scholar]
- 31.Van Bossuyt H, De Zanger R B, Wisse E. Cellular and subcellular distribution of injected lipopolysaccharide in rat liver and its inactivation by bile salts. J Hepatol. 1988;7:325–337. doi: 10.1016/s0168-8278(88)80005-9. [DOI] [PubMed] [Google Scholar]
- 32.Van Vliet E, Melis M, Van Ewijk W. Marginal zone macrophages in the mouse spleen identified by a monoclonal antibody. J Histochem Cytochem. 1985;33:40–44. doi: 10.1177/33.1.3880783. [DOI] [PubMed] [Google Scholar]
- 33.Young L S. Sepsis syndrome. In: Mandell G L, Bennett J E, Dolin R, editors. Principles and practice of infectious diseases. New York, N.Y: Churchill Livingstone, Ltd.; 1995. pp. 690–705. [Google Scholar]