

Abstract
Background
Average backfat thickness (BFT) is a critical complex trait in pig and an important indicator for fat deposition and lean rate. Usually, genome-wide association study (GWAS) was used to discover quantitative trait loci (QTLs) of BFT in a single population. However, the power of GWAS is limited by sample size in a single population. Alternatively, meta-analysis of GWAS (metaGWAS) is an attractive method to increase the statistical power by integrating data from multiple breeds and populations. The aim of this study is to identify shared genetic characterization of BFT across breeds in pigs via metaGWAS.
Results
In this study, we performed metaGWAS on BFT using 15,353 pigs (5,143 Duroc, 7,275 Yorkshire, and 2,935 Landrace) from 19 populations. We detected 40 genome-wide significant SNPs (Bonferroni corrected P < 0.05) and defined five breed-shared QTLs in across-breed metaGWAS. Markers within the five QTL regions explained 7 ~ 9% additive genetic variance and showed strong heritability enrichment. Furthermore, by integrating information from multiple bioinformatics databases, we annotated 46 candidate genes located in the five QTLs. Among them, three important (MC4R, PPARD, and SLC27A1) and seven suggestive candidate genes (PHLPP1, NUDT3, ILRUN, RELCH, KCNQ5, ITPR3, and U3) were identified.
Conclusion
QTLs and candidate genes underlying BFT across breeds were identified via metaGWAS from multiple populations. Our findings contribute to the understanding of the genetic architecture of BFT and the regulating mechanism underlying fat deposition in pigs.
Supplementary Information
The online version contains supplementary material available at 10.1186/s12864-022-09036-z.
Keywords: Pig, GWAS, meta-analysis, Backfat thickness, QTL
Background
Human obesity has become an increasingly common social phenomenon in the past decades related to both quality of life and life expectancy [1]. Pig was often chosen as a model animal for scientific research due to its similar physiology architecture with human. Up to date, PigQTLdb (release 48) released 35,846 quantitative trait loci (QTLs), expression quantitative trait loci (eQTLs), and associations [2], of which 3,608 associations are related to fatness traits. Average backfat thickness (BFT) is an indicator for fat deposition and directly linked to fat content and lean rate in pig [3, 4]. Uncovering the genetic characteristics of BFT can be useful for pig genetic improvement.
Previous studies showed that BFT is a complex trait [5–7] with moderate or high heritability ranged from 0.2 ~ 0.6 [5–8]. Genome-wide association study (GWAS) in single population has been widely used to mine the potential QTLs and genes that associated with BFT [9–11]. And Gozalo‑Marcilla1 et al. [12] performed large scale GWAS of pig BFT from eight lines to highlight the genes involved in pathways for fat deposition. However, the power of single population GWAS is usually limited by sample size. Meta-analysis of GWAS summary statistics (metaGWAS) is an attractive method to increase the statistical power by integrating data from multiple breeds and populations. In recent years, metaGWAS has gradually become a popular method to study the genetic architecture of pig complex trait. For instance, Cai et al. [13] performed a large-scale metaGWAS in pig to identify the candidate gene of average daily gain across three breeds. Zhou et al. [14] conducted both GWAS and metaGWAS in pig to reveal new insights into the genetic architecture of average daily gain and lean meat percentage. Although, several candidate genes associated with BFT (e.g., MC4R [15, 16], PPARD [17], and LEPR [18, 19]) have been reported via GWAS and followed by experimental validations, the potential shared QTLs and genes of BFT across breeds are rarely reported.
The objectives of this study were to identify QTLs and candidate genes via metaGWAS and to explore the genetic architecture of BFT across pig breeds. In this study, we performed GWAS for BFT in 19 different populations genotyped with Porcine SNP BeadChip. Further, metaGWAS was performed for within-breed and across-breed strategies. Our findings contribute to the understanding of the genetic architecture of BFT and the regulating mechanism underlying fat deposition in pigs.
Results
Data summary and population stratification
The detail information (i.e., breed, sample size, genotypes, and phenotypes) of 19 populations used in this study were presented in Table 1. In total, we used data from 15,353 pigs consisting of 5,143 Duroc in seven populations, 7,275 Yorkshire in seven populations, and 2,935 Landrace in five populations. The average values (± standard deviation) of BFT for Duroc, Yorkshire, and Landrace were 10.87 (± 2.20 mm), 12.07 (± 3.36 mm), and 12.60 (± 4.28 mm), respectively.
Table 1.
Summary of experimental data
| Population | Breed | Number of Sample | Number of SNPs | Mean (mm) |
Minimum (mm) |
Maximum (mm) |
SD (mm) |
CV (%) |
Chip |
|---|---|---|---|---|---|---|---|---|---|
| PP1 | Duroc | 1,993 | 39,311 | 9.82 | 4.58 | 17.45 | 1.66 | 16.94 | a |
| PP2 [8] | Duroc | 1,071 | 23,766 | 12.45 | 6.68 | 21.02 | 2.10 | 16.91 | ab |
| PP3 | Duroc | 1,048 | 40,139 | 10.90 | 5.18 | 22.88 | 2.27 | 20.81 | a |
| PP4 | Duroc | 353 | 39,082 | 12.47 | 8.98 | 16.59 | 1.45 | 11.64 | b |
| PP5 | Duroc | 328 | 31,680 | 10.07 | 6.27 | 20.81 | 1.51 | 15.02 | c |
| PP6 | Duroc | 190 | 39,927 | 12.07 | 8.06 | 19.71 | 2.02 | 16.70 | a |
| PP7 | Duroc | 160 | 37,074 | 9.86 | 5.05 | 15.72 | 1.93 | 19.61 | a |
| PP8 | Yorkshire | 2,179 | 41,314 | 11.14 | 3.02 | 23.35 | 2.51 | 22.55 | ad |
| PP9 | Yorkshire | 1,794 | 41,360 | 10.74 | 4.29 | 29.09 | 2.73 | 25.41 | ad |
| PP10 | Yorkshire | 1,259 | 45,002 | 11.77 | 5.03 | 22.01 | 2.42 | 20.56 | a |
| PP11 | Yorkshire | 1,146 | 44,434 | 13.13 | 6.61 | 26.06 | 2.92 | 22.20 | a |
| PP12 | Yorkshire | 406 | 44,200 | 16.25 | 7.17 | 22.76 | 2.36 | 14.51 | a |
| PP13 | Yorkshire | 314 | 31,564 | 18.76 | 8.02 | 29.76 | 4.51 | 24.01 | c |
| PP14 | Yorkshire | 177 | 40,070 | 12.02 | 7.53 | 17.33 | 1.66 | 13.83 | c |
| PP15 | Landrace | 1,094 | 43,304 | 10.31 | 5.30 | 19.52 | 2.20 | 21.30 | a |
| PP16 | Landrace | 556 | 30,705 | 18.89 | 8.66 | 29.69 | 4.51 | 23.86 | c |
| PP17 | Landrace | 554 | 45,132 | 12.17 | 7.20 | 22.20 | 2.29 | 18.86 | a |
| PP18 | Landrace | 552 | 43,644 | 12.00 | 5.75 | 22.87 | 2.71 | 22.59 | a |
| PP19 | Landrace | 179 | 33,809 | 9.69 | 6.20 | 14.41 | 1.37 | 14.14 | c |
Chip a: GeneSeek GGP-Porcine Beadchip (Neogen Corporation, Lansing, MI, USA) with 50 K
Chip b: Illumina PorcineSNP60 BeadChip (Illumina, San Diego, CA, USA) with 60 K
Chip c: “ZhongxinI” Porcine Breeding Chip (Beijing Compass Agritechnology Co., Ltd., Beijing, China) with 50 K
Chip d: GeneSeek GGP-Porcine Beadchip (Neogen Corporation, Lansing, MI, USA) with 80 K
Principal component analysis (PCA) based on genotypes of all pigs (Fig. 1) and each of three breeds (Additional file 1: Figure S1) showed that samples from three breeds were clustered clearly. The first two genotype principal components (PCs) explained for 14.66% and 7.49% of total population variance, respectively.
Fig. 1.
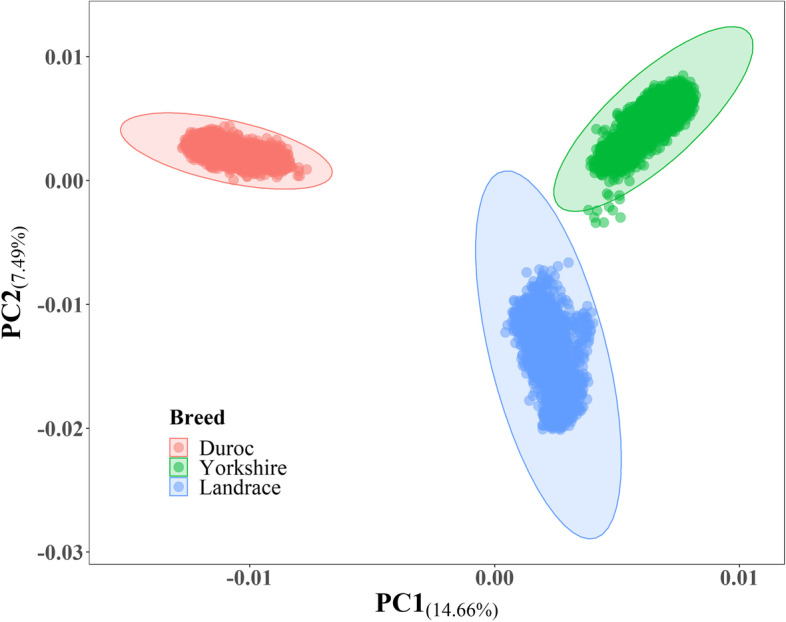
Population structure for nineteen populations. PC1 = the first principal component, PC2 = the second principal component. The number in brackets on axis represented the proportion of eigenvalues among all components
Meta‑analysis within breed
To overcome the limitation of power of single population GWAS, we then performed metaGWAS by integrating GWAS summaries from multiple populations with the same breed. As a basis for metaGWAS, we first conducted GWAS in each of 19 populations separately (Additional file 2: Table S1). In single population GWAS, the number of significant SNPs (P < 1 × 10− 5) ranged from 0 to 43. 105 SNPs were significant (P < 1 × 10− 5) only in one of 19 single population GWAS. 85 SNPs were significant (P < 1 × 10− 5) only in one of four metaGWAS (within-breed and across-breed), and 49 SNPs were significant in at least one metaGWAS. 28 SNPs were significant both in single population GWAS and metaGWAS (Additional file 3: Figure S2). The genomic inflation factor () values for within-breed metaGWAS of Duroc, Yorkshire, and Landrace were 1.07, 1.05, 1.00, respectively (Table 2) and were within the normal range.
Table 2.
Summary of QTLs from metaGWAS.
| Breed | Lambda (λ) |
Lead SNP | SSC | Position (bp) |
-log10 (P-value) |
QTL region | Number of significant SNPs within QTL in metaGWAS | Significant in single population GWAS (P < 1 × 10− 5) | Reported frequency in pigQTLdb |
|---|---|---|---|---|---|---|---|---|---|
| Duroc | 1.07 | rs81284646 | 1 | 161,824,864 | 7.70 | 155,986,286 ~ 161,824,864 | 10 | Yes | 21 |
| rs80936157 | 7 | 30,356,985 | 9.28 | 29,356,985 ~ 31,356,985 | 13 | Yes | 21 | ||
| rs81236473 | 18 | 10,555,467 | 6.29 | 9,555,467 ~ 11,555,467 | 1 | No | 1 | ||
| Yorkshire | 1.05 | rs337892438 | 1 | 160,513,631 | 6.96 | 159,513,631 ~ 161,513,631 | 5 | No | 14 |
| rs81358998 | 2 | 49,246,142 | 6.86 | 48,246,142 ~ 50,246,142 | 2 | Yes | 0 | ||
| Landrace | 1.00 | rs81359965 | 2 | 80,229,065 | 6.24 | 79,229,065 ~ 81,229,065 | 1 | No | 2 |
| All breeds | 1.07 | rs319638368 | 1 | 53,666,889 | 6.80 | 52,666,889 ~ 54,666,889 | 20 | No | 0 |
| rs80877507 | 1 | 160,347,188 | 11.84 | 158,589,475 ~ 162,192,627 | 2 | Yes | 21 | ||
| rs81359652 | 2 | 60,697,443 | 7.56 | 59,697,443 ~ 61,697,443 | 1 | No | 2 | ||
| rs80962638 | 7 | 30,716,800 | 10.13 | 29,476,173 ~ 31,569,645 | 16 | Yes | 20 | ||
| rs81236473 | 18 | 10,555,467 | 8.16 | 9,555,467 ~ 11,555,467 | 1 | No | 1 |
In Duroc within-breed metaGWAS, a total of 24 significant SNPs were detected (Additional file 4: Table S2). Further, we identified three QTLs on sus scrofa chromosome (SSC) 1, 7, and 18 respectively (Fig. 2a, Additional file 5: Figure S3a). The QTL on SSC7 was identified by the most significant lead SNP rs80936157 (7:30356985, -log10P = 9.28) which was located in the gene body of NUDT3. The QTL on SSC1 included the second most significant lead SNP rs81284646 (1:161824864, -log10P = 7.70) which was located in the gene body of ENSSSCG00000004911. And the QTL on SSC18 was detected via the lead SNP (18:10555467, -log10P = 6.29) which was located in the gene body of ZC3HAV1.
Fig. 2.

Manhattan plots of metaGWAS within breed (a, b, c) and across breeds (d). The red lines indicate the 5% genome-wide Bonferroni corrected thresholds as -log10P equal to 6.14, 6.14, 6.12, and 6.20, respectively
In Yorkshire within-breed metaGWAS, we identified seven significant SNPs (Additional file 4: Table S2) and discovered two QTLs on SSC1 and SSC2, respectively (Fig. 2b, Additional file 5: Figure S3b). The QTL on SSC1 was defined by the most significant lead SNP rs337892438 (1:160513631, -log10P = 6.96) which was located in the downstream of ENSSSCG00000045579 with distance equal to 69,671 bp. The QTL on SSC2 included the second most significant lead SNP rs81358998 (2:49246142, -log10P = 6.86) which was located in the gene body of SBF2.
In Landrace within-breed metaGWAS, we only identified one significant SNP (Additional file 4: Table S2) and one QTL on SSC2 (Fig. 2c, Additional file 5: Figure S3c). The lead SNP rs81359965 (2:80229065, -log10P = 6.24) was located in the gene body of RMND5B.
Meta‑analysis across breeds
To maximize the sample size to discover the significant QTLs associated with BFT, we conducted an across-breed metaGWAS combining GWAS summary statistics of all 19 populations. The inflation factor () value was 1.07 for this across-breed metaGWAS (Table 2). A total of 40 significant SNPs (Additional file 4: Table S2) and five QTLs on SSC1, 2, 7, and 18 (Fig. 2d, Additional file 5: Figure S3d) were detected.
Five QTLs were detected in across-breed metaGWAS. Two out of the five QTLs (SSC1, 52,666,889 ~ 54,666,889 bp; SSC2, 59,697,443 ~ 61,697,443 bp) were not observed in within-breed metaGWAS, while the remaining three QTLs were overlapped with those in within-breed metaGWAS.
The QTL on SSC1 was defined by the most significant lead SNP rs80877507 (1:160347188, -log10P = 11.84) which was located in the gene body of ENSSSCG00000048538. The QTL on SSC7 was identified by the second most significant lead SNP rs80962638 (7:30716800, -log10P = 10.13) which was located in the gene body of SNRPC. The QTL on SSC18 included the third most significant lead SNP rs81236473 (18:10555467, -log10P = 8.16) which was located in the gene body of ZC3HAV1. The QTL on SSC2 included the lead SNP rs81359652 (2:60697443, -log10P = 7.56) which was located in the downstream of ENSSSCG00000013869 with distance equal to 14,538 bp. The remaining QTL was detected on SSC1 by the lead SNP (1:53666889, -log10P = 6.80) which was located in the downstream of CEP162 with distance equal to 224,490 bp.
We also examined the linkage disequilibrium (LD) blocks across breed for each of the five QTLs (Additional file 6: Figure S4). And the two QTLs located on SSC1 showed high regional LD while the QTL located on SSC18 showed the low LD. Obviously, the LD pattern showed a high similarity across breeds.
Post‑GWAS analysis
Genomic heritability estimation and predictive ability of five across-breed QTLs.
To verify the reliability of metaGWAS results, we conducted heritability estimation (Table 3) and predictive ability evaluation (Fig. 3). Given that the standard error of heritability estimates is influenced by sample size, we used the largest population in each breed for heritability estimation to obtain a reliable estimate of heritability.
Table 3.
Estimated heritability of the five QTLs in across-breed metaGWAS.
| Population | Breed | Number of SNPs | (SE) | (SE) | Proportion (/) |
Enrichment |
|---|---|---|---|---|---|---|
| PP1 | Duroc | 157 | 0.01(0.01) | 0.20(0.03) | 0.07 | 18.15 |
| PP8 | Yorkshire | 169 | 0.02(0.01) | 0.21(0.03) | 0.09 | 21.63 |
| PP15 | Landrace | 174 | 0.02(0.02) | 0.22(0.05) | 0.09 | 22.43 |
: Estimated heritability of SNPs in the five QTLs in our study; : Estimated heritability of all SNPs; : Genetic variance of SNPs in the five QTLs in our study; : Genetic variance of all SNPs
Fig. 3.

The correlation between predicted values and average phenotypes. Each point represented one genotype combination in five lead SNPs. Shaded part represented the 95% confidence interval
The estimated heritability for SNPs within the five QTL regions was 0.01 for Duroc, 0.02 for Yorkshire, and 0.02 for Landrace. These SNPs explained 7.25%, 8.85% and 9.01% additive genetic variance for Duroc, Yorkshire, and Landrace respectively. It should be noted that the five QTL regions were 18.15, 21.63, and 22.43 folds enriched for heritability in Duroc, Yorkshire, and landrace, respectively.
To evaluate whether the five QTLs are effective across-breed, we conducted genomic prediction with the lead SNPs within these QTLs, and calculated the Pearson’s correlation coefficient between predicted genetic value and average phenotype value in all populations. Finally, we observed an significant predicting ability across breed for these QTLs (Pearson’s correlation coefficient = 0.22, P < 3.98 × 10− 2).
Candidate genes mapping
To investigate the gene that potentially regulates BFT, we conducted candidate gene mapping from total 138 positional candidate genes via four external biological annotations (Table 4, Additional file 7: Table S3). Within the five QTLs, we identified 46 candidate genes based on functional annotations in Gene Ontology (GO) and Kyoto Encyclopedia of Genes and Genomes (KEGG), PigQTLdb and NHGRI-EBI. Several genes were identified in three annotations such as MC4R (SSC1, ~ 160.8 Mb), PPARD (SSC7, ~ 31.2 Mb), RELCH (SSC1, ~ 159.2 Mb), and ITPR3 (SSC7, ~ 29.8 Mb), of which MC4R was widely reported in both pig (11 evidence in backfat) and human (24 evidence in body mass index). SLC27A1 (SSC2, ~ 60.2 Mb) was the gene annotated in five fat metabolism processes in GO and KEGG including lipid, fatty acid, and triglyceride metabolic pathways, insulin signaling pathway and homeostasis regulation pathway. Two genes, ILRUN (SSC7 at ~ 30.6 Mb) and KCNQ5 (SSC1 at ~ 52.4 Mb) were not previously reported in pig but reported in human research.
Table 4.
Candidate genes in the five QTLs in across-breed metaGWAS
| SSC | QTL Position | Candidate Genes |
|---|---|---|
| 1 | 52,666,889 ~ 54,666,889 | KCNQ5 c, CYB5R4c, CEP162c, NT5Eb |
| 1 | 155,986,286 ~ 161,824,864 | PHLPP1 bc, RELCHabc, RNF152b, CDH20b, MC4Rabc, CCBE1b, GRPc, SEC11Cc, ALPK2b |
| 2 | 59,697,443 ~ 61,697,443 | MAST3 c, JAK3a, B3GNT3a, SLC27A1a, NXNL1a, U3c, KLF2a, KCNN1a |
| 7 | 29,476,173 ~ 31,569,645 | B3GALT4 a, ITPR3abc, IP6K3a, LEMD2c, GRM4c, NUDT3c, PACSIN1c, SPDEFc, ILRUNc, SNRPCc, UHRF1BP1c, ANKS1Ac, TCP11c, SCUBE3c, PPARDabc, TEAD3c, TULP1c, FKBP5c |
| 18 | 9,555,467 ~ 11,555,467 | KDM7A c, TBXAS1a, ZC3HAV1c, TRIM24c, AKR1D1a, CREB3L2ab, FMC1a |
Superscript numbers: aGO/KEGG annotation; bPigQTLdb annotation; cNHGRI-EBI annotation
Discussion
In this research, we conducted metaGWAS for BFT by combining 19 populations across three commercial breeds and identified shared QTLs across breeds. The BFT is a complex trait with heritability of 0.20 ~ 0.22 in our study, which was agreed with previous studies [7, 8, 20–22]. On one hand, additive genetics variance explained by SNPs among the defined QTL regions ranged from 7.25 to 9.01% for the three breeds. These SNPs also showed high enrichment in heritability for all breeds, which indicated a shared genetic architecture of BFT across breeds at these QTL regions. On the other hand, across-breed metaGWAS showed a greater power to detect potential QTLs compared with within-breed metaGWAS. For instance, QTLs detected by across-breed metaGWAS on SSC1 (~ 160 Mb) and SSC7 were significant in both Duroc and Yorkshire within-breed metaGWAS, but not significant in Landrace. Similarly, QTLs detected by across-breed metaGWAS on SSC1 (~ 50 Mb) were significant in both Yorkshire and Landrace within-breed metaGWAS, but not significant in Duroc. Finally, five QTLs were identified with a high enrichment of heritability and a moderate predictive ability for across breeds. These results indicate that across-breed metaGWAS could identify shared QTLs for BFT across breeds.
Analysis method is crucial for association study, as complicated population structure and relatedness have the potential to lead to false signals [23–25]. Recent studies have also shown that inclusion of candidate maker in the SNP-derived genetic relationship matrix (GRM) can lead to a loss of power [26–28]. Therefore, we used GCTA-LOCO for single locus regression in order to avoid “proximal contamination” [27, 29] and corrected population stratification and relatedness by the mixed linear model and the first five PCs [30–33]. In addition, considering the data inconsistency among each population, we unified them into summary statistics independently by single population GWAS. Then we used summary statistics to performed metaGWAS to enhance the power of detecting significant SNPs.
Among the discovered candidate genes, three genes associated with BFT were well studied. First, MC4R (SSC1, ~ 160 Mb) was associated with fatness, growth, as well as meat quality traits reported in recent decades [12, 15, 34–36]. The reliability of MC4R had been validated by several studies in pigs [37–39] and in human [40–42]. In this study, MC4R was located in the QTL on SSC1 (~ 160 Mb) and was annotated with several homeostasis regulation pathways in GO biological processes. Second, PPARD is recognized as an important gene associated with fat deposition traits in gene and gene expression layers [17, 43, 44], which was detected in our study in QTL on SSC7 and annotated by all the databases. Third, SLC27A1 was another detected important genes that showed multiple evidence associated with fat deposition [45–47]. In addition, we identified seven promising candidate genes, PHLPP1, NUDT3, ILRUN, RELCH, KCNQ5, ITPR3, and U3. RELCH, the candidate gene near MC4R, belonged to sterol and lipid metabolism pathway in GO database and was also identified in previous researches [14, 48]. ITPR3, the candidate gene near PPARD, belonged to insulin secretion pathway in KEGG database and both annotated in pig [49] and human. PHLPP1 (SSC1, ~ 160 Mb) and NUDT3 (SSC7), two candidate genes near major genes MC4R and PPARD respectively, were found to be associated with backfat in pig [8, 14, 50–53]. Three of them, ILRUN, KCNQ5, and U3 were reported in human associations studies in BMI and body weight for 15, 7, and 9 times respectively but not reported as the candidate genes in pig.
The sample size is a crucial factor to the power of GWAS. Generally, there are two strategies to enlarge the sample size. One is to combine individual level data (genotypes and phenotypes) from multiple populations, then to conduct GWAS analysis. The other is metaGWAS, in which the GWAS summary statistics from multiple populations are analyzed jointly, rather than put individual data together. In this study, we aimed at testing the metaGWAS strategy, in which individual data from different populations are not allowed to be combined together. The other strategy would be an interesting solution deserve further attempt in our further investigation. Integrating data from multiple populations could further increase the power of association analysis. However, the weakness of metaGWAS still could not be neglected. For example, the available SNPs were different for different populations, hence the number of common SNPs across all populations were usually too small to perform GWAS effectively. Therefore, a large pig genotype imputation reference panel was necessary for unifying the SNP maps for different populations.
Conclusion
In this study, we conducted metaGWAS for BFT on 15,353 pigs from diverse genetic backgrounds. We identified 40 genome-wide significant SNPs located in five QTL regions and annotated 46 candidate genes based on across-breed metaGWAS. Among the candidate genes, MC4R, PPARD, and SLC27A1, had been well studied. Additionally, seven candidate genes, PHLPP1, NUDT3, ILRUN, RELCH, KCNQ5, ITPR3, and U3, were identified to be promising candidate genes associated with BFT. Our results provided useful reference for understanding the genetic architecture of BFT and the regulating mechanism underlying fat deposition in pigs.
Materials and methods
Population and data
A total of 15,353 pigs (5,143 Duroc, 7,275 Yorkshire, and 2,935 Landrace) from 19 populations in 12 Chinese pig farms were used in this analysis. The average age of pigs was 156, 172, and 164 days for Duroc, Yorkshire, and Landrace respectively. The backfat thickness was measured by living B-ultrasonic within each population at the end of test. The measurements were adjusted to body weight at 100 kg using within farm genetic evaluation system. Details about the populations, phenotypes, and genotypes were shown in Table 1.
All pigs were genotyped with one of the following SNP chips, GeneSeek GGP-Porcine Beadchip 50 and 80 K (Neogen Corporation, Lansing, MI, USA), Illumina PorcineSNP60 BeadChip 60 K (Illumina, San Diego, CA, USA), “ZhongxinI” Porcine Breeding Chip 50 K (Beijing Compass Agritechnology Co., Ltd., Beijing, China). The annotation of all SNPs has been unified to Ensemble Sus scrofa 11.1 reference genome version. And the missing genotypes were phased within each population using Beagle 5.1 [54]. Quality control on genotypes were carried out within each population using PLINK v1.9 [55] with the criterion of minor allele frequency > 0.01 and P > 1 × 10− 6 for Hardy-Weinberg equilibrium test. SNPs with no position information or located on sex SSC were removed. After quality control, the number of SNPs within each population was shown in Table 1.
Population structure analysis
To correct the population structure in GWAS model and investigate the population structure, we performed PCA using GCTA-PCA (1.94.0beta) [30, 56] in single population and mixed population, respectively. After that we visualized the result using R package ggplot2 [57].
Single population GWAS
We performed GWAS in each single population separately using GCTA-LOCO [29, 56] with the following mixed linear model:
where is an vector of phenotypic values; is the SNP genotype indicator variable coded as 0, 1 or 2 with its additive effect ; is the incidence matrix of fixed covariates (test farm, test year, test season, sex and the first five principal components) with their corresponding coefficient ; is the accumulated genetics effect captured by all SNPs except those on the chromosome where the candidate SNP is located via , where is the SNP-derived genetic relationship matrix follows the formula [58]: , where was the minor allele frequency (MAF) adjusted genotype matrix with elements (), (), and (); is a vector of residuals with . The variance-covariance matrix of is .
Meta-analysis of GWAS
To perform meta-analysis of GWAS, we utilized the GWAS summary statistics from single population GWAS as input for METAL (released on 2011-03-25) [59], in which inverse-variance weighted fixed-effects model was implemented with parameters “SCHEME STDERR” and “GENOMICCONTROL ON”. We set the genome-wide significant threshold of metaGWAS via Bonferroni correction (0.05/number of SNPs). The Manhattan plots and Quantile-Quantile (Q-Q) plots as well as [60] were performed using R program. To detect the QTLs within and across breed, we performed metaGWAS with GWAS summary statistics from a single breed or all breeds, which were defined as within-breed and across-breed metaGWAS, separately. To fairly compare single population GWAS with metaGWAS, we used the same genome-wide significance level (1 × 10− 5) for both method in the comparative analysis.
Definition of QTL and LD analysis
In this study, we defined QTL as the genomic region containing a set of significant SNPs, where physical distance of each neighbor pair was less than 5 Mb. If the length of a QTL region was less than 2 Mb, the region was defined as the 1 Mb on the either side of the lead SNP. A lead SNP was defined as the most significant SNP within a QTL.
To assess the LD event in each QTLs across breeds, we computed the coefficient of LD (r2) in the largest population within each breed (PP1, PP8, PP15) using PLINK v1.9 [55], and displayed LD blocks using R package LDheatmap [61]. In addition, to compare the result in this study with that in previous studies, we summarized each QTL overlapped frequency between this study and pigQTLdb on 25 March 2022 [2] with trait “backfat” and distance shorter than 5 Mb.
Estimation of genomic heritability explained by SNPs
To estimate the genomic heritability explained by SNPs within identified QTLs, SNPs were partitioned into two sets, within QTLs and outside QTLs. Then they were imported as two variance components in mixed linear model to estimate heritability using GCTA-GREML [56, 62]:
where is an vector of phenotypic values; is the incidence matrix of fixed covariates (test farm, test year, test season and the first five PCs) with their corresponding coefficient ; and represent additive effects explained by the SNPs within or outside all QTL regions via , where is the SNP-derived genetic relationship matrix follows the formula [58]: , where was the minor allele frequency (MAF) adjusted genotype matrix with elements (), (), and (). and represented incidence matrix explained by the SNPs within or outside identified QTLs. is a vector of residuals with . The heritability proportion for the SNPs within QTLs was defined as .
Predictive ability of genotype combination of five lead SNPs across populations
To evaluate the predictive ability, we computed prediction value of phenotype from five lead SNPs (detected in across-breed metaGWAS) genotype combinations via the formula [21]:
where was the prediction value of phenotype; denotes the estimate of marginal genetic effect in association summary statistics; denotes the number of effect alleles for each genetic variant coded as 0, 1, or 2. The Person’s correlation between predicted values and average phenotypes every genotype combination (sample size > 30) was calculated as predictive ability.
Identification candidate genes by functional annotation
To identify the candidate genes from QTLs, we firstly retrieved them based on physical location (within QTLs detected in across-breed metaGWAS) and multi external biological annotations. Second, we matched genes within QTLs with those belong to fat regulation biology process (“lipid/fatty acid/sterol/triglyceride/bile acid metabolic pathways”, “insulin signal pathways”, “homeostasis regulation pathways”) in GO [63, 64] and KEGG [65, 66]. In this step, enrichment test was calculated based on hypergeometric distribution using R-package clusterProfiler [67] with commands “enrichGO” and “enrichKEGG”. Third, we matched genes within QTLs with those associated with pig traits (“backfat”, “obesity index”, “body weight”, “body mass index”, “intramuscular fat content”, lean meat percentage”, “feed intake”) in pigQTLdb [2]. Finally, we matched genes within QTLs with whose associated with human obesity-related traits (“body mass index”, “body weight”, “obesity”, “energy intake”) in NHGRI-EBI Database [68].
Supplementary Information
Additional file 1: Figure S1. PCA plot of each of three breeds.
Additional file 2: Table S1. Summary information of 19 single population GWAS.
Additional file 3: Figure S2. Venn diagram of the number of significant SNPs (P<10-5) between 19 single population GWAS and four metaGWAS.
Additional file 4: Table S2. Summary information of significant SNPs in four metaGWAS.
Additional file 5: Figure S3. Q-Q plot of four metaGWAS.
Additional file 6: Figure S4. LD blocks of each of the five QTLs in three breeds.
Additional file 7: Table S3 (Sheet1). Annotation of candidate genes belongs to fat regulation biology process in GO and KEGG. Table S3 (Sheet2). Annotation of candidate genes in pigQTLdb. Table S3 (Sheet3). Annotation of candidate genes in human NHGRI-EBI Catalog.
Acknowledgements
The authors thank all the editors and reviewers for their insightful comments and constructive suggestions, and thank National Supercomputer Center in Guangzhou for supporting computing platform.
Abbreviations
- BFT
average backfat thickness
- CV
coefficient of variation
- eQTL
expression quantitative trait locus
- GO
Gene Ontology
- GRM
SNP-derived genetic relationship matrix
- GWAS
genome-wide association study
- KEGG
Kyoto Encyclopedia of Genes and Genomes
- LD
linkage disequilibrium
- LOCO
leave-one-chromosome-out
- metaGWAS
meta-analysis of genome-wide association studies
- MLM
mixed linear model
- PCA
principal component analysis
- PC
principal component
- QTL
quantitative trait locus
- SD
standard deviation
- SE
standard error
- SNP
single-nucleotide polymorphism
- SSC
sus scrofa chromosome
- λ
genomic inflation factor (lambda)
Authors’ contributions
Zhe Zhang and Jiaqi Li conceived and designed the experiment. Xiangdong Ding, Zhiting Xu, and Chen Wei collected the samples, recorded the phenotypes. Haonan Zeng analyzed the data and wrote the manuscript. Zhanming Zhong, Jinyan Teng, and Zitao Chen contributed to the materials. Zhe Zhang, Jinyan Teng, Zhiting Xu, Zitao Chen, and Wenjing Zhang revised the manuscript. All authors reviewed and approved the manuscript.
Funding
This work was supported by the earmarked fund for China Agriculture Research System (CARS-35), and the National Natural Science Foundation of China (32022078).
Availability of data and materials
All annotation information was obtained from a publicly available source (http://www.ensembl.org). The genotype and phenotype used and/or analyzed during the current study are available from the corresponding author on reasonable request.
Declarations
Ethics approval and consent to participate
Not applicable since no biological samples were collected and no animal handling was performed for this study. All data was collected from existing databases provided by South China Agricultural University (Guangzhou 510642, China) and China Agricultural University (Beijing 100193, China).
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
Footnotes
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
References
- 1.Blüher M. Obesity: global epidemiology and pathogenesis. Nat Rev Endocrinol. 2019;15:288–98. doi: 10.1038/s41574-019-0176-8. [DOI] [PubMed] [Google Scholar]
- 2.Hu ZL, Park CA, Reecy JM. Bringing the animal QTLdb and CorrDB into the future: meeting new challenges and providing updated services. Nucleic Acids Res. 2022;50:D956–61. doi: 10.1093/nar/gkab1116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Blaj I, Tetens J, Preuß S, Bennewitz J, Thaller G. Genome-wide association studies and meta-analysis uncovers new candidate genes for growth and carcass traits in pigs. PLoS ONE. 2018;13:e0205576. doi: 10.1371/journal.pone.0205576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Qiu Y, Ding R, Zhuang Z, Wu J, Yang M, Zhou S, et al. Genome-wide detection of CNV regions and their potential association with growth and fatness traits in Duroc pigs. BMC Genomics. 2021;22:332. doi: 10.1186/s12864-021-07654-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Li X, Kennedy BW. Genetic parameters for growth rate and backfat in canadian Yorkshire, Landrace, Duroc, and Hampshire pigs. J Anim Sci. 1994;72:1450–4. doi: 10.2527/1994.7261450x. [DOI] [PubMed] [Google Scholar]
- 6.Guo X, Christensen OF, Ostersen T, Wang Y, Lund MS, Su G. Genomic prediction using models with dominance and imprinting effects for backfat thickness and average daily gain in danish duroc pigs. Genet Sel Evol. 2016;48:67. doi: 10.1186/s12711-016-0245-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Lopes MS, Bastiaansen JWM, Janss L, Knol EF, Bovenhuis H. Estimation of Additive, Dominance, and Imprinting Genetic Variance using genomic data. G3 (Bethesda) 2015;5:2629–37. doi: 10.1534/g3.115.019513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Zhang Z, Chen Z, Diao S, Ye S, Wang J, Gao N, et al. Identifying the complex genetic architecture of growth and fatness traits in a Duroc pig population. J Integr Agric. 2021;20:1607–14. doi: 10.1016/S2095-3119(20)63264-6. [DOI] [Google Scholar]
- 9.Chen D, Wu P, Yang Q, WANG K, ZHOU J, Yang X, et al. Genome-wide association study for backfat thickness at 100 kg and loin muscle thickness in domestic pigs based on genotyping by sequencing. Physiol Genomics. 2019;51:261–6. doi: 10.1152/physiolgenomics.00008.2019. [DOI] [PubMed] [Google Scholar]
- 10.Fabbri MC, Zappaterra M, Davoli R, Zambonelli P. Genome-wide association study identifies markers associated with carcass and meat quality traits in italian large White pigs. Anim Genet. 2020;51:950–2. doi: 10.1111/age.13013. [DOI] [PubMed] [Google Scholar]
- 11.Crespo-Piazuelo D, Criado-Mesas L, Revilla M, Castelló A, Noguera JL, Fernández AI, et al. Identification of strong candidate genes for backfat and intramuscular fatty acid composition in three crosses based on the Iberian pig. Sci Rep. 2020;10:13962. doi: 10.1038/s41598-020-70894-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Gozalo-Marcilla M, Buntjer J, Johnsson M, Batista L, Diez F, Werner CR, et al. Genetic architecture and major genes for backfat thickness in pig lines of diverse genetic backgrounds. Genet Sel Evol. 2021;53:76. doi: 10.1186/s12711-021-00671-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Cai Z, Christensen OF, Lund MS, Ostersen T, Sahana G. Large-scale association study on daily weight gain in pigs reveals overlap of genetic factors for growth in humans. BMC Genomics. 2022;23:133. doi: 10.1186/s12864-022-08373-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Zhou S, Ding R, Meng F, Wang X, Zhuang Z, Quan J, et al. A meta-analysis of genome-wide association studies for average daily gain and lean meat percentage in two Duroc pig populations. BMC Genomics. 2021;22:12. doi: 10.1186/s12864-020-07288-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Kim KS, Larsen N, Short T, Plastow G, Rothschild MF. A missense variant of the porcine melanocortin-4 receptor (MC4R) gene is associated with fatness, growth, and feed intake traits. Mamm Genome. 2000;11:131–5. doi: 10.1007/s003350010025. [DOI] [PubMed] [Google Scholar]
- 16.Bruun CS, Jørgensen CB, Nielsen VH, Andersson L, Fredholm M. Evaluation of the porcine melanocortin 4 receptor (MC4R) gene as a positional candidate for a fatness QTL in a cross between Landrace and Hampshire. Anim Genet. 2006;37:359–62. doi: 10.1111/j.1365-2052.2006.01488.x. [DOI] [PubMed] [Google Scholar]
- 17.Zhang Y, Gao T, Hu S, Lin B, Yan D, Xu Z, et al. The functional SNPs in the 5’ Regulatory Region of the Porcine PPARD Gene have Significant Association with Fat Deposition Traits. PLoS ONE. 2015;10:e0143734. doi: 10.1371/journal.pone.0143734. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Uemoto Y, Kikuchi T, Nakano H, Sato S, Shibata T, Kadowaki H, et al. Effects of porcine leptin receptor gene polymorphisms on backfat thickness, fat area ratios by image analysis, and serum leptin concentrations in a Duroc purebred population. Anim Sci J. 2012;83:375–85. doi: 10.1111/j.1740-0929.2011.00963.x. [DOI] [PubMed] [Google Scholar]
- 19.Muñoz G, Ovilo C, Silió L, Tomás A, Noguera JL, Rodríguez MC. Single- and joint-population analyses of two experimental pig crosses to confirm quantitative trait loci on Sus scrofa chromosome 6 and leptin receptor effects on fatness and growth traits. J Anim Sci. 2009;87:459–68. doi: 10.2527/jas.2008-1127. [DOI] [PubMed] [Google Scholar]
- 20.Bergamaschi M, Maltecca C, Fix J, Schwab C, Tiezzi F. Genome-wide association study for carcass quality traits and growth in purebred and crossbred pigs1. J Anim Sci. 2020;98(1):skz360. doi: 10.1093/jas/skz360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Liu W, Zhuang Z, Wang W, Huang T, Liu Z. An Improved genome-wide polygenic score model for Predicting the risk of type 2 diabetes. Front Genet. 2021;12:632385. doi: 10.3389/fgene.2021.632385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Yang W, Wu J, Yu J, Zheng X, Kang H, Wang Z, et al. A genome-wide association study reveals additive and dominance effects on growth and fatness traits in large white pigs. Anim Genet. 2021;52:749–53. doi: 10.1111/age.13131. [DOI] [PubMed] [Google Scholar]
- 23.Astle W, Balding DJ. Population structure and cryptic relatedness in Genetic Association Studies. Statist Sci. 2009;24:451–71. doi: 10.1214/09-STS307. [DOI] [Google Scholar]
- 24.Cardon LR, Palmer LJ. Population stratification and spurious allelic association. The Lancet. 2003;361:598–604. doi: 10.1016/S0140-6736(03)12520-2. [DOI] [PubMed] [Google Scholar]
- 25.Ding R, Yang M, Wang X, Quan J, Zhuang Z, Zhou S, et al. Genetic Architecture of feeding behavior and feed efficiency in a Duroc Pig Population. Front Genet. 2018;9:220. doi: 10.3389/fgene.2018.00220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Lippert C, Listgarten J, Liu Y, Kadie CM, Davidson RI, Heckerman D. FaST linear mixed models for genome-wide association studies. Nat Methods. 2011;8:833–5. doi: 10.1038/nmeth.1681. [DOI] [PubMed] [Google Scholar]
- 27.Listgarten J, Lippert C, Kadie CM, Davidson RI, Eskin E, Heckerman D. Improved linear mixed models for genome-wide association studies. Nat Methods. 2012;9:525–6. doi: 10.1038/nmeth.2037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Mathieson I, McVean G. FaST-LMM-Select for addressing confounding from spatial structure and rare variants reply. Nat Genet. 2013;45:471. doi: 10.1038/ng.2619. [DOI] [PubMed] [Google Scholar]
- 29.Yang J, Zaitlen NA, Goddard ME, Visscher PM, Price AL. Advantages and pitfalls in the application of mixed-model association methods. Nat Genet. 2014;46:100–6. doi: 10.1038/ng.2876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Price AL, Patterson NJ, Plenge RM, Weinblatt ME, Shadick NA, Reich D. Principal components analysis corrects for stratification in genome-wide association studies. Nat Genet. 2006;38:904–9. doi: 10.1038/ng1847. [DOI] [PubMed] [Google Scholar]
- 31.Kang HM, Zaitlen NA, Wade CM, Kirby A, Heckerman D, Daly MJ, Eskin E. Efficient control of population structure in model organism association mapping. Genetics. 2008;178:1709–23. doi: 10.1534/genetics.107.080101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Zhang Z, Ersoz E, Lai C-Q, Todhunter RJ, Tiwari HK, Gore MA, et al. Mixed linear model approach adapted for genome-wide association studies. Nat Genet. 2010;42:355–60. doi: 10.1038/ng.546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Zhou X, Stephens M. Genome-wide efficient mixed-model analysis for association studies. Nat Genet. 2012;44:821–4. doi: 10.1038/ng.2310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Hao H, Lin R, Li Z, Shi W, Huang T, Niu J, et al. MC4R deficiency in pigs results in hyperphagia and ultimately hepatic steatosis without high-fat diet. Biochem Biophys Res Commun. 2019;520:651–6. doi: 10.1016/j.bbrc.2019.08.016. [DOI] [PubMed] [Google Scholar]
- 35.Jae-Hoon KYong-Jun,KSang-Geum,KSu-Yeon,SMoon-Cheol,W, Nam-Young K, et al. Association of Melanocortin 4 receptor (MC4R) genotypes with fatty acid compositions in an Intercross Population between Duroc and Jeju native Pigs. [Association of Melanocortin 4 receptor (MC4R) genotypes with fatty acid compositions in an Intercross Population between Duroc and Jeju native Pigs] J Life Sci. 2020;30:58–63. [Google Scholar]
- 36.Onteru SK, Gorbach DM, Young JM, Garrick DJ, Dekkers JCM, Rothschild MF. Whole Genome Association Studies of residual feed intake and related traits in the Pig. PLoS ONE. 2013;8:e61756. doi: 10.1371/journal.pone.0061756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Chen J, Xiong Y, Zuo B, Zheng R, Li F, Lei M, et al. New Evidences of Effect of Melanocortin-4 receptor and insulin-like growth factor 2 genes on Fat deposition and carcass traits in different pig populations. Asian Australas J Anim Sci. 2005;18:1542–7. doi: 10.5713/ajas.2005.1542. [DOI] [Google Scholar]
- 38.Fan B, Onteru SK, Du Z-Q, Garrick DJ, Stalder KJ, Rothschild MF. Genome-wide association study identifies loci for body composition and structural soundness traits in pigs. PLoS ONE. 2011;6:e14726. doi: 10.1371/journal.pone.0014726. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Houston RD, Cameron ND, Rance KA. A melanocortin-4 receptor (MC4R) polymorphism is associated with performance traits in divergently selected large White pig populations. Anim Genet. 2004;35:386–90. doi: 10.1111/j.1365-2052.2004.01182.x. [DOI] [PubMed] [Google Scholar]
- 40.Chami N, Preuss M, Walker RW, Moscati A, Loos RJF. The role of polygenic susceptibility to obesity among carriers of pathogenic mutations in MC4R in the UK Biobank population. PLoS Med. 2020;17:e1003196. doi: 10.1371/journal.pmed.1003196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Clément K, van den Akker E, Argente J, Bahm A, Chung WK, Connors H, et al. Efficacy and safety of setmelanotide, an MC4R agonist, in individuals with severe obesity due to LEPR or POMC deficiency: single-arm, open-label, multicentre, phase 3 trials. The Lancet Diabetes & Endocrinology. 2020;8:960–70. doi: 10.1016/S2213-8587(20)30364-8. [DOI] [PubMed] [Google Scholar]
- 42.Williams DM, Nawaz A, Evans M. Drug therapy in obesity: a review of current and emerging treatments. Diabetes Ther. 2020;11:1199–216. doi: 10.1007/s13300-020-00816-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Meidtner K, Schwarzenbacher H, Scharfe M, Severitt S, Blöcker H, Fries R. Haplotypes of the porcine peroxisome proliferator-activated receptor delta gene are associated with backfat thickness. BMC Genet. 2009;10:76. doi: 10.1186/1471-2156-10-76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Yao G, Wang Y, Zhang Y, Wu T, Liu J, Wang Y. Postnatal growth changes of myoglobin and its gene expression level in M. longissimus Dorsi muscle of Jinhua and Landrace Barrows. J of Animal and Veterinary Advances. 2011;10:2493–8. doi: 10.3923/javaa.2011.2493.2498. [DOI] [Google Scholar]
- 45.Melo C, Gallardo D, Quintanilla R, Zidi A, Castelló A, Díaz I, et al. An association analysis between polymorphisms of the pig solute carrier family 27A (SLC27A), member 1 and 4 genes and serum and muscle lipid traits. Livest Sci. 2013;152:143–6. doi: 10.1016/j.livsci.2013.01.004. [DOI] [Google Scholar]
- 46.Villaplana-Velasco A, Noguera JL, Pena RN, Ballester M, Muñoz L, González E, et al. Comparative Transcriptome Profile between Iberian Pig Varieties provides New Insights into their distinct Fat deposition and fatty acids content. Animals (Basel) 2021;11(3):627. doi: 10.3390/ani11030627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Wang H, Wang J, Yang D, Liu Z, Zeng Y, Chen W. Expression of lipid metabolism genes provides new insights into intramuscular fat deposition in Laiwu pigs. Asian Australas J Anim Sci. 2020;33:390–7. doi: 10.5713/ajas.18.0225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Kogelman LJA, Pant SD, Fredholm M, Kadarmideen HN. Systems genetics of obesity in an F2 pig model by genome-wide association, genetic network, and pathway analyses. Front Genet. 2014;5:214. doi: 10.3389/fgene.2014.00214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Zhang H, Zhuang Z, Yang M, Ding R, Quan J, Zhou S, et al. Genome-wide detection of genetic loci and candidate genes for body conformation traits in duroc × landrace × Yorkshire crossbred Pigs. Front Genet. 2021;12:664343. doi: 10.3389/fgene.2021.664343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Ding R, Zhuang Z, Qiu Y, Ruan D, Wu J, Ye J, et al. Identify known and novel candidate genes associated with backfat thickness in Duroc pigs by large-scale genome-wide association analysis. J Anim Sci. 2022;100(2):skac012. doi: 10.1093/jas/skac012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Liu X, Wang L, Liang J, Yan H, Zhao K, Li N, et al. Genome-wide Association study for certain carcass traits and Organ Weights in a large White×Minzhu Intercross Porcine Population. J Integr Agric. 2014;13:2721–30. doi: 10.1016/S2095-3119(14)60787-5. [DOI] [Google Scholar]
- 52.Wang L, Zhang L, Yan H, Liu X, Li N, Liang J, et al. Genome-wide association studies identify the loci for 5 exterior traits in a large White × Minzhu pig population. PLoS ONE. 2014;9:e103766. doi: 10.1371/journal.pone.0103766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Guo Y, Qiu H, Xiao S, Wu Z, Yang M, Yang J, et al. A genome-wide association study identifies genomic loci associated with backfat thickness, carcass weight, and body weight in two commercial pig populations. J Appl Genet. 2017;58:499–508. doi: 10.1007/s13353-017-0405-6. [DOI] [PubMed] [Google Scholar]
- 54.Browning SR, Browning BL. Rapid and accurate haplotype phasing and missing-data inference for whole-genome association studies by use of localized haplotype clustering. Am J Hum Genet. 2007;81:1084–97. doi: 10.1086/521987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Chang CC, Chow CC, Tellier LC, Vattikuti S, Purcell SM, Lee JJ. Second-generation PLINK: rising to the challenge of larger and richer datasets. Gigascience. 2015;4:7. doi: 10.1186/s13742-015-0047-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Yang J, Lee SH, Goddard ME, Visscher PM. GCTA: a tool for genome-wide complex trait analysis. Am J Hum Genet. 2011;88:76–82. doi: 10.1016/j.ajhg.2010.11.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Ginestet C. ggplot2: Elegant Graphics for Data Analysis. J Royal Stat Society: Ser (Statistics Society) 2011;174:245–6. doi: 10.1111/j.1467-985X.2010.00676_9.x. [DOI] [Google Scholar]
- 58.VanRaden PM. Efficient methods to compute genomic predictions. J Dairy Sci. 2008;91:4414–23. doi: 10.3168/jds.2007-0980. [DOI] [PubMed] [Google Scholar]
- 59.Willer CJ, Li Y, Abecasis GR. METAL: fast and efficient meta-analysis of genomewide association scans. Bioinformatics. 2010;26:2190–1. doi: 10.1093/bioinformatics/btq340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Yang J, Weedon MN, Purcell S, Lettre G, Estrada K, Willer CJ, et al. Genomic inflation factors under polygenic inheritance. Eur J Hum Genet. 2011;19:807–12. doi: 10.1038/ejhg.2011.39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Shin J-H, Blay S, Graham J, McNeney B. LDheatmap: an R function for graphical Display of pairwise linkage Disequilibria between single nucleotide polymorphisms. J Stat Softw. 2006;16:1–9. doi: 10.18637/jss.v016.c03. [DOI] [Google Scholar]
- 62.Yang J, Benyamin B, McEvoy BP, Gordon S, Henders AK, Nyholt DR, et al. Common SNPs explain a large proportion of the heritability for human height. Nat Genet. 2010;42:565–9. doi: 10.1038/ng.608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Ashburner M, Ball CA, Blake JA, Botstein D, Butler H, Cherry JM, et al. Gene ontology: tool for the unification of biology. The Gene Ontology Consortium. Nat Genet. 2000;25:25–9. doi: 10.1038/75556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Carbon S, Douglass E, Good BM, Unni DR, Elser J. The Gene Ontology resource: enriching a GOld mine. Nucleic Acids Res. 2021;49:D325–34. doi: 10.1093/nar/gkaa1113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Minoru K, Miho F, Yoko S, Mari IW, Mao T. KEGG: integrating viruses and cellular organisms. Nucleic Acids Res. 2021;49:D545–51. doi: 10.1093/nar/gkaa970. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Kanehisa M. Toward pathway engineering: a new database of genetic and molecular pathways. Sci Technol Japan. 1996;59:34–8. [Google Scholar]
- 67.Yu G, Wang L, Han Y, He Q. clusterProfiler: an R package for comparing biological themes among gene clusters. OMICS. 2012;16:284–7. doi: 10.1089/omi.2011.0118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.MacArthur J, Bowler E, Cerezo M, Gil L, Hall P, Hastings E, et al. The new NHGRI-EBI catalog of published genome-wide association studies (GWAS catalog). Nucleic Acids Res. 2017;45:D896–901. [DOI] [PMC free article] [PubMed]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Additional file 1: Figure S1. PCA plot of each of three breeds.
Additional file 2: Table S1. Summary information of 19 single population GWAS.
Additional file 3: Figure S2. Venn diagram of the number of significant SNPs (P<10-5) between 19 single population GWAS and four metaGWAS.
Additional file 4: Table S2. Summary information of significant SNPs in four metaGWAS.
Additional file 5: Figure S3. Q-Q plot of four metaGWAS.
Additional file 6: Figure S4. LD blocks of each of the five QTLs in three breeds.
Additional file 7: Table S3 (Sheet1). Annotation of candidate genes belongs to fat regulation biology process in GO and KEGG. Table S3 (Sheet2). Annotation of candidate genes in pigQTLdb. Table S3 (Sheet3). Annotation of candidate genes in human NHGRI-EBI Catalog.
Data Availability Statement
All annotation information was obtained from a publicly available source (http://www.ensembl.org). The genotype and phenotype used and/or analyzed during the current study are available from the corresponding author on reasonable request.