

Abstract
Cereblon (CRBN) is a ubiquitin ligase (E3) substrate receptor protein co-opted by CRBN E3 ligase modulatory drug (CELMoD) agents that target therapeutically relevant proteins for degradation. Prior crystallographic studies defined the drug-binding site within CRBN’s thalidomide-binding domain (TBD), but the allostery of drug-induced neosubstrate binding remains unclear. We performed cryo–electron microscopy analyses of the DNA damage-binding protein 1 (DDB1)–CRBN apo complex and compared these structures with DDB1-CRBN in the presence of CELMoD compounds alone and complexed with neosubstrates. Association of CELMoD compounds to the TBD is necessary and sufficient for triggering CRBN allosteric rearrangement from an open conformation to the canonical closed conformation. The neosubstrate Ikaros only stably associates with the closed CRBN conformation, illustrating the importance of allostery for CELMoD compound efficacy and informing structure-guided design strategies to improve therapeutic efficacy.
Eukaryotic proteins are targeted for degradation through covalent attachment of ubiquitin moieties to specific residues, primarily lysine side chains (1). Ubiquitination is facilitated by ubiquitin ligases (E3s), which coordinate and position targeted substrates for ubiquitin ligation, often through adaptor proteins and interchangeable receptor modules. Substrate specificity of one such E3 complex, the Cullin-4 RING ligase (CRL4), is mediated by the cereblon (CRBN) substrate receptor. CRBN facilitates the transfer of ubiquitin to substrate proteins by reversibly interacting with the CRL4 core complex, which consists of the Cullin-4 scaffold, the DNA damage-binding protein 1 (DDB1) adaptor protein, and RING finger protein ROC1 (fig. S1A). CRBN is also the cellular receptor for a class of drugs known as CELMoD (CRBN E3 ligase modulatory drug) agents, which bind to a conserved hydrophobic pocket in CRBN to create a molecular surface capable of recruiting neosubstrates, that is, cellular proteins that are targeted only in response to drug. Such ligand-induced CRL4-CRBN recruitment facilitates neosubstrate ubiquitination and subsequent proteasomal degradation, thereby dictating therapeutic efficacy (2). The CELMoD agent lenalidomide (Revlimid) has been used as a first-line therapy for multiple myeloma and other hematological malignancies for more than a decade, and next-generation CELMoD compounds markedly improve patient outcomes in clinical trials (3). Although crystallographic structural snapshots of CRBN-DDB1 bound to a variety of drugs and neosubstrates have facilitated a molecular description of the stable quaternary complex, the allostery associated with substrate targeting remains unknown, and the structural features of the unliganded complex have yet to be described.
CRBN is a 50-kDa protein containing three folded domains: an N-terminal Lon protease-like domain (hereafter, Lon domain), an intermedial helical bundle (HB), and a C-terminal thalidomide-binding domain (TBD) that harbors the drug-binding pocket. CRBN is recruited to the Cullin-4–ROC1 ligase module by the adaptor protein DDB1, which consists of three WD-40 beta propeller domains (BPA, BPB, and BPC). The HB of CRBN docks into a central hydrophobic cleft formed at the interface of BPA and BPC of DDB1, while the mobile BPB domain interacts with Cullin-4, positioning CRBN-bound substrates for ubiquitination. Over a dozen crystal structures of the CRBN-DDB1 complex show the Lon domain and TBD of CRBN tightly interacting with one another in what can be described as a closed conformation (CRBNclosed) (2, 4–10). These structures of the closed conformer bound to small molecules have provided the basis for structure-guided drug design of next-generation CELMoD agents with enhanced neosubstrate specificity and potency, such as CC-92480 (mezigdomide). Although it is thought that association of the TBD and Lon domains is concomitant with drug and neosubstrate binding, this assumption is challenged by structures of the isolated TBD bound to ligands, demonstrating that the TBD is competent for drug binding in the absence of the Lon domain (11).
Two crystal structures have captured the Lon domain and TBD in an open conformation (CRBNopen) where the two domains are separated and positioned at a ~45° angle relative to one another. These reports raise questions about the assumed constraints associated with drug accessibility and binding, as well as the mechanisms by which ligands modify the surface chemistry of CRBN to enable neosubstrate recruitment and subsequent ubiquitination and degradation (4, 5). If CRBNopen represents a physiological conformer (i.e., not an artifact of crystallization), we must reconsider recruitment of drug and neosubstrates in the context of these two conformers. Additionally, the dynamics of interconversion between the open and closed conformers, the effect that ligands and neosubstrates have on this process, and how these dynamics affect neosubstrate positioning and subsequent degradation may have profound effects on the design, and ultimately the therapeutic impact, of next-generation CELMoD compounds.
To better understand the spectrum of CRBN conformations, we used cryo–electron microscopy (cryo-EM) to examine the population ensemble of CRBN-DDB1 conformers present in solution under various conditions. To overcome preferred orientation and partial denaturation or dissociation of the complex during cryo-EM grid preparation, a combination of grid pretreatment with a CRBN-agnostic protein, incorporation of mild amphiphilic detergent, and sample dilution immediately before plunging improved particle homogeneity and distribution in ice (see methods in the supplementary materials). Unexpectedly, whether copurified with either full-length DDB1 or DDB1 lacking BPB (CRBN-DDB1ΔBPB, a construct commonly used for x-ray crystallography), apo-CRBN exclusively adopts an open conformation, with the TBD and Lon domains separated from one another (Fig. 1A and fig. S1B).
Fig. 1. CRBNopen is allosterically modulated to CRBNclosed by pomalidomide.
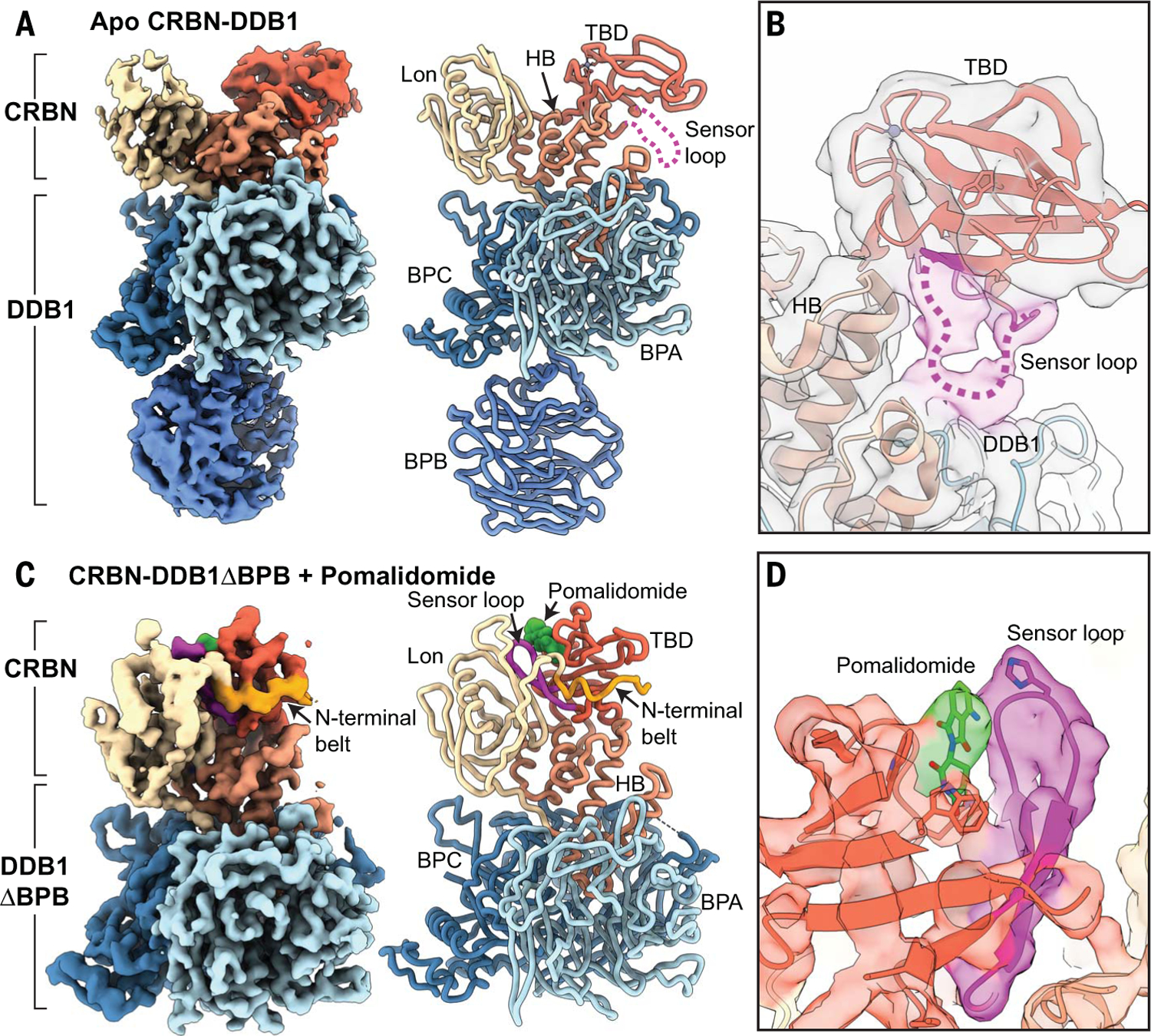
(A) ~3.5-Å-resolution cryo-EM reconstruction of CRBN-DDB1 in the unliganded apo form. The Lon domain (tan) is separated from the TBD (red), while the helical bundle (HB, light red) mediates interaction with DDB1, composed of beta propeller A (BPA, light blue), beta propeller B (BPB, medium blue), and beta propeller C (BPC, steel blue). CRBN and DDB1 are thresholded independently to illustrate important features. To the right, a ribbon representation of the CRBN-DDB1 complex modeled from density is shown (same color scheme as on the left). The sensor loop is denoted as a dotted magenta line extending away from the TBD domain. (B) The unsharpened cryo-EM map shows the path of the sensor loop, which interacts with the HB and BPA of DDB1. (C) Surface representation of the ~3.9-Å-resolution cryo-EM reconstruction of CRBN-DDB1 in the closed form in complex with pomalidomide. The N-terminal belt (orange) wraps around the TBD domain. To the right, a ribbon representation of CRBN-DDB1ΔBPB in complex with pomalidomide is shown, with the sensor loop (magenta) adopting a β-hairpin organization that is tightly packed between the TBD and Lon domains. (D) A detailed view of the density corresponding to pomalidomide (green) and the sensor loop (magenta) in CRBNclosed.
Our ~3.5-Å-resolution cryo-EM structure confirms the physiological existence of the open conformer and provides the first opportunity to examine this CRBN conformation in the absence of ligand or substrate. Notably, we do not observe density for the “sensor loop,” a beta-insert hairpin within CRBN’s TBD (residues ~341 to 361) that has been shown to directly bind immunomodulatory drugs and neosubstrates in prior structures (2, 12), at its canonical position adjacent to the thalidomide-binding pocket. Instead, we observe strong, low-resolution density corresponding to the sensor loop engaged in previously unobserved interactions with a helix in CRBN’s HB (residues ~210 to 220) and a loop of BPC in DDB1 (residues ~776 to 780) (Fig. 1B). These regions of the HB and DDB1 are usually disordered in crystal structures (5, 10, 12, 13), consistent with the flexibility of this region observed in our cryo-EM density. However, the clearly observed association between the sensor loop and this HB-DDB1 interface in our cryo-EM structures suggests that interaction between these flexible loops plays a role in maintaining the TBD in the CRBNopen conformer.
To better understand how CELMoD agents interact with and influence this open CRBN conformation, we added saturating amounts of different compounds to CRBN-DDB1 for cryo-EM analyses. Addition of pomalidomide was sufficient to induce conformational rearrangement within CRBN for only ~20% of the particles. The ~3.9-Å-resolution structure of CRBNclosed arising from pomalidomide-bound particles shows the ~15-kDa TBD positioned adjacent to the Lon domain and unambiguous density within the ligand-binding pocket consistent with pomalidomide association (Fig. 1C). Necessarily, the sensor loop in CRBNclosed is observed in the canonical β-hairpin fold interfacing the TBD and Lon domain, and a portion of the CRBN N terminus (residues 48 to 63) that is disordered in the CRBNopen becomes ordered, extending from the Lon domain, trussing the TBD, and supporting the closed conformation (Fig. 1C). To better define the role of these N-terminal residues, hereafter referred to as the N-terminal belt, in CRBN closure, we truncated 63 residues from the CRBN N terminus (CRBNΔNTD) and incubated the CRBNΔNTD-DDB1 with saturating pomalidomide concentrations. Deletion of the N terminus all but abolishes CRBN closure, with less than ~2% of the complexes adopting a closed conformation with a poorly ordered sensor loop, underscoring the importance of the CRBN N-terminal belt in stabilizing the CRBNclosed conformer (fig. S2A).
We expected to observe the pomalidomide ligand in the binding pocket of only the CRBNclosed conformer, but we were surprised to find strong density corresponding to pomalidomide within the binding pocket of CRBNopen as well (fig. S3A). Even with the ligand bound—and in stark contrast to prior crystal structures of the CRBNopen conformer—the sensor loop of our cryo-EM CRBNopen conformer remains engaged in interactions with the CRBN HB and DDB1 as we observe in the apo-CRBN conformer (fig. S3B). These results indicate that limitations in neosubstrate recruitment, which directly translate to drug efficacy, may not solely correlate with CRBN-binding kinetics. Rather, conversion of the CRBNopen to the CRBNclosed conformation may be a key mechanistic bottleneck in the allosteric transformation of apo-CRBN to a neosubstrate-binding conformation after ligand association.
We were intrigued by the observation that saturating concentrations of pomalidomide did not induce widespread closure of the CRBN domains, despite high occupancy. We rationalized that next-generation CELMoD agents with improved binding affinity may also have a more pronounced allosteric effect through enhanced interactions with the sensor loop, contributing to adoption of the sensor loop to the CRBNclosed–competent β-hairpin conformation. Indeed, an analysis of CRBN-DDB1 in the presence of CC-220 (iberdomide), a molecule with ~20-fold improved affinity and ~24-fold enhanced Ikaros degradation (13), shows that nearly 50% of particles have shifted to the CRBNclosed conformer (Fig. 2A). As with pomalidomide, in the presence of excess ligand, we observe density consistent with iberdomide within the glutarimide-binding pocket of both the CRBNclosed and CRBNopen conformers, suggesting that drug occupancy within CRBN is not responsible for the increased allosteric transition. Two prior crystal structures of the iberdomide-bound CRBN show that the additional phenyl and morpholino moieties of the iberdomide molecule can arch over the top of the sensor loop β-hairpin (13, 14), a conformation that may aid in the stabilization of this structural motif. These data support our proposed allosteric model whereby enhanced ligand interaction with the sensor loop stabilizes the β-hairpin conformation, which promotes the closure of the TBD and Lon domains and ultimately translates to improved neosubstrate ubiquitination and subsequent degradation efficacy. We note, however, that these extended moieties are limited in resolution in our cryo-EM structure (fig. S3C) and have high B-factors in published x-ray structures. This observation, coupled with recently described alternative positions of these extended ligand groups (14), suggests that iberdomide’s interaction with the sensor loop may be dynamic. It is likely that a more stable interaction with the sensor loop could be attained with next-generation CELMoD agents.
Fig. 2. Improved CELMoD compounds drive CRBNclosed and recruit Ikaros.
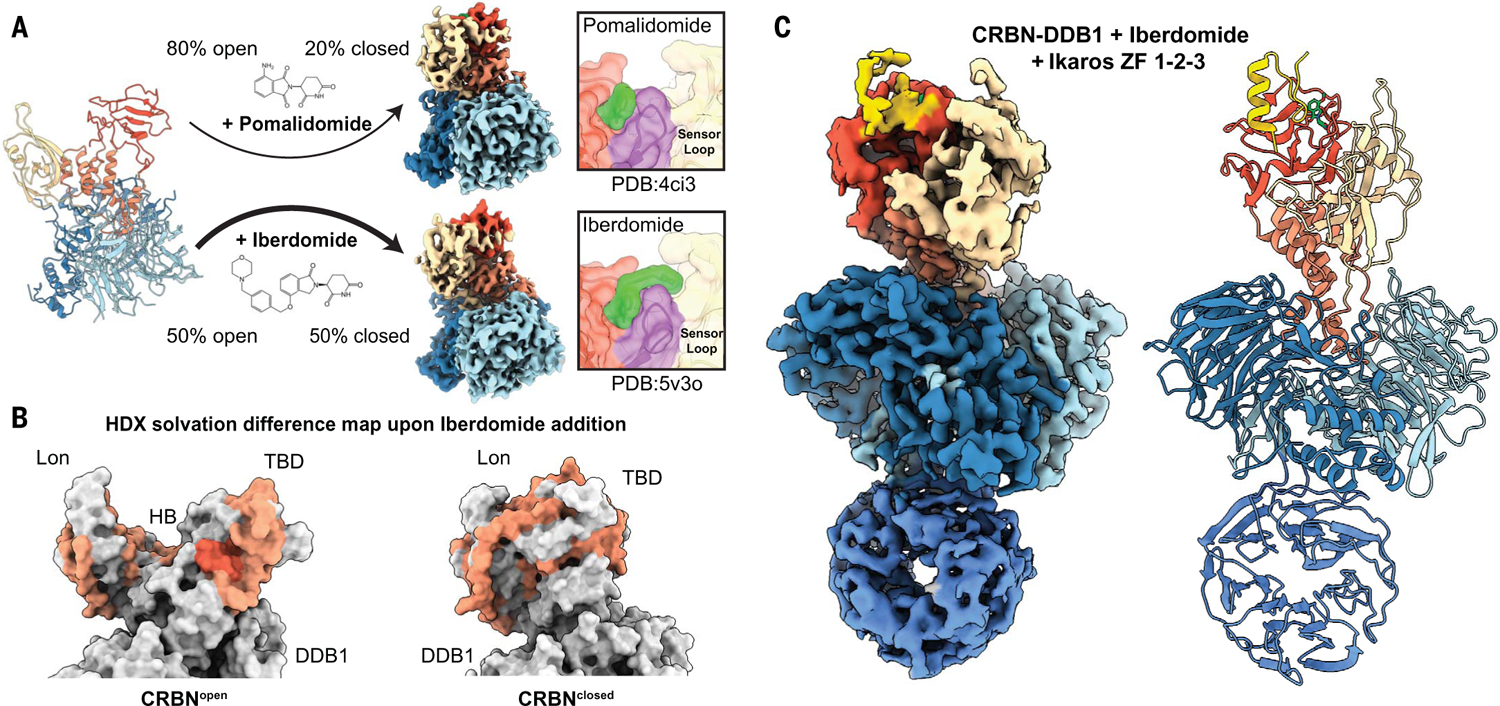
(A) The CRBNopen transition to CRBNclosed is differentially regulated between pomalidomide (~20% of particles adopt CRBNclosed) and the CELMoD agent iberdomide (~50% of particles adopt CRBNclosed). (Top right) The ~3.9-Å-resolution cryo-EM structure of pomalidomide-induced CRBNclosed. (Bottom right) The ~3.7-Å-resolution reconstruction of iberdomide-induced CRBNclosed. The inset panels on the far right depict surface representations of published crystal structures illustrating the ligand (green) interactions with sensor loop (magenta). (B) Colored space-filling representation of CRBN models with residue-specific coloring according to changes in solvency upon addition of iberdomide as detected by HDX-MS. Orange residues correspond to peptides with mild HDX differential, and red residues correspond to peptides with large HDX differential. Mixed conformations are expected during the experiment, so CRBNopen and CRBNclosed are both shown. (C) The ~3.6-Å-resolution reconstruction of iberdomide-induced CRBNclosed in complex with Ikaros ZF1-ZF2-ZF3 (gold). DDB1 and CRBN are thresholded independently to illustrate important features.
To further investigate the allosteric influence of iberdomide on CRBN closure, we used hydrogen-deuterium exchange mass spectrometry (HDX-MS) to profile changes in solvent accessibility of CRBN-DDB1 peptides in the presence of iberdomide. Compared with unliganded CRBN-DDB1, addition of drug substantially reduced solvation for regions within the Lon, TBD, and HB domains found at the domain-domain interface of the closed conformation, consistent with transition from CRBNopen to CRBNclosed (Fig. 2B and fig. S4A), whereas the DDB1 surfaces were unchanged (fig. S4B). As expected, the most extreme change is observed for the sensor loop itself, consistent with formation of the buried hairpin we observe in CRBNclosed (Fig. 1C). These findings offer a potential new modality for drug development, wherein properties influencing not only binding kinetics for CRBN and neosubstrates (15) but also the capacity to stabilize the β-hairpin conformation of the sensor loop as a means of both initiating and maintaining the closed conformation should be considered. The CRBNclosed conformation may in turn be a prerequisite for substrate recruitment.
Indeed, the substantial population of ligand-bound CRBNopen conformers in our datasets indicates that ligand binding is not concurrent with adoption of the CRBNclosed conformation and that CRBN closure is dependent on subsequent allosteric triggers. We posit that ligand binding is involved in inducing the sensor loop’s adoption of the β-hairpin arrangement, thereby untethering the TBD from the CRBN HB to enable the CRBNclosed conformation for substrate interaction. Despite extensive focused classification of the tether region, we were unable to identify a population of liganded CRBNopen conformers where the sensor loop is untethered from the HB in any of our datasets. This finding indicates that ligand-mediated detachment of the sensor loop from the HB is concomitant with adoption of the structured β-hairpin and triggers immediate closure of the CRBN TBD and Lon domains.
We next aimed to probe the role of drug binding in neosubstrate recruitment and positioning in the context of the CRBNopen and CRBNclosed conformers. Prior crystallographic studies yielded structures of CRBNopen conformers bound to neosubstrates (4, 9), although it was speculated that these conformations may have resulted from crystallization conditions and lattice contacts. To investigate drug-mediated CRBN-DDB1 recruitment of neosubstrates, we used the zinc finger (ZF) transcription factor Ikaros, a cellular target of pomalidomide and iberdomide. We generated different constructs containing tandem Ikaros ZF1, ZF2, and ZF3 domains (fig. S5A) for increased recruitment efficiency (4) and incubated each individually with CRBN-DDB1 in the presence of pomalidomide or iberdomide. Regardless of drug identity or Ikaros ZF construct design, our single-particle analyses reveal density consistent with the location and positioning of a single Ikaros ZF motif associated with the TBD domain of the CRBNclosed conformer (Fig. 2C and fig. S5B), again confirmed in HDX-MS analyses showing even more pronounced changes for residues within the sensor loop that contact the known Ikaros recruitment motif (fig. S4, C and D). We were unable to detect any Ikaros association with the CRBNopen conformer, consistent with a mechanism whereby drug-induced CRBN closing occurs before substrate recruitment. This result further demonstrates the importance of an allosteric control mechanism, whereby drug-induced CRBN closing occurs prior to neosubstrate recruitment.
The Cullin-4–ROC1 ligase core recruits the DDB1-CRBNclosed-Ikaros complex by associating with the second beta propeller of DDB1 (called BPB), which is located distally to CRBN. BPB extends from the DDB1 core as a mobile element, and this flexibility has been implicated in the mechanism of DDB1 as a promiscuous adaptor that bridges Cullin-4 with structurally diverse substrate-binding modules. Correspondingly, BPB has been captured in a variety of conformations in several prior studies (16). We sought to describe the motion of BPB and its potential relevance to Ikaros positioning for ubiquitination in the context of the CRL4-CRBN complex assembly. Our image analyses of the BPB domain within our apo CRBN data reveal that, rather than demonstrating a full continuum of motion, the BPB position is limited to three metastable positions. Approximately 65% of the particles adopt a “linear” orientation, in which the broad face of BPB is in a similar orientation as BPC (Fig. 3A, left). A ~70° rotated “hinged” orientation, where the BPB face more closely matches BPA, is the second major class observed, with ~20 to 30% of particles (Fig. 3A, middle), and only a minor population contain BPB in the third, “twisted” orientation, rotated ~140° from the linear orientation (Fig. 3A, right). Further classification of particles contributing to each of these states reveals only minor positional variability for BPB, as has been previously described (16), centered around these discrete conformations.
Fig. 3. DDB1 and next-generation CELMoD agents further poise CRBN substrates for ubiquitination in disease contexts.
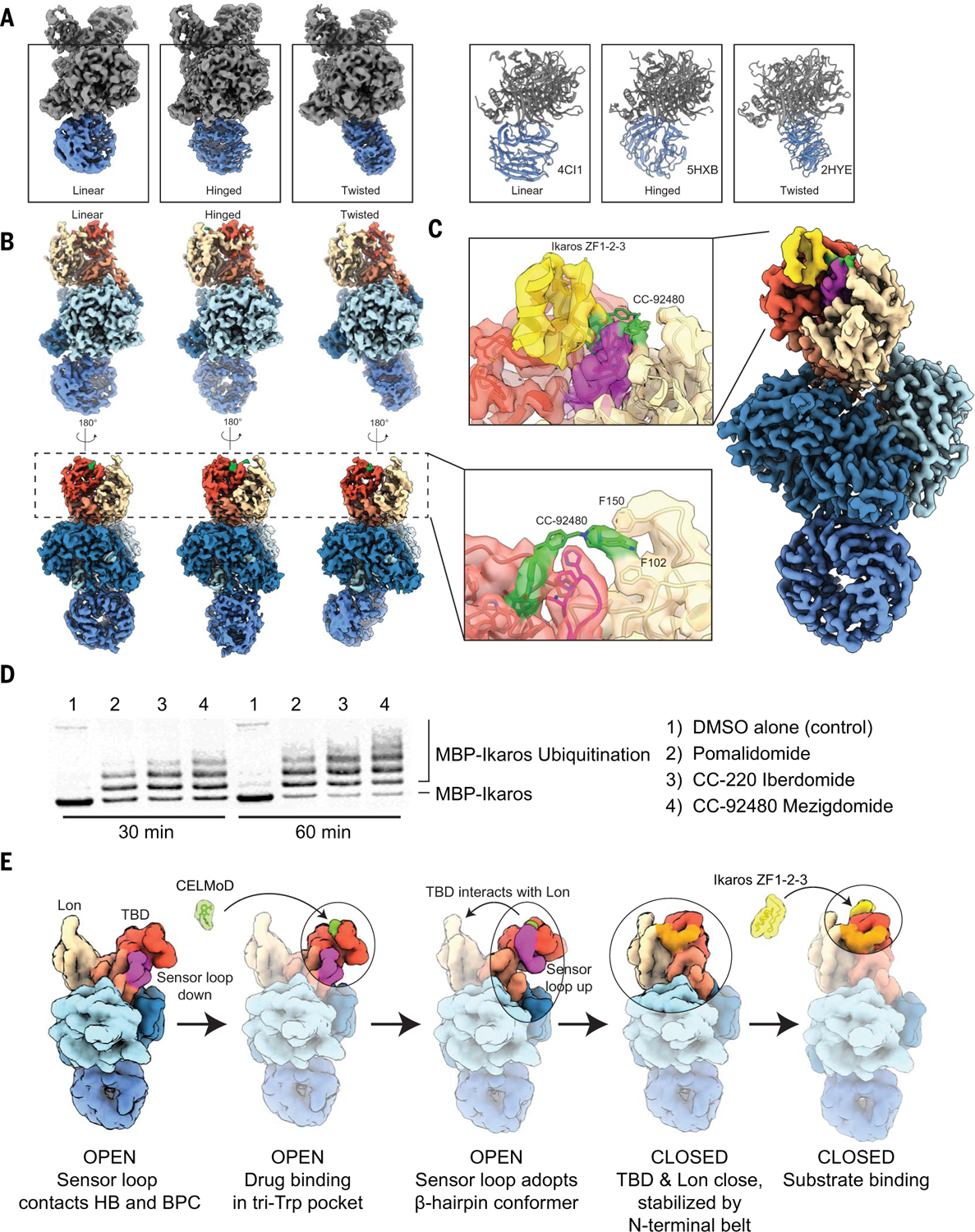
(A) Three-dimensional classification of unliganded CRBN-DDB1 yields three discrete positions of DDB1’s mobile BPB propeller (colored blue). (Left) ~3.5-Å-resolution cryo-EM reconstructions of CRBN-DDB1 with BPB in a linear (left), hinged (middle), or twisted (right) position. (Right) Structural models in similar orientations for reference, adopting a linear (PDB ID 4CI1, left), hinged (PDB ID 5HXB, middle), or twisted (PDB ID 2HYE, right) position. (B) The ~3.2-Å-resolution cryo-EM reconstructions of CRBN-DDB1 complexed with mezigdomide with a linear (left), hinged (middle), or twisted (right) position of BPB. (Inset) ~3.1-Å-resolution focused refinement of particles from all three orientations reveal a connection between mezigdomide and the Lon domain, “stapling” the CRBNclosed conformation. (C) The ~3.1-Å-resolution reconstruction of mezigdomide-induced CRBNclosed bound to Ikaros ZF1-ZF2-ZF3. The inset shows a focused view of connection between mezigdomide and the Lon domain, stapling CRBNclosed in the presence of Ikaros. (D) In vitro ubiquitination assay tracking MBP-Ikaros ubiquitination in the presence of different molecules used in this study (labeled on the right). (E) Mechanistic model illustrating pathway of assembly. First, unliganded CRBN is open with the sensor loop attached to HB. Second, ligand is added and associates with TBD in the hydrophobic pocket. Third, binding of ligand in the tri-Trp pocket stabilizes the sensor loop as a β-hairpin detached from the HB. Fourth, sensor loop refolding promotes transition of CRBNopen to CRBNclosed without an observed intermediate, and the N-terminal belt is seated (orange). Fifth, substrate is recruited to the CRBNclosed conformation for subsequent ubiquitination by CRL4.
These three principal orientations were observed in recent low-resolution cryo-EM structures of DDB1-DCAF1 (DDB1 and Cullin-4 associated factor 1) bound to Cullin-4 (17), and our structures demonstrate that the BPB likely switches between these three energetic minima irrespective of the substrate adaptor module or interaction with Cullin. Although we were unable to discern allosteric coordination upon ligand or Ikaros binding, the population distribution of the states varies greatly compared with those observed for the Cullin-bound DDB1 (17). Notably, the twisted orientation that we observe in <10% of our particles is the majority conformer when bound to Cullin (17), suggesting that association with Cullin may substantially modulate BPB positioning between these three states, thereby influencing proximity and positioning of neosubstrates for ubiquitination.
Given the potential clinical relevance of the observed CRBN allostery, we considered the mechanisms of action responsible for the improved efficacy of next-generation CELMoD molecules. Mezigdomide, for example, was recently shown to display efficient, rapid neosubstrate degradation kinetics and is considerably more efficacious in the treatment of relapse or refractory patients who no longer respond to primary treatment with lenalidomide and/or pomalidomide (18). To investigate the structural features responsible for the enhanced efficacy of this compound, we incubated mezigdomide with CRBN-DDB1 for cryo-EM analyses. One hundred percent of the complexes adopted the CRBNclosed conformer. Density corresponding to mezigdomide is clearly observed in the canonical binding site in the ~3.2-Å-resolution structures of CRBN-DDB1 with BPB in the three discrete orientations (Fig. 3B). We also observe protruding drug density that extends to the distal Lon domain, a feature that had not been observed in the structures of any other liganded CRBN (Fig. 3B, right). We were able to unambiguously fit mezigdomide into the density to reveal that the benzonitrile moiety is likely stabilized by aromatic or cation-pi interactions with CRBN Phe102 and Phe150 of the Lon domain. Association of the glutarimide and the benzonitrile moieties to distinct domains within CRBN, that is, the TBD and Lon domains, respectively, both provides avidity for improved ligand affinity and serves to staple the domains together in the closed conformation, which is traditionally accomplished by the CRBN N-terminal belt (Fig. 1C). Ikaros is recruited to mezigdomide-bound CRBN-DDB1 (Fig. 3C), and its ubiquitination efficiency is enhanced in assays with Cullin-4–ROC1 (Fig. 3D). Within the mezigdomide-bound dataset, we observe a substantial population of CRBNclosed particles that lack discernible density for the stabilizing N-terminal belt (fig. S2B). We thus propose that there is functional redundancy in securing the closed conformer and that mezigdomide further promotes neosubstrate recruitment and subsequent ubiquitination by prolonging the mechanistic cycle of closure, neosubstrate binding, N-terminal engagement, and stepwise reversal to the open conformation. These data elucidate a distinctive, previously unexplored compound design modality and describe the structural mechanism for enhanced therapeutic benefit of mezigdomide in patients unresponsive to pomalidomide, further illustrating the therapeutic value of allosteric control. Although not tested directly, we speculate that incorporation of these improved design elements to other molecular glues and heterobifunctional molecules would similarly improve their efficacy.
Our results provide mechanistic insights into the therapeutic efficacy of CELMoD compounds, a class of molecules that epitomize molecular glues and are central players in the field of targeted protein degradation. In so doing, we highlight previously unappreciated allosteric effects for consideration in the design of CRBN-directed molecular glue therapeutics (Fig. 3E). By characterizing the conformational rearrangements inherent to the CRBN-DDB1 system, and showing how three distinct degrader molecules affect allostery and neosubstrate-binding capacity, we reveal how conformational control of the mobile drug-binding TBD within CRBN has cryptically driven the therapeutic success of neosubstrate-targeting agents.
Supplementary Material
ACKNOWLEDGMENTS
We thank J. C. Ducom at Scripps Research High Performance Computing and C. Bowman at Scripps Research for computational support, as well as B. Anderson at the Scripps Research Electron Microscopy Facility for microscopy support. Computational analyses of EM data were performed using shared instrumentation funded by NIH S10OD021634 to G.C.L.
Funding:
This work was supported by National Institutes of Health grant S10OD021634 (G.C.L.).
Competing interests:
M.E.M., P.P.C., A.H.d.l.P., J.Z., E.T., and I.E.W. are or were employees of Bristol-Myers Squibb and have received Bristol-Myers Squibb stock. G.C.L. has received research funding from Bristol-Myers Squibb. A provisional patent regarding aspects of this work has been filed under US Provisional Patent Application no. 63/293,402. E.R.W. has received consulting fees for service rendered in cryo-EM method applications. P.P.C. is a founder, employee, and equity holder of Neomorph Inc. M.E.M. and A.H.d.l.P. are employees and stockholders of Neomorph Inc.
Footnotes
SUPPLEMENTARY MATERIALS
Data and materials availability:
Atomic models have been deposited to the Protein Data Bank (PDB) under IDs 8CVP, 8D7U, 8D7V, 8D7W, 8D7X, 8D7Y, 8D7Z, 8D80, and 8D81, as described in table S2. Electron microscopy reconstructions have been deposited to the Electron Microscopy Databank (EMDB) under accession codes EMDB-27012, EMDB-27234, EMDB-27235, EMDB-27236, EMDB-27237, EMDB-27238, EMDB-27239, EMDB-27240, EMDB-27241, and EMDB-27242, as described in table S1.
REFERENCES AND NOTES
- 1.Swatek KN, Komander D, Cell Res. 26, 399–422 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Chamberlain PP et al. , Nat. Struct. Mol. Biol 21, 803–809 (2014). [DOI] [PubMed] [Google Scholar]
- 3.Richardson PG et al. , Blood 138 (suppl. 1), 2731 (2021).34499707 [Google Scholar]
- 4.Sievers QL et al. , Science 362, eaat0572 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Petzold G, Fischer ES, Thomä NH, Nature 532, 127–130 (2016). [DOI] [PubMed] [Google Scholar]
- 6.Matyskiela ME et al. , Nat. Struct. Mol. Biol 27, 319–322 (2020). [DOI] [PubMed] [Google Scholar]
- 7.Surka C et al. , Blood 137, 661–677 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Fischer ES et al. , Nature 512, 49–53 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Nowak RP et al. , Nat. Chem. Biol 14, 706–714 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Wang ES et al. , Nat. Chem. Biol 17, 711–717 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Hartmann MD, Boichenko I, Coles M, Lupas AN, Hernandez Alvarez B, PLOS ONE 10, e0128342 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Matyskiela ME et al. , Nature 535, 252–257 (2016). [DOI] [PubMed] [Google Scholar]
- 13.Matyskiela ME et al. , J. Med. Chem 61, 535–542 (2018). [DOI] [PubMed] [Google Scholar]
- 14.Heim C, Hartmann MD, Acta Crystallogr. D Struct. Biol 78, 290–298 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Kong NR, Liu H, Che J, Jones LH, ACS Med. Chem. Lett 12, 1861–1865 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Bai N et al. , J. Biol. Chem 298, 101653 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Banchenko S et al. , PLOS Pathog. 17, e1009775 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Hansen JD et al. , J. Med. Chem 63, 6648–6676 (2020). [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Atomic models have been deposited to the Protein Data Bank (PDB) under IDs 8CVP, 8D7U, 8D7V, 8D7W, 8D7X, 8D7Y, 8D7Z, 8D80, and 8D81, as described in table S2. Electron microscopy reconstructions have been deposited to the Electron Microscopy Databank (EMDB) under accession codes EMDB-27012, EMDB-27234, EMDB-27235, EMDB-27236, EMDB-27237, EMDB-27238, EMDB-27239, EMDB-27240, EMDB-27241, and EMDB-27242, as described in table S1.