

Abstract
Our previous studies demonstrated increased serum levels of macrophage migration inhibitory factor (MIF-1) and its homologue, MIF-2, in males during MS progression; and that genetically high-MIF-expressing male subjects with relapsing multiple sclerosis (MS) had a significantly greater risk of conversion to progressive MS than lower-MIF-expressing males and females. However, female MS subjects with severe disease expressed higher levels of CD74, the common MIF-1/MIF-2 receptor, on blood cells. In the murine model of MS, experimental autoimmune encephalomyelitis (EAE), both male and female mice lacking MIF-1 and/or MIF-2 were clinically improved during development of moderately severe disease, thus implicating both homologs as co-pathogenic contributors. The current study using MIF-deficient mice with severe acute EAE revealed a highly significant reduction of EAE scores in MIF-1-deficient females, in contrast to only minor and delayed reduction of clinical signs in MIF-1-deficient males. However, clinical EAE scores and factor expression were strongly suppressed in males and further reduced in females after treatment of WT and MIF-1-, MIF-2- and MIF-1/2-DUAL-deficient female and male mice with a MHCII DRα1-MOG-35-55 molecular construct that competitively inhibits MIF-1 & MIF-2 signaling through CD74 as well as T cell activation. These results suggest sex-dependent differences in which the absence of the MIF-1 and/or MIF-2 genotypes may permit stronger compensatory CD74-dependent EAE-inducing responses in males than in females. However, EAE severity in both sexes could still be reduced nearly to background (a “near cure”) with DRα1-MOG-35-55 blockade of compensatory MIF and CD74-dependent factors known to attract peripheral inflammatory cells into the spinal cord tissue.
Keywords: Multiple sclerosis (MS); Eperimental autoimmune encephalomyelitis (EAE); Sex differences; Inflammation; Cytokines/chemokines; Central nervous system (CNS); Macrophage migration inhibitory factors (MIF-1 & MIF-2); DRα1-MOG-35-55 construct, CD74
1. Introduction
It is now recognized that the immunopathologic processes that drive progression of multiple sclerosis (MS) are influenced by both genetic and environmental factors [1–4]. The predominant genetic effect has been attributed to the major histocompatibility complex (MHC) that encodes polymorphic Class I and Class II proteins that present “self” myelin peptides to activate encephalitogenic CD4+ and CD8+ T cells [3,4]. Other contributing non-polymorphic MHC proteins include the class II invariant chain (CD74, II) and the HLA-DRα chain. CD74 not only chaperones peptide-loaded MHC class II molecules from intracellular compartments to the surface of antigen-presenting cells (APC), but also functions as the cognate receptor for macrophage migration inhibitory factor (MIF or MIF-1) and its homologue MIF-2 (MIF-2 = d-dopachrome tautomerase or D-DT) when expressed on the cell surface [5,6] or secreted in a soluble form [7]. MIF-1 and MIF-2 are pleiotropic innate cytokines that function as key mediators of both acute and chronic inflammatory diseases such as septic shock, rheumatoid arthritis, atherosclerosis, and MS [8–10]. Increased MIF levels have been reported in MS blood and cerebrospinal fluid during clinical relapses [11–13] and also were found in the rim of active MS white matter lesions [14]. Binding of MIF to CD74 on macrophages and monocytes potentiated up-regulation of CD44 and CXCR2, as part of the MIF receptor signaling complex that initiates MAPK activation and increased cell migration and survival [15–18]. MIF-deficient mice exhibited acute signs of experimental autoimmune encephalomyelitis (EAE) but no further progression of clinical disease [19], supporting its role in promoting chronic leukocyte infiltration into the central nervous system (CNS). Moreover, neutralizing anti-MIF antibodies block homing of pathogenic T cells to the CNS, thus reducing EAE severity [20]. These foundational studies provided support for MIF’s role in promoting disease-enhancing cellular infiltration into the CNS. Furthermore, we reported an early increase in expression of the MIF receptor, CD74, on monocytes in mice developing clinical signs of EAE, indicating its possible use as a biomarker for disease induction [21].
Our studies in MS subjects and EAE mice revealed that mechanisms that drive disease progression may differ in males vs. females: males have a marked increase in serum and CNS levels of MIF-1 and MIF-2 that are enhanced in those with high-expression −798 CATT7,-8 MIF alleles, whereas females expressed higher levels of the MIF-1 and MIF-2 receptor, CD74 [22]. To more fully address the role of these factors in the EAE model, we posited that EAE clinical severity and inflammatory cytokine expression would decrease in MIF-1-deficient, MIF-2-deficient and MIF-1/2-DUAL-deficient mice compared to WT mice and that treatment with our CD74 blocker, DRα1-MOG-35-55, would further reduce disease severity [23]. These predictions regarding the role of MIF-1 and MIF-2 were strongly supported by our prior experiments in mice with moderately severe EAE, in which we demonstrated that loss of MIF-1 or MIF-2 resulted in less-severe disease progression compared with WT mice, with the MIF-2-KO and MIF-1/2-DUAL-KO male mice demonstrating delayed onset of disease but both sexes experiencing higher maximal disease severity compared with MIF-1-KO mice [22,23]. However, the results suggested that MIF-1 and MIF-2 do not have entirely redundant effects on EAE, but that both of these factors contribute to disease progression. Our analyses also indicated that mice lacking MIF-2 had reduced migration of inflammatory cells into the CNS, but increased frequencies of activated monocytes and memory T cells in spleen, due perhaps to residual MIF-1-dependent inflammatory responses. Residual MIF-2 activity likely accounts for a similar pattern of responses in MIF-1-KO mice [22]. To this point, Honigman et al. demonstrated that MIF-2, like MIF-1, is widely expressed in the brain and parenchyma of adult mice [24]. Taken together with our observation that both MIF-1 and MIF-2 were expressed in white matter tissue of SPMS subjects, these findings indicate that both MIF homologs have a neuroinflammatory role and similarly affect cell migration during chronic and progressive disease [22]. However, additional studies are needed to more precisely clarify the roles of MIF-1 and MIF-2 in MS and EAE, especially with respect to the mechanistic role of their common receptor CD74, and its associated non-cognate MIF receptors, CXCR2 and CXCR4 [15,16].
We have shown previously that partial (p)MHC class II constructs comprised of the extracellular α1 domain of MHC class II linked covalently to myelin oligodendroglial cell glycoprotein (MOG)-35-55 peptide (e.g. DRα1-MOG-35-55) can bind to cell-expressed CD74 and thereby competitively inhibit MIF binding and signaling [25]. This unique construct can also bind to T cell receptors and can effectively reverse established clinical signs of EAE and promote neuroprotection [26]. In a Phase 1 clinical trial, a prototypic construct, RTL1000, was found to be safe and well tolerated at a single dose ≤ 60 mg in males and females with relapsing remitting-, primary progressive- and secondary progressive-MS [27]. These findings provided a strong rationale for more fully characterizing the therapeutic potential of DRα1-MOG-35-55, which has now been shown to reverse inflammation, promote remyelination and axonal recovery, and limit EAE disease progression [28–30]. Notably, DRα1-MOG-35-55 treatment could also reverse clinical deficits in murine models of stroke, methamphetamine abuse, and traumatic brain injury [31].
In a recently published study [23], we utilized MIF-1-, MIF-2-, and MIF-1/2-DUAL-deficient male mice to quantify the respective contributions of these genotypes to EAE disease severity and the ability of DRα1-MOG-35-55 to reverse the EAE disease process in the presence and absence of MIF-1 and/or MIF-2. We found that the modulating effects of MIF-1 and MIF-2 observed in a previous study in moderately severe EAE were neither additive nor compensatory but were displaced in part by other inflammatory mechanisms that prevailed in mice with severe EAE. Moreover, treatment of male mice with severe EAE using DRα1-mMOG-35-55 was more effective in the absence of MIF-1 than MIF-2, but exceeded the disease-enhancing effects of both factors, thus indicating its ability to target additional disease-enhancing mechanisms beyond MIF-1 and MIF-2.
In the current study, we evaluated disease severity and cytokine expression patterns in female WT, MIF-1-KO, MIF-2-KO and MIF-1/2-DUAL-KO C57BL/6 mice with severe acute EAE and compared these responses to those reported for males. We found that unlike males, vehicle-treated MIF-1-KO female mice had significantly reduced EAE severity and a different cytokine expression pattern, indicating a significant contribution of the MIF-1 genotype to severe acute EAE in females. Moreover, both female and male mice benefitted from treatment with DRa1-MOG-35-55 to reach “near cure” EAE scores, thus providing strong support for use of this new class of treatment for MS in both sexes.
2. Materials and methods
2.1. Mice
C57BL/6J wild type (WT) female mice were obtained from The Jackson Laboratory (Sacramento, CA) at 6–7 weeks of age and used in experiments between 8 and 12 weeks of age. Mice deficient in MIF-1, MIF-2, and MIF-1 & 2, all on the C57BL/6 background, were from Yale University [32,33] and bred and maintained in the Animal Resource Facility at the VA Portland Health Care System. Mice were on a 12-hour light/dark cycle with access to food and water ad libitum. This study was carried out in strict accordance with Federal, NIH, and Institutional guidelines using a protocol approved by the Portland VA Animal Care and Use Committee.
2.2. Induction and treatment of acute severe EAE
Female mice were categorized in eight independent experiments into four genetically distinct subgroups (10–15/group; cf. 13–24/group for the comparator male mice from [23]) consisting of wild-type (WT), MIF-1 knockout (KO), MIF-2-KO and MIF-1/2-DUAL-KO (dual knockout). To induce acute severe EAE, mice were immunized in the flanks at four sites with 200 μl total emulsion containing 200 μg of MOG-35-55 peptide and 400 μg of CFA containing 4 mg/ml of heat-killed Mycobacterium tuberculosis [23]. Mice were also given injections of 75 ng and 200 ng of Pertussis toxin (Ptx) intraperitoneally (i.p.) on days 0 and 2 respectively, relative to immunization. The mice were assessed for signs of EAE according to the following scale: 0 = no signs; 1 = limp tail or mild hind limb weakness; 2 = moderate hind limb weakness or mild ataxia; 3 = moderately severe hind limb weakness; 4 = severe hind limb weakness or mild forelimb weakness or moderate ataxia; 5 = paraplegia with no more than moderate forelimb weakness; 6 = paraplegia with severe forelimb weakness or severe ataxia or moribund condition. Mice were monitored daily for changes in disease score and weight changes and were treated after onset of clinical signs of EAE at a score > 2.0 daily for 5 days with either vehicle (20 mM TRIS/HCl, pH 8.5) or 100 μg DRα1-mMOG-35-55 peptide in vehicle and scored daily until Day 20 post-immunization when they were euthanized for ex vivo analyses. The mean sum of daily scores from each mouse in each group from days 8–20 post-immunization was represented as the cumulative disease index (CDI) for the group (numerical integration of the EAE score curve over the entire experiment to represent total disease load).
2.3. Statistical analysis
For nonparametric comparison between WT C57BL/6J, MIF-1-KO, MIF-2-KO, and DUAL-KO mice, daily EAE response values for each mouse were organized into a time curve for the mouse’s follow-up and preprocessed using the discrete Fourier transform to decorrelate the values obtained on successive days; the sets of curves for each group of mice were then averaged in the frequency domain, and the group mean curves compared using Fan and Lin’s adaptive Neyman test [34]. Mean CDIs were compared for each group vs. WT using Welch’s one-sided t test after augmenting the within-group variances to include the CDI estimation error [35]. P-values are reported numerically as calculated and were not subjected to significance thresholds, but values < 0.05, <0.01, or < 0.001 are noted on figures. Spans of time where KO- or treatment-group curves are confidently separated from the WT curve were identified using a Bayesian approach based on noninformative reference priors [36]. Specifically, all groups at all time points were evaluated in a global two-way model of mean EAE daily scores at each unique group-time point combination, allowing for different residual variance of each mean [i.e. a fully interacted model of both means and variances, where all mean parameters were based on the same normal reference prior distribution (prior mean = 0, prior standard deviation = 10) and all variance parameters were based on the same inverse gamma reference prior distribution (both prior shape parameters = 1/100)]. Adaptive Markov-chain Monte Carlo (MCMC) using random-walk Metropolis-Hastings sampling was performed for a single run of 100,000 posterior samples after a burn-in period of 25,000 (discarded) samples. Convergence of MCMC was visually verified using trace plots. Based on the posterior samples, we estimated the probability that a non-WT curve was separated from the WT curve jointly across some contiguous or noncontiguous span of time for each non-WT curve and every visually coherent set of time points. (For example, for Fig. 1 we examined day spans 11–20, 11–15 paired with 18–20, and so on for many different visually plausible combinations). Spans where the posterior probability was >95% that the curves being compared were separated across the entire span were noted on the figures. All analyses were conducted using Stata (version 16.1, StataCorp, College Station, TX).
Fig. 1. Depletion of MIF-1 but not MIF-2, or both MIF-1&2 in female mice with severe acute EAE resulted in significantly reduced clinical scores. Additionally, all groups, especially MIF-1-KO mice, were treated successfully with DRα1-MOG-35-55.
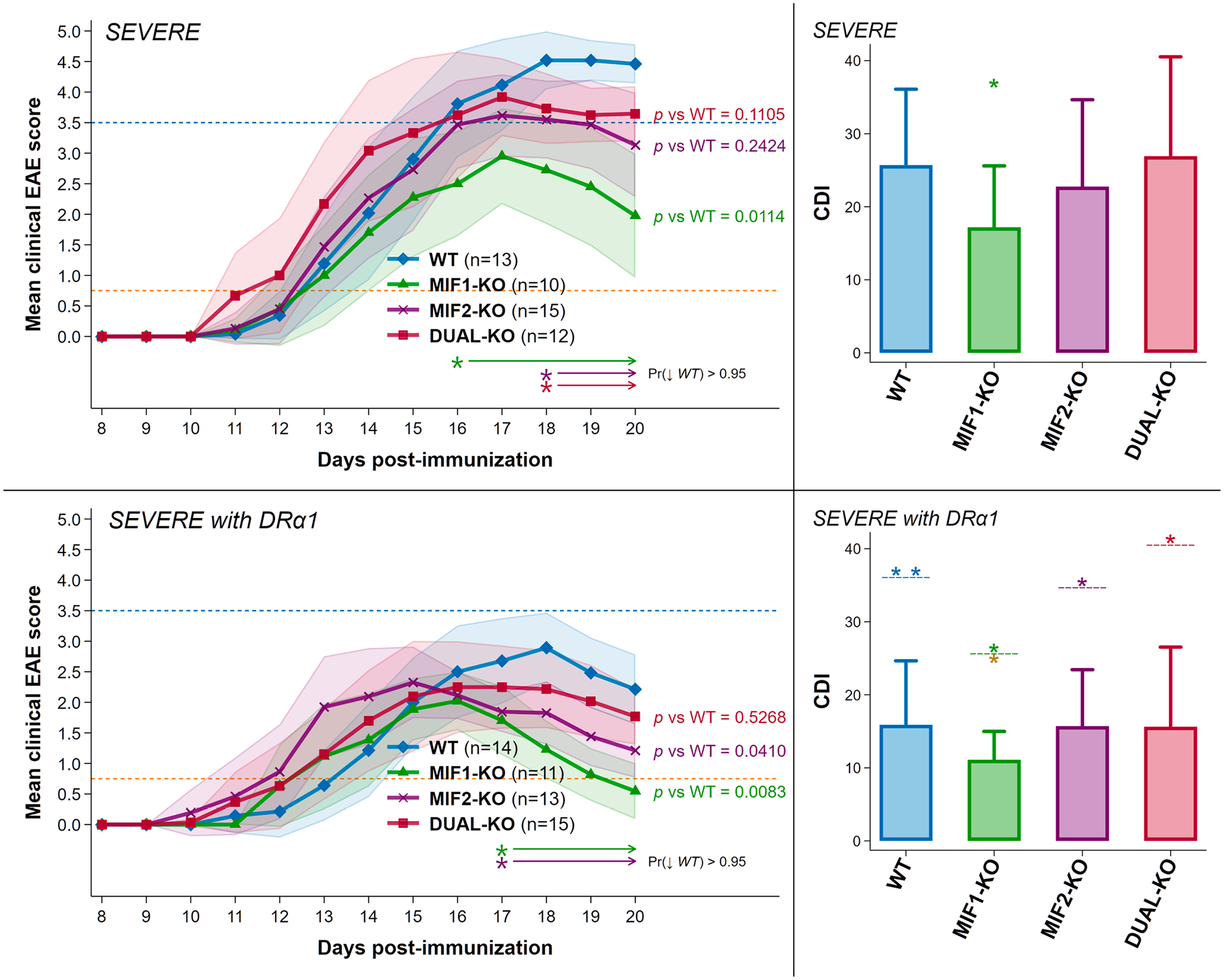
Upper Panels: Depletion of MIF-1 significantly reduced the mean final daily EAE score from 4.5 in WT to 2.0 in MIF-1-KO mice and the cumulative disease index (CDI) score from 26 in WT mice to 17 in MIF-1-KO mice with nominal changes in MIF-2-KO and DUAL-KO mice. Lower panels: Treatment of severe EAE with 5 daily doses of 100 μg DRα1-MOG-35-55 further reduced mean final daily scores for all groups to average (and even maximum) scores < 3.0 and for MIF-1-KO mice to ~ 0.75 (see blue and orange reference lines at EAE scores of 3.5 and 0.75) and EAE CDI scores (WT = 16; MIF-1-KO = 11; MIF-2-KO = 16; DUAL-KO = 16). The matched-color arrow-tipped lines underneath each set of curves [Pr (↓WT)] indicate spans of time where the joint probability that the corresponding curves lie strictly below the WT curve for all indicated spans exceeds 95% according to a Bayesian calculation based on noninformative reference priors. In CDI plots: *p ≤ 0.05 **p ≤ 0.01 for CDI scores in all WT vs. non-WT; and in all DRα1-mMOG-35-55 vs. group-coded Vehicle-treated groups (above dashed line); and for DRα1-MOG-35-55-treated MIF-1-KO mice vs. all other treated groups (below dashed line for MIF-1-KO CDI).
2.4. RNA expression normalization and analysis
Stata software was used to interrogate the Ct values reported from the per-protocol RT-PCR analysis of the Qiagen PAMM-022ZA array used to measure chemokine expression in the pooled murine spinal cord tissue samples from 3 to 5 representative mice from each experimental group. Data were monitored thoroughly for artifacts and subjected to extensive quality-control (QC) rules based on the performance of control probes on the array to monitor genomic DNA contamination, PCR efficiency, and RT failures. All QC go/no-go criteria recommended by Qiagen [37] were met for each array card. The profile plots showed very strong amplification curves for each of the 84 chemokines on the panel, with no evidence of genomic DNA contamination. After evaluating amplification performance and counts, we excluded 4 chemokines from further analysis for having variant or too-low expression, including Ccl20, Ccr1l1, Cmtm2a, and Cxcr1. Thus, we retained data from 80 chemokines for the study.
3. Results
3.1. Disease-enhancing effects of MIF-1 and MIF-2 in female mice with severe acute EAE
To induce severe EAE, female WT, MIF-1-KO, MIF-2-KO and MIF-1/2-DUAL-KO mice were immunized with an adjuvant mixture that included 200 μg mMOG-35-55 peptide emulsified in CFA containing 400 μg heat-killed Mtb followed by Ptx on Days 0 and 2, as in previous studies [23,38]. Disease-enhancing effects of MIF-1 and MIF-2 are quantified as the reduction in EAE severity scores in mice lacking the MIF-1 gene, the MIF-2 gene, or both MIF-1 and MIF-2 genes compared to WT mice with EAE that express both the MIF-1 and MIF-2 genes. As shown in Fig. 1 Upper Panels and Table S1, WT female mice developed a severe clinical score of ~ 4.5 with an EAE cumulative disease index (CDI) score of ~ 26 by Day 20 after disease induction. Comparatively, the MIF-1-KO mice developed a lower clinical score of ~ 2.0 over the same time period with a significantly lower CDI of ~ 17 vs. WT mice (p = 0.02), thus indicating that MIF-1 contributed ~ 35% of the total EAE disease load. The disease-enhancing effect of MIF-2 in females with severe EAE was weaker, with MIF-2-KO mice reaching a score of ~ 3.0 and a CDI of ~ 23 representing a non-significant overall contribution (p = 0.25) of ~ 12% of total disease load but with a downward trend over Days 18–20 [Pr(↓ WT) > 0.95]. Similarly, the MIF-1/2-DUAL-KO mice reached a clinical score of ~ 3.7 and a CDI of ~ 27, representing a non-significant contribution to the female EAE CDI score (p = 0.60), also despite a late downward trend over days 18–20 [Pr(↓ WT) > 0.95]. The pronounced disease-enhancing effect of MIF-1 on severe EAE in females differed from the limited MIF-1 contribution in males in which MIF-1-KO mice did not have a reduced CDI vs. WT mice (~30 in both groups, p = 0.44, Table S1). However, there was a significant downward trend on Days 18–20 [Pr(↓ WT) > 0.95] as shown in Supplementary Figure S1 Upper Panels, published previously [23] and included here for comparison. Also shown in Figure S1 is the lack of an effect on CDIs in the male MIF-2-KO and MIF-1/2-DUAL-KO mice, and again a downward trend in the DUAL-KO males but not MIF-2-KO males over Days 18–20 of disease. These data indicate a significantly stronger contribution to EAE severity of MIF-1 in females vs. males with severe acute EAE. A summary of the statistical analyses of clinical changes in EAE scores and curve shapes for females and males is provided in Supplementary Table S1.
3.2. Treatment effects of DRα1-MOG-35-55 in WT and MIF-KO female mice with severe EAE
We have previously demonstrated the ability of DRα1-MOG-35-55, a molecular construct designed to competitively block the MIF-1 and MIF-2 receptor, CD74, to reverse severity of EAE in WT male and female mice with EAE [22]. The differing contributions of MIF-1 and MIF-2 to EAE severity in females vs. males raised the key question as to whether treatment with DRα1-MOG-35-55 would be more or less effective in the absence of MIF-1, MIF-2, or both homologues. As shown in Fig. 1 Lower Panels and Table S1, treatment of WT and all MIF-deficient females with DRα1-MOG-35-55 significantly reduced EAE CDI scores for all groups to daily scores of < 3.0 and CDI scores to ≤ 20. Notably, treatment with DRα1-MOG-35-55 of MIF-1-KO mice reduced the final daily group score to < 0.6 and the CDI score to ~ 11.0, a striking ~ 57% total reduction vs. untreated WT female mice, a ~ 35% reduction vs. untreated MIF-1-KO mice, and a ~ 30% reduction vs. DRα1-MOG-35-55-treated WT mice. The MIF-2-KO and the DUAL-KO female mice with EAE also benefitted from DRα1-MOG-35-55 treatment, with CDI scores (~16, 39% reduction vs. untreated WT) similar to that of treated WT females (Fig. 1 Lower Panels). EAE disease curves for the DRα1-MOG-35-55-treated groups appeared to be similar in females vs. males, with the rank order of best- to least-treated groups being MIF-1-KO > MIF-2-KO = MIF-1/2-DUAL-KO = WT groups (Figures 1 & S1 Lower Panels). Overall, treatment of females vs. males with DRα1-MOG-35-55 was very similar in MIF-1-KO mice (~57% reduction of EAE CDI in females vs. ~ 60% reduction in males compared to corresponding untreated WT), but somewhat better in female vs. male WT, MIF-2-KO, and DUAL-KO groups (~39% reduction in EAE CDI in females vs. ~ 33% reduction in males, respectively, with essentially similar reductions seen in all non-MIF-1-KO groups).
3.3. Quantitative assessment of inhibitory effects of MIF-1, MIF-2, and MIF-1&2 deletion as well as DRα1-MOG-35-55 treatment on expression of EAE- and adjuvant-associated factors in female mice with severe acute EAE
3.3.1. EAE-associated factor expression
In a previous study, we identified an array of cytokines, chemokines and cell-associated receptors that were increased significantly in spinal cord tissue during the course of severe acute EAE in males vs. females [38]. This array included subsets of factors that were EAE associated, those associated with adjuvant effects (CFA + Ptx), and a few that were associated with both EAE and adjuvant effects. Data from that study constituted a baseline comparator for assessing the degree of reduction of expression for each factor in Vehicle-treated and DRα1-MOG-35-55-treated WT, MIF-1-KO, MIF-2-KO, and MIF-1/2-DUAL-KO mice with severe acute EAE. Results from female mice in this study are highlighted and contrasted with a comparable data set from males reported earlier [23].
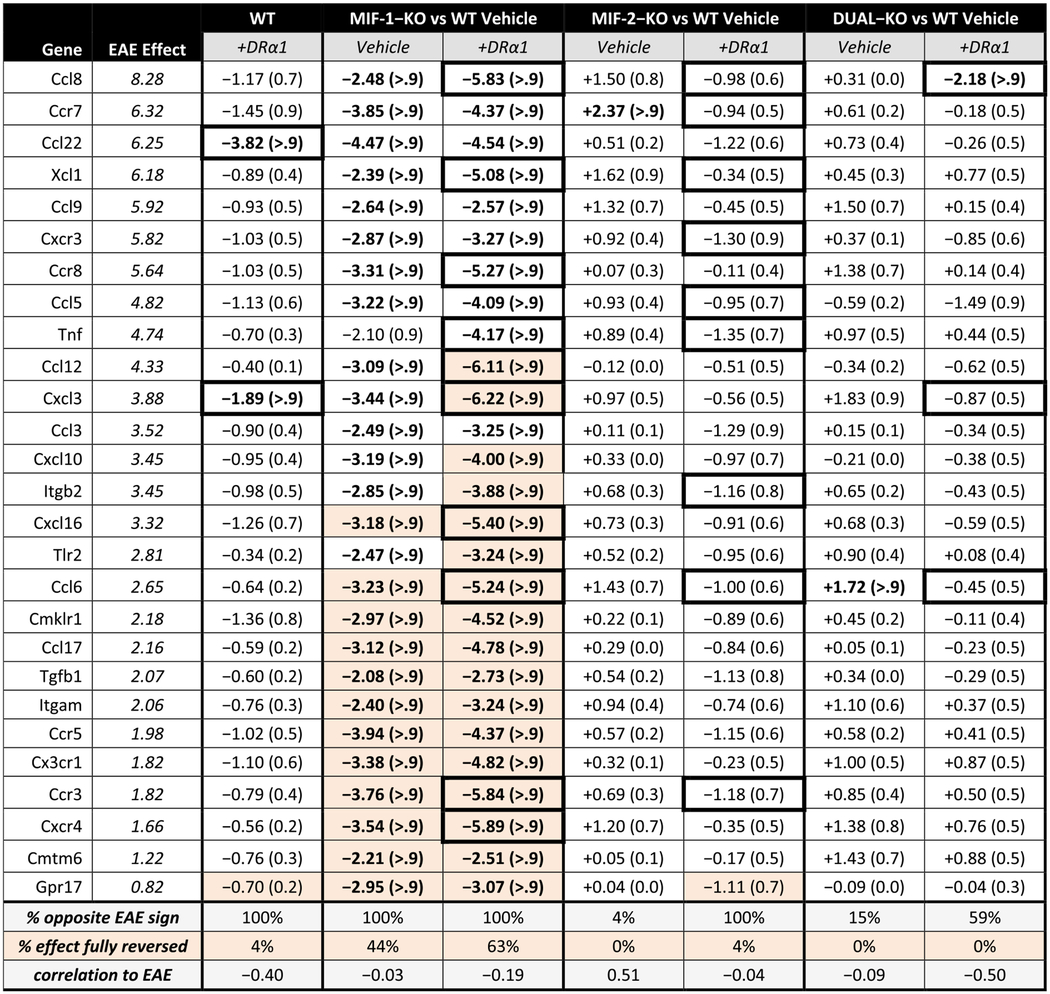
Results from females showed major reductions in expression from baseline values of 27 EAE-associated factors (4 unique for females) mainly associated with the MIF-1-KO genotype, with reductions in expression also observed in WT and MIF-1-KO mice after treatment with DRα1-MOG-35-55. As shown in Table 1 (two far left columns), the EAE effect magnitude (ΔCT scores in log2 units) in WT female mice with severe EAE (compared with baseline ΔCT scores from EAE-naïve healthy female mice) obtained in the previous baseline study [38] ranged in descending order from + 8.28 for Ccl8 to + 0.82 for Gpr17 (illustrated in Fig. 2). The profiles of all unique pairwise expression ratios observed under this EAE effect in the previous baseline study had a rank correlation of 0.94 with the profiles of the same expression ratios obtained for Vehicle-treated WT female EAE mice in the current study, strongly supporting the reliability of measurements of the observed chemokine expression patterns under EAE induction, and thus the basic comparability of the ΔCT scores obtained in each experiment. The third column from the left in Table 1 shows ΔCT score changes after treatment of the female WT EAE mice with DRα1-MOG-35-55. As indicated in the shaded boxes at the bottom of the table, treatment with DRα1-MOG-35-55 changed the expression pattern from 100% positive ΔCT scores to 100% negative ΔCT scores in WT female EAE mice, indicating across-the-board inhibitory effects of the treatment, with −0.4 correlation of ΔCT treatment magnitudes compared with the rank-order sorted EAE effect magnitudes. The value in parentheses after each ΔCT value is the Bayesian posterior probability that the estimated change is > 1 in magnitude (ΔCT units). According to this analysis, changes observed for Ccl22 (−3.82 ΔCT units) and Cxcl3 (−1.89 ΔCT units) were the only 2 EAE-associated factors with a probability > 0.9 that the observed changes from treatment were > 1 ΔCT unit in WT female EAE mice. These results suggest that the level of inhibition induced by DRα1-MOG-35-55 treatment on the expression of this set of factors in these mice was consistent but relatively minor, with the exception of only Ccl22 and Cxcl3 being most likely to be inhibited by at least 1 ΔCT unit. However, essentially none of the EAE-associated factor expression levels were fully corrected back to levels found in naïve healthy mice (highlighted boxes in Table 1). These changes in ΔCT scores in DRα1-MOG-35-55-treated female WT mice (2/27 = 7% of factors with > 0.9 probability of reduced expression > 1 ΔCT unit) were far lower than in DRα1-MOG-35-55-treated male WT mice (30/36 = 83% of factors with > 0.9 probability of reduced expression > 1 ΔCT unit, with 6/36 = 17% of factors fully corrected; Table S2 and Figure S2).
Table 1. Female EAE-associated factors.
Changes in expression of female EAE-associated factors for DRα1-MOG-35-55-treated WT and each knockout genotype with and without DRα1-MOG-35-55 treatment, compared to untreated WT mice. All groups are female mice with the severe EAE immunization regimen. Columns are sorted in descending order of EAE effect magnitude, representing the change in expression caused by inducing EAE in similar mice in a previous experiment [38]. The EAE effect estimates are listed in italics for reference; an experimental effect of opposite sign and equal or greater magnitude would indicate reversal of the effect of EAE induction on the factor. Under each column are listed percentage of expression changes that are in the opposite direction to the EAE effect and the correlation of change magnitudes to the EAE effect magnitudes. Expression changes are represented by the ΔCT score in log2 units, and Bayesian posterior probabilities that the estimated change is greater than 1 in magnitude appear in parentheses; entries with posterior probability >90% are bolded. Cells that show complete reversal (to within −0.1) of the EAE effect are highlighted, and cells where DRα1-MOG-35-55 treatment boosts suppression by ~2 or better are noted by thicker borders.
|
Fig. 2. Heatmap of female EAE-associated factors.
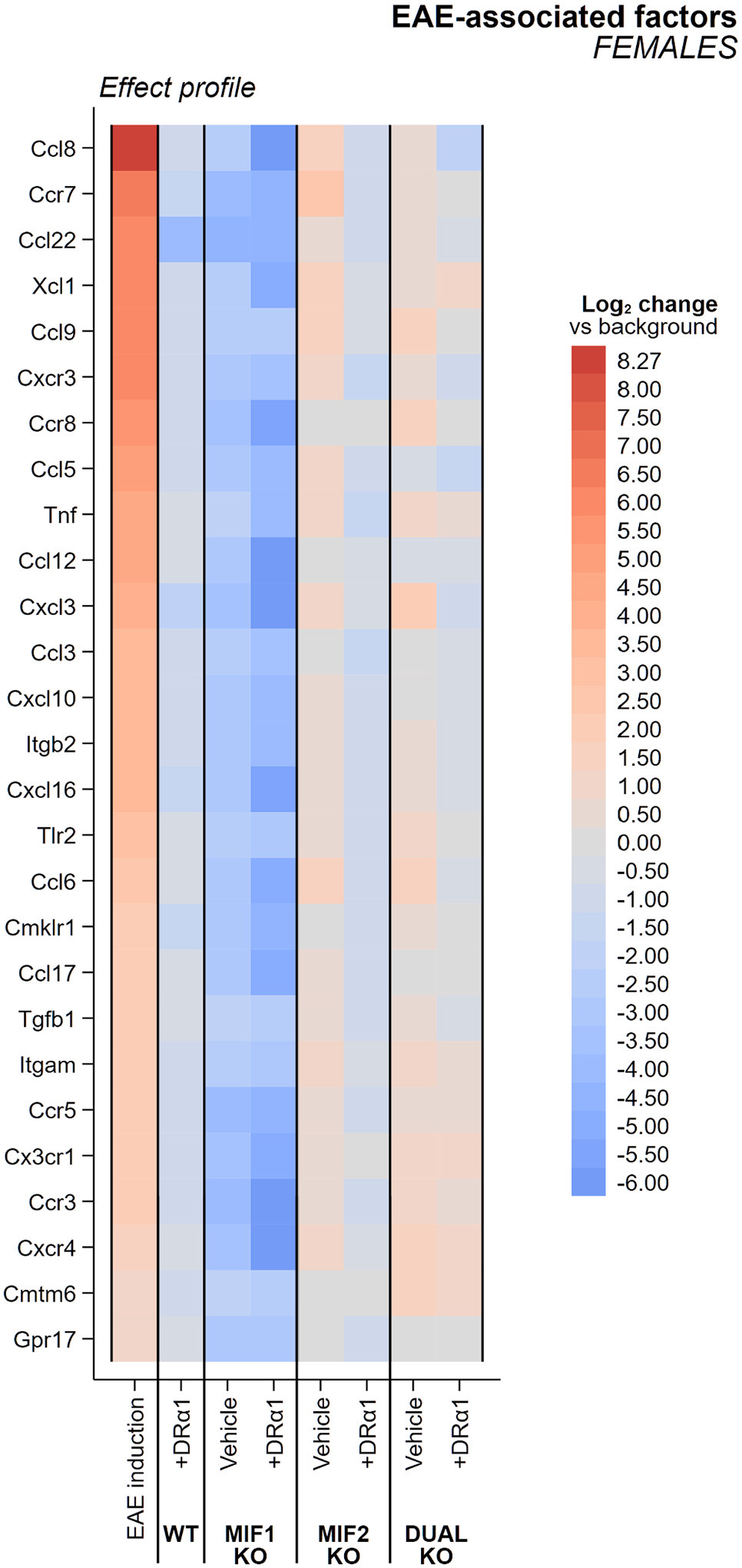
Changes in expression of female EAE-associated factors for WT and each knockout genotype with and without DRα1-MOG-35-55 treatment are shown in comparison to untreated WT mice. All groups are female mice with the severe EAE immunization regimen. Columns are sorted in descending order of EAE effect magnitude, representing the change in factor expression vs. that in disease-naïve mice evaluated in a previous experiment [38]. An experimental effect of opposing color and equal or greater intensity would indicate reversal of the effect of EAE induction on the factor.
Deletion of the MIF-1 gene had a much greater inhibitory effect on EAE-associated gene expression, again changing 100% of factors from positive to negative values, but with 26/27 = 96% of factors having a probability > 0.9 of reduced expression > 1 ΔCT unit, and full correction of 12/27 factors (44%) (Table 1 and Fig. 2). However, the strongest effect in all of the genotype female groups was DRα1-MOG-35-55 treatment of the MIF-1-KO mice, with 100% of factors with > 0.9 probability of reduced expression > 1 ΔCT units and 17/27 factors (63%) fully corrected (Table 1 and Fig. 2). Unlike MIF-1 deletion, deletion of MIF-2 or both MIF-1 & 2 genes in Vehicle-treated females had essentially no effects on factor expression, showing changes from positive to negative values in only 4–15% of EAE-associated genes, none having a probability > 0.9 of reduced expression > 1 ΔCT unit (1/27 of factors in each case even showing significant increase in expression relative to Vehicle-treated WT), and no factors that were fully corrected (Table 1 and Fig. 2). Moreover, DRα1-MOG-35-55 treatment of MIF-2-KO mice and MIF-1/2-DUAL-KO mice with DRα1-MOG-35-55 had only weak effects, changing expression levels from positive to negative in 59–100% of the factors but with only 1/27 factors (4%) having a probability > 0.9 of reduced expression > 1 ΔCT unit (in DUAL-KO only) and only 1/27 factors (in MIF-2-KO only) that were fully corrected (Table 1 and Fig. 2). (Note in addition that the sole fully-corrected factor in DRα1-MOG-35-55-treated MIF-2-KO females, Gpr17, was the factor least impacted by EAE induction.) More generally, both MIF genotype and DRα1-MOG-35-55 treatment effects on factor expression were less pronounced in females than males, which had a much higher percentage of factors having a probability > 0.9 of reduced expression > 1 ΔCT unit, and also higher percentage of factors that were fully corrected after treatment with Vehicle or DRα1-MOG-35-55 (Table S2 and Figure S2). It should be noted for both sexes that none of the genes with lowered levels of expression in Vehicle-treated MIF-2-KO or MIF-1/2-DUAL-KO mice exceeded the lowering of their expression in Vehicle-treated MIF-1-KO mice, suggesting that there were no unique inflammatory factors in our arrays that were associated with lack of MIF-2 or MIF-1/2 expression. Moreover in both sexes, the degree of changes in factor expression generally correlated respectively with the pattern of clinical changes shown in Fig. 1 and Figure S1 above, with MIF-1 being the dominant EAE-enhancing factor and only a subset of factors being further inhibited after DRα1-MOG-35-55 treatment.
3.3.2. Factor expression due to adjuvant (CFA + Ptx) effects
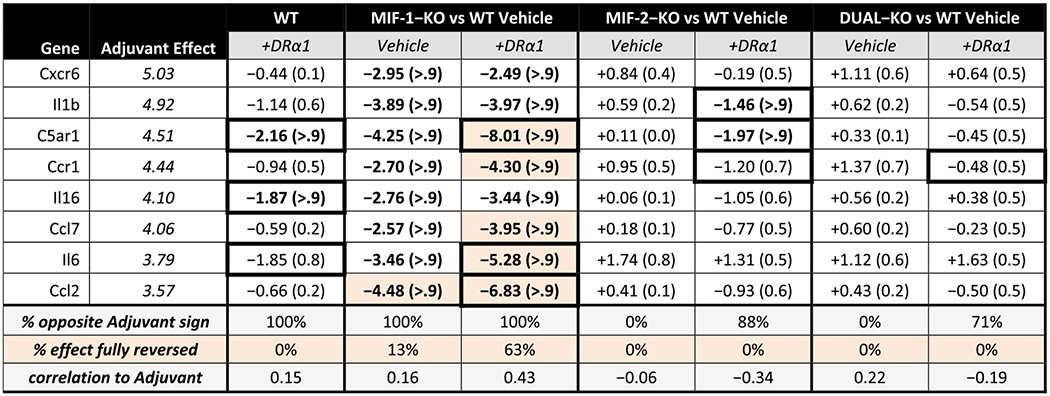
In our baseline study [38], we identified the 11 most reactive adjuvant-associated genes as those showing a significant contribution of CFA + Ptx to the EAE effect [38]. These genes included Cxcr6, Il1b & Il6 in both sexes, C5ar1, Ccr1, Il16, Ccl7 & Ccl2 in females only and Xcl1, Cxcl13 & Ccr2 in males only, resulting in a total of 8 such genes in females and 6 such genes in males. These significant adjuvant-associated genes were defined as those where the posterior probabilities that ΔEAE > 0.5 Ct and ΔCFA > 2 Ct (both strictly positive) are each at least 2/3 (i.e. minimum 2:1 odds in favor of the adjuvant effect being large, positive, and contributory to a probably positive EAE effect). Changes in expression of the above-listed adjuvant effect genes are shown for females (Table 2 and Fig. 3) and compared with males (Table S3 and Figure S3) by strain and treatment.
Table 2. Female adjuvant-associated factors.
Changes in expression of adjuvant (CFA + Ptx)-associated factors for DRα1-MOG-35-55-treated WT and each knockout genotype with and without DRα1-MOG-35-55 treatment, compared to untreated WT mice. All groups are female mice with the severe EAE immunization regimen. Columns are sorted in descending order of adjuvant effect magnitude, representing the change in expression caused by priming similar mice in a previous experiment [38] with CFA + Ptx alone that induced cellular infiltration into the spinal cord but no demyelination or clinical signs of EAE. The adjuvant effect estimates are listed in italics for reference; an experimental effect of opposite sign and equal or greater magnitude would indicate reversal of the effect of adjuvant immunization on the factor. Under each column are listed percentage of expression changes that are in the opposite direction to the adjuvant effect and the correlation of change magnitudes to the adjuvant effect magnitudes. Expression changes are represented by the ΔCT score in log2 units, and Bayesian posterior probabilities that the estimated change is greater than 1 in magnitude appear in parentheses; entries with posterior probability >90% are bolded. Cells that show complete reversal (to within −0.1) of the adjuvant effect are highlighted, and cells where DRα1-MOG-35-55 treatment boosts suppression by ~2 or better are noted by thicker borders.
|
Fig. 3. Heatmap of female adjuvant-associated factors.
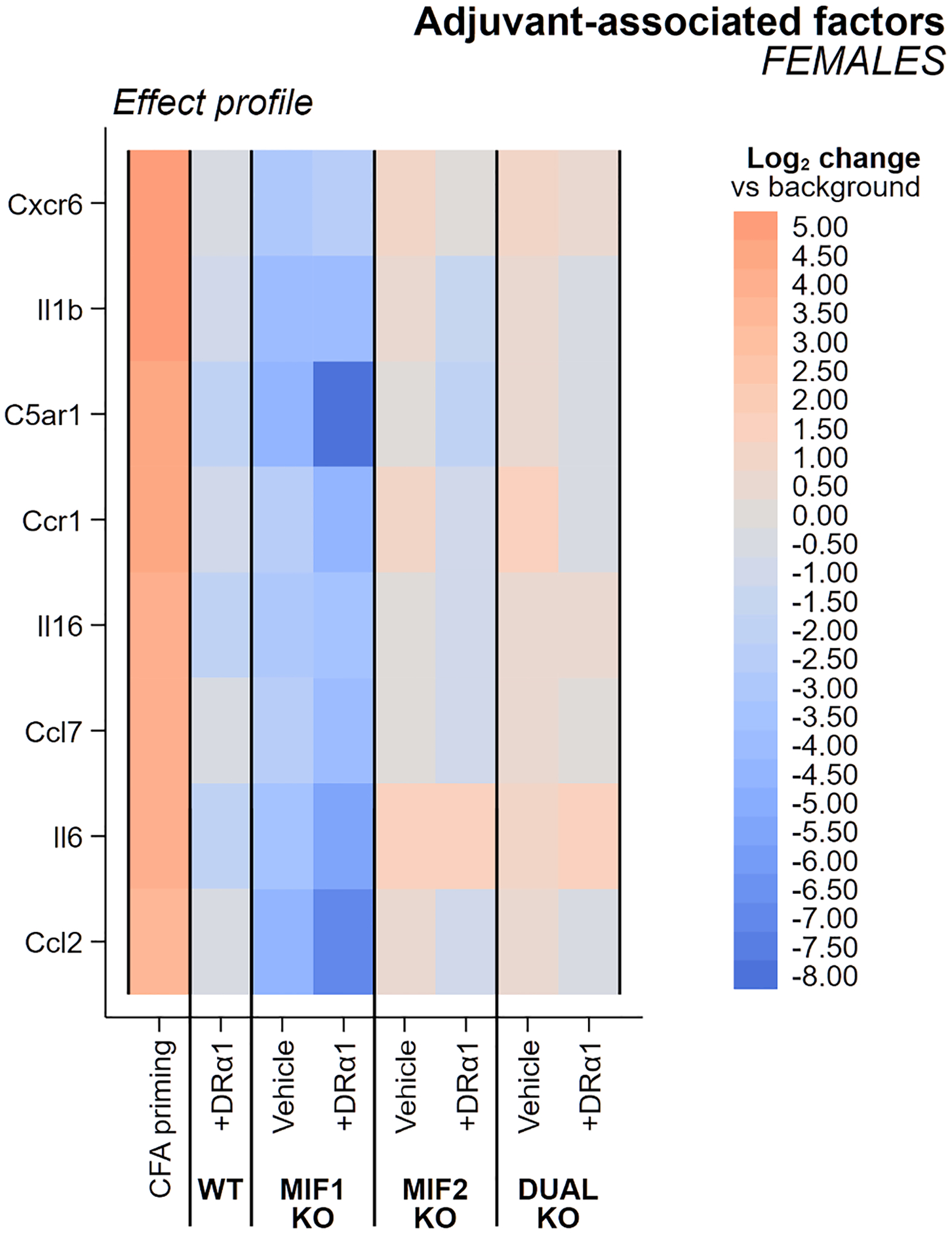
Changes in expression of adjuvant-associated factors (CFA + Ptx) in WT and each and knockout genotype with and without DRα1-MOG-35-55 treatment are shown in comparison to untreated WT mice. All groups are female mice with the severe EAE immunization regimen. Columns are sorted in descending order of adjuvant effect magnitude, representing the change in factor expression seen in CFA + Ptx adjuvant-immunized mice evaluated in a previous study [38] that exhibited cellular infiltration into the spinal cord but not demyelination or any clinical signs of EAE. An experimental effect of opposing color and equal or greater intensity would indicate reversal of the effect of adjuvant immunization on the factor.
The EAE female baseline adjuvant-associated ΔCT scores (compared to naïve WT females) ranged from + 5.03 for Cxcr6 to + 3.57 for Ccl2, values somewhat higher than for EAE males with baseline adjuvant-associated ΔCT scores ranging from + 4.46 for Cxcr6 to + 2.61 for Ccr2. However, although treatment with DRα1-MOG-35-55 further reduced nearly all adjuvant-associated factor expression values in Vehicle-treated WT mice of both sexes, the effects were less pronounced in females than males (2/8 vs. 4/6 factors with > 0.9 probability of reduced expression > 1 and 0/8 vs 2/6 factors with fully corrected ΔCT scores). Generally, deletion of the MIF-1 genotype had the greatest inhibitory effects on expression of adjuvant-associated genes in Vehicle-treated EAE females and males, with 100% of factors changing from positive to negative ΔCT scores in both sexes, 8/8 (females) vs. 6/6 (males) factors with > 0.9 probability of reduced expression > 1, and 1/8 vs. 5/6 factors with fully corrected ΔCT scores. Moreover, DRα1-MOG-35-55 treatment also differed between MIF-1-KO females and males, respectively with 5/8 vs. 5/6 factors with fully corrected ΔCT scores (and the less-than-fully-corrected factor in DRα1-MOG-35-55-treated MIF-1-KO males, Cxcr6, was nearly there at −4.24 vs. the adjuvant effect of + 4.46).
In contrast to the moderate factor inhibition in Vehicle- and DRα1-MOG-35-55-treated female MIF-1-KO mice, Vehicle-treated MIF-2- and MIF-1/2-deleted female mice had 0/8 factors with significantly reduced ΔCT scores and DRα1-MOG-35-55 treatment only nominally reduced ΔCT scores for 2/8 factors in MIF-2- and 0/8 factors in MIF-1/2-deleted mice. This lack of effect of MIF-2 or MIF-1/2 deletions on adjuvant factor expression in females was markedly different than in comparably treated males. In males, the lack of MIF-2 or both MIF-1 & 2 respectively reduced expression of 4/6 and 5/6 adjuvant associated factors with > 0.9 probability of reduced expression > 1 in Vehicle-treated mice, but with little further reductions after treatment with DRα1-MOG-35-55. It is noteworthy that MIF-1 deletion (but not MIF-2 or MIF-1 & 2 deletions) in females reduced the adjuvant ΔCT scores beyond that of DRα1-MOG-35-55-treated WT mice and that treatment of the MIF-2-KO and MIF-1/2-DUAL-KO groups with DRα1-MOG-35-55 had little further impact (± ~1 ΔCT unit) on adjuvant-associated gene expression. The changes induced by DRα1-MOG-35-55 beyond genotype effects can be observed in Fig. 4 for females and Figure S4 for males.
Fig. 4. Heatmap of DRα1 effects in females.
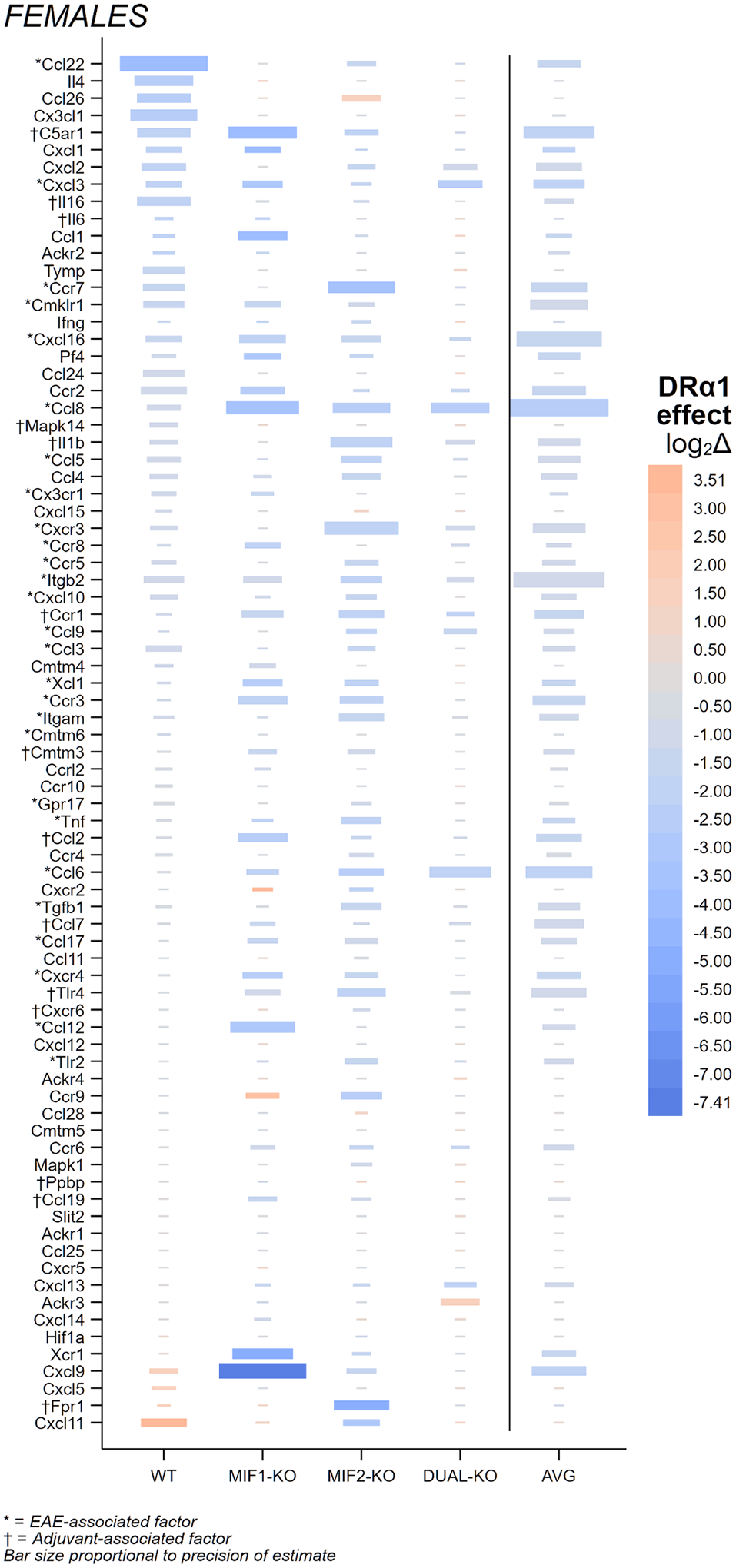
Changes attributable to DRα1-MOG-35-55 treatment in female mice are shown as colored bars with size of the bar proportional to the log posterior Bayesian odds (i.e. the log Bayes factor) in favor of the hypothesis that the treatment effect is >1 in magnitude; larger bars reflect higher confidence that the intensity of the color (representing the effect size) is trustworthy. Each main column of the plot presents a genotype used in the study, and the far right (‘AVG’) column (separated from the others by a thin black line) is the average size of effect over all the genotypes; deeper intensity of color in this column reflects greater consistency of effect direction across genotypes. Names of EAE-associated factors as identified previously [38] in females are marked with an asterisk (*), names of adjuvant-associated factors in females are marked with a dagger (†), and all unmarked names correspond to factors not judged to be associated with EAE induction in females.
3.3.3. Summary of genotype and DRα1-MOG-35-55 treatment on factor expression
The distribution and overlap of numbers in subgroups of these factors and their sensitivity to interventions considered in this study (i.e. MIF-1 deletion and DRα1-MOG-35-55 treatment) are illustrated in a Venn diagram for females vs. males (Fig. 5), color-coded by association intersection region and intervention sufficient for suppression (DRα1-MOG-35-55 treatment in WT vs. MIF-1-KO genotype vs. both), listing total counts and names of EAE- and adjuvant-associated factors that were shown to be important for EAE in our previous study [38] and observed to be suppressed under one of these conditions in the current study. The top panel shows the divisions of counts along each axis, and the bottom panel provides annotation of gene names for each region of the diagram, organized by color. (Note that a small number of genes appear twice, in separate cells of the Venn diagram, once as an adjuvant effect for one sex and once as an EAE effect for the other sex. An example is C5ar1, which has a significant effect in EAE males but not EAE females, and a significant effect in adjuvant-treated females but not adjuvant-treated males.) A noteworthy takeaway from this presentation is that suppression of these factors in EAE mice appears to happen more easily for males, where DRα1-MOG-35-55 treatment alone was usually sufficient (less so for male-specific adjuvant-associated factors); in contrast, our female EAE mice minimally required absence (or would mere aggressive reduction have been enough?) of MIF-1 to reduce activity of most of these factors. Note also that all of the important factors for males are successfully suppressed in the MIF-1-KO genotype without need for DRα1-MOG-35-55, so either intervention would appear to succeed in males, whereas females may require at least very aggressive MIF-1 suppression to reduce most factors, and in one case (Tnf) appear to need even more suppression, as observed with DRα1-MOG-35-55 treatment.
Fig. 5. Female and male factor summary Venn diagram.
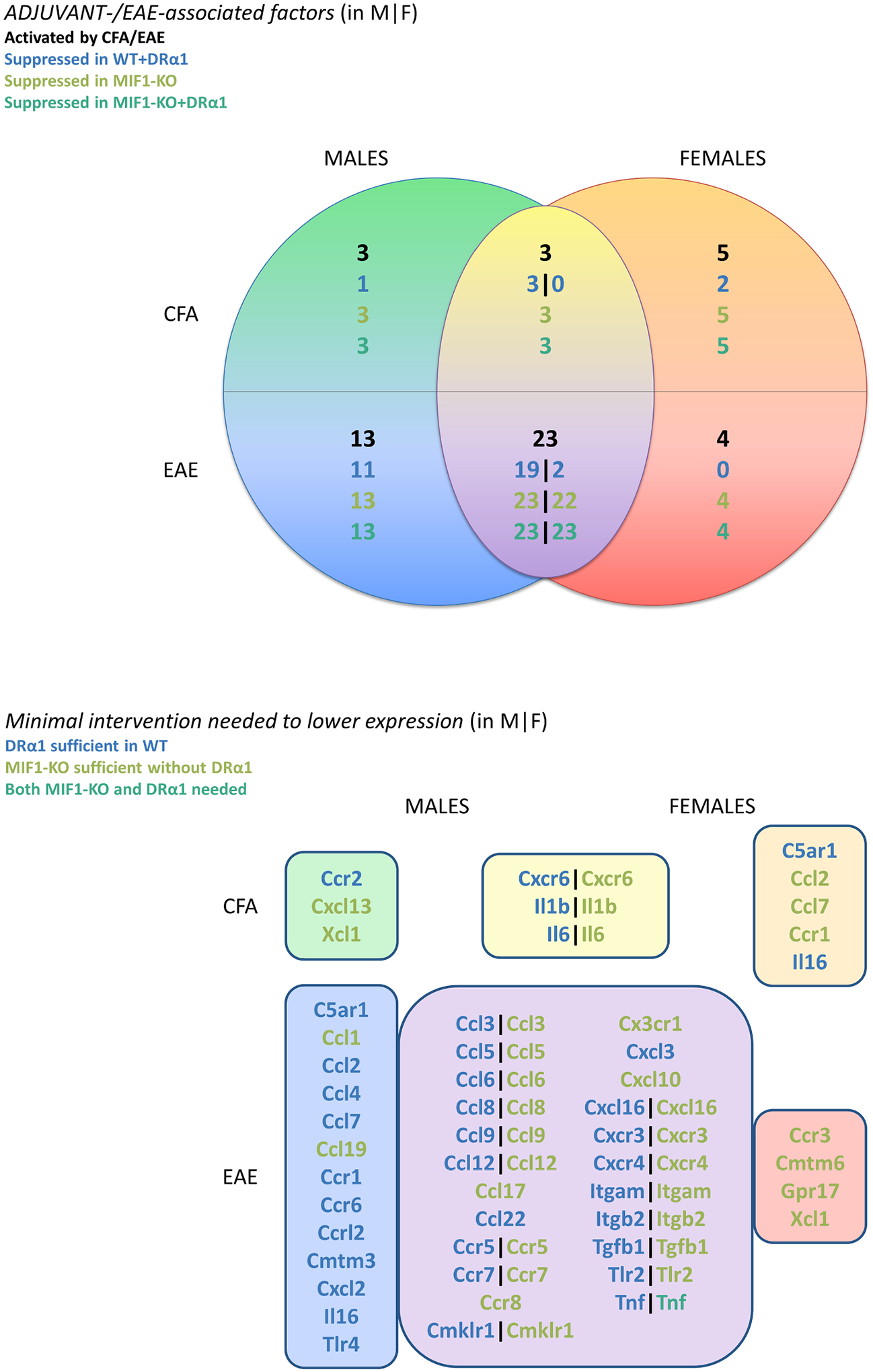
This Venn diagram of significant EAE- and adjuvant-associated genes by sex recapitulates a figure presented in our previous study (Fig. 6 from [38]) showing the large overlap in genes important for EAE (and a few associated with adjuvant effects, albeit with little overlap between sexes), with additional annotation summarizing how many and which of the genes in each region were observed in the current study to be suppressed in expression by minimally (in order): treatment of the corresponding sex of WT mice by DRα1-MOG-35-55, deletion of MIF-1 in the corresponding sex, or requiring both MIF-1 deletion and DRα1-MOG-35-55 to achieve reliable suppression. The determination of suppression or not was decided by whether the gene showed sex-specific reduction of>1 ΔCT (log2) unit in magnitude with Bayesian posterior probability of > 90% (i.e. these are the genes appearing in bold in Tables 1 and 2 for females, and in Tables S2 and S3 for males). The diagram divides the significant genes along two axes: sex and whether the gene is primarily associated with adjuvant or EAE effects. The top panel shows the divisions of total counts for each region (numerals in black), followed by sub-total counts for genes that: can be suppressed by DRα1-MOG-35-55 treatment in WT mice (blue); or failing that, can be suppressed by MIF-1 deletion alone (olive); or failing that, can be suppressed by DRα1-MOG-35-55 treatment in MIF-1-KO mice (teal). (Note that none of the genes were unaffected both by treatment and MIF-1 deletion, so all fell into one of these classes.) Where the counts in regions of male-female intersection are split by a vertical bar (|), the left-hand count is for males and the right-hand count for females. The bottom panel of the figure provides annotation of gene names for each cell of the Venn diagram in the top panel, with the containing boxes color-coded to match the corresponding regions in the Venn diagram. Note that a few genes appear in two cells because their role with respect to adjuvant and EAE effects appears to differ by sex (e.g. C5ar1, which is associated with adjuvant and not EAE in females only and associated with EAE and not adjuvant in males only). Color-coding of gene names in the bottom panel follows the same convention as the counts in the top panel, indicating which genes can be suppressed in the specific sex by DRα1-MOG-35-55 in WT, by MIF-1 deletion (with no need for DRα1-MOG-35-55 treatment), or that required both deletion of MIF-1 and treatment with DRα1-MOG-35-55.
Figures S5 (females) and S6 (males) further illustrate fold changes in terms of the overall distribution of effects of genotype and treatment and their sum on the entire set of EAE- and adjuvant-associated factors compared to baseline expression in Vehicle-treated WT mice with EAE. For females, the MIF-1-KO peak effect is dominantly suppressive, whereas MIF-2-KO and DUAL-KO genotypes tend to promote factor expression. The DRα1-MOG-35-55 treatment effect is mostly suppressive and further potentiates suppression especially in the MIF-1-KO female mice. In males, the patterns show markedly increased suppression for all genotypes and higher fold changes for MIF-2-KO and DUAL-KO mice treated with DRα1-MOG-35-55 compared with female patterns. Similar patterns are evident when expressing the total (aggregated) mean expression changes caused by DRα1-MOG-35-55 as a fraction of total mean log2 expression of all EAE- and adjuvant-associated factors in the Vehicle-treated conditions (Fig. 6): females show approximately constant suppression fractions due to DRα1-MOG-35-55 across all strains; whereas males see most suppression from DRα1-MOG-35-55 in WT, a somewhat lesser amount in MIF-2-KO, and smaller-to-zero amounts in MIF-1-KO and MIF-1/2-DUAL-KO.
Fig. 6. Total mean DRα1-MOG-35-55 effects on female vs. male factor expression as a fraction of total mean Vehicle-treated EAE levels.
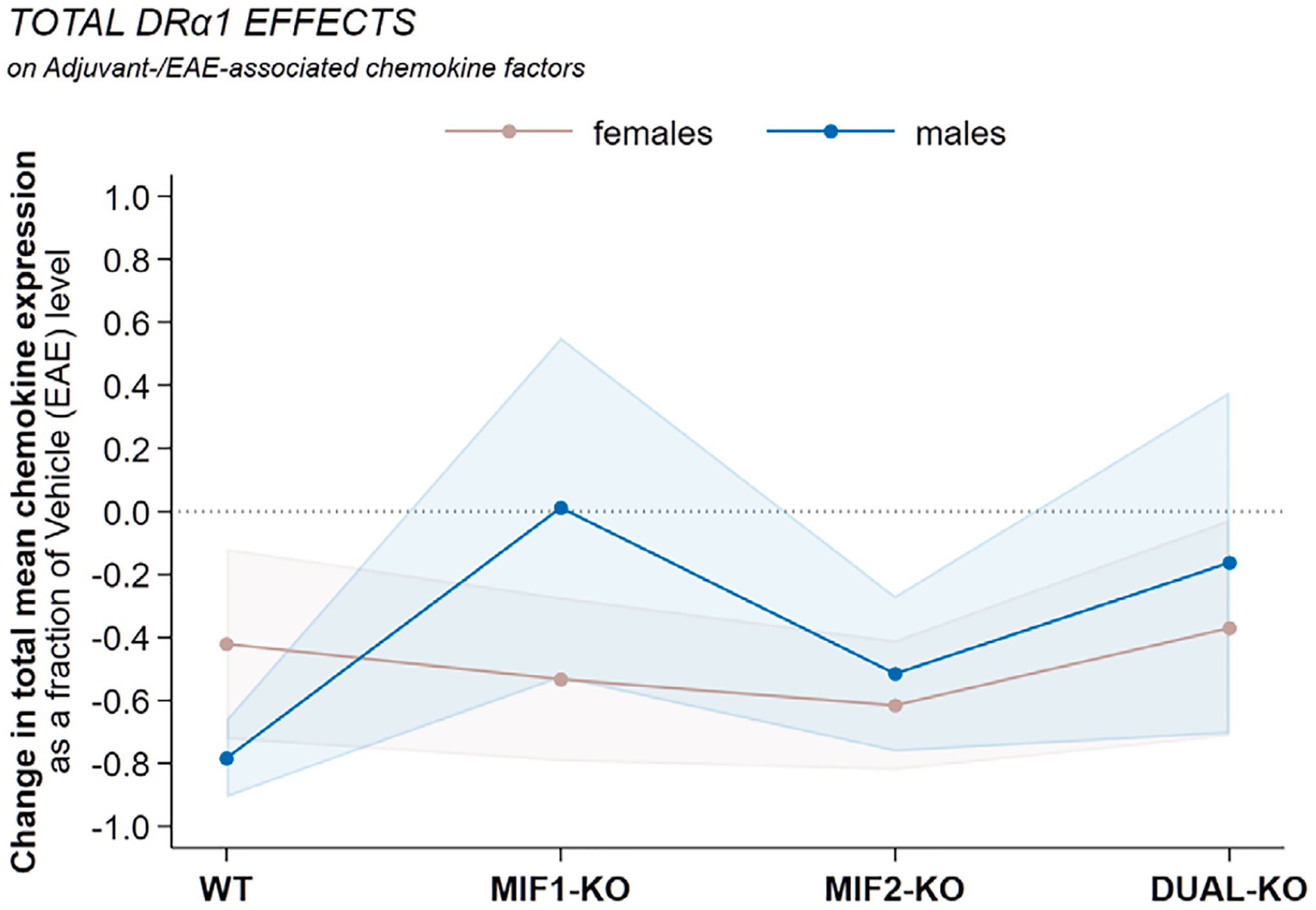
Mean chemokine expression levels for DRα1-MOG-35-55-treated mice were summed within sex and genotype and compared to the same aggregated mean levels in the sex-and-genotype-matched Vehicle-treated (EAE) mice. The combined effects are expressed as relative changes with respect to the Vehicle-treated EAE levels, with 95% confidence intervals based on Taylor-series linearization of the variance of the ratio.
3.3.4. Comparison of EAE-associated factor/receptor combinations in females vs. males with severe acute EAE
Given that individual EAE-associated factors require ligation of sometimes highly specific cell-associated receptor(s) to mediate their effects in vivo, we sought to identify the most pronounced EAE-associated factor/receptor combinations in the context of WT and MIF-KO genotypes and DRα1-MOG-35-55 treatment of these combinations. Thus, we noted EAE effects and corresponding reductions > 5 ΔCT units in expression of EAE-associated factors alone or > 10 ΔCT units when combined with their known receptors for females vs. males (Tables 3 and 4).
Table 3. EAE effects on chemokines and receptors in females.
EAE effects on chemokines and receptors compared to the corresponding changes in expression for WT and each knockout genotype vs. WT, with and without DRα1-MOG-35-55 treatment, in females. Expression changes are represented by the ΔCT score in log2 units. Effects in bold font are those >5 (log2 units) in magnitude individually or for sums of linked receptors, or >10 summed over matched ligands and receptors (the latter noted by thick borders around the cell). Cells that show complete reversal (to within −0.1) of the EAE and adjuvant effects in either the ligand or matched (sum of) receptors are highlighted.
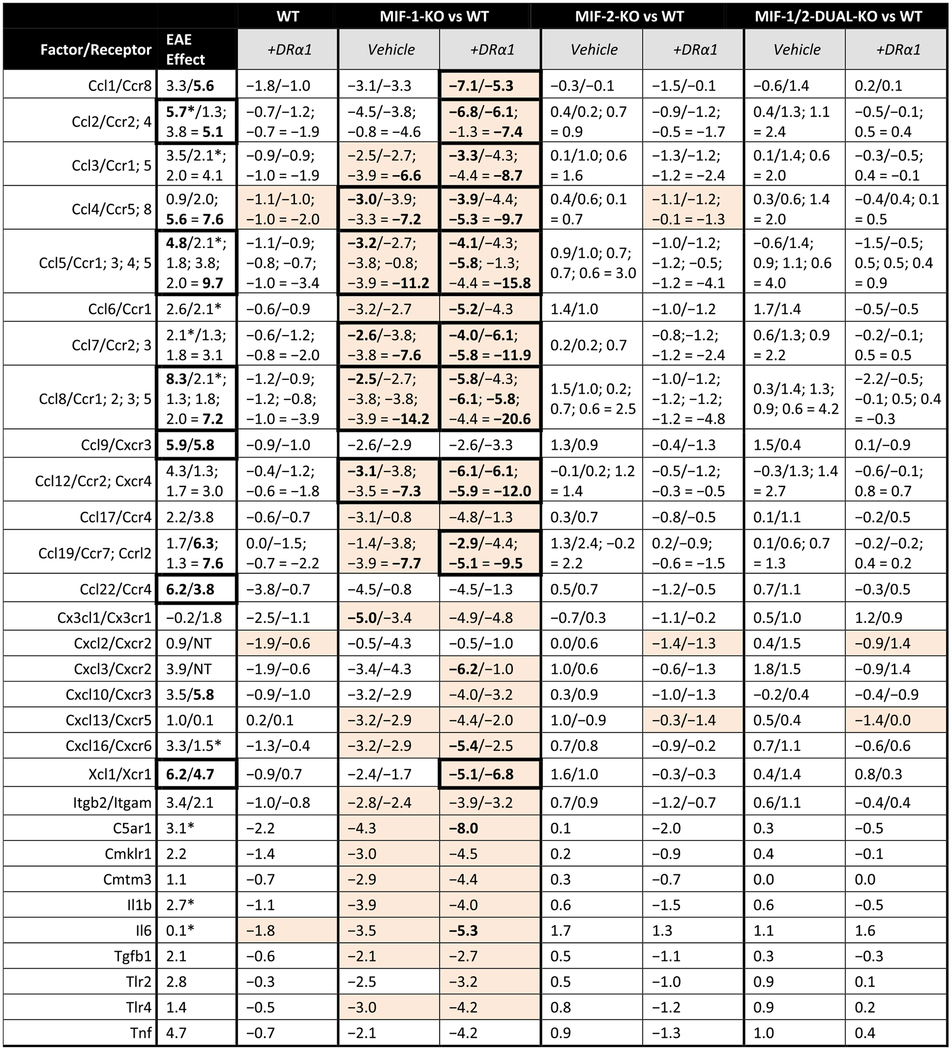
|
Also contributed to adjuvant effect. NT = Not Tested.
Table 4. EAE effects of chemokines and receptors in males.
EAE effects on chemokines and receptors compared to the corresponding changes in expression for WT and each knockout genotype vs. WT, with and without DRα1-MOG-35-55 treatment, in males. Expression changes are represented by the ΔCT score in log2 units. Effects in bold font are those >5 (log2 units) in magnitude individually or for sums of linked receptors, or >10 summed over matched ligands and receptors (the latter noted by thick borders around the cell). Cells that show complete reversal (to within −0.1) of the EAE and adjuvant effects in either the ligand or matched (sum of) receptors are highlighted.
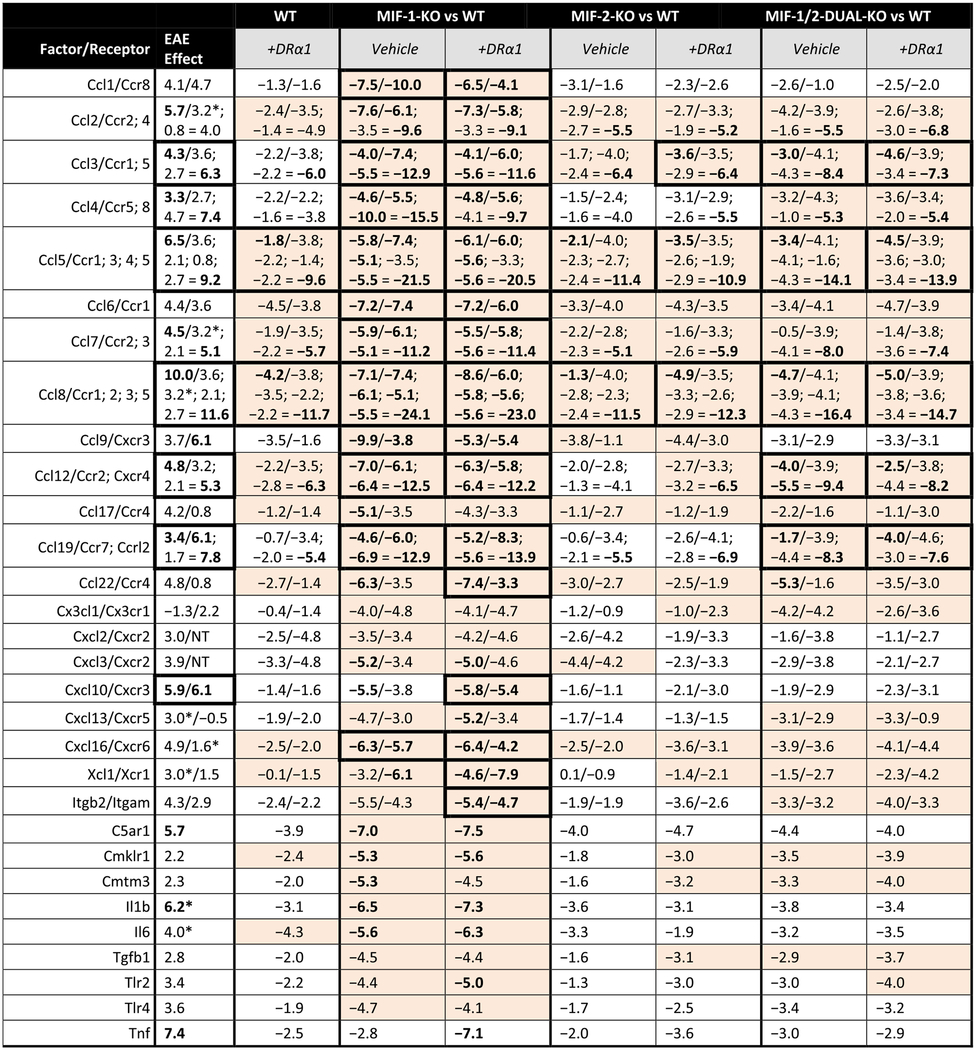
|
Also contributed to adjuvant effect. NT = Not Tested.
In females, there were 6 strong EAE-associated combinations with > 10 ΔCT unit increases, including Ccl2/Ccr2,4 (+10.8), Ccl5/Ccr1,3,4,5 (+14.5), Ccl8/Ccr1,2,3,5 (+15.5), Ccl9/Cxcr3 (+11.7), Ccl22/Ccr4 (+10.0) and Xcl1/Xcr1 (+10.9) (Table 3). Of these combinations, the corresponding (Vehicle-treated) MIF-1-KO genotype combinations for Ccl5/Ccr1,3,4,5 (−14.4) and Ccl8/Ccr1,2,3,5 (−16.7) could fully correct the EAE-associated inflammatory combinations. Moreover, adding DRα1-MOG-35-55 treatment could fully correct the Ccl2/Ccr2,4 (−14.2) and the Xcl1/Xcr1 (−11.9) combinations, as well as the Ccl5/Ccr1,3,4,5 (−19.9) and Ccl8/Ccr1,2,3,5 (−26.4) combinations. None of the MIF-2-KO or MIF-1/2-DUAL-KO Vehicle-treated combinations or the WT, MIF-2-KO or MIF-1/2-DUAL-KO DRα1-MOG-35-55-treated combinations had any factors with > 5 ΔCT unit reductions or combination > 10 ΔCT unit reductions.
In males, there were 7 strong EAE-associated combinations with > 10 ΔCT unit increases, including Ccl3/Ccr1,5 (+10.6), Ccl4/Ccr5,8 (+10.7), Ccl5/Ccr1,3,4,5 (+15.7), Ccl8/Ccr1,2,3,5 (+21.6), Ccl12/Ccr2,Cxcr4 (+10.1), Ccl19/Ccr7,Ccr12 (+11.2) and Cxcl10/Cxcr3 (+12.0) (Table 4). As shown in the table, both Vehicle-treated and DRα1-MOG-35-55-treated combinations in the MIF-1-KO group could fully correct all of the above EAE-associated combinations with > 10 positive ΔCT unit changes except Cxcl10/Cxcr3 in Vehicle-treated mice (but combined suppression was still substantial at −9.3). Interestingly, several factor/receptor combinations in the MIF-1/2-DUAL-KO group (sometimes requiring DRα1-MOG-35-55), but none in the MIF-2-KO group (with or without DRα1-MOG-35-55), could also nearly or fully correct both the ligand and receptor levels simultaneously in 4 of the 7 EAE-associated combinations with > 10 ΔCT unit increases.
It should be noted that only 2 EAE-associated combinations with > 10 ΔCT unit increases, Ccl5/Ccr1,3,4,5 (+14.5F; +15.7 M) and Ccl8/Ccr1,2,3,5 (+15.5F; +21.6 M) were shared between females and males. This finding might suggest that complete blocking of inflammatory activity of these combinations may contribute to improvement in clinical outcomes especially in DRα1-MOG-35-55-treated MIF-1-KO female and male mice.
4. Discussion
The results of this study provide new perspectives on the roles of MIF-1 and MIF-2 as major contributors to the underlying processes (i.e. attraction of inflammatory mononuclear cells from periphery into the CNS), involved in the induction of severe acute EAE in C57BL/6J mice. Our approach was to identify significant differences in EAE clinical severity and changes in expression of a ranked set of EAE- and CFA + Ptx adjuvant-associated factors in spinal cord tissue from female and male WT mice [38] vs. MIF-1-KO, MIF-2-KO, and MIF-1/2-DUAL-KO mice treated with Vehicle or DRα1-MOG-35-55, a competitive inhibitor of MIF-1 and MIF-2 binding to their common CD74 receptor. These studies revealed that the loss of MIF-1 alone resulted in a significantly better clinical outcome in females than males, with reductions in CDI of ~ 35% vs. ~ 2% respectively, but with male MIF-1-KO and MIF-1/2-DUAL-KO mice showing a trend of improved daily clinical scores over the last 3 days of observation. The delayed tendency to achieve reduced clinical EAE scores in MIF-deleted males with severe acute EAE was not expected since significant reductions occurred in MIF-deficient males and females with moderately severe EAE [22,23]. However, DRα1-MOG-35-55 treatment significantly improved CDI scores of not only WT but also MIF-deficient females and males with severe acute EAE. Treatment of MIF-1-KO females reduced EAE scores by ~ 57% vs. ~ 60% in males, with treatment of WT, MIF-2-KO, and DUAL-KO groups being less impactful but similar in effect across these other genotypes, and nominally better in females (~39% average reduction in EAE CDI scores in females vs. ~ 33% average reduction in males, respectively).
Remarkably, the final mean daily EAE scores in DRα1-MOG-35-55-treated MIF-1-KO mice were very small and nearly identical (~0.5 EAE score units) in females and males, thereby approaching a “cure”. This result suggests that other potentially pathogenic factors which may become activated in the absence of MIF-1 might still be silenced by treatment with DRα1-MOG-35-55. One example of this possibility in males is CCR6, a known proinflammatory chemokine [39] that we found associated with EAE induction in males but not females in our baseline paper [38]. As shown in Table 5, Ccr6 expression can be mildly reduced by DRα1-MOG-35-55 treatment of females but only in MIF-deleted genotypes (no effect in WT), whereas in males (where CCR6 may contribute to EAE pathogenesis) DRα1-MOG-35-55 treatment is conversely most impactful in MIF-1-KO (and effective in WT). The effect of treatment on Ccr6 expression is weaker in MIF-2-deleted (including DUAL-KO) male mice and similar in magnitude to that seen in MIF-2-deleted females, hinting at the possibility of a complex sex-moderated compensation mechanism involving CCR6 (and perhaps assisted by MIF-2) when MIF-1 is absent. Moreover, as reported previously for males [23], there was no compensation evident between the two MIF homologues during severe EAE in females. These findings would suggest that the MIF-1 and MIF-2 promoters in both sexes were not linked or synchronized in mice with severe acute EAE.
Table 5. CCR6 as a DRα1-MOG-35-55-treatable compensation factor in MIF-deleted mice.
Ccr6 gene expression levels were measured by sex and treatment for each MIF genotype in the study. EAE-naïve WT baseline values represent the healthy control group in a previous study [38] where we examined the effects of inducing EAE in similar mice and are listed in italics for reference. CCR6 is a known proinflammatory chemokine [39] that we found associated with EAE induction in males (but not females) in our baseline paper [38], and in the current experiment we also observed that Ccr6 expression is much higher than baseline for WT EAE males but about the same as baseline (even a bit lower) for WT EAE females. Expression is somewhat dampened in males by deleting either MIF-1 or MIF-2 or both (more for MIF-1 alone) but only appreciably dampened in females under MIF-1 deletion. Moreover, clear sex differences in sensitivity to DRα1-MOG-35-55 treatment are visible in both WT and MIF-1-KO mice but not in mice without MIF-2. That is, it cannot be suppressed by DRα1-MOG-35-55 treatment alone in WT females, and the amount of suppression is weakest in females for the MIF-1-KO genotype, whereas in males suppression is possible from DRα1-MOG-35-55 treatment alone and strongest in MIF-1-KO; the effect of treatment in MIF-2-KO or DUAL-KO mice is similar for females and males, but this effect is about the same as the dampening effect in MIF-1-KO females, versus much less than the dampening effect in MIF-1-KO (or WT) males. Thus, CCR6 seems to play an active role in EAE for males, and possibly a compensatory role in the absence of either MIF gene. It is nearly always dampened with DRα1-MOG-35-55 treatment, but especially sensitive to this treatment in the absence of MIF-1. Our calculated Bayesian posterior probability that the DRα1-MOG-35-55 treatment effect in a particular background is larger than 1 Ct in magnitude is at least 70% in every case in this table (DRα1-treated vs. Vehicle-treated WT males, MIF-1-KO males, and MIF-2-KO and DUAL-KO females and males) where the nominal effect is at least that large.
| Ccr6 | WT baseline (EAE-naïve) | WT | MIF-1–KO | MIF-2–KO | MIF-1/2-DUAL–KO | ||||
|---|---|---|---|---|---|---|---|---|---|
| Vehicle | +DRα1 | Vehicle | +DRα1 | Vehicle | +DRα1 | Vehicle | +DRα1 | ||
| Females | 12.06 | 11.30 | 11.81 | 9.17 | 8.31 | 12.44 | 10.84 | 12.26 | 10.96 |
| Males | 9.42 | 13.81 | 11.89 | 10.45 | 8.26 | 12.08 | 10.80 | 12.11 | 10.57 |
These clinical sex-associated changes attributed to MIF genotypes were best reflected by the increased numbers of EAE- and adjuvant-associated factors as well as differences in factor composition in males vs. females (16/42 male-only factors vs. 9/35 female-only factors) and factor/receptor combinations (2 in common had higher total ΔCT unit increases in males vs. females). Moreover, highly salient EAE-associated single-factor (i.e. Tnf) and factor/receptor combinations that were not fully reversed by the MIF-deletion genotype in males (e.g. Cxcl10/Cxcr3 in MIF-1-KO mice, all 7 in MIF-2-KO mice, and Ccl19/Ccr7;Ccrl2 in DUAL-KO mice) may also have contributed to the delayed clinical improvement in males. One caveat not addressed in the current study is that the factors being followed were all attributed to induction of EAE in WT mice. To further address the question of factor composition effects, we are currently assessing rank order expression of all array factor changes in Vehicle and DRα1-MOG-35-55-treated MIF-KO strains compared to baseline expressions in naïve MIF-KO strains without EAE.
For EAE- and adjuvant-associated inflammatory factors, both sexes showed the greatest reduction in factor expression in MIF-1-KO mice. Lesser reduction was observed in WT, MIF-2-KO and MIF-1/2-DUAL-KO males, whereas females showed no benefit (and possibly disease-promoting effects) from MIF-2 or MIF-1/2 deletions. Treatment of WT mice with DRα1-MOG-35-55 provided mild to moderate correction of baseline EAE-associated factor expression in WT females (Table 1, bolded) but aggressive reduction (Table S2, thick border) and often full reversal (Table S2, highlighted) of EAE-associated factors in males. Moreover, the few factors that failed to fully reverse in DRα1-MOG-35-55-treated MIF-1-KO male mice were in fact extremely close to that line (often much <1 log2 unit away) in nearly all cases, whereas this was much less often true for MIF-1-KO females. As a rule, DRα1-MOG-35-55 treatment more frequently reversed factors with lower ΔCT values than those with higher ΔCT values.
In addition to genotype-driven factors that were fully corrected in comparison to baseline inflammatory effects (Vehicle-treated groups), there were sex differences noted in smaller subsets of factors for each group that were reduced ~ 2 ΔCT values or more (cells with thick borders in Tables 1, 2, S2, and S3) beyond the genotype effects after treatment with DRα1-MOG-35-55. For MIF-1-KO female mice there were 10 such EAE-associated factors and 3 adjuvant-associated factors, but for MIF-1-KO males only 3 such EAE-associated factors and no adjuvant-associated factors. For MIF-2 and MIF-1/2-DUAL-depleted females, the majority (96% and 85% respectively) of EAE-associated factors showed surprisingly increased expression levels in untreated mice, but treatment with DRα1-MOG-35-55 reversed all of the increases in MIF-2-KO and a majority in DUAL-KO mice. MIF-2 deleted male mice showed only modest and sporadic additional suppression of factors by DRα1-MOG-35-55 treatment.
Factors that were reduced by treatment with DRα1-MOG-35-55 beyond genotype effects might qualify as potential factors linked to a better clinical outcome. As mentioned above, DRα1-MOG-35-55 treatment of MIF-1-KO females substantially reduced levels of 3/8 adjuvant-associated genes beyond genotype effects compared to none in males, suggesting that these factors may contribute to EAE inhibition in females but are able to operate outside of the MIF:CD74 axis in males. This assertion is supported by increased cellular infiltration into the CNS and increased demyelination in females vs. males with EAE reported previously [26]. Additional studies will be needed to directly evaluate effects of neutralizing the above EAE- and adjuvant-associated factors on EAE clinical outcomes using factor KO mice or factor-specific antibodies in combination with the respective anti-MIF antibodies. However, it is also possible that the clinical improvements could be mediated through non-MIF-associated compensatory mechanisms targeted by DRα1-MOG-35-55 such as inhibition of IL-2 secretion by activated T cells or other key factors not included in our factor array.
While it is beyond the scope of the current study to determine, in view of the impressive dampening of clinical EAE disease by DRα1-MOG-35-55 treatment in MIF-1-KO mice of both sexes, the question of what MIF-independent pathways are affected by the (p)MHC class II DRα1-MOG-35-55 construct invites some speculation. (We thank an anonymous reviewer for raising this point.) We believe that the DRα1-MOG-35-55 effect on T cells is dependent on the effects of the linked MOG-35-55 peptide moiety on CD74 loading of the CLIP region peptide and replacement of CLIP with antigenic peptides into MHC class II molecules in APC prior to T cell activation. DRα1-MOG-35-55 fundamentally alters the function of CD74 in both the antigen presentation pathway (we think by competitively blocking CD74 interactions with MHC class II) and the MIF/CD74 axis (as a competitive blocker of MIF binding and signaling). The DRα1 component binds to and down-regulates CD74, and the addition of the MOG-35-55 peptide to the construct enhances this CD74 inhibitory activity [25,28]. Remarkably, the complete DRα1-MOG-35-55 construct undergoes changes in secondary structure, including increased α-helix and β-sheet content that are not present in the DRa1 or the MOG-35-55 peptide moieties alone. Studies testing the necessity of the MOG-35-55 moiety for MHCII binding are needed (and currently in progress by our group), but we predict that the increased ability of DRα1-MOG-35-55 to modulate CD74 expression and to interfere with CD74-dependent peptide loading into MHCII represent two separable CD74-dependent mechanisms that could account for its ability to inhibit activation and EAE severity of both cognate (MOG-35-55-specific) and non-cognate (e.g. MBP-85–99) encephalitogenic T cells [28], the latter mechanism being MIF-independent.
On a larger scale, the current study addresses in detail the dominant enhancing effects of the MIF-1 genotype on expression of essentially all of the EAE-associated factors in our array deemed to be critical for EAE induction in both females and males [38,40], with some additional contribution of the MIF-2 genotype in males only (Figure S7). These MIF-1-associated factors included IL-6, IL-1β, IL-23, and TGFβ needed to differentiate autoreactive Th17 cells to an encephalitogenic CCR2+, CCR6+ phenotype. Upon local reactivation by monocytes and dendritic cells within the CNS, the CCR2+ Th17 cells and monocytes release IL-17, GM-CSF, TNFα, and CCL2 that activate microglial cells and further recruit other CCR2, CCR6, and CXCR2 (SJL/J mice only) expressing leukocytes across a CCR2+ vascular endothelial cell barrier into the CNS where they release a variety of inflammatory factors that cause demyelination, axonal damage, and clinical signs of EAE. Although our array did not include IL-2, IL-17, IL-23, or GM-CSF, it did clearly implicate IL-6, IL-1β, and a third factor, CXCR6 (aka CD186, an upstream marker on DC and NK cells) as adjuvant-induced contributors to EAE in both males and females. Our data also implicated the critical-for-EAE CCR2 axis involving CCL2, CCL7, and CCL8 (monocyte chemoattractant proteins = MCP-1, 3, and 2 respectively) demonstrating both adjuvant-assisted and non-adjuvant-associated effects. Of particular interest is CCL8, our highest-ranked factor not yet implicated in EAE that activates mast cells that release vasoactive amines, the key EAE-enhancing components induced by Ptx [41]. Interestingly, there were strong adjuvant-induced increases in C5ar1 in females that implicate the complement cascade in EAE induction. Additionally, TNFα and TGFβ1 were strongly upregulated EAE-associated genes in the CNS of both male and female mice.
In addition to CCL2 mentioned above, a number of other known chemokines and receptors were reported to be highly expressed in the CNS of mice with EAE, including CCL5 (showed a correlation with increased clinical signs in C57BL/6 mice) [42]; macrophage CXCR3 [43]; macrophage CCR4 [44,45]; and macrophage CXCR7 [46]. In both our previous [38] and current studies, CCL5 and CXCR3 were found to be associated with EAE or adjuvant in both females and males. CCR4 was marginally increased in males but not females as an adjuvant effect and could be subsequently reduced in males by MIF deletion and/or DRα1-MOG-35-55 treatment. CXCR7 was not EAE- or adjuvant-associated in our study in either sex, but was significantly reduced by deletion of MIF-1 (both sexes) and MIF-2 (mainly males) and by DRα1-MOG-35-55 treatment (WT males only). The critical CCR2-CCL2 (MCP1) axis also involved other CCR2-family ligands strongly associated with MIF-1 and MIF-2 in our study, including CCL7 (MCP3) that guides Th17 cells to lymph nodes rather than the CNS, and CCL8 (MCP2), our top candidate in both males and females that activates mast cells, all of which were reduced by MIF deletion and DRα1-MOG-35-55 treatment.
Results from our studies also shed some light on female predominance of MS. As shown previously [38], females developing EAE had increased cellular infiltration, demyelination, adjuvant effects and CD74 expression in spinal cord compared to males (the last a confirmation of increased CD74 expression in blood from MS females [22]. Here we demonstrate greater impact of MIF-1 on EAE in females than males. These findings suggest that females may intrinsically be closer to the line for disease expression mediated by MIF-1 and thus at more risk for getting MS from factors that promote MIF-1 expression, including adjuvant effects. Together, these data support the contention that the immunopathogenic mechanisms for female predominance in MS could be expressed, in part, through MIF/CD74 directed pathways.
Finally, as discussed in detail above, treatment of WT, MIF-1-KO, MIF-2-KO, and MIF-1/2-DUAL-KO mice with DRα1-MOG-35-55 stabilized and in many cases enhanced EAE-protective genotype effects on reducing both clinical severity and the above-specified EAE- and adjuvant-associated factors. These remarkable widespread suppressive effects of DRα1-MOG-35-55 in both males and females portends well for its use in clinical trials involving MIF-dependent and MIF-independent CD74 interactions.
5. Declarations
5.1. Ethics approval and consent to participate
All applicable international, national and/or institutional guidelines for the care and use of animals were followed. All procedures performed in studies involving animals were in accordance with the ethical standards of the institution or practice at which the studies were conducted. This article does not contain any studies with human participants performed by any of the authors.
Supplementary Material
Acknowledgements
The authors would like to thank Dr. Denesa Lockwood for assistance in preparing the manuscript and the VAPORHCS Veterinarian, Dr. Samuel Dehlinger and staff for oversight, housing and maintaining mouse environments.
Funding
This work was funded by the Department of Veterans Affairs, Veterans Health Administration, Office of Research and Development, Biomedical Laboratory Research and Development Merit Review Award 2I01 BX000226 (AV), BLR&D Merit Review for Pre-IND studies of Drugs and Biologics Award 5I01 BX005112 (AV), Senior Research Career Scientist Award 1IK6BX004209 (AV), the National Institute of Allergy and Infectious Diseases awards 2R42AI122574 (AV) and R21 AI148409 (HO), and the National Institute of Arthritis and Musculoskeletal and Skin Diseases award 1R01AR078334 (RB). The contents do not represent the views of the Department of Veterans Affairs or the US Government.
Abbreviations:
- C57BL/6J
wild type mouse strain
- cDNA
Complementary deoxynucleic acid
- CDI
Cumulative Disease Index
- CNS
Central Nervous System
- CFA
Complete Freund’s adjuvant
- therapeutic molecular construct
DRα1-MOG-35-55
- EAE
Experimental autoimmune encephalomyelitis
- IFNγ
Interferon gamma
- i.p
Intraperitoneal
- LN
Lymph nodes
- MHC
Major histocompatibility complex
- MS
Multiple sclerosis
- Mtb
Mycobacterium tuberculosis
- MOG-35-55
Myelin oligodendrocyte glycoprotein 35-55
- PBS
Phosphate-buffered saline
- Ptx
Pertussis toxin
- RNA
Ribonucleic acid
- SC
Spinal Cord
- SEM
Standard error of the mean
- TNF
Tumor necrosis factor
- VA
Veterans’ Affairs
- WT
Wild type
Footnotes
Declaration of Competing Interest
The authors declare the following financial interests/personal relationships which may be considered as potential competing interests: Drs. Vandenbark, Offner, Meza-Romero, and OHSU have a significant financial interest in Artielle ImmunoTherapeutics, Inc., a company that may have a commercial interest in the results of this research and technology. This potential conflict of interest has been reviewed and managed by the OHSU and VA Portland Health Care System Conflict of Interest in Research Committees.
Appendix A. Supplementary material
Supplementary data to this article can be found online at https://doi.org/10.1016/j.cellimm.2022.104561.
6. Availability of data and materials
The datasets used and/or analyzed during the current study are available from the corresponding author on reasonable request.
References
- [1].Dutta R, Trapp BD, Relapsing and progressive forms of multiple sclerosis: insights from pathology, Curr. Opin. Neurol 27 (2014) 271–278, 10.1097/WCO.0000000000000094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2].Steinman L, Multiple sclerosis: a two-stage disease, Nat Immunol 2 (9) (2001) 762–764, 10.1038/ni0901-762. [DOI] [PubMed] [Google Scholar]
- [3].Didonna A, Oksenberg JR, The Genetics of Multiple Sclerosis, in: Zagon IS, McLaughlin PJ (Eds.), Multiple Sclerosis: Perspectives in Treatment and Pathogenesis, Codon Publications, Brisbane (AU), 2017. http://www.ncbi.nlm.nih.gov/books/NBK470155/ (accessed January 31, 2022. [Google Scholar]
- [4].Oksenberg JR, Baranzini SE, Sawcer S, Hauser SL, The genetics of multiple sclerosis: SNPs to pathways to pathogenesis, Nat. Rev. Genet 9 (7) (2008) 516–526, 10.1038/nrg2395. [DOI] [PubMed] [Google Scholar]
- [5].Leng L, Metz CN, Fang Y, Xu J, Donnelly S, Baugh J, Delohery T, Chen Y, Mitchell RA, Bucala R, MIF signal transduction initiated by binding to CD74, J. Exp. Med 197 (2003) 1467–1476, 10.1084/jem.20030286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Cresswell P, Invariant chain structure and MHC class II function, Cell 84 (4) (1996) 505–507. [DOI] [PubMed] [Google Scholar]
- [7].Assis DN, Leng L, Du X, Zhang CK, Grieb G, Merk M, Garcia AB, McCrann C, Chapiro J, Meinhardt A, Mizue Y, Nikolic-Paterson DJ, Bernhagen J, Kaplan MM, Zhao H, Boyer JL, Bucala R, The role of macrophage migration inhibitory factor in autoimmune liver disease, Hepatology 59 (2) (2014) 580–591, 10.1002/hep.26664. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Calandra T, Roger T, Macrophage migration inhibitory factor: a regulator of innate immunity, Nat. Rev. Immunol 3 (10) (2003) 791–800, 10.1038/nri1200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Akcali A, Pehlivan S, Pehlivan M, Sever T, Neyal M, Association of macrophage migration inhibitory factor gene promoter polymorphisms with multiple sclerosis in Turkish patients, J. Int. Med. Res 38 (1) (2010) 69–77, 10.1177/147323001003800108. [DOI] [PubMed] [Google Scholar]
- [10].Kang I, Bucala R, The immunobiology of MIF: function, genetics and prospects for precision medicine, Nat. Rev. Rheumatol 15 (7) (2019) 427–437, 10.1038/s41584-019-0238-2. [DOI] [PubMed] [Google Scholar]
- [11].Niino M, Ogata A, Kikuchi S, Tashiro K, Nishihira J, Macrophage migration inhibitory factor in the cerebrospinal fluid of patients with conventional and optic-spinal forms of multiple sclerosis and neuro-Behçet’s disease, J. Neurol. Sci 179 (1–2) (2000) 127–131. [DOI] [PubMed] [Google Scholar]
- [12].Rinta S, Kuusisto H, Raunio M, Paalavuo R, Levula M, Lehtimäki T, Elovaara I, Apoptosis-related molecules in blood in multiple sclerosis, J. Neuroimmunol 205 (1–2) (2008) 135–141, 10.1016/j.jneuroim.2008.09.002. [DOI] [PubMed] [Google Scholar]
- [13].Hagman S, Raunio M, Rossi M, Dastidar P, Elovaara I, Disease-associated inflammatory biomarker profiles in blood in different subtypes of multiple sclerosis: prospective clinical and MRI follow-up study, J. Neuroimmunol 234 (1–2) (2011) 141–147, 10.1016/j.jneuroim.2011.02.009. [DOI] [PubMed] [Google Scholar]
- [14].Cox GM, Kithcart AP, Pitt D, Guan Z, Alexander J, Williams JL, Shawler T, Dagia NM, Popovich PG, Satoskar AR, Whitacre CC, Macrophage migration inhibitory factor potentiates autoimmune-mediated neuroinflammation, J. Immunol 191 (3) (2013) 1043–1054, 10.4049/jimmunol.1200485. [DOI] [PubMed] [Google Scholar]
- [15].Bernhagen J, Krohn R, Lue H, Gregory JL, Zernecke A, Koenen RR, Dewor M, Georgiev I, Schober A, Leng L, Kooistra T, Fingerle-Rowson G, Ghezzi P, Kleemann R, McColl SR, Bucala R, Hickey MJ, Weber C, MIF is a noncognate ligand of CXC chemokine receptors in inflammatory and atherogenic cell recruitment, Nat. Med 13 (5) (2007) 587–596, 10.1038/nm1567. [DOI] [PubMed] [Google Scholar]
- [16].Schwartz V, Lue H, Kraemer S, Korbiel J, Krohn R, Ohl K, Bucala R, Weber C, Bernhagen J, A functional heteromeric MIF receptor formed by CD74 and CXCR4, FEBS Lett 583 (2009) 2749–2757, 10.1016/j.febslet.2009.07.058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Shi X, Leng L, Wang T, Wang W, Du X, Li J.i., McDonald C, Chen Z, Murphy JW, Lolis E, Noble P, Knudson W, Bucala R, CD44 is the signaling component of the macrophage migration inhibitory factor-CD74 receptor complex, Immunity 25 (4) (2006) 595–606, 10.1016/j.immuni.2006.08.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Yoo S-A, Leng L, Kim B-J, Du X, Tilstam PV, Kim KH, Kong J-S, Yoon H-J, Liu A, Wang T, Song Y, Sauler M, Bernhagen J, Ritchlin CT, Lee P, Cho C-S, Kim W-U, Bucala R, MIF allele-dependent regulation of the MIF coreceptor CD44 and role in rheumatoid arthritis, Proc. Natl. Acad. Sci. USA 113 (2016) E7917–E7926, 10.1073/pnas.1612717113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Powell ND, Papenfuss TL, McClain MA, Gienapp IE, Shawler TM, Satoskar AR, Whitacre CC, Cutting edge: macrophage migration inhibitory factor is necessary for progression of experimental autoimmune encephalomyelitis, J. Immunol 175 (9) (2005) 5611–5614, 10.4049/jimmunol.175.9.5611. [DOI] [PubMed] [Google Scholar]
- [20].Denkinger CM, Denkinger M, Kort JJ, Metz C, Forsthuber TG, In vivo blockade of macrophage migration inhibitory factor ameliorates acute experimental autoimmune encephalomyelitis by impairing the homing of encephalitogenic T cells to the central nervous system, J. Immunol 170 (3) (2003) 1274–1282, 10.4049/jimmunol.170.3.1274. [DOI] [PubMed] [Google Scholar]
- [21].Benedek G, Meza-Romero R, Andrew S, Leng L, Burrows GG, Bourdette D, Offner H, Bucala R, Vandenbark AA, Partial MHC class II constructs inhibit MIF/CD74 binding and downstream effects, Eur. J. Immunol 43 (5) (2013) 1309–1321, 10.1002/eji.201243162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Benedek G, Meza-Romero R, Jordan K, Zhang Y, Nguyen H, Kent G, Li J, Siu E, Frazer J, Piecychna M, Du X, Sreih A, Leng L, Wiedrick J, Caillier SJ, Offner H, Oksenberg JR, Yadav V, Bourdette D, Bucala R, Vandenbark AA, MIF and D-DT are potential disease severity modifiers in male MS subjects, Proc. Natl. Acad. Sci. USA 114 (2017) E8421–E8429, 10.1073/pnas.1712288114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Vandenbark AA, Meza-Romero R, Wiedrick J, Gerstner G, Headrick A, Kent G, Seifert H, Benedek G, Bucala R, Offner H, Brief report: Enhanced DRα1-mMOG-35-55 treatment of severe EAE in MIF-1-deficient male mice, CellImmunol 370 (2021) 104439, 10.1016/j.cellimm.2021.104439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Honigman JS, DiGregorio KM, Dedkov EI, Leheste JR, Leng L, Bucala R, Torres G, Distribution maps of D-dopachrome tautomerase in the mouse brain, Neuroscience 226 (2012) 382–387, 10.1016/j.neuroscience.2012.09.009. [DOI] [PubMed] [Google Scholar]
- [25].Vandenbark AA, Meza-Romero R, Benedek G, Andrew S, Huan J, Chou YK, Buenafe AC, Dahan R, Reiter Y, Mooney JL, Offner H, Burrows GG, A novel regulatory pathway for autoimmune disease: binding of partial MHC class II constructs to monocytes reduces CD74 expression and induces both specific and bystander T-cell tolerance, J. Autoimmun 40 (2013) 96–110, 10.1016/j.jaut.2012.08.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Wang C, Gold BG, Kaler LJ, Yu X, Afentoulis ME, Burrows GG, Vandenbark AA, Bourdette DN, Offner H, Antigen-specific therapy promotes repair of myelin and axonal damage in established EAE, J. Neurochem 98 (2006) 1817–1827, 10.1111/j.1471-4159.2006.04081.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Yadav V, Bourdette DN, Bowen JD, Lynch SG, Mattson D, Preiningerova J, Bever CT, Simon J, Goldstein A, Burrows GG, Offner H, Ferro AJ, Vandenbark AA, Recombinant T-Cell Receptor Ligand (RTL) for Treatment of Multiple Sclerosis: A Double-Blind, Placebo-Controlled, Phase 1, Dose-Escalation Study, Autoimmune Dis 2012 (2012) 1–11, 10.1155/2012/954739. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Meza-Romero R, Benedek G, Yu X, Mooney JL, Dahan R, Duvshani N, Bucala R, Offner H, Reiter Y, Burrows GG, Vandenbark AA, HLA-DRα1 constructs block CD74 expression and MIF effects in experimental autoimmune encephalomyelitis, J. Immunol 192 (9) (2014) 4164–4173, 10.4049/jimmunol.1303118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Benedek G, Meza-Romero R, Jordan K, Keenlyside L, Offner H, Vandenbark AA, HLA-DRα1-mMOG-35-55 treatment of experimental autoimmune encephalomyelitis reduces CNS inflammation, enhances M2 macrophage frequency, and promotes neuroprotection, J. Neuroinflammation 12 (2015) 123, 10.1186/s12974-015-0342-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Benedek G, Chaudhary P, Meza-Romero R, Calkins E, Kent G, Offner H, Bourdette D, Vandenbark AA, Sex-dependent treatment of chronic EAE with partial MHC class II constructs, J. Neuroinflammation 14 (2017) 100, 10.1186/s12974-017-0873-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Vandenbark AA, Meza-Romero R, Benedek G, Offner H, A novel neurotherapeutic for multiple sclerosis, ischemic injury, methamphetamine addiction, and traumatic brain injury, J. Neuroinflammation 16 (2019) 14, 10.1186/s12974-018-1393-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Fingerle-Rowson G, Petrenko O, Metz CN, Forsthuber TG, Mitchell R, Huss R, Moll U, Müller W, Bucala R, The p53-dependent effects of macrophage migration inhibitory factor revealed by gene targeting, Proc. Natl. Acad. Sci. USA 100 (16) (2003) 9354–9359, 10.1073/pnas.1533295100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Tilstam PV, Schulte W, Holowka T, Kim B-S, Nouws J, Sauler M, Piecychna M, Pantouris G, Lolis E, Leng L, Bernhagen J, Fingerle-Rowson G, Bucala R, MIF but not MIF-2 recruits inflammatory macrophages in an experimental polymicrobial sepsis model, J. Clin. Invest 131 (2021) e127171, 10.1172/JCI127171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Fan J, Lin S-K, Test of Significance When Data Are Curves, J. Am. Stat. Assoc 93 (1998) 1007–1021, 10.2307/2669845. [DOI] [Google Scholar]
- [35].Jaki T, Wolfsegger MJ, A Theoretical Framework for Estimation of AUCs in Complete and Incomplete Sampling Designs, Stat. Biopharm. Res 1 (2) (2009) 176–184, 10.1198/sbr.2009.0025. [DOI] [Google Scholar]
- [36].Gelman A, Carlin J, Stern H, Dunson D, Vehtari A, Rubin D, Bayesian Data Analysis, Chapman and Hall/CRC, Third, 2013. [Google Scholar]
- [37].Qiagen, RT2 RNA QC PCR Array Handbook, (2012). www.qiagen.com.
- [38].Wiedrick J, Meza-Romero R, Gerstner G, Seifert H, Chaudhary P, Headrick A, Kent G, Maestas A, Offner H, Vandenbark AA, Sex differences in EAE reveal common and distinct cellular and molecular components, CellImmunol 359 (2021) 104242, 10.1016/j.cellimm.2020.104242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [39].Yamazaki T, Yang XO, Chung Y, Fukunaga A, Nurieva R, Pappu B, Martin-Orozco N, Kang HS, Ma L.i., Panopoulos AD, Craig S, Watowich SS, Jetten AM, Tian Q, Dong C, CCR6 Regulates the Migration of Inflammatory and Regulatory T Cells, J. Immunol 181 (12) (2008) 8391–8401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [40].Karpus WJ, Cytokines and Chemokines in the Pathogenesis of Experimental Autoimmune Encephalomyelitis, J. Immunol 204 (2) (2020) 316–326, 10.4049/jimmunol.1900914. [DOI] [PubMed] [Google Scholar]
- [41].Linthicum DS, Munoz JJ, Blaskett A, Acute experimental autoimmune encephalomyelitis in mice. I. Adjuvant action of Bordetella pertussis is due to vasoactive amine sensitization and increased vascular permeability of the central nervous system, CellImmunol 73 (2) (1982) 299–310, 10.1016/0008-8749(82)90457-9. [DOI] [PubMed] [Google Scholar]
- [42].Juedes AE, Hjelmström P, Bergman CM, Neild AL, Ruddle NH, Kinetics and cellular origin of cytokines in the central nervous system: insight into mechanisms of myelin oligodendrocyte glycoprotein-induced experimental autoimmune encephalomyelitis, J. Immunol 164 (1) (2000) 419–426, 10.4049/jimmunol.164.1.419. [DOI] [PubMed] [Google Scholar]
- [43].Ni J, Zhu Y-N, Zhong X-G, Ding Y, Hou L-F, Tong X-K, Tang W, Ono S, Yang Y-F, Zuo J-P, The chemokine receptor antagonist, TAK-779, decreased experimental autoimmune encephalomyelitis by reducing inflammatory cell migration into the central nervous system, without affecting T cell function, Br. J. Pharmacol 158 (2009) 2046–2056, 10.1111/j.1476-5381.2009.00528.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [44].Moriguchi K, Miyamoto K, Tanaka N, Ueno R, Nakayama T, Yoshie O, Kusunoki S, C-C chemokine receptor type 4 antagonist Compound 22 ameliorates experimental autoimmune encephalomyelitis, J. Neuroimmunol 291 (2016) 54–58, 10.1016/j.jneuroim.2015.12.011. [DOI] [PubMed] [Google Scholar]
- [45].Othy S, Topçu S, Kaveri SV, Bayry J, Effect of CC chemokine receptor 4 antagonism on the evolution of experimental autoimmune encephalomyelitis, PNAS 109 (37) (2012), 10.1073/pnas.1209124109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [46].Cruz-Orengo L, Chen Y-J, Kim JH, Dorsey D, Song S-K, Klein RS, CXCR7 antagonism prevents axonal injury during experimental autoimmune encephalomyelitis as revealed by in vivo axial diffusivity, J. Neuroinflammation 8 (2011) 170, 10.1186/1742-2094-8-170. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The datasets used and/or analyzed during the current study are available from the corresponding author on reasonable request.