

Abstract
Cissus is the largest genus in Vitaceae and is mainly distributed in the tropics and subtropics. Crassulacean acid metabolism (CAM), a photosynthetic adaptation to the occurrence of succulent leaves or stems, indicates that convergent evolution occurred in response to drought stress during species radiation. Here we provide the chromosomal level assembly of Cissus rotundifolia (an endemic species in Eastern Africa) and a genome-wide comparison with grape to understand genome divergence within an ancient eudicot family. Extensive transcriptome data were produced to illustrate the genetics underpinning C. rotundifolia’s ecological adaption to seasonal aridity. The modern karyotype and smaller genome of C. rotundifolia (n = 12, 350.69 Mb/1C), which lack further whole-genome duplication, were mainly derived from gross chromosomal rearrangements such as fusions and segmental duplications, and were sculpted by a very recent burst of retrotransposon activity. Bias in local gene amplification contributed to its remarkable functional divergence from grape, and the specific proliferated genes associated with abiotic and biotic responses (e.g. HSP-20, NBS-LRR) enabled C. rotundifolia to survive in a hostile environment. Reorganization of existing enzymes of CAM characterized as diurnal expression patterns of relevant genes further confer the ability to thrive in dry savannas.
Introduction
The plant family Vitaceae is well known for its economically important fruit crop, the grape (Vitis vinifera). It comprises 16 genera with >950 species and is classified into five tribes [1]. Many species in the family are dominant climbers in tropical/temperate forests, savannas, and mountains [2], representing one of the earliest diverged lineages in the major rosid clade of eudicot plants (Fig. 1a and b) [3, 4]. The grapevine (PN40024) was the first fruit crop species whose genome was decoded [5].
Figure 1.
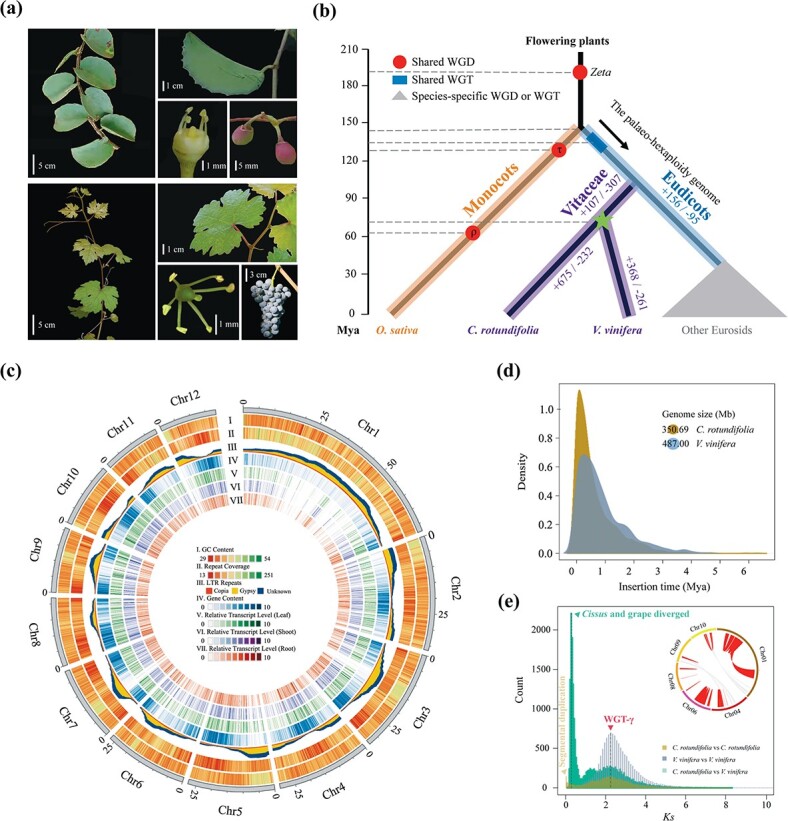
Morphological features and genome evolution of C. rotundifolia. a The succulent leaf, leaf abaxial surface, flower (without petals), and fruit of C. rotundifolia (top). Correspondingly, the vegetative and reproductive organs of grape (bottom). b Divergency history between Cissus and grape within the phylogeny of flowering plants. Age estimates of each node are based on 342 single-copy genes from 13 representative plant species. WGD or WGT is indicated on the corresponding branches. The numbers of gene family expansions and contractions are indicated along the related branches. c Distribution of genomic features of the Cissus genome. Each track shows the GC content, repetitive sequence distribution, gene density, and gene expression profile in different tissues from outside to inside. d Estimation of LTR activity shows a very recent burst event in Cissus <90.77 kya and a much more severe invasion of LTR than in grape. e Distribution of Ks for the whole paranome of Cissus and cross-comparison between Cissus and grape. Right corner of the image shows the segmental duplications within the chromosomes.
Cissus L. is the largest genus in Vitaceae, comprising >300 species [1], and the only genus of the tribe Cisseae Rchb. Unlike grapevines, which are mostly distributed in temperate regions, Cissus mainly occurs in the seasonal arid regions of the tropics and subtropics [6]. Species in this genus exhibit considerable variation in both chromosome number (2n = 24–66) and genome size (1C = 0.38–1.03 pg) [7]. Morphological modifications such as succulent leaves or stems have arisen in some Cissus species inthe face of drought stress (Fig. 1a) [8]. Therefore, these groups provide an opportunity to investigate the strategies of plant adaptive evolution with respect to drought tolerance. Crassulacean acid metabolism (CAM) is a water-use-efficient adaptation of photosynthesis that has evolved independently many times in diverse lineages of flowering plants [9]. The genomes of CAM plants, including pineapple (Ananas comosus), orchid (Phalaenopsis equestris), Kalanchoe fedtschenkoi, Dendrobium catenatum, Dendrobium officinale, and Sedum album, are available [3, 10–14]. Comparative analyses between K. fedtschenkoi and non-CAM species identified convergence in protein sequence in nocturnal CO2 fixation and carbohydrate metabolism [14]. CAM is also widespread in Cissus [6, 15–17], enabling us to dissect the convergent evolution of CAM in the plant kingdom.
Cissus rotundifolia Lam. is mainly distributed in the tropical savannas of Eastern Africa. The leaves are consumed as a local traditional food [18]. It has a relatively small genome with 1C = 0.38 pg. [7] To understand the adaptive strategies of the genus Cissus in the harsh climate, we generated and compared the draft genome of C. rotundifolia with that of V. vinifera to uncover how the genome has evolved and to identify the genes underpinning its arid adaptation. Further, we conducted extensive transcriptome comparisons to characterize the evolution of CAM in C. rotundifolia.
Results
Genome assembly, annotation, and repetitive sequence characterization
We assembled a highly heterozygous (1.19%) genome of C. rotundifolia, by combining the 39.38 Gb of PacBio Sequel sequences (~106×) and 28.31 Gb of Illumina paired-end reads (~77×) (Supplementary Data Fig. S1, Supplementary Data Table S5). We arranged 3289 contigs (contig N50 = 186 kb) based on the spatial relationship deduced from 130.44 Gb of Hi-C assay data (~362×) (Supplementary Data Table S6). A total length of 350.69 Mb scaffolds was ordered and anchored onto 12 pseudo-chromosomes with scaffold N50 up to 27.6 Mb, covering 94.53% of the assembled genome (Fig. 1c, Supplementary Data Fig. S1, Supplementary Data Table S7). We identified 169 723 homozygous mutation bases representing 0.045% of the assembled genomes (one error per 2.22 kb).
A total of 30 824 protein-coding genes were predicted by using a combination of ab initio, transcript evidence, and homology-based methods. We used the Swiss-Prot, NCBI, GO, KEGG, and eggNOG databases to annotate ~82.15% of the coding genes (Supplementary Data Table S8). Moreover, Benchmarking Universal Single-Copy Orthologs analysis suggested that 92.4% of the genes could be recovered (Supplementary Data Table S9). In addition, we identified 692 transfer RNAs, 128 microRNAs, 232 ribosomal RNAs (18S, 28S, 5.8S, and 5S), and 971 small nucleolar RNAs (Supplementary Data Fig. S2).
Repetitive sequences dominated 47.41% of the genome, of which 31.07% were LTR elements (Supplementary Data Table S10). Estimates of sequence divergence times between the adjacent 5′ and 3′ LTRs of the same retrotransposon suggested a very recent burst of activity <90.77 thousand years ago (kya) and much severe invasion than in grape (Fig. 1d, Supplementary Data Table S10). Further, we found 584 679 (12.90 Mb) SSRs with six as the most abundant unit size, slightly less than that in V. vinifera (PN40024, 930 680, 23.05 Mb) (Supplementary Data Table S11).
Gross chromosomal shuffling reassembled the C. rotundifolia genome
We collected a total of 342 single-copy genes (61 639 homologous amino acids) among 13 representative angiosperms to clarify the divergence of C. rotundifolia (hereafter Cissus) and V. vinifera (hereafter grape) (Supplementary Data Fig. S3, Supplementary Data Table S1). Reconstruction of the phylogeny indicated these two species had separated as early as 60.19–84.68 million years ago (Fig. 1b), coinciding with the distribution pattern of synonymous substitutions per synonymous site (Ks) (Ks = 0.33) (Fig. 1e). The subsequent Ks analysis of all paralogous genes in the genomes of Cissus and grape and syntenic regions support a shared whole-genome triplication, namely WGT-γ, the ‘pivot’ paleo-hexaploidy event that occurred in the most recent common ancestors (MRCAs) of all eudicots (Fig. 1b and e) [5, 52]. No more signatures of WGD were observed in Cissus and grape genomes. Nevertheless, in Cissus, there is another small peak of duplicated genes with Ks = ~0.1, and the majority of the paired genes were devoid of interchromosomal regions (Fig. 1e, Supplementary Data Fig. S4). Approximately 236 duplication events occurred inside their chromosomes and were characterized as segmental duplications. Such recent local gene cluster duplication burst finally accounted for 8.31% of Cissus functional genome profiles (4.75% in grape; Supplementary Data Table S12).
Interestingly, gene ontology (GO) analysis of the segmental duplication gene clusters in two species revealed similar function enrichments mostly associated with basic biological processes, such as phosphorus metabolic process and cellular protein metabolic process. Meanwhile, specific biochemical pathways (e.g. brassinosteroid homeostasis) and chromosome dynamics (e.g. meiotic chromosome condensation and meiotic sister chromatid cohesion) are only enriched in Cissus (Supplementary Data Table S12). The role of the ‘connected’ gene cluster as a module of function during speciation and the retention of duplicated segments with gene dosage relationship preserved would be worthy of further investigation [72].
Considerable high collinearity was observed between Cissus and grape chromosomes, presenting a pattern of a combination of each two of the 19 chromosomes in grape often corresponding to one chromosome in Cissus, leading to fewer monoploid chromosome numbers in the latter (n = 12) (Fig. 2a). To search for genomic features that might contribute to Cissus’s modern 12 chromosomes, we compared the ancestral eudicot karyotype (AEK) reconstructed from an integration of the Vitales (grape), Malvales (cacao), and Rosales (peach) major subfamilies to uncover that at least five fusions occurred in Cissus after inheriting 21 AEK post-γ chromosomes from the MRCA of eudicots (Fig. 2b) [58]. Specifically, pairwise comparisons among Cissus, grape, and AEK post-γ revealed that 82.59% of grape genomic regions were linked to AEK post-γ, higher than the number (71.24%) of Cissus (Supplementary Data Table S2). This may be partly due to the ancestral reference derived from a comparison of grape–cacao–peach, particularly when the grape preserved more ancestral genomic organizations. Alternatively, the Cissus genome may have lost more ancestral gene arrays than the grape, probably attributable to a higher frequency of chromosomal rearrangement and recombination [73, 74]. Beyond that, 17.7% of grape genes (4757 genes) were embedded in syntenic blocks with a 3:1 relationship to each Amborella trichopoda [52] region that resulted from WGT-γ, higher than syntenic block genes in Cissus genome (13.7%, 3687 genes) (Fig. 2c and d, Supplementary Data Table S3). Likewise, the number of syntenic genes was lower in Cissus (19.3%, 4188 genes) than in grape (23.4%, 5089 genes) when aligned with Aristolochia fimbriata (a species that is similar to Amborella in lacking further WGDs since the origin of extant angiosperms) [75]. Moreover, 76% genes (2419 genes) in chromosome 1 were specific to Cissus (Fig. 2b, Supplementary Data Fig. S5, Supplementary Data Table S13). Together with the above, it would imply a more diverged genome of Cissus reshaped after long-term separation from Vitis.
Figure 2.
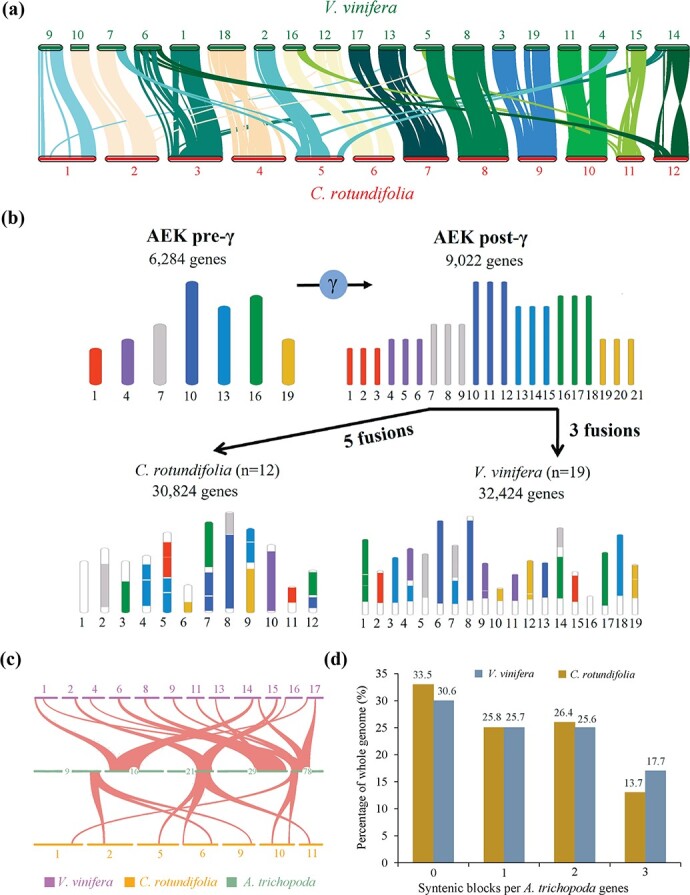
Gross chromosomal rearrangement underlying 12 modern C. rotundifolia chromosomes. a Macro-synteny patterns between C. rotundifolia and V. vinifera. The C. rotundifolia chromosomes are numbered according to their physical length from long to short. b Seven colored codes are used according to the earlier prediction of seven ancient chromosomes of AEK pre-γ, the schematic representation of paralogous regions derived from a grape–cacao–peach comparison (Freeling, 2009) [72]. The karyotypes of C. rotundifolia and V. vinifera were derived from syntenic comparison with AEK pre-γ and were defined by the occurrence of the syntenic regions as linked clusters in AEK pre-γ, independently of intrachromosomal rearrangements. The evolutionary events were predicted according to the more parsimonious model of evolution. c The syntenic relationship among Cissus, grape, and Amborella. Each A. trichopoda scaffold region aligns with up to three regions in either Cissus or grape, which are highlighted in red. Shades represent matching gene pairs. d Statistics of syntenic regions among A. trichopoda, V. vinifera, and C. rotundifolia. The subset of the proportion of genes in syntenic blocks to the whole genome is indicated on the histograms.
Functional divergence enabled Cissus adaptation to aridity
In Cissus, 675 orthogroups were remarkably expanded (P < .05) and 232 were diminished (P < .05) compared with other representative eudicots (Supplementary Data Fig. S3). The expanded orthogroups were mainly enriched in the abiotic/biotic stress-responsive pathways, metabolism of carbohydrates, and hormone biosynthesis (Fig. 3a). Cytochrome P450, found in dramatic proliferation (Supplementary Data Table S14), could contribute to the foliar wax deposition in Cissus [76]. Some polysaccharide-related genes, such as pectate lyase, pectinesterase, and polysaccharide biosynthesis genes, also displayed an increased paralogous number, probably attributable to the succulent leaf formation through the modification of pectin and other polysaccharides in cells [8]. Apart from the genes that directly contributed to the leaf character, transcription factors like MYB, WRKY, AP2/ERF, GRAS, and LEA were strongly expanded (Fig. 3a), suggesting the probability of enhanced abiotic stress resistance and secondary metabolism in Cissus [77–79]. To further address the functional divergence of Cissus referring to adaptation, we compared its gene repertoire with that of grape, which indicated that selective amplification of genes belonging to plant immunity had occurred in these two species (Supplementary Data Fig. S6, Supplementary Data Table S15). Among respective orthogroups, nucleotide-binding site leucine-rich repeat (NBS-LRR) genes were found in favor of expansion in both species but showed preference for different subclasses (e.g. orthogroup 12 in the grape; orthogroup 4,7 in Cissus). The co-abundance of R genes would represent the basic objective of an organism to protect itself against surging threats from microbial pathogens [80, 81]. The significant copy number variation of paralogous genes (orthogroup 13, terpenoid cyclase; orthogroup 2, TMV resistance protein N-like) likely suggested the different responses to pathogen induction [82, 83]. Additionally, heat shock protein like 20 (HSP20-like) were found particularly amplified in Cissus and upregulated in its shoot and leaves compared with root (Supplementary Data Fig. S6, Supplementary Data Table S16). This would fairly reflect the increased ability of Cissus’s vegetative organs to deal with heat shock and promote resistance to environmental stress factors [84, 85]. The enrichment pattern of the gene family in Cissus led us to investigate if a similar preference for gene proliferation occurred in other succulent species. To this end, we took another four typical succulent plants (A. comosus, H. undatus, K. fedtschenkoi, and K. laxiflora) into account in the gene family comparison. We found that 88 of the 97 335 orthogroups demonstrated succulent-specific expansion, which were significantly (P-adjust <.05) enriched in ‘terpene synthase’ and ‘HSP20’ (Fig. 3b, Supplementary Data Tables S17–S19). However, 178 orthogroups that were GO-termed mainly as serine/threonine-protein kinase receptor precursor (SKR), cysteine-rich receptor-like protein kinase (CRK), and wall-associated receptor kinase (RLK) were observed in co-expansion in the other 13 non-succulent plant genomes investigated (Fig. 3b, Supplementary Data Tables S17, S18, and S20). The diverged preference for functional gene families would reflect a specialized convergent mechanism in succulent plants dealing with high temperatures and water deficiency [8]. On the other hand, we identified 1878 tandemly duplicate (TD) arrays of two or more genes in Cissus, and the total number of genes in such arrays is 4746, slightly higher than the 3958 genes in 1524 TD arrays in grape (Supplementary Data Table S21). There are 2582 TD genes shared in two species, whose functional classification is mainly enriched in 134 GO terms (e.g. oxidoreductase activity, oxidation–reduction process, and response to auxin), and a total of 2164 TD genes are species-specific in Cissus (Supplementary Data Table S22). Functional bias in TD retention was observed encompassing different periods of evolution in Cissus (Fig. 3c). An overrepresented number of genes in the Cissus lineage were enriched in cell wall-related pathways (e.g. cell wall modification, cell wall organization, and xyloglucan metabolic activity), probably conferring its succulent leaves or stems [86]. In contrast, functional categories specific to grapes were mainly associated with stress responses (Fig. 3c, Supplementary Data Table S23). The result is consistent with the notion that TD genes would have a lineage-specific selection [72]. Nevertheless, earlier studies in Arabidopsis and rice demonstrated that an elevated probability of retention of stress-responsive TD is preferential for adaptive evolution after speciation [87, 88]. The functional bias of TD in Cissus indicates that genes referred to as morphological innovation for adaptation might be particularly selected and expanded via local duplication. It would be interesting to check if a similar profile of lineage-specific TD is exhibited in other morphology-specialized plants. We found that lineage-TD genes categorized as ‘cellular component-related’ and ‘resistance’ were overrepresented in gross tandem duplicated genes in succulent species, in contrast to the discrete pattern that occurred in the other non-succulent plants (P = .03 and P = .007) (Fig. 3d, Supplementary Data Table S24). Local gene amplification with a high frequency of gene birth/death plays a critical role in plants’ adaptive responses to environmental stimuli and is mostly attributable to gene copy number and allelic variation within a population [79, 87]. The fashion of TD expansion in succulent plants observed here would suggest another pattern of functional bias in TD retention during seed plant evolution. We speculated that the intense environment change provided multiple options for plants on morphological innovation and rapid expansion of resistance genes.
Figure 3.

Evolutionary history of functional profiles of C. rotundifolia genome. a Heat map showing categorized orthogroups that have significantly increased paralogous numbers in Cissus compared with other angiosperms analyzed. b Scatter plot displaying the expanded orthogroups in 5 succulent plants and 13 other non-succulent plants. Numbers in square brackets associated with circle sizes stand for -log (P-adjust), where P-adjust is the P-value of the binomial test adjusted for multiple testing. 1–18 are terpene synthase, plant self-incompatibility protein S1, hsp20/α crystallin family protein, aspartic proteinase nepenthesin-1 precursor, eukaryotic aspartyl protease family protein, leucine rich repeat protein, F-box domain and LRR-containing protein, MATE efflux family protein, nuclear transcription factor Y subunit, UDP-glycosyltransferase, serine/threonine-protein kinase receptor precursor, retrotransposon protein, disease resistance RPP13-like protein 1, cysteine-rich receptor-like protein kinase, leucine-rich repeat receptor-like protein kinase family protein, retrotransposon protein, wall-associated receptor kinase, and F-box family protein. c GO categories with an overrepresented number of tandemly duplicated genes in expanded orthogroups encompassing different evolutionary periods of Cissus (upper) and the functional bias of tandem duplicate gene retention in grape (lower). The number of TD events is indicated on the branches. d Percentage of GO categories from expanded lineage-specific TD in succulent plants and other non-succulent plants. Cellular component and resistance categories in two subgroups were tested by a two-sample t-test.
CAM photosynthesis in C. rotundifolia
CAM photosynthesis is a recurrently evolved strategy for high water use efficiency (WUE), enabling plants to survive in water-limited environments [89]. In CAM plants, carbon dioxide (CO2) is fixed in the cytosol and stored as malic acid in the vacuole during the night (Fig. 4c). The stomata remained closed during the daytime to decrease water loss by evapotranspiration, and the stored malic acid is decarboxylated to release CO2 that could be re-fixed through the Calvin–Benson cycle [90]. Such a feature of CO2 uptake was ubiquitous in the Cissus lineage, which may have facilitated the spread of the genus from wet into arid tropics [6].
Figure 4.

The CAM pathway in C. rotundifolia. a Diurnal variation of titratable acidity in C. rotundifolia leaves.b Expression patterns and cis-regulatory elements of CAM-related genes across the diurnal variation. The expression level of each gene is shown using the log10-transformed method. The numbers of five circadian clock-related motifs, including G-box element, evening element (EE), morning element (MOM), CIRCADIAN CLOCK ASSOCIATED 1 (ACC1) binding site, and TCP15, are shown in the 2-kb promoter region of each gene. c Overview of the CAM pathway. The carboxylation process (dark period) is shown on the left and the decarboxylation process (light period) on the right. Enzymes are marked in blue and green, respectively, with corresponding expression profiles. A network was constructed for Cluster 4 (d) and Cluster5 (e) using ARACNE. The top 1% of each network is highlighted with a yellow circle, and blue nodes with more than 10 edges were CAM-related genes in C. rotundifolia. The yellow and blue nodes are annotated in Supplementary Data Table S29.
To investigate CAM evolution in Cissus, we determined the pattern of diurnal oscillation of titratable acidity in the leaves of C. rotundifolia in growth chambers with a climate close to the dry seasons in Kenya (Fig. 4a). The amount of titratable acid reached its maximum (150 μeq g−1 FW) early in the dawn (~6:00) and dropped to its minimum (20 μeq g−1 FW) later in the day (~18:00), which qualified C. rotundifolia as a CAM species [16, 91]. We identified 47 candidate CAM pathway genes based on their orthologs in pineapple (A. comosus L. Merr., CAM plant) [11], maize (Z. mays L., C4 plant) [65], rice (O. sativa L., C3 plant), K. fedtschenkoi (CAM plant) [14], and P. equestris (CAM plant) [10]. Further, these genes were well categorized into nine gene families that characterized the core network of carboxylation and decarboxylation pathways (Supplementary Data Table S25) [11]. These gene families showed no significant expansions in C. rotundifolia compared with other plants, as shown in Supplementary Data Table S26, implying that CAM photosynthesis might evolve through the re-organization of existing enzymes [69].
The diurnal expression patterns of these CAM genes were interrogated by transcriptome comparison of leaves during 3-hour intervals over a 24-hour period. In general, the expression of 21 genes showed typical circadian patterns as defined via a polynomial regression (Fig. 4b). The transcripts of enzymes involved in carbon assimilation, such as carbonic anhydrase (CA), phosphoenolpyruvate carboxylase kinase (PPCK), and malate dehydrogenase (MDH), were highly accumulated at night. As the core genes involved in CO2 fixation, four PEPC genes of Cissus have extremely high expression in the daytime rather than at nighttime (Supplementary Data Fig. S7). Similar expression patterns were also found in other CAM plants, such as Kaladp0095s0055.1 in Kalanchoe and Sal_001109 in S. album [3, 12, 14, 92]. Correspondingly, the enzymes that participate in decarboxylation processes, such as MDH, ME-NADP, and phosphoenolpyruvate carboxykinase (PEPCK) were highly expressed during the day (Fig. 4b). Interestingly, as a major protein for carbon fixation, previous studies in pineapple have shown that only the βCA subfamily is expressed at nighttime and early morning in green leaf tissues [11]. We observed all five CAs, including α (1), β (3), and γ (1) expressed at night in C. rotundifolia (Fig. 4b). The expression of βCA1 in Cissus and pineapple (Rcr_ac > .8) increased during the night, and a peak occurred at 9:00 in the morning, while its orthologs in Arabidopsis (Rcr_at < .5) showed stable and lower expression during the diurnal cycle (Supplementary Data Fig. S8, Supplementary Data Table S4) [68]. Beyond that, members of MDH also showed diverged expression patterns as MDH2 was more active at night while the other four MDHs were upregulated during the day, consistent with other CAM plants (Fig. 4b) [11, 14, 93]. This may be associated with their different roles in decarboxylation processes since MDH catalyzes the reversible reaction between oxaloacetic acid and malic acid.
We constructed the gene co-expression network based on the transcriptome data from nine mature leaf samples collected every 3 hours over a 24-hour period. Among 27 modules identified, MEbrown2 (2020 genes that were highly expressed during the night) was significantly (P < .05) related to the night period (Supplementary Data Fig. S9). We found that βCA2, βCA3, and γCA were also found in this MEbrown2 module. Pathways such as response to organonitrogen compound and root meristem growth in this module were significantly enriched in this module (Supplementary Data Table S27). The MEdarkorange2 module (311 genes that were highly expressed during the day) was found to be significantly associated with the day period. We found PEPCK, PPDK, MDH6, and ALMTs in this module. Biological processes such as response to abiotic stimulus were enriched in this module (Supplementary Data Table S27).
Moreover, transcripts in leaf with time-course diel expression patterns were classified into nine clusters (Supplementary Data Fig. S10, Supplementary Data Table S28). The highly connected hub genes identified by network construction for each cluster were associated with CAM genes. For example, Cluster 4 contained PPCK2 (CRGY0218762) and γCA (CRGY0214246) and had patatin-like phospholipase as the hub (Fig. 4d, Supplementary Data Table S29). Heat shock protein, which played important roles during stress responses in many plants, was the hub in Cluster5 and connected with PEPC1 and PEPC5 (Fig. 4e, Supplementary Data Table S29).
The promoters of the diurnally expressed photosynthetic genes were enriched in circadian clock-related cis elements (Fig. 4b) [69, 94]. Comparative analysis between Cissus, pineapple, rice, maize, and sorghum showed that only βCA1 with typical circadian patterns in Cissus had one evening element (EE) (Supplementary Data Table S30) [11], suggesting its contribution to CO2 fixation via a combination EE motif during nighttime [12]. Additional comparison within CAM genes indicated that EE and G-box elements were mainly enriched in the subgroups of highly expressed genes at night (Supplementary Data Table S30).
The higher WUE in CAM plants relied on the appropriate control of stomatal movement during day and night. We identified the stomata open/close-related genes in the C. rotundifolia genome based on their homologs in Arabidopsis (Supplementary Data Table S31) [69]. A subset of genes that are responsible for the stomata opening or closing were uniquely expressed either at night or during the daytime, which implied the coincidental organization of stomata movement and CAM genes (Supplementary Data Fig. S11). The expression patterns of stomata movement genes were compared with their orthologs in Arabidopsis (Supplementary Data Table S4). We identified 86 out of 141 stomata movement genes with diurnal expression patterns in Cissus (Supplementary Data Table S31). The diurnal expression of 64 genes showed a low correlation with its orthologs in Arabidopsis (Supplementary Data Fig. S11), suggesting their putative roles during stomatal movement in Cissus. OST1 (Stomatal opening factor1), which plays a vital role in abscisic acid (ABA)-triggered stomatal closure [95], was found to be highly expressed at 9 a.m. (Supplementary Data Fig. S11), compared with accumulated transcription of its orthologs at night in Arabidopsis. The result was also consistent with diel expression patterns of OST1 in Agave americana, K. laxiflora, and Kalanchoe [92, 96, 97]. Interestingly, the MOE and G-box motifs were enriched in the promoter of OST1 in C. rotundifolia but not in V. vinifera and Arabidopsis (Supplementary Data Table S31). These results indicate the contribution of coordinated transcriptional regulation of circadian in CAM pathway in C. rotundifolia.
Discussion
Vitaceae is a sister to most of the rosids in the highly diverse rosid clade of the flowering plants [98]. Grape (V. vinifera) was believed to be one of the most slowly evolved species, representing a more conserved ancestral structure of the genome that can be used to unravel the evolution and genome duplication history of other eudicots [58]. Here, we present another genome in Vitaceae, C. rotundifolia, to show a probable diverse evolutionary history considering distinct ecological niches. It is exciting to observe the evidence of the paleo-hexaploidy event shared by Cissus and grape. Moreover, the lack of any other WGDs suggests Cissus also might hold a relatively ancestral state of genome organization after divergence from the common ancestors [75, 99, 100]. This would be reflected by ~13.7% of the total genes in Cissus (~17.7% of grape) were located in the syntenic regions which exhibited ratio of 3:1 to the genomic region of Amborella. Besides, we propose that more frequent chromosome shuffling, including at least five independent fusion events, might have occurred in Cissus after its origin (three fusions in grape). The recent segmental duplications probably further accounted for the increased genetic and biological complexity [84], together with chromosome fusions serving as a prelude to the modern karyotype configuration of Vitaceae.
The very recent burst of activity in LTRs (90.77 kya) detected in Cissus could probably be explained by the severe climate transition from arid to wet that occurred in the past 100 kya in eastern Africa, where substantial ecological habitat turnover was recorded (Fig. 1d) [101]. However, such severe invasion of retrotransposons left a smaller genome (350.69 Mb) than grape (475 Mb). Indeed, we found much higher gene density in Cissus than in grape, indicating the lower frequency of repeats in intergenic regions where LTR insertion is usually preferred (Supplementary Data Table S32). Moreover, Cissus probably had experienced a fair loss of ancient genomic arrays compared with grape. Together, transposable element removal and sequence elimination accompanied by chromosomal rearrangement (e.g. chromosomal fusion and recombination) could contribute to the selected, size-reduced genome of Cissus [73, 74]. Nevertheless, a small genome size could be particularly advantageous for plants to enhance water use efficiency through increased stomatal responsiveness of smaller cells [102, 103].
Seasonal drought is one of the biggest challenges for agriculture in East Africa. The evolution of the water storage tissue of plants is the most common adaptive strategy in arid and semi-arid regions [104]. The leaves of C. rotundifolia are succulent, which exemplifies a convergent evolution with plants from dry habitats, like agave [105]. We found gene families of enzymes responsible for polysaccharide synthesis, such as pectate lyase and pectinesterase, were remarkably expanded in Cissus. Therefore, modified pectin and other polysaccharides in cells may confer the occurrence of succulent leaves [106]. The noticeable proliferation of gene families associated with biotic and abiotic responses (i.e. P450, LEA, and LRR) would play key roles in the objective arms race against pathogens and unfavorable environments [80, 107, 108].
A clear pattern of selective amplification of immunity genes in Cissus and Vitis was present, indicating a potential functional divergence related to adaptations. Further, succulent-specific expansion in a certain gene family (e.g. terpene synthase, HSP20-like) suggested a convergent mechanism in such a morphologically modified group. Interestingly, the fashion of TD expansion in succulent plants was also correlated to morphological innovation, which might unveil another functional bias pattern of TD content in the face of rapid and intense environmental change during seed plant evolution.
The innovation of the CAM photosynthetic pathway in Cissus further contributes to its adaptation in the dry savannas by enhancing WUE [109]. The decarboxylation in Cissus is likely induced in two ways: one is driven by ME and PPDK enzymes, and another is catalyzed by PEPCK enzyme (Fig. 4c, Supplementary Data Fig. S8). For decarboxylase process, ME and PPDK enzymes were used in K. fedtschenkoi and PEPCK enzyme was utilized in pineapple [11, 14]. The genes had undergone convergent evolution in K. fedtschenkoi, which included PEPC, nucleosome assembly protein 1-like 4 (NAP1L4), transcription factor hy5-like protein (HY5), and chloroplast-localized glucose-6-phosphate isomerase (GPI). However, no amino acids showed convergent evolution patterns for CAM and stoma-related genes in Cissus by a similar analysis [14], suggesting that the evolution of the characteristics may be derived from multiple modifications. In S. album, the number variation of cis elements between C3 and CAM-cycling status showed a phase shift during the daytime [12], while cis elements of CAM cycling genes in Isoetes howellii are not strongly associated with transcript expression, additionally lacking ME and G-box on promoters of CAM genes [93]. The EE is over-presented in the promoter of evening-phased genes [110]. Compared with other plants [11], βCA1 with one EE in Cissus may contribute to CO2 fixation during nighttime (Supplementary Data Table S30). The identification of cis-regulatory elements in the promoter of CAM genes in C. rotundifolia would help to explain the evolution of CAM from C3 plants and provide valuable information for breeding drought-tolerant crops.
Materials and methods
Plant materials
Stem cuttings of C. rotundifolia were collected from Endau hill, Kitui County, Kenya (1°17′49″ S, 38°31′59″ E). Voucher specimens (JAJIT-MU0128) were deposited in the Wuhan Botanical Garden herbarium (HIB). Young leaves of C. rotundifolia were collected for genome size evaluation and DNA isolation. Root tips were used for chromosome number determination. Mature leaves, stems, and young roots were collected for RNA isolation and tissue-specific transcriptome analysis.
C. rotundifolia individuals were grown in a glasshouse under artificial conditions (16 hours light/8 hours dark, 25°C) in 3-l pots. After being well watered, plants were cultivated for 1 week and transferred to an incubator with 12 hours light (from 6:00 to 18:00) at 30°C and 12 hours dark (18:00 to 6:00) at 20°C (30% humidity). After 3 days, leaves were collected from 18:00 on 9 January 2020to 18:00 on 10 January 2020 at 3-hour intervals. We selected nine time points including 18:00, 21:00, 24:00, 3:00, 6:00, 9:00,12:00, 15:00, and 18:00 for the study. Three biological replicates were collected for each sample and immediately frozen in liquidnitrogen. Samples were stored at −80°C for RNA extraction and titratable acid measurement.
Estimation of genome size
About 28.31 Gb of Illumina reads were used to evaluate the genome size of C. rotundifolia by K-mer analysis. The K-mer frequency distribution was calculated using Jellyfish (v2.2.6) [19] with the K-mer length of 19. Genomic heterozygosity was estimated by GenomeScope (v2.0) [20]. Flow cytometry was used to determine the nuclear DNA content using hand-chopped materials as described by Galbraith et al. [21], with minor modifications. Woody plant buffer was used instead of Tris–MgCl2 buffer to isolate the nuclei [22]. Raphanus sativus cv. ‘Saxa’ was used as the reference standard. Young leaves of C. rotundifolia and R. sativus were collected, and the protocols were as described in detail by Gichuki et al. [24].
Chromosome count
Root tips were treated with a saturated solution of 1-bromonaphthalene for 3 hours at room temperature (25–28°C) to halt cell division. Microscope slides were prepared from the treated root tips using the protocol developed by Kirov et al. [23] and Gichuki et al. [24]. The prepared microscopic slides were stained with 4′,-6-diamidino-2-phenylindole (DAPI), and images were captured using a fluorescence microscope (Leica DMi8) fitted with a camera (Leica DFC 550).
Genome sequencing
Genomic DNA of C. rotundifolia was extracted using the CTAB-based protocol described by Doyle and Doyle [25], with minor modifications. Briefly, a washing step was included before CTAB extraction to exclude secondary metabolites. The washing buffer contained 50 mM Tris–HCL, 5 mM EDTA-2Na, 0.35 M d-sorbitol, 1% (w/v) polyvinyl pyrrolidone (PVP-K 30), and 1% 2-hydroxy-1-ethanethiol. Illumina libraries with 450-bp insertions were constructed according to the Illumina standard protocol, and the paired-end libraries were sequenced to ~77 × coverage on the Illumina Hiseq platform. Approximate 20 kb SMRTbell libraries were prepared and sequenced on the PacBio Sequel system following standard protocols (Berry Genomics Corporation, Beijing, China).
Hi-C library construction and sequencing
Young leaves from the same C. rotundifolia plant used for genome sequencing were collected for Hi-C analysis. The Hi-C library construction and sequencing for chromosome-level assembly were implemented by Biomarker Technologies Corporation (Beijing, China) using a previously published method [26]. Briefly, shoot tips of C. rotundifolia plants were covered with a black box, and the etiolated leaves were fixed with formaldehyde and lysed. The cross-linked DNA was digested overnight using HindIII. Digested fragments were biotinylated and ligated to form chimeric junctions that were enriched, sheared, and processed. Then libraries were produced on the Illumina Hiseq platform. The paired-end Hi-C reads were uniquely mapped onto the contigs using Juicer [27], and the non-duplicate mapped results were used as the input for the 3D-DNA pipeline [28] to construct the genome sequence. Two rounds (−r 2) provided the best results with an assembly of 12 pseudo-chromosomes and N50 of ~28 Mb. Juicebox was used to fine-tune the assembled genome in a graphic and interaction matrix [29].
Genome assembly
The contig-level assembly of the C. rotundifolia genome was processed by combining ~100× PacBio long reads and ~ 76× Illumina short reads. The raw PacBio reads were initially corrected and trimmed by CANU-1.8 with the parameters of genomeSize = 600 m, useGrid = false, maxMemory = 200 g, ovsMemory = 16G, ovbConcurrency = 15, ovsConcurrency = 15 and batOptions = -dg 3 -db 3 -dr 1 -ca 500 -cp 50 [30]. Then, the corrected PacBio reads were assembled by two widely used PacBio assemblers, CANU-1.8 [30] and WTDBG2 [31]. We used N50 and the genome size of each assembler to inspect assembly quality. The Canu assembled results were adopted in the end. Illumina reads were used to further polish the PacBio assembly using the Pilon program with the following parameters: -verbose -mindepth 4 -fix snps, indels -vcf [32]. Then, the redundancy of the assembled sequences was removed to improve the continuity of the assembled contigs using Redundans with the parameters -identity 0.5 -overlap 0.66 and Purge Haplotigs with the parameters -l 15 -m 60 -h 190 [33, 34].
Evaluation of the genome assembly
The genome assembly quality of C. rotundifolia was evaluated using two methods. Firstly, the error rate considering the homozygous mutation was estimated by mapping the 28.31-Gb whole-genome sequence (WGS) reads onto this assembly by BWA software. Compared with the eudicotyledons_odb10 database, the single-copy orthologs in the assembled genome were identified and the completeness of the assembly was evaluated using Benchmarking Universal Single-Copy Orthologs v2 with the -long and default parameter, respectively [35].
Repeat sequence annotation
Repeat structures were analyzed by a combined strategy of de novo prediction and homology-based prediction. A de novo repeat library of the C. rotundifolia genome was built by RepeatModeler (v1.0.11, http://www.repeatmasker.org/RepeatModeler/) with the -engine ncbi parameter. Using this library, we processed repetitive sequences to annotate, classify, and mark by RepeatMasker (v4.0.7, http://www.repeatmasker.org/). Two built libraries were combined with the Repbase (v20170127, https://www.girinst.org/) [36] and Dfam (v20170127, http://www.dfam.org/) databases with default parameters [37]. Simple sequence repeats (SSRs) were identified using MISA (http://pgrc.ipk-gatersleben.de/misa/misa.html) [38], with unit length ranging from 1 to 7 and min-length set to 10 bp. Long terminal repeats (LTRs) were identified by LTR_retriever according to the method of Ou and Jiang [39].
Gene model prediction
Three approaches were combined to annotate protein-coding genes. Firstly, Illumina RNA-seq data from three representative tissues were assembled with two different strategies (de novo or genome-guided assembly) by Trinity (v2.2.0) [40]. The assembled RNA-seq data were then aligned to the assembled genome and its evidence-based prediction was performed using PASA (v2.0.2) [41]. Secondly, the ab initio methods AUGUSTUS (v3.3) [42], SNAP [43], and GeneMarkHMM (v 4.32) [44] with default parameters were used to predict gene models with training of the best candidate genes obtained from PASA [41]. Thirdly, protein sequences from closely related species, including V. vinifera (PN40024) and other Cissus species downloaded from NCBI, were used to annotate protein homologs of C. rotundifolia by GenomeThreader (https://genomethreader.org/). Finally, the annotation results generated from evidence-based prediction, ab initio prediction, and homologous mapping were combined by EVM (v 1.1.1) [45] to integrate the consensus gene model, and genes were renamed according to their position in the genome sequence with the prefix CRGY (C. rotundifolia genome).
Phylogenomic tree construction and gene family analyses
The protein-coding sequences from C. rotundifolia and 13 other representative species were used to identify orthologous groups, including those of Arabidopsis thaliana, Oryza sativa, V. vinifera, Actinidia chinensis, Coffea arabica, Solanum lycopersicum, Populus trichocarpa, Theobroma cacao, Carica papaya, Citrus sinensis, Fragaria ananassa, Malus domestica, and Prunus persica. All-vs-all BLASTP [46] with an e-value cutoff of 1e−05 was performed, and orthologous genes were clustered using OrthoMCL [47]. Single-copy genes were extracted from the clustering results and multiple sequence alignments were performed using MUSCLE (v 3.8.31) [48]. After removing low-quality alignment or divergent regions by Gblocks, high-quality aligned protein sequences remained. All aligned sequences were concatenated to one long sequence for each species, and these sequences were used to construct a phylogenetic tree by RAxML (v2.5.1) with the PROTGAMMAJTTF model and bootstrap of 1000 [49]. MCMCtree (4.8a) from the PAML package [50] was adopted to estimate the species divergence time according to TimeTree (http://www.timetree.org). Four divergence times were used in this analysis, including those of C. arabica and S. lycopersicum, A. thaliana and C. papaya, P. persica and M. domestica, and F. × ananassa and P. persica. The divergence times of V. vinifera and C. rotundifolia in TimeTree were also referenced in this study. The Markov chain Monte Carlo (MCMC) process analysis was set for 50 000 generations and 50 000 burn-in iterations. The OrthMCL results and time divergence tree were used as the input for the CAFÉ (v 3.1) program [51], which was used to identify expansions and contractions of gene families across 15 plant genomes. Family expansion and contraction were analyzed by Count, and the methods and parameters were according to a study of the Amborella genome [52]. Multi-species orthologous clusters with gene numbers >0 in V. vinifera and C. rotundifolia were considered orthologous groups between these two species. Expanded orthogroups were defined according to P-value <.05 and a gene number greater than the average value of multiple species. Dot plot representation of orthologous groups was performed with the R package ggplot2 (https://ggplot2.tidyverse.org/) [53].
Tandem duplication analysis
Four additional typical succulent species, including A. comosus, Hylocereus undatus, K. fedtschenkoi, and Kalanchoe laxiflora, were added to the gene family clustering (Supplementary Data Table S1). Multi-species orthologous clusters with a gene number >0 in V. vinifera and C. rotundifolia were used to identify lineage-specific expansion. Expanded orthogroups were identified with a P-value <.05 and gene number greater than the average value of multiple species. The tandem genes were identified by MCScanX [54], which was consistent with the method described in our whole-genome duplication (WGD) analysis. The gained tandemly duplicate (TD) genes of 18 species were obtained from the Count results and were further categorized into either co-expanded or lineage-specific expanded ones. The TD genes were gene ontology (GO)-termed by agriGO database (http://systemsbiology.cau.edu.cn/agriGOv2/index.php). Further, four succulent plants were annotated both by the GO database and the agriGO database.
Synteny analyses
All-vs-all BLASTP [46] (e-value 1e−05) and MCScanX [54] were used to predict the collinear relationships and positional features between C. rotundifolia and V. vinifera (PN40024). Blocks of >10 genes and gaps of <5 genes were obtained. The synteny map and dot plot were processed by MCScan and drawn by the python scripts in MCScan packages [55].
The segment duplication events were predicted using self-vs-self BLASTP [46] (e-value 1e−05) and MCScanX [54] among the C. rotundifolia genome, requiring at least five genes per collinear block. Subsequently, the pairwise sequences from the synteny blocks and segment duplication pairs were processed by ParaAT (v2.0) [56]. Values of the non-synonymous mutation rate (Ka) and synonymous mutation rate (Ks) were calculated using the NG estimation method in Kaks_Calculator (v2.0) [57]. The visualization plots of the Ks distribution were made using a custom R script. Additionally, WGD events were determined by the distribution of Ks of segment duplication pairs and identified by comparisons with the events of V. vinifera (PN40024). The ancestral eudicot karyotype (AEK) was inferred from the genomes of eudicot species with the smallest numbers of historical polyploidization events, including grape, cacao, and peach. Further, the AEK was refined as a post-τ ancestral monocot karyotype (AMK) with 10 protochromosomes and 13 916 ordered protogenes, a pre-τ AMK with five protochromosomes and 6707 ordered protogenes [58]. In the current study, the reconstructions of the karyotypes of V. vinifera and C. rotundifolia were advised by a previous study by Murat et al. [58], and the genes and gene orders were used to construct the 7 chromosomes and 21 chromosomes of AEK. To cover as many genes as possible, we used version 2.1 of the grape assembly, which anchored 32 424 coding genes (Supplementary Data Table S1). MCScanX [54], in a BLASTP and dot plot-based approach, was used to detect the syntenic blocks between C. rotundifolia versus AEK and V. vinifera versus AEK with default parameters. The protein of the pre-γAEK and post-γAEK was compared with Cissus and grape by BLASTP. The syntenic blocks were ordered according to the gene order of C. rotundifolia and V. vinifera. Some small syntenic blocks and small gaps were abandoned or closed to make the syntenic segments more complete. On the base of dot plot illustrations of the synteny between these two species, the karyotypic structures of the ancestral eudicots were explained by taking into account the lowest number of genomic rearrangements, which may have occurred between the AEK and modern eudicot genomes (Supplementary Data Fig. S5, Supplementary Data Tables S2 and S3).
Detection of significant expansion and contraction in succulent plants
To investigate the significant expansion or contraction of gene families, we divided 18 species into two categories, including five succulent plants (S5) and 13 non-succulent/other plants (O13). Five succulent plants, including C. rotundifolia, A. comosus, K. laxiflora, H. undatus, K. fedtschenkoi, and other 13 plants are described in Supplementary Data Table S1. The average number of genes per orthogroup between two categories was available to evaluate the significant events. For S5 plants, a binomial test with a probability of success of P(W) = 5/18 was used. The criteria of significant expansion or contraction are as follows: (i) a statistically false discovery rate-adjusted P-value <0.05 from the initial set of 97 344 orthogroups; (ii) the minimal contribution of about three for S5 and seven for O13 species; (iii) the ratio of contribution to per orthogroup satisfied with (S5n/5)/(O13n/13) > 1. We found that 88 orthogroups were expanded (corresponding to 5696 genes), and 178 were contracted in succulent plants relative to non-succulent plants.
Measurement of titratable acidity
The diurnal changes of titratable acid in leaves of C. rotundifolia were measured as described by Chen and Black [59]. The samples were collected as mentioned above. A total of 0.5 g of leaves of each sample were cut into pieces, placed in centrifuge tubes, and boiled for 30 minutes after adding 10 ml CO2-free distilled water. The supernatant after centrifugation was retained. An additional 10 ml CO2-free water was added to the pellet to and extraction and centrifugation were repeated. Total supernatants obtained by the two-stage extraction process were titrated to pH 8.3 with 0.01 mol/l NaOH, and the acidity of the leaf was represented in microequivalents of acid per gram fresh weight (μeq g−1 FW).
RNA extraction and RNA-seq library preparation
Total RNA was extracted from the samples using the Universal Plant Total RNA Fast Extraction Kit (BioTeke Corporation, Beijing, China). RNase-free DNase I was used to remove DNA from the extracted RNA. The purity and concentration of RNA were determined by a Nanodrop and Agilent 2100 bioanalyzer (Thermo Fisher Scientific, MA, USA). Subsequently, mRNA enriched by oligo(dT)-attached magnetic beads was randomly fragmented into short pieces with an additional fragmentation buffer. Then, first-strand cDNA was synthesized by random hexamer-primed reverse transcription, followed by second-strand cDNA synthesis. A-tailing mix and RNA index adapters were added. The obtained cDNA fragments were amplified by PCR, and then products were purified using Ampure XP Beads. An Agilent Technologies 2100 bioanalyzer was used for quality control of products. Finally, the cDNA library was constructed, and the MGISEQ-2000 platform was used for paired-end sequencing (2 × 150 bp). Approximately 40 000 000 bp were generated for each sample.
Transcriptome analysis
The quality of paired-end raw transcriptome data was checked by FastQC v 0.11.8 and trimmed using Trimmomatic (v 0.36) [60]. Then the trimmed reads were mapped onto the latest assembled genome of C. rotundifolia using TopHat (v 2.1.1) [61]. Using the gene model of C. rotundifolia, the expression levels of genes represented by FPKM (fragments per kilobase million) for each sample were calculated by Cufflinks (v 2.2.1) [61] with default parameters. The genes involved in the stomatal movement process and CAM pathway were picked to show their expression patterns using the pheatmap package in R.
Identification of CAM pathway and stomatal movement process-related genes
The genomes of A. comosus, V. vinifera (PN40024), O. sativa, Zea mays, and Phalaenopsis equestris were downloaded from the Pineapple Genomics Database [62], the Phytozome database (https://phytozome.jgi.doe.gov/pz/portal.html) [63], the Rice Genome Annotation Project (http://rice.plantbiology.msu.edu/) [64], MaizeGDB (https://maizegdb.org/) [65], and NCBI (https://www.ncbi.nlm.nih.gov/), respectively. Further, the CAM gene list of Kalanchoe was obtained from a supplementary table of its genome [14]. The list of gene families, which included carbonic anhydrase (CA), phosphoenolpyruvate carboxylase (PEPC), phosphoenolpyruvate carboxylase kinase (PEPCK), malate dehydrogenase (MDH), malic enzyme (ME), phosphoenolpyruvate carboxykinase (PPCK), and pyruvate phosphate dikinase regulatory protein (PPDKRP), was obtained from PGD [62]. All given gene sequences in each family from pineapple, O. sativa, and Z. mays, P. equestris and Kalanchoe were used as queries to search corresponding family members in C. rotundifolia and V. vinifera (PN40024) by BLASTP. The genes with alignment length >100 bp and e-value <1e−05 were considered as potential members. Then online software CD-search (https://www.ncbi.nlm.nih.gov/cdd) [66] and PFAM (https://pfam.xfam.org/) [67] were used to detect the specific domain. The genes without a unique domain of gene family were abandoned. Then the remaining genes were defined as candidate members and used for further analyses. Diel expression datasets of Arabidopsis C3 leaf [68] and pineapple CAM leaf were used to compare with CAM genes shown in Fig. 4b in C. rotundifolia, whose orthologs were identified by BLASTP based on sequence similarity, and then gene pairs between two species were used to calculate their relationship (Pearson and Spearman) with respect to transcript expression (Supplementary Data Table S4). On the basis of satisfying two correlation coefficients (Pearson and Spearman), genes (Rcr-at < .5) were determined as not having correlative expression patterns during a day/night cycle between Cissus and Arabidopsis. Gene pairs (Rcr-at < .5 and Rcr-ac > .8) were defined as strongly CAM genes. The genes for stomatal movement were identified using BLASTP with an e-value cutoff of 1e−5 based on orthology in Arabidopsis as described by Chen et al. [69].
Co-expression network and cluster analysis
Transcripts with average FPKM >1 (calculated from three biological replicates) in at least one of the nine samples were used to construct a weighted gene co-expression network by the R package WGCNA. The transcript expression was log2-transformed. Modules were constructed using the following parameters: power = 16, networkType = ‘signe’, mergeCutHeight = 0.18, corType = ‘bicor’, minModuleSize = 30. All the nine time-point transcripts with three replicates were used to perform cluster analysis with the maSigPro package. The parameters were as following: degree = 3, counts = F, MT.adjust = ‘BH’. Transcripts were marked as influential by the T.fit() function. Genes with ‘non-flat’ significantly changed across the nine time points. Nine clusters were displayed using the ‘see.genes’ function with cluster.method = ‘hclust’, k = 9 in maSigPro. The network of each cluster was constructed by the ARACNE algorithm with ‘Discovery’ mode and ‘Naïve Bayes’ mutual information (MI) algorithm type in Cytoscape software. The P-value was calculated based on MI, in which <.05 was selected in each cluster. One percent of genes with at least 10 edges in each network were selected by cytoHubba, and CAM genes also were chosen based on a minimum of 10 directed edges.
Cis-element annotation and enrichment analysis of CAM-related genes
Promoter sequences in 2 kb upstream of genes involved in CAM were extracted from the C. rotundifolia genome. Of all the promoter sequences, the cis-element enrichment of light, circadian, temperature, and drought in CAM-related and stomatal movement-related promoters were implemented by the FIMO [70] program with P-value <.0002 in MEME. Enrichment analysis of about five known cis-elements, including the morning element (CCACAC), the evening element (AAAATATCT), the CCA1-binding site (AAAAATCT), the TCP15 element (NGGNCCCAC), and the G-box element (G-box; CACGTG) [71], was performed by the FIMO program [70].
Acknowledgements
This work was supported by the National Science Foundation of China (31961143026) and the Scientific Research Program of Sino-Africa Joint Research Center (SAJC201614 and SAJL201607). We would like to thank the Institute of Experimental Botany, Czech Republic, for kindly providing the seeds of R. sativus cv. ‘Saxa’ as the standard for flow cytometry.
Author contributions
H.P.X. and Q.F.W. initiated the study of the C. rotundifolia genome sequencing project. H.P.X., Y.W., Q.Y.L., and T.W. are joint first authors. D.K.G. and Z.F.Z. confirmed the genome size and chromosome numbers with help of B.L. D.K.G. and H.M.Z. isolated DNA. Y.W. and Q.Y.L. carried out the genome assembly, annotation, and transcriptome analysis with the help of J.S.Z. Y.W. carried out the phylogenomic analyses with the help of Y.D.Z., B.L., T.W., and Z.D.C. Y.J.H., Y.S.L., and C.X. detected the diel acid fluctuation in the leaves of C. rotundifolia. Y.S.L., R.J.L, Z.M.L., and Q.Y.L. identified the CAM pathway-related genes. Y.J.H. isolated the total RNAs, and Q.Y.L. performed the expression pattern analysis of CAM pathway related genes with the help of H.S.J. H.P.X., Y.W., Q.Y.L., J.W. and Q.F.W. wrote the initial manuscript. J.W., J.N.W., Z.D.C., Z.C.L., L.M.L., G.W.H., R.J.L., R.A. and R.W.G. contributed to the discussion of project at different stages. All authors revised and contributed to the final version of the text.
Data availability
All data used and generated in this study have been deposited in the National Genomics Data Center (NGDC, https://ngdc.cncb.ac.cn/) with the project number PRJCA005006. The final assembled genome and annotation files were also deposited in www.grapeworld.cn/ggh/cis.html. All data is available from the corresponding author upon reasonable request.
Conflict of interest
The authors declare no competing interests.
Supplementary data
Supplementary data is available at Horticulture Research online.
Supplementary Material
Contributor Information
Haiping Xin, Core Botanical Gardens/Wuhan Botanical Garden, Chinese Academy of Sciences, Wuhan 430074, China; CAS Key Laboratory of Plant Germplasm Enhancement and Specialty Agriculture, Wuhan Botanical Garden, Chinese Academy of Sciences, Wuhan 430074, China; Sino-Africa Joint Research Center, Chinese Academy of Sciences, Wuhan 430074, China.
Yi Wang, CAS Key Laboratory of Plant Resources, Institute of Botany, Chinese Academy of Science, Beijing 100093, China.
Qingyun Li, Core Botanical Gardens/Wuhan Botanical Garden, Chinese Academy of Sciences, Wuhan 430074, China; CAS Key Laboratory of Plant Germplasm Enhancement and Specialty Agriculture, Wuhan Botanical Garden, Chinese Academy of Sciences, Wuhan 430074, China; Sino-Africa Joint Research Center, Chinese Academy of Sciences, Wuhan 430074, China; University of Chinese Academy of Sciences, Beijing 100049, China.
Tao Wan, Core Botanical Gardens/Wuhan Botanical Garden, Chinese Academy of Sciences, Wuhan 430074, China; Sino-Africa Joint Research Center, Chinese Academy of Sciences, Wuhan 430074, China; Key Laboratory of Southern Subtropical Plant Diversity, Fairy Lake Botanical Garden, Shenzhen & Chinese Academy of Science, Shenzhen 518004, China.
Yujun Hou, Core Botanical Gardens/Wuhan Botanical Garden, Chinese Academy of Sciences, Wuhan 430074, China; University of Chinese Academy of Sciences, Beijing 100049, China.
Yuanshuang Liu, Core Botanical Gardens/Wuhan Botanical Garden, Chinese Academy of Sciences, Wuhan 430074, China; University of Chinese Academy of Sciences, Beijing 100049, China.
Duncan Kiragu Gichuki, Core Botanical Gardens/Wuhan Botanical Garden, Chinese Academy of Sciences, Wuhan 430074, China; University of Chinese Academy of Sciences, Beijing 100049, China.
Huimin Zhou, Core Botanical Gardens/Wuhan Botanical Garden, Chinese Academy of Sciences, Wuhan 430074, China; University of Chinese Academy of Sciences, Beijing 100049, China.
Zhenfei Zhu, Core Botanical Gardens/Wuhan Botanical Garden, Chinese Academy of Sciences, Wuhan 430074, China; University of Chinese Academy of Sciences, Beijing 100049, China.
Chen Xu, Core Botanical Gardens/Wuhan Botanical Garden, Chinese Academy of Sciences, Wuhan 430074, China.
Yadong Zhou, Core Botanical Gardens/Wuhan Botanical Garden, Chinese Academy of Sciences, Wuhan 430074, China; CAS Key Laboratory of Plant Germplasm Enhancement and Specialty Agriculture, Wuhan Botanical Garden, Chinese Academy of Sciences, Wuhan 430074, China; Sino-Africa Joint Research Center, Chinese Academy of Sciences, Wuhan 430074, China.
Zhiming Liu, Key Laboratory of Southern Subtropical Plant Diversity, Fairy Lake Botanical Garden, Shenzhen & Chinese Academy of Science, Shenzhen 518004, China.
Rongjun Li, Core Botanical Gardens/Wuhan Botanical Garden, Chinese Academy of Sciences, Wuhan 430074, China; CAS Key Laboratory of Plant Germplasm Enhancement and Specialty Agriculture, Wuhan Botanical Garden, Chinese Academy of Sciences, Wuhan 430074, China; Sino-Africa Joint Research Center, Chinese Academy of Sciences, Wuhan 430074, China.
Bing Liu, Sino-Africa Joint Research Center, Chinese Academy of Sciences, Wuhan 430074, China; State Key Laboratory of Systematic and Evolutionary Botany, Institute of Botany, Chinese Academy of Science, Beijing 100093, China.
Limin Lu, Sino-Africa Joint Research Center, Chinese Academy of Sciences, Wuhan 430074, China; State Key Laboratory of Systematic and Evolutionary Botany, Institute of Botany, Chinese Academy of Science, Beijing 100093, China.
Hongsheng Jiang, Core Botanical Gardens/Wuhan Botanical Garden, Chinese Academy of Sciences, Wuhan 430074, China.
Jisen Zhang, Center for Genomics and Biotechnology, Haixia Institute of Science and Technology, Fujian Agriculture and Forestry University, Fuzhou 350002, China.
Junnan Wan, Core Botanical Gardens/Wuhan Botanical Garden, Chinese Academy of Sciences, Wuhan 430074, China; CAS Key Laboratory of Plant Germplasm Enhancement and Specialty Agriculture, Wuhan Botanical Garden, Chinese Academy of Sciences, Wuhan 430074, China; Sino-Africa Joint Research Center, Chinese Academy of Sciences, Wuhan 430074, China.
Rishi Aryal, Department of Horticultural Science, North Carolina State University, Raleigh, NC 27695, USA.
Guangwan Hu, Core Botanical Gardens/Wuhan Botanical Garden, Chinese Academy of Sciences, Wuhan 430074, China; Sino-Africa Joint Research Center, Chinese Academy of Sciences, Wuhan 430074, China.
Zhiduan Chen, Sino-Africa Joint Research Center, Chinese Academy of Sciences, Wuhan 430074, China; State Key Laboratory of Systematic and Evolutionary Botany, Institute of Botany, Chinese Academy of Science, Beijing 100093, China.
Robert Wahiti Gituru, Department of Botany, Jomo Kenyatta University of Agriculture and Technology, 62000-00200, Nairobi, Kenya.
Zhenchang Liang, Sino-Africa Joint Research Center, Chinese Academy of Sciences, Wuhan 430074, China; CAS Key Laboratory of Plant Resources, Institute of Botany, Chinese Academy of Science, Beijing 100093, China.
Jun Wen, Department of Botany, National Museum of Natural History, Smithsonian Institution, Washington DC 20013-7012, USA.
Qingfeng Wang, Core Botanical Gardens/Wuhan Botanical Garden, Chinese Academy of Sciences, Wuhan 430074, China; CAS Key Laboratory of Plant Germplasm Enhancement and Specialty Agriculture, Wuhan Botanical Garden, Chinese Academy of Sciences, Wuhan 430074, China; Sino-Africa Joint Research Center, Chinese Academy of Sciences, Wuhan 430074, China.
References
- 1. Wen J, Lu LM, Nie ZLet al. A new phylogenetic tribal classification of the grape family (Vitaceae). J Syst Evol. 2018;56:262–72. [Google Scholar]
- 2. Kubitzki K, Bayer C, Stevens PF. Flowering Plants. Eudicots: Berberidopsidales, Buxales, Crossosomatales, Fabales p.p., Geraniales, Gunnerales, Myrtales P.P., Proteales, Saxifragales, Vitales, Zygophyllales, Clusiaceae Alliance, Passifloraceae Alliance, Dilleniaceae, Huaceae, Picramniaceae, Sabiaceae. Berlin: Springer; 2007. [Google Scholar]
- 3. Zhang N, Wen J, Zimmer EA. Another look at the phylogenetic position of the grape order Vitales: chloroplast phylogenomics with an expanded sampling of key lineages. Mol Phylogenet Evol. 2016;101:216–23. [DOI] [PubMed] [Google Scholar]
- 4. Zeng LP, Zhang N, Zhang QAet al. Resolution of deep eudicot phylogeny and their temporal diversification using nuclear genes from transcriptomic and genomic datasets. New Phytol. 2017;214:1338–54. [DOI] [PubMed] [Google Scholar]
- 5. Jaillon O, Aury JM, Noel Bet al. The grapevine genome sequence suggests ancestral hexaploidization in major angiosperm phyla. Nature. 2007;449:463–7. [DOI] [PubMed] [Google Scholar]
- 6. DeSanto AV, Bartoli G. Crassulacean acid metabolism in leaves and stems of Cissus quadrangularis. In: Winter K, JAC S, eds. Crassulacean Acid Metabolism. Springer: Berlin, 1996,216–29. [Google Scholar]
- 7. Chu ZF, Wen J, Yang YPet al. Genome size variation and evolution in the grape family Vitaceae. J Syst Evol. 2018;56:273–82. [Google Scholar]
- 8. Griffiths H, Males J. Succulent plants. Curr Biol. 2017;27:R890–6. [DOI] [PubMed] [Google Scholar]
- 9. Niechayev NA, Paula NP, John CC. Understanding trait diversity associated with crassulacean acid metabolism (CAM). Curr Opin Plant Biol. 2019;49:74–85. [DOI] [PubMed] [Google Scholar]
- 10. Cai J, Liu X, Vanneste Ket al. The genome sequence of the orchid Phalaenopsis equestris. Nat Genet. 2015;47:65–72. [DOI] [PubMed] [Google Scholar]
- 11. Ming R, VanBuren R, Wai CMet al. The pineapple genome and the evolution of CAM photosynthesis. Nat Genet. 2015;47:1435–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Wai CM, Weise SE, Ozersky Pet al. Time of day and network reprogramming during drought induced CAM photosynthesis in Sedum album. PLoS Genet. 2019;15:e1008209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Yan L, Wang X, Liu Het al. The genome of Dendrobium officinale illuminates the biology of the important traditional Chinese orchid herb. Mol Plant. 2015;8:922–34. [DOI] [PubMed] [Google Scholar]
- 14. Yang XH, Hu RB, Yin HFet al. The Kalanchoë genome provides insights into convergent evolution and building blocks of crassulacean acid metabolism. Nat Commun. 2017;8:1899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Olivares E, Urich R, Montes Get al. Occurrence of crassulacean acid metabolism in Cissus trifoliata L. (Vitaceae). Oecologia. 1984;61:358–62. [DOI] [PubMed] [Google Scholar]
- 16. Sayed OH. Crassulacean acid metabolism 1975-2000, a check list. Photosynthetica. 2001;39:339–52. [Google Scholar]
- 17. Ting IP, Sternberg LO, Deniro MJ. Variable photosynthetic metabolism in leaves and stems of Cissus quadrangularis L. Plant Physiol. 1983;71:677–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Al-Bukhaiti WQ, Noman A, Mahdi AAet al. Proximate composition, nutritional evaluation and functional properties of a promising food: Arabian wax Cissus (Cissus rotundifolia Forssk) leaves. J Food Sci Technol. 2019;56:4844–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Marcais G, Kingsford C. A fast, lock-free approach for efficient parallel counting of occurrences of k-mers. Bioinformatics. 2011;27:764–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Ranallo-Benavidez TR, Jaron KS, Schatz MC. GenomeScope 2.0 and Smudgeplot for reference-free profiling of polyploid genomes. Nat Commun. 2020;11:1432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Galbraith DW, Harkins KR, Maddox JMet al. Rapid flow cytometric analysis of the cell cycle in intact plant tissues. Science. 1983;220:1049–51. [DOI] [PubMed] [Google Scholar]
- 22. Loureiro J, Rodriguez E, Dolezel Jet al. Two new nuclear isolation buffers for plant DNA flow cytometry: a test with 37 species. Ann Bot. 2007;100:875–88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Kirov I, Divashuk M, Van Laere Ket al. An easy "SteamDrop" method for high quality plant chromosome preparation. Mol Cytogenet. 2014;7:21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Gichuki DK, Ma L, Zhu Zet al. Genome size, chromosome number determination, and analysis of the repetitive elements in Cissus quadrangularis. PeerJ. 2019;7:e8201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Doyle JJ, Doyle JL. A rapid DNA isolation procedure for small quantities of fresh leaf tissue. Phytochem Bull. 1987;19:11–5. [Google Scholar]
- 26. Xie T, Zheng JF, Liu Set al. De novo plant genome assembly based on chromatin interactions: a case study of Arabidopsis thaliana. Mol Plant. 2015;8:489–92. [DOI] [PubMed] [Google Scholar]
- 27. Durand NC, Shamim MS, Machol Iet al. Juicer provides a one-click system for analyzing loop-resolution Hi-C experiments. Cell Syst. 2016;3:95–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Dudchenko O, Batra SS, Omer ADet al. De novo assembly of the Aedes aegypti genome using Hi-C yields chromosome-length scaffolds. Science. 2017;356:92–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Durand NC, Robinson JT, Shamim MSet al. Juicebox provides a visualization system for Hi-C contact maps with unlimited zoom. Cell Syst. 2016;3:99–101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Koren S, Walenz BP, Berlin Ket al. Canu: scalable and accurate long-read assembly via adaptive k-mer weighting and repeat separation. Genome Res. 2017;27:722–36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Ruan J, Li H. Fast and accurate long-read assembly with wtdbg2. Nat Methods. 2019;17:155–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Wang J, Walker BJ, Abeel Tet al. Pilon: an integrated tool for comprehensive microbial variant detection and genome assembly improvement. PLoS One. 2014;9:e112963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Pryszcz LP, Gabaldón T. Redundans: an assembly pipeline for highly heterozygous genomes. Nucleic Acids Res. 2016;44:e113–3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Roach MJ, Schmidt SA, Borneman AR. Purge Haplotigs: allelic contig reassignment for third-gen diploid genome assemblies. BMC Bioinformatics. 2018;19:460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Simão FA, Waterhouse RM, Ioannidis Pet al. BUSCO: assessing genome assembly and annotation completeness with single-copy orthologs. Bioinformatics. 2015;31:3210–2. [DOI] [PubMed] [Google Scholar]
- 36. Jurka J, Kapitonov VV, Pavlicek Aet al. Repbase update, a database of eukaryotic repetitive elements. Cytogenet Genome Res. 2005;110:462–7. [DOI] [PubMed] [Google Scholar]
- 37. Hubley R, Finn RD, Clements Jet al. The Dfam database of repetitive DNA families. Nucleic Acids Res. 2016;44:D81–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Thiel T, Michalek W, Varshney RKet al. Exploiting EST databases for the development and characterization of gene-derived SSR-markers in barley (Hordeum vulgare L.). Theor Appl Genet. 2003;106:411–22. [DOI] [PubMed] [Google Scholar]
- 39. Ou S, Jiang N. LTR_retriever: a highly accurate and sensitive program for identification of long terminal repeat retrotransposons. Plant Physiol. 2018;176:1410–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Haas BJ, Papanicolaou A, Yassour Met al. De novo transcript sequence reconstruction from RNA-seq using the Trinity platform for reference generation and analysis. Nat Protoc. 2013;8:1494–512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Haas BJ, Delcher AL, Mount SMet al. Improving the Arabidopsis genome annotation using maximal transcript alignment assemblies. Nucleic Acids Res. 2003;31:5654–66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Stanke M, Keller O, Gunduz Iet al. AUGUSTUS: ab initio prediction of alternative transcripts. Nucleic Acids Res. 2006;34:W435–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Korf I. Gene finding in novel genomes. BMC Bioinformatics. 2004;5:59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Lukashin AV, Borodovsky M. GeneMark.Hmm: new solutions for gene finding. Nucleic Acids Res. 1998;26:1107–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Haas BJ, Salzberg SL, Zhu Wet al. Automated eukaryotic gene structure annotation using EVidenceModeler and the program to assemble spliced alignments. Genome Biol. 2008;9:R7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Camacho C, Coulouris G, Avagyan Vet al. BLAST+: architecture and applications. BMC Bioinformatics. 2009;10:21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Li L, Stoeckert CJ Jr, Roos DS. OrthoMCL: identification of ortholog groups for eukaryotic genomes. Genome Res. 2003;13:2178–89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Edgar RC. MUSCLE: multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Res. 2004;32:1792–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Stamatakis A. RAxML-VI-HPC: maximum likelihood-based phylogenetic analyses with thousands of taxa and mixed models. Bioinformatics. 2006;22:2688–90. [DOI] [PubMed] [Google Scholar]
- 50. Yang Z. PAML 4: phylogenetic analysis by maximum likelihood. Mol Biol Evol. 2007;24:1586–91. [DOI] [PubMed] [Google Scholar]
- 51. Han MV, Thomas GW, Lugo-Martinez Jet al. Estimating gene gain and loss rates in the presence of error in genome assembly and annotation using CAFE 3. Mol Biol Evol. 2013;30:1987–97. [DOI] [PubMed] [Google Scholar]
- 52. Albert VA, Barbazuk WB, dePamphilis CWet al. The Amborella genome and the evolution of flowering plants. Science. 2013;342:1241089. [DOI] [PubMed] [Google Scholar]
- 53. Wickham H. ggplot2. Wiley interdisciplinary reviews: computational statistics. 2011;3:180–5. [Google Scholar]
- 54. Wang YP, Tang HB, DeBarry JDet al. MCScanX: a toolkit for detection and evolutionary analysis of gene synteny and collinearity. Nucleic Acids Res. 2012;40:e49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Tang H, Wang X, Bowers JEet al. Unraveling ancient hexaploidy through multiply-aligned angiosperm gene maps. Genome Res. 2008;18:1944–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Zhang Z, Xiao J, Wu Jet al. ParaAT: a parallel tool for constructing multiple protein-coding DNA alignments. Biochem Biophys Res Commun. 2012;419:779–81. [DOI] [PubMed] [Google Scholar]
- 57. Wang D, Zhang Y, Zhang Zet al. KaKs_Calculator 2.0: a toolkit incorporating gamma-series methods and sliding window strategies. Genomics Proteomics Bioinformatics. 2010;8:77–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Murat F, Armero A, Pont Cet al. Reconstructing the genome of the most recent common ancestor of flowering plants. Nat Genet. 2017;49:490–6. [DOI] [PubMed] [Google Scholar]
- 59. Chen SS, Black CC. Diurnal changes in volume and specific tissue weight of crassulacean acid metabolism plants. Plant Physiol. 1983;71:373–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Bolger AM, Lohse M, Usadel B. Trimmomatic: a flexible trimmer for Illumina sequence data. Bioinformatics. 2014;30:2114–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Trapnell C, Roberts A, Goff Let al. Differential gene and transcript expression analysis of RNA-seq experiments with TopHat and cufflinks. Nat Protoc. 2012;7:562–78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Xu H, Yu Q, Shi Yet al. PGD: pineapple genomics database. Hortic Res. 2018;5:66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Goodstein DM, Shu S, Howson Ret al. Phytozome: a comparative platform for green plant genomics. Nucleic Acids Res. 2012;40:D1178–86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Kawahara Y, Bastide M, Hamilton JPet al. Improvement of the Oryza sativa Nipponbare reference genome using next generation sequence and optical map data. Rice. 2013;6:4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Portwood JL, Woodhouse MR, Cannon EKet al. MaizeGDB 2018: the maize multi-genome genetics and genomics database. Nucleic Acids Res. 2019;47:D1146–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Marchler-Bauer A, Bryant SH. CD-search: protein domain annotations on the fly. Nucleic Acids Res. 2004;32:W327–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. El-Gebali S, Mistry J, Bateman Aet al. The Pfam protein families database in 2019. Nucleic Acids Res. 2019;47:D427–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Mockler TC, Michael TP, Priest HDet al. The DIURNAL project: DIURNAL and circadian expression profiling, model-based pattern matching, and promoter analysis. Cold Spring Harb Symp Quant Biol. 2007;72:353–63. [DOI] [PubMed] [Google Scholar]
- 69. Chen LY, Xin Y, Wai CMet al. The role of cis-elements in the evolution of crassulacean acid metabolism photosynthesis. Hortic Res. 2020;7:5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Grant CE, Bailey TL, Noble WS. FIMO: scanning for occurrences of a given motif. Bioinformatics. 2011;27:1017–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Michael TP, McClung CR. Phase-specific circadian clock regulatory elements in Arabidopsis. Plant Physiol. 2002;130:627–38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Freeling M. Bias in plant gene content following different sorts of duplication: tandem, whole-genome, segmental, or by transposition. Annu Rev Plant Biol. 2009;60:433–53. [DOI] [PubMed] [Google Scholar]
- 73. Wan T, Liu ZM, Leitch IJet al. The Welwitschia genome reveals a unique biology underpinning extreme longevity in deserts. Nat Commun. 2021;12:4247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Xiong ZY, Gaeta RT, Pires JC. Homoeologous shuffling and chromosome compensation maintain genome balance in resynthesized allopolyploid Brassica napus. Proc Natl Acad Sci USA. 2011;108:7908–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Qin LY, Hu YH, Wang JPet al. Insights into angiosperm evolution, floral development and chemical biosynthesis from the Aristolochia fimbriata genome. Nat Plants. 2021;7:1239–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Shepherd T, Wynne GD. The effects of stress on plant cuticular waxes. New Phytol. 2006;171:469–99. [DOI] [PubMed] [Google Scholar]
- 77. Dubos C, Stracke R, Grotewold Eet al. MYB transcription factors in Arabidopsis. Trends Plant Sci. 2010;15:573–81. [DOI] [PubMed] [Google Scholar]
- 78. Gao J, Lan T. Functional characterization of the late embryogenesis abundant (LEA) protein gene family from Pinus tabuliformis (Pinaceae) in Escherichia coli. Sci Rep. 2016;6:19467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79. Jiang CK, Rao GY. Insights into the diversification and evolution of R2R3-MYB transcription factors in plants. Plant Physiol. 2020;183:637–55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80. Plomion C, Aury JM, Amselem Jet al. Oak genome reveals facets of long lifespan. Nat Plants. 2018;4:440–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81. Tobias PA, Guest DI. Tree immunity: growing old without antibodies. Trends Plant Sci. 2014;19:367–70. [DOI] [PubMed] [Google Scholar]
- 82. Mestre P, Baulcombe DC. Elicitor-mediated oligomerization of the tobacco N disease resistance protein. Plant Cell. 2006;18:491–501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83. Warren RL, Keeling CI, Yuen MMet al. Improved white spruce (Picea glauca) genome assemblies and annotation of large gene families of conifer terpenoid and phenolic defense metabolism. Plant J. 2015;83:189–212. [DOI] [PubMed] [Google Scholar]
- 84. Bondino HG, Valle EM, Ten Have A. Evolution and functional diversification of the small heat shock protein/α-crystallin family in higher plants. Planta. 2012;235:1299–313. [DOI] [PubMed] [Google Scholar]
- 85. Guo LM, Li J, He Jet al. A class I cytosolic HSP20 of rice enhances heat and salt tolerance in different organisms. Sci Rep. 2020;10:1383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86. Ahl LI, Mravec J, Jorgensen Bet al. Dynamics of intracellular mannan and cell wall folding in the drought responses of succulent aloe species. Plant Cell Environ. 2019;42:2458–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87. Hanada K, Zou C, Lehti-Shiu MDet al. Importance of lineage-specific expansion of plant tandem duplicates in the adaptive response to environmental stimuli. Plant Physiol. 2008;148:993–1003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88. Rizzon C, Ponger L, Gaut BS. Striking similarities in the genomic distribution of tandemly arrayed genes in Arabidopsis and rice. PLoS Comput Biol. 2006;2:989–1000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89. Silvera K, Neubig KM, Whitten WMet al. Evolution along the crassulacean acid metabolism continuum. Funct Plant Biol. 2010;37:995–1010. [Google Scholar]
- 90. Borland AM, Hartwell J, Weston DJet al. Engineering crassulacean acid metabolism to improve water-use efficiency. Trends Plant Sci. 2014;19:327–38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91. Nelson EA, Sage RF. Functional constraints of CAM leaf anatomy: tight cell packing is associated with increased CAM function across a gradient of CAM expression. J Exp Bot. 2008;59:1841–50. [DOI] [PubMed] [Google Scholar]
- 92. Abraham PE, Castano NH, Cowan-Turner Det al. Peeling back the layers of crassulacean acid metabolism: functional differentiation between Kalanchoë fedtschenkoi epidermis and mesophyll proteomes. Plant J. 2020;103:869–88. [DOI] [PubMed] [Google Scholar]
- 93. Wickell D, Kuo LY, Yang HPet al. Underwater CAM photosynthesis elucidated by Isoetes genome. Nat Commun. 2021;12:6348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94. Michael TP, Mockler TC, Breton Get al. Network discovery pipeline elucidates conserved time-of-day-specific cis-regulatory modules. PLoS Genet. 2008;4:e14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95. Mustilli AC, Merlot S, Vavasseur Aet al. Arabidopsis OST1 protein kinase mediates the regulation of stomatal aperture by abscisic acid and acts upstream of reactive oxygen species production. Plant Cell. 2002;14:3089–99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96. Boxall SF, Kadu N, Dever LVet al. Kalanchoë PPC1 is essential for crassulacean acid metabolism and the regulation of core circadian clock and guard cell signaling genes. Plant Cell. 2020;32:1136–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97. Abraham PE, Yin H, Borland AMet al. Transcript, protein and metabolite temporal dynamics in the CAM plant agave. Nat Plants. 2016;2:16178. [DOI] [PubMed] [Google Scholar]
- 98. Soltis DE, Soltis PS, Chase MWet al. Angiosperm phylogeny inferred from 18S rDNA, rbcL, and atpB sequences. Bot J Linn Soc. 2000;133:381–461. [Google Scholar]
- 99. Chanderbali AS, Jin LL, Xu QJet al. Buxus and Tetracentron genomes help resolve eudicot genome history. Nat Commun. 2022;13:1–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100. Van de Peer Y, Maere S, Meyer A. The evolutionary significance of ancient genome duplications. Nat Rev Genet. 2009;10:725–32. [DOI] [PubMed] [Google Scholar]
- 101. Alzohairy AM, Yousef MA, Edris Set al. Detection of LTR retrotransposons reactivation induced by in vitro environmental stresses in barley (Hordeum vulgare) via RT-qPCR. Life Sci J. 2012;9:5019–26. [Google Scholar]
- 102. Drake PL, Froend RH, Franks PJ. Smaller, faster stomata: scaling of stomatal size, rate of response, and stomatal conductance. J Exp Bot. 2013;64:495–505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103. Roddy AB, Théroux-Rancourt G, Abbo Tet al. The scaling of genome size and cell size limits maximum rates of photosynthesis with implications for ecological strategies. Int J Plant Sci. 2020;181:75–87. [Google Scholar]
- 104. Eggli U, Nyffeler R. Living under temporarily arid conditions – succulence as an adaptive strategy. Bradleya. 2009;27:13–36. [Google Scholar]
- 105. Newton DJ, Chan J. South Africa's trade in Southern African succulent plants. Johannesburg: TRAFFIC East/Southern Africa,1998. [Google Scholar]
- 106. Morse SR. Water-balance in Hemizonia luzulifolia – the role of extracellular polysaccharides. Plant Cell Environ. 1990;13:39–48. [Google Scholar]
- 107. Hundertmark M, Hincha DK. LEA (late embryogenesis abundant) proteins and their encoding genes in Arabidopsis thaliana. BMC Genomics. 2008;9:118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108. Rai A, Singh R, Shirke PAet al. Expression of rice CYP450-like gene (Os08g01480) in Arabidopsis modulates regulatory network leading to heavy metal and other abiotic stress tolerance. PLoS One. 2015;10:e0138574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109. Bloom AJ, Troughton JH. High productivity and photosynthetic flexibility in a CAM plant. Oecologia. 1979;38:35–43. [DOI] [PubMed] [Google Scholar]
- 110. Huang H, Nusinow DA. Into the evening: complex interactions in the Arabidopsis circadian clock. Trends Genet. 2016;32:674–86. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
All data used and generated in this study have been deposited in the National Genomics Data Center (NGDC, https://ngdc.cncb.ac.cn/) with the project number PRJCA005006. The final assembled genome and annotation files were also deposited in www.grapeworld.cn/ggh/cis.html. All data is available from the corresponding author upon reasonable request.