

Abstract
Aims
Orchestrating the transition from reversible medial hypertrophy to irreversible plexiform lesions is crucial for pulmonary arterial hypertension related to congenital heart disease (CHD‐PAH). Transgelin is an actin‐binding protein that modulates pulmonary arterial smooth muscle cell (PASMC) dysfunction. In this study, we aimed to probe the molecular mechanism and biological function of transgelin in the pathogenesis of CHD‐PAH.
Methods and results
Transgelin expression was detected in lung tissues from both CHD‐PAH patients and monocrotaline (MCT)‐plus aortocaval (AV)‐induced PAH rats by immunohistochemistry. In vitro, the effects of transgelin on the proliferation, migration, and apoptosis of human PASMCs (HPASMCs) were evaluated by the cell count and EdU assays, transwell migration assay, and TUNEL assay, respectively. And the effect of transgelin on the expression of HPASMC phenotype markers was assessed by the immunoblotting assay. (i) Compared with the normal control group (n = 12), transgelin expression was significantly overexpressed in the pulmonary arterioles of the reversible (n = 15) and irreversible CHD‐PAH group (n = 4) (reversible group vs. control group: 18.2 ± 5.1 vs. 13.6 ± 2.6%, P < 0.05; irreversible group vs. control group: 29.9 ± 4.7 vs. 13.6 ± 2.6%, P < 0.001; irreversible group vs. reversible group: 29.9 ± 4.7 vs. 18.2 ± 5.1, P < 0.001). This result was further confirmed in MCT‐AV‐induced PAH rats. Besides, the transgelin expression level was positively correlated with the pathological grading of pulmonary arteries in CHD‐PAH patients (r = 0.48, P = 0.03, n = 19). (ii) Compared with the normal control group (n = 12), TGF‐β1 expression was notably overexpressed in the pulmonary arterioles of the reversible (n = 15) and irreversible CHD‐PAH group (n = 4) (reversible group vs. control group: 14.8 ± 4.4 vs. 6.0 ± 2.5%, P < 0.001; irreversible group vs. control group: 20.1 ± 4.4 vs. 6.0 ± 2.5%, P < 0.001; irreversible group vs. reversible group: 20.1 ± 4.4 vs. 14.8 ± 4.4, P < 0.01). The progression‐dependent correlation between TGF‐β1 and transgelin was demonstrated in CHD‐PAH patients (r = 0.48, P = 0.04, n = 19) and MCT‐AV‐induced PAH rats, which was further confirmed at sub‐cellular levels. (iii) Knockdown of transgelin diminished proliferation, migration, apoptosis resistance, and phenotypic transformation of HPASMCs through repressing the TGF‐β1 signalling pathway. On the contrary, transgelin overexpression resulted in the opposite effects.
Conclusions
These results indicate that transgelin may be an indicator of CHD‐PAH development via boosting HPASMC dysfunction through positive regulation of the TGF‐β1 signalling pathway, as well as a potential therapeutic target for the treatment of CHD‐PAH.
Keywords: Congenital heart disease (CHD), Pulmonary arterial hypertension (PAH), Pulmonary artery smooth muscle cells (PASMCs), Transgelin
Introduction
Pulmonary arterial hypertension (PAH) is a relatively common complication of congenital heart disease (CHD). It is characterized by progressive narrowing of the small pulmonary arteries due to abnormal cellular proliferation, fibrosis, and in situ thrombosis, ultimately resulting in successive elevation of pulmonary vascular resistance and subsequent right ventricular failure. 1 In the European registry, 4–28% of CHD patients had various degrees of PAH, while 30% of unrepaired CHD patients have PAH. 2 In CHD‐PAH patients, the arteriopathy is trigged by increased pulmonary blood flow resulting from a left‐to‐right shunt due to an intracardiac or extracardiac defect. Closing the shunt in time can reverse this arteriopathy in the early disease phase, but salutary effects diminish considerably once the disease state is established. 3 , 4 After the correction of complete atrioventricular septal defects, 14% of CHD patients still reported severe PAH, and their 1 year mortality rate was 18.5%. 5 Furthermore, due to sudden hemodynamic change, the 10 year postoperative survival rates of persistent post‐operative PAH patients were worse than those of Eisenmenger syndrome patients. 6 This implies that the pulmonary vascular lesion changes become irreversible and the progressive hemodynamic disorder cannot be surgically reversed. Accurate early detection of this ‘window for reversibility’ is therefore extremely important before surgery in patients with CHD‐PAH. Increased research on identifying key molecules and pathways participating in maladaptive arterial remodelling will have important implications in further elucidating the progression of irreversible PAH and improving the identification of reversibility in CHD‐PAH.
Phenotypic transformation of vascular smooth muscle cells (VSMCs) plays a major role in the initial stage of vascular proliferative diseases, such as PAH, atherosclerosis, and restenosis. 7 Transgelin, a transforming growth factor‐beta 1 (TGF‐β1)‐inducible protein, is characterized by its specificity to smooth muscle and phenotype‐specific expression in VSMCs. 8 , 9 It functions as an actin‐associated protein that participates in cytoskeleton remodelling and modulation of proliferation, migration, apoptosis, and oxidative stress in VSMCs. 10 Growing evidence has shown a close correlation between transgelin and the progression of PAH. Suppression of transgelin attenuated the phenotypic transformation of pulmonary artery smooth muscle cells (PASMCs) from contractile to synthetic cells, resulting in reduced proliferation, migration, resistance to apoptosis in vitro, and prevention of PAH and pulmonary vascular remodelling in vivo. 11 , 12 Recently, our group demonstrated that transgelin was markedly elevated in irreversible CHD‐PAH patients compared with reversible CHD‐PAH and normal control groups, and its expression was positively correlated with pathological grading of pulmonary arterioles, 13 which implied that transgelin may be involved in the progression of CHD‐PAH.
TGF‐β1, a member of the TGF‐β family, has been well‐studied and is recognized as a key regulatory cytokine that participates in the pathogenesis of vascular remodelling. 14 The expression of TGF‐β1 is highly up‐regulated in pre‐clinical experimental models of PAH, which results in PASMC proliferation and migration, thus indicating that TGF‐β1 contributes to the progression of PAH and pulmonary vascular remodelling; however, the underlying molecular mechanism has not been thoroughly elucidated. Chen et al. reported the progression‐dependent correlation between TGF‐β1 and transgelin was found at both the tissue and the sub‐cellular levels in bladder cancer, while TGF‐β1‐mediated migration was completely inhibited by the suppression of transgelin. 15 Nevertheless, little is known about how transgelin participates in the TGF‐β1 signalling pathway during the progression of CHD‐PAH.
In this study, we demonstrate that transgelin and TGF‐β1 were overexpressed in lung tissues of CHD‐PAH patients and MCT‐AV‐induced PAH rats, and there was a significant correlation between them. Transgelin expression was significantly positively correlated with the pulmonary vascular remodelling in CHD‐PAH patients. Importantly, TGF‐β1 up‐regulated transgelin expression, whereas transgelin overexpression promoted TGF‐β1‐induced proliferation, migration, and apoptosis resistance of human PASMCs (HPASMCs). Overall, these results may contribute to an improved understanding of maladaptive arterial remodelling in CHD‐PAH.
Methods
Human lung tissue collection
This study protocol adhered to the principles of the Helsinki Declaration and was confirmed by the Medical Ethics Committee of Fuwai Hospital. Informed consent was obtained from all subjects before the study. Lung tissues were derived from patients with CHD‐PAH who met the standard entry requirements as determined using right heart catheterization (RHC) and underwent repair surgery. According to today's recommendations, operability is determined by the level of pulmonary vascular resistance (PVR) in congenital heart disease‐associated pulmonary arterial hypertension (CHD‐PAH). From the strict hemodynamic point of view from the 2015 ESC/ERS Guidelines for the diagnosis and treatment of pulmonary hypertension, closure of the defect is contraindicated when PVR index >8 WU m2, but permitted when PVR index is <4 WU m2. Patients with borderline PVR index between 4 and 8 WU m2 (in the ‘grey zone’) should be evaluated in tertiary centres with expertise in CHD‐PAH (European Heart Journal. 2016 Jan 1;37 (1):67–119). In our study, the patient who fulfilled the surgical criteria were enrolled to further study (Table 1 ). One year post‐surgery, the patients were divided into reversible (mPAP < 25 mmHg, n = 15) and irreversible CHD‐PAH groups (mPAP ≥ 25 mmHg, n = 4). Normal lung tissue specimens were collected from 12 patients who underwent lung surgery. To ensure normal tissues, these lung tissues were sufficiently distal to the tumour. All specimens were processed in paraffin and stored at −20°C.
Table 1.
The clinical features of the enrolled reversible and irreversible CHD‐PAH patients
| No. | Age (year) | Gender | Aetiology | Pre‐sPAP (mmHg) | Pre‐dPAP (mmHg) | Pre‐mPAP (mmHg) | PVR index (WU m2) | PG | Post‐sPAP (mmHg) | Post‐dPAP (mmHg) | Post‐mPAP (mmHg) |
|---|---|---|---|---|---|---|---|---|---|---|---|
| Reversible group | |||||||||||
| 1 | 45 | F | ASD | 40 | 23 | 31 | 3.5 | I | 23 | 11 | 16 |
| 2 | 45 | F | ASD | 56 | 37 | 28 | 3.8 | II | 27 | 13 | 17 |
| 3 | 5 | M | VSD | 47 | 22 | 27 | 2.6 | I | 23 | 14 | 17 |
| 4 | 54 | F | ASD | 88 | 24 | 48 | 3.2 | I | 31 | 12 | 21 |
| 5 | 59 | M | VSD | 42 | 22 | 30 | 2.4 | 0 | 26 | 15 | 18 |
| 6 | 55 | F | ASD | 50 | 17 | 28 | 2.6 | II | 29 | 16 | 21 |
| 7 | 15 | M | ASD | 64 | 22 | 36 | 2.5 | I | 32 | 15 | 22 |
| 8 | 15 | M | VSD | 35 | 15 | 25 | 2.9 | 0 | 25 | 10 | 15 |
| 9 | 16 | F | ASD | 39 | 20 | 26 | 1.6 | 0 | 25 | 10 | 15 |
| 10 | 4 | F | VSD | 106 | 54 | 70 | 3.1 | I | 35 | 8 | 17 |
| 11 | 9 | F | VSD | 55 | 23 | 35 | 1.2 | I | 39 | 12 | 21 |
| 12 | 41 | M | ASD | 54 | 21 | 35 | 0.9 | 0 | 30 | 12 | 23 |
| 13 | 19 | F | ASD | 70 | 23 | 41 | 1.9 | I | 17 | 5 | 8 |
| 14 | 21 | F | ASD | 69 | 27 | 40 | 4.5 | I | 29 | 6 | 14 |
| 15 | 13 | M | VSD | 88 | 49 | 62 | 2.4 | I | 29 | 6 | 14 |
| Irreversible group | |||||||||||
| 1 | 65 | M | ASD | 63 | 29 | 40 | 3.5 | I | 40 | 20 | 26 |
| 2 | 49 | F | ASD | 80 | 16 | 30 | 2.6 | III | 50 | 25 | 32 |
| 3 | 40 | M | VSD | 104 | 34 | 60 | 2.5 | III | 63 | 27 | 35 |
| 4 | 31 | F | Other | 126 | 61 | 83 | 2.3 | II | 70 | 35 | 47 |
ASD, atrial septal defect; dPAP, diastolic pulmonary artery pressure; F, female; M, male; mPAP, mean pulmonary artery pressure; other, complete endocardial cushion defect; PG, pathological grading; Post, postoperative; PVR, pulmonary vascular resistance; Pre, preoperative; sPAP, systolic pulmonary artery pressure; VSD, ventricular septal defect.
Shunt‐related pulmonary arterial hypertension rat model
All animal experiments were conducted by the Institutional Animal Care and Use Committee. Sixty‐six adult male Sprague–Dawley rats (aged 8 weeks and weighing 260–280 g) purchased from Vital River Laboratory Animal Technology Co., Ltd. (Beijing, China) were randomized into three groups as follows: (i) Control (n = 8); (ii) Monocrotaline plus aortocaval shunt (MCT‐AV; n = 50); and (iii) Sham (n = 8). Shunt‐related PAH was induced in rats by injection of MCT (60 mg/kg) combined with an abdominal aortocaval shunt 1 week later. 16 , 17 The patency of the shunt was determined visually based on swelling of the vena cava and admixture of arterial and venous blood. Baseline body weight and hemodynamic parameters of rats were recorded at 0, 1, 2, 3, and 4 weeks post‐surgery, and lung tissues were obtained.
Immunohistochemistry (IHC) staining
Paraffin sections were used for immunohistochemical detection of transgelin and TGF‐β1 localization using rabbit polyclonal antibodies (Supporting Information, Table S1 ). The average optical density (AOD) of transgelin or TGF‐β1 in each pulmonary arteriole was quantified by the ratio of optical density (IOD) to the positive area (AOD% = IOD/positive area). Ten representative pulmonary arterioles with diameters between 50 and 200 μm were randomly chosen in each section.
Lentivirus infection in human pulmonary arterial smooth muscle cells
HPASMCs (ScienCell Research Laboratories, Carlsbad, CA, USA) were infected with lentiviruses in the presence of smooth muscle cell medium (SMCM; ScienCell) containing 2% fetal bovine serum. Lentivirus/GV248‐siTAGLN (hU6‐MCS‐Ubiquitin‐EGFP‐IRES‐puromycin) and lentivirus/GV358‐TAGLN (Ubi‐MCS‐3FLAG‐SV40‐EGFPIRES‐puromycin), and their corresponding control lentiviruses, lentivirus/GV248, and lentivirus/GV358 were synthesized by GeneChem (Shanghai, China).
Cell proliferation detection by cell count
Transduced HPASMCs were seeded into cell culture plates for 24 h. Cells were resuspended in 1 mL SMCM and counted with a haemocytometer (Countess™, ThermoFisher Scientific, Waltham, MA, USA) under a microscope.
Cell proliferation detected by EdU assay
EdU (5‐ethynyl‐2′‐deoxyuridine) (Invitrogen, ThermoFisher Scientific) was used to detect the proliferation of transduced HPASMCs. Sections were counterstained with Hoechst 33342 for 30 min to mark cell nuclei. Cells were observed and counted under a fluorescence microscope. Proliferation was assessed by the ratio of proliferative cells to the total number of cells.
Transwell migration assay
A transwell chamber apparatus was used to measure the migration of transduced HPASMCs. Transduced HPASMCs were harvested in serum‐free SMCM and added to the upper compartment of the transwell chamber, whereas SMCM containing 2% FBS was added to the lower chamber. After 16 h, the membranes were fixed with 4% paraformaldehyde for 90 min at room temperature and then stained with toluidine blue for 30 min. The number of migratory cells was counted five times in random fields under a microscope. Data are presented as the fold change of the migrated cells.
Apoptosis detection by TUNEL assay
Transduced HPASMCs were cocultured with TdT‐mediated dUTP Nick End Labeling (TUNEL) reaction mixture (Roche, Basel, Switzerland) for 1 h. Cell nuclei were stained with DAPI solution for 5 min and visualized under a fluorescence microscope. Apoptosis was calculated as the ratio of the percentage of positive cells in the experimental and control groups.
Immunoblotting assay
Lung tissues were lysed in radioimmunoprecipitation (RIPA) lysis buffer for 30 min on ice. Protein concentration was quantified by Bradford assay. Immunoblotting was carried out using sodium dodecyl sulfate‐polyacrylamide gel electrophoresis (SDS‐PAGE) followed by protein transfer to a PVDF membrane (Millipore, St. Louis, MO, USA). Specific primary antibodies (Supporting Information, Table S1 ) were incubated overnight followed by incubation with secondary antibodies for 1 h. Protein expression is reported as fold change relative to GAPDH.
Statistical analysis
Data analysis was conducted using SPSS Version 23 statistical software. Data are represented as the mean ± standard error of the mean (SEM) of three independent experiments. One‐way analysis of variance (ANOVA) statistical test was applied followed by Bonferroni's multiple comparison test to identify individual differences among groups. Spearman's rank‐order correlation was applied to determine the relationship between transgelin expression in lung tissues and clinical parameters or pathological grading. Differences were considered significant at P < 0.05.
Results
Characteristics of the pulmonary arterial hypertension related to congenital heart disease patients
The detailed clinical characteristics of CHD‐PAH patients are described in Table 1 . After 1 year of follow‐up, 19 CHD‐PAH patients were enrolled, 42% (8/19) of which were men. The major aetiologies of these CHD‐PAH patients were atrial septal defect (ASD) and ventricular septal defect (VSD), accounting for 57% (11/19) and 42% (8/19) of cases, respectively. Of these, 15 were defined as reversible CHD‐PAH, while four patients were confirmed to have irreversible CHD‐PAH. There were no significant age differences between reversible and irreversible CHD‐PAH patients (27.7 ± 19.7 and 46.3 ± 14.5 years, respectively, P > 0.05). In the reversible group, four patients showed normal pulmonary arterioles, nine patients had pathological grade I, and two patients showed grade II, while all patients in the irreversible group had abnormal pathologic changes: one in grade I, one in grade II, and two in grade III.
Expression of transgelin and transforming growth factor‐beta 1 in lung tissues from pulmonary arterial hypertension related to congenital heart disease patients
Previous proteomic analysis revealed that transgelin expression was higher in cases of irreversible CHD‐PAH compared with reversible. 13 Compared with the normal control group (n = 12), transgelin expression was significantly overexpressed in the pulmonary arterioles of the reversible (n = 15) and irreversible CHD‐PAH group (n = 4) (reversible group vs. control group: 18.2 ± 5.1 vs. 13.6 ± 2.6%, P < 0.05; irreversible group vs. control group: 29.9 ± 4.7 vs. 13.6 ± 2.6%, P < 0.001; irreversible group vs. reversible group: 29.9 ± 4.7 vs. 18.2 ± 5.1, P < 0.001) (Figure 1 A ). Notably, the transgelin expression level was positively correlated with the pathological grading of pulmonary arteries in 19 CHD‐PAH patients (r = 0.48, P = 0.03, n = 19) (Figure 1 A ).
Figure 1.
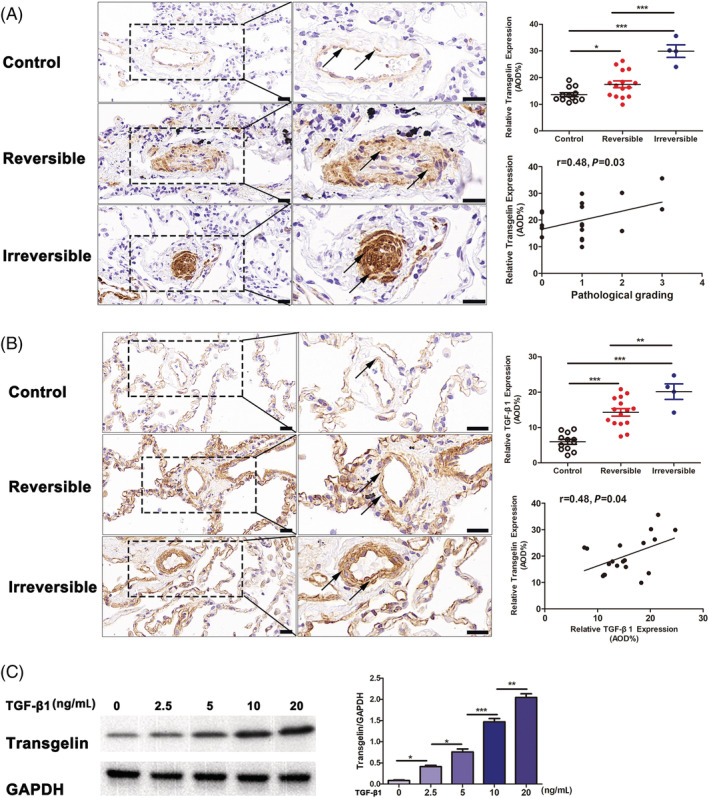
Expression of transgelin and TGF‐β1 in lung tissues from CHD‐PAH patients. (A) IHC staining for transgelin expression in lung tissues from CHD‐PAH patients. The black arrows indicate transgelin+ cells located in pulmonary arterioles. Scar bars, 20 μm. (B) IHC staining for TGF‐β1 expression in lung tissues from CHD‐PAH patients. The black arrows indicate TGF‐β1+ cells located in pulmonary arterioles. Scar bars, 20 μm. (C) Immunoblotting for TGF‐β1‐induced transgelin expression in HPASMCs. Data are represented as mean ± SEM from three independent experiments. *P < 0.05, **P < 0.01, ***P < 0.001.
The TGF‐β1 signalling pathway is a critical pathway associated with inflammation and excessive proliferation. In CHD‐PAH, this pathway is associated with reduced apoptosis of lung vascular cells. 18 Given the known role of TGF‐β1 signalling in the regulation of transgelin expression, 8 , 9 we subsequently explored the expression of transgelin in lung tissues from CHD‐PAH patients and in HPASMCs cultured with TGF‐β1. Compared with the normal control group (n = 12), TGF‐β1 expression was notably overexpressed in the pulmonary arterioles of the reversible (n = 15) and irreversible CHD‐PAH group (n = 4) (reversible group vs. control group: 14.8 ± 4.4 vs. 6.0 ± 2.5%, P < 0.001; irreversible group vs. control group: 20.1 ± 4.4 vs. 6.0 ± 2.5%, P < 0.001; irreversible group vs. reversible group: 20.1 ± 4.4 vs. 14.8 ± 4.4, P < 0.01) (Figure 1 B ). In addition, significant positive correlations were found between TGF‐β1 and transgelin expression in CHD‐PAH patients (r = 0.48, P = 0.04, n = 19) (Figure 1 B ). Importantly, dose–response experiments demonstrate induction of transgelin 48 h after treatment by TGF‐β1 at concentrations of 0–20 ng/mL (Figure 1 C ).
Time course of transgelin and TGF‐β1 expression in lung tissues from MCT‐AV‐induced PAH rats
To further explore the changes of transgelin in the progression of CHD‐PAH, a shunt‐related PAH rat model was developed by AV shunt surgery following MCT injection. 16 , 17 The MCT‐AV‐induced PAH rats displayed progressive pulmonary vascular remodelling typical of CHD‐PAH, which was characterized by the increased medial wall thickness of the small pulmonary arteries and abnormal muscular extension to peripheral pulmonary arteries as well as increased pulmonary blood flow. 16 , 19 , 20 IHC showed transgelin mainly localized in the PASMCs of the middle pulmonary arterioles (Figure 2 A ). Finally, immunoblotting indicated that transgelin expression was dramatically increased between 1 and 4 weeks post‐surgery, peaking at 3 weeks (Figure 2 B ). This observation is comparable with the progress of morphological signs of pulmonary vascular remodelling. Expression of TGF‐β1 in these tissues changed along with a similar trend as transgelin resulting in a significant increase in TGF‐β1 expression 1 to 2 weeks after shunt establishment (Figure 2 B ).
Figure 2.
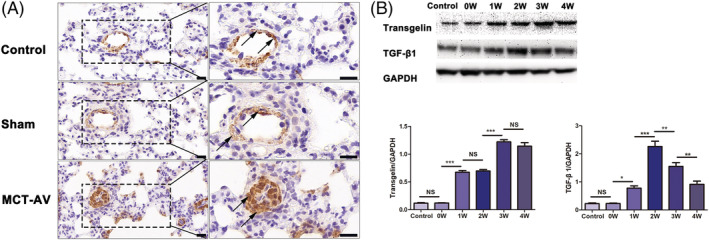
Time course of transgelin and TGF‐β1 expression in lung tissues from MCT‐AV‐induced PAH rats. (A) IHC staining for transgelin expression in lung tissues from MCT‐AV‐induced PAH rats. The black arrows indicate transgelin+ cells located in pulmonary arterioles. Scar bars, 20 μm. (B) Immunoblotting for expression of transgelin and TGF‐β1 in lung tissues from MCT‐AV‐induced PAH rats. Data are represented as mean ± SEM (n = 8). *P < 0.05, **P < 0.01, ***P < 0.001, NS = no significance.
Transgelin promotes human pulmonary arterial smooth muscle cell dysfunction via the transforming growth factor‐beta 1 signalling pathway
Based on the known involvement of transgelin in HPASMC dysfunction and that TGF‐β1 regulation of transgelin has been reported, 21 , 22 we sought to determine if the expression of HPASMC phenotype markers was changed in the presence of TGF‐β1. HPASMCs were transduced with lentiviral RNA vectors to regulate transgelin expression (LV‐siTAGLN). Immunoblotting showed that LV‐siTAGLN inhibited the expression of transgelin while LV‐TAGLN enhanced expression (Figure 3 A ). As expected, the expression of HPASMC synthetic phenotype markers [osteopontin (OPN), vimentin (VIM), and proliferating cell nuclear antigen (PCNA)] was increased by TGF‐β1 stimulation, accompanied by decreased expression of contractile phenotype markers (α‐SMA, calponin [CALP]) and caspase‐3 (CASP3) (Figure 3 B ). The proliferation and migration ability of HPASMCs were dramatically enhanced following TGF‐β1 stimulation, but not antiapoptotic ability (Figure 4 ). And these effects were abolished by transgelin knockdown (Figures 3 B and 4 C ), while enhanced by transgelin overexpression. As described above, TGF‐β1 stimulates transgelin expression in HPASMCs (Figure 1 C ). Thus, these observations suggest that transgelin modulates the phenotypic transformation, proliferation, migration, and apoptosis resistance in HPASMCs through the TGF‐β1 signalling pathway, thereby contributing to pulmonary vascular remodelling in CHD‐PAH.
Figure 3.
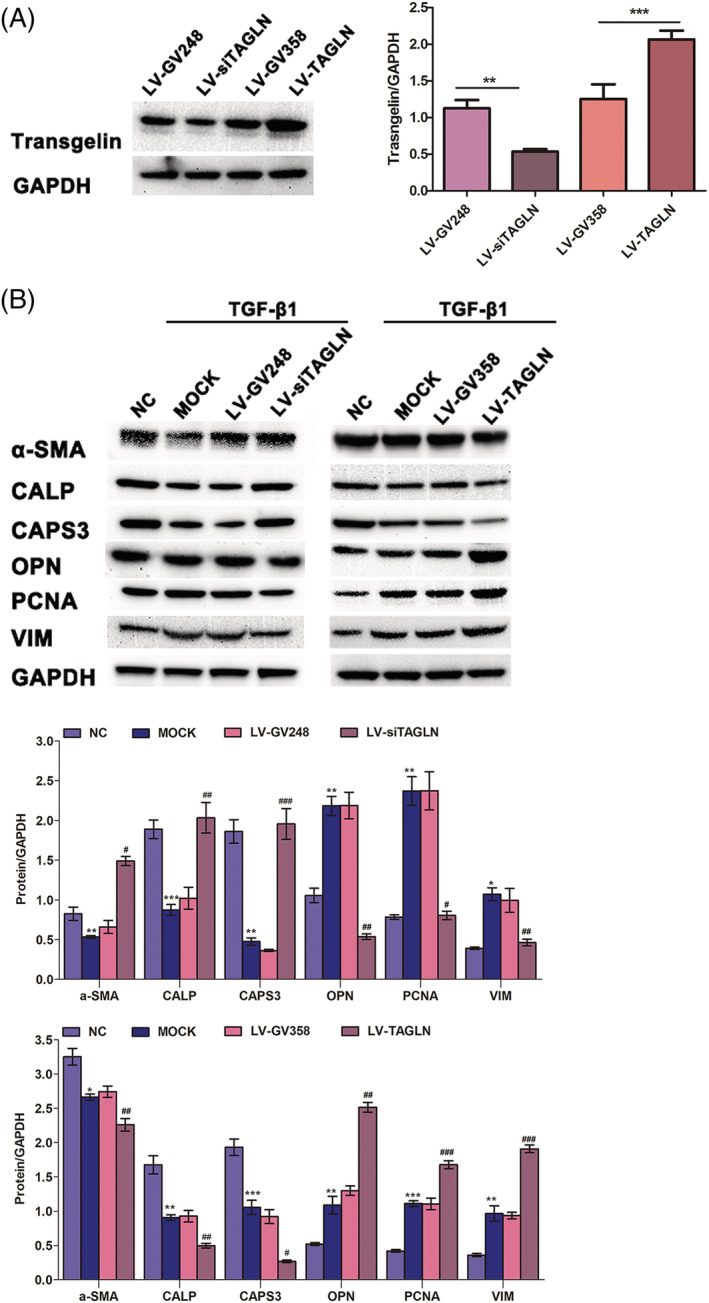
Transgelin promotes the phenotypic transformation of HPASMCs via TGF‐β1 signalling pathway. (A) HPASMCs were transfected with lentivirus to regulate transgelin expression. The efficiency of virus transfection was evaluated by immunoblotting. Data are represented as mean ± SEM (n = 3). ** P < 0.01; *** P < 0.001. (B) Effects of transgelin on the TGF‐β1‐induced expression of HPASMC phenotype markers, as determined by immunoblotting assay. Data are represented as mean ± SEM (n = 3). *: MOCK vs. negative control (NC); #: LV‐siTAGLN vs. LV‐GV248; #: LV‐TAGLN vs. LV‐GV358. * P < 0.05, ** P < 0.01, ***P < 0.001; # P < 0.05, ## P < 0.01, ### P < 0.001.
Figure 4.
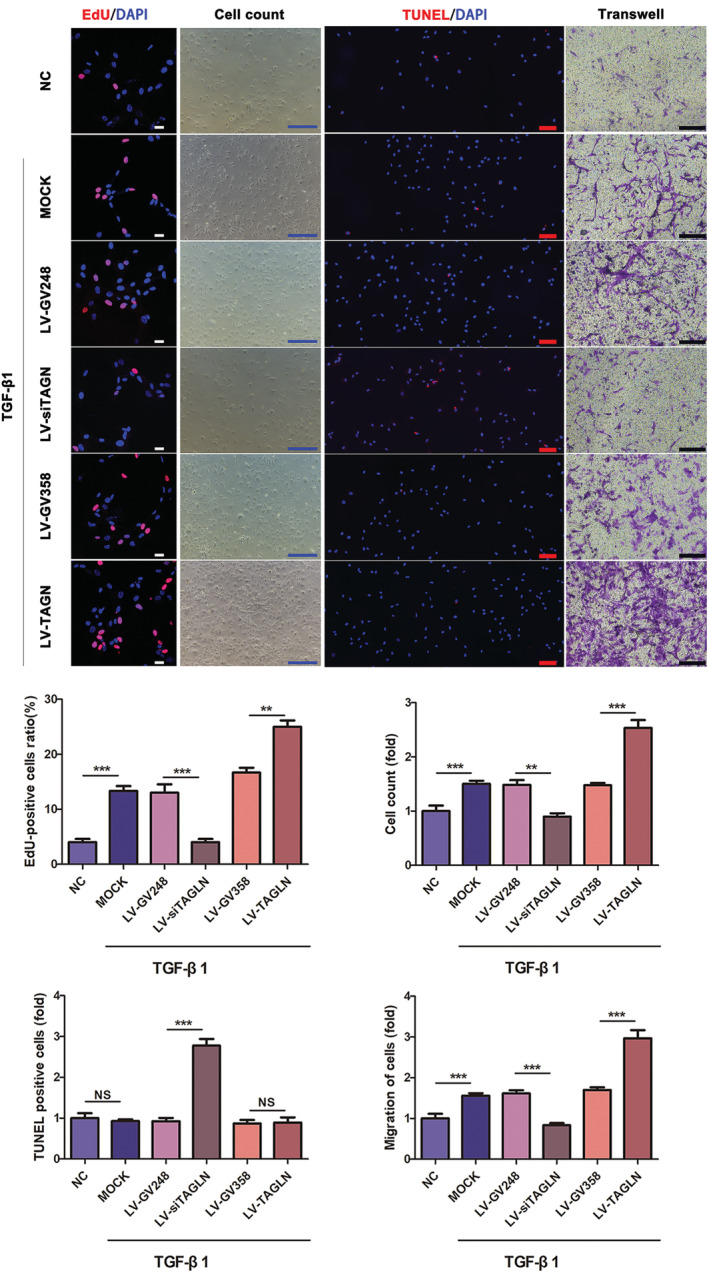
Transgelin enhances the proliferation, migration, and apoptosis resistance of HPASMCs via TGF‐β1 signalling pathway. Effects of transgelin on the TGF‐β1‐induced proliferation, apoptosis and migration of HPASMCs, as determined by EdU assay (or cell count), TUNEL assay and transwell assay, respectively. Data are represented as mean ± SEM (n = 3). ** P < 0.01, ***P < 0.001, NS = no significance. Scar bars (white: 25 μm; blue: 500 μm; red: 100 μm; black: 200 μm).
Discussion
In this study, we investigated transgelin expression in lung tissues of CHD‐PAH patients and MCT‐AV‐induced PAH rats, the molecular functions of transgelin in HPASMCs, and the role of transgelin in the progression of PAH and pulmonary vascular remodelling in MCT‐AV‐induced PAH rats. The results showed that transgelin was overexpressed in lung tissues from CHD‐PAH patients and MCT‐AV‐induced PAH rats, and positively correlated with the pathological grading of pulmonary arteries in CHD‐PAH patients. The positive correlation between TGF‐β1 and transgelin expression during the progression of CHD‐PAH was confirmed. Interestingly, TGF‐β1 promoted transgelin overexpression, which in turn enhanced TGF‐β1‐induced HPASMC dysfunction. Additionally, knockdown of transgelin significantly suppressed TGF‐β1‐induced HPASMC dysfunction. Overall, these results provide further insight into the molecular mechanisms of maladaptive arterial remodelling in CHD‐PAH, indicating that transgelin may represent a potential therapeutic target for pulmonary vascular remodelling in CHD‐PAH.
Changes of transgelin in lung tissue of shunt‐related pulmonary arterial hypertension and its significance
In CHD‐PAH, remodelling of the pulmonary vasculature reaches an irreversible phenotype comparable with all types of end‐stage PAH. However, it can be completely reversible if an early reversible stage is identified. Thus, recognition of the transition from reversible medial hypertrophy into irreversible plexiform lesions is central to the clinical evaluation of CHD‐PAH progression. Transgelin is an abundant protein expressed in the smooth muscle of mammals. In this study, we demonstrated that transgelin expression is enhanced in PASMCs of remodelled pulmonary arterioles of CHD‐PAH patients, especially in those with irreversible CHD‐PAH. This was further substantiated by the observation that transgelin was also expressed in PASMCs of the pulmonary artery media of MCT‐AV‐induced PAH rats. Transgelin expression was increased between 1 and 4 weeks post‐surgery, peaking at 3 weeks, in MCT‐AV‐induced PAH rats. A previous study showed that the proliferative activity of PASMCs is maximized 1 week after exposure to hypoxic conditions, and then mild proliferative activity was sustained for the following 2 to 4 weeks. 23 Therefore, we speculate that the dynamic expression of transgelin may be involved in phenotypic changes in PASMCs during the progression of pulmonary vascular remodelling in shunt‐related PAH. Moreover, transgelin expression was positively correlated with the degree of pulmonary vascular remodelling in CHD‐PAH patients, and comparable with the morphological signs of pulmonary vascular remodelling in lung tissues of MCT‐AV‐induced PAH rats, suggesting that transgelin may be an indicator of pulmonary vascular remodelling in shunt‐related PAH.
These findings demonstrate the importance of exploring the factors that affect the expression of transgelin in the progression of CHD‐PAH. TGF‐β1 induction of transgelin has been reported in various systems, but this has not been adequately studied in CHD‐PAH. Several lines of evidence have confirmed the presence of a TGF‐β1 control element (TCE) in the transgelin promoter, suggesting a genetic mechanism of transgelin regulation by TGF‐β1. 8 , 9 Increasing evidence demonstrates that TGF‐β1 is up‐regulated and plays a central role in pulmonary vascular remodelling of shunt‐related PAH. 18 , 24 Accordingly, TGF‐β1 was highly expressed in lung tissues of CHD‐PAH patients and showed a strong positive correlation with increased transgelin levels. Likewise, the expression of TGF‐β1 changed along with a similar trend as transgelin over time. In HPASMCs, TGF‐β1 strongly stimulated transgelin expression. Based on these results, we suggest that increased pulmonary blood flow dysregulates TGF‐β1 signalling, leading to a sharp increase in transgelin, which may be important in vascular remodelling in shunt‐related PAH.
Crucial roles for transgelin in human pulmonary arterial smooth muscle cell dysfunction
TGF‐β1 is a multifunctional cytokine involved in the regulation of proliferation, differentiation, migration, and apoptosis in PASMCs in PAH. Blockade of the TGF‐β1 signalling pathway is effective in many pre‐clinical models of experimental PAH including experimental hypoxia, MCT, and exposure to the parasite Schistosoma mansoni. 18 , 25 , 26 Therefore, we explored the role of TGF‐β1 in HPASMC dysfunction. Consistent with previous work, TGF‐β1 enhanced the phenotypic transformation, and proliferative, migratory, and antiapoptotic capability of HPASMCs, 27 , 28 , 29 and these effects were enhanced by transgelin overexpression. Based on our observations, we may conclude that transgelin patriciates in enhancing HPASMC dysfunction via the TGF‐β1 signalling pathway. These results are in line with prior studies that showed increased expression of transgelin accelerated cell proliferation and migration through the TGF‐β1 signalling pathway. 11 , 15 , 30 , 31 In contrast, transgelin overexpression has been reported to suppress VSMC proliferation and neointimal formation. 32 These paradoxical roles may be the result of an organ‐specific mechanism and/or variable pathological conditions. It has been shown that TGF‐β1 can modulate cell contraction by altering actin cytoskeletal components. Exposure of cells in culture to TGF‐β1 promoted stress fibre formation, which can affect the phenotype marker expression of smooth muscle cells (SMCs), implying a functional interaction between the actin cytoskeleton and the smooth muscle contractile apparatus. Transgelin is a member of the calponin family of actin‐binding proteins and is localized in the cytoskeleton. 33 , 34 Cytoskeletal remodelling may be involved in the mechanism by which transgelin enhances HPASMC phenotypic transformation. Further studies focusing on the effects of transgelin on the cytoskeletal organization may help to elucidate its mechanisms in these discrepant roles.
Conclusions
In summary, this study has identified that transgelin may be an indicator of CHD‐PAH development, due to its function in promoting HPASMC dysfunction through the TGF‐β1 signalling pathway. Transgelin was also shown to be induced by TGF‐β1 in HPASMCs. Based on our findings, transgelin may serve as a promising factor for assessing the degree of vascular remodelling in CHD‐PAH and represent a potential therapeutic target for treatment.
Conflict of interest
No conflict of interests, financial or otherwise, are declared by the authors.
Funding
This study was provided by the National Natural Science Foundation of China (No. 8177021278), the National Key Research and Development Program of China (No. 2016YFC1304400), and CAMS Innovation Fund for Medical Sciences (CIFMS; No. 2017‐12M‐1‐009).
Supporting information
Table S1. Antibodies information for immunohistochemistry (IHC), immunofluorescent (IF) and immunoblotting.
Acknowledgements
We thank all study subjects for participating in the study.
Zhou, J. , Yang, J. , Li, L. , Quan, R. , Chen, X. , Qian, Y. , Huang, L. , Wang, P. , Li, Y. , Meng, X. , Chen, X. , Gu, Q. , and He, J.‐G. (2022) Transgelin exacerbates pulmonary artery smooth muscle cell dysfunction in shunt‐related pulmonary arterial hypertension. ESC Heart Failure, 9: 3407–3417. 10.1002/ehf2.14080.
Contributor Information
Qing Gu, Email: guqingfw@126.com.
Jian‐Guo He, Email: hejianguofw@163.com.
References
- 1. Pascall E, Tulloh RM. Pulmonary hypertension in congenital heart disease. Future Cardiol. 2018; 14: 343–353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. D'Alto M, Mahadevan VS. Pulmonary arterial hypertension associated with congenital heart disease. Eur Respi Rev Off J Eur Respi Soc. 2012; 21: 328–337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Wagenvoort CA, Wagenvoort N, Draulans‐Noe Y. Reversibility of plexogenic pulmonary arteriopathy following banding of the pulmonary artery. J Thorac Cardiovasc Surg. 1984; 87: 876–886. [PubMed] [Google Scholar]
- 4. Heath D, Edwards JE. The pathology of hypertensive pulmonary vascular disease; a description of six grades of structural changes in the pulmonary arteries with special reference to congenital cardiac septal defects. Circulation. 1958; 18: 533–547. [DOI] [PubMed] [Google Scholar]
- 5. Lindberg L, Olsson AK, Jogi P, Jonmarker C. How common is severe pulmonary hypertension after pediatric cardiac surgery? J Thorac Cardiovasc Surg. 2002; 123: 1155–1163. [DOI] [PubMed] [Google Scholar]
- 6. Manes A, Palazzini M, Leci E, Bacchi Reggiani ML, Branzi A, Galie N. Current era survival of patients with pulmonary arterial hypertension associated with congenital heart disease: A comparison between clinical subgroups. Eur Heart J. 2014; 35: 716–724. [DOI] [PubMed] [Google Scholar]
- 7. Wang D, Uhrin P, Mocan A, Waltenberger B, Breuss JM, Tewari D, Mihaly‐Bison J, Huminiecki L, Starzynski RR, Tzvetkov NT, Horbanczuk J, Atanasov AG. Vascular smooth muscle cell proliferation as a therapeutic target. Part 1: Molecular targets and pathways. Biotechnol Adv. 2018; 36: 1586–1607. [DOI] [PubMed] [Google Scholar]
- 8. Hernandez‐Saavedra D, Sanders L, Freeman S, Reisz JA, Lee MH, Mickael C, Kumar R, Kassa B, Gu S, Alessandro A, Stenmark KR, Tuder RM, Graham BB. Stable isotope metabolomics of pulmonary artery smooth muscle and endothelial cells in pulmonary hypertension and with TGF‐beta treatment. Sci Rep. 2020; 10: 413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Elsafadi M, Manikandan M, Almalki S, Mahmood A, Shinwari T, Vishnubalaji R, Mobarak M, Alfayez M, Aldahmash A, Kassem M, Alajez NM. Transgelin is a poor prognostic factor associated with advanced colorectal cancer (CRC) stage promoting tumor growth and migration in a TGFbeta‐dependent manner. Cell Death Dis. 2020; 11: 341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Shapland C, Hsuan JJ, Totty NF, Lawson D. Purification and properties of transgelin: A transformation and shape change sensitive actin‐gelling protein. J Cell Biol. 1993; 121: 1065–1073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Zhang R, Shi L, Zhou L, Zhang G, Wu X, Shao F, Ma G, Ying K. Transgelin as a therapeutic target to prevent hypoxic pulmonary hypertension. Am J Physiol Lung Cell Mol Physiol. 2014; 306: L574–L583. [DOI] [PubMed] [Google Scholar]
- 12. Huang L, Li L, Yang T, Li W, Song L, Meng X, Gu Q, Xiong C, He J. Transgelin as a potential target in the reversibility of pulmonary arterial hypertension secondary to congenital heart disease. J Cell Mol Med. 2018; 22: 6249–6261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Huang L, Li L, Hu E, Chen G, Meng X, Xiong C, He J. Potential biomarkers and targets in reversibility of pulmonary arterial hypertension secondary to congenital heart disease: An explorative study. Pulmonary circulation. 2018; 8():2045893218755987: 1–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Bartram U, Speer CP. The role of transforming growth factor beta in lung development and disease. Chest. 2004; 125: 754–765. [DOI] [PubMed] [Google Scholar]
- 15. Chen Z, He S, Zhan Y, He A, Fang D, Gong Y, Li X, Zhou L. TGF‐beta‐induced transgelin promotes bladder cancer metastasis by regulating epithelial‐mesenchymal transition and invadopodia formation. EBioMedicine. 2019; 47: 208–220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Zhou JJ, Li H, Qian YL, Quan RL, Chen XX, Li L, Li Y, Wang PH, Meng XM, Jing XL, He JG. Nestin represents a potential marker of pulmonary vascular remodeling in pulmonary arterial hypertension associated with congenital heart disease. J Mol Cell Cardiol. 2020; 18: 41–53. [DOI] [PubMed] [Google Scholar]
- 17. Zhou JJ, Li H, Li L, Li Y, Wang PH, Meng XM, He JG. CYLD mediates human pulmonary artery smooth muscle cell dysfunction in congenital heart disease‐associated pulmonary arterial hypertension. J Cell Physiol. 2021; 236: 6297–6311. [DOI] [PubMed] [Google Scholar]
- 18. Kumar R, Mickael C, Kassa B, Gebreab L, Robinson JC, Koyanagi DE, Sanders L, Barthel L, Meadows C, Fox D, Irwin D, Li M, McKeon BA, Riddle S, Dale Brown R, Morgan LE, Evans CM, Hernandez‐Saavedra D, Bandeira A, Maloney JP, Bull TM, Janssen WJ, Stenmark KR, Tuder RM, Graham BB. TGF‐beta activation by bone marrow‐derived thrombospondin‐1 causes Schistosoma‐ and hypoxia‐induced pulmonary hypertension. Nat Commun. 2017: 15494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Dickinson MG, Bartelds B, Molema G, Borgdorff MA, Boersma B, Takens J, Weij M, Wichers P, Sietsma H, Berger RM. Egr‐1 expression during neointimal development in flow‐associated pulmonary hypertension. Am J Pathol. 2011; 179: 2199–2209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Garcia R, Diebold S. Simple, rapid, and effective method of producing aortocaval shunts in the rat. Cardiovasc Res. 1990; 24: 430–432. [DOI] [PubMed] [Google Scholar]
- 21. Chen S, Kulik M, Lechleider RJ. Smad proteins regulate transcriptional induction of the SM22alpha gene by TGF‐beta. Nucleic Acids Res. 2003; 31: 1302–1310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Qiu P, Feng XH, Li L. Interaction of Smad3 and SRF‐associated complex mediates TGF‐beta1 signals to regulate SM22 transcription during myofibroblast differentiation. J Mol Cell Cardiol. 2003; 35: 1407–1420. [DOI] [PubMed] [Google Scholar]
- 23. Aoshima D, Murata T, Hori M, Ozaki H. Time‐dependent phenotypic and contractile changes of pulmonary artery in chronic hypoxia‐induced pulmonary hypertension. J Pharmacol Sci. 2009; 110: 182–190. [DOI] [PubMed] [Google Scholar]
- 24. Zhang N, Dong M, Luo Y, Zhao F, Li Y. Danshensu prevents hypoxic pulmonary hypertension in rats by inhibiting the proliferation of pulmonary artery smooth muscle cells via TGF‐β‐smad3‐associated pathway. Eur J Pharmacol. 2018; 820: 1–7. [DOI] [PubMed] [Google Scholar]
- 25. Zaiman AL, Podowski M, Medicherla S, Gordy K, Xu F, Zhen L, Shimoda LA, Neptune E, Higgins L, Murphy A, Chakravarty S, Protter A, Sehgal PB, Champion HC, Tuder RM. Role of the TGF‐beta/Alk5 signaling pathway in monocrotaline‐induced pulmonary hypertension. Am J Respir Crit Care Med. 2008; 177: 896–905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Ma W, Han W, Greer PA, Tuder RM, Toque HA, Wang KK, Caldwell RW, Su Y. Calpain mediates pulmonary vascular remodeling in rodent models of pulmonary hypertension, and its inhibition attenuates pathologic features of disease. J Clin Invest. 2011; 121: 4548–4566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Haga S, Tsuchiya H, Hirai T, Hamano T, Mimori A, Ishizaka Y. A novel ACE2 activator reduces monocrotaline‐induced pulmonary hypertension by suppressing the JAK/STAT and TGF‐beta cascades with restored caveolin‐1 expression. Exp Lung Res. 2015; 41: 21–31. [DOI] [PubMed] [Google Scholar]
- 28. Roger I, Milara J, Montero P, Cortijo J. The role of JAK/STAT molecular pathway in vascular remodeling associated with pulmonary hypertension. Int J Mol Sci. 2021; 22: 4980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Liu RY, Zeng Y, Lei Z, Wang L, Yang H, Liu Z, Zhao J, Zhang HT. JAK/STAT3 signaling is required for TGF‐β‐induced epithelial‐mesenchymal transition in lung cancer cells. Int J Oncol. 2014; 44: 1643–1651. [DOI] [PubMed] [Google Scholar]
- 30. Mitarai H, Wada N, Hasegawa D, Yoshida S, Sonoda M, Tomokiyo A, Hamano S, Serita S, Mizumachi H, Maeda H. Transgelin mediates transforming growth factor‐beta1‐induced proliferation of human periodontal ligament cells. J Periodontal Res. 2017; 52: 984–993. [DOI] [PubMed] [Google Scholar]
- 31. Zhou HM, Fang YY, Weinberger PM, Ding LL, Cowell JK, Hudson FZ, Ren M, Lee JR, Chen QK, Su H, Dynan WS, Lin Y. Transgelin increases metastatic potential of colorectal cancer cells in vivo and alters expression of genes involved in cell motility. BMC Cancer. 2016: 55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Dong LH, Wen JK, Liu G, McNutt MA, Miao SB, Gao R, Zheng B, Zhang H, Han M. Blockade of the Ras‐extracellular signal‐regulated kinase 1/2 pathway is involved in smooth muscle 22 alpha‐mediated suppression of vascular smooth muscle cell proliferation and neointima hyperplasia. Arterioscler Thromb Vasc Biol. 2010; 30: 683–691. [DOI] [PubMed] [Google Scholar]
- 33. Lv P, Miao SB, Shu YN, Dong LH, Liu G, Xie XL, Gao M, Wang YC, Yin YJ, Wang XJ, Han M. Phosphorylation of smooth muscle 22alpha facilitates angiotensin II‐induced ROS production via activation of the PKCdelta‐P47phox axis through release of PKCdelta and actin dynamics and is associated with hypertrophy and hyperplasia of vascular smooth muscle cells in vitro and in vivo. Circ Res. 2012; 111: 697–707. [DOI] [PubMed] [Google Scholar]
- 34. Elsafadi M, Manikandan M, Dawud RA, Alajez NM, Hamam R, Alfayez M, Kassem M, Aldahmash A, Mahmood A. Transgelin is a TGFbeta‐inducible gene that regulates osteoblastic and adipogenic differentiation of human skeletal stem cells through actin cytoskeleston organization. Cell Death Dis. 2016; 7: e2321. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Table S1. Antibodies information for immunohistochemistry (IHC), immunofluorescent (IF) and immunoblotting.