

Abstract
B-cell lymphoma 6 (BCL6) is a protooncogene in adult and pediatric cancers, first identified in diffuse large B-cell lymphoma (DLBCL) where it acts as a repressor of the tumor suppressor TP53, conferring survival, protection, and maintenance of lymphoma cells. BCL6 expression in normal B cells is fundamental in the regulation of humoral immunity, via initiation and maintenance of the germinal centers (GC). Its role in B cells during the production of high affinity immunoglobins (that recognize and bind specific antigens) is believed to underpin its function as an oncogene. BCL6 is known to drive the self-renewal capacity of leukemia-initiating cells (LIC), with high BCL6 expression in acute lymphoblastic leukemia (ALL), acute myeloid leukemia (AML), and glioblastoma (GBM) associated with disease progression and treatment resistance. The mechanisms underpinning BCL6-driven therapy resistance are yet to be uncovered; however, high activity is considered to confer poor prognosis in the clinical setting. BCL6’s key binding partner, BCL6 corepressor (BCOR), is frequently mutated in pediatric cancers and appears to act in concert with BCL6. Using publicly available data, here we show that BCL6 is ubiquitously overexpressed in pediatric brain tumors, inversely to BCOR, highlighting the potential for targeting BCL6 in these often lethal and untreatable cancers. In this review, we summarize what is known of BCL6 (role, effect, mechanisms) in pediatric cancers, highlighting the two sides of BCL6 function, humoral immunity, and tumorigenesis, as well as to review BCL6 inhibitors and highlight areas of opportunity to improve the outcomes of patients with pediatric cancer.
Introduction
BCL6 encodes a zinc finger transcription repressor that is frequently translocated in diffuse large B-cell lymphomas (DLBCL; ref. 1). A master regulator of humoral immunity, the BCL6 protein plays a critical role in the initiation and maintenance of the germinal centers (GC). GCs are microstructures formed in secondary lymphoid tissues (such as lymph nodes, tonsils, and spleen) and are the sites of antibody diversification and affinity maturation. Reiterative cycles of somatic hypermutation (SHM) in immunoglobulin gene (Ig) variable regions produce high-affinity antibodies in response to T-cell dependent antigen presentation, and hence, are vital to humoral immunity (2).
Transcription of tumor suppressors, high-fidelity DNA repair genes, and genes implicated in cell differentiation are repressed by BCL6. Simultaneously, BCL6 promotes the expression of genes linked with proliferation, immune avoidance, antiapoptosis, cell-cycle arrest, and cell differentiation, hallmarks of cancer cell biology. The many cancer-associated functions linked to BCL6 activity promote a “perfect oncostorm”, deregulated transcriptional programs with increased tolerance to genome insult and instability. BCL6 acts similarly in B cells, as an engine of cell turnover, where it promotes SHM and mediates immune cell metamorphosis in response to encountering foreign antigens (3).
The oncogenic functions of BCL6 have been characterized in lymphoma as early as 1993 (4). Notably, BCL6 has since been implicated in pediatric cancers, including acute lymphoblastic leukemia (ALL; refs. 5, 6), acute myeloid leukemia (AML; ref. 7), and solid cancers such as high-grade glioma (HGG; refs. 8, 9) and sarcoma (discussed in Section BCL6 in Pediatric Cancers). With BCL6 playing an important role in transcriptional repression, BCL6 rearrangement (10) and/or overexpression (11) is associated with poor overall survival, and resistance to chemotherapies (5, 12).
Despite the important roles outlined above, targeting BCL6 as an anticancer approach is yet to be effectively executed in the clinic. Drawing on what is known of BCL6 (role, effect, mechanisms) in pediatric cancers, and humoral immunity, we review the utility of BCL6-targeted therapies in the hope of improving future outcomes in the pediatric cancer setting.
BCL6 Structure
The BCL6 gene is located on chromosome 3q27 and encodes a 95 kDa protein that acts as a master regulator, necessary for the formation of GCs in B-cell follicles of lymphoid tissues in response to antigen encounter (13, 14). The BCL6 protein harbors a trimodular structure consisting of (i) an N-terminus Broad-complex, Tramtrack and Bric‐à‐brac/poxvirus and zinc finger (BTB/POZ) domain, a docking site for corepressor proteins; (ii) a central transcriptional repressor (RD2) domain containing a proline (P), glutamic acid (E), serine (S), and threonine (T) – PEST region, important in the control of protein half-life and interactions with auto regulatory proteins (15); and (iii) a series of C-terminal Krüppel-like C2H2-type zinc fingers that bind DNA (Fig. 1; ref. 16).
Figure 1.

Structure and function of BCL6. BCL6 harbors a trimodular structure consisting of a N-terminus Broad-complex, Tramtrack and Bris‐à-brac/poxvirus and zinc finger (BTB/POZ) domain for protein interactions such as corepressor proteins (darker orange). A central transcriptional repressor domain (RD2) containing a proline (P), glutamic acid (E), serine (S), and threonine (T) – PEST region, which dictates the proteins activity and stability by binding regulatory proteins that determine its half-life (darker blue). A series of C-terminal Krüppel-like C2H2-type zinc fingers which bind DNA to repressed gene expression (darker green).
The BCL6 homodimer binds to target gene promoters to influence transcriptional programs during the formation of GCs. This is regulated following recruitment of corepressor proteins that bind to an exposed surface at the interface of the two chains of the BCL6 dimer, referred to as the lateral groove BTB domain (Fig. 1; ref. 17). BCL6 corepressor (BCOR), nuclear receptor corepressor 1 and 2 (NCOR1, NCOR2/SMRT) form a multifunctional ternary corepressor complex - BCL6 binds at gene promoters, with BCOR bound to polycomb repressive complex 1 (PRC1) and NCOR2/SMRT bound to histone deacetylase 3 (HDAC3; Fig. 2; refs. 18, 19). Not only does this drive the deacetylation of histone H3 at lysine 27 (H3K27), but also recruits the H3K27 methyltransferase Enhancer of zeste homlog 2 (EZH2) through tethering to noncanonical PRC1–BCOR–CBX8 complexes. EZH2 catalyzes the trimethylation of H3K27 (H3K27me3) and the formation of bivalent chromatin, resulting in epigenetic silencing of the cell-cycle arrest gene, cyclin-dependent kinase inhibitor 1A (CDKN1A), and retinoblastoma RB/E2F1 tumor suppressor pathway (Fig. 2; ref. 20).
Figure 2.

BCL6 role in transcriptional regulation: BCL6 homodimer binds to target gene promoters to repress the transcriptional programs required for tumor suppression and apoptosis during the formation of the GCs. This is regulated via the recruitment of BCL6 corepressor (BCOR), nuclear receptor corepressor (NCOR1), and nuclear receptor corepressor 2 (NCOR2) to form a multifunctional ternary corepressor complex. BCL6 represses or poises transcription through BCOR and NCOR2 binding to histone deacetylase 3 (HDAC3), resulting in deacetylation of H3K27. When BCOR binds and recruits noncanonical polycomb repressive complex 1 (PRC1)-CBX8, the polycomb repressive complex 2 (PRC2) complex subunit enhancer of zester homolog 2 (EZH2) catalyzes H3K27 trimethylation resulting in epigenetic silencing. Ac = acetylation, Me3 = trimethylation. (Created with BioRender.com)
It is of little wonder that BCL6 regulatory BTB domain has become an attractive target for the development of BCL6 inhibitors. The unique lateral groove at the surface of the BTB domain differentiates BCL6 from other BTB proteins (Fig. 1; refs. 14, 21) and hence, competitive inhibitors have the potential to be BCL6 specific (Table 1). Peptomimetic inhibitors and competitive peptide aptamers of the BTB have been shown to compete with corepressor binding, and effectively eliminate the repressor functions of BCL6, and increase expression of BCL6 target tumor suppressor genes (discussed in Section BCL6 as a Therapeutic Target; ref. 22).
Table 1.
BCL6 targeted inhibitors.
| Type | Name | MW g/mol | Malignancy | IC50 | KD | In vivo model | In vivo dose | In vivo outcome | Challenges for translation | Reference |
|---|---|---|---|---|---|---|---|---|---|---|
| Peptide Inhibitors | BPI | 4,550.19 | DLBCL | 14.2 μmol/L | 20 μmol/L | NR | NR | NR | Easily degraded by proteases | (93) |
| F1324 | 1,765.04 | NR | 1 nmol/L | 0.57 nmol/L | NR | NR | NR | No inhibitory effects in vitro | (95) | |
| Apt48 | 1,536.74 | BL | NR | NR | NR | NR | NR | No cytotoxic effects, requires cytokines | (22) | |
| Reverse peptide inhibitors | RI-BPI | 4,510.12 | DLBCL | 16.5 μmol/L | 16 μmol/L | SU-DHL4 SU-DHL6 Toledo SCID mice | 150 & 500 μmol/L/day | ↓ TV ↑survival SU-DHL4/6 | Weak affinity + large MW | (94) |
| Glioma | 5–25 μmol/L | NR | JM94 | 50 mg/kg/day | ↓ TS ↑ response with EGFR I | (9) | ||||
| Small molecules | 79-6 | 457.28 | DLBCL | 24–93.6 μmol/L | 138 μmol/L | OCI-Ly7 SU-DHL6 Toledo SCID mice | 0.5–50 mg/kg/day | ↓TV OCI-Ly7 SU-DHL6 | Non apoptotic, BCL6 lymphoma specific | (95) |
| FX1 | 368.82 | NSCLC | 35 μmol/L | 4 μmol/L | OCI-Ly7 SU-DHL6 Toledo SCID mice | 25 & 50 mg/kg/day | ↓TV OCI-Ly7 SU-DHL6 | BCL6 lymphoma specific | (101) | |
| GSK137 | 374 | DLBCL | 10 nmol/L | 10 nmol/L | NR | NR | NR | Non cytotoxic | (101) | |
| BI-3802 | 485 | DLBCL | 20 nmol/L | <3 nmol/L | NR | NR | NR | Degrades BCL6 | (98,99) | |
| BI-3812 | 558 | DLBCL | 3 nmol/L | <3 nmol/L | NR | NR | NR | Bioavailability | (98,99) |
Abbreviations: IC50, inhibitory concentration 50%; KD, disassociation constant; NR, not reported in cancer models; TS, tumor size; TV, tumor volume.
BCL6 in Humoral Immunity
Regulation of the GCs
The formation of GCs in secondary lymphoid tissues (lymph nodes, tonsils, spleen, Peyer's patches, and mucosa-associated lymphoid tissues) is critical in the production of long-lived antibody-secreting plasma cells and memory B cells in response to infection, and as such, underpin the function of the humoral immune system (13). In lymphoid organs, upon antigen presentation, B cells undergo SHM, followed by Ig class switch recombination (CSR), resulting in mass proliferation of mature B cells with foreign antigen affinity. BCL6 is known to play an essential role in the maturation of B cells and the formation of GCs, as BCL6−/− knockout mice fail to form GCs (13). Activated B cells are called “centroblasts” and form the dark zone of the GCs, upregulating BCL6 following interactions with T cells/antigens (23). Once differentiated, nondividing activated GC B cells derived from centroblasts are called “centrocytes” and migrate to the light zone of the GCs, where the affinity of their antibody receptors is evaluated with the help of T follicular helper cells (TFH) through interactions with follicular dendritic cells (FDCs; ref. 24).
BCL6 in SHM and CSR
SHM and CSR occur in B cells following immunization or infection through T-cell–dependent (Td) activation of follicular B cells. A consequential and continuous elevation in BCL6 expression is seen during formation of GCs (25). This rise in BCL6 expression is essential to direct transcriptional programs that promote SHM while repressing expression of tumor surveillance and antiapoptotic gene signatures, allowing CSR to occur. Not only does BCL6 target the promoters of protein coding genes, but also microRNAs (miRNA), including miR-155 and miR-361, which target activation-induced cytidine deaminase (AID; ref. 26). AID is part of a family of cytidine deaminases that deaminate deoxycytosine to deoxyuracil (dCs to dUs) on both single-stranded DNA (ssDNA) substrates, and ssDNA generated by the formation of RNA–DNA hybrids (27). Repression of miR-155 and miR-361 drives AID expression, which introduces point mutations into the DNA at a relatively high rate (up to 10−4 to 10−3 per base per division) in both Ig and non-Ig genes (26, 28). AID is a key factor driving CSR and therefore an adaptive immune response.
Central to sustained DNA damage, antiapoptosis, and increased proliferation of mature B cells during SHM and CSR, BCL6 represses DNA repair and tumor suppressor gene expression. Repression of multiple genes including CHEK1, ATR, ARF, TP53, RB, and CDKN1A switch off high-fidelity DNA repair and apoptosis, driving the cell cycle (Fig. 2). These mechanisms have evolved to evade premature apoptosis and to reprogram B cells to allow for the selective specificity necessary to generate antibodies for the infinite number of possibly encountered foreign antigens (29).
Regulation of BCL6 expression in B cells
Inhibition of BCL6 function is a fundamental process in the normal immune response, occurring once GC B cells have matured with the correct antigen affinity, and is controlled via several mechanisms (30). The Myocyte enhancer-binding factor 2B (MEF2B) is thought to be the master transcription factor for BCL6 in B cells, binding to the promoter approximately 1 kilobase upstream of the BCL6 gene following T-cell activation (31). Activity of MEF2B is regulated by interactions with its corepressor phosphatase CABIN1 (Calcineurin binding protein 1; Fig. 3; ref. 32). Mutations within the C-terminus of MEF2B are associated with escape from phosphorylation-mediated negative regulation (31), with MEF2B deletion reducing the formation of GCs (33). MEF2B is mutated in approximately 10% to 12% of DLBCLs and follicular lymphomas (FL; ref. 31). MEF2B binds predominantly to chromatin at histone marks suggestive of active promotors, enhancers or superenhancers (H3K4me3+ and H3K27ac+) to regulate transcription of genes influencing DNA replication and repair, cell cycle, apoptosis, and GC B-cell confinement (Figs. 2 and 3; ref. 34). Interestingly, like BCL6, MEF2B plays an important role in regulating epigenetic modifiers, chromatin marks that are also modulated by the formation of BCL6 corepressor complex (Figs. 2 and 3, discussed in BCL6 in Humoral Immunity).
Figure 3.

BCL6 Regulation: BCL6 expression in B cells during GC formation occurs once B cells have matured with correct antigen affinity. T-cell activation and IL4 stimulation via cytokines drives metabolic tricarboxylic acid (TCA) cycle reprogramming through IDH2 and IDH3 and hence the production of alpha ketoglutarate (αKG), which acts as a substrate for Lysine-specific demethylase 6A (KDM6A) leading the loss of repressive H3K27me3 marks. KDM6A is recruited by phosphorylated Signal transducer and activator of transcription (STAT6), to the BCL6 locus causing the demethylation of H3K27me3, leading to euchromatin and active transcription of BCL6. Myocyte enhancer-binding factor 2B (MEF2B), the master regulator of BCL6 expression is regulated via its corepressor phosphatase, CABIN 1 (Calcineurin Binding Protein 1) and binds to BCL6 promoter, following T-cell activation. MEF2B binds predominantly to chromatin with histone marks, H3K4me3 (trimethylation, Me3) and H3K27ac (acetylation, Ac) suggestive of active marks. Following IL2 stimulation, CD40 activation and B-cell receptor activation, NF-kB is translocated to the nucleus where it suppresses the formation of the BCL6–NCOR complex, alleviating the negative regulation of the c-MYC promoter by BCL6. NF-kB activation promotes IRF4 to bind to the BCL6 promoter to negative regulate its transcription. (Created with BioRender.com)
Recent studies have shown an indirect mechanism of BCL6 expression in GCs during B-cell differentiation. Following IL4 stimulation, metabolic reprogramming sees the accumulation of α-ketoglutarate (αKG), produced by its conversion in the tricarboxylic acid (TCA) cycle from isocitrate. Isocitrate is converted to αKG using the Isocitrate dehydrogenase (IDH) enzymes (IDH2 and IDH3; Fig. 3). αKG acts as a substrate for epigenetic modifier Lysine-specific demethylase 6A (KDM6A), also known as Ubiquitously transcribed tetratricopeptide repeat, X chromosome (UTX). Signal transducer and activator of transcription 6 (STAT6) recruits KDM6A to the BCL6 locus, reducing methylation of repressive histone mark H3K27me3, leading to euchromatin and active transcription of BCL6 and B-cell differentiation in the developing GCs (35).
Following an immune response, transient activation of the B-cell receptor (BCR), cluster of differentiation 40 (CD40), and IL2 receptors drive increased NF-κB activity and the dissociation of the BCL6–NCOR complex, alleviating the negative regulation of c-MYC by BCL6, driving the expression of cell-cycle genes (34). MYC activates both Cyclin D2 (CCND2) and Cyclin D3 (CCND3) resulting in a CCND2-dependent proliferation (36). Transient activation of surface receptors (such as CD40) lead to the nuclear translocation of NF-κB and induction of Interferon regulatory factor 4 (IRF4; ref. 34). IRF4 binds to the promoter region of BCL6 to suppress its expression (37) and is a member of the interferon-regulatory factor family of transcription factors (required for plasma cell differentiation). Hence, the expression level and activity of CD40, their respective TFH, promotes memory B-cell differentiation from GC B cells, ending the requirement for BCL6’s repressor activity (38). CD40 stimulation has also been reported to dysregulate BCL6 transcriptional repression of the DNA repair gene Serine/threonine-protein kinase (ATR; Fig. 2), responsible for the detection of DNA damage to be repaired, or in the case of genotoxicity, to trigger apoptosis (30). A subset of DLBCLs harboring BCL6 translocations or mutations (depending on the position of the chromosomal breakpoints) have been reported to be refractory to CD40-IRF4–induced BCL6 repression (34). Mutations in the IRF4-responsive region of the BCL6 promoter are identified in 60% of DLBCLs harboring BCL6 translocations (approximately 30% of all DLBCL diagnoses). With this mutation directly linked to pathogenesis and malignant growth of DLBCL subsets (34), it is therefore important to understand the regulation of BCL6 if we are to develop strategies to improve the treatment of BCL6-driven neoplasms.
Members of the STAT family are promiscuous transcription factors that are phosphorylated and activated by receptor-associated tyrosine kinases (39), causing them to dimerize and cross the nuclear membrane to bind to promoters and enhancers of regulatory genes, including BCL6. IL2, IL4, and IL10 induce growth of B cells through activation of JAK/STAT signaling pathways, whereas IL4 drives STAT6 activity to promote CSR (40) and upregulate BCL6 expression by enhancing RNA polymerase II recruitment and initiating transcription (41). The IL4/IL4R axis is known to activate STAT3, STAT5, and STAT6 (discussed in Regulation of BCL6 expression in B Cells), and is further enhanced by IL4-dependent metabolic reprogramming, driving KDM6A activity and demethylation of repressive H3K27me3 marks, increasing STAT6-dependent BCL6 expression (Fig. 3; ref. 35). STAT5 is a potent oncogenic transcription factor in acute hematologic malignancies that has been shown to outcompete STAT3 for binding to the enhancers of BCL6 at one of the two shared regulatory sites (41), highlighting the potential of targeting BCL6 in malignancies characterized by high STAT5 activity.
BCL6 in Pediatric Cancers
Over the past four decades, mortality rates for children diagnosed with cancer have reduced by more than 50% (42). This is underpinned by sophisticated improvements to protocol-driven clinical trials that utilize combinations of chemotherapies that attempt to address the somatic and epigenetic heterogeneity of these cancers. However, for some of the most aggressive types of childhood malignancies, there remains no effective pharmacologic intervention and children succumb to their disease within a short timeframe (39, 43–45). As 35% to 40% of children's malignancies originate in cells of the lymphoid linage where BCL6 plays a critical role in their maturation, there is a potential for targeting BCL6 in combination with therapies focused on the recurring somatic alterations responsible for disease initiation and progression. Furthermore, the opportunity to increase p53 activity (and hence tumor suppression) through BCL6 inhibition is an attractive paradigm in the pediatric setting, as somatic loss-of-function mutations in TP53 are only seen in 4% of cases, compared with adults where TP53 mutations are the most common mutation (>50% of unselected sporadic tumors; ref. 46). However, this is not to say that p53 shows ubiquitous tumor suppression in pediatric cancers. Rather, p53 dysregulation occurs via the altered activity of cell cycle regulators (CDKN2A or CDKN2B) and through mutations in genes implicit in modulating the pediatric cancer epigenome (TP53, H3K27M, IDH2/3, SETD2), with these recognized as some of the most recurring mutations in children's cancers (47). Furthermore, mutations in the direct regulators of BCL6 activity, that is, BCOR, are common in pediatric cancers of the central nervous system (CNS; discussed in Pediatric central nervous system cancers). BCOR interacts selectively with the POZ domain of BCL6, and interestingly, these mutations lead to reduced affinity for BCL6 which drives cell proliferation, Protein kinase B (AKT) phosphorylation, the expression of IL2, and importantly, inhibition of TP53 through gene repression.
Using publicly available pediatric cancer genome and RNA expression data from the St. Jude Cloud (48), we analyzed the expression and mutation of BCL6 and BCOR across the three main types of pediatric cancer categories: hematopoietic malignancies (HM), brain tumors (BT), and solid tumors (ST), in the de novo and relapse setting (Fig. 4). Interestingly, there is a significant increase in mRNA expression of BCL6 in BT at diagnosis and relapse compared with HM or ST (Fig. 4A). The reciprocal trend was seen for BCOR mRNA expression with higher expression in HM and ST compared with BT (Fig. 4B). This is not surprising given that high-grade pediatric cancers of the CNS are often caused by epigenetic alterations, driving hypomethylation of repressive chromatin marks resulting in the formation of euchromatin and potentially high expression of BCL6 (43, 45).
Figure 4.

BCL6 and BCOR shows hyperexpression in pediatric brain cancers. A,BCL6 and B,BCOR mRNA expression data from 1,853 patients was downloaded from the St Jude cloud database (48) and grouped into three pediatric tumor types, HM, BT, and ST. Healthy bone marrow (BM) mRNA expressions from the BEAT AML dataset (102) was used as a control for the analysis. Within each cancer type diagnosis (HM_D, n = 1173; BT_D, n = 232; ST_D, n = 453) and relapse (HM_R, n = 96; BT_R, n = 22; ST_R, n = 9) data are identified. BCL6 mRNA expression showed increased expression in BTs both at diagnosis and relapse compared with other cancer types. BCOR mRNA expression showed an inverse expression correlation, and significantly reduced in BT both at D and R compared with other tumor types. In both BT and ST, BCL6 mRNA expression showed a nonstatistical trend toward increased expression in the relapse setting. BCL6 mRNA expression in HM showed no difference to the BM control. No significant difference was seen between HM_D and HM_R in both BCL6 and BCOR expression (one-way ANOVA performed and corrected for multiple comparison using Tukey statistical hypothesis testing; *, P < 0.05; **, P < 0.01; ***, P < 0.001; ****, P < 0.0001).
Lymphomas
Non-Hodgkin lymphoma
Accounting for an estimated 7% of childhood cancers in the developed world, pediatric non-Hodgkin lymphoma (NHL) diagnoses are the fifth most common in children aged 15 years and under (49). The majority of B-cell non-Hodgkin lymphomas derive from the dysregulation of GC or post-GC B cells (14). B cells transitioning to GCs manifest many of the hallmark features of cancer: rapid proliferation, clonal expansion, inactivation of tumor suppressors, genome instability, and inhibition of DNA damage repair pathways (discussed in BCL6 in Humoral Immunity); hence, it is of no surprise that the development of cancer occurs in theses lymphoid organs.
DLBCL
DLBCLs makes up 10% to 20% of pediatric non-Hodgkin lymphomas and comprises about 8% of childhood malignancies (50). Although DLBCL shows epigenetic and somatic heterogeneity, most recurringly, 45% of patients harbor 3q27 translocations in the BCL6 gene (51), which does not appear to alter BCL6 mRNA expression (52). The first line of treatment for DLBCL is an immunochemotherapy regimen consisting of a short 9-week course of rituximab, cyclophosphamide, doxorubicin, vincristine, and prednisone (R-CHOP; ref. 53). R-CHOP achieves a complete response (CR) in 65% of cases; however, relapse is seen in 30% to 40% of patients, particularly those deemed to have “high-risk” or “high-grade” B-cell lymphoma (HGBL) at diagnosis (53). Two other genes frequently mutated in DLBCL (either singularly or concurrently) are the MYC (2%–20%) and BCL2 (20%) oncogenes. Patients harboring MYC rearrangements along with concurrent BCL2 and/or BCL6 mutations are classified as having HGBL; these combinations are commonly referred to as “double-hit” (DHL) or “triple-hit” lymphomas (THL; ref. 51). Conjecture persists about the prognosis for patients harboring either BCL2 or BCL6 translocations. One study showed patients with DLBCL harboring MYC/BCL6 rearrangements experienced reduced overall survival (OS) compared with MYC/BCL2 rearrangements; 4.5 months for those with BCL6 aberrations compared with 34.6 months for patients with BCL2 mutations (54). Early-stage clinical trials have evaluated the toxicity and benefit of adding a BCL2 inhibitor (venetoclax) to standard-of-care treatment in the adult population, citing promising preliminary results in patients with HGBL; however, clinical trials introducing BCL6 inhibitors are yet to be established (55).
Follicular lymphoma
FL accounts for 35% of all NHLs, and 70% of indolent lymphomas, portending a 10-year OS of approximately 80% (56). In adult FL, the vast majority of cases contain a t(14;18) translocation at diagnosis that leads to overexpression of the antiapoptotic protein BCL2 (56). However, pediatric FL lack abnormal BCL2 protein expression or the BCL2 gene rearrangements that characterize the majority of adult FL cases (57). Rather, BCL2-independent lymphogenesis is driven by Bcl-2–like protein 1 (BCL2L1 also known as BCL-XL), BCL2-associated agonist of cell death (BAD), and BCL6 (58). Patients with FL harboring BCL6 translocations are reported to experience earlier transformation to DLBCL and face a less favorable prognosis (59). One proposed mechanism of early progression is BCL6-driven antagonism of NOTCH signaling (60). Notch receptor 2 (NOTCH2) is preferentially expressed in mature B cells and is inversely correlated with BCL6 expression following terminal B-cell differentiation in the GCs. This is also seen in FLs that are dependent on BCL6 expression. Hence, pharmacologic or genetic inhibition of BCL6 drives NOTCH2 expression to suppress growth of human FL xenografts in vivo, and primary human FL specimens ex vivo.
Burkitt lymphoma
Burkitt lymphomas are rare and aggressive cancers primarily diagnosed in children. Burkitt lymphoma represents 30% of childhood NHL compared with only 1% of adult NHL diagnoses. Although multiple genetic abnormalities are implicated in the pathogenesis of Burkitt lymphoma, chromosomal translocations affecting the MYC oncogene family are considered a biological hallmark of the disease (61). A retrospective karyotype study of 34 MYC-immunoglobulin rearranged lymphomas (including 24 Burkitt lymphoma cases) concluded that BCL6 expression and noncomplex karyotype were independent predictors of better overall survival (62), contrary to what we have seen in other cancers.
Primary central nervous system lymphoma
This particularly rare form of NHL is highly aggressive with the tumor confined to the CNS (63). Primary central nervous system lymphoma (PCNSL) accounts for approximately 1% of all lymphoma diagnoses (64) and of this, around 1% are pediatric cases (65). PCNSLs harbor similar morphology to DLBCLs; BCL6 rearrangements occur in 20% to 40% of cases (66). Prognosis for PCNSL patients that harbor BCL6 rearrangements are suggested to be better than for PCNSL patients with BCL2, human GC-associated lymphoma (HGAL), or LIM domain only-2 (LMO2) mutations (67); however, this study was conducted using adult patients samples and with a small cohort size of 69.
Leukemias
ALL
Five-year survival rates for patients diagnosed with ALL have dramatically increased following the inclusion of multimodal treatment regimens; 89% of patients live >5 years if diagnosed under the age of 20. However, for patients diagnosed >20 years, 5-year survival rate is only 38%. Interestingly, BCL6 appears to be an oncogene in patients that are either refractory to treatment, or relapse with an aggressive and resistant form of ALL. The standard-of-care (SOC) treatment for ALL patients includes combinations of chemotherapies, including tyrosine kinase inhibitors (TKI). TKIs are an effective treatment, however, they are unable to eradicate the rare population of leukemia-initiating cells (LIC), which often lead to relapse even after an initial period of response. BCL6 expression is increased in ALL cells treated with TKIs and drives transcriptional inactivation of the TP53 pathway. This aids in leukemic cell protection from DNA-damaging chemotherapies (5).
LICs originate in the bone marrow. The bone marrow plays pivotal roles in leukemogenesis, but also may promote the activity of BCL6, in disease initiation and during the development of treatment resistance (6). Clonal evolution drives treatment resistance through the acquisition of additional genetic lesions caused by DNA damage. ALLs harboring molecular deletion of BCL6 harbor reduced mutational profiles compared to ALL overexpressing BCL6, suggesting that increased sensitivity to DNA damage limits clonal evolution in the absence of BCL6 (5).
AML
AML is the second most common acute leukemia in children, with a 5-year survival rate of 67% (68). Currently, there is limited research on the role of BCL6 in AML. A recent study using cell lines and primary patient samples demonstrated variable, but often, high-level expression of BCL6 in different subtypes of AML (particularly those with a less differentiated phenotype) compared with either hematopoietic stem cells or granulocyte–monocyte progenitor cells (69). Interestingly, BCL6 expression was induced by treatment with the SOC chemotherapy cytarabine, both at the mRNA and protein level. This resulted in the development of drug resistance, suggested to be due to the increased blast cell tolerance to DNA-damaging chemotherapies (69). Furthermore, inhibition of BCL6 decreased the number of leukemic stem cells (LSC). Knockdown of BCL6 in primary cells resulted in a significant reduction of leukemia-initiating capacity in mice, while BCL6 inhibition had no effect on normal cells of hematopoietic phenotype (69). As BCL6 plays a role in leukemia-initiating capacity in both ALL and AML (5), it is interesting to consider whether combining SOC and pharmacologic inhibition of BCL6 is a potential therapeutic strategy that may reduce leukemia repopulation and improve outcomes.
Pediatric central nervous system cancers
The role of BCL6 in pediatric brain tumors is also largely unknown, although a correlation between increased BCL6 expression and poor survival has been reported in adult studies (70). BCL6 plays an important role in neurogenesis, a key regulator of differentiation of progenitor cells to neurons (71). In somatic transgenic glioma models, BCL6 expression is also required for glioblastoma cell-cycle progression by silencing TP53 (9). As discussed above, BCL6 requires homodimerization and the formation of a complex with its cofactors, including the BCOR. Recurring mutations in BCOR are found in pediatric CNS neuroepithelial tumors (72), and diffuse midline gliomas (DMG), including those diagnosed in the pontine region, diffuse intrinsic pontine glioma (DIPG; ref. 73). However, uncertainty remains regarding the genomic landscape and significance of BCOR alterations in pediatric CNS malignancies and their influence on BCL6 function.
Pediatric HGG
Glioblastoma.
The most frequently diagnosed HGGs in children are anaplastic astrocytomas (WHO grade 3) followed by glioblastoma (WHO grade 4). These highly aggressive tumors are often resistant to treatment; median OS survival estimated at just 15 months (74). BCL6 translocations have been identified in patients diagnosed with glioblastoma (36.6% n = 11/30; ref. 8). BCL6 overexpression at both the transcript and protein level is also seen in patients harboring BCL6 translocations, with expression correlating with decreased apoptotic processes (8). More recent studies showed that molecular inhibition of BCL6 expression in glioblastoma cell lines decreased expression of BCL2, and Cyclin D1 (CCND1), and proteins influencing the diffuse migratory and invasive growth features of these tumors influenced by matrix metalloproteinases- MMP2 and MMP9. Commensurate, BCL6 knockdown increased the expression of proapoptotic protein BAX and CDKN1A to potentiate p53 activity in response to radiation and decreased Extracellular signal-regulated kinase (ERK) signaling (9, 75). The Tyrosine-protein kinase receptor (AXL) is a transcriptional target of BCL6 and responsible for the decreased ERK activity following BCL6 or AXL inhibition in glioblastoma. The coupling of BCL6 inhibition with SOC radiation and chemotherapeutic temozolomide enhanced the therapeutic effect of these therapies in vitro and in vivo (12), highlighting the potential for BCL6 inhibition as a strategy to improve treatment outcomes for this patient population facing such poor prognoses.
The BCL6 transcriptional target miR-361–5p (76) is also repressed in high-grade glioma cells and in tumor tissue, when compared with normal tissue and low-grade gliomas (77). Overexpression of miR-361–5p in glioblastoma cell lines significantly inhibited glioma cell migration, invasion, and epithelial–mesenchymal transition (EMT). The transcription factor Twist-related protein 1 (TWIST1) is a target of miR-361–5p, with overexpression rescuing the inhibitory effect of miR-361–5p on EMT. TWIST1 is a STAT3 target gene directly required for the development of TH cells and is involved in the regulation of BCL6 expression, resulting in a negative feedback loop (78).
DMG.
DMGs, including DIPG, are pediatric CNS tumors recognized as the most lethal of all children's cancers (44, 45). Palliative radiotherapy is the only approved treatment, with median survival just 9 to 11 months postdiagnosis (79). DMG is an immunologically cold tumor (80) characterized by recurring somatic mutations in H3 genes including, HIST1H3B/C (H3.1K27M) or H3F3A (H3.3K27M), or through overexpression of EZH inhibitory protein (EZHIP) EZHIP in patients harboring wildtype H3 – all of which lead to hypomethylation of H3K27 (45). TP53 is the second most recurring lesion in H3.3K27M+ DMG (60%–80%; ref. 43); however, it is only seen in 13% of H3.1K27M and 11% of EZHIP DMGs (73). Both H3.1K27M+ and EZHIP+ DMG harbor recurring mutations to BCOR and BCORL1 (45). Although yet to be fully characterized in DMG, these recurring aberrations may act comparably with TP53 mutations in H3.3K27M+ DMG, leading to increased BCL6 activity, and hence, decreased p53-driven tumor suppression and poor outcomes.
Medulloblastoma
Medulloblastoma is the most common malignant brain tumor in children, accounting for over 20% of all pediatric brain tumors with 70% of medulloblastoma cases occurring in children under the age of 10 (81). Treatment regimens for medulloblastoma are complex and usually include a mixed modality approach: surgical resection, craniospinal irradiation, combination chemotherapies. Interestingly, TP53 mutations are rarely seen in cases of medulloblastoma (82). However, recurring cytogenetic mutations in isochromosome 17q resulting from the loss of the short arm (p) cause a gain of genetic material from the long arm (q). In addition, recurring deletions in the short arm are also seen, leading to loss of heterozygosity of 17p (83).
Interestingly, medulloblastoma is characterized by sonic hedgehog (SHH) pathway activation where BCL6 acts as a tumor suppressor. Indeed, transient overexpression of BCL6 in vivo delays or inhibits medulloblastoma formation and tumor growth (84), through suppression of NOTCH signaling and epigenetic transcriptional silencing of the glioma-associated oncogenes, Zinc finger protein GLI1 and GLI2. BCL6 recruits BCOR and SIRT1 (to achieve transcriptional repression), which in turn promotes neurogenic conversion (84). Although recurring mutations in BCL6 have not been identified in patients with medulloblastoma, recurrent BCOR mutations have been identified and may play a role in medulloblastoma formation (85).
CNS high-grade neuroepithelial tumor
CNS high-grade neuroepithelial tumors (HGNET) with BCOR alteration (CNS HGNET-BCOR) are rare pediatric tumors that occur mostly in the supratentorial region of the brain (less frequently in the infratentorial region), suggested to harbor inferior survival outcomes compared with non BCOR altered HGNETs (72). HGNET-BCOR are considered a certain type of neuroepithelial tumor, like a glioma, due to the HGNET-BCOR tumors exhibiting glial cell morphology, ependymoma-like perivascular pseudorosettes and palisading necrosis (86). A recurring somatic internal tandem duplication in the 3′ end of BCOR gives rise to CNS HGNET-BCOR tumors. These mutations are also seen in clear cell sarcomas of the kidney (CCSK) and soft tissue undifferentiated round cell sarcomas/primitive myxoid mesenchymal tumors of infancy (discussed in Pediatric non-CNS solid tumors; Sarcoma) and are reported to share similar pathological features (86). Immunohistopathologic analysis identified CNS HGNET-BCOR to harbor positive expression of glial fibrillary acidic protein (GFAP) and oligodendrocyte transcription factor 2 (OLIG2), which are indicative of glial malignancy. Although the functional consequences of expression of BCL6 in CNS HGNET-BCOR is yet to be determined, other tumors that harbor BCOR internal tandem duplications coexpress BCL6 in most of their nuclei, suggestive of cooperating oncogenic roles.
Pediatric non-CNS solid tumors
Sarcoma
Sarcomas represent over 20% of all pediatric solid malignancies (87), including over 50 different subtypes identified to date. They originate in the mesenchymal tissue and therefore can arise almost anywhere in the body. Almost 80% of all sarcomas originate from soft tissue with the remaining 20% originating in bone (88). Rhabdomyosarcoma, a soft tissue located in the striated muscle, account for 50% of pediatric cases, while the reaming soft tissue malignancies are commonly classified according to the normal tissue types where they are derived.
The broad spectrum of sarcoma subtypes makes diagnosis, treatment, and research complex; it is difficult to gain significance in small population subtypes in this sense. In the soft tissue sarcoma primitive myxoid mesenchymal tumor of infancy (PMMTI), Santiago and colleagues reported BCOR-internal tandem duplications (ITD) and high BCOR and BCL6 immunoreactivity in 11 of 12 cases studied (89). In addition, in 9 of the 11 pediatric clear cell sarcomas (CCS) cases, BCOR immunoreactivity was again strongly detected (90). BCOR-ITD mutations appear to be chromosome X-linked, with cases diagnosed in males two times more frequently than in females (89, 91, 92). Recurring, BCOR-ITD mutations are also seen in up to 85% of small cell carcinoma of the kidney (SCCK) cases, CNS high-grade neuroepithelial tumors and undifferentiated round cell sarcomas of the bone. These results highlight not only the potential for improved diagnosis of these rare tumors, but also enables future informed targeted treatment that may improve reduce relapse rates and increase overall survival.
BCL6 as a Therapeutic Target
BCL6 plays an oncogenic role across a variety of cancer types, highlighting the potential for targeted inhibition to improve outcomes. BCL6’s unique structure, its crucial involvement in the repression of tumor suppressors, and its role in the activation of oncogenes, highlights a unique opportunity to develop novel and selective anticancer therapies. Several peptidomimetic and small molecule inhibitors have been developed and explored in cellular and animal models of cancer, but none have yet progressed into the clinic or been investigated under clinical trial settings.
Peptide inhibitors
The first anticancer therapeutics to be developed targeting BCL6 were peptide inhibitors. In 2004, Polo and colleagues (93), published data on the first BCL6 lateral groove inhibitor. Developing a wild-type BCL6-targeted peptide and mutant-targeted peptide that would compete for corepressor binding at the BTB/POZ binding domain (BBD) (Table 1). The wild-type peptide contained the SMRT BTB motif sequence along with a polyhistidine-tag, pTAT protein transduction domain and a hemagglutinin epitope tag. The mutant peptide incorporated point mutations in the SMRT binding motif, which reduced its affinity to BCL6. Although the wild-type peptide demonstrated the ability to penetrate cells and selectively bind BCL6, inhibit corepressor recruitment, and reactive expression of BCL6 target genes such as CD80 and CD69 (93), it also resulted in complete loss of biological BCL6 function, which mimicked the BCL6-null phenotype in vivo. BCL6 knockout mice develop a lethal inflammatory condition that causes fierce migration of macrophage and T cells into tissues, as well as an inability to form GCs (93). Thus, the need for specific peptide inhibitors, that explicitly target the BTB domain of BCL6, while ensuring BCL6 is still active, would minimize unwarranted immune side effects, while still promoting anticancer activity by inhibiting BCL6 repression of antitumor genes.
In 2006, Chattopadhyay and colleagues (22) developed a peptide aptamer (small peptides with both terminal ends anchored to a scaffold protein), Apt 48, which binds selectively to the POZ domain of BCL6, although outside the known corepressor and dimerization interface (Table 1). In Ramos B lymphocyte cell lines, Apt 48 successfully reinstated gene expression of BLIMP1 (PR domain zinc finger protein 1, PRDM1) and CCND2 (known targets of BCL6 repression); however, in vivo data on Apt 48 is yet to be published.
Following these studies, research then focused on inverting the original 17 amino acid sequence of the NCOR2-BBD peptide inhibitor, resulting in the development of the “retro-inverso” BCL6 peptide inhibitors (RI-BPIs) (Table 1). This category of BCL6 inhibitor showed success across multiple cancers types, with activity characterized similarly to their predecessor BBD peptides, with the advantage of RI-BPIs lacking immunomodulatory side effects (14, 21). Using BCL6-dependant DLBCL patient-derived xenograft mouse models (SU-DHL6, SU-DHL4), RI-BPI effectively induced cell death and growth arrest and was deemed nontoxic as treatment was continued for over a year in immunocompromised mice with no reported side effects (94). RI-BPI were also tested in multiple glioblastoma models (U87, U251, DA66, DBTRG, JM94, SN172) all of which harbored high BCL6 expression, and showed in vitro and in vivo efficacy (9). Promisingly, RI-BPI's sensitivity in glioblastoma was similar to DLBCL (OCI-Ly3 and SU-DHL-4) both in vitro and in vivo (9). RI-BPI alone, and in combination with STAT3 and tyrosine kinase inhibitors, has also been shown to be effective in other cancer models such as FL, ALL, and breast cancer (3, 5, 60). Phage-display technologies identified the peptide inhibitor F1234, showing higher affinity and potency than all other peptide inhibitors (Table 1); however, to our knowledge, the anticancer effects of F1324 have not been tested (95).
Small-molecule inhibitors
This series of BCL6 inhibitors were discovered using computer-assisted, small-molecule drug screen of the BTB domain lateral groove to identify low molecular weight, potent, BCL6 inhibitors (96).
79–6 is a small-molecule compound that was first developed in response to this computer-aided drug discovery approach, and elicits its inhibitory effect by binding to the lateral groove, once again inhibiting recruitment of NCOR and SMRT to the BTB domain of BCL6 (Table 1; ref. 21). 79–6 has had success in inducing apoptosis and reducing EMT in aggressive triple-negative breast cancer cell lines (SK-MDA-MB-468 and BT-549), as well as showing selective killing of BCL6-dependant DLBCL cells (OCI-ly7), in vitro (Table 1; refs. 3, 14, 96). Unfortunately, 79–6 is predicted to show limited utility for the treatment of brain tumors based on CNS multiparametric optimization desirability (CNS-MPO; Table 2; ref. 97).
Table 2.
Brain penetration prediction for BCL6 small-molecule inhibitors.
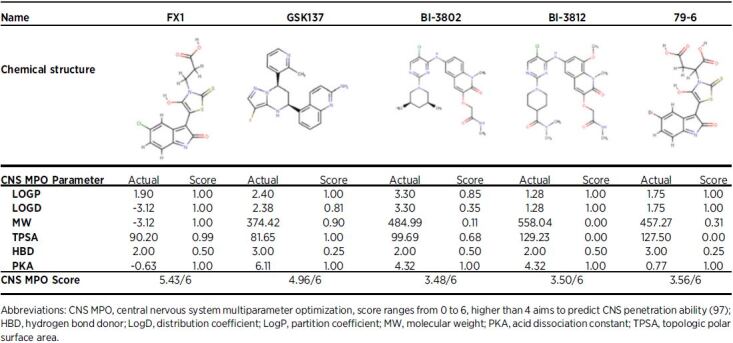
FX1 was developed in response to 79–6 with the view of identifying a drug with a higher affinity to specific areas of the BTB domain lateral groove than endogenous corepressors (21). FX1 was shown to out-compete every other BCL6 inhibitor based on affinity for the lateral groove of BTB domain and in pharmacodynamic assays (14, 21). In immunocompromised mice, FX1 inhibited the growth of BCL6 dependent DLBCL cells (OCI-ly7), and inhibited proliferation of non–small cell lung cancer in cells (H1299 and H383) engineered to express high BCL6 mRNA levels (Table 1; ref. 21). Interestingly, CNS-MPO (97) predicts FX1 to harbor the highest potential to show activity in the brain (Table 2) of all BCL6 inhibitors described (Table 1). This is potentially important information for the choice of BCL6 inhibitor to test in preclinical brain tumor models.
BI-3802 and BI-3812 were developed using structure-based drug design methods and high-throughput screening, using high-affinity binding to the BTB domain as the selection criterion (98). BI-3802 inhibits corepressor binding to BCL6 PTP/POZ domain and promotes rapid BCL6 degradation by the formation of BCL6 filaments, to drive ubiquitination by the SIAH1 E3 ubiquitin ligase (99). BI-3812 is a structural analog of BI-3802, however, inhibits rather than degrades BCL6. Although both compounds BI-3802 and BI-3812 demonstrated BCL6 inhibition and reestablishment of tumor suppressor gene transcription such as BLIMP1 in DLBCL cell lines (SU-DHL-4, OCI-ly7, OCI-Ly-1, and Farage), poor bioavailability has limited their use in preclinical animal models (Table 1; ref. 98). Furthermore, neither of these two therapies are predicted to show activity in the brain when delivered systemically (Table 2).
GSK137, was recently discovered using a high-throughput screening approach, again selected based on affinity for the BCL6 BTB-POZ domain (100). GSK137 binds the lateral groove to antagonize corepressor binding, including NCOR2/SMRT; however, unlike BI-3802, it does not induce BCL6 degradation. Unfortunately, GSK137 caused little change to the viability or proliferation of BCL6-expressing B-cell lymphoma lines (Farage, Karpas422, ULA, VAL) in vitro (Table 1). Despite this, in vivo studies were still conducted to determine GSK137’s pharmacokinetic profile and determined that it suppressed GC-dependent antibody response (male CD1 mice). Promising in vivo studies demonstrated that GSK137 suppressed immunoglobulin G responses following immunization of naïve mice with the hapten trinitrophenol, reducing the numbers of GCs and GC B cells, suggesting an on-target effect (100). The oral bioavailability and pharmacokinetic profile of GSK137, is the most promising agent to date, and therefore, although it is ineffective at driving apoptosis in cancer cells as a single agent, it may prove to be a promising drug when used in combination with SOC (as described in Sections Leukemias, AML and Pediatric HGG, Glioblastoma, DMG). GSK137 is predicted to show some activity in the brain, warranting further investigation in preclinical models of brain tumors (Table 2).
Conclusion
BCL6’s role in transcriptional silencing of tumor suppressor and checkpoint regulators during the formation of GCs is also fundamental in the way it acts as an oncogene in cancer. BCL6 suppresses DNA repair genes and antiapoptotic signatures to promote B cell SHM and CSR, leading to the production of high-affinity immunoglobulins necessary to recognize and bind to specific antigens, triggering their destruction. This is largely coordinated by the recruitment of repressor proteins, including NCOR1, NCOR2, BCOR, PRC1, and HDAC3, critical regulators of epigenetic signaling and gene silencing. BCL6 translocations were originally described as the driver of the lymphomagenesis of DLBCL. However, when we take a closer look at BCL6 in pediatric cancers (St Jude Pediatric Cancer database, n = 1,985 samples), it is clear that BCL6 translocations/mutations are not necessarily the main driver of increased BCL6 expression and oncogenesis in the majority of cases. 36.6% of glioblastoma cases harbor a BCL6 translocation, corresponding with increased mRNA and protein expression; however, 45% of DLBCL cases harbor a BCL6 translocation, with no change in mRNA expression. Sarcomas and medulloblastoma are rarely BCL6 mutant: however, BCOR mutations are highly prevalent in both cancer types, which can also drive BCL6 expression. Overall, increased expression of BCL6 mRNA appears a predictor of worse prognosis in most pediatric cancers; however, survival studies using protein expression have not been widely conducted. In two rare cancers including medulloblastoma and Burkitt lymphoma, overexpression of BCL6 conferred better prognosis, albeit in only a small sample size. The mechanisms underpinning the role of BCL6 in the pediatric cancer population remains poorly understood, but as it appears to be a notable player in many cancer subtypes, highlighting the importance of more research in this area. Molecular inhibition of BCL6in vivo and in vitro decreased leukemia-initiating cell capacity, increased chemotherapy sensitivity, and increased expression of the master tumor suppressor p53, rarely mutated in pediatric cancers (except DMG; ref. 45), as well as promoted expression of cell-cycle checkpoint genes such as CDKN1A (9). These observations highlight the potential for targeting BCL6, particularly in combination with DNA-damaging therapies including chemotherapies and radiotherapy. It is becoming even clearer that combinations of treatments are necessary to achieve long-term positive outcomes for many aggressive pediatric cancers, and we propose targeting/evaluation of BCL6 warrants consideration. Development of nonimmunosuppressive BCL6 therapies may even open the door to new combinations, including radiotherapy and immunotherapy. Radiotherapy promotes the expression of checkpoint proteins that underpin the efficacy of immune checkpoint inhibitors, the latter having revolutionized the treatment of aggressive adult cancers including melanoma, non–small cell lung cancer and Hodgkin lymphoma (80). In the context of pediatric tumors such as DMG, for which radiotherapy is the only recognized treatment (43, 45), BCL6 inhibition may serve to enhance the therapeutic benefit and increase the survival of this poor outcome patient cohort.
As it stands, BCL6 remains a challenging target; however, the field is moving closer to the development of specific anti-BCL6, nonimmunosuppressive therapies. Over the coming years, we hope that new trials will open to test the efficacy of the next generation of BCL6 inhibitors.
Acknowledgments
This study was supported by Cancer Institute NSW Fellowships (M.D. Dun). M.D. Dun is supported by an NHMRC Investigator Grant – GNT1173892. The contents of the published material are solely the responsibility of the research institutions involved or individual authors and do not reflect the views of NHMRC. E.R. Jackson is a Josephine Dun Scholar, supported by the Miette Skiller Scholarship Fund, a sub-fund of the Australian Communities Foundation. I.J. Findlay is supported by the RUN DIPG “Moving Towards a Cure” HDR Scholarship and CureCell ExCELLerate Award. M.L. Persson is supported by the RUN DIPG International HDR Scholarship. Grants from the Hunter Medical Research Institute, Hunter Children's Research Foundation, Jurox Animal Health, Zebra Equities, Hunter District Hunting Club and Ski for Kids, The Estate of James Scott Lawrie grants, the Little Legs Foundation in partnership with the Charlie Teo Foundation, Strategic Group, Blackjack Pastoral Company, Vinva Foundation, Edie's Kindness Project, Hirsch Family Funderpants, Yuvaan Tiwari Foundation via the Pacific Pediatric Neuro-Oncology Consortium Foundation, Kiriwina Investments via the Hunter Medical Research Institute, The Cure Starts Now Foundation, The Cure Starts Now Australia, Brooke Healey Foundation, Wayland Villars Foundation, ChadTough Foundation, Aidan's Avengers, Austin Strong, Cure Brain Cancer, Jeffrey Thomas Hayden Foundation, Laurie's Love Foundation, Love Chloe Foundation, Musella Foundation, Pray Hope Believe, Reflections Of Grace, Storm the Heavens Fund, Aubreigh's Army, Whitley's Wishes, Ryan's Hope, Benny's World, The Isabella and Marcus Foundation, Lauren's Fight for Cure, Robert Connor Dawes Foundation, The Gold Hope Project, Julia Barbara Foundation, Lily Larue Foundation, American Childhood Cancer Organization, RUN DIPG, Gabriella's Smile Foundation, The DIPG Collaborative, and Snapgrant.com, Fight on the Beaches, Tour de Cure, Kids’ Cancer Project, Michael Mosier Defeat DIPG Foundation, CureCell, Maitland Cancer Appeal and the Liv Like A Unicorn Foundation. We acknowledge and pay our respects to all children and their families diagnosed with cancer. Figures 2 and 3 were created with the help of Biorender.com
Authors' Disclosures
M.D. Dun reports being the founder and director of the non-for-profit charity RUN DIPG, Ltd. No disclosures were reported by the other authors.
References
- 1. Ye BH, Lista F, Lo Coco F, Knowles DM, Offit K, Chaganti RS, et al. Alterations of a zinc finger-encoding gene, BCL-6, in diffuse large-cell lymphoma. Science 1993;262:747–50. [DOI] [PubMed] [Google Scholar]
- 2. Victora GD, Nussenzweig MC. Germinal centers. Annu Rev Immunol 2012;30:429–57. [DOI] [PubMed] [Google Scholar]
- 3. Walker SR, Liu S, Xiang M, Nicolais M, Hatzi K, Giannopoulou E, et al. The transcriptional modulator BCL6 as a molecular target for breast cancer therapy. Oncogene 2015;34:1073–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Kerckaert J-P, Deweindt C, Tilly H, Quief S, Lecocq G, Bastard C. LAZ3, a novel zinc–finger encoding gene, is disrupted by recurring chromosome 3q27 translocations in human lymphomas. Nat Genet 1993;5:66–70. [DOI] [PubMed] [Google Scholar]
- 5. Duy C, Hurtz C, Shojaee S, Cerchietti L, Geng H, Swaminathan S, et al. BCL6 enables Ph+ acute lymphoblastic leukaemia cells to survive BCR-ABL1 kinase inhibition. Nature 2011;473:384–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Slone WL, Moses BS, Hare I, Evans R, Piktel D, Gibson LF. BCL6 modulation of acute lymphoblastic leukemia response to chemotherapy. Oncotarget 2016;7:23439–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Madapura HS, Nagy N, Ujvari D, Kallas T, Kröhnke MCL, Amu S, et al. Interferon γ is a STAT1-dependent direct inducer of BCL6 expression in imatinib-treated chronic myeloid leukemia cells. Oncogene 2017;36:4619–28. [DOI] [PubMed] [Google Scholar]
- 8. Ruggieri S, Tamma R, Marzullo A, Annese T, Marinaccio C, Errede M, et al. Translocation of the proto-oncogene Bcl-6 in human glioblastoma multiforme. Cancer Lett 2014;353:41–51. [DOI] [PubMed] [Google Scholar]
- 9. Xu L, Chen Y, Dutra-Clarke M, Mayakonda A, Hazawa M, Savinoff SE, et al. BCL6 promotes glioma and serves as a therapeutic target. Proc Natl Acad Sci U S A 2017;114:3981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Li S, Wang Z, Lin L, Wu Z, Yu Q, Gao F, et al. BCL6 rearrangement indicates poor prognosis in diffuse large B-cell lymphoma patients: a meta-analysis of cohort studies. J Cancer 2019;10:530–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Fernando TM, Marullo R, Pera Gresely B, Phillip JM, Yang SN, Lundell-Smith G, et al. BCL6 evolved to enable stress tolerance in vertebrates and is broadly required by cancer cells to adapt to stress. Cancer Discov 2019;9:662–79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Fabre MS, Stanton NM, Slatter TL, Lee S, Senanayake D, Gordon RMA, et al. The oncogene BCL6 is up-regulated in glioblastoma in response to DNA damage, and drives survival after therapy. PLoS One 2020;15:e0231470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Ye BH, Cattoretti G, Shen Q, Zhang J, Hawe N, de Waard R, et al. The BCL-6 proto-oncogene controls germinal-centre formation and Th2-type inflammation. Nat Genet 1997;16:161–70. [DOI] [PubMed] [Google Scholar]
- 14. Leeman-Neill RJ, Bhagat G. BCL6 as a therapeutic target for lymphoma. Expert Opin Ther Targets 2018;22:143–52. [DOI] [PubMed] [Google Scholar]
- 15. Mendez LM, Polo JM, Yu JJ, Krupski M, Ding BB, Melnick A, et al. CtBP is an essential corepressor for BCL6 autoregulation. Mol Cell Biol 2008;28:2175–86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Stogios PJ, Downs GS, Jauhal JJ, Nandra SK, Prive GG. Sequence and structural analysis of BTB domain proteins. Genome Biol 2005;6:R82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Miles RR, Crockett DK, Lim MS, KS E-J. Analysis of BCL6-interacting proteins by tandem mass spectrometry. Mol Cell Proteomics 2005;4:1898–909. [DOI] [PubMed] [Google Scholar]
- 18. Huynh KD, Bardwell VJ. The BCL-6 POZ domain and other POZ domains interact with the co-repressors N-CoR and SMRT. Oncogene 1998;17:2473–84. [DOI] [PubMed] [Google Scholar]
- 19. Wong CW, Privalsky ML. Components of the SMRT corepressor complex exhibit distinctive interactions with the POZ domain oncoproteins PLZF, PLZF-RARalpha, and BCL-6. J Biol Chem 1998;273:27695–702. [DOI] [PubMed] [Google Scholar]
- 20. Beguelin W, Rivas MA, Calvo Fernandez MT, Teater M, Purwada A, Redmond D, et al. EZH2 enables germinal centre formation through epigenetic silencing of CDKN1A and an Rb-E2F1 feedback loop. Nat Commun 2017;8:877. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Cardenas MG, Oswald E, Yu W, Xue F, MacKerell AD Jr, Melnick AM. The expanding role of the BCL6 oncoprotein as a cancer therapeutic target. Clin Cancer Res 2017;23:885–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Chattopadhyay A, Tate SA, Beswick RW, Wagner SD, Ko Ferrigno P. A peptide aptamer to antagonize BCL-6 function. Oncogene 2006;25:2223–33. [DOI] [PubMed] [Google Scholar]
- 23. Hatzi K, Melnick A. Breaking bad in the germinal center: how deregulation of BCL6 contributes to lymphomagenesis. Trends Mol Med 2014;20:343–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Allen CD, Okada T, Cyster JG. Germinal-center organization and cellular dynamics. Immunity 2007;27:190–202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Phan RT, Saito M, Kitagawa Y, Means AR, Dalla-Favera R. Genotoxic stress regulates expression of the proto-oncogene Bcl6 in germinal center B cells. Nat Immunol 2007;8:1132–9. [DOI] [PubMed] [Google Scholar]
- 26. Phan RT, Saito M, Basso K, Niu H, Dalla-Favera R. BCL6 interacts with the transcription factor Miz-1 to suppress the cyclin-dependent kinase inhibitor p21 and cell cycle arrest in germinal center B cells. Nat Immunol 2005;6:1054–60. [DOI] [PubMed] [Google Scholar]
- 27. Chaudhuri J, Tian M, Khuong C, Chua K, Pinaud E, Alt FW. Transcription-targeted DNA deamination by the AID antibody diversification enzyme. Nature 2003;422:726–30. [DOI] [PubMed] [Google Scholar]
- 28. Ranuncolo SM, Polo JM, Dierov J, Singer M, Kuo T, Greally J, et al. Bcl-6 mediates the germinal center B cell phenotype and lymphomagenesis through transcriptional repression of the DNA-damage sensor ATR. Nat Immunol 2007;8:705–14. [DOI] [PubMed] [Google Scholar]
- 29. Bian L, Meng Y, Zhang M, Li D. MRE11-RAD50-NBS1 complex alterations and DNA damage response: implications for cancer treatment. Mol Cancer 2019;18:169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Ci W, Polo JM, Melnick A. B-cell lymphoma 6 and the molecular pathogenesis of diffuse large B-cell lymphoma. Curr Opin Hematol 2008;15:381–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Ying CY, Dominguez-Sola D, Fabi M, Lorenz IC, Hussein S, Bansal M, et al. MEF2B mutations lead to deregulated expression of the oncogene BCL6 in diffuse large B cell lymphoma. Nat Immunol 2013;14:1084–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Rogers S, Wells R, Rechsteiner M. Amino acid sequences common to rapidly degraded proteins: the PEST hypothesis. Science 1986;234:364–8. [DOI] [PubMed] [Google Scholar]
- 33. Brescia P, Schneider C, Holmes AB, Shen Q, Hussein S, Pasqualucci L, et al. MEF2B instructs germinal center development and acts as an oncogene in B cell lymphomagenesis. Cancer Cell 2018;34:453–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Saito M, Gao J, Basso K, Kitagawa Y, Smith PM, Bhagat G, et al. A signaling pathway mediating downregulation of BCL6 in germinal center B cells is blocked by BCL6 gene alterations in B cell lymphoma. Cancer Cell 2007;12:280–92. [DOI] [PubMed] [Google Scholar]
- 35. Haniuda K, Fukao S, Kitamura D. Metabolic reprogramming induces germinal center B cell differentiation through Bcl6 locus remodeling. Cell Rep 2020;33:108333. [DOI] [PubMed] [Google Scholar]
- 36. Nguyen L, Papenhausen P, Shao H. The role of c-MYC in B-cell lymphomas: diagnostic and molecular aspects. Genes 2017;8:116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Lossos IS. The endless complexity of lymphocyte differentiation and lymphomagenesis: IRF-4 downregulates BCL6 expression. Cancer Cell 2007;12:189–91. [DOI] [PubMed] [Google Scholar]
- 38. Koike T, Harada K, Horiuchi S, Kitamura D. The quantity of CD40 signaling determines the differentiation of B cells into functionally distinct memory cell subsets. Elife 2019;8:e44245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Staudt D, Murray HC, McLachlan T, Alvaro F, Enjeti AK, Verrills NM, et al. Targeting oncogenic signaling in mutant FLT3 acute myeloid leukemia: the path to least resistance. Int J Mol Sci 2018;19:3198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Kaplan MH, Schindler U, Smiley ST, Grusby MJ. Stat6 is required for mediating responses to IL-4 and for development of Th2 cells. Immunity 1996;4:313–9. [DOI] [PubMed] [Google Scholar]
- 41. Walker SR, Nelson EA, Frank DA. STAT5 represses BCL6 expression by binding to a regulatory region frequently mutated in lymphomas. Oncogene 2007;26:224–33. [DOI] [PubMed] [Google Scholar]
- 42. Pui CH, Gajjar AJ, Kane JR, Qaddoumi IA, Pappo AS. Challenging issues in pediatric oncology. Nat Rev Clin Oncol 2011;8:540–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Duchatel RJ, Jackson ER, Alvaro F, Nixon B, Hondermarck H, Dun MD. Signal transduction in diffuse intrinsic pontine glioma. Proteomics 2019;19:e1800479. [DOI] [PubMed] [Google Scholar]
- 44. Duchatel RJ, Mannan A, Woldu AS, Hawtrey T, Hindley PA, Douglas AM, et al. Preclinical and clinical evaluation of German-sourced ONC201 for the treatment of H3K27M-mutant diffuse intrinsic pontine glioma. Neurooncol Adv 2021;3:vdab169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Findlay IJ, De Iuliis GN, Duchatel RJ, Jackson ER, Vitanza NA, Cain JE, et al. Pharmaco-proteogenomic profiling of pediatric diffuse midline glioma to inform future treatment strategies. Oncogene 2022;41:461–75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Rivlin N, Brosh R, Oren M, Rotter V. Mutations in the p53 tumor suppressor gene: important milestones at the various steps of tumorigenesis. Genes Cancer 2011;2:466–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Grobner SN, Worst BC, Weischenfeldt J, Buchhalter I, Kleinheinz K, Rudneva VA, et al. The landscape of genomic alterations across childhood cancers. Nature 2018;555:321–7. [DOI] [PubMed] [Google Scholar]
- 48. McLeod C, Gout AM, Zhou X, Thrasher A, Rahbarinia D, Brady SW, et al. St. Jude Cloud: a pediatric cancer genomic data-sharing ecosystem. Cancer Discov 2021;11:1082–99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Kaatsch P. Epidemiology of childhood cancer. Cancer Treat Rev 2010;36:277–85. [DOI] [PubMed] [Google Scholar]
- 50. Sandlund JT, Downing JR, Crist WM. Non-Hodgkin's lymphoma in childhood. N Engl J Med 1996;334:1238–48. [DOI] [PubMed] [Google Scholar]
- 51. Huang S, Nong L, Wang W, Liang L, Zheng Y, Liu J, et al. Prognostic impact of diffuse large B-cell lymphoma with extra copies of MYC, BCL2 and/or BCL6: comparison with double/triple hit lymphoma and double expressor lymphoma. Diagnostic Pathology 2019;14:81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Lossos IS, Akasaka T, Martinez-Climent JA, Siebert R, Levy R. The BCL6 gene in B-cell lymphomas with 3q27 translocations is expressed mainly from the rearranged allele irrespective of the partner gene. Leukemia 2003;17:1390–7. [DOI] [PubMed] [Google Scholar]
- 53. Coiffier B, Thieblemont C, Van Den Neste E, Lepeu G, Plantier I, Castaigne S, et al. Long-term outcome of patients in the LNH-98.5 trial, the first randomized study comparing rituximab-CHOP to standard CHOP chemotherapy in DLBCL patients: a study by the Groupe d'Etudes des Lymphomes de l'Adulte. Blood 2010;116:2040–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Landsburg DJ, Petrich AM, Abramson JS, Sohani AR, Press O, Cassaday R, et al. Impact of oncogene rearrangement patterns on outcomes in patients with double-hit non-Hodgkin lymphoma. Cancer 2016;122:559–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Zelenetz AD, Salles G, Mason KD, Casulo C, Le Gouill S, Sehn LH, et al. Venetoclax plus R- or G-CHOP in non-Hodgkin lymphoma: results from the CAVALLI phase 1b trial. Blood 2019;133:1964–76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Freedman A, Jacobsen E. Follicular lymphoma: 2020 update on diagnosis and management. Am J Hematol 2020;95:316–27. [DOI] [PubMed] [Google Scholar]
- 57. Agrawal R, Wang J. Pediatric follicular lymphoma: a rare clinicopathologic entity. Arch Pathol Lab Med 2009;133:142–6. [DOI] [PubMed] [Google Scholar]
- 58. Zha H, Raffeld M, Charboneau L, Pittaluga S, Kwak LW, Petricoin E III, et al. Similarities of prosurvival signals in Bcl-2-positive and Bcl-2-negative follicular lymphomas identified by reverse phase protein microarray. Lab Invest 2004;84:235–44. [DOI] [PubMed] [Google Scholar]
- 59. Akasaka T, Lossos IS, Levy R. BCL6 gene translocation in follicular lymphoma: a harbinger of eventual transformation to diffuse aggressive lymphoma. Blood 2003;102:1443–8. [DOI] [PubMed] [Google Scholar]
- 60. Valls E, Lobry C, Geng H, Wang L, Cardenas M, Rivas M, et al. BCL6 antagonizes NOTCH2 to maintain survival of human follicular lymphoma cells. Cancer Discov 2017;7:506–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Aukema SM, Kreuz M, Kohler CW, Rosolowski M, Hasenclever D, Hummel M, et al. Biological characterization of adult MYC-translocation-positive mature B-cell lymphomas other than molecular Burkitt lymphoma. Haematologica 2014;99:726–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Seegmiller AC, Garcia R, Huang R, Maleki A, Karandikar NJ, Chen W. Simple karyotype and bcl-6 expression predict a diagnosis of Burkitt lymphoma and better survival in IG-MYC rearranged high-grade B-cell lymphomas. Mod Pathol 2010;23:909–20. [DOI] [PubMed] [Google Scholar]
- 63. Cai Q, Fang Y, Young KH. Primary central nervous system lymphoma: molecular pathogenesis and advances in treatment. Translational Oncology 2019;12:523–38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Ge L, Lu S, Xu L, Yan H. MYC, BCL2, and BCL6 expression as prognostic indicators in primary central nervous system lymphoma: A systematic review and meta-analysis. Clin Neurol Neu 2021;208:106838. [DOI] [PubMed] [Google Scholar]
- 65. Abla O, Weitzman S, Blay J-Y, O'Neill BP, Abrey LE, Neuwelt E, et al. Primary CNS lymphoma in children and adolescents: a descriptive analysis from the international primary CNS lymphoma collaborative group (IPCG). Clin Cancer Res 2011;17:346–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Cady FM, O'Neill BP, Law ME, Decker PA, Kurtz DM, Giannini C, et al. Del(6)(q22) and BCL6 rearrangements in primary CNS lymphoma are indicators of an aggressive clinical course. J Clin Oncol 2008;26:4814–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Lossos C, Bayraktar S, Weinzierl E, Younes SF, Hosein PJ, Tibshirani RJ, et al. LMO2 and BCL6 are associated with improved survival in primary central nervous system lymphoma. Br J Haematol 2014;165:640–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Roboz GJ. Current treatment of acute myeloid leukemia. Curr Opin Oncol 2012;24:711–9. [DOI] [PubMed] [Google Scholar]
- 69. Kawabata KC, Zong H, Meydan C, Wyman S, Wouters BJ, Sugita M, et al. BCL6 maintains survival and self-renewal of primary human acute myeloid leukemia cells. Blood 2021;137:812–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Hutterer M, Knyazev P, Abate A, Reschke M, Maier H, Stefanova N, et al. Axl and growth arrest-specific gene 6 are frequently overexpressed in human gliomas and predict poor prognosis in patients with glioblastoma multiforme. Clin Cancer Res 2008;14:130–8. [DOI] [PubMed] [Google Scholar]
- 71. Bonnefont J, Tiberi L, van den Ameele J, Potier D, Gaber ZB, Lin X, et al. Cortical neurogenesis requires Bcl6-mediated transcriptional repression of multiple self-renewal-promoting extrinsic pathways. Neuron 2019;103:1096–108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Sturm D, Orr BA, Toprak UH, Hovestadt V, Jones DTW, Capper D, et al. New brain tumor entities emerge from molecular classification of CNS-PNETs. Cell 2016;164:1060–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Mackay A, Burford A, Carvalho D, Izquierdo E, Fazal-Salom J, Taylor KR, et al. Integrated molecular meta-analysis of 1,000 pediatric high-grade and diffuse intrinsic pontine glioma. Cancer Cell 2017;32:520–37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Efremov L, Abera S, Bedir A, Vordermark D, Medenwald D. Patterns of glioblastoma treatment and survival over a 16-years period: pooled data from the German Cancer Registries. J Cancer Res Clin Oncol 2021;147:3381–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Song W, Wang Z, Kan P, Ma Z, Wang Y, Wu Q, et al. Knockdown of BCL6 inhibited malignant phenotype and enhanced sensitivity of glioblastoma cells to TMZ through AKT pathway. Biomed Res Int 2018;2018:6953506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Basso K, Schneider C, Shen Q, Holmes AB, Setty M, Leslie C, et al. BCL6 positively regulates AID and germinal center gene expression via repression of miR-155. J Exp Med 2012;209:2455–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Zhang X, Wei C, Li J, Liu J, Qu J. MicroRNA-361–5p inhibits epithelial-to-mesenchymal transition of glioma cells through targeting Twist1. Oncol Rep 2017;37:1849–56. [DOI] [PubMed] [Google Scholar]
- 78. Pham D, Walline CC, Hollister K, Dent AL, Blum JS, Firulli AB, et al. The transcription factor Twist1 limits T helper 17 and T follicular helper cell development by repressing the gene encoding the interleukin-6 receptor alpha chain. J Biol Chem 2013;288:27423–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79. Hoffman LM, Veldhuijzen van Zanten SEM, Colditz N, Baugh J, Chaney B, Hoffmann M, et al. Clinical, radiologic, pathologic, and molecular characteristics of long-term survivors of diffuse intrinsic pontine glioma (DIPG): a collaborative report from the international and european society for pediatric oncology DIPG registries. J Clin Oncol 2018;36:1963–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80. Persson ML, Douglas AM, Alvaro F, Faridi P, Larsen MR, Alonso MM, et al. The intrinsic and microenvironmental features of diffuse midline glioma; implications for the development of effective immunotherapeutic treatment strategies. Neuro Oncol 2022;24:1408–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81. Rossi A, Caracciolo V, Russo G, Reiss K, Giordano A. Medulloblastoma: from molecular pathology to therapy. Clin Cancer Res 2008;14:971–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82. Adesina AM, Nalbantoglu J, Cavenee WK. p53 gene mutation and mdm2 gene amplification are uncommon in medulloblastoma. Cancer Res 1994;54:5649–51. [PubMed] [Google Scholar]
- 83. Cogen PH, McDonald JD. Tumor suppressor genes and medulloblastoma. J Neurooncol 1996;29:103–12. [DOI] [PubMed] [Google Scholar]
- 84. Tiberi L, Bonnefont J, van den Ameele J, Le Bon S-D, Herpoel A, Bilheu A, et al. A BCL6/BCOR/SIRT1 complex triggers neurogenesis and suppresses medulloblastoma by repressing sonic hedgehog signaling. Cancer Cell 2014;26:797–812. [DOI] [PubMed] [Google Scholar]
- 85. Kutscher LM, Okonechnikov K, Batora NV, Clark J, Silva PBG, Vouri M, et al. Functional loss of a noncanonical BCOR-PRC1.1 complex accelerates SHH-driven medulloblastoma formation. Genes Dev 2020;34:1161–76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86. Yoshida Y, Nobusawa S, Nakata S, Nakada M, Arakawa Y, Mineharu Y, et al. CNS high-grade neuroepithelial tumor with BCOR internal tandem duplication: a comparison with its counterparts in the kidney and soft tissue. Brain Pathol 2018;28:710–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87. Burningham Z, Hashibe M, Spector L, Schiffman JD. The epidemiology of sarcoma. Clinical Sarcoma Research 2012;2:14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88. Lahat G, Lazar A, Lev D. Sarcoma epidemiology and etiology: potential environmental and genetic factors. Surg Clin North Am 2008;88:451–81. [DOI] [PubMed] [Google Scholar]
- 89. Santiago T, Clay MR, Allen SJ, Orr BA. Recurrent BCOR internal tandem duplication and BCOR or BCL6 expression distinguish primitive myxoid mesenchymal tumor of infancy from congenital infantile fibrosarcoma. Mod Pathol 2017;30:884–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90. Argani P, Pawel B, Szabo S, Reyes-Múgica M, Timmons C, Antonescu CR. Diffuse strong BCOR immunoreactivity is a sensitive and specific marker for clear cell sarcoma of the kidney (CCSK) in pediatric renal neoplasia. Am J Surg Pathol 2018;42:1128–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91. Kao YC, Sung YS, Zhang L, Huang SC, Argani P, Chung CT, et al. Recurrent BCOR internal tandem duplication and YWHAE-NUTM2B fusions in soft tissue undifferentiated round cell sarcoma of infancy: overlapping genetic features with clear cell sarcoma of kidney. Am J Surg Pathol 2016;40:1009–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92. Aldera AP, Pillay K. Clear cell sarcoma of the kidney. Arch Pathol Lab Med 2020;144:119–23. [DOI] [PubMed] [Google Scholar]
- 93. Polo JM, Dell'Oso T, Ranuncolo SM, Cerchietti L, Beck D, Da Silva GF, et al. Specific peptide interference reveals BCL6 transcriptional and oncogenic mechanisms in B-cell lymphoma cells. Nat Med 2004;10:1329–35. [DOI] [PubMed] [Google Scholar]
- 94. Cerchietti LC, Yang SN, Shaknovich R, Hatzi K, Polo JM, Chadburn A, et al. A peptomimetic inhibitor of BCL6 with potent antilymphoma effects in vitro and in vivo. Blood 2009;113:3397–405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95. Sakamoto K, Sogabe S, Kamada Y, Sakai N, Asano K, Yoshimatsu M, et al. Discovery of high-affinity BCL6-binding peptide and its structure-activity relationship. Biochem Biophys Res Commun 2017;482:310–6. [DOI] [PubMed] [Google Scholar]
- 96. Cerchietti LC, Ghetu AF, Zhu X, Da Silva GF, Zhong S, Matthews M, et al. A small-molecule inhibitor of BCL6 kills DLBCL cells in vitro and in vivo. Cancer Cell 2010;17:400–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97. Wager TT, Hou X, Verhoest PR, Villalobos A. Central nervous system multiparameter optimization desirability: application in drug discovery. ACS Chem Neurosci 2016;7:767–75. [DOI] [PubMed] [Google Scholar]
- 98. Kerres N, Steurer S, Schlager S, Bader G, Berger H, Caligiuri M, et al. Chemically induced degradation of the oncogenic transcription factor BCL6. Cell Rep 2017;20:2860–75. [DOI] [PubMed] [Google Scholar]
- 99. Słabicki M, Yoon H, Koeppel J, Nitsch L, Roy Burman SS, Di Genua C, et al. Small-molecule-induced polymerization triggers degradation of BCL6. Nature 2020;588:164–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100. Pearce AC, Bamford MJ, Barber R, Bridges A, Convery MA, Demetriou C, et al. GSK137, a potent small-molecule BCL6 inhibitor with in vivo activity, suppresses antibody responses in mice. J Biol Chem 2021;297:100928. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101. Cardenas MG, Yu W, Beguelin W, Teater MR, Geng H, Goldstein RL, et al. Rationally designed BCL6 inhibitors target activated B cell diffuse large B cell lymphoma. J Clin Invest 2016;126:3351–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102. Tyner JW, Tognon CE, Bottomly D, Wilmot B, Kurtz SE, Savage SL, et al. Functional genomic landscape of acute myeloid leukaemia. Nature 2018;562:526–31. [DOI] [PMC free article] [PubMed] [Google Scholar]